Chapter 5 Biologics and Their Interactions with Radiation
Over the past decade, radiation oncology researchers have rapidly developed the capability of delivering “targeted” therapy with the evolution of intensity-modulated radiation therapy (IMRT) and incorporation of image-guided motion tracking. During this same decade, advances in molecular pathology have resulted in powerful predictive biomarkers such as KRAS and BRAF that have ushered in a new era in cancer therapy, that of personalized cancer therapeutics. Improved understanding of the molecular mechanisms underlying malignant processes has allowed the development of novel therapeutic agents that target specific cellular processes. Molecularly targeted therapeutic agents, or biologics, is a class of agents that have been developed to specifically interfere with functions central to the pathophysiology of the malignant phenotype. Several biologic modifiers have now been approved for clinical use (Table 5-1).
Epidermal Growth Factor Receptor Family Inhibitors
Epidermal Growth Factor Receptor Family Biology
The epidermal growth factor receptor (EGFR) family regulates mesenchymal-epithelial interactions during growth and development, transmitting extracellular cues to intracellular signaling cascades.1–3 The family has four known members: EGFR, HER2 (erbB2), HER3 (erbB3), and HER4 (erbB4). These membrane-spanning tyrosine kinase receptors contain an extracellular ligand-binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain. These normally quiescent receptors are activated when ligand binds to the extracellular domain of a receptor monomer. Ligand binding induces a structural change that favors dimerization with the same member (homodimer) or a different member (heterodimer) of the family. When dimerized, the tyrosine kinase domains are activated and they phosphorylate key tyrosine residues in the intracellular domain, resulting in activation of several downstream signaling cascades.
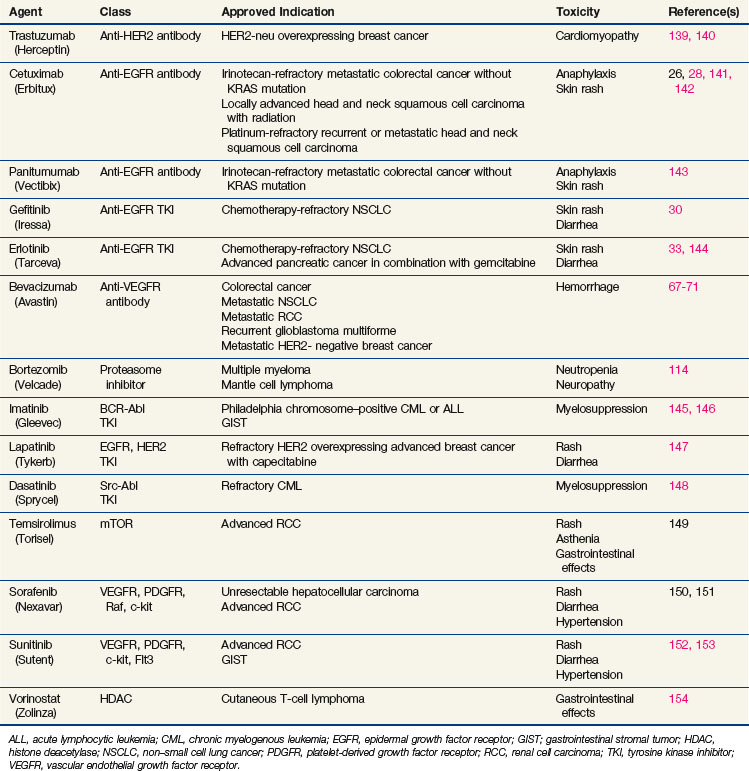
The end result of receptor activation, proliferation, differentiation, migration, or survival signaling depends on many factors, including which receptor pairs are formed and for how long they are activated.4 This in turn depends on which receptors are predominantly present in the cell and which ligand is involved in activation. There are two ligand families that activate the EGFR family receptors, the EGF-like and heregulin families.5,6 The EGF-like family includes EGF, TGF-α, amphiregulin, betacellulin, and HB-EGF. The heregulin (neuregulin) family includes many proteins resulting from splice variations of two different genes, all designated as heregulin with different subtypes. The ligands exhibit preference for particular receptors and induce different receptor combinations (Fig. 5-1A). HER2 has no known ligand. Instead, HER2 is the favored partner of the other receptors when ligand binds to either EGFR, HER3, or HER4.7 This complex interplay of receptors is important for understanding and interpreting the effect of an inhibitor of a single member of the family.
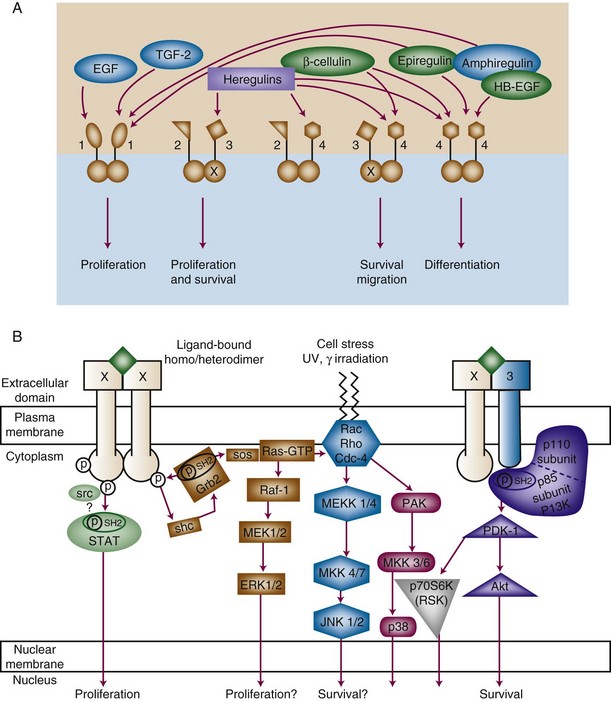
The effect of receptor activation also depends on which downstream signals are activated. EGFR family members signal via a diverse network of signal transduction pathways, including the protein kinase C (PKC), Ras-Raf-ERK, PI3K-Akt, and STAT pathways8 (Figure 5-1B). Furthermore, various receptor pairs recruit different downstream effectors. For instance, HER3 contains multiple PI3K-binding motifs, resulting in strong signaling via PI3K, which plays a role in cell survival, invasion, and proliferation. Interestingly, HER3 alone among the receptors has an inefficient kinase domain, requiring heterodimerization with other family members to become phosphorylated. The need for heterodimerization juxtaposes the PI3K signal emanating from HER3 with the Ras-Raf-ERK or STAT signal emanating from EGFR, HER2, or HER4.
EGFR Family and Tumor Pathogenesis
In 1986, the Nobel Prize was awarded to Stanley Cohen for the discovery of growth factors, resulting from his work in identifying EGF and its receptor.9 EGFR was first identified as a proto-oncogene because of its homology to the avian erythroblastosis (v-erb) oncogene.10 Aberrant function of EGFR or HER2 occurs frequently in human tumors; gene amplification results in massive overexpression in a proportion of gliomas and breast cancers.11–13 Alternatively, dysregulation occurs at more modest levels of expression when the receptor is activated as the result of autocrine stimulation, in which the tumor produces its own ligand to activate the receptor. This type of dysregulation occurs frequently in cancers of the head and neck, gastrointestinal system, and prostate gland.14–17 Another mechanism of dysregulation is the development of mutations in the kinase domain that render the kinase activity more potent, most clearly demonstrated in lung cancer.18 Likewise, mutations in the ligand-binding domain can cause the receptor to be constitutively active even in the absence of ligand, as occurs in a significant proportion of gliomas.19 Dysregulation via mechanisms other than amplification may not always result in overexpression as detected by standard immunohistochemical techniques, raising the issue of how best to identify all tumors in which EGFR dysregulation promotes tumor proliferation and resistance to therapy.
EGFR Family Inhibitors
The frequent dysregulation of the EGFR family in tumors makes the family an attractive target for exploitation. Remarkable progress has been made in the development of EGFR family inhibitors.20 Antibodies directed against the extracellular domains of EGFR and HER2 and small-molecule tyrosine kinase inhibitors have both been approved by the Food and Drug Administration (FDA) for clinical application, and several more are in various stages of development.21 Table 5-2 lists selected examples of EGFR family inhibitors that have undergone clinical testing.
| Generic and Trade Names of EGFR Inhibitors | Type of Inhibitor | EGFR Targets |
|---|---|---|
| Cetuximab (Erbitux,C225) | Antibody | EGFR |
| Trastuzumab (Herceptin) | Antibody | HER2 |
| Panitumumab (Vectibix, ABX-EGF) | Antibody | EGFR |
| Necitimumab (IMC-11F8) | Antibody | EGFR |
| Zalutumumab | Antibody | EGFR |
| Nimotuzumab (hR3) | Antibody | EGFR |
| MAb-425 | Antibody | EGFR |
| RO5083945 | Antibody | EGFR |
| Gefitinib (Iressa, ZD1839) | TKI | EGFR |
| Erlotinib HCl (Tarceva, OSI-774) | TKI | EGFR |
| Lapatinib (Tykerb, GW572016) | TKI | EGFR/HER2 |
| Pazopanib (GW786034) | TKI | EGFR |
| PKI-166 | TKI | EGFR/HER2 |
| CI-1033 | TKI | EGFR/HER2/HER3/HER4 |
| BIBW 2992 | TKI | EGFR |
| AVI-412 | TKI | EGFR/HER2 |
| CUDC-101 | TKI | EGFR/HER2/HDAC |
EGFR, epidermal growth factor receptor.
