[level-membership-for-pathology-category]
Chapter 16 Liver, biliary system and exocrine pancreas
COMMON CLINICAL PROBLEMS FROM LIVER AND BILIARY SYSTEM DISEASE
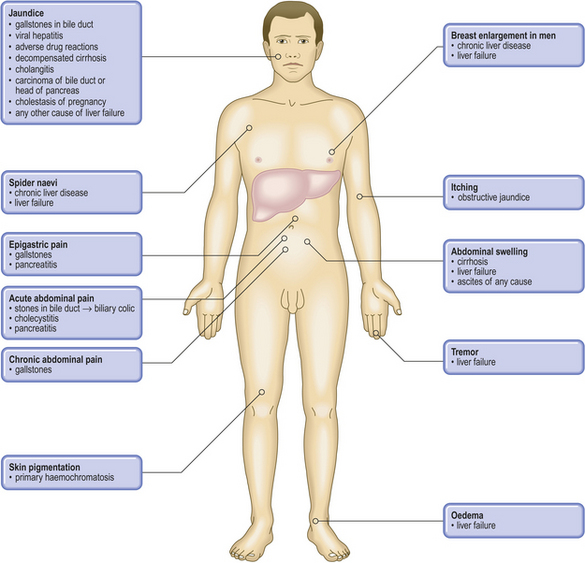
Pathological basis of hepatic signs and symptoms
| Sign or symptom | Pathological basis |
|---|---|
| Jaundice | Haemolysis (increased formation of bilirubin), liver disease (impaired conjugation and/or excretion) or biliary obstruction |
| Dark urine | Conjugated hyperbilirubinaemia (water-soluble) |
| Pale faeces | Biliary obstruction causing lack of bile pigments |
| Spider naevi | Secondary to hyperoestrogenism |
| Gynaecomastia | |
| Oedema | Reduced plasma oncotic pressure due to hypoalbuminaemia |
| Xanthelasma | Cutaneous lipid deposits due to hypercholesterolaemia in chronic biliary obstruction |
| Steatorrhoea | Malabsorption of fat due to lack of bile (e.g. biliary obstruction) |
| Pruritus | Biliary obstruction resulting in bile salt accumulation |
| Ascites | Combination of hypoalbuminaemia, portal hypertension and secondary hyperaldosteronism |
| Bruising or bleeding | Impaired hepatic synthesis of clotting factors |
| Hepatomegaly | Increased size of liver due to inflammation (e.g. hepatitis), infiltration (e.g. amyloid, fat) or tumour (primary or secondary) |
| Haematemesis | Ruptured oesophageal varices due to portal hypertension |
| Encephalopathy | Failure of liver to remove exogenous or endogenous substances mimicking or altering balance of neurotransmitters |
LIVER
NORMAL STRUCTURE AND FUNCTION
Forming the interface between the gastrointestinal tract and the rest of the body, the liver is of crucial importance in metabolising, storing or excreting the absorbed products of digestion. The liver has numerous other vital functions; therefore, the clinical consequences of liver disease are often wide-ranging and, if severe, life-threatening. However, considerable functional reserve and reparative capacity enables many patients to tolerate cellular injury or losses that, in other organs, would imperil their survival.
This wedge-shaped organ, weighing approximately 1.5 kg in the adult, is situated in the right hypochondrial region of the abdominal cavity. It has four lobes: the right is larger than the left; the smaller caudate lobe is situated posteriorly and the quadrate lobe is more anterior. The liver receives blood from two sources:
Blood leaves the liver through the hepatic veins, which drain into the inferior vena cava.
Bile is formed in the liver and drains from it into the right and left hepatic ducts; these fuse to form the common bile duct to be joined by the cystic duct, which communicates with the gallbladder where the bile is stored and concentrated.
Most of the liver comprises liver cells (hepatocytes). These are arranged in plates one cell thick, bordering the vascular sinusoids through which flows hepatic arterial and portal venous blood. The blood flowing through the vascular sinusoids is separated from the liver cells by a thin fenestrated (porous) barrier of cells (endothelial cells and phagocytic Kupffer cells) and the space of Disse. Within the space of Disse the basement membrane is interrupted, thus allowing free exchange of molecules at the liver cell membrane. Blood flowing through the vascular sinusoids drains into hepatic vein branches (central veins or terminal hepatic venules). Bile formed by the liver cells is secreted from them into minute canaliculi which run along the centre of the liver cell plates to drain into the bile duct branches in the portal tracts. Close to the vascular sinusoids in the vicinity of the terminal hepatic venules are stellate cells called the perisinusoidal cells of Ito; these are involved in hepatic fibrosis by synthesising collagen.
The portal tracts each contain three tubular structures, which are branches of:
These constitute the portal triad and are supported by collagen-rich connective tissue.
The microanatomy of the liver can be regarded conceptually as either acinar or lobular (Fig. 16.1):
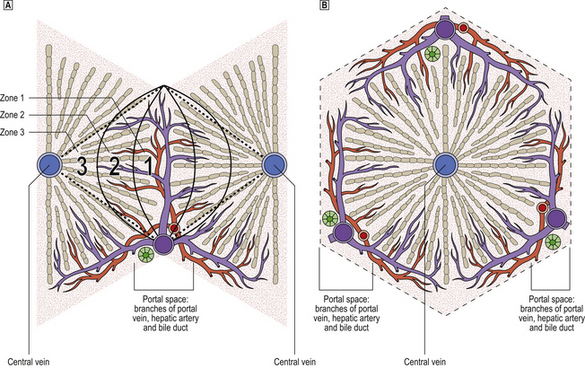
Fig. 16.1 Comparison of acinar and lobular concepts of the microanatomical units of the liver.  The acinar concept explains better the pathophysiology of the liver. Cells in acinar zone 3, being the most remote from the vascular supply in the hilum of the acinus, are consequently the most vulnerable to injury. Death of liver cells in zone 3 results in the observed necrosis bridging between portal tracts and central veins in severe liver injury.
The acinar concept explains better the pathophysiology of the liver. Cells in acinar zone 3, being the most remote from the vascular supply in the hilum of the acinus, are consequently the most vulnerable to injury. Death of liver cells in zone 3 results in the observed necrosis bridging between portal tracts and central veins in severe liver injury.  Lobular units are, however, often easier to perceive in histological sections.
Lobular units are, however, often easier to perceive in histological sections.
Of the two microanatomical concepts—acinar or lobular—the ‘acinar concept’ is now considered to be more useful because it explains better many of the pathophysiological disturbances in liver disease. The zone of liver cells most remote from the axial vessels in the centre of the acinus (acinar zone 3) is the most susceptible to injury resulting from vascular insufficiency, as in circulatory shock or cardiac failure. As adjacent zones 3 are contiguous, liver cell death in this zone is often confluent.
The portal tracts are circumscribed by a boundary of liver cells, known as the limiting plate, which is breached in some forms of chronic liver inflammation; disruption of the limiting plate, when seen in biopsies, denotes that progression to cirrhosis is likely. The liver cells at the portal tract boundary can, in response to bile duct injury or obstruction, undergo a metaplastic change and proliferate to form new bile ductules.
Liver cells are rich in organelles, including numerous mitochondria, lysosomes, peroxisomes (microbodies), and rough and smooth endoplasmic reticulum, reflecting their wide range of metabolic functions. The cytoplasm is also laden with glycogen; this glycogen can be excessive in diabetes and in congenital deficiencies of glycogen debranching enzymes (the glycogenoses).
Liver cells synthesise albumin, clotting factors including fibrinogen, some complement components, alpha-1 antitrypsin, etc., and remove from the body many waste products and potentially toxic substances. Through the expression of specific receptors on the liver cells, the liver—the site of action of statins, the cholesterol-lowering drugs—has a major role in the uptake and metabolism of the low-density lipoproteins involved in atheroma (Ch. 13). Liver cells also metabolise or activate many other drugs. Extensive disease of the liver therefore affects many vital functions and has profound effects on the body.
Liver cells contain many enzymes, some of which are diagnostically important. Their release from damaged or dying liver cells into the blood, where their activity can be measured, indicates the presence and severity of liver disease (Table 16.1). These enzymes include:
Table 16.1 Diagnostic usefulness of serum analyses in liver disease
| Test | Deviation from normal | Interpretation |
|---|---|---|
| AlbuminNormal 35–50 g/l | ↓ | Liver failure |
| Prothrombin timeNormal < 15 s | ↑ | Liver failure |
| Alanine aminotransferase (ALT)Normal < 40 IU/l | ↑ | Hepatocellular injury |
| Aspartate aminotransferase (AST)Normal < 40 IU/l | ↑ | Hepatocellular injury |
| Gamma-glutamyltransferase (GGT)Normal < 50 IU/l | ↑ | Hepatocellular injury (centrilobular) |
| Alkaline phosphataseNormal < 100 IU/l | ↑ |
BilirubinNormal 5–12 μmol/l↑
IgM anti-HAV antibodyPresentHepatitis AHBsAgPresentHepatitis B or carrierHBeAgPresentActive hepatitis B infectionAnti-HCV antibodyPresentHepatitis C virus exposureHCV RNAPresentActive hepatitis C infectionCaeruloplasmin↓Wilson’s diseaseIgA↑Alcoholic cirrhosisIgG↑Autoimmune hepatitisIgM↑Primary biliary cirrhosisAnti-mitochondrial antibodyPresentPrimary biliary cirrhosisAnti-smooth muscle, antinuclear or anti-LKM antibodiesPresentAutoimmune hepatitisFerritin↑HaemochromatosisAlpha-1 antitrypsin↓Alpha-1 antitrypsin deficiencyAlpha-fetoprotein (AFP) (normally undetectable)↑Liver cell carcinoma
HAV, hepatitis A virus; HBsAg, hepatitis B surface antigen; HCV, hepatitis C virus; LKM, liver and kidney microsomal antigen.
All cells in the liver are capable of regeneration. The liver cells are classified as stable—that is, they are not normally replicating but will do so if the liver is injured. This regenerative capacity is vital in the recovery of patients with liver damage due to viruses, drugs or trauma, but if the damage is persistent or occurs repeatedly, it can result in loss of the normal acinar or lobular structure and its replacement by regenerative liver cell nodules which are functionally inefficient. This is the condition called cirrhosis.
Some changes occur naturally in the liver with age. In the fetus, the liver is a relatively larger organ compared to the rest of the body. It is a major site of haemopoiesis and the adult liver can revert to this activity in some haematological disorders. The fetal liver synthesises alpha-fetoprotein, a fetal serum protein, and this is replaced by albumin towards the end of gestation. Alpha-fetoprotein synthesis by the adult liver usually denotes the presence of a primary liver cell carcinoma. With advancing age, the liver shrinks and becomes dark brown due to an increased amount of lipofuscin pigment in the liver cells (‘brown atrophy’).
INVESTIGATION OF LIVER DISEASE
The investigation of a patient with liver disease commonly includes:
These investigations complement careful history-taking and a thorough clinical examination.
Biochemistry
Bilirubin
Bilirubin pigment is a breakdown product of the haem moiety of haemoglobin (Fig. 16.2). It is produced at sites of red cell destruction (e.g. spleen) and circulates in the blood in an unconjugated water-insoluble form bound to albumin. In the liver it is conjugated to glucuronic acid by the enzyme glucuronyl transferase. Conjugated bilirubin is water-soluble and can therefore appear in the urine if the outflow of bile from the liver is interrupted; the patient’s urine then becomes stained with conjugated bilirubin. Bilirubin is converted by bacteria in the intestine to faecal urobilinogen (stercobilinogen), some of which is absorbed and then excreted, mostly in the bile to complete its enterohepatic circulation or, in only trace amounts normally, by the kidneys to appear in the urine as urobilinogen. Stercobilinogen is oxidised to stercobilin (faecal urobilin), the principal faecal pigment.
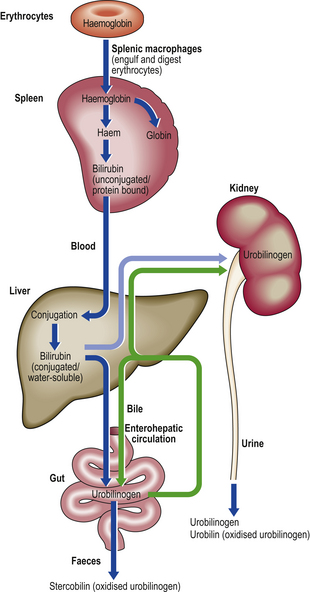
Fig. 16.2 Simplified pathways of bilirubin metabolism. Excessive breakdown of haemoglobin, as in haemolytic anaemias, will lead to increased biliary excretion of bilirubin. Biliary obstruction will cause regurgitation of conjugated water-soluble bilirubin into the blood which is then excreted in the urine, causing it to darken. Liver cell damage in hepatitis will cause impaired biliary excretion of urobilinogen and conjugated bilirubin; these are excreted in the urine, causing it to darken. The enterohepatic circulation, which involves urobilinogen, also returns cholic acid and chenodeoxycholic acid to the liver; this enhances bile secretion.
In early or recovering viral hepatitis, impaired biliary excretion results in preformed stercobilinogen appearing in the urine in excess as urobilinogen; this is one sensitive marker of early liver injury. In well-established biliary obstruction, the urinary urobilinogen concentration falls, because the cessation of biliary excretion into the gut results in sustained absence of synthesis of faecal urobilinogen.
Enzymes
In liver cell injury, damage to the membranes of cells and their organelles allows intracellular enzymes to leak into the blood, where the elevated concentrations can be measured. Examples include ALT, AST and GGT. Their diagnostic usefulness is summarised in Table 16.1.
The enzyme alkaline phosphatase is normally present in bile. Obstruction to the flow of bile, by gallstones for example, causes regurgitation of alkaline phosphatase into the blood, resulting in increased serum concentrations.
Many of these enzymes are not exclusively specific to the liver; therefore the results of diagnostic serum assays need careful interpretation.
Albumin
Albumin is a major serum protein synthesised by the liver cells. It has a relatively long half-life, compared to that of clotting factors (see below), so liver damage has to persist before decreased serum levels are found. In chronic liver disease, such as cirrhosis, a low serum albumin concentration is an important manifestation of liver failure, which results in peripheral oedema and contributes to the presence of ascites due to a reduction in plasma oncotic pressure.
Clotting factors
Liver cells synthesise the vitamin K-dependent clotting factors, deficiency of which results in a bleeding tendency. This can be detected in the laboratory by measuring the prothrombin time. A prolonged bleeding and prothrombin time is a further manifestation of liver failure and, because these clotting factors have a relatively short half-life, deficiency may be found quite early in the course of the illness. The prothrombin time should be measured before performing a liver biopsy or undertaking surgery on a patient with liver disease to avoid the risk of unexpected haemorrhage. These clotting factor deficiencies can be corrected by administration of high doses of vitamin K or of the clotting factors themselves.
Immunology
Although insignificant amounts of immunoglobulins are synthesised in the liver, immunological abnormalities often accompany liver disease and are useful diagnostic markers. These abnormalities include the appearance in the patient’s blood of auto-antibodies to normal tissue antigens. The antibodies are not responsible for the tissue damage in the liver diseases with which they are associated. Examples include:
Polyclonal immunoglobulin elevations also occur:
Antibodies to hepatitis viruses are, in some instances (e.g. hepatitis A), clinically useful markers of these infections. Viral antigens or nucleic acids can also be tested for.
Imaging
Techniques used to visualise the liver and detect lesions within it include:
Biopsy
The two common types of liver biopsy are:
Both procedures carry a small but significant risk of haemorrhage and biliary leakage from the biopsy site. Bile duct obstruction is a contraindication to liver biopsy because of the increased risk of biliary peritonitis from bile leakage from the biopsy site. The risk must be outweighed by the likely therapeutic benefit to the patient resulting from an accurate assessment of their liver disease.
Most liver diseases produce diffuse abnormalities in the organ; a biopsy from any part of it will therefore be representative. Focal lesions such as tumours may be missed, particularly by percutaneous needle sampling, but the biopsy needle can be guided to them by using imaging techniques such as ultrasonography.
Liver biopsies are examined by light microscopy after sectioning and staining. Unlike renal biopsies, little additional clinically useful information is obtained by examining liver biopsies with the electron microscope.
JAUNDICE
Jaundice (or icterus) is the name given to yellowing of the skin and mucosal surfaces due to the presence of bilirubin. Usually jaundice is observable when the serum bilirubin concentration exceeds 40 micromol/l. Note, however, that:
The accumulation of bilirubin in the skin may cause some embarrassment to the patient and, often if due to biliary obstruction, discomfort due to pruritus.
Jaundice in infants
Physiological neonatal jaundice is relatively common, particularly in premature infants. Although it causes understandable parental anxiety, the jaundice is rarely severe and it fades as liver function matures. However, high bilirubin levels in infancy can be directly harmful. Because the neonatal blood–brain barrier is relatively permeable, unconjugated bilirubin can accumulate in the lipid-rich brain tissue, causing bilirubin encephalopathy or kernicterus; this can be avoided by phototherapy or, in severe cases, exchange transfusion.
Worsening jaundice may be one of the clinical features alerting to the presence of a congenital abnormality within the hepato-biliary system. Such abnormalities may be:
Structural congenital abnormalities include:
Functional abnormalities include congenital metabolic defects involving the liver (see Ch. 7) and congenital hyperbilirubinaemias.
Classification of jaundice
Jaundice may be classified as pre-hepatic, intrahepatic or post-hepatic, depending on the site of the lesion, or conjugated and unconjugated, based on chemical analysis of the bilirubin in the blood or by deduction from the colour of the patient’s urine. Only conjugated bilirubin is sufficiently water soluble to be excreted in the urine.
Pre-hepatic jaundice
The main cause of pre-hepatic jaundice is haemolysis, due for example to hereditary spherocytosis or autoimmune red cell destruction (see Ch. 23). In these conditions there is excessive production of bilirubin from the haemoglobin released from lysed red cells. Because the excess bilirubin is unconjugated, it is not excretable in the urine; the urine colour is normal (hence the synonym ‘acholuric jaundice’). The bile, however, may contain so much bilirubin that there is a risk of pigment gallstone formation.
Intrahepatic jaundice
Hepatic disorders in which jaundice may be a feature include:
In these conditions there is accumulation of bilirubin within the liver (intrahepatic cholestasis), often histologically evident in biopsies as plugs of bile pigment distending canaliculi. The excess bilirubin is predominantly conjugated, is therefore water soluble and is excreted in the urine, causing darkening; this is a simple but diagnostically useful observation.
Post-hepatic jaundice
Obstruction of the extrahepatic bile ducts is an important cause of jaundice necessitating urgent investigation and alleviation in order to prevent serious damage to the liver. Important causes are:
As with intrahepatic causes, some of which also directly interfere with biliary drainage (e.g. primary biliary cirrhosis, sclerosing cholangitis), the excess bilirubin is conjugated and darkens the urine. Conversely, the patient’s faeces are pale. Pruritus is a common and troublesome symptom, probably due to bile salt accumulation.
ACUTE LIVER INJURY



