Biology of Cervical Squamous Neoplasia
Introduction
In the past half-century, cytologic screening, particularly in high-resource settings, has dramatically decreased the burden of squamous cervical cancers, virtually all of which are caused by tissue-specific, persistent infection with human papillomavirus (HPV). The elucidation of the infectious etiology of this disease led to a Nobel Prize in 2008, and to development of preventative vaccines that became commercially available in 2006. This was a public health milestone; worldwide, chronic infections initiate approximately 20% of human cancers,1 with HPVs causing more malignancies than any other virus. However, because primary prevention strategies are cumbersome and expensive, cervical cancer is still the third most common cause of cancer death in women.2 Moreover, the incidence of HPV-associated cancers in other anatomic sites, for which no screening algorithms have been developed, is increasing rapidly.
HPV disease is not restricted to the cervix. A growing body of evidence links persistent HPV infection to squamous cancers of the vagina, vulva, anus, and oropharynx.3 Almost all vaginal and anal squamous cancers4 and approximately 20% of squamous cancers of the vulva5 are caused by HPV. The incidence of preinvasive vulvar disease attributed to HPV has increased by 411% over the past three decades, primarily in women under the age 65.6 HPVs have been identified in one in four squamous carcinomas of the head and neck; to date, the incidence of HPV-associated oropharyngeal cancers is greater in men than in women.7 Indeed, in the United States, the incidence of HPV-associated cancers of the oropharynx is approximately equal to that of cervical cancer, and is likely to surpass it soon.8 Because prophylactic vaccines are not administered to the full target population, cohort-appropriate strategies for preventing and treating HPV disease are needed. A better understanding of the biology of intraepithelial, preinvasive HPV disease will inform strategies for screening, secondary prevention, and treatment.
HPV Infection
Like other viruses that cause human cancers, most HPV infections are asymptomatic, eventually cleared, and do not harm the host (Figure 9.1). HPV is an epitheliotropic virus that is essentially endemic. In unvaccinated cohorts, the lifetime risk of acquiring genital HPV at least once is greater than 80%.9 Over 40 genotypes of HPV can infect the genital tract, 15 of which have been classified as carcinogenic (Table 9.1). These include HPV 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68, and 72.10 Together, HPV 16 and HPV 18 are the most common carcinogenic genotypes worldwide (Figures 9.2 and 9.3).11
Table 9.1
Classification of HPV Types Associated with Genital Lesions
| Classification | HPV Type |
| Oncogenic | 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 73, 82 |
| Putatively oncogenic | 26, 53, 66, 70 |
| Non-oncogenic | 6, 11, 40, 42, 54, 55, 57, 84 |
Adapted from Muñoz et al.13
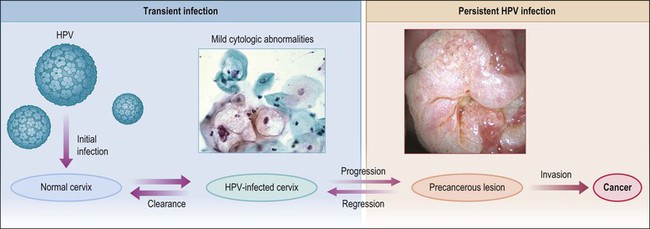
Figure 9.1 Model of HPV-induced cervical pathogenesis, proceeding from HPV infection of the normal cervix to mild cytologic abnormalities, progression to persistent HPV infection and precancerous lesions, and, finally, invasion. Of significance is the ability for precancerous lesions to regress, and the ability of most HPV-infected cervices to clear infection. (Reproduced with permission from Wright and Schiffman.118)
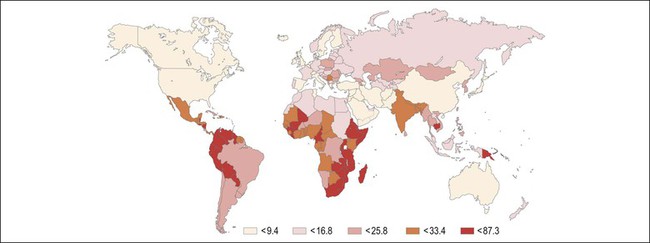
Figure 9.2 Age-standardized incidence rate of cervix uteri per 100,000 women worldwide in the year 2002. The lowest disease burden is largely found among regions with effective screening programs (e.g., North America) and regions where HPV prevalence is low (e.g., China). The greatest disease burden is denoted in red and is found in those countries with high HPV prevalence and without an effective screening program (e.g., East Africa). (Reproduced with permission from Ferlay et al.119)
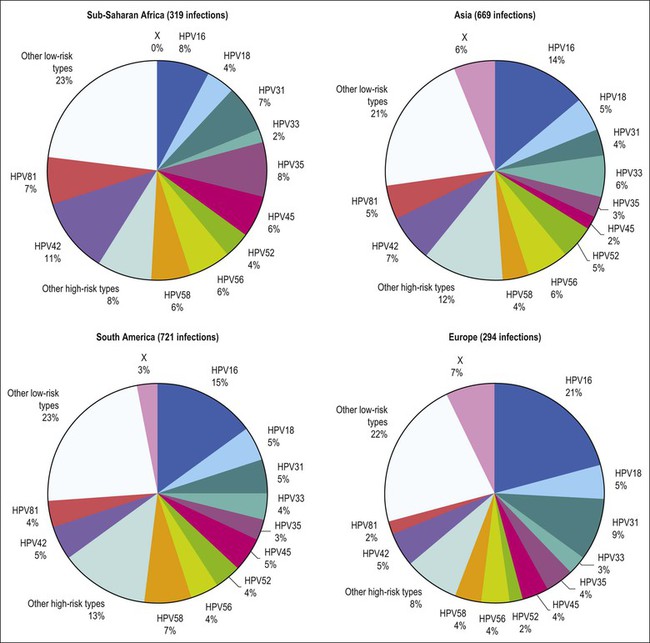
Figure 9.3 Prevalence of HPV types in cytologically normal women worldwide, by geographic region. HPV type-specific burden varies by geography, with HPV 16 prevalence highest (21%) in Europe but HPV 18 prevalence equivalent in all regions. (Reproduced from Clifford et al,120 with permission from Elsevier)
HPV infects cervical epithelium shortly after sexual debut, and can be detected in up to 50% of young women who have initiated sexual intercourse in the preceding 36 months.12 Most women clear an incident HPV infection within 1–2 years. Several host cofactors that have been associated with progression of HPV infection to carcinoma are shown in Table 9.2. Older women take longer to clear infections, as do smokers, and women with underlying immunosuppression.14 In the cervix, persistent infection is the proximate cause of squamous cancers and their precursors, high-grade squamous intraepithelial lesion (HSIL), or cervical intraepithelial neoplasia (CIN) grade 2 or 3 (CIN 2, CIN 3).