The first EGFR family–targeted agent to be approved for clinical use was the HER2-specific antibody trastuzumab (Herceptin). Trastuzumab binds to the extracellular domain of HER2, enhancing receptor down-regulation, but its major benefit may result from immune-mediated cytotoxicity, perhaps by “tagging” HER2 overexpressing cells.22,23 Trastuzumab is approved as single-agent therapy or in combination with paclitaxel for metastatic, HER2-overexpressing breast cancer and as adjuvant therapy for localized HER2-overexpressing breast cancer. Trastuzumab is only effective when HER2 is highly overexpressed because of gene amplification; it is not indicated for treatment of tumors that do not overexpress HER2.
The primary toxicity of trastuzumab was unanticipated. Initial studies demonstrated low toxicity rates, but when trastuzumab was investigated in combination with anthracycline-based chemotherapy, an unexpectedly high incidence of congestive heart failure was observed.24 Subsequent studies have not clearly elucidated the mechanism, but one hypothesis is that HER2 may be involved in recovery from anthracycline-induced cardiac stress. Although trastuzumab is commonly used with irradiation, there is little information regarding the efficacy of this combination compared with that of irradiation alone in patients with breast cancer.
Cetuximab (Erbitux), an anti-EGFR monoclonal antibody that binds to the extracellular domain of EGFR, interferes with ligand binding and, hence, dimerization and activation.25 Like trastuzumab, cetuximab has modest activity as a single agent but gives more impressive results when it is combined with cytotoxic therapy. Cetuximab is given intravenously on a weekly schedule. When combined with radiation therapy, a loading dose is given the week before initiation of radiation treatment. Cetuximab has gained FDA approval for use in treating colorectal cancer and head and neck cancer. Initial approval for cetuximab in treating metastatic colorectal cancer was based largely on the results of a trial including 329 patients randomized to receive either cetuximab and irinotecan or cetuximab alone.26 The response rates were higher with the combination therapy than with the monotherapy, as was median time to progression (4.1 months vs. 1.5 months) and median survival time (8.6 months vs. 6.9 months). Further molecular analysis demonstrated that patients with KRAS mutations in codons 12 or 13 do not respond to cetuximab27; this is presumably related to alternate activation of signal transduction pathways. This finding underscores the complexity of cancer biogenetics and marks an important turning point toward an era of personalized cancer therapy.
The benefit of combining cetuximab and conventional systemic therapy has also been demonstrated in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. The EXTREME trial confirmed results of previous phase II trials with survival benefits seen in this cohort of patients with combination cetuximab and cisplatin therapy or carboplatin-based chemotherapy.28 Cetuximab has also demonstrated activity, albeit quite modest activity, in advanced non–small cell lung cancer (NSCLC), which, like head and neck squamous cell carcinoma and colorectal cancer, commonly overexpresses EGFR. The FLEX trial included 1125 patients with metastatic or pleural effusion cytology–positive NSCLC expressing EGFR. Patients were randomized to cisplatin 80 mg/m2 and vinorelbine 25 mg/m2 on days 1 and 8 with cycles delivered every three weeks for up to six cycles with or without cetuximab. Patients who received chemotherapy with cetuximab lived a modestly short time longer than patients who received chemotherapy alone (median, 11.3 months vs. 10.1 months; hazard ratio for death, 0.87).29
Gefitinib is approved for use in locally advanced or metastatic NSCLC after failure of both platinum-based and docetaxel chemotherapy, based on an objective response rate of 10.6% with a median response duration of 7 months in a setting where no drug has demonstrated efficacy.30 Combined therapy with cytotoxic chemotherapy has not been shown to be better than monotherapy.31,32 The decision to approve gefitinib was based in part on significant response rates in a small subset of patients. Further investigation elucidated that these patients’ tumors contained mutations in the kinase domain of the EGFR, rendering them highly susceptible to inhibition by gefitinib.18,19 The mutations were found predominantly in female nonsmokers with adenocarcinoma. It remains to be proven whether screening for kinase domain mutations will be an effective means of selecting patients likely to benefit from gefitinib.
Erlotinib is approved for the treatment of locally advanced NSCLC after failure of at least one prior chemotherapy regimen and for treatment of unresectable pancreatic cancer when given in combination with gemcitabine. Initial approval for treatment of NSCLC was based on the results of a randomized trial comparing erlotinib with placebo. The median overall survival time was 6.7 months in the erlotinib group compared with 4.7 months in the placebo group (p <.001). The 1-year survival rate was 31.2% with erlotinib versus 21.5% with placebo (p <.001), with a median response duration of 34 weeks versus 16 weeks.33 Symptomatic improvement was also demonstrated in a phase II trial.34 Conversely, no clinical benefit was observed in another trial testing the efficacy of erlotinib with platinum-based chemotherapy.35
EGFR Family and Radiation Response
EGFR family members play an important role in radiation response. Preclinical studies showed that cells made to express v-erb were rendered radio resistant.36 Similarly, breast cancer cell lines become more radioresistant when made to overexpress HER2.37 Likewise, radioresistance correlates with EGFR expression levels in head and neck cancer cells.38–40
Clinical studies also suggest that EGFR family dysregulation influences radiation response. A study of 170 gliomas treated with primary radiotherapy demonstrated lower response rates in tumors that overexpressed EGFR; the response rate was 33% in EGFR-negative tumors, 18% in EGFR-intermediate tumors, and 9% in EGFR-positive tumors.41 In smaller series of head and neck cancer patients, locoregional recurrence after radiotherapy was associated with EGFR overexpression.42,43 In breast cancer, a case-control series of patients with in-breast tumor recurrence after breast-conserving surgery and radiotherapy found that the proportion of patients with HER2 overexpression was higher in the recurrence group than in the controls.44
Preclinical Studies of EGFR Family Inhibitors as Radiosensitizers
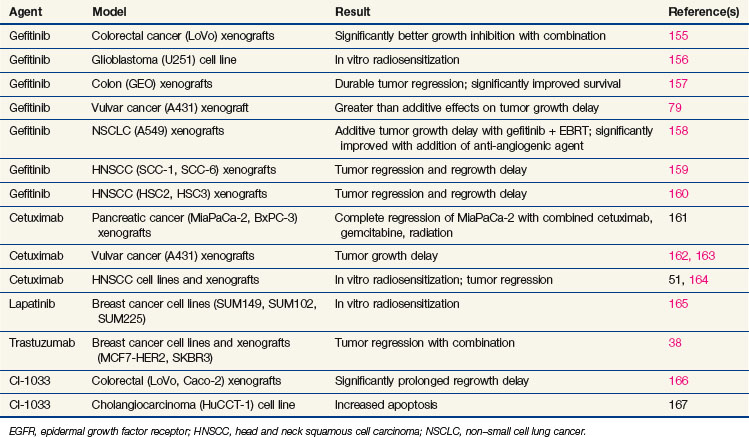
The role of EGFR family members in radiation response was further clarified by studies using newly developed EGFR family inhibitors (Table 5-3). In virtually every study, EGFR or HER2 inhibitors demonstrated modest radiosensitization.45–47 Radiosensitization is more pronounced in vivo than in vitro and with fractionated-dose than with single-dose irradiation. The understanding of the mechanisms underlying enhanced radiosensitization is evolving.48–50 In every case, the combination of EGFR or HER2 inhibitors and radiation resulted in increased cell cycle arrest, predominately in the G1 phase, but with a substantial decrease in S phase, which translates in vivo to decreased proliferation. Radiosensitization with EGFR family inhibitors also causes decreased angiogenesis. It is not yet clear whether this is an additive result of combined anti-angiogenesis effects from both radiation and EGFR inhibitors, or whether the EGFR inhibitors further increase the susceptibility of the vascular elements of the tumor to radiation. The combination of EGFR inhibitors and radiation increases apoptosis in some, but not all, models. Finally, EGFR inhibitors may directly interfere with repair of radiation-induced DNA damage from DNA-PK inhibition.51,52
Clinical Studies of EGFR Family Inhibitors as Radiosensitizers
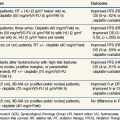
Promising preclinical studies with EGFR family inhibitors have translated into improved patient outcomes in randomized controlled trials. The most mature is a phase III trial that compared the efficacy of standard radiotherapy with standard radiotherapy plus the anti-EGFR antibody cetuximab (Erbitux). In this study, 424 patients with stage III or IV squamous cell carcinoma of the oropharynx, hypopharynx, or larynx were stratified by T stage, nodal status, and performance score and then were randomly assigned to receive radiotherapy alone or radiotherapy plus weekly, concurrent cetuximab. Radiotherapy was delivered using one of three fractionation regimens (stratified): a once-daily regimen (2 Gy × 35 fractions over 7 weeks), a twice-daily regimen (1.2 Gy × 60 to 64 fractions over 5 to 5.5 weeks), or a concomitant-boost regimen (1.8 Gy × 30 fractions with a second daily fraction of 1.5 Gy for the last 12 treatment days over 6 weeks). Concurrent chemotherapy was not allowed. Two-year local control was 56% in the radiotherapy-plus-cetuximab arm versus 48% in the radiotherapy-alone arm, with a median duration of local control of 36 months versus 19 months, respectively (p = .02).53 Overall survival was also significantly enhanced with combined therapy. A recent trial update reported 45.6% 5-year overall survival in the radiotherapy-plus-cetuximab arm versus 36.4% in the radiotherapy-alone arm, with median survival of 49 months versus 29 months, respectively (p = .018).54 The improvement in outcome was associated with an increase in acute skin, but not mucosal, toxicity. Furthermore, there were no differences between the groups on standardized quality of life assessment scores.55 Interestingly, a more severe (grade 2 or greater) acneiform rash actually correlated with better survival rates. Although they do not necessarily define the appropriateness of adding cetuximab to the current standard of chemoradiotherapy (as we await results from RTOG 0522) for head and neck cancer, the results of this trial prove that EGFR inhibitors can effectively radiosensitize at least a subset of head and neck cancers. Results of RTOG 0234, a phase II randomized trial comparing radiation plus cetuximab and either weekly cisplatin or weekly docetaxel, suggest that both are feasible regimens with outcomes superior to results from RTOG 9501 that used high-dose cisplatin on days 1, 22, and 43.56 Grade 3 to 4 myelosuppression was observed in 28% (cisplatin) and 14% (docetaxel) of patients. Dermatitis was seen in 39% of patients in each group. The rates of grade 3 or higher mucositis were 37% and 33% in the cisplatin and docetaxel arms; these rates seem somewhat low but are encouraging compared with historical controls. The 2-year distant metastasis rate was 13% in the group that received cetuximab and docetaxel versus around 26% with cisplatin and cetuximab. One of the conclusions from this trial is that perturbing growth factor signaling may allow us, under the right circumstances, to reduce administration of high doses of standard chemotherapy and reduce morbidity to the patient. Further work is needed to begin to cull out which patients benefit from targeting a particular pathway such as epidermal growth factor signaling and which patients, based on specific mutations, may need interference with Akt, mTOR, or DNA repair pathways instead.
