CHAPTER 11 Biochemical Genetics
In this chapter, we consider single-gene biochemical or metabolic diseases, including mitochondrial disorders. The range of known disorders is vast, so only an overview is possible, but it is hoped that the reader will gain a flavor of this fascinating area of medicine. At the beginning of the twentieth century, Garrod introduced the concept of ‘chemical individuality’, leading in turn to the concept of the inborn error of metabolism (IEM). Beadle and Tatum later developed the idea that metabolic processes, whether in humans or any other organism, proceed by steps. They proposed that each step was controlled by a particular enzyme and that this, in turn, was the product of a particular gene. This was referred to as the one gene–one enzyme (or protein) concept.
Inborn Errors of Metabolism
In excess of 200 IEMs are known that can be grouped by either the metabolite, metabolic pathway, function of the enzyme, or cellular organelle involved (Table 11.1). Most follow autosomal recessive or X-linked recessive inheritance, with only a few being autosomal dominant. This is because the defective protein in most cases is a diffusible enzyme, and there is usually sufficient residual activity in the heterozygous state (loss-of-function, see p. 26) for the enzyme to function normally in most situations. If, however, the reaction catalysed by an enzyme is rate limiting (haploinsufficiency, see p. 26) or the gene product is part of a multimeric complex (dominant-negative, see p. 26), the disorder can manifest in the heterozygous state and follow dominant inheritance (p. 109).

Disorders of Amino Acid Metabolism
Phenylketonuria
Children with phenylketonuria (PKU), if untreated, are severely intellectually impaired and often develop seizures. There is a deficiency of the enzyme required for the conversion of phenylalanine to tyrosine, phenylalanine hydroxylase (PAH)—causing a ‘genetic block’ in the metabolic pathway (Figure 11.1).
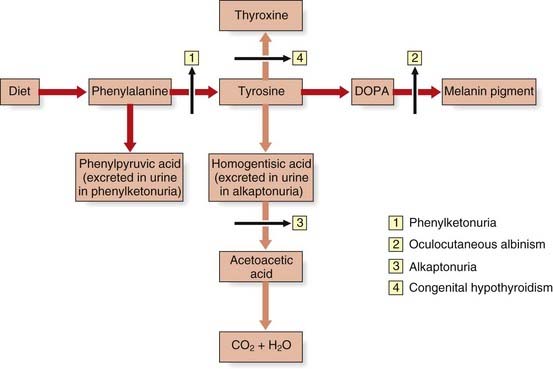
PKU was the first genetic disorder in humans shown to be caused by a specific enzyme deficiency, by Jervis in 1953. As a result of the enzyme defect, phenylalanine accumulates and is converted into phenylpyruvic acid and other metabolites that are excreted in the urine. The enzyme block leads to a deficiency of tyrosine, with a consequent reduction in melanin formation, and children therefore often have blond hair and blue eyes (Figure 11.2). In addition, areas of the brain that are usually pigmented, such as the substantia nigra, may also lack pigment.

Treatment of PKU
An obvious method of treating children with PKU would be to replace the missing enzyme, but this is not simply achieved (p. 349). Bickel, just 1 year after the enzyme deficiency had been identified, suggested that PKU could be treated by removal of phenylalanine from the diet and this has proved effective. If PKU is detected early enough in infancy, intellectual impairment can be prevented by giving a phenylalanine restricted diet. Phenylalanine is an essential amino acid and therefore cannot be removed entirely from the diet. By monitoring the level of phenylalanine in the blood, it is possible to supply sufficient amounts to meet normal requirements but avoiding toxic levels, resulting in mental retardation. After brain development is complete, dietary restriction can be relaxed—from adolescence onward.
Diagnosis of PKU
PKU affects approximately 1 in 10,000 people in western Europe and was the first IEM routinely screened for in newborns. The test detects the presence of the metabolite of phenylalanine, phenylpyruvic acid, in the urine by its reaction with ferric chloride, or through increased levels of phenylalanine in the blood. The latter, known as the Guthrie test, involved analysing blood from newborns and comparing the amount of growth induced by the sample, against standards, in a strain of the bacterium Bacillus subtilis, which requires phenylalanine for growth. This has been replaced by the use of a variety of biochemical assays of phenylalanine levels.
Mutational Basis of PKU
More than 450 different mutations in the PAH gene have been identified. Certain mutations are more prevalent in specific population groups and, in western Europeans with PKU, they are found on the background of a limited number of DNA haplotypes. Interestingly though, a variety of different individual mutations has been found in association with some of these haplotypes.