Table 9.2
Summary of Evaluated HPV Cofactors for Squamous Cell Carcinoma and Adenocarcinoma of the Cervix
| HPV Cofactor | Squamous Cell Carcinoma | Adenocarcinoma |
| Smoking | ++ Increased | No association |
| Parity | ++ Increased | No association |
| Oral contraceptives/hormones | ++ Increased | + Increased |
| Chlamydia | Increased, but current evidence not significant | No association |
| Herpes simplex virus 2 | Increased, but current evidence not significant | No association |
| Antioxidants | Decreased, but current evidence not significant | Current evidence not significant |
| Obesity | No association | + Increased |
HPV infections are limited to the suprabasal compartment of nonsterile barrier epithelia in which the immunologic contributions of the local microbiome are incompletely understood. Viral replication, assembly, and release of virions occur in the context of host cellular maturation and desquamation, without cell lysis, and without systemic viremia. Persistent HPV infections increase susceptibility to malignant transformation by a variety of mechanisms, including enhancing genetic instability and cell proliferation, inducing angiogenesis, interfering with intrinsic and extrinsic apoptotic pathways, downregulating expression of adhesion molecules, abrogating DNA damage responses, and interfering with both innate and adaptive immune responses in the lesion microenvironment. Known virally mediated immune-suppressive mechanisms include interfering with type I interferon responses and Toll-like receptor (TLR) signaling, downregulating cell surface MHC class I expression, enhancing secretion of immunosuppressive cytokines, recruitment of immune cells with suppressive function, and downregulating expression of adhesion molecules in lesional neovascular endothelium.15–17 Indeed, as we learn more about the immunobiology of persistent infection it becomes less surprising that immune-based therapeutic strategies to date, which have focused solely on eliciting an effector T-cell response to HPV antigens, have failed.
Low-Grade Squamous Intraepithelial Lesions (CIN1)
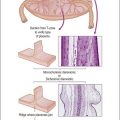
HPV infects keratinocytes at the basal epithelial layer of the squamocolumnar junction.18 A productive infection leads to the production of whole infectious virions, and is limited to epithelia that undergo maturation. As HPV does not encode DNA polymerase, host cellular DNA polymerase must be expressed for viral replication. In normal physiologic conditions in stratified squamous epithelium, DNA is copied only in the basal layer. In contrast, in virally infected epithelium, HPV drives DNA replication and cell division in the upper layers of differentiating epithelium, where HPV DNA is packaged into viral capsids, resulting in infectious virions (Figure 9.4).19 Infectious virions are released not by cell lysis, but in the course of host cell maturation and desquamation, without eliciting an inflammatory response.
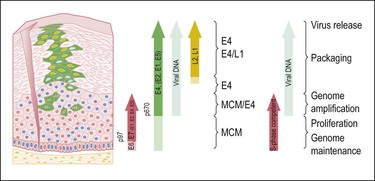
Figure 9.4 HPV gene expression during its life cycle in the genital squamous mucosa, from HPV-infected basal cell and maturation to surface of epithelium. Upon infection, the viral genome is maintained as a low copy number episome. During epithelial differentiation, the p97 promoter directs E6 and E7 expression for S-phase entry (red) and viral replication proteins (E1, E2, E4, E5) increase in abundance (green), facilitating amplification of viral genomes (blue). E4 persists in upper epithelial layers where viral capsid proteins (L1, L2; yellow) are found. (Reproduced with permission from Doorbar.121)
HPV infection produces a distinctive cytopathic effect that is recognizable both cytologically and histologically as a sharply demarcated cytoplasmic halo surrounding an enlarged, irregular, hyperchromatic nucleus with uneven chromatin distribution, termed ‘koilocytic atypia.’20 Other findings include multinucleation, parakeratosis, and hyperkeratosis. Histologically, HPV infections can be distinguished by koilocytotic changes in differentiating cells, with increased ratios of nuclear to cytoplasmic area, and mitotically active cells above the basal layer. Clinically, the behavior of cytology interpreted as ‘atypical squamous cells of undetermined significance’ with a concurrent positive test for oncogenic HPV DNA, cytology interpreted as ‘low-grade squamous intraepithelial lesion,’ or a tissue biopsy diagnosis of ‘low-grade squamous intraepithelial lesion’ or ‘cervical intraepithelial neoplasia grade I’ (CIN 1) is essentially the same; most eventually regress without intervention. Most CIN 1 lesions maintain HPV as an episome, sustain a complete viral replication cycle, and can be thought of as HPV infection.
High-Grade Squamous Intraepithelial Lesions (CIN 2/3)
HSILs, or CIN grade 2 or 3 (CIN 2/3), are associated with integration of the viral genome into the host genome, and subsequent constitutive expression of the E6 and E7 viral proteins (Figure 9.5). These proteins are functionally required for initiation and persistence of disease.21,22 Viral integration sites, although randomly distributed within the human genome,23 occur principally at sites where human DNA is prone to breakage (e.g., fragile sites), and appear to affect only the expression of the HPV genome itself. Specifically, during integration, E1 and/or E2 are frequently disrupted, while the E6 and E7 viral oncogenes are retained, resulting in constitutive expression. Morphologically at the cellular level, HSILs are characterized by a high nuclear to cytoplasmic ratio. Histologically, high-grade lesions display disarray of the basal layer itself, full thickness lack of cell maturation, and are mitotically active.
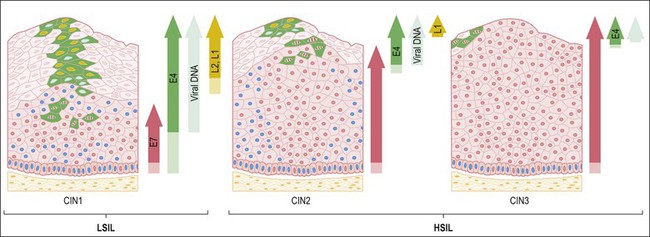
Figure 9.5 HPV early and late gene expression from initial HPV infection through maturation, from CIN 1–3, by evolutionary HPV type. The productive cycle begins close to the basal layer and viral genome amplification begins in the parabasal cell layers. (Reproduced with permission from Doorbar.121)
Although all squamous cervical cancers arise from untreated HSIL, not all HSILs progress to invasive cancer. Approximately 35% of HSILs undergo complete regression in a time frame of 4–6 months.24,25 Lesions associated with HPV 16 are less likely to regress than lesions associated with other HPV types. Because it is not possible to distinguish HSILs that are likely to regress from those that are not, the standard of care is excision or ablation. Excisional approaches include loop electrocautery excision procedure or cold knife conization. The rate of recurrence is 10%.26 Iterative excisional procedures are associated with cervical incompetence and subsequent preterm birth.27,28
Mechanisms of Oncogenesis
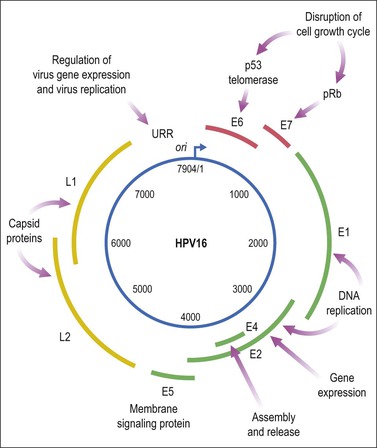
HPV is a small, double-stranded DNA virus with an 8 kb genome comprised of three domains (Figure 9.6): a noncoding upstream regulatory region, an early region containing six genes (E1–E7), and a late region encoding capsid proteins L1 (major capsid) and L2 (minor capsid).29 The E6 and E7 viral proteins enhance cell proliferation in the face of abrogated DNA damage responses and genetic instability. Additionally, E6 mediates immortalization by activating telomerase. Finally, these oncoproteins compromise both innate and adaptive immune responses in the tissue microenvironment.