Although concern for unwanted normal tissue effects when combining targeted drugs against EGFR and radiation therapy is justifiable, the additional morbidity of this approach appears minimal based on the experiences in the clinic to date. Concurrent administration of adjuvant radiotherapy with trastuzumab in patients with early-stage breast cancer does not increase the incidence of acute radiation toxicity.57 At a median follow-up of 3.7 years, there were no significant differences in the incidence of skin reaction, pneumonitis, dyspnea, cough, dysphagia, or neutropenia between those patients who received adjuvant radiation with or without trastuzumab concurrently. There was a higher incidence of leukopenia in the arm that received trastuzumab and radiotherapy. Radiotherapy did not increase the frequency of cardiac events, and there was no difference in cardiac events between left-sided and right-sided tumors. These results must be interpreted cautiously until more clinical data are accumulated. Longer follow-up is also warranted to evaluate whether acute reversible reactions could be predictive for late sequelae. Multiple clinical trials testing EGFR family inhibitors as radiosensitizers are ongoing. Results of these trials are eagerly awaited.
Angiogenesis Inhibitors
Angiogenesis and Tumor Pathogenesis
All tumors require development (or expansion) of blood vessels to promote further tumor growth and nutritional support beyond a 2-mm diameter.58 Molecules such as vascular endothelial growth factor (VEGF) mediate stimulation of angiogenic signaling and neovascularization. Elevated levels of specific isoforms of VEGF and other indirect markers predict for a worse prognosis in many types of cancer, including those of the gastrointestinal tract such as pancreatic and esophageal cancers.59,60 VEGF expression is affected by both the genetic aberrancies of the particular cancer as well as the microenvironmental changes, including hypoxia.
Angiogenesis Inhibitors
Currently, there are two VEGFR-TKIs that have gained FDA approval. Sorafenib (Nexavar) and sunitinib (Sutent) are indicated for treatment of patients with unresectable hepatocellular carcinoma, advanced renal cell carcinoma, and gastrointestinal stromal tumors based on positive results of phase III randomized studies. ZD6474 (Vandetanib) is one of several VEGFR-TKIs that appeared to hold early promise; however, phase III trials in stage IV lung cancer were disappointing. ZD6474 has dual anti-EGFR and anti-VEGFR properties as well as activity against RET kinase.58 This particular drug may have a niche role in the treatment of medullary thyroid cancers based on its activity against RET kinase mutations.61,62
Agents that target the microtubule formation of intratumoral vasculature, thus destabilizing vessels and causing rapid necrosis within the central part of the tumor, might be effective compounds to combine with radiation. The rationale for pursuing agents that attack intratumoral vessels is based on the premise that endothelial cells in tumors display a different growth pattern than those in normal tissues; it is a chaotic, rapidly expanding pattern. VTAs were developed to take advantage of this differential to selectively occlude or destroy tumor blood vessels, and the validity of this approach was demonstrated a decade ago.63 In general, two classes of agents are used to target the tumor vasculature: (1) VTAs, which use ligand-directed strategies to deliver toxins, for example, to the intratumoral vasculature, and (2) small-molecule VTAs, which exploit the differences between the disorganized immature vessels within the tumor and the surrounding mature endothelial matrix. Either approach is designed to establish an occlusive, ischemic pattern within tumors within a short time period (<24 hr), resulting in extensive necrosis within the central core of the tumor. Several studies have confirmed the validity of this targeting strategy. ZD6126, a prodrug of N-acetylcolchinol, binds beta tubulin, resulting in tumor vessel occlusion and central necrosis in a variety of human tumor models.64,65 Most studies evaluating VTAs and radiation have been in the preclinical setting. Little progress has been achieved in clinical trials with VTAs, in part because of issues related to cardiac toxicity; however, this still remains a potentially promising approach.
An alternative approach, inhibiting vascular endothelial growth factor (VEGF) using an anti-VEGF monoclonal antibody is akin to scooping up the keys rather than blocking the lock. Antibodies against VEGF have shown anticancer activity in preclinical colorectal models.66 Bevacizamab (Avastin) is a recombinant humanized version of the murine anti-human VEGF monoclonal antibody rhuMAb VEGF. Bevacizumab is FDA-approved for use in patients with metastatic colorectal cancer when used in combination with 5-fluorouracil (5-FU). Two separate clinical trials demonstrated superior response rates, progression-free survival (PFS), and overall survival in patients treated with combined 5-FU–based therapy with bevacizumab compared with others treated with 5-FU–based therapy alone in either the first- or second-line setting.67,68 The addition of bevacizumab to carboplatin and paclitaxel also improved overall survival in chemotherapy-naïve patients with metastatic or recurrent, nonsquamous NSCLC69 and has been approved for use in patients with recurrent glioblastoma multiforme (GBM), based on trials demonstrating good rates of radiographic response and stable to decreased corticosteroid requirement.70 Improvements in PFS have also been demonstrated in patients with renal cell carcinoma and breast cancer receiving combinations of traditional systemic therapy and bevacizumab.71 These promising clinical results have led to further investigation with bevacizumab and other anti-angiogenic agents in various clinical populations and settings.
Angiogenesis Inhibitors as Radiosensitizers
Because hypoxia induces pro-angiogenic factors that contribute in part to radioresistance, is it rational to inhibit angiogenic signaling to enhance radiotherapeutic response? Early work by Teicher and colleagues72 demonstrated enhanced radiation response to TNP-470, an anti-angiogenic compound. Later studies designed to inhibit angiogenic signaling in the surrounding endothelium confirmed the results of Teicher, improving radiation cytotoxic effects in a variety of models.73–75 Recent comprehensive reviews of combination anti-angiogenic inhibitors and radiation provide pertinent information regarding the different strategies under investigation.76,77 Anti-angiogenic agents may enhance the effect of radiation by stabilizing tumor vasculature,78 thereby enhancing tumor oxygenation. By inhibiting pro-angiogenic signaling, regulation of anti-apoptotic proteins such as amplified NFκB may be improved. Many of these molecules, including VEGF, are activated by radiation, so it would seem logical to reverse this process. This in turn may prevent development of radioresistance.
The VTAs leave a viable oxic rim of tumor remaining in the periphery. This surviving rim of tumor provides a rationale for combining these types of agents with radiation. In combination with radiation, cooperative effects were reported.79–81 PTK787/ZK 222584 induced marked tumor growth inhibition in radiation-resistant p53-dysfuctional SW480 colon adenocarcinoma xenografts when combined with ionizing radiation.82 SU6668 inhibited tumor growth alone in C3H mice bearing SCC VII carcinomas known to express VEGF, FGF, PDGF, and their cognate receptors and enhanced the efficacy of irradiation.75 ZD6474 sequencing with radiation was investigated in a xenograft model bearing EGFR-TKI–insensitive NSCLC Calu-6 tumors.83 Two combined treatment schedules were examined: (1) a concurrent schedule using ZD6474 (50 mg/kg) dosing given 2 hours before the first dose of radiation, and (2) a sequential schedule using ZD6474 dosing given 30 minutes after the last dose of radiotherapy. The sequential approach was felt to be superior in terms of the time for treated tumors to quadruple in volume (RTV4) from their pretreatment size (p <.0001). Importantly, the reduced RTV4 (30 ± 1 day) in the concurrent schedule was also significantly better than either ZD6474 or radiation alone (p <.02). Current National Cancer Institute (NCI)–sponsored clinical trials84 with this compound and radiation include a phase II trial with temozolomide for patients with glioblastoma multiforme, several trials with radiation alone or with carboplatin/paclitaxel in patients with medically or surgically inoperable NSCLC, a phase I study with hypofractionated radiation in patients with recurrent gliomas, and a phase I trial with concurrent chemoradiation in patients with locally advanced esophageal cancer. The compound was being investigated in combination with weekly cisplatin and radiotherapy in a randomized phase II RTOG study (RTOG 0619) compared with high-dose cisplatin (100 mg/m2) and radiotherapy from arm 2 of RTOG 9501. However, the trial was closed to accrual because of changes in support from the sponsor. Agents that block VEGFR-2 selectively are also under clinical investigation. Cederinib (ZD2171) is being investigated in a randomized phase II setting within the RTOG (RTOG 0837) given as an adjuvant with temozolomide after chemoradiation for patients with high-grade gliomas.85
Similar to studies showing enhanced radiation effects with VEGFR signaling interference, studies combining anti-VEGF antibodies with radiation confirmed that although exposure of human tumor xenografts to radiation promoted induction of VEGF expression, inhibiting VEGF with anti-VEGF antibodies supplanted this effect and resulted in increased endothelial cell killing and synergized antitumor effects in murine tumor model systems.86 Several early-phase studies have been reported combining anti-angiogenics and radiation therapy.87–89,90–92 A multi-institutional phase III trial investigating postoperative temozolomide and radiation with or without bevacizumab is accruing for patients with newly diagnosed glioblastoma or gliosarcoma. Recent studies have suggested that bevacizumab may even reverse radiation necrosis in patients with central nervous system tumors.93
Are there biomarkers that might be helpful in determining response to anti-VEGF antibodies? In a phase I/II trial combining bevacizumab with chemoradiation for patients with locally advanced rectal cancer, plasma-soluble VEGFR-1 appeared to correlate with tumor regression and toxicity and may be used in future studies for stratification in anti-VEGF antibody studies with radiation.94
We continue to accumulate toxicity data with bevacizumab and radiation. This combination did not increase acute locoregional toxicity in breast cancer patients in comparison with matched control patients who received RT alone.95 Considering the importance of sequencing with respect to concerns about hypoxia when inhibiting angiogenesis, McGee and associates96 demonstrated in U87 glioma orthotopic models that bevacizumab delivered before radiation was more effective in improving oxygenation than when it was delivered during or after radiation. Importantly, the effect was transient, being lost after 5 days.