Alkaptonuria
Alkaptonuria was the original autosomal recessive IEM described by Garrod. Here there is a block in the breakdown of homogentisic acid, a metabolite of tyrosine, because of a deficiency of the enzyme homogentisic acid oxidase (see Figure 11.1). As a consequence, homogentisic acid accumulates and is excreted in the urine, which then darkens on exposure to air. Dark pigment is also deposited in certain tissues, such as the ear wax, cartilage, and joints, where it is known as ochronosis, which in joints can lead to arthritis later in life.
Oculocutaneous Albinism
Oculocutaneous albinism (OCA) is an autosomal recessive disorder resulting from a deficiency of the enzyme tyrosinase, which is necessary for the formation of melanin from tyrosine (see Figure 11.1). In OCA there is a lack of pigment in the skin, hair, iris, and ocular fundus (Figure 11.3), and the lack of eye pigment results in poor visual acuity and uncontrolled pendular eye movements—nystagmus. Reduced fundal pigmentation leads to underdevelopment of part of retina for fine vision—the fovea—and abnormal projection of the visual pathways to the optic cortex.
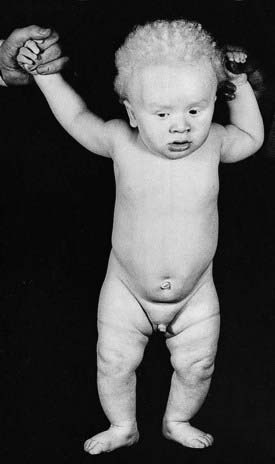
FIGURE 11.3 Oculocutaneous albinism in a child of Afro-Caribbean origin.
(Courtesy of Dr V. A. McKusick.)
Homocystinuria
Homocystinuria is a recessively inherited sulfur amino-acid IEM characterized by learning disability, seizures, thrombophilia, osteoporosis, scoliosis, pectus excavatum, long fingers and toes (arachnodactyly), and a tendency to dislocation of the lenses. The somatic features therefore resemble the autosomal dominant disorder Marfan syndrome (p. 300).
Urea Cycle Disorders
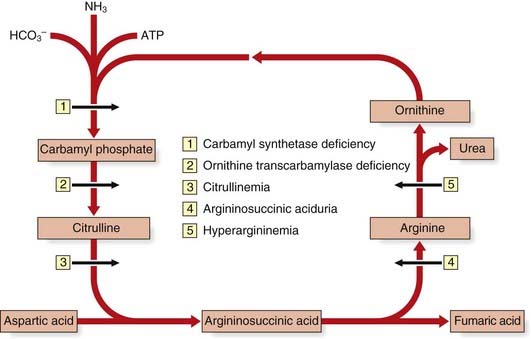
The urea cycle is a five-step metabolic pathway that takes place primarily in liver cells for the removal of waste nitrogen from the amino groups of amino acids arising from the normal turnover of protein. It converts two molecules of ammonia and one of bicarbonate into urea (Figure 11.4). Deficiencies of enzymes in the urea cycle result in intolerance to protein from the accumulation of ammonia in the body—hyperammonemia. Increased ammonia levels are toxic to the central nervous system and can lead to coma and, with some untreated urea cycle disorders, death. They are collectively and individually rare and, with the exception of X-linked ornithine transcarbamylase deficiency, inherited as autosomal recessive traits.
Disorders of Carbohydrate Metabolism
Glycogen Storage Diseases
Glycogen Storage Diseases that Primarily Affect Liver
Glycogen Storage Diseases that Primarily Affect Muscle
Pompe disease (GSD-II)
Infants with Pompe disease usually present in the first few months of life with floppiness (hypotonia) and delay in the gross motor milestones because of muscle weakness. They then develop an enlarged heart and die from cardiac failure in the first or second year. Voluntary and cardiac muscle accumulates glycogen because of a deficiency of the lysosomal enzyme α-1,4-glucosidase, which is needed to break down glycogen. The diagnosis can be confirmed by enzyme assay of white blood cells or fibroblasts. Early reports of enzyme replacement therapy appear promising.
Disorders of Steroid Metabolism
The disorders of steroid metabolism include a number of autosomal recessive inborn errors of the biosynthetic pathways of cortisol. Virilization of a female fetus may occur together with salt loss in infants of either sex from a deficiency of the hormone aldosterone. In addition, defects of the androgen receptor result in lack of virilization of chromosomally male individuals (Figure 11.5).