Viral persistence is mediated in part by dysregulated innate immunity involving pattern recognition receptor, particularly alterations in expression and function of TLRs in dendritic cells and macrophages. However, as TLRs sense pathogen-associated molecular patterns, including viral genetic material, they are also found in normal cervical epithelial cells.30,31 While less is known about TLR function in epithelial cells, evidence of dysregulated TLR3 signaling, including dampened expression of genes encoding antimicrobial molecules, chemotactic and pro-inflammatory cytokines, and proteins involved in antigen presentation, has been identified in cervical keratinocytes containing episomal HPV.32
The HPV E6 and E7 Oncoproteins
E6 binds to an endogenous E3 ubiquitin ligase, E6 associated protein (E6AP).33 The E6/E6AP heterodimer binds p53, a tumor suppressor protein that also regulates apoptosis, and targets it for rapid proteasome-mediated degradation.34 p53 can be stimulated either by genotoxic or cytotoxic damage, or by dysregulated DNA synthesis. These signals would be triggered in an HPV infection, particularly in a persistent infection in which cell cycling continues despite cellular damage.
In infected cells, E6 blocks both the intrinsic and extrinsic apoptosis pathways.35 The extrinsic apoptosis pathway is triggered by viral infections and activates death receptors on the cell surface. E6 blocks death receptors from interacting with death-inducing signaling complexes, and also degrades signaling proteins, such as FADD and caspase-8.36,37 The intrinsic apoptosis pathway is activated by DNA damage and oxidative stress. The intrinsic pathway starts in the mitochondria with Bak, and Bak expression in the mitochondrial membrane is blocked by E6.38 Downstream proteins, such as additional caspases, are degraded by E6, and inhibitors of apoptosis, including NF kappa B, survivin, and c-IAP, are stabilized by E6.39,40
E6 also has direct roles in chromosomal instability and DNA damage. Minichromosome maintenance 7 (MCM7) insures there is only a single round of DNA replication in a cell cycle. E6 degrades MCM7, allowing multiple copies of its own DNA to be made.41 A byproduct of HPV DNA amplification is the risk of excessive cellular chromatin duplication. This aneuploidy normally would be sensed in G2 by p53, triggering cell cycle arrest and apoptosis, but p53 is removed by E6/E6AP. E6 also degrades XRCC1 and O6MGMT.42,43 Both are involved in single-stranded DNA repair, and, without them, stochastic errors that drive cancer development can accrue.
E6 disrupts replicative senescence signals in keratinocytes. With each copy, linear chromosomes lose 100–200 bases of repetitive telomeric DNA. Thus, the age of a cell is inversely proportional to the length of its telomeres. Replicative senescence occurs when somatic cells recognize their intrinsic age.44 Stem cells express telomerase, a ribonucleoprotein, to extend this DNA, and the catalytic subunit of telomerase, hTERT, is rate determining.45 By lengthening telomeric DNA, stem cells avoid cellular senescence. Almost all cancers express telomerase to also avoid replicative senescence signals.46 E6/E6AP enables replicative immortality through hTERT activation. E6/E6AP removes repressors and recruits activators to the normally constitutively repressed promoter of hTERT, and E6 also affects the stability of hTERT mRNA to increase protein production and telomerase activity.47–52
E6 degrades proteins involved in cell–cell adhesion and apicobasal polarity. It degrades hScrib, a tight-junction protein, and many postsynaptic density protein 95, drosophila disc large tumor suppressor, zonula occludens-1 protein (PDZ) domain-containing proteins.53–57 Finally E6 binds proteins in the extracellular matrix.58 These changes are important to allow viral release from cells as they slough off the upper layers of stratified squamous epithelium. They are also important in invasion and metastases.59,60
Finally, E6 interferes with innate immune responses. Interferon regulatory factor 3 (IRF-3) is normally activated by a viral infection, but is inhibited by E6.61 TLR9 is activated by viral DNA motifs and induces cytokine production, but E6 blocks its expression.62
The second crucial HPV oncoprotein, E7, destabilizes retinoblastoma protein (pRb) and other pocket proteins by targeting them for proteasome-mediated degradation, resulting in cell cycle progression, as these proteins normally hold cells in G1, not allowing DNA synthesis.63–65 In E7-driven proliferation, S-phase gene expression ensues, and viral DNA is copied in addition to cellular DNA. E7 drives cell division gene activation and increases the open chromatin structure at gene promoters. E7 also binds histone deacetylases and represses polycomb group complexes, some of which are histone methyltransferases.66,67
E7 not only pushes cells to continue to divide, but also helps them to evade growth suppressors. E7 blocks transforming growth factor-beta (TGF-β) and tumor necrosis factor (TNF)-alpha G1.68,69 TNF-α G1 is a cytokine that induces programmed cell death, and is produced by cytotoxic T-cells to eliminate virally infected cells. Activation of IRFs is also blocked by E7, thereby compromising activation of innate immunity.70
E7 also binds to p600, a pRb-associated factor critical for anchorage-dependent growth.71 E7 increases expression of vascular endothelial growth factor and IL-8, both of which are angiogenic, and enhances transcriptional activity of hypoxia inducible factor 1α (HIF 1α), and disrupts interaction with histone deacetylases.72–74 Finally E7, like E6, can induce DNA damage at the chromosomal level. E7 increases centrosome amplification early in the viral life cycle.75,76 This increase in centrosomes can lead to DNA damage, and increases the frequency of DNA integration. As has been demonstrated in cell lines and mouse model systems, E6 and E7 (or equivalently inactivation of p53 and Rb) cooperate to produce full-fledged malignancy.77
Naturally Occurring Immune Responses to HPV Antigens
In immune-competent hosts, over 90% of genital HPV infections become undetectable without intervention.78 However, naturally occurring systemic humoral and adaptive responses to HPV antigens, even in cohorts with documented type-specific mucosal infections that have become undetectable, are hard to detect in peripheral blood.79–81 Type-specific serum antibodies to capsid proteins are detectable in less than half of women in whom cervical HPV infections of known serotype have cleared. Nonetheless, data from cohorts undergoing prophylactic vaccination demonstrate an anamnestic response to a single dose of virus-like particle vaccine in previously infected subjects.82 Dysplasia is associated with ineffective immune responses to viral nonstructural proteins. Antibody to E7 can be measured in persons with invasive cancer, but not in earlier stage disease.83
HSIL (CIN 2/3) is a lesion that should be susceptible to an HPV-specific effector immune response. Expression of viral, non-‘self’ proteins is functionally required to initiate and maintain the transformed phenotype, thereby providing true tumor-associated antigenic targets.21,22 Not all cervical HSILs progress to cancer; 25% of established CIN 2/3 lesions caused by HPV 16, the genotype most commonly associated with malignancy, undergo complete regression.25
However, women with squamous intraepithelial HPV lesions rarely have systemic T-cell responses to HPV E6 or E7 that can be detected directly ex vivo, likely reflecting the tissue compartmentalization of early disease. In contrast to immune responses to other viral infections, the frequency of systemic memory CD8+ T-cells in individuals with a known prior cervical HPV infection, which has subsequently become undetectable, is vanishingly low. For example, using direct ex vivo assays, the frequency of systemic virus-specific CD8+ T-cells after primary infection with cytomegalovirus or hepatitis C virus (HCV) can be up to 5%.84,85 In contrast, in subjects with CIN, the frequency of HPV-specific T-cells is two to three orders of magnitude lower, in the range of 0.1–0.01%.86 Detection of systemic HPV-specific T-cell responses in subjects with intraepithelial neoplasia requires ex vivo sensitization.87–89 After in vitro stimulation with HPV antigens, peptide-specific T-cell frequencies increase in subjects with concurrent disease at the time of blood sampling, compared with subjects with no evidence of disease.86,90 While amplification can identify qualitative responses to HPV antigens, it is likely to have limited use in accurately distinguishing quantitative differences between individual subjects, either in the course of a natural infection or in subjects with intraepithelial disease. Responses identified only after prolonged in vitro restimulation likely represent expansion of previously induced memory immune responses, rather than an ongoing response at the time of sampling.