Each disease entity and site should be taken into consideration when evaluating angiogenic inhibitors with radiation. Serious concerns have emerged when combining bevacizumab with radiation or chemoradiation in patients with lung cancer. The FDA has recently issued warning statements based on final reports from two small trials that were halted, showing that bevacizumab and chemoradiotherapy were associated with a concerning incidence of tracheoesophageal fistula formation in both small cell lung cancer and NSCLC patients treated in 2006 and 2007.97 Provencio and colleagues98 discuss the current use of targeted agents with radiation on lung cancer in a recent review, providing the reader with a comprehensive discussion of current and future avenues.
Survival Factor Inhibitors: Focus on Nuclear Factor Kappa B (NFκB)
NFκB Biology
Nuclear factor kappa B (NFκB) is an anti-apoptotic transcription regulator constitutively active in many cancers and may be the principal cause of de novo treatment resistance. NFκB was first described as an important controller of immune and inflammatory responses, a property that likely stems from the ability of NFκB to inhibit apoptosis. Study of NFκB in malignancy found to be a potentially important inhibitor of apoptosis initiated by cancer therapies, including radiation. NFκB is a collection of dimers, both homodimers and heterodimers, comprised of a family of five subunits: p65 (RelA), RelB, c-Rel, p50, and p52.99 The heterodimer comprised of the p50 and p65 subunits has been implicated in the ability of NFκB to inhibit apoptosis resultant from therapeutic damage to cancer cells.
The pathway that regulates NFκB—not NFκB itself—comprises the potential therapeutic target. NFκB is likely present in all cells but is activated when released from a constitutively expressed repressor protein known as IκB (Fig. 5-2). IκB is targeted for degradation by the proteasome (the proteinase complex through which most cellular proteins are “recycled” into amino acids) on specific phosphorylation and subsequent polyubiquitylation.100 The phosphorylation of IκB is at least in part accomplished by a kinase complex, IκB kinase (IKK), which is formed of three subunits. On release from IκB, NFκB dimers translocate from the cytoplasm to the nucleus, where they bind a large number of promoter elements. Important genes that are up-regulated by NFκB are anti-apoptotic effectors, including Bcl-2 family members Bcl-xL and A1/Bfl-1, the inhibitors of apoptosis XIAP, c-IAP1 and c-IAP2, and the apical caspase inhibitors TRAF1 and TRAF2.101
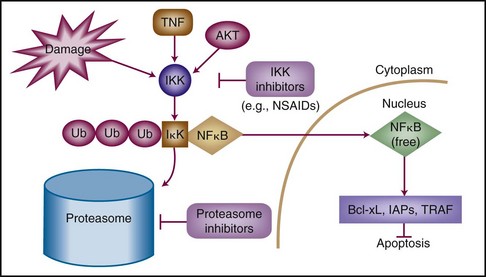
Although various stimuli can activate IKK to phosphorylate IκB and thereby activate NFκB, most of the mechanisms by which these stimuli activate IKK are still unknown. Additionally, some signals may activate NFκB independently of IKK, a finding with significant relevance in the development of NFκB-inhibiting drugs.102,103 Stimuli that can cause NFκB activation classically involved exposure to tumor necrosis factor–alpha (TNF-α), but they also include DNA damage (by ionizing radiation or chemotherapeutic agents) as well as signaling through the TNF-related apoptosis-inducing ligand pathway (TRAIL).
NFκB and Tumor Pathogenesis
Although there are no known mutations of members of the NFκB pathway in cancer, certain malignancies, particularly multiple myeloma, appear to be exquisitely dependent on NFκB activity. Myeloma and some other lymphoid malignancies are dozens to hundreds of times more sensitive to loss of NFκB function than their normal cellular counterparts.104–106 The reason for this dependence on NFκB activity is unclear but appears to reflect a distinction between normal cells and tumor cells.
Solid tumors may also be partially dependent on NFκB activation (particularly in comparison with most normal tissue) because of constitutive activation of signaling pathways that activate NFκB, such as Akt, which directly phosphorylates IKK. In a recent study of human gliomas, the activation status of Akt and NFκB was strongly correlated.107 Akt is frequently dysregulated in tumors by a variety of mechanisms. Chief among these is PTEN, a tumor suppressor that acts by preventing Akt activation via the phosphoinositide-3-kinase (PI3K) pathway. PTEN loss is common in malignancy and is seen with particular frequency in cancers of the prostate gland, breast, and colorectal region and in gliomas. Akt activation is also noted in cancers that contain RAS mutations or deregulated EGFR, as discussed above. PI3K itself has been described as being mutated in a potentially activating way in almost a third of colorectal cancers.108
NFκB Inhibitors
Specific inhibition of NFκB can be achieved via the introduction of an altered, nondegradable IκB molecule known as a “superrepressor” of NFκB. Introduction of superrepressor IκB into myeloma cells results in massive apoptosis. Proteasome inhibitors are another, albeit less selective, means of inhibiting NFκB. Inhibition of the proteasome by molecule inhibitors results in cellular accumulation of IκB by preventing its targeted degradation. The response of myeloma cells to proteasome inhibitors mimics that achieved with superrepressor IκB.104,109,110 COX inhibitors, thalidomide, and curcumin also are potential NFκB inhibitors.111–113
The proteasome inhibitor bortezomib (PS-341, Velcade Millenium Pharmaceuticals, Cambridge, Mass.) was approved as single-agent therapy for multiple myeloma based on striking activity in chemotherapy-refractory disease.114,115 Toxicities include modest cytopenias, diarrhea (or constipation), mild nausea, and occasionally neuropathy, particularly in patients with myeloma who have neuropathy at baseline.114 Trials investigating bortezomib in combination with cytotoxic chemotherapy are under way for a variety of solid tumors.
NFκB Inhibitors as Radiosensitizers
In solid tumor models, inhibition of NFκB (or the proteasome) alone results in little effect in many cell types; however, NFκB is strongly activated by certain therapeutic stimuli, including ionizing radiation. Inhibition of NFκB in conjunction with radiation has been shown preclinically to result at times in a dramatic increase in apoptosis in tumors relatively resistant to therapy. For example, glioma cells transfected with a superrepressor IκB were more radiosensitive than parental cell lines.116 Similarly, transfection with a superrepressor IκB is able to radiosensitize both HT29 and HCT15 colorectal cancer cells.117 The human immunodeficiency virus (HIV) protease inhibitor saquinavir, which also inhibits the proteasome, can radiosensitize cancer cell lines with demonstration of NFκB inhibition.118 In a LoVo colorectal cancer xenograft model, proteasome inhibition by bortezomib dramatically radiosensitized tumors.119
Radiosensitization trials of bortezomib are under way. Van Waes and colleagues120 at NCI have completed a phase I trial of bortezomib in conjunction with EBRT for squamous cell cancers of the head and neck. In that trial, patients were treated with doses of bortezomib ranging from 0.6 mg/m2 up to a maximally tolerated dose (MTD) of 0.9 mg/m2. In that study, increases in mucositis were not noted, but hyponatremia and dehydration were dose-limiting, possibly reflecting the patient population. Several additional phase I trials have been reported combining escalating doses of bortezomib with radiation or chemoradiation, including a once-weekly dosing with palliative radiation to gain a sense of interactions with radiation in different areas of the body.121 In patients with locally advanced rectal cancer, bortezomib was administered on days 1, 4, 8, and 11 every 21 days for two cycles, with 5-FU at 225 mg/m2/day continuously and 50.4 Gy of radiation. Bortezomib was given using a standard 3 + 3 dose escalation design. Diarrhea was the principal dose-limiting toxicity in the small cohort of patients entered into this trial (9 patients) and occurred at the 1.0-mg/m2 dose level. When feasible, patients underwent serial tumor biopsies for correlative studies. In contrast to preclinical findings, NFκB target gene expression was unaffected in these biopsy samples, so further work is needed to better understand just what targets (if any, at the doses administered) are being affected by bortezomib.122
Although proteasome inhibition by bortezomib is relatively well tolerated, the drug does have the relatively narrow therapeutic index of a chemotherapeutic agent, unlike the other biologic agents discussed above. In mice, whole-body ionizing radiation results in NFκB activation in a variety of tissues, including nodal and spleen tissues, but not in other normal tissues. However, NFκB may play a role in normal tissue radioprotection, as evidenced by studies indicating that normal intestinal mucosa is protected from radiation damage by NFκB,123 and that NFκB inhibition markedly increases epithelial cell apoptosis. At least in the case of the NCI trial studying head and neck tumors, there did not appear to be a significant increase in mucositis.