Liver injury is conveniently divided into acute and chronic for the purposes of description and clinical management. However, in practice, the same cause may produce either an acute or a chronic illness, in the latter event not necessarily with any preceding clinically evident acute phase. For example, viral hepatitis is considered here under the heading of acute liver injury, but it can lead to chronic liver damage.
Aetiology
The major causes of acute liver injury are:
Direct physical injury to the liver, such as laceration in a road traffic accident, is another important form of acute liver injury, but the focal nature of the injury contrasts with the diffuse injury produced by the agents listed above. Recovery from acute liver injury, focal or diffuse, is attributable to the capacity of the organ for cellular regeneration.
Clinicopathological features
The clinical and laboratory manifestations of acute liver injury are:
Most of the signs and symptoms of acute liver damage are predictable from the known functions of the liver. The best known is jaundice (or icterus) due to failure of the liver to secrete bile at the rate at which it is formed in the body from the destruction of red cells. Severe acute liver damage can lead to bruising and haemorrhage, due to clotting factor deficiency, and coma due to the accumulation of toxic metabolites that mimic neurotransmitters (‘false neurotransmitters’).
Laboratory investigations
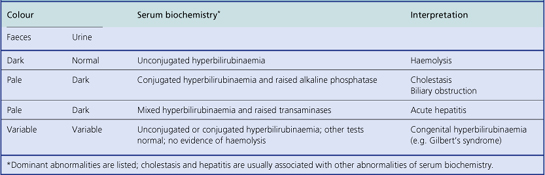
Laboratory investigations will reveal evidence of liver cell damage, in that there will be elevated levels of serum enzymes, particularly the transaminases, and bilirubin. Liver cell damage results in some impairment of bilirubin conjugation, but also failure to excrete conjugated bilirubin and any stercobilinogen absorbed from the gut. Consequently, the urine is darkened by the presence of excess conjugated bilirubin and urobilin (derived by oxidation from urobilinogen) that cannot be excreted by the liver (Fig. 16.2). Eventually, as the liver damage persists, urobilinogen disappears from the urine because little or no bilirubin is being excreted by the liver. Jaundice due to bile duct obstruction—commonly by gallstones—also results in dark urine due to excess conjugated bilirubin that cannot be excreted by the liver; urobilinogen is usually absent, unless the obstruction is of very recent onset or intermittent, because no bilirubin reaches the intestine. Examination of urine and faeces (for colour) can therefore assist in the differential diagnosis of jaundice (Table 16.2).
Histology
In almost all cases of acute liver injury there will be liver cell degeneration or death and an inflammatory reaction. Superimposed on this uniform reaction to acute injury are, in many cases, diagnostic changes specific to the causative agent.
Also evident in liver biopsies will be the pattern of cell damage, from which the prognosis can be deduced (Fig. 16.3):
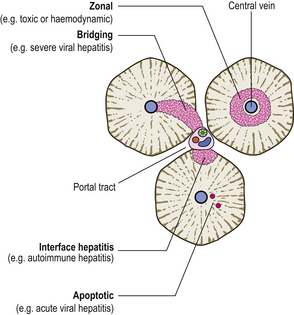
Fig. 16.3 Patterns of liver cell death and their clinicopathological significance. Death of the liver cells immediately surrounding central veins denotes cardiac failure, some other impediment to venous drainage, or some toxic cause (e.g. paracetamol overdose). Bridging necrosis (in acinar zone 3) is a feature of severe hepatitis. Interface hepatitis is death of liver cells at the margin of the portal tracts; this is a feature of chronic hepatitis due to a variety of causes. Apoptotic death of single cells is typical of acute viral hepatitis.
Viral hepatitis