In immune-competent persons with undetectable systemic HPV T-cell responses, dense immune cell infiltrates nonetheless localize at the lesion site, and, in persistent disease, fail to access the lesional epithelium. In contrast, dysplastic lesions that ‘permit’ CD8+ T-cell access to the epithelial compartment are likely to regress.17 This observation suggests that, even in the clinical setting of preinvasive, incipient neoplasia, factors in the lesions mitigate the ability of recruited CD8+ T-cells to eliminate disease.
Tissue-Resident Immune Cells
Antigen-experienced T-lymphocytes display preferential tropism for the tissue in which they encountered their cognate antigen.91–94 Constitutive trafficking of antigen-experienced T-lymphocytes in a tissue-specific manner is mediated by distinct sets of chemokines and adhesion molecules. Tissue homing occurs via sequential tethering of leukocyte selectins to vascular endothelium, followed by leukocyte activation by locally expressed chemokines, conformational changes in surface integrins, and subsequent interactions with tissue-localized ligands to mediate firm adhesion and extravasation. This compartmentalization provides a mechanism that is essential for rapid immune recognition of pathogens at the most probable sites of encounter. To date, it has not been possible to study cervical resident T-cell populations in detail, in part because tissues obtained during routine care are small, and because conventional methods of isolating T-cells from tissue biopsies yield only low numbers of cells. Above all, studies on clinical specimens must not compromise the ability to make an accurate diagnosis, or place a study subject at risk from excessive tissue procurement.
The recent development of methods for isolating tissue-resident T-cells from normal and dysplastic primary tissue explants has opened up opportunities to quantitate tissue-specific immunologic parameters. Small tissue cervical mucosal explants cultured short term without cytokine support yield sufficient numbers of tissue T-cells to permit limited characterization of immune cell subset phenotype, and their functional polarization. Like other tissue-resident T-cell populations, cervical T-cells in normal mucosa have an effector memory phenotype (CD45RO + CD62L–CCR7–), and are slightly activated (CD25lo/CD69lo). Virtually all T-cells in both normal and dysplastic cervical mucosa express the α4β7 surface integrin, and the intensity of lesional epithelial CD8+ T-cell infiltrates correlates directly with expression of MAdCAM-1 in lesional epithelial vascular endothelium.17 Downregulated vascular endothelial expression of adhesion molecules is a common mechanism by which invasive or metastatic human solid tumors evade locally recruited immune effector cells; this finding suggests that endothelial activation is a reasonable therapeutic target in HSILs. Together, this constellation of observations suggests that methods to monitor biologically plausible end points in the target tissue should be validated, as they are likely to provide insights as to the success or failure of immune-based therapies.
HPV Vaccines
Primary Prevention: Early Detection and Prophylactic Vaccines
The goal of cervical cancer screening is to identify patients at risk for the development of disease, that is, those with the immediate precursor lesion, HSIL. In high-resource settings, routine screening includes repeated cytologic and HPV screening over the course of a lifetime. Current guidelines issued by the U.S. Preventive Services Task Force recommend age-specific screening strategies; screening should not begin until age 21 regardless of sexual activity. Women aged 21–29 years should be screened every 3 years by cytology alone. In ‘older’ women aged 30–65 years, co-testing with cytology and HPV testing is recommended at 5 year intervals. More frequent screening is recommended for immunosuppressed patients; women infected with human immunodeficiency virus (HIV); women exposed to diethylstilbestrol in utero; and women previously treated for CIN 2, CIN 3, or cancer. Screening may be discontinued in women age 65–70 with three prior consecutively normal Pap smears, and no abnormal Pap smears over a period of 10 years.95
Despite the effectiveness of using cytology and HPV DNA testing to detect disease, it is expensive and cumbersome. Many women undergo repetitive Pap smears and colposcopy for evaluation of low-grade dysplastic lesions that are likely to resolve over time. Repetitive clinic visits and testing not only places a psychological burden on the patient, but also places economic strain upon the society providing the screening. In the United States alone, it has been estimated that 6 billion dollars per year are spent on evaluation of low-grade lesions.96 Recently, the long-term predictive value of HPV testing in a cohort of women followed for up to 18 years has been reported.97 Women with a baseline prevalent infection, as well as women with incident infections detected over the course of the study were included in this analysis. HPV detection was confirmed using a commercially available diagnostic test that is used routinely in clinical practice. HPV detection stratified risk more accurately than cytology; persons with a baseline negative HPV test were less likely to have a CIN 3 or squamous carcinoma (‘CIN 3+’) diagnosed than persons who had a normal Pap smear. Conversely, detectable HPV DNA identified women at elevated risk of subsequent CIN 3 or cancer. In the face of a normal Pap smear, detection of either HPV 16 or HPV 18 conferred a higher risk of CIN 3 or cancer than detection of other HPV genotypes. This work is important because it provides data to support risk-based screening, which can be done using tools that are already available as part of routine practice. The implication of using a test that is quantitative and not subjective as a primary screening tool is that it does not need to be performed by a provider with specialized training.
Currently available preventative vaccines, Gardasil (Merck & Co) (HPV 4) and Cervarix (GlaxoSmithKline) (HPV 2), are virtually 100% effective in prevention of high-grade cervical intraepithelial lesions associated with HPV 16 and HPV 18. However, rates of vaccine administration vary widely. For example, in Australia, administration in eligible girls is >80%.98 In the United States, the Centers for Disease Control reported that, in 2009, less than half (44%) of eligible children had been vaccinated.99 Rates of vaccine completion, defined as undergoing the full series of three prophylactic vaccinations, is dismal. Among vaccine initiators in the United States, less than one in four complete vaccination.99 As HPV 4 and HPV 2 elicit type-specific antibody responses to capsid (L1), they have no therapeutic effect whatsoever.
Secondary Prevention: Immune-Based Therapies
Development of immune therapeutic strategies presents another strategy to decrease the global burden of HPV disease. HPV-infected epithelial cells express viral proteins that should, in theory, provide antigenic targets for therapeutic vaccines. Many therapeutic HPV vaccine constructs, including peptide, protein, recombinant viral vector-based, and DNA vaccines, have elicited antigen-specific effector immune responses that are curative, in preclinical murine models using transplanted E7-expressing epithelial tumor cell lines.100 To date, clinical success has not been nearly as robust. Several factors are likely to play a role in the pace of translation that has been incremental until relatively recently. Broadly, these include the immunogenicity in humans of vaccines tested, and clinical trial design, including cohort selection, length of study window, and end points assessed.
Vaccine Immunogenicity
Three broad categories of therapeutic vaccines have been tested to date in subjects with preinvasive HPV disease. These include DNA vaccines, recombinant viral vector-based vaccines, and adjuvanted protein or peptide vaccines. The antigen specificity of DNA vaccines can be engineered precisely, and production can be accomplished relatively simply on a large scale with high purity. Multicomponent vaccines can be engineered to include specific immunogens to amplify immunologic responses. DNA vaccines can be used for repeated administration, as the immunogenicity of plasmid vectors is not influenced by pre-existing neutralizing antibodies.101 DNA vaccination has been shown to cause minimal if any toxicities or side effects in humans. In fact, to date, the major drawback of DNA vaccine approaches in clinical translation has been their relatively low potency. The immunogenicity of conventional intramuscular administration is likely to be limited by inefficient uptake of DNA by host cells. Delivery via electroporation, a technology that applies short electrical pulses in situ, increases uptake by temporarily permeabilizing cell membranes,102 which in nonhuman primate models increases immunogenicity of DNA vaccination by 100-fold.103 Currently, several clinical trials testing electroporation-mediated delivery of DNA vaccines are underway, targeting HIV, HCV, and solid tumor antigens (prostate, carcinoembryonic antigen (CEA), HER2/neu, melanoma), and HPV.104
While recombinant viral vector-based vaccines are more immunogenic in humans than DNA vaccines, they cannot be administered sequentially, as immune responses to the vector are likely to outstrip responses to engineered antigenic targets. However, a growing body of preclinical as well as clinical evidence suggests that sequential vaccination consisting of a DNA priming vaccination, followed by a recombinant viral vector-based boost vaccination, is more immunogenic than either strategy used alone.105 Heterologous prime-boost trials targeting malarial and HIV antigens have demonstrated proof of principle, and studies testing this strategy in HPV disease are underway (National Cancer Institute funded trial NCT00788164).