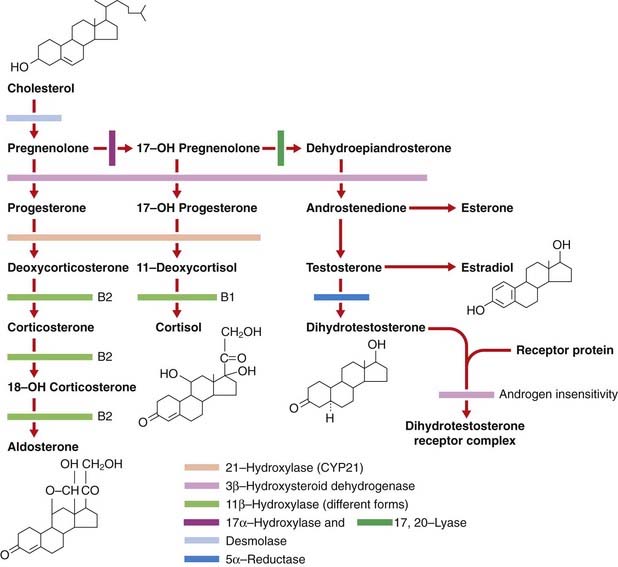
FIGURE 11.5 Steroid biosynthesis indicating the site of the common inborn errors of steroid biosynthesis.
Congenital Adrenal Hyperplasia (Adrenogenital Syndrome)
The diagnosis of congenital adrenal hyperplasia (CAH) should be considered in any newborn female infant presenting with virilization of the external genitalia, because this is the most common cause of ambiguous genitalia in female newborns (p. 288) (Figure 11.6). 21-Hydroxylase deficiency accounts for more than 90% of cases. Approximately 25% have the salt-losing form, presenting in the second or third week of life with circulatory collapse, hyponatremia, and hyperkalemia. Less commonly, CAH is a result of deficiency of the enzymes 11β-hydroxylase or 3β-dehydrogenase, and very rarely occurs as a result of deficiencies of enzymes 17α-hydroxylase and 17,20-lyase. Desmolase deficiency is very rare, with all pathways blocked, causing a reversed phenotype of ambiguous genitalia in males, and severe addisonian crises. Males with the rare 5α-reductase deficiency are significantly under-masculinized but do not suffer other metabolic problems and are likely to be raised as females. At puberty, however, the surge in androgen production is sufficient to stimulate growth of the phallus, making gender identity and assignment problematic.
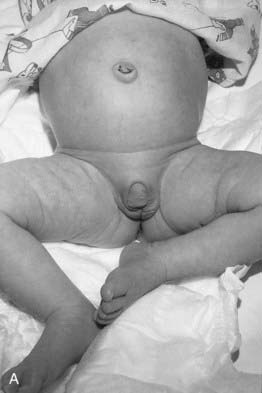
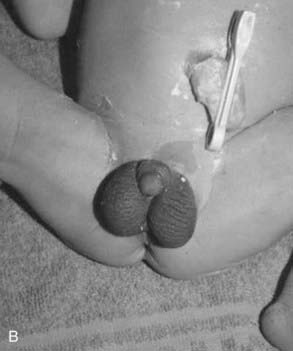
Affected females with classic CAH are virilized from accumulation of the adrenocortical steroids proximal to the enzyme block in the steroid biosynthetic pathway, many of which have testosterone-like activity (see Figure 11.5). However, they have normal müllerian-derived internal organs. The possibility of CAH should not be forgotten, of course, in male infants presenting with circulatory collapse in the first few weeks of life.
Androgen Insensitivity Syndrome
Individuals with the androgen insensitivity syndrome have female external genitalia and undergo breast development in puberty (p. 288). They classically present either with primary amenorrhea or with an inguinal hernia containing a gonad that turns out to be a testis. Inguinal hernia is uncommon in girls and if present, especially if bilateral, androgen insensitivity syndrome should be considered. There is often scanty secondary sexual hair and investigation of the internal genitalia reveals an absent uterus and fallopian tubes with a blind-ending vagina. Chromosome analysis reveals a normal male karyotype, 46,XY.
Androgen production by the testes is normal but androgen does not bind normally because of an abnormal androgen receptor (see Figure 11.5)—the androgen receptor gene on the X chromosome is mutated. This can be functionally assayed in skin fibroblasts. Some individuals have incomplete or partial androgen insensitivity, and under-virilization is variable. Affected subjects may have a female sexual orientation and are sterile. Testes must be removed because of an increased malignancy risk, and estrogen given for secondary sexual development and prevention of long-term osteoporosis.