Hepatitis viruses
The main hepatitis viruses (Table 16.3) are:
Table 16.3Hepatitis viruses: their characteristics and associated diseases (delta agent, a defective virus, is not included)
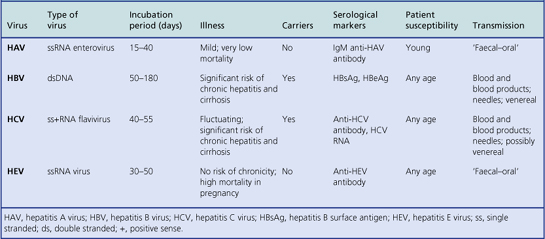
These hepatitis viruses are immunologically distinct. Infection usually confers life-long immunity to the infecting virus but not to the others.
The clinical features range from a trivial illness without jaundice (anicteric hepatitis) which may escape detection (this is a common result of HAV infection) to a more significant illness with jaundice and other clinical evidence of disturbed liver function. Sometimes the illness is dominated by jaundice, with little elevation of serum transaminases (cholestatic hepatitis). Severe infection leads to overt liver failure.
Yellow fever, caused by a group B arbovirus, shares many clinical and histological features with the illness usually designated viral hepatitis, but it is not normally included within this group for the purposes of description, mainly because its geographical distribution is very restricted.
The liver may also become infected by many other viruses, but these are not regarded as ‘hepatitis viruses’ because the infection is not confined to the liver. Examples include:
Hepatitis A virus
The main characteristics of hepatitis A are:
Infection by HAV used to be called ‘infectious hepatitis’ because of its common occurrence in epidemics, though it also occurs sporadically. In most countries, infection by the virus is common, usually in youth; the resulting illness is often very mild and jaundice absent or so slight that it escapes notice. Overt jaundice and clinical recognition of the infection is less common. Hepatitis sufficiently severe to warrant hospital admission is unusual, and long-term sequelae or death are exceptional rarities. It is therefore a relatively benign infection.
HAV passes from one individual to another by ‘faecal– oral’ transmission—usually indirectly, such as by the contamination of food and drinking water with sewage. Because the virus is excreted in the faeces before jaundice appears, thus leading to the recognition of the illness and isolation of the patient, many other individuals can be rapidly exposed to the hazard of infection. The incubation period is relatively short. HAV produces liver cell damage by a direct cytopathic effect.
Specific diagnosis is made by seeking an IgM-class antibody to HAV in the patient’s serum; this indicates recent infection. A carrier state does not exist.
Hepatitis B virus
The main characteristics of hepatitis B are:
Infection by HBV used to be called ‘serum hepatitis’ because it was known to be transmitted by blood and blood products. This is because infected, but apparently healthy, individuals can carry the virus in their blood and pass it on to others by the transfusion of blood or its products. This mode of transmission is much less common now that blood donors are screened by testing for the presence of the virus. However, the term ‘serum hepatitis’ has been abandoned because it misleadingly excludes transmission of the virus by other methods, notably venereally; the disease is often transmitted between homosexual males. HBV can also be transmitted by contaminated needles, such as may be used for tattooing or by drug addicts. There is a relatively high incidence of the carrier state in underdeveloped countries and the virus can be transmitted vertically from mother to child—in utero, during delivery or through intimate post-natal contact.
Specific diagnosis is made by seeking the hepatitis B surface antigen (HBsAg, formerly known as ‘Australia antigen’ because it was first detected in the serum of an Australian aborigine). The presence of the ‘e’ antigen (HBeAg) in the patient’s serum indicates active viral replication.
HBV produces liver cell damage not by a direct cytopathic effect but by causing viral antigens to appear on the cell surface (HBsAg); these are then recognised by the body’s immune system and the infected liver cells are destroyed (Fig. 16.4). Thus, if immunity is generally impaired or there is specific tolerance to the antigen, the virus can survive in the liver cells without causing damage; such patients become asymptomatic carriers of the virus and their body fluids are a hazard to other individuals. Liver biopsies of HBV-infected carriers show that the liver cells have a ground-glass texture to their cytoplasm due to the abundance of virus particles.
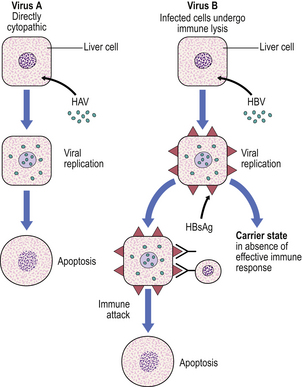
Fig. 16.4 Comparison of the pathogenesis of HAV and HBV hepatitis. The A virus, in contrast to the B virus, appears to be directly cytopathic. The B virus evokes liver cell injury by causing viral antigens to be expressed on the liver cell surface (HBsAg, hepatitis B surface antigen). The infected cells are eliminated immunologically, but an asymptomatic carrier state can ensue in the absence of specific immunity. The pathogenesis of other hepatitis viral infections is less certain.
HBV infection is much more serious than HAV. Infection is more likely to produce a clinical illness and jaundice, and it is more likely to result in long-term sequelae such as chronic hepatitis and cirrhosis, or even death due to fulminating acute infection causing extensive hepatic necrosis. HBV is also involved in the pathogenesis of liver cell carcinoma.
Hepatitis C virus
Hepatitis C virus was characterised in the late 1980s. Its main features are:
HCV has been an important cause of hepatitis following blood transfusion and the administration of clotting factor concentrates. Indeed, its existence was first suspected after HBV had been excluded as a possible cause by blood donor screening; it used to be known as ‘non-A, non-B’ hepatitis. In many countries, transmission of HCV by blood transfusion and blood product administration is much less common now that donors are screened for HCV.
The initial illness is often asymptomatic and the abnormalities of liver biochemistry (e.g. raised serum transaminases) are usually fluctuant. However, despite these misleadingly benign signals, the infection is prone to chronicity and cirrhosis is a frequent consequence.
Hepatitis E virus and other ‘non-A, non-B’ viruses
There are other authentic hepatitis viruses. The best characterised is a water-borne agent, distinct from HAV; it has been designated hepatitis E virus (HEV). Fortunately, the disease rarely progresses to chronicity and, as with HAV, full recovery is usual except in pregnancy, when it is associated with a high mortality rate.
Other ‘non-A, non-B’ viruses associated with blood transfusions and blood products include hepatitis G virus and the TT virus. Their role in liver disease appears to be relatively minor.
Histology
Histological features of acute viral hepatitis are:
Although the pathogenesis of the liver cell damage resulting from HAV and HBV infection is different, the changes in the liver in a typical case are very similar (Fig. 16.5). The principal features are:
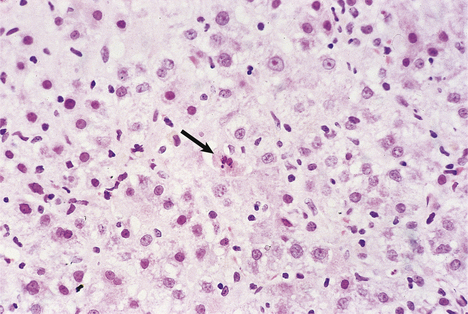
Fig. 16.5 Liver histology in acute viral hepatitis. There is disarray of the liver cell plates, accompanied by an inflammatory infiltrate and liver cell apoptosis (arrowed).
The swelling of liver cells, portal oedema and the infiltration by inflammatory cells are responsible for hepatomegaly in viral hepatitis.
In severe viral hepatitis there may be confluent liver cell death, resulting in liver failure. At autopsy the liver will be small, have a wrinkled instead of a smooth capsule, and show extensive necrosis on the cut surfaces.
HCV infection often results in numerous lymphocytes within the vascular sinusoids and fatty change in the hepatocytes, sometimes with relatively little evidence of active liver cell death. This combination of features is unusual in HAV or HBV infection.
Alcoholic liver injury