The third broad category of therapeutic HPV vaccines tested to date includes the use of adjuvanted peptide and proteins. Like DNA vaccines, antigen specificity can be engineered precisely. However, the use of individual peptide targets is restricted by the assumption of immunodominant epitopes. Vaccination with protein or with long peptides has the advantage of allowing antigen processing in the host. Recently, the use of adjuvanted long peptides spanning the length of HPV 16 E6 and E7 has sparked a great deal of interest, as clinical responses were observed in 47% of vaccinated subjects with vulvar intraepithelial neoplasia (VIN).106
Several painstaking studies of immune responses to HPV early antigens, in clinically annotated cohorts, provide important insights for the design and interpretation of clinical trials to test therapeutic HPV vaccines. First, it is clear that weak virus-specific systemic effector responses measured in the peripheral blood in otherwise healthy subjects with preinvasive lesions can be associated with lesion regression.81,107–109 In vaccinated as well as unvaccinated cohorts, systemic effector immune responses to HPV E6 and E7, even in healthy subjects, have required ex vivo expansion for detection.108,110,111 The fact that women with pre-existing detectable systemic T-cell responses to HPV antigens are more likely to respond clinically to directed manipulations creating a local proinflammatory microenvironment suggests that virus-specific adaptive responses do play a role in lesion clearance.112
Mechanisms of immune evasion that have already been identified in invasive or metastatic solid tumors are also likely to play a role in preinvasive HPV disease. The study of effector and homeostatic immune cell infiltrates in HPV-infected epithelia has begun to provide insights that are likely to be useful in directing future clinical investigations. CIN 2/3 lesions are associated with accumulation of Foxp3+ and IDO+ immune-suppressive T-cells, compared with normal cervical tissue.113,114 Conversely, immune-active CD4 and CD8 T-cell infiltrates in lesional epithelium are identified in regressing lesions.107,108,112 Lesional stroma is also likely to play a role in immune suppression, particularly in the functional polarization of recruited immune cells, via secretion of immune-suppressive cytokines.115 Most analyses of clinical tissue samples, however, have been case–control studies, and it is worth stating that preinterventional and postinterventional studies of immunologic parameters in the target tissues should be incorporated into clinical trial design, when possible.
Recent therapeutic trials in healthy cohorts with VIN illustrate the concept of testing combinatorial regimens that include both therapeutic vaccination as well as manipulation of the lesion microenvironment. VIN lesions, although indolent, are also clinically recalcitrant. Currently, the mainstay of treatment for VIN is wide local excision. Recurrence is common, particularly in multifocal disease, requiring additional surgery in approximately 15% of subjects with negative resection margins, 40% of subjects with multifocal disease, and nearly half of subjects with positive margins.116 Recently, two approaches have demonstrated significant clinical responses that are associated with immune readouts: systemic immunization with long peptides spanning E6 and E7, administered with adjuvant, and local manipulation of the lesion site using topical application of imiquimod, a TLR7 agonist, followed by therapeutic HPV vaccination.
In subjects with HPV 16+ VIN, therapeutic vaccination with long peptides spanning HPV 16 E6 and E7, adjuvanted with montanide, achieved a complete response rate of 45%.106 This result is remarkable because, unvaccinated, the expected rate of regression in this cohort is well under 5%. In this study, clinical responders had smaller lesions at study entry than nonresponders. Responders also had peripheral blood proliferative responses to vaccine antigens that were broader and of greater magnitude than nonresponders. In contrast, nonresponders had a detectable increase in CD4+CD25+Foxp3+ cells post vaccination in the peripheral blood. However, the magnitude of effector responses in responders overlapped with that identified in nonresponders, which suggests that factors in the lesions themselves may have mitigated the ability of vaccine-induced immune responses to eliminate disease.
Subjects enrolled in a different trial that tested the sequence of lesion manipulation with imiquimod, a topical TLR7 agonist, followed by therapeutic vaccination, achieved a complete response rate of 63%.112 Similar to previous therapeutic vaccine studies, peripheral blood responses of responders overlapped with those detected in nonresponders. However, responders were nonetheless more likely than nonresponders to have had detectable pre-existing proliferative responses to HPV antigens in the peripheral blood. Similar to subjects in the long peptide trial, responders had postvaccination proliferative responses to vaccine antigens that were broader and of greater magnitude than nonresponders. The fact that, in both of these trials, sophisticated methods using ex vivo manipulation were required to detect immune responses suggests that HPV-specific cells identified in the blood were not effector memory cells, which respond immediately, but were more likely to have been central memory cells, which have a longer time frame for response. Quantitative analyses of immune cell subsets in lesional tissue identified increased aggregates of both CD4 and CD8 T-cells in responders, and increased infiltrates of regulatory T-cells in nonresponders.
In both of these studies, clinical responses ‘accumulated’ over study time frames that extended well beyond the last study vaccination. In both studies, subjects with VIN were monitored for 52 weeks before definitive surgical excision. Although subjects whose lesions underwent regression tended to do so earlier rather than later, these trials suggest that, in subjects whose disease can be monitored safely, allowing immune responses to access lesions may require patience. Second, although both local manipulation to enhance acute inflammation as well as systemic vaccination enhanced the rate of lesion regression, immunologic parameters that could reliably distinguish or predict ‘responders’ from ‘nonresponders’ at the outset have yet to be identified. These studies are consistent with animal studies in which clearance of HPV disease requires enabling HPV-specific immune effector cells via tissue-localized acute inflammation.117 Together these studies emphasize the need to include immunologic parameters in the target tissues as study end points. They also suggest that interventional clinical trials testing systemically administered therapeutic vaccines should be designed to allow a sufficient time frame for responses to accumulate.
References
1. Parkin, DM, Bray, F, Ferlay, J, Pisani, P. Global cancer statistics, 2002. CA Cancer J Clin. 2005; 55(2):74–108.
2. Ferlay, J, Shin, HR, Bray, F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2008; 127(12):2893–2917.
3. De Vuyst, H, Clifford, GM, Nascimento, MC, et al. Prevalence and type distribution of human papillomavirus in carcinoma and intraepithelial neoplasia of the vulva, vagina and anus: a meta-analysis. Int J Cancer. 2009; 124(7):1626–1636.
4. Parkin, DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006; 118(12):3030–3044.
5. van de Nieuwenhof, HP, van Kempen, LC, de Hullu, JA, et al. The etiologic role of HPV in vulvar squamous cell carcinoma fine tuned. Cancer Epidemiol Biomarkers Prev. 2009; 18(7):2061–2067.
6. Jones, RW, Rowan, DM, Stewart, AW. Vulvar intraepithelial neoplasia: aspects of the natural history and outcome in 405 women. Obstet Gynecol. 2005; 106(6):1319–1326.