Disorders of Lipid Metabolism
Familial hypercholesterolemia is the most common autosomal dominant single-gene disorder in Western society and is associated with high morbidity and mortality rates through premature coronary artery disease (p. 241).
Familial Hypercholesterolemia
Persons with familial hypercholesterolemia (FH) have raised cholesterol levels with a significant risk of developing early coronary artery disease (p. 241). They can present in childhood or adolescence with subcutaneous deposition of lipid, known as xanthomata (Figure 11.7). Starting with families who presented with early coronary artery disease, Brown and Goldstein unraveled the biology of the low-density lipoprotein (LDL) receptor (p. 241) and the pathological basis of FH.
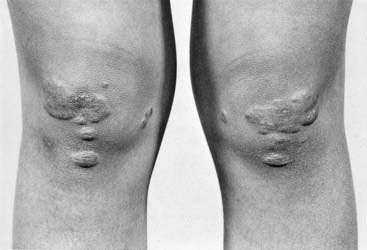
FIGURE 11.7 Legs of a person homozygous for familial hypercholesterolemia, showing multiple xanthomata.
(Courtesy Dr. E. Wraith, Royal Manchester Children’s Hospital, Manchester, UK.)
High cholesterol levels in FH are due to deficient or defective function of the LDL receptors leading to increased levels of endogenous cholesterol synthesis. Four main classes of mutation in the LDL receptor have been identified: (1) reduced or defective biosynthesis of the receptor; (2) reduced or defective transport of the receptor from the endoplasmic reticulum to the Golgi apparatus; (3) abnormal binding of LDL by the receptor; and (4) abnormal internalization of LDL by the receptor (Figure 11.8). Specific mutations are more prevalent in certain ethnic groups because of founder effects (p. 133).
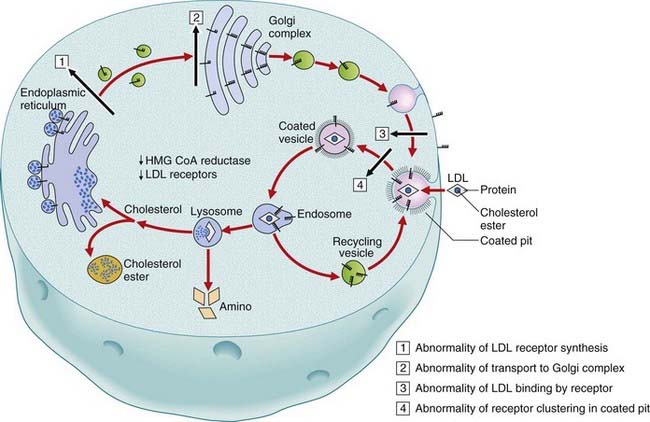
Lysosomal Storage Disorders
In addition to the IEMs in which an enzyme defect leads to deficiency of an essential metabolite and accumulation of intermediate metabolic precursors, there are a number of disorders in which deficiency of a lysosomal enzyme involved in the degradation of complex macromolecules leads to their accumulation. This accumulation occurs because macromolecules are normally in a constant state of flux, with a delicate balance between their rates of synthesis and breakdown. Children born with lysosomal storage diseases are usually normal initially but with the passage of time commence a downhill course of variable duration owing to the accumulation of one or more of a variety or type of macromolecules.
Mucopolysaccharidoses
Hunter Syndrome (MPS-II)
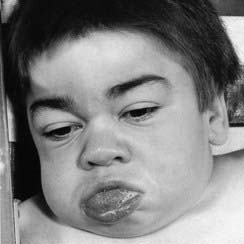
Males with Hunter syndrome usually present between 2 and 5 years with hearing loss, recurrent infections, diarrhea, and poor growth. Facial features are characteristic with coarsening (Figure 11.9), the liver and spleen are enlarged, and joint stiffness occurs. Spinal radiographs show abnormal shape of the vertebrae. Progressive physical and mental deterioration occurs with death usually in adolescence.
Sphingolipidoses (Lipid Storage Diseases)
Tay-Sachs Disease
This well-known sphingolipidosis has an incidence of ~1:3600 in Ashkenazi Jews (pp. 313–314). Infants usually present by 6 months of age with poor feeding, lethargy, and floppiness. Developmental regression usually becomes apparent in late infancy, feeding becomes increasingly difficult, and the infant progressively deteriorates, with deafness, visual impairment, and spasticity, which progresses to rigidity. Death usually occurs by the age of 3 years from respiratory infection. Less severe juvenile, adult, and chronic forms are reported.