Alcohol (ethyl alcohol) is a common cause of acute and chronic liver injury. The spectrum of alcoholic liver injury includes:
Histology
The fatty change (steatosis) is evident as fat globules within the cytoplasm of the liver cells; those in the centrilobular or acinar zone 3 areas are usually most severely affected. Fatty change by itself is a relatively non-specific event because it is seen in many disorders. More specific, but not exclusive, to alcoholic liver injury is Mallory’s hyalin; this is an intracytoplasmic aggregate of ubiquinated intermediate filaments in the liver cells (Fig. 16.6). This is usually accompanied by acute inflammation and, in contrast to pure fatty change, denotes an appreciable risk of progression to irreversible architectural disturbance and possibly cirrhosis.
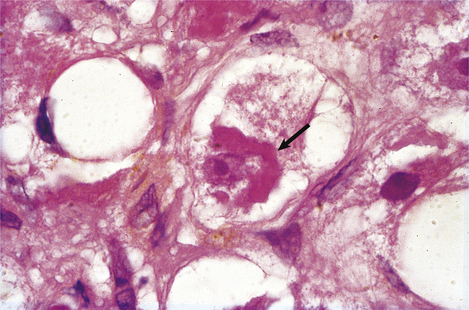
Pathogenesis
Alcohol produces liver injury by a variety of mechanisms (Fig. 16.7):
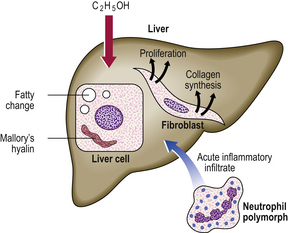
Fig. 16.7 Pathogenesis of alcoholic liver disease. There is increased peripheral release of fatty acids, and increased synthesis of fatty acids and triglycerides within the liver cells. Acetaldehyde, a product of alcohol metabolism, is probably responsible for liver cell injury, manifested by the formation of Mallory’s hyalin. There is increased collagen synthesis by fibroblasts and by the perisinusoidal cells of Ito.
Sustained alcoholic liver injury results in irreversible architectural disturbance, initially the linking of portal tracts and/or terminal hepatic venules by fibrous tissue, and ultimately nodular regeneration of the liver cells; this is alcoholic cirrhosis.
Recommended safe limits for alcohol continue to be debated. However, the current consensus is that, in men, up to 21 units of alcohol per week is unlikely to be harmful. In women, the safe limit is usually regarded as 14 units of alcohol per week. (One unit of alcohol is 10 ml by volume, equivalent to: half a pint, or c. 300 ml, of beer; or 25 ml of spirits. A small glass of wine contains 1.5 units.)
Drug-induced liver injury



Approximately 10% of all adverse reactions to drugs involve the liver. This is not surprising in view of the central role played by the liver in metabolism and in the conjugation and elimination of toxic substances from the body. A full drug history should therefore be taken from any patient presenting with liver disease, and any suspected or proven association reported to the appropriate body (in the UK, the Commission on Human Medicines).
Adverse drug reactions may be predictable or unpredictable (Ch. 2). They may be caused by injury to the liver cells (hepatocellular) that is pathologically indistinguishable from viral hepatitis, or to bile production or excretion (cholestatic). Predictable reactions will occur in any individual if exposed to a sufficient dose; examples include coagulative centrilobular necrosis due to paracetamol overdose and cholestatic jaundice due to methyl testosterone. Unpredictable reactions include an idiosyncratic response to a drug and are not necessarily dose-related; examples include cholestatic jaundice due to chlorpromazine.
Acute biliary obstruction




Acute obstruction of the main bile ducts is most commonly due to gallstones. Clinically, it usually results in colicky pain and jaundice. If there is superimposed infection of the biliary tract, the ducts become inflamed (cholangitis) and the patient develops a fever. Cholangitis can lead to the formation of liver abscesses.
Bile accumulates within the liver, initially in the canaliculi (Fig. 16.8) and later within the intrahepatic bile ducts. Rupture of these may result in extravasation of bile into the adjacent liver tissue; the resulting necrosis has been misnamed as a ‘bile infarct’. The portal tracts are oedematous and infiltrated with neutrophil polymorphs. The hepatocytes at the edge of the portal tract undergo ductular metaplasia. Cholangitis is recognised histologically by the presence of neutrophil polymorphs in the bile ducts.
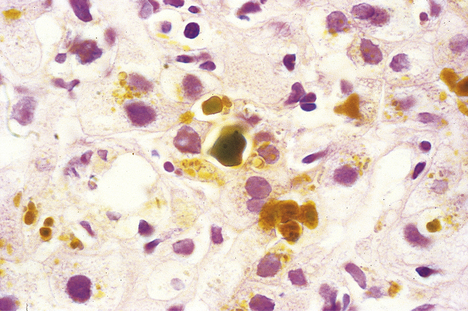
Fig. 16.8 Histology of intrahepatic cholestasis in biliary obstruction. The bile canaliculi are stuffed with stagnant brown bile that cannot be discharged from the liver because the common bile duct is blocked by a gallstone. Similar appearances result from viral hepatitis and some adverse drug reactions.
Repeated episodes of biliary obstruction lead to portal tract fibrosis and nodular regeneration of the liver cells—secondary biliary cirrhosis.
CHRONIC LIVER DISEASES
Chronic liver diseases, of which there are many, are a common clinical problem. Some follow a clinically evident episode of acute liver injury; others present insidiously and may be asymptomatic until the later stages. Many forms of chronic liver disease culminate in hepatic cirrhosis.
Chronic hepatitis



Chronic hepatitis is generally defined as inflammation of the liver lasting at least 6 months without evidence of resolution. The inflammation and consequent liver cell injury cause a sustained elevation of the serum transaminases, but confirmation of the diagnosis and precise classification of the disease in an individual patient usually requires a liver biopsy.
Classification
Classification of chronic hepatitis involves assessment of:
Clues to the aetiological type of chronic hepatitis can be discovered from examination of the liver biopsy (Table 16.4), but in most cases the cause of the liver disease is already known from previous investigations (e.g. markers of HBV or HCV infection). The main purpose of the biopsy is to assess the severity of the liver damage and thereby to guide decisions about the patient’s clinical management.
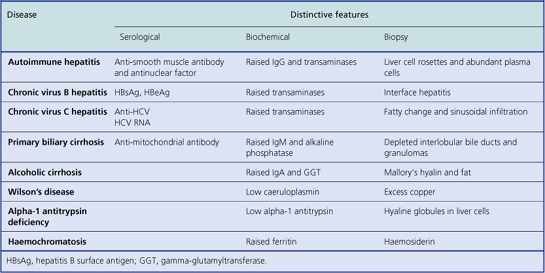
The grade of severity of the liver cell damage and inflammation is based on assessment of:
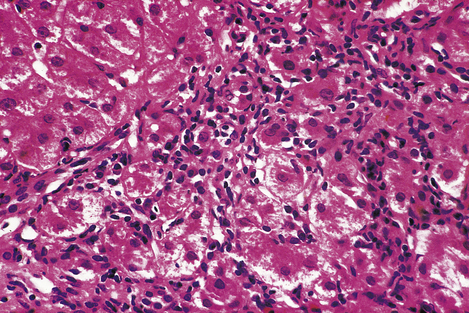
Fig. 16.9 Histology of interface hepatitis. Degenerate liver cells adjacent to the edge of a portal tract are associated with dense inflammatory cell infiltration. This leads to progressive erosion of liver architecture, frequently culminating in cirrhosis.
Each of these features is scored using published protocols, such as those devised by Ishak and others, to derive a total histological activity index. This numerical score can then be translated into a named grade of severity, which may be minimal, mild, moderate or severe chronic hepatitis (Fig. 16.10). The greater the severity, the greater the risk of progression to significant architectural disturbance or, eventually, cirrhosis.
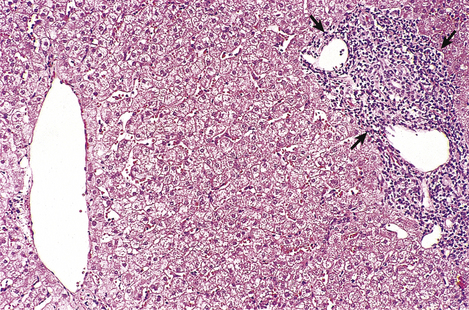
Fig. 16.10 Histology of mild chronic hepatitis. The dense lymphocytic infiltrate is confined to the portal tract (arrowed) and there is no erosion of hepatic architecture. This grade of activity progresses only rarely to cirrhosis.
The stage of the architectural disturbance is also assessed histologically and ranges from normal architecture, fibrous enlargement of portal tracts, bridging fibrosis, to cirrhosis. The greater the disturbance, the less likely it is to be reversible when the inflammatory activity has subsided.
Iron overload and the liver