7. Kreimer, AR, Clifford, GM, Boyle, P, Franceschi, S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol Biomarkers Prev. 2005; 4(2):467–475.
8. Chaturvedi, AK, Engels, EA, Pfeiffer, RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011; 29(32):4294–4301.
9. Baseman, JG, Koutsky, LA. The epidemiology of human papillomavirus infections. J Clin Virol. 2005; 32(Suppl 1):S16–S24.
10. Muñoz, N, Franco, EL, Herrero, R, et al. Recommendations for cervical cancer prevention in Latin America and the Caribbean. Vaccine. 2008; 26(Suppl 11):L96–107.
11. de Sanjose, S, Diaz, M, Castellsague, X, et al. Worldwide prevalence and genotype distribution of cervical human papillomavirus DNA in women with normal cytology: a meta-analysis. Lancet Infect Dis. 2007; 7(7):453–459.
12. Woodman, CB, Collins, S, Winter, H, et al. Natural history of cervical human papillomavirus infection in young women: a longitudinal cohort study. Lancet. 2001; 357(9271):1831–1836.
13. Muñoz, N, Bosch, FX, de Sanjose, S, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003; 348:518–527.
14. Frazer, IH, Lowy, DR, Schiller, JT. Prevention of cancer through immunization: prospects and challenges for the 21st century. Eur J Immunol. 2007; 37(Suppl 1):S148–S155.
15. Visser, J, Nijman, HW, Hoogenboom, BN, et al. Frequencies and role of regulatory T cells in patients with (pre)malignant cervical neoplasia. Clin Exp Immunol. 2007; 150(2):199–209.
16. Cromme, FV, Snijders, PJ, van den Brule, AJ, et al. MHC class I expression in HPV 16 positive cervical carcinomas is post-transcriptionally controlled and independent from c-myc overexpression. Oncogene. 1993; 8(11):2969–2975.
17. Trimble, CL, Clark, RA, Thoburn, C, et al. Human papillomavirus 16-associated cervical intraepithelial neoplasia in humans excludes CD8 T cells from dysplastic epithelium. J Immunol. 2010; 185(11):7107–7114.
18. Herfs, M, Yamamoto, Y, Laura, A, et al. A discrete population of squamocolumnar junction cells implicated in the pathogenesis of cervical cancer. Proc Natl Acad Sci USA. 2012; 109(26):10516–10521.
19. Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci (Lond). 2006; 110(5):525–541.
20. Koss, LG, Durfee, GR. Unusual patterns of squamous epithelium of the uterine cervix: cytologic and pathologic study of koilocytotic atypia. Ann NY Acad Sci. 1956; 63(6):1245–1261.
21. Werness, B, Levine, A, Howley, P. Association of human papillomavirus types 16 and 18 proteins with p53. Science. 1990; 248:76–79.
22. Hudson, JB, Bedell, MA, McCance, DJ, Laiminis, LA. Immortalization and altered differentiation of human keratinocytes in vitro by the E6 and E7 open reading frames of human papillomavirus type 18. J Virol. 1990; 64(2):519–526.
23. Wang, SS, Hildesheim, A, Chapter 5: Viral and host factors in human papillomavirus persistence and progression. J Natl Cancer Inst Monogr, 2003;(31):35–40.
24. Melnikow, J, Nuovo, J, Willan, AR, et al. Natural history of cervical squamous intraepithelial lesions: a meta- analysis. Obstet Gynecol. 1998; 92(4 Pt 2):727–735.
25. Trimble, CL, Piantadosi, S, Gravitt, P, et al. Spontaneous regression of high-grade cervical dysplasia: effects of human papillomavirus type and HLA phenotype. Clin Cancer Res. 2005; 11(13):4717–4723.
26. Ramchandani, SM, Houck, KL, Hernandez, E, Gaughan, JP. Predicting persistent/recurrent disease in the cervix after excisional biopsy. MedGenMed. 2007; 9(2):24.
27. Bevis, KS, Biggio, JR. Cervical conization and the risk of preterm delivery. Am J Obstet Gynecol. 2011; 205(1):19–27.
28. Sadler, L, Saftlas, A, Wang, W, et al. Treatment for cervical intraepithelial neoplasia and risk of preterm delivery. JAMA. 2004; 291(17):2100–2106.
29. Laimins, LA. The biology of human papillomaviruses: from warts to cancer. Infect Agents Dis. 1993; 2(2):74–86.
30. Andersen, JM, Al-Khairy, D, Ingalls, RR. Innate immunity at the mucosal surface: role of toll-like receptor 3 and toll-like receptor 9 in cervical epithelial cell responses to microbial pathogens. Biol Reprod. 2006; 74(5):824–831.
31. DeCarlo, CA, Rosa, B, Jackson, R, et al. Toll-like receptor transcriptome in the HPV-positive cervical cancer microenvironment. Clin Dev Immunol. 2012; 2012:785825.
32. Karim, R, Meyers, C, Backendorf, C, et al. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLoS One. 2011; 6(3):e17848.
33. Huibregtse, JM, Scheffner, M, Howley, PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991; 10(13):4129–4135.
34. Huibregtse, JM, Scheffner, M, Howley, PM. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol Cell Biol. 1993; 13(2):775–784.
35. Howie, HL, Katzenellenbogen, RA, Galloway, DA. Papillomavirus E6 proteins. Virology. 2009; 384(2):324–334.
36. Filippova, M, Johnson, MM, Bautista, M, et al. The large and small isoforms of human papillomavirus type 16 E6 bind to and differentially affect procaspase 8 stability and activity. J Virol. 2007; 81(8):4116–4129.
37. Tungteakkhun, SS, Filippova, M, Neidigh, JW, et al. The interaction between human papillomavirus type 16 and FADD is mediated by a novel E6 binding domain. J Virol. 2008; 82(19):9600–9614.
38. Thomas, M, Banks, L. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene. 1998; 17(23):2943–2954.
39. James, MA, Lee, JH, Klingelhutz, AJ. Human papillomavirus type 16 E6 activates NF-kappaB, induces cIAP-2 expression, and protects against apoptosis in a PDZ binding motif-dependent manner. J Virol. 2006; 80(11):5301–5307.
40. Borbely, AA, Murvai, M, Konya, J, et al. Effects of human papillomavirus type 16 oncoproteins on survivin gene expression. J Gen Virol. 2006; 87(Pt 2):287–294.
41. Kuhne, C, Bank, L. E3-ubiquitin ligase/E6-AP links multicopy maintenance protein 7 to the ubiquitination pathway by a novel motif, the L2G box. J Biol Chem. 1998; 273(51):34302–34309.
42. Iftner, T, Elbel, M, Schopp, B, et al. Interference of papillomavirus E6 protein with single-strand break repair by interaction with XRCC1. EMBO J. 2002; 21(17):4741–4748.
43. Srivenugopal, KS, Ali-Osman, F. The DNA repair protein, O(6)-methylguanine-DNA methyltransferase is a proteolytic target for the E6 human papillomavirus oncoprotein. Oncogene. 2002; 21(38):5940–5945.
44. Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965; 37:614–636.
45. Counter, CM, Meyerson, M, Eaton, EN, et al. Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene. 1998; 16(9):1217–1222.
46. Shay, JW, Bacchetti, S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997; 33(5):787–791.
47. Veldman, T, Horikawa, I, Barrett, JC, Schlegel, R. Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J Virol. 2001; 75(9):4467–4472.