Disorders of Purine/Pyrimidine Metabolism
Immunodeficiency Diseases Caused By Defects in Purine Metabolism
Two inherited immunodeficiency disorders (pp. 203–204), perhaps surprisingly, are inborn errors of purine metabolism.
Adenosine Deaminase Deficiency
About half of all children with the autosomal recessive form of severe combined immunodeficiency with impaired B- and T-cell function (p. 203) have deficiency of the enzyme adenosine deaminase. Presentation is in infancy with recurrent viral and bacterial infections which, if untreated, soon cause death from overwhelming infection. The diagnosis is confirmed by deficient red blood cell adenosine deaminase activity. Bone marrow transplantation has been successful—even for the fetus in utero.
Disorders of Porphyrin Metabolism
There are several different disorders of porphyrin metabolism that are from a deficiency of enzymes in the biosynthetic pathway of the iron-containing group in hemoglobin—heme (p. 155). They all follow autosomal dominant inheritance, with the exception of autosomal recessive congenital erythropoietic porphyria. This is because the enzymes are rate limiting (p. 26), so that haploinsufficiency results in clinical disease.
Hepatic Porphyrias
These include acute intermittent porphyria, hereditary co-proporphyria, and porphyria variegata.
Porphyria Variegata
People with this form of porphyria, particularly prevalent in South African Afrikaans (p. 109), have variable skin photosensitivity with neurological and visceral features that can also be triggered by drugs. Increased fecal excretion of the porphyrin precursors protoporphyrin and co-proporphyrin can be demonstrated and the disorder has been shown to be due to deficiency of the enzyme protoporphyrinogen oxidase.
Erythropoietic Porphyrias
Erythropoietic Protoporphyria
Erythropoietic protoporphyria is from a deficiency of the enzyme ferrochelatase, which is responsible for the insertion of ferrous iron into the porphyrin precursor to form heme. Affected persons have photosensitivity and sometimes develop chronic liver disease. Successful treatment of the photosensitivity has been reported with β-carotene.
Disorders of Copper Metabolism
There are two inborn errors of copper metabolism: Menkes disease and Wilson disease.
Menkes Disease
Menkes disease is an X-linked recessive disorder in which affected males present in the first few months of life with feeding difficulties, vomiting, and poor weight gain. Subsequently, hypotonia, seizures, and progressive neurological deterioration ensue, with death from recurrent respiratory infection usually occurring by the age of 3 years. A characteristic feature is the hair, which lacks pigment, is kinky, and is brittle. This was noted to resemble the wool of sheep suffering from copper deficiency. Serum copper and ceruloplasmin levels are very low. Cloning of the gene for Menkes disease was facilitated through an affected female with an X-autosome translocation (p. 116) and revealed it to code for an ATPase cation transport protein for copper. Treatment regimens with different exogenous copper sources have had limited benefit to date.
Peroxisomal Disorders
Zellweger Syndrome
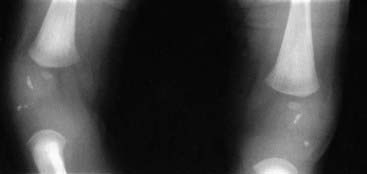
Newborn infants with Zellweger syndrome present with hypotonia and weakness and have mildly dysmorphic facial features (Figure 11.10), consisting of a prominent forehead and a large anterior fontanelle (‘soft spot’). They may also have cataracts and an enlarged liver. They generally go on to have fits with developmental regression and usually die by 1 year of age. Investigations can reveal renal cysts and abnormal calcification in the cartilaginous growing ends of the long bones (Figure 11.11). There is a range of severity of this disorder, with different clinical diagnoses being given to the less severe types. The diagnosis can be confirmed by raised levels of plasma long-chain fatty acids. It is genetically heterogeneous, due to any one of several genes crucial to peroxisome biogenesis.
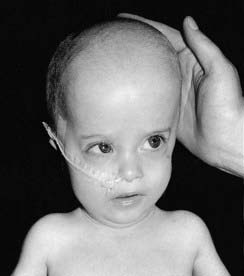
It is unusual for IEMs to give rise to a dysmorphic syndrome (p. 249), but another is Smith-Lemli-Opitz syndrome, an inborn error of cholesterol biosynthesis from a mutation in the sterol delta-7-reductase (DHCR7) gene.