In haemosiderosis (or ‘siderosis’ of the liver) and haemochromatosis, the liver is dark brown due to the deposition of excess iron in the form of haemosiderin (an iron-rich protein). The haemosiderin is visible in histological sections as light brown granules; its identity can be confirmed by Perls’ stain for iron.
The distinction between these two entities is as follows:
Haemosiderosis
Haemosiderosis usually results from parenteral iron overload, as in the case of a patient with aplastic anaemia treated with blood transfusions. In this instance, the iron liberated from eventual lysis of the transfused blood cells cannot be reutilised in the patient for haemoglobin synthesis because of absence of haemopoiesis, so it accumulates in various organs, notably in the liver where it is stored as haemosiderin, mainly in Kupffer cells. Haemosiderosis is not commonly associated with significant liver damage or progression to cirrhosis; however, if cirrhosis does develop as a result of massive iron overload by this mechanism, the condition is referred to as secondary haemochromatosis.
Haemosiderosis can also occur in alcoholic liver disease because alcohol enhances iron absorption from the gut. Any hepatic architectural disturbance is more likely to be due to the alcohol than the iron in these cases.
Primary (genetic) haemochromatosis
Primary haemochromatosis is the most common form of iron overload. It is an inherited autosomal recessive (except for a rare autosomal dominant type) disease usually due to HFE gene defects (most commonly a C282Y mutation) on chromosome 6 near the HLA-A locus. Heterozygotes have increased absorption of iron, but only in homozygotes does this reach dangerous levels. The defect causes excessive absorption of iron in the small intestine even when transferrin, the iron-binding protein in the blood, is fully saturated. By adult life the total body iron stores may reach as high as 40–60 g (normal about 4 g). The disease is clinically manifested more commonly in men than women; pre-menopausal women compensate for the excessive iron absorption through natural iron loss due to menstrual bleeding.
For many years the iron is deposited, as haemosiderin, in the hepatocytes without any clinical effects. Eventually, however, the iron deposition becomes more extensive, involving Kupffer cells, bile duct epithelium and portal tract connective tissue (Fig. 16.11). Hepatic fibrosis ensues, followed by cirrhosis. In advanced cases the haemosiderin is deposited in other tissues, notably in endocrine organs; the clinical syndrome of ‘bronze diabetes’ is due to concomitant iron-induced damage to the pancreatic islets (resulting in diabetes) and the effect of raised melanotrophin levels on the skin (resulting in bronze coloration). Cardiac failure and impotence may also result.
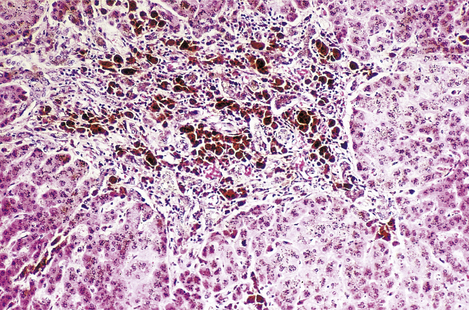
Fig. 16.11 Histology of haemochromatosis. The portal tract and the liver cells contain brown granules of haemosiderin. This has caused portal fibrosis and, if untreated, will eventually lead to cirrhosis.
If the condition is diagnosed in the pre-cirrhotic phase, the process may be arrested by depleting the body’s iron stores by regular venesection or by the administration of desferrioxamine, a chelating agent. The genetic aetiology of primary haemochromatosis means that it is important to screen first-degree relatives to identify those at risk from this potentially treatable liver disease.
Secondary haemochromatosis
Unlike primary haemochromatosis, secondary haemochromatosis is not a single discrete disease entity. It may be due to increased iron in the diet (e.g. excessive medicinal iron tablets) or to parenteral iron loading. For example, secondary haemochromatosis may be seen in patients with aplastic anaemia and haemoglobinopathies who have received multiple transfusions. It is possible, however, that the liver damage in some cases is due to co-existent post-transfusion viral hepatitis rather than iron overload.
Wilson’s disease (hepatolenticular degeneration)




Wilson’s disease is a rare but treatable inherited autosomal recessive disorder in which copper accumulates in the liver and in the basal ganglia of the brain. The underlying defect is failure of the liver to excrete copper in the bile. Copper accumulation in the liver causes chronic hepatitis and, ultimately, cirrhosis. Copper deposition in the brain causes severe progressive neurological disability. Surplus copper released into the blood may cause episodes of haemolysis.
Clinically Wilson’s disease is recognised by the combination of hepatic and neurological abnormalities and from the presence of characteristic brown Kayser–Fleischer rings at the corneal limbus. Diagnosis is confirmed by finding a low concentration of caeruloplasmin (a copper-containing protein) in the serum and an excess of copper in the liver biopsy. It is treated with penicillamine, a chelating agent that binds the copper and eliminates it in the urine.
Alpha-1 antitrypsin deficiency



Alpha-1 antitrypsin is a serum protein with alpha mobility on electrophoresis. It is normally synthesised in the liver and is immediately secreted into the blood, where it has antiproteolytic properties. Several phenotypes occur in the population. The normal phenotype is referred to as MM.
Phenotypes of alpha-1 antitrypsin associated with low serum levels are not uncommon. They are characterized in the laboratory by their unusually fast or slow electrophoretic mobility. Heterozygous states, such as MZ or MS, are not considered to have any significance, but homozygous states, such as ZZ, are associated with a predisposition to pulmonary emphysema and hepatic cirrhosis. These unusual phenotypes of alpha-1 antitrypsin are not readily released from the liver cell after synthesis; low serum levels are therefore found and the unreleased protein accumulates in the cytoplasm of periportal hepatocytes as hyaline intracytoplasmic globules.
Autoimmune liver disease
There are two main chronic liver diseases with an autoimmune basis:
In most cases these autoimmune diseases are distinguishable on investigation, including liver biopsy. However, in a small proportion, there appears to be some overlap and a clear-cut distinction may not be possible. Like almost all other autoimmune diseases, they are more common in females.
Autoimmune hepatitis



Autoimmune hepatitis occurs more commonly in females and is characterised histologically by the appearance of chronic hepatitis, often dominated by numerous plasma cells and rosette-like arrangements of swollen liver cells. Formerly called ‘lupoid’ hepatitis, it is not related to systemic lupus erythematosus, although it shares the presence of antinuclear antibodies in the serum with that condition. Auto-antibodies to smooth muscle antigens or to nuclear DNA or to liver–kidney microsomal (LKM) antigens are also often present. Associated biochemical factors include raised serum IgG and transaminases. Patients often benefit from treatment with steroids.
Primary biliary cirrhosis



Primary biliary cirrhosis is misleadingly named because cirrhosis is a late manifestation of the disease and many patients have the condition diagnosed before this stage is reached.
The stages in the development of the disease are:
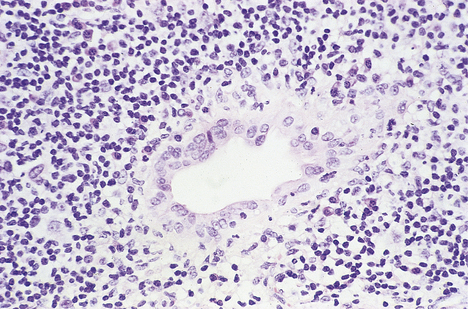
Fig. 16.12 Histology of primary biliary cirrhosis. Lymphocytes surround a bile duct, the epithelium of which is damaged as a result of the autoimmune process.
Copper accumulates in the liver cells because it can no longer be adequately excreted in the bile.
In addition to the biopsy appearances, which may not be absolutely diagnostic in the later stages, other important features of primary biliary cirrhosis include:
Sclerosing cholangitis
Primary sclerosing cholangitis is a chronic inflammatory process affecting intrahepatic, and sometimes extrahepatic, bile ducts. Initially, the ducts are surrounded by a mantle of chronic inflammatory cells, but this is eventually replaced by fibrosis and obliteration of the ducts. In c.10% of patients, cholangiocarcinoma (bile duct carcinoma) develops.
There is an association with chronic inflammatory bowel disease, particularly ulcerative colitis.
Non-alcoholic fatty liver disease
The precise diagnosis of fatty liver disease is a common problem. Alcohol is a major cause of a fatty liver, the fat accumulating as globules within the cytoplasm of the liver cells. Non-alcoholic fatty liver disease (NAFLD) occurs in the metabolic syndrome that is characterised by the presence of any three of the following conditions: central obesity, type 2 diabetes mellitus, low levels of HDL cholesterol, hypertriglyceridaemia, and hypertension (Ch. 7). In such patients, if a liver biopsy reveals fat within hepatocytes accompanied by inflammation, the liver lesion can be described as primary non-alcoholic steatohepatitis (NASH). Secondary NASH is due to a wide range of other causes, such as drugs (e.g. methotrexate).
Acute fatty liver of pregnancy is a rare but serious condition occurring in the third trimester or in the immediate post-natal period.
CIRRHOSIS