48. Katzenellenbogen, RA, Vilet-Grett, P, Xu, M, Galloway, D. NFX1-123 increases hTERT expression and telomerase activity posttranscriptionally in human papillomavirus type 16 E6 keratinocytes. J Virol. 2009; 83(13):6446–6456.
49. James, MA, Lee, JH, Klingelhutz, AJ. HPV16-E6 associated hTERT promoter acetylation is E6AP dependent, increased in later passage cells and enhanced by loss of p300. Int J Cancer. 2006; 119(8):1878–1885.
50. Liu, X, Yuan, H, Fu, B, et al. The E6AP ubiquitin ligase is required for transactivation of the hTERT promoter by the human papillomavirus E6 oncoprotein. J Biol Chem. 2005; 280(11):10807–10816.
51. McMurray, HR, McCance, DJ. Human papillomavirus type 16 E6 activates TERT gene transcription through induction of c-Myc and release of USF-mediated repression. J Virol. 2003; 77(18):9852–9861.
52. Oh, ST, Kyo, S, Laimins, LA. Telomerase activation by human papillomavirus type 16 E6 protein: induction of human telomerase reverse transcriptase expression through Myc and GC-rich Sp1 binding sites. J Virol. 2001; 75(12):5559–5566.
53. Nakagawa, S, Huibregtse, JM. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol Cell Biol. 2000; 20(21):8244–8253.
54. Lee, SS, Glaunsinger, B, Mantovani, F, et al. Multi-PDZ domain protein MUPP1 is a cellular target for both adenovirus E4-ORF1 and high-risk papillomavirus type 18 E6 oncoproteins. J Virol. 2000; 74(20):9680–9693.
55. Massimi, P, Gammoh, N, Thomas, M, Banks, L. HPV E6 specifically targets different cellular pools of its PDZ domain-containing tumour suppressor substrates for proteasome-mediated degradation. Oncogene. 2004; 23(49):8033–8039.
56. Spanos, WC, Hoover, A, Harris, GF, et al. The PDZ binding motif of human papillomavirus type 16 E6 induces PTPN13 loss, which allows anchorage-independent growth and synergizes with ras for invasive growth. J Virol. 2008; 82(5):2493–2500.
57. Kranjec, C, Banks, L. A systematic analysis of human papillomavirus (HPV) E6 PDZ substrates identifies MAGI-1 as a major target of HPV type 16 (HPV-16) and HPV-18 whose loss accompanies disruption of tight junctions. J Virol. 2011; 85(4):1757–1764.
58. Du, M, Fan, X, Hong, E, Chen, JJ. Interaction of oncogenic papillomavirus E6 proteins with fibulin-1. Biochem Biophys Res Commun. 2002; 296(4):962–969.
59. Watson, RA, Thomas, M, Banaks, L, et al. Activity of the human papillomavirus E6 PDZ-binding motif correlates with an enhanced morphological transformation of immortalized human keratinocytes. J Cell Sci. 2003; 116(Pt 24):4925–4934.
60. Nguyen, ML, Nguyen, MM, Lee, D, et al. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6’s induction of epithelial hyperplasia in vivo. J Virol. 2003; 77(12):6957–6964.
61. Ronco, LV, Karpova, AY, Vidal, M, Howley, PM. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998; 12(13):2061–2072.
62. Hasan, UA, Bates, E, Takeshita, F, et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J Immunol. 2007; 178(5):3186–3197.
63. Dyson, N, Howley, PM, Munger, K, Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989; 243(4893):934–937.
64. Balsitis, S, Dick, F, Dyson, N, Lambert, PF. Critical roles for non-pRb targets of human papillomavirus type 16 E7 in cervical carcinogenesis. Cancer Res. 2006; 66(19):9393–9400.
65. Huh, K, Zhou, X, Kayakawa, H, et al. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol. 2007; 81(18):9737–9747.
66. Longworth, MS, Wilson, R, Laimins, LA. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J. 2005; 24(10):1821–1830.
67. Burgers, WA, Blanchon, L, Pradhan, S, et al. Viral oncoproteins target the DNA methyltransferases. Oncogene. 2007; 26(11):1650–1655.
68. De Geest, K, Bergman, CA, Turyk, ME, et al. Differential response of cervical intraepithelial and cervical carcinoma cell lines to transforming growth factor-beta 1. Gynecol Oncol. 1994; 55(3 Pt 1):376–385.
69. Pietenpol, JA, Stein, RW, Moran, E, et al. TGF-beta 1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell. 1990; 61(5):777–785.
70. Park, JS, Kim, EJ, Kwon, HJ, et al. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem. 2000; 275(10):6764–6769.
71. Huh, KW, DeMasi, J, Ogawa, H, et al. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc Natl Acad Sci USA. 2005; 102(32):11492–11497.
72. Nakamura, M, Bodily, JM, Beglin, M, et al. Hypoxia-specific stabilization of HIF-1alpha by human papillomaviruses. Virology. 2009; 387(2):442–448.
73. Chen, W, Li, F, Mead, L, et al. Human papillomavirus causes an angiogenic switch in keratinocytes which is sufficient to alter endothelial cell behavior. Virology. 2007; 367(1):168–174.
74. Toussaint-Smith, E, Donner, DB, Roman, A. Expression of human papillomavirus type 16 E6 and E7 oncoproteins in primary foreskin keratinocytes is sufficient to alter the expression of angiogenic factors. Oncogene. 2004; 23(17):2988–2995.
75. Duensing, S, Duensing, A, Crum, CP, et al. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001; 61(6):2356–2360.
76. Korzeniewski, N, Treat, B, Duensing, S. The HPV-16 E7 oncoprotein induces centriole multiplication through deregulation of Polo-like kinase 4 expression. Mol Cancer. 2011; 10:61.
77. Hawley-Nelson, P, Vousden, KH, Hubbert, NL, et al. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 1989; 8(12):3905–3910.
78. Bosch, FX, Burchell, AN, Schiffman, M, et al. Epidemiology and natural history of human papillomavirus infections and type-specific implications in cervical neoplasia. Vaccine. 2008; 26(Suppl 10):K1–16.
79. Frazer, IH. Interaction of human papillomaviruses with the host immune system: a well evolved relationship. Virology. 2009; 384(2):410–414.
80. Konya, J, Dillner, J. Immunity to oncogenic human papillomaviruses. Adv Cancer Res. 2001; 82:205–238.
81. Carter, JJ, Koutsky, LA, Hughes, JP, et al. Comparison of human papillomavirus types 16, 18, and 6 capsid antibody responses following incident infection. J Infect Dis. 2000; 181(6):1911–1919.
82. Villa, LL, Ault, KA, Giuliano, AR, et al. Immunologic responses following administration of a vaccine targeting human papillomavirus Types 6, 11, 16, and 18. Vaccine. 2006; 24(27–28):5571–5583.
83. Jochmus-Kudielka, I, Schneider, A, Braun, R, et al. Antibodies against the human papillomavirus type 16 early proteins in human sera: correlation of anti-E7 reactivity with cervical cancer. J Natl Cancer Inst. 1989; 81(22):1698–1704.
84. Lechner, F, Gruener, NH, Urbani, S, et al. CD8+ T lymphocyte responses are induced during acute hepatitis C virus infection but are not sustained. Eur J Immunol. 2000; 30(9):2479–2487.
85. Sester, M, Sester, U, Gartner, BC, et al. Dominance of virus-specific CD8 T cells in human primary cytomegalovirus infection. J Am Soc Nephrol. 2002; 13(10):2577–2584.