Future Directions
Several other classes of agents have not yet received FDA approval but have significant potential as radiosensitizers. The central role of signal transduction, DNA repair, and cell cycle control in radiation response leads to obvious interest in investigating agents that selectively perturb these processes in tumor cells as radiosensitizers.124,125
One of the most important pathways related to cancer cell survival, the PI3K/Akt/mTOR kinase pathway (Fig. 5-3) is a central regulator of cell metabolism, proliferation, and survival by preventing apoptosis. PI3K/Akt/mTOR is up-regulated in many tumors, including head and neck tumors.126 One relevant fact is that radiation can up-regulate many survival pathways, including activation of PI3K/Akt/mTOR, and, therefore, may be a prime target to inhibit during radiation therapy. Agents under development and in clinical trials to block this pathway include rapamycin analogs such as CCI-779 and RAD001, which block signal transduction downstream of mTOR and facilitate apoptosis, suggesting that rapamycin analogs might be effective as radiosensitizing agents. The radiosensitizing effects of mTOR inhibitors have been shown in a variety of preclinical cancer cell models.127 CCI-779 is as effective as cisplatin in radiosensitizing head and neck squamous cell carcinoma (HNSCC) lines, and triple-combination therapy did not provide additional cooperative effects over CCI-779 with radiation.128 The in vivo effects were more dramatic partly because of an anti-angiogenic by-product of mTOR inhibition. Temsirolimus is a rapamycin analog in clinical trials with radiation versus radiation and temozolomide in a phase II study through the EORTC (EORTC 26082) for patients lacking methylation of the MGMT promoter with gliomas. This study also incorporates maintenance temsirolimus versus temozolomide. Additional studies in lung cancer and in a preoperative approach with rectal cancer in phase I settings are also under way.
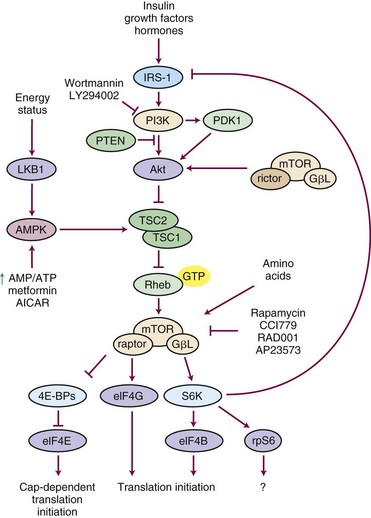
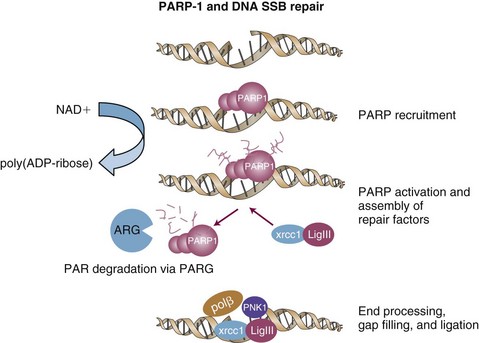
Another area of excitement and promise involves the use of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. The PARP family of proteins is responsible for posttranslational modification of proteins and plays an important role in single-strand homologous DNA repair (Fig. 5-4). PARP-1 is activated by base damage, single-strand DNA breaks, and double-strand DNA breaks caused by insults, including chemotherapy and ionizing radiation.129 PARP-1 also helps regulate cell cycle arrest after radiation.130 In addition, it has activity in repair of DNA double-strand breaks.131 Remarkable activity has been observed in patients with BRCA1, BRCA2 mutations in a phase I study using olaparib (AZD2281), an orally bioavailable PARP inhibitor.132 Importantly, minimal toxicity was observed in doses up to 600 mg twice a day. Objective antitumor activity was reported only in mutation carriers (22/60 patients entered, all of whom had refractory ovarian, breast, or prostate cancer). Preclinical studies have demonstrated cooperative effects with PARP inhibitors and chemotherapy in models such as breast cancer133 and with radiation in models such as glioma and lung cancer.134 The RTOG is planning a clinical trial combining PARP inhibitors with radiation and temozolomide for patients with gliomas. In addition, clinical trials are under development with radiation in head and neck cancer and lung cancer.
Insulin growth factor signaling has been implicated as an important prognostic survival biomarker in patients with breast cancer.135 Inhibiting this kinase pathway, associated with the regulation of cancer cell growth, transformation, and anti-apoptosis, may be another way of bringing about a differential improvement in cancer cell kill. Emerging evidence suggests it may interact favorably with radiation using anti-IGFR antibodies; however, it is important to discover whether this approach makes sense for all cancers or if activity relates at all to IGFR expression. The University of Wisconsin group136 has worked with the A12 anti-IGFR antibody that has been under development over the last decade and has shown activity in a variety of models. In vivo, A12 antibody inhibited in a dose-dependent manner xenograft growth in NSCLC lines. In the H460 xenograft model, combining A12 and radiation significantly enhanced antitumor efficacy compared with either modality alone. It was suggested that these cooperative effects might be mediated by promotion of radiation-induced, double-stranded DNA damage and apoptosis.137 Iwasa and associates138 reported on the effectiveness of an anti-IGFR antibody (CP-751,871) to increase the sensitivity of NSCLC cells to ionizing radiation in standard clonogenic survival assays. CP-751,871 inhibited radiation-induced IGF-IR signaling and increased caspase-3 expression as well as histone gamma-H2AX and Rad51, related to DNA double-strand breaks. If the underlying mechanism for action is related to DNA repair inhibition then perhaps DNA repair inhibitors might have wider applicability. It would be interesting to evaluate the relationship or correlation of IGFR expression and PARP expression or BRCA mutations. Under discussion with CTEP and the RTOG is a study evaluating the A12 antibody with radiation and possibly cetuximab for patients with human papillomavirus (HPV)–negative head and neck cancers.
1 Earp HS3rd, Calvo BF, Sartor CI. The EGF receptor family—multiple roles in proliferation, differentiation, and neoplasia with an emphasis on HER4. Trans Am Clin Climatol Assoc. 2007;114:315-333. discussion 333-334
2 Yarden Y. The EGFR family and its ligands in human cancer. Signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2004;37(Suppl 4):S3-S8.
3 Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203-212.
4 Klapper LN, Kirschbaum MH, Sela M, Yarden Y. Biochemical and clinical implications of the ErbB/HER signaling network of growth factor receptors. Adv Cancer Res. 2000;77:25-79.
5 Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network. Receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159-3167.
6 Riese DJ2nd, Stern DF. Specificity within the EGF family/ErbB receptor family signaling network. Bioessays. 1998;20:41-48.
7 Klapper LN, Glathe S, Vaisman N, et al. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc Natl Acad Sci U S A. 1999;96:4995-5000.
8 Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211-225.
9 Cohen S, Carpenter G, King LJr. Epidermal growth factor-receptor-protein kinase interactions. Co-purification of receptor and epidermal growth factor-enhanced phosphorylation activity. J Biol Chem. 1980;255:4834-4842.
10 Downward J, Yarden Y, Mayes E, et al. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature. 1984;307:521-527.
11 Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A. 1982;89:4309-4313.
12 Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer. Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177-1782.
13 Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma. A population-based study. Cancer Res. 2004;64:6892-6899.
14 Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53:3579-3584.
15 Yamanaka Y, Friess H, Kobrin MS, et al. Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressiveness. Anticancer Res. 1993;13:565-569.
16 Spano JP, Lagorce C, Atlan D, et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann Oncol. 2005;16:102-108.
17 Scher HI, Sarkis A, Reuter V, et al. Changing pattern of expression of the epidermal growth factor receptor and transforming growth factor alpha in the progression of prostatic neoplasms. Clin Cancer Res. 1995;1:545-550.
18 Lynch TJ, Bell DW, Sordelia R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129-2139.
19 Wong AJ, Ruppert JM, Bigner SH, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992;89:2965-2969.
20 Arteaga C. Targeting HER1/EGFR. A molecular approach to cancer therapy. Semin Oncol. 2003;30(3 Suppl 7):3-14.
21 Fry DW. Mechanism of action of erbB tyrosine kinase inhibitors. Exp Cell Res. 2003;284:131-139.
22 Sliwkowski MX, Lofgren JA, Lewis GD, et al. Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin). Semin Oncol. 1999;26(4 Suppl 12):60-70.
23 Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443-446.
24 Perez EA, Rodehefferr. Clinical cardiac tolerability of trastuzumab. J Clin Oncol. 2004;22:322-329.
25 Mendelsohn J. Epidermal growth factor receptor inhibition by a monoclonal antibody as anticancer therapy. Clin Cancer Res. 1997;3(12 Pt 2):2703-2707.
26 Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337-345.
27 Lièvre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374-379.
51 Huang SM, Harari PM. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas. Inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin Cancer Res. 2000;6:2166-2174.