The liver has considerable powers of regeneration such that quite severe loss of liver cells can be restored and normal architecture retained. However, if the loss of liver cells is recurrent or takes place against a background of severe architectural disturbance (e.g. bridging necrosis) then cirrhosis can result.
Cirrhosis is not a specific disease; it is the end result of a variety of diseases causing chronic liver injury. It is an irreversible disturbance of hepatic architecture, affecting the entire liver, and is characterised by:
The amount of fibrous tissue greatly exceeds that in the normal liver and the liver cells are no longer arranged in acini or lobules, but regenerate after various forms of injury in a nodular pattern (Fig. 16.13).
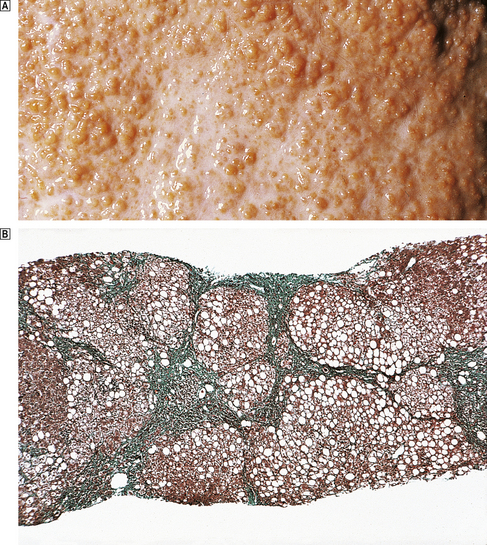
Fig. 16.13 Cirrhotic liver.  External surface of a cirrhotic liver studded with regeneration nodules about 2 mm in diameter.
External surface of a cirrhotic liver studded with regeneration nodules about 2 mm in diameter.  Histology of a needle biopsy of a cirrhotic liver revealing regeneration nodules surrounded by dense connective tissue. (Masson trichrome stain in which connective tissue is green.)
Histology of a needle biopsy of a cirrhotic liver revealing regeneration nodules surrounded by dense connective tissue. (Masson trichrome stain in which connective tissue is green.)
The regeneration nodules lack the well-organised zonal structure of the normal liver lobules or acini. The blood perfuses them in a haphazard fashion, resulting in a relatively inefficient organ that is prone to failure.
Classification
Cirrhosis is classified in two ways:
The two classification systems are complementary and not mutually exclusive. The aetiological classification has much greater clinical importance.
Morphological classification
Classified by the average size of the regeneration nodules, cirrhosis may be:
A cirrhotic liver intermediate between these two categories is described as ‘mixed’.
One of the commonest causes of micronodular cirrhosis is alcoholic liver disease. The significance of macronodular cirrhosis, irrespective of cause, is that it is believed to carry a greater risk of complication by liver cell carcinoma. Macronodular cirrhosis may also be difficult to diagnose with certainty in needle biopsies because the abnormal architecture is not often revealed fully in such small tissue samples.
Aetiological classification
The aetiological classification of a cirrhotic liver can often be deduced from clinical, biochemical, immunological or biopsy features (Fig. 16.14). Important causes include:
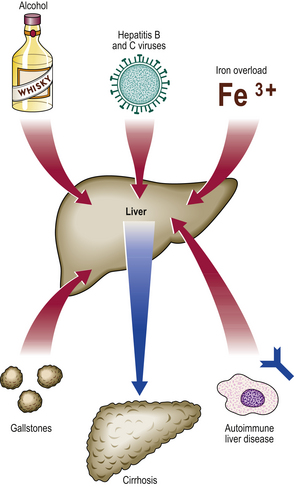
In countries such as the UK, alcohol is one of the commonest causes. If the cause is unknown, then the cirrhosis is labelled cryptogenic (hidden cause) or idiopathic, although with modern investigations the proportion of cases thus designated is falling.
Complications
The major complications of cirrhosis are:
Liver failure
Cirrhosis may be functionally compensated or decompensated. Indeed, if the disease process that led to the cirrhosis is now inactive, then there may be no detectable abnormalities of liver function. Liver failure is a manifestation of decompensation (Table 16.5) and is characterised clinically by:
Table 16.5 Pathophysiological basis of clinical features of chronic liver disease
| Clinical feature | Explanation |
|---|---|
| Oedema | Reduced albumin synthesis resulting in hypoalbuminaemia |
| Ascites | Hypoalbuminaemia, secondary hyperaldosteronism, portal hypertension |
| Haematemesis | Ruptured oesophageal varices due to portal hypertension |
| Spider naevi | Hyperoestrogenism |
| Gynaecomastia | |
| Purpura and bleeding | Reduced clotting factor synthesis |
| Coma | Failure to eliminate toxic gut bacterial metabolites (‘false neurotransmitters’) |
| Infection | Reduced Kupffer cell number and function |
Hepatic encephalopathy is due to the failure of the liver to eliminate toxic nitrogenous products of gut bacteria; some of these mimic the effect of neurotransmitters (i.e. they are ‘false neurotransmitters’). Renal failure may also occur with hepatic failure (hepato-renal syndrome). The patient’s breath has a characteristic odour (fetor hepaticus).
Failure to eliminate endogenous steroid hormones results in secondary hyperaldosteronism, causing sodium and water retention and, in the male, loss of secondary sexual characteristics and gynaecomastia due to hyperoestrogenism. ‘Spider naevi’ are small vascular lesions on the skin, commonly seen in pregnancy, associated with hyperoestrogenism in cirrhosis.
Defective Kupffer cell function may be responsible for the increased incidence of bacteraemia in patients with cirrhosis, sometimes in the absence of other manifestations of liver failure.
Portal hypertension
Cirrhosis is the commonest cause of portal hypertension (Fig. 16.15). In cirrhosis, the increased blood pressure (>7 mmHg) in the hepatic portal vein is due to a combination of:
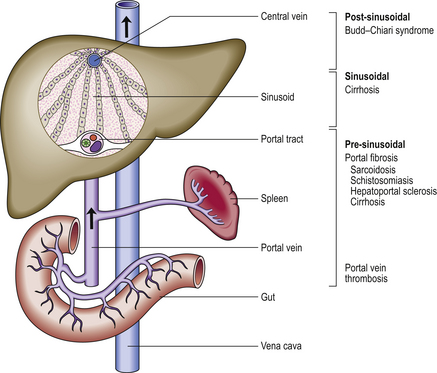
Fig. 16.15 Causes and pathogenesis of portal hypertension. Portal hypertension may be due to haemodynamic abnormalities proximal or distal to the sinusoids or at the sinusoidal level. Increased portal vascular resistance and intrahepatic shunting between high-pressure hepatic arterial and low-pressure portal venous channels are postulated explanations for portal hypertension in cirrhosis.
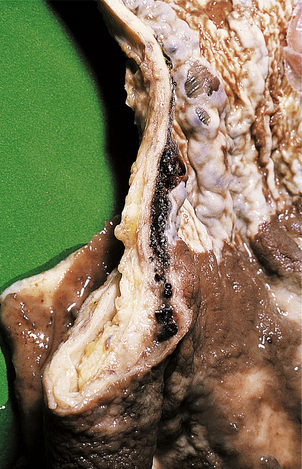
Portal hypertension leads to oesophageal varices (Fig. 16.16) and haemorrhoids (because normal anastomoses between the portal and systemic venous systems at these sites are enlarged) and also contributes to the development of ascites. Oesophageal varices are a particularly serious complication because these thin-walled dilated veins are prone to rupture, causing massive haematemesis which can be fatal. Other manifestations of portal hypertension include the less common ‘caput medusae’ around the umbilicus. Portal hypertension may be further complicated by portal vein thrombosis; this complication can lead to sudden clinical deterioration.
Liver cell carcinoma
Cirrhosis is a premalignant condition; it is associated with liver cell dysplasia and an increased risk of liver cell carcinoma. The tumour often appears to be multifocal, arising at multiple sites within the liver. The risk is greatest in macronodular cirrhosis and applies to all aetiological types. Liver cell carcinoma is considered in more detail below.
TUMOURS OF THE LIVER



Benign tumours
Benign tumours of the liver rarely give rise to serious clinical problems, except when they cause confusion with their malignant counterparts. Benign tumours of the liver include:
Liver cell adenoma
Liver cell adenoma is a benign, well-differentiated neoplasm of liver cells. It forms a well-circumscribed nodule, with a texture and colour resembling that of the normal liver. Adenomas may arise spontaneously, but an increased incidence occurs in patients taking anabolic, androgenic or oestrogenic steroids. They may be clinically silent or cause hepatomegaly. Occasionally haemoperitoneum due to spontaneous rupture may be the first manifestation.
Angioma
Angioma, a benign vascular neoplasm, is sometimes multiple, rarely exceeding a few centimetres in diameter. Angiomas are rarely of clinical significance, but they may be mistaken for something more sinister when found unexpectedly during a laparotomy.
Bile duct hamartoma
Bile duct hamartomas are not strictly tumours but are tumour-like congenital malformations. They are usually small and often multiple, sometimes referred to as von Meyenberg complexes. They are often seen in association with congenital hepatic fibrosis.
Focal nodular hyperplasia
Focal nodular hyperplasia is neither a true neoplasm nor, despite its name, a hyperplastic disorder; it is thought to be hamartomatous. It is usually solitary and up to 50 mm in diameter. At its centre is a stellate mass of fibrous connective tissue in which there are numerous small bile ducts. The rest of the lesion comprises liver cells and vascular sinusoids. These lesions are usually clinically silent, but they can become abnormally vascular, enlarge and rupture in patients receiving oestrogenic steroids such as oral contraceptives.
Malignant tumours
Malignant tumours in the liver often present with jaundice and weight loss. Most often they are metastases from other organs.
Primary malignant tumours of the liver include:
Metastases
The commonest malignant neoplasms in the liver are metastases from primary lesions in another organ. These metastases usually form multiple deposits with central necrosis, causing an umbilicated appearance when they are visible on the liver surface. They are usually white unless they are derived from a malignant melanoma, in which case they may be dark brown or black. Common primary origins for hepatic metastases include the entire gastrointestinal tract, including pancreas and bowel, the lung and the breast.
Metastases receive their vascular supply from the hepatic arterial system. This is sometimes selectively perfused with cytotoxic drugs or artificially embolised to induce infarction.
Liver cell carcinoma
Liver cell carcinoma (hepatocellular carcinoma) is one of the commonest tumours in some parts of the world (Fig. 16.17). Known or suspected aetiological factors include:
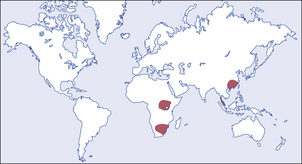
Fig. 16.17 Geographic areas of high incidence of liver cell carcinoma. The tumour occurs in all countries, particularly in patients with cirrhosis. Regions with a well-documented, very high incidence are shaded.
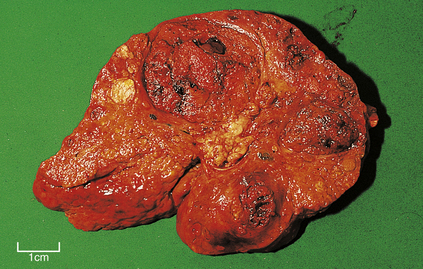
Fig. 16.18 Liver cell carcinoma arising in a cirrhotic liver. Cirrhosis is a pre-neoplastic condition. Several tumour nodules are present, perhaps reflecting the multifocal origin in cirrhotic livers.
Various growth patterns are recognised, resembling to a variable extent the normal trabecular arrangement of liver cells. If the tumour is sufficiently well differentiated it retains the capacity to secrete bile, so that the tumour and any metastases from it appear bile-stained. In cirrhotic livers, liver cell carcinomas are often multifocal.
A type of liver cell carcinoma with specific features is the fibrolamellar variant in which the neoplastic liver cells are arranged in broad bands or lamellae separated by dense fibrous tissue. This variant occurs most often in young women, without cirrhosis as a predisposing cause.
Liver cell carcinomas often produce alpha-fetoprotein. This is a normal serum protein of the fetus, synthesis of which declines towards the end of gestation when it is replaced by albumin. Alpha-fetoprotein is secreted by the tumour cells into the patient’s blood, where it is a useful diagnostic marker.
Cholangiocarcinoma
Cholangiocarcinoma is an adenocarcinoma of bile duct epithelium. In liver biopsies this gland-forming neoplasm can be extremely difficult to distinguish from metastatic adenocarcinoma from some other organ. Known aetiological factors include infestation with the Chinese liver fluke Clonorchis sinensis and with Opisthorchis viverrini, both of which induce recurrent pyogenic cholangitis (‘oriental cholangitis’). There is also an increased incidence of cholangiocarcinoma in patients with ulcerative colitis, particularly in those who have developed sclerosing cholangitis.
Angiosarcoma
This highly malignant neoplasm originates from the endothelium of the vascular sinusoids and infiltrates the liver by spreading along the vascular sinusoids. Known aetiological factors include vinyl chloride, used in the manufacture of polyvinyl chloride (PVC), and administration of the now-obsolete alpha-emitting radiological contrast medium Thorotrast (Ch. 11).
Hepatoblastoma
Hepatoblastoma is a rare malignant neoplasm of the liver occurring almost exclusively in children below the age of 5 years. In a significant proportion of cases it is associated with various developmental abnormalities. Histologically, its growth pattern resembles some features of the embryonic liver, but it is not uncommon to find unexpected tissues such as muscle. The prognosis is poor.
LIVER CYSTS
Liver cysts can often be distinguished from solid tumours by modern imaging techniques, although some malignant tumours may be so necrotic in the centre that they mimic cysts.
Simple cysts
Simple cysts are common, relatively small (10–20 mm in diameter), and often multiple. Sometimes they are associated with lesions elsewhere, as in the rare von Hippel–Lindau syndrome. They have little or no intrinsic clinical importance.
Hydatid cysts
Hydatid cysts are due to the parasite Echinococcus granulosus. They are usually many centimetres in diameter, have a fibrous laminated wall and contain numerous daughter cysts (Fig. 16.19). Great care must be taken during surgical removal, because spillage of the cyst fluid into the peritoneum may precipitate anaphylactic shock due to the presence of hydatid antigens in a patient already sensitised to them.