Disorders Affecting Mitochondrial Function
Mitochondrial disease was first identified in 1962 in a patient whose mitochondria showed structural abnormalities and loss of coupling between oxidation and phosphorylation, although it was not until 20 years later that the relevance of mutated mitochondrial DNA (mtDNA) to human disease began to be appreciated. The small circular double-stranded mtDNA (see Figure 2.7, p. 18) contains genes coding for ribosomal RNA (rRNA) production and various transfer RNAs (tRNA) required for mitochondrial protein biosynthesis, as well as some of the proteins involved in electron transport. There are 5523 codons and a total of 37 gene products. Guanine and cytosine nucleotides are asymmetrically distributed between the two mtDNA strands—the guanine-rich strand being called the heavy (H) strand and the cytosine-rich the light (L) strand. Replication and transcription is controlled by a 1122-bp sequence of mtDNA known as the displacement loop (D-loop). Oxidative phosphorylation (OXPHOS) is the biochemical process responsible for generating much of the ATP required for cellular energy. The process is mediated by five intramitochondrial enzyme complexes, referred to as complexes I–V, and the mtDNA encodes 13 OXPHOS subunits, 22 tRNAs, and 2 rRNAs.
The ‘complexes’ are aptly named. Analysis of complex I, for example, has revealed approximately 41 different subunits, of which 7 are polypeptides encoded by mtDNA genes known as ND1, ND2, ND3, NDL4, ND4, ND4L, ND5, and ND6, with the remaining 34 subunits encoded by nuclear DNA genes. Complex V comprises 12 or 13 subunits, of which two, ATPase 6 and 8, are encoded by mtDNA. Maximal activity of complex V appears to require tight linking with cardiolipin (see Barth syndrome, p. 182), encoded by nuclear DNA.
Because most mitochondrial proteins, including subunits involved in electron transport, are encoded by nuclear genes, these most often follow autosomal recessive inheritance. As with other metabolic autosomal recessive diseases, disorders resulting from mutations in these genes tend to breed true. However, the disorders resulting from mutations in mtDNA are extremely variable owing to the phenomenon of heteroplasmy (see Figure 7.30, p. 126). The clinical features are mainly a combination of neurological signs—encephalopathy, dementia, ataxia, dystonia, neuropathy, and seizures—and myopathic signs—hypotonia, weakness, and cardiomyopathy with conduction defects. Other symptoms and signs may include deafness, diabetes mellitus, retinal pigmentation, and acidosis may occur. The clinical manifestations are so variable that a mitochondrial cytopathy should be considered as a possibility at any age when the presenting illness has a neurological or myopathic component. Several distinct clinical entities have been determined and, although some of them overlap considerably, there is a degree of genotype–phenotype correlation.
Prenatal Diagnosis of Inborn Errors of Metabolism
For the majority of inborn errors of metabolism in which an abnormal or deficient gene product can be identified, prenatal diagnosis is possible. Biochemical analysis of cultured amniocytes obtained at mid-trimester amniocentesis is possible but has largely given way to earlier testing using direct or cultured chorionic villi (CV), which allows a diagnosis to be made by 12 to 14 weeks’ gestation (p. 325). For many conditions a biochemical analysis on cultured CV tissue is the appropriate test but, increasingly, direct mutation analysis is possible. This avoids the inherent delay of culturing CV tissue and is of particular value for inborn errors for which the biochemical basis is not clearly identified, or where the enzyme is not expressed in amniocytes or CV.
Benson PF, Fensom AH. Genetic biochemical disorders. Oxford: Oxford University Press; 1985.
A good reference source for detailed basic further information on the inborn errors of metabolism.
Clarke JTR. A clinical guide to inherited metabolic diseases. Cambridge: Cambridge University Press; 1996.
A good basic text, problem based and clinically oriented.
Cohn RM, Roth KS. Metabolic disease: a guide to early recognition. Philadelphia: WB Saunders; 1983.
Garrod AE. Inborn errors of metabolism. Lancet. 1908;ii:1-7. 73–79, 142–148, 214–220
Reports of the first inborn errors of metabolism.
Nyhan WL, Ozand PT. Atlas of metabolic diseases. London: Chapman & Hall; 1998.
A detailed text but very readable and full of excellent illustrations and clinical images.
Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Principles and practice of medical genetics, 4th edn, Edinburgh: Churchill Livingstone, 2001.
Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic basis of inherited disease, 8 edn, New York: McGraw Hill, 2000.
Elements