53 Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354(6):567-578.
54 Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer. 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21-28.
55 Curran D, Giralt J, Harari PM, et al. Quality of life in head and neck cancer patients after treatment with high-dose radiotherapy alone or in combination with cetuximab. J Clin Oncol. 2007;25:2191-2197.
56 Kies MS, Holsinger FC, Lee JJ, et al. Induction chemotherapy and cetuximab for locally advanced squamous cell carcinoma of the head and neck. Results from a phase II prospective trial. J Clin Oncol. 2010;28:8-14.
57 Halyard MY, Pisansky TM, Dueck AC, et al. Radiotherapy and adjuvant trastuzumab in operable breast cancer. Tolerability and adverse event data from the NCCTG Phase III Trial N9831. J Clin Oncol. 2009;27:2638-2644.
58 Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645-4655.
90 Seiwert TY, Haraf DJ, Cohen EE, et al. Phase I study of bevacizumab added to fluorouracil- and hydroxyurea-based concomitant chemoradiotherapy for poor-prognosis head and neck cancer. J Clin Oncol. 2008;26:1732-1741.
91 Willett CG, Duda DG, di Tomaso E, et al. Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer. A multidisciplinary phase II study. J Clin Oncol. 2009;27:3020-3026.
92 Crane CH, Winter K, Regine WF, et al. Phase II study of bevacizumab with concurrent capecitabine and radiation followed by maintenance gemcitabine and bevacizumab for locally advanced pancreatic cancer: Radiation Therapy Oncology Group RTOG 0411. J Clin Oncol. 2009;27:4096-4102.
93 Levin VA, Bidaut L, Hou P, et al. Randomized double-blind placebo-controlled trial of bevacizumab therapy for radiation necrosis of the central nervous system. Int J Radiat Oncol Biol Phys. 2010. (Epub ahead of print) Apr 15
117 Mukogawa T, Koyama F, Tachibana M, et al. Adenovirus-mediated gene transduction of truncated I kappa B alpha enhances radiosensitivity in human colon cancer cells. Cancer Sci. 2003;94:745-750.
118 Pajonk F, Pajonk K, McBride WH. Inhibition of NF-kappaB, clonogenicity, and radiosensitivity of human cancer cells. J Natl Cancer Inst. 1999;91:1956-1960.
119 Russo SM, Tepper JE, Baldwin ASJr, et al. Enhancement of radiosensitivity by proteasome inhibition. Implications for a role of NF-kappaB. Int J Radiat Oncol Biol Phys. 2001;50:183-193.
120 Van Waes C, Chang AA, Lebowitz PF, et al. Inhibition of nuclear factor-kappaB and target genes during combined therapy with proteasome inhibitor bortezomib and reirradiation in patients with recurrent head-and-neck squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2005;63:1400-1412.
121 Pugh TJ, Chen C, Rabinovitch R, et al. Phase I trial of bortezomib and concurrent external beam radiation in patients with advanced solid malignancies. Int J Radiat Oncol Biol Phys. 2010;78(2):521-526.
122 O’Neil BH, Raftery L, Calvo BF, et al. A phase I study of bortezomib in combination with standard 5-fluorouracil and external-beam radiation therapy for the treatment of locally advanced or metastatic rectal cancer. Clin Colorectal Cancer. 2010;9:119-125.
123 Egan LJ, Eckmann L, Grefen FR, et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci U S A. 2004;101:2452-2457.
149 Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271-2281.
150 Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125-134.
151 Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-390.
161 Buchsbaum DJ, Bonner JA, Grizzle WE, et al. Treatment of pancreatic cancer xenografts with Erbitux (IMC-C225) anti-EGFR antibody, gemcitabine, and radiation. Int J Radiat Oncol Biol Phys. 2002;54:1180-1193.
167 Murakami M, Sasaki T, Yamasaki S, et al. Induction of apoptosis by ionizing radiation and CI-1033 in HuCCT-1 cells. Biochem Biophys Res Commun. 2004;319:114-119.
1 Earp HS3rd, Calvo BF, Sartor CI. The EGF receptor family—multiple roles in proliferation, differentiation, and neoplasia with an emphasis on HER4. Trans Am Clin Climatol Assoc. 2007;114:315-333. discussion 333-334
2 Yarden Y. The EGFR family and its ligands in human cancer. Signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2004;37(Suppl 4):S3-S8.
3 Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203-212.
4 Klapper LN, Kirschbaum MH, Sela M, Yarden Y. Biochemical and clinical implications of the ErbB/HER signaling network of growth factor receptors. Adv Cancer Res. 2000;77:25-79.
5 Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network. Receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159-3167.
6 Riese DJ2nd, Stern DF. Specificity within the EGF family/ErbB receptor family signaling network. Bioessays. 1998;20:41-48.
7 Klapper LN, Glathe S, Vaisman N, et al. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc Natl Acad Sci U S A. 1999;96:4995-5000.
8 Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211-225.
9 Cohen S, Carpenter G, King LJr. Epidermal growth factor-receptor-protein kinase interactions. Co-purification of receptor and epidermal growth factor-enhanced phosphorylation activity. J Biol Chem. 1980;255:4834-4842.
10 Downward J, Yarden Y, Mayes E, et al. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature. 1984;307:521-527.
11 Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A. 1982;89:4309-4313.
12 Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer. Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177-1782.
13 Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma. A population-based study. Cancer Res. 2004;64:6892-6899.
14 Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53:3579-3584.
15 Yamanaka Y, Friess H, Kobrin MS, et al. Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressiveness. Anticancer Res. 1993;13:565-569.
16 Spano JP, Lagorce C, Atlan D, et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann Oncol. 2005;16:102-108.
17 Scher HI, Sarkis A, Reuter V, et al. Changing pattern of expression of the epidermal growth factor receptor and transforming growth factor alpha in the progression of prostatic neoplasms. Clin Cancer Res. 1995;1:545-550.
18 Lynch TJ, Bell DW, Sordelia R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129-2139.
19 Wong AJ, Ruppert JM, Bigner SH, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992;89:2965-2969.
20 Arteaga C. Targeting HER1/EGFR. A molecular approach to cancer therapy. Semin Oncol. 2003;30(3 Suppl 7):3-14.
21 Fry DW. Mechanism of action of erbB tyrosine kinase inhibitors. Exp Cell Res. 2003;284:131-139.
22 Sliwkowski MX, Lofgren JA, Lewis GD, et al. Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin). Semin Oncol. 1999;26(4 Suppl 12):60-70.
23 Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443-446.
24 Perez EA, Rodehefferr. Clinical cardiac tolerability of trastuzumab. J Clin Oncol. 2004;22:322-329.
25 Mendelsohn J. Epidermal growth factor receptor inhibition by a monoclonal antibody as anticancer therapy. Clin Cancer Res. 1997;3(12 Pt 2):2703-2707.
26 Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337-345.
27 Lièvre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374-379.
28 Vermorken JB, Trigo J, Hitt R, et al. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol. 2007;25(16):2171-2177.
29 Pirker R, Pereira JR, Szczesna A, et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet. 2009;373:1525-1531.
30 Cohen MH, et al. United States Food and Drug Administration Drug Approval summary. Gefitinib (ZD1839; Iressa) tablets. Clin Cancer Res. 2004;10(4):1212-1218.
31 Herbst RS, et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer. A phase III trial—INTACT 2. J Clin Oncol. 2004;22:785-794.
32 Giaccone G, et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer. A phase III trial—INTACT 1. J Clin Oncol. 2004;22:777-784.
33 Shepherd FA, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123-132.
34 Perez-Soler R, et al. Determinants of tumor response and survival with erlotinib in patients with non–small-cell lung cancer. J Clin Oncol. 2004;22:3238-3247.
35 Gatzemeier U, et al. Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer. The Tarceva Lung Cancer Investigation Trial. J Clin Oncol. 2007;25:1545-1552.
36 FitzGerald TJ, et al. Expression of transfected recombinant oncogenes increases radiation resistance of clonal hematopoietic and fibroblast cell lines selectively at clinical low dose rate. Radiat Res. 1990;122:44-52.
37 Pietras RJ, et al. Monoclonal antibody to HER-2/neureceptor modulates repair of radiation-induced DNA damage and enhances radiosensitivity of human breast cancer cells overexpressing this oncogene. Cancer Res. 1999;59:1347-1355.
38 Miyaguchi M, et al. Correlation of epidermal growth factor receptor and radiosensitivity in human maxillary carcinoma cell lines. Acta Otolaryngol. 1998;118:428-431.
39 Sheridan MT, West CM. Ability to undergo apoptosis does not correlate with the intrinsic radiosensitivity (SF2) of human cervix tumor cell lines. Int J Radiat Oncol Biol Phys. 2001;50:503-509.
40 Akimoto T, et al. Inverse relationship between epidermal growth factor receptor expression and radiocurability of murine carcinomas. Clin Cancer Res. 1999;5:2884-2890.
41 Barker FG2nd, et al. EGFR overexpression and radiation response in glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2001;51:410-418.
42 Gupta AK, et al. Local recurrence in head and neck cancer. Relationship to radiation resistance and signal transduction. Clin Cancer Res. 2002;8:885-892.
43 Ang KK, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62:7350-7356.
44 Haffty BG, et al. Evaluation of HER-2 neu oncoprotein expression as a prognostic indicator of local recurrence in conservatively treated breast cancer. A case-control study. Int J Radiat Oncol Biol Phys. 1996;35:751-757.
45 Sartor CI. Epidermal growth factor family receptors and inhibitors. Radiation response modulators. Semin Radiat Oncol. 2003;13:22-30.