LIVER INVOLVEMENT BY SYSTEMIC DISEASE
The liver is commonly affected by disease primarily arising in other organs or systems; this often causes hepatomegaly. Examples include:
Liver biopsy is often done in the diagnosis and investigation of systemic diseases such as suspected sarcoidosis or carcinomatosis. The biopsy may be diagnostic of the systemic problem.
The commonest liver involvement by systemic disease occurs in cardiac failure. At autopsy in such cases, the liver appears to have a finely mottled surface due to central acinar congestion surrounded by fatty change. This is the so-called ‘nutmeg liver’.
TRANSPLANTATION AND THE LIVER
The pathology of transplantation and the liver is important for two reasons. First, although liver transplants are relatively well tolerated, immunological rejection of the grafted organ can occur. Second, the liver is frequently affected by graft-versus-host disease following allogeneic bone marrow transplantation.
Liver transplants
Liver transplants have been performed successfully since the 1960s. Common indications include congenital abnormalities, advanced hepatitis C and primary biliary cirrhosis. The 5-year survival in experienced centres exceeds 60%.
Although the immunogenicity of the liver is relatively low, for example compared with the kidney, in clinical practice there is still a significant risk of rejection. The most vulnerable cells are biliary epithelium and vascular endothelium; these express MHC class II antigens. Rejection may be:
Fortunately, close monitoring of liver transplants and improvements in immunosuppressive therapy are gradually reducing the probability of graft loss by rejection.
Graft-versus-host disease
Patients with leukaemia or lymphoma may be treated by allogeneic bone marrow transplantation after whole-body irradiation to eliminate the neoplastic cells. Although every effort is made to find a closely matched donor by histocompatibility testing, there is a risk that the lymphocytes in the marrow allograft will recognise and react to the normal antigens on the host’s tissues; the result is graft-versus-host disease. Skin, gut epithelium and the liver are especially vulnerable. In the liver, the principal target for the immune reaction is biliary epithelium. If the condition is untreated, many bile ducts will be destroyed, resulting in jaundice.
BILIARY SYSTEM
NORMAL STRUCTURE AND FUNCTION
The biliary system comprises the intra- and extrahepatic bile ducts and the gallbladder. The system is lined by a glandular mucus-secreting epithelial cell layer. Bile is secreted by the liver into the right and left hepatic ducts which fuse to form the common hepatic duct. The bile consists of micelles of cholesterol, phospholipid and bile salts, and of course bilirubin.
Bile enters the gallbladder through the cystic duct; it is then stored and concentrated in the gallbladder. In response to the ingestion of food, particularly with a high fat content, the gallbladder contracts by stimulation with cholecystokinin and expels the concentrated bile into the second part of the duodenum, through the ampulla of Vater as the sphincter of Oddi relaxes.
CONGENITAL ABNORMALITIES
Malformations of the biliary system include:
Intrahepatic malformations of the biliary system are inaccessible to surgical correction and, if life-threatening, may be an indication for liver transplantation.
In addition to these malformations, the liver is often affected by the production of abnormally viscous bile in patients with cystic fibrosis (mucoviscidosis) (Ch. 7).
DISEASES OF THE GALLBLADDER
Gallbladder disease is extremely common and in almost every case it is associated with or due to the presence of gallstones.
Cholelithiasis (gallstones)



Cholelithiasis is the name given to the common condition in which gallstones form within the biliary system. Risk factors for cholesterol-rich stones include female gender and obesity (hence ‘fat, fair, forty, fertile, female’, an alliterative description of the typical patient) and diabetes mellitus. Stones are prone to occur if there is a relative excess of cholesterol in the bile. Gallstones are usually composed of a mixture of cholesterol and bile pigment (Fig. 16.20), although almost pure cholesterol or pigment stones are occasionally found. Pure pigment gallstones occur notably in patients with haemolytic anaemia where there is consequent excessive excretion of bilirubin. Calcium carbonate stones are also found rarely.
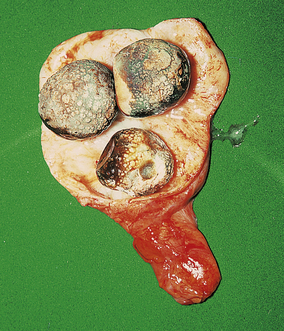
Fig. 16.20 Gallstones and chronic cholecystitis. The thickened gallbladder has been opened to reveal several large cholesterol-rich stones.
The stones often have a laminated internal structure and, if multiple (as they commonly are), have faceted surfaces.
Pathogenesis
Cholesterol stones may form if there is an imbalance between the ratio of cholesterol and bile salts; normally, the latter form micelles which have a hydrophilic exterior enclosing the hydrophobic cholesterol. Thus, gallstones can result from:
Pathological effects
The pathological effects of gallstones include (Fig. 16.21):
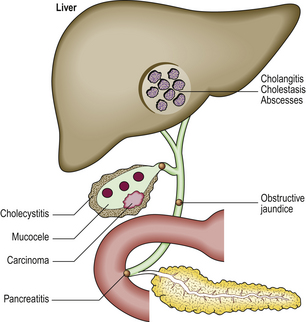
Cholesterosis
Cholesterosis is the name given to the clinically unimportant occurrence of cholesterol-laden macrophages in the lamina propria of the gallbladder mucosa. This occurrence gives the mucosa a yellow-speckled appearance known as ‘strawberry gallbladder’.
Cholecystitis
Cholecystitis is an inflammatory condition of the gallbladder. It is almost always associated with gallstones and occurs as an acute or chronic condition. It is a common cause of abdominal pain in the right hypochondrium.
Acute cholecystitis



Acute cholecystitis is usually due to obstruction of the outflow from the gallbladder by a gallstone. The initial inflammatory reaction is due to the irritant effects of bile and is therefore usually sterile at this stage. However, stasis of bile predisposes to infection, which then stimulates a more vigorous and often pyogenic acute inflammatory response. The gallbladder wall becomes oedematous, due to increased vascular permeability, and infiltrated with acute inflammatory cells. The lumen distends with pus, and stretching of the wall already weakened by inflammation leads to a risk of perforation and peritonitis. Alternatively, a fistula may form with the second part of the duodenum and allow stones to enter the bowel lumen. Large stones may occasionally lodge at the ileocaecal valve and cause intestinal obstruction (gallstone ileus).
An inflamed gallbladder grossly distended with pus is called an empyema.
Chronic cholecystitis


Chronic cholecystitis may develop insidiously or after repeated episodes of acute cholecystitis.
The gallbladder wall is thickened by fibrosis and is relatively rigid. Thus obstructive jaundice due to gallstones is not usually associated with a palpable gallbladder because the stones will be associated with chronic cholecystitis and therefore a rigid gallbladder. Conversely, obstructive jaundice due to carcinoma of the head of the pancreas often results in a palpable distended gallbladder; this is the pathological basis of Courvoisier’s law.
The thick gallbladder wall has within it Aschoff–Rokitansky sinuses, mucosal herniations (diverticula) often containing inspissated bile or even small stones. The wall bears an infiltrate of chronic inflammatory cells and the blood vessels often show endarteritis obliterans (Fig. 16.22). A stone is often found in Hartmann’s pouch, a pathological dilatation in the neck of the gallbladder formed by increased intraluminal pressure or impaction of the stone.
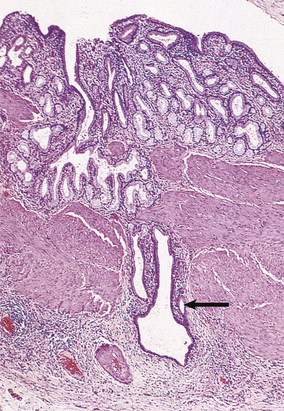
Fig. 16.22 Histology of chronic cholecystitis. A thickened gallbladder with diffuse chronic inflammatory infiltration and Aschoff–Rokitansky sinuses (arrowed).
A rare variant is xanthogranulomatous cholecystitis in which lipid-laden macrophages and giant cells accumulate in large numbers; this mimics a neoplasm grossly and, specifically, a clear-cell carcinoma histologically. The lesion, like its more common renal counterpart, xanthogranulomatous pyelonephritis, is prone to cause fistulae.
Mucocele
A mucocele of the gallbladder is the result of sterile obstruction of the neck by a gallstone. The lack of inflammation permits the gallbladder to distend with mucus without rupturing. The mucocele has a thin wall and demands careful handling during surgical removal to avoid the risk of spillage of mucus into the peritoneal cavity and thus the risk of pseudomyxoma peritonei, a rare complication in which the peritoneum becomes seeded with mucus-producing epithelial cells and the cavity fills with mucus.
Carcinoma of the gallbladder


Carcinoma of the gallbladder is almost always associated with the presence of gallstones; this relationship may be causal. The tumour is most often an adenocarcinoma, although squamous cell carcinoma is also seen. As the gallbladder is not a vital organ, the tumour is often advanced at the time of clinical presentation, and invasion of the liver and other adjacent structures defeats attempts at operative removal. It therefore has a poor prognosis.
Carcinoma of the bile duct