86. Youde, SJ, Dunbar, PR, Evans, EM, et al. Use of fluorogenic histocompatibility leukocyte antigen-A*0201/HPV 16 E7 peptide complexes to isolate rare human cytotoxic T-lymphocyte- recognizing endogenous human papillomavirus antigens. Cancer Res. 2000; 60(2):365–371.
87. Kadish, AS, Timmins, P, Wang, Y, et al. Regression of cervical intraepithelial neoplasia and loss of human papillomavirus (HPV) infection is associated with cell-mediated immune responses to an HPV type 16 E7 peptide. Cancer Epidemiol Biomarkers Prev. 2002; 11(5):483–488.
88. Nimako, M, Flander, AN, Wilkinson, GW, et al. Human papillomavirus-specific cytotoxic T lymphocytes in patients with cervical intraepithelial neoplasia grade III. Cancer Res. 1997; 57(21):4855–4861.
89. van der Burg, SH, Ressing, ME, Kwappenberg, KM, et al. Natural T-helper immunity against human papillomavirus type 16 (HPV16) E7-derived peptide epitopes in patients with HPV16-positive cervical lesions: identification of 3 human leukocyte antigen class II-restricted epitopes. Int J Cancer. 2001; 91(5):612–618.
90. Bontkes, HJ, de Gruijl, TD, van den Muysenberg, AJ, et al. Human papillomavirus type 16 E6/E7-specific cytotoxic T lymphocytes in women with cervical neoplasia. Int J Cancer. 2000; 88(1):92–98.
91. Campbell, DJ, Butcher, EC. Rapid acquisition of tissue-specific homing phenotypes by CD4(+) T cells activated in cutaneous or mucosal lymphoid tissues. J Exp Med. 2002; 195(1):135–141.
92. Kupper, TS, Fuhlbrigge, RC. Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol. 2004; 4(3):211–222.
93. Clark, RA, Chong, B, Mirchandani, N. The vast majority of CLA+ T cells are resident in normal skin. J Immunol. 2006; 76(7):4431–4439.
94. Koelle, DM, Liu, Z, McClurkan, CM, et al. Expression of cutaneous lymphocyte-associated antigen by CD8(+) T cells specific for a skin-tropic virus. J Clin Invest. 2002; 110(4):537–548.
95. Moyer, VA. Screening for cervical cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2012; 156(12):880–891.
96. Follen, M, Richards-Kortum, R. Emerging technologies and cervical cancer. J Natl Cancer Inst. 2000; 92(5):363–365.
97. Castle, PE, Glass, AG, Rush, BB, et al. Clinical human papillomavirus detection forecasts cervical cancer risk in women over 18 years of follow-up. J Clin Oncol. 2012; 30(25):3044–3050.
98. Smith, MA, Lew, JB, Walker, RJ, et al. The predicted impact of HPV vaccination on male infections and male HPV-related cancers in Australia. Vaccine. 2011; 29(48):9112–9122.
99. Ioannidis, V, Beermann, F, Clevers, H, Held, W. The beta-catenin—TCF-1 pathway ensures CD4(+)CD8(+) thymocyte survival. Nat Immunol. 2001; 2(8):691–697.
100. Frazer, IH. Prevention of cervical cancer through papillomavirus vaccination. Nat Rev Immunol. 2004; 4(1):46–54.
101. Chattergoon, M, Boyer, J, Weiner, DB. Genetic immunization: a new era in vaccines and immune therapeutics. FASEB J. 1997; 11(10):753–763.
102. Aihara, H, Miyazaki, J. Gene transfer into muscle by electroporation in vivo. Nat Biotechnol. 1998; 16(9):867–870.
103. Luckay, A, Sidhu, MK, Kjeken, R, et al. Effect of plasmid DNA vaccine design and in vivo electroporation on the resulting vaccine-specific immune responses in rhesus macaques. J Virol. 2007; 81(10):5257–5269.
104. van Drunen Littel-van den Hurk, Hannaman, D. Electroporation for DNA immunization: clinical application. Expert Rev Vaccines. 2010; 9(5):503–517.
105. Hanke, T, Goonetilleke, N, McMichael, AJ, Dorrell, L. Clinical experience with plasmid DNA- and modified vaccinia virus Ankara-vectored human immunodeficiency virus type 1 clade A vaccine focusing on T-cell induction. J Gen Virol. 2007; 88(Pt 1):1–12.
106. Kenter, GG, Welters, MJ, Valentijn, AR, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009; 361(19):1838–1847.
107. Coleman, N, Birley, HD, Renton, AM, et al. Immunological events in regressing genital warts. Am J Clin Pathol. 1994; 102(6):768–774.
108. Trimble, CL, Peng, S, Thoburn, C, et al. Naturally occurring systemic immune responses to HPV antigens do not predict regression of CIN2/3. Cancer Immunol Immunother. 2010; 59(5):799–803.
109. Farhat, S, Nakagawa, M, Moscicki, AB. Cell-mediated immune responses to human papillomavirus 16 E6 and E7 antigens as measured by interferon gamma enzyme-linked immunospot in women with cleared or persistent human papillomavirus infection. Int J Gynecol Cancer. 2009; 19(4):508–512.
110. Nakagawa, M, Stites, DP, Patel, S, et al. Persistence of human papillomavirus type 16 infection is associated with lack of cytotoxic T lymphocyte response to the E6 antigens. J Infect Dis. 2000; 182(2):595–598.
111. Frazer, IH, Leggatt, GR, Mattarollo, SR. Prevention and treatment of papillomavirus-related cancers through immunization. Annu Rev Immunol. 2011; 29:111–138.
112. Daayana, S, Elkord, E, Winters, U, et al. Phase II trial of imiquimod and HPV therapeutic vaccination in patients with vulval intraepithelial neoplasia. Br J Cancer. 2010; 102(7):1129–1136.
113. Kobayashi, A, Weinberg, V, Darragh, T, Smith-McCune, K. Evolving immunosuppressive microenvironment during human cervical carcinogenesis. Mucosal Immunol. 2008; 1(5):412–420.
114. Nakamura, T, Shima, T, Saeki, A, et al. Expression of indoleamine 2, 3-dioxygenase and the recruitment of Foxp3-expressing regulatory T cells in the development and progression of uterine cervical cancer. Cancer Sci. 2007; 98(6):874–881.
115. El-Sherif, AM, Seth, R, Tighe, PJ, Jenkins, D. Quantitative analysis of IL-10 and IFN-gamma mRNA levels in normal cervix and human papillomavirus type 16 associated cervical precancer. J Pathol. 2001; 195(2):179–185.
116. Lanneau, GS, Argenta, PA, Lanneau, MS, et al. Vulvar cancer in young women: demographic features and outcome evaluation. Am J Obstet Gynecol. 2009; 200(6):645 e1–5.
117. Frazer, IH. Measuring serum antibody to human papillomavirus following infection or vaccination. Gynecol Oncol. 2010; 118(1 Suppl):S8–11.
118. Wright, TC, Jr., Schiffman, M. Adding a test for human papillomavirus DNA to cervical-cancer screening. N Engl J Med. 2003; 348(6):489–490.
119. Ferlay, JB, Pisani, FG, Parkin, DM. GLOBOCAN 2000: Cancer incidence, mortality and prevalence worldwide. IARC Cancer Base No.5. 2006.
120. Clifford, GM, Gallus, S, Herrero, R, et al. Worldwide distribution of human papillomavirus types in cytologically normal women in the International Agency for Research on Cancer HPV prevalence surveys: a pooled analysis. Lancet. 2005; 366(9490):991–998.
121. Doorbar, J. The papillomavirus life cycle. J Clin Virol. 2005; 32(Suppl 1):S7–15.