46 Harari PM, Huang SM. Radiation response modification following molecular inhibition of epidermal growth factor receptor signaling. Semin Radiat Oncol. 2001;11:281-289.
47 Raben D, Helfrich B, Bunn PAJr. Targeted therapies for non-small-cell lung cancer. Biology, rationale, and preclinical results from a radiation oncology perspective. Int J Radiat Oncol Biol Phys. 2004;59(2 Suppl):27-38.
48 Sartor CI. Mechanisms of disease. Radiosensitization by epidermal growth factor receptor inhibitors. Nat Clin Pract Oncol. 2004;1:80-87.
49 Raben D, et al. Understanding the mechanisms of action of EGFR inhibitors in NSCLC. What we know and what we do not know. Lung Cancer. 2003;41(Suppl 1):S15-S22.
50 Schmidt-Ullrich RK, et al. ERBB receptor tyrosine kinases and cellular radiation responses. Oncogene. 2003;22:5855-5865.
51 Huang SM, Harari PM. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas. Inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin Cancer Res. 2000;6:2166-2174.
52 Dittmann K, et al. The radioprotector O-phospho-tyrosine stimulates DNA-repair via epidermal growth factor receptor- and DNA-dependent kinase phosphorylation. Radiother Oncol. 2007;84:328-334.
53 Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354(6):567-578.
54 Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer. 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21-28.
55 Curran D, Giralt J, Harari PM, et al. Quality of life in head and neck cancer patients after treatment with high-dose radiotherapy alone or in combination with cetuximab. J Clin Oncol. 2007;25:2191-2197.
56 Kies MS, Holsinger FC, Lee JJ, et al. Induction chemotherapy and cetuximab for locally advanced squamous cell carcinoma of the head and neck. Results from a phase II prospective trial. J Clin Oncol. 2010;28:8-14.
57 Halyard MY, Pisansky TM, Dueck AC, et al. Radiotherapy and adjuvant trastuzumab in operable breast cancer. Tolerability and adverse event data from the NCCTG Phase III Trial N9831. J Clin Oncol. 2009;27:2638-2644.
58 Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645-4655.
59 Millikan KW, et al. Do angiogenesis and growth factor expression predict prognosis of esophageal cancer? Am Surg. 2000;66:401-405. discussion, 405-406
60 Niedergethmann M, et al. High expression of vascular endothelial growth factor predicts early recurrence and poor prognosis after curative resection for ductal adenocarcinoma of the pancreas. Pancreas. 2002;25:122-129.
61 Wells SAJr, Gosnell JE, Gagel RF, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28:767-772.
62 Carlomagno F, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62:7284-7290.
63 Burrows FJ, Thorpe PE. Vascular targeting—a new approach to the therapy of solid tumors. Pharmacol Ther. 1994;64:155-174.
64 Blakey DC, et al. Antitumor activity of the novel vascular targeting agent ZD6126 in a panel of tumor models. Clin Cancer Res. 2002;8:1974-1983.
65 Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res. 2004;10:415-427.
66 Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4-25.
67 Hurwitz H, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335-2342.
68 Giantonio BJ, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer. Results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007;25:1539-1544.
69 Sandler A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542-2550.
70 Friedman HS, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733-4740.
71 Miller KD, et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005;23:792-799.
72 Teicher BA, et al. TNP-470/minocycline/cytotoxic therapy. A systems approach to cancer therapy. Eur J Cancer. 1996;32A:2461-2466.
73 Mauceri HJ, et al. Combined effects of angiostatin and ionizing radiation in antitumour therapy. Nature. 1998;394:287-291.
74 Kozin SV, et al. Vascular endothelial growth factor receptor-2-blocking antibody potentiates radiation-induced long-term control of human tumor xenografts. Cancer Res. 2001;61:39-44.
75 Ning S, et al. The antiangiogenic agents SU5416 and SU6668 increase the antitumor effects of fractionated irradiation. Radiat Res. 2002;157:45-51.
76 Wachsberger P, Burd R, Dicker AP. Tumor response to ionizing radiation combined with antiangiogenesis or vascular targeting agents. Exploring mechanisms of interaction. Clin Cancer Res. 2003;9:1957-1971.
77 Raben D, Helfrich B. Angiogenesis inhibitors. A rational strategy for radiosensitization in the treatment of non-small-cell lung cancer? Clin Lung Cancer. 2004;6:48-57.
78 Lee CG, et al. Anti-vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. 2000;60:5565-5570.
79 Solomon B, et al. EGFR blockade with ZD1839 (“Iressa”) potentiates the antitumor effects of single and multiple fractions of ionizing radiation in human A431 squamous cell carcinoma. Epidermal growth factor receptor. Int J Radiat Oncol Biol Phys. 2003;55:713-723.
80 Horsman MR, Murata R. Vascular targeting effects of ZD6126 in a C3H mouse mammary carcinoma and the enhancement of radiation response. Int J Radiat Oncol Biol Phys. 2003;57:1047-1055.
81 Siemann DW, Rojiani AM. Enhancement of radiation therapy by the novel vascular targeting agent ZD6126. Int J Radiat Oncol Biol Phys. 2002;53:164-171.
82 Hess C, et al. Effect of VEGF receptor inhibitor PTK787/ZK222584 (correction of ZK222548) combined with ionizing radiation on endothelial cells and tumour growth. Br J Cancer. 2001;85:2010-2016.
83 Williams KJ, et al. ZD6474, a potent inhibitor of vascular endothelial growth factor signaling, combined with radiotherapy. Schedule-dependent enhancement of antitumor activity. Clin Cancer Res. 2004;10:8587-8593.
84 National Cancer Institute Clinical Trial Database. (Cited 2010 July 12.) Available from http://www.cancer.gov/search/ViewClinicalTrials
85 Radiation Therapy Oncology Group. (Cited 2010 July 12.) Available from www.rtog.org
86 Gorski DH, et al. Blockage of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res. 1999;59:3374-3378.
87 Czito BG, et al. Bevacizumab, oxaliplatin, and capecitabine with radiation therapy in rectal cancer. Phase I trial results. Int J Radiat Oncol Biol Phys. 2007;68:472-478.
88 Crane CH, et al. Phase I trial evaluating the safety of bevacizumab with concurrent radiotherapy and capecitabine in locally advanced pancreatic cancer. J Clin Oncol. 2006;24:1145-1151.
89 Lai A, et al. Phase II pilot study of bevacizumab in combination with temozolomide and regional radiation therapy for up-front treatment of patients with newly diagnosed glioblastoma multiforme. Interim analysis of safety and tolerability. Int J Radiat Oncol Biol Phys. 2008;71:1372-1380.
90 Seiwert TY, Haraf DJ, Cohen EE, et al. Phase I study of bevacizumab added to fluorouracil- and hydroxyurea-based concomitant chemoradiotherapy for poor-prognosis head and neck cancer. J Clin Oncol. 2008;26:1732-1741.
91 Willett CG, Duda DG, di Tomaso E, et al. Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer. A multidisciplinary phase II study. J Clin Oncol. 2009;27:3020-3026.
92 Crane CH, Winter K, Regine WF, et al. Phase II study of bevacizumab with concurrent capecitabine and radiation followed by maintenance gemcitabine and bevacizumab for locally advanced pancreatic cancer: Radiation Therapy Oncology Group RTOG 0411. J Clin Oncol. 2009;27:4096-4102.
93 Levin VA, Bidaut L, Hou P, et al. Randomized double-blind placebo-controlled trial of bevacizumab therapy for radiation necrosis of the central nervous system. Int J Radiat Oncol Biol Phys. Apr 15, 2010. (Epub ahead of print)
94 Duda DG, Willett CG, Ancukiewicz M, et al. Plasma soluble VEGFR-1 is a potential dual biomarker of response and toxicity for bevacizumab with chemoradiation in locally advanced rectal cancer. Oncologist. 2010;15:577-583.
95 Goyal S, Rao MS, Khan A, et al. Evaluation of acute locoregional toxicity in patients with breast cancer treated with adjuvant radiotherapy in combination with bevacizumab. Int J Radiat Oncol Biol Phys. 2011;79(2):408-413.
96 McGee MC, Hamner JB, Williams RF, et al. Improved intratumoral oxygenation through vascular normalization increases glioma sensitivity to ionizing radiation. Int J Radiat Oncol Biol Phys. 2010;76:1537-1545.
97 Spigel DR, Hainsworth JD, Yardley DA, et al. Tracheoesophageal fistula formation in patients with lung cancer treated with chemoradiation and bevacizumab. J Clin Oncol. 2010;28:43-48.
98 Provencio M, Sánchez A, Garrido P, Valcárcel F. New molecular targeted therapies integrated with radiation therapy in lung cancer. Clin Lung Cancer. 2010;11:91-97.
99 Orlowski RZ, Baldwin ASJr. NF-kappaB as a therapeutic target in cancer. Trends Mol Med. 2002;8:385-389.
100 Voorhees PM, et al. The proteasome as a target for cancer therapy. Clin Cancer Res. 2003;9:6316-6325.
101 Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest. 2001;107:241-246.
102 Russell JS, Tofilon PJ. Radiation-induced activation of nuclear factor-kappaB involves selective degradation of plasma membrane-associated I(kappa)B(alpha). Mol Biol Cell. 2002;13:3431-3440.
103 Lin YC, Brown K, Siebenlist U. Activation of NF-kappa B requires proteolysis of the inhibitor I kappa B-alpha. Signal-induced phosphorylation of I kappa B-alpha alone does not release active NF-kappa B. Proc Natl Acad Sci U S A. 1995;92:552-556.
104 Hideshima T, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61:3071-3076.
105 Masdehors P, et al. Increased sensitivity of CLL-derived lymphocytes to apoptotic death activation by the proteasome-specific inhibitor lactacystin. Br J Haematol. 1999;105:752-757.
106 Guzman ML, et al. Preferential induction of apoptosis for primary human leukemic stem cells. Proc Natl Acad Sci U S A. 2002;99:16220-16225.
107 Wang H, et al. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas. Lab Invest. 2004;84:941-951.
108 Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3:1221-1224.
109 Ni H, et al. Analysis of expression of nuclear factor kappa B (NF-kappa B) in multiple myeloma. Downregulation of NF-kappa B induces apoptosis. Br J Haematol. 2001;115:279-286.
110 Feinman R, et al. Role of NF-kappaB in the rescue of multiple myeloma cells from glucocorticoid-induced apoptosis by bcl-2. Blood. 1999;93:3044-3052.
111 Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265:956-959.
112 Keifer JA, et al. Inhibition of NF-kappa B activity by thalidomide through suppression of IkappaB kinase activity. J Biol Chem. 2001;276:22382-22387.
113 Chendil D, et al. Curcumin confers radiosensitizing effect in prostate cancer cell line PC-3. Oncogene. 2004;23:1599-1607.
114 Richardson PG, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348:2609-2617.
115 Orlowski RZ, et al. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J Clin Oncol. 2002;20:4420-4427.
116 Yamagishi N, Miyakoshi J, Takebe H. Enhanced radiosensitivity by inhibition of nuclear factor kappa B activation in human malignant glioma cells. Int J Radiat Biol. 1997;72:157-162.
117 Mukogawa T, Koyama F, Tachibana M, et al. Adenovirus-mediated gene transduction of truncated I kappa B alpha enhances radiosensitivity in human colon cancer cells. Cancer Sci. 2003;94:745-750.
118 Pajonk F, Pajonk K, McBride WH. Inhibition of NF-kappaB, clonogenicity, and radiosensitivity of human cancer cells. J Natl Cancer Inst. 1999;91:1956-1960.
119 Russo SM, Tepper JE, Baldwin ASJr, et al. Enhancement of radiosensitivity by proteasome inhibition. Implications for a role of NF-kappaB. Int J Radiat Oncol Biol Phys. 2001;50:183-193.
120 Van Waes C, Chang AA, Lebowitz PF, et al. Inhibition of nuclear factor-kappaB and target genes during combined therapy with proteasome inhibitor bortezomib and reirradiation in patients with recurrent head-and-neck squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2005;63:1400-1412.
121 Pugh TJ, Chen C, Rabinovitch R, et al. Phase I trial of bortezomib and concurrent external beam radiation in patients with advanced solid malignancies. Int J Radiat Oncol Biol Phys. 2010;78(2):521-526.
122 O’Neil BH, Raftery L, Calvo BF, et al. A phase I study of bortezomib in combination with standard 5-fluorouracil and external-beam radiation therapy for the treatment of locally advanced or metastatic rectal cancer. Clin Colorectal Cancer. 2010;9:119-125.
123 Egan LJ, Eckmann L, Grefen FR, et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci U S A. 2004;101:2452-2457.
124 Ma BB, et al. Combined-modality treatment of solid tumors using radiotherapy and molecular targeted agents. J Clin Oncol. 2003;21:2760-2776.
125 Maity A, et al. Potential molecular targets for manipulating the radiation response. Int J Radiat Oncol Biol Phys. 1997;37:639-653.
126 Molinolo AA, et al. Dissecting the Akt/mammalian target of rapamycin signaling network. Emerging results from the head and neck cancer tissue array initiative. Clin Cancer Res. 2007;13:4964-4973.
127 Shinohara ET, et al. Enhanced radiation damage of tumor vasculature by mTOR inhibitors. Oncogene. 2005;24:5414-5422.
128 Ekshyyan O, et al. Comparison of radiosensitizing effects of the mammalian target of rapamycin inhibitor CCI-779 to cisplatin in experimental models of head and neck squamous cell carcinoma. Mol Cancer Ther. 2009;8:2255-2265.
129 Miwa M, Masutani M. PolyADP-ribosylation and cancer. Cancer Sci. 2007;98:1528-1535.
130 Nozaki T, et al. Suppression of G1 arrest and enhancement of G2 arrest by inhibitors of poly(ADP-ribose) polymerase: possible involvement of poly(ADP-ribosyl)ation in cell cycle arrest following gamma-irradiation. Jpn J Cancer Res. 1994;85:1094-1098.
131 Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897-1905.
132 Fong PC, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123-134.
133 Hay T, et al. Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res. 2009;69:3850-3855.
134 Albert JM, et al. Inhibition of poly(ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin Cancer Res. 2007;13:3033-3042.
135 Taunk NK, Goyal S, Moran MS, et al. Prognostic significance of IGF-1R expression in patients treated with breast-conserving surgery and radiation therapy. Radiother Oncol. 2010;96:204-208.
136 Burtrum D, et al. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res. 2003;63:8912-8921.
137 Allen GW, et al. Insulin-like growth factor-I receptor signaling blockade combined with radiation. Cancer Res. 2007;67:1155-1162.
138 Iwasa T, et al. Inhibition of insulin-like growth factor 1 receptor by CP-751,871 radiosensitizes NSCLC cells. Clin Cancer Res. 2009;15:5117-5125.
139 Slamon DJ, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783-792.
140 Piccart-Gebhart MJ, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353(16):1659-1672.
141 Burtness B, et al. Phase III randomized trial of cisplatin plus placebo compared with cisplatin plus cetuximab in metastatic/recurrent head and neck cancer. An Eastern Cooperative Oncology Group study. J Clin Oncol. 2005;23(34):8646-8654.
142 Vermorken JB, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359(11):1116-1127.
143 Van Cutsem E, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25(13):1658-1664.
144 Moore MJ, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer. A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960-1966.
145 Kantarjian H, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346:645-652.
146 O’Brien SG, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994-1004.
147 Geyer CE, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733-2743.
148 Kantarjian H, et al. Dasatinib or high-dose imatinib for chronic-phase chronic myeloid leukemia after failure of first-line imatinib: a randomized phase 2 trial. Blood. 2007;109:5143-5150.
149 Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271-2281.
150 Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125-134.
151 Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-390.
152 Motzer RJ, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115-124.
153 Demetri GD, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib. A randomised controlled trial. Lancet. 2006;368:1329-1338.
154 Duvic M, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109:31-39.
155 Williams KJ, et al. ZD1839 (“Iressa”), a specific oral epidermal growth factor receptor-tyrosine kinase inhibitor, potentiates radiotherapy in a human colorectal cancer xenograft model. Br J Cancer. 2002;86:1157-1161.
156 Stea B, et al. Time and dose-dependent radiosensitization of the glioblastoma multiforme U251 cells by the EGF receptor tyrosine kinase inhibitor ZD1839 (“Iressa”). Cancer Lett. 2003;202:43-51.
157 Bianco C, et al. Enhancement of antitumor activity of ionizing radiation by combined treatment with the selective epidermal growth factor receptor-tyrosine kinase inhibitor ZD1839 (Iressa). Clin Cancer Res. 2002;8:3250-3258.
158 Raben D, et al. Antitumor activity of ZD6126, a novel vascular-targeting agent, is enhanced when combined with ZD1839, an epidermal growth factor receptor tyrosine kinase inhibitor, and potentiates the effects of radiation in a human non-small cell lung cancer xenograft model. Mol Cancer Ther. 2004;3:977-983.
159 Huang SM, et al. Modulation of radiation response and tumor-induced angiogenesis after epidermal growth factor receptor inhibition by ZD1839 (Iressa). Cancer Res. 2002;62:4300-4306.
160 Shintani S, et al. Enhancement of tumor radioresponse by combined treatment with gefitinib (Iressa, ZD1839), an epidermal growth factor receptor tyrosine kinase inhibitor, is accompanied by inhibition of DNA damage repair and cell growth in oral cancer. Int J Cancer. 2003;107:1030-1037.
161 Buchsbaum DJ, Bonner JA, Grizzle WE, et al. Treatment of pancreatic cancer xenografts with Erbitux (IMC-C225) anti-EGFR antibody, gemcitabine, and radiation. Int J Radiat Oncol Biol Phys. 2002;54:1180-1193.
162 Milas L, et al. In vivo enhancement of tumor radioresponse by C225 antiepidermal growth factor receptor antibody. Clin Cancer Res. 2000;6:701-708.
163 Milas L, et al. Epidermal growth factor receptor and tumor response to radiation. In vivo preclinical studies. Int J Radiat Oncol Biol Phys. 2004;58:966-971.
164 Harari PM, Huang SM. Head and neck cancer as a clinical model for molecular targeting of therapy. Combining EGFR blockade with radiation. Int J Radiat Oncol Biol Phys. 2001;49:427-433.
165 Zhou H, et al. Effects of the EGFR/HER2 kinase inhibitor GW572016 on EGFR- and HER2-overexpressing breast cancer cell line proliferation, radiosensitization, and resistance. Int J Radiat Oncol Biol Phys. 2004;58:344-352.
166 Nyati MK, et al. Radiosensitization by pan ErbB inhibitor CI-1033 in vitro and in vivo. Clin Cancer Res. 2004;10:691-700.
167 Murakami M, Sasaki T, Yamasaki S, et al. Induction of apoptosis by ionizing radiation and CI-1033 in HuCCT-1 cells. Biochem Biophys Res Commun. 2004;319:114-119.