Carcinoma of the bile duct is most commonly an adenocarcinoma. There is an increased incidence in patients with chronic ulcerative colitis. It tends to present at a relatively early stage with obstructive jaundice.
Biliary obstruction
Bile duct obstruction is a fairly common event and may be due to:
The patient becomes jaundiced, deeply so if the obstruction is not relieved, with a raised conjugated serum bilirubin, pale stools and dark urine. A raised serum alkaline phosphatase with only modest elevation of transaminases is usual.
If the biliary obstruction persists, there is a risk that the static bile becomes infected, causing acute cholangitis and eventually liver abscesses. Lack of bile in the small intestine compromises the absorption of fat and fat-soluble substances (e.g. some vitamins).
EXOCRINE PANCREAS
NORMAL STRUCTURE AND FUNCTION
The pancreas is a retroperitoneal organ, the head and uncinate process lying within the duodenal loop, the body crossing the aorta and inferior vena cava, and the tail abutting onto the splenic hilum.
The pancreas is a mixed exocrine and endocrine organ. Scattered through the gland are the islets of Langerhans consisting of endocrine cells producing peptide hormones, the most important of which are insulin and glucagon (Ch. 17); their secretion drains directly into the blood and ultimately into the liver through the hepatic portal vein.
The exocrine pancreas comprises the bulk of the organ and is composed of glands and ducts with a lobular arrangement, the latter fusing to form the pancreatic and accessory ducts which convey the exocrine secretions into the duodenum. The exocrine glands contain numerous zymogen granules and produce trypsin, lipase, phospholipase, amylase and elastase; these enzymes require activation, normally in the duodenum. The pancreas also secretes a bicarbonate-rich alkaline medium.
Some of the exocrine glands are perfused with blood that has already perfused islets in the vicinity (i.e. they have a portal blood supply). This almost certainly provides some physiological advantages, but it does mean that these glands are especially vulnerable to ischaemia if the circulation is impaired.
INVESTIGATION OF PANCREATIC DISEASE
Disorders of the exocrine pancreas can be investigated radiologically by the technique of ERCP (endoscopic retrograde cholangiopancreatography) or MRCP (magnetic resonance cholangiopancreatography). These techniques reveal any deformities due to inflammatory fibrosis or tumours. Pancreatic juice can be collected through the cannula and examined biochemically for enzymes, or cytologically for abnormal cells such as may be shed from a carcinoma.
Operative biopsies of the pancreas are hazardous because there is a significant risk of precipitating acute pancreatitis (inflammation of the pancreas) due to leakage of exocrine secretions. Nevertheless, an intra-operative frozen section diagnosis may be required in cases of suspected pancreatic carcinoma before attempting to remove the lesion, a procedure with a relatively high post-operative complication rate. Fine-needle aspiration cytology is possible through the unopened abdomen under ultrasonographic or computed axial tomography (CAT) guidance.
Serum amylase is an important marker of pancreatic inflammation. The concentration is greatly elevated in acute pancreatitis; lesser elevations may occur following a perforated peptic ulcer.
CONGENITAL ABNORMALITIES
Congenital abnormalities of the pancreas include:
In addition, the pancreas is severely affected in cystic fibrosis (mucoviscidosis), a congenital disorder of exocrine secretions in which they are abnormally viscous (Ch. 7). The mucus plugs the pancreatic ducts, resulting in retention of secretions and damage to the exocrine glands.
DISEASES OF THE PANCREAS
Pancreatitis
Pancreatitis (inflammation of the pancreas) can be classified into acute and chronic forms. There is, however, overlap in that patients with chronic pancreatitis may have acute exacerbations.
Acute pancreatitis




Aetiology
Acute pancreatitis (Fig. 16.23) may be due to:
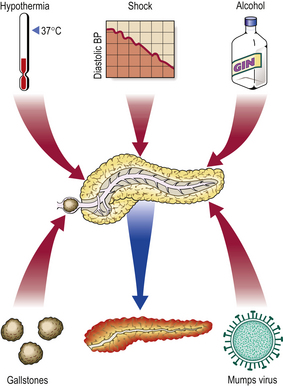
Although many cases are mild, acute pancreatitis is often a serious disorder with a high mortality. It is more common in adults than in children. The condition is serious because the gland, once injured, releases its lytic enzymes into the blood, contributing to the severe shock, and into the surrounding tissue, causing tissue digestion.
Clinical features
Patients present with a sudden onset of severe abdominal pain, often radiating into the back, and nausea and vomiting. The upper abdomen is tender. The clinical deterioration may be rapid, the patient becoming severely shocked. Diagnosis is made by finding a greatly elevated serum amylase concentration.
Pathogenesis
The pathogenesis of the early stages varies according to the aetiology. For example, when the pancreatic duct is obstructed by a gallstone at the ampulla of Vater, or where there is biliary reflux into the pancreatic duct for any other reason, the duct epithelium is damaged, particularly if the bile is infected or admixed with trypsin. The damage extends into the gland and results in the leakage and activation of pancreatic enzymes. In contrast, when pancreatitis is attributable to vascular insufficiency, the hypoxic injury due to reduced blood flow occurs first in the acini at the periphery of the lobules; they are precariously remote from the vascular supply and some are fed by portal vessels from islets in the vicinity. Irrespective of the initiating event, the liberation of lytic enzymes causes further damage and diffuse pancreatitis develops rapidly.
The gland becomes swollen and often haemorrhagic if the inflammation is severe. Proteases digest the walls of blood vessels, causing extravasation of blood. Amylase is released into the blood, where measurement of its concentration is an important diagnostic marker of the condition.
Lipolytic action causes fat necrosis, which can be quite extensive within the abdomen and subcutaneous tissue. Sometimes the necrosis extends anteriorly around the abdominal wall to produce discoloration of the skin (Grey Turner’s sign). The released fatty acids bind calcium ions, forming white precipitates in the necrotic fat; severe fat necrosis can bind so much calcium that hypocalcaemia results, sometimes causing tetany.
Concomitant destruction of the adjacent islets can result in hyperglycaemia.
Other complications include the formation of abscesses and cysts within the pancreas or adjacent tissues. These often necessitate surgical drainage.
Chronic pancreatitis



Aetiology
Chronic pancreatitis is a relapsing disorder that may either result from repeated episodes of clinically evident acute pancreatitis or develop insidiously without previous symptoms of pancreatic disease. The commonest cause is chronic excessive alcohol consumption. A much less common, but increasingly recognised, autoimmune pancreatitis also occurs. Chronic pancreatitis is also a feature of cystic fibrosis (mucoviscidosis). There is also a rare familial pancreatitis inherited as an autosomal dominant trait, in some cases associated with aminoaciduria or hyperparathyroidism.
Clinicopathological features
Chronic pancreatitis is more common in adults than in children, and presents with intermittent upper abdominal and back pain and weight loss. A plain X-ray of the upper abdomen often reveals flecks of calcification due to previous fat necrosis. ERCP or MRCP often shows that the pancreatic ducts are distorted by scar tissue resulting from the chronic inflammatory process.
The exocrine tissue is eventually replaced by fibrosis (Fig. 16.24) and, if localised, its hard texture mimics that of a carcinoma when the gland is palpated during laparotomy. The endocrine component of the gland is relatively unaffected except at an advanced stage.
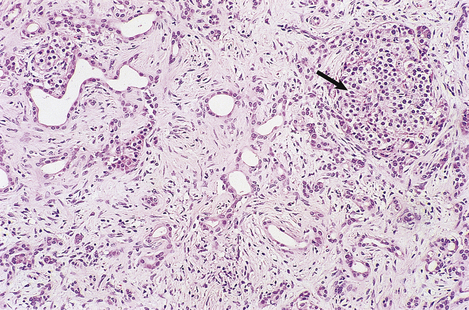
Pancreatic malabsorption
Chronic pancreatitis, with loss of exocrine secretions, results in malabsorption of fat because of the relative lack of lipases. The patient’s faeces contain abnormally high quantities of fat (steatorrhoea), and absorption of fat-soluble substances such as vitamins A, D, E and K is impaired.
Carcinoma of the pancreas



Aetiology
Pancreatic carcinoma is increasing in incidence in many countries. Over 7000 cases occur annually in the United Kingdom. There is an association with cigarette smoking and with diabetes mellitus. There appears to be an increased risk in the rare entity of familial pancreatitis. The prognosis is poor even in operable cases.
Weight loss is a common presenting feature; other symptoms are attributable to the precise location of the tumour. Some cases develop flitting venous thromboses (thrombophlebitis migrans): this is Trousseau’s sign.
Clinicopathological features
Most pancreatic carcinomas are adenocarcinomas with a marked desmoplastic stromal reaction (Fig. 16.25), making them very firm on palpation during surgery. They arise most commonly in the head of the organ, where they tend to compress the common bile duct and cause obstructive jaundice. Elsewhere in the gland, they can present at a relatively late stage because they cause few signs or symptoms. Extensive replacement of the gland by carcinoma can lead to diabetes mellitus due to destruction of the islets of Langerhans.
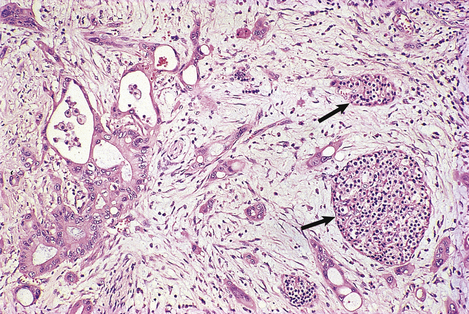
Fig. 16.25 Histology of pancreatic adenocarcinoma. The neoplastic glands with pleomorphic atypical nuclei are invested by dense fibrous connective tissue, thus mimicking the texture of chronic pancreatitis when the gland is palpated during surgery. Islets of Langerhans (arrowed) survive at the edge of the tumour.
Pancreatic adenocarcinomas spread by direct invasion, by lymphatics to lymph nodes and by the blood stream to the liver (Fig. 16.26). Prognosis is relatively poor because metastases are often present at the time of surgery. If surgical removal is attempted, it involves excision of at least part of the pancreas, the duodenum and regional nodes with anastomotic restoration of intestinal, biliary and residual pancreatic flow; this procedure is associated with a high risk of operative complications and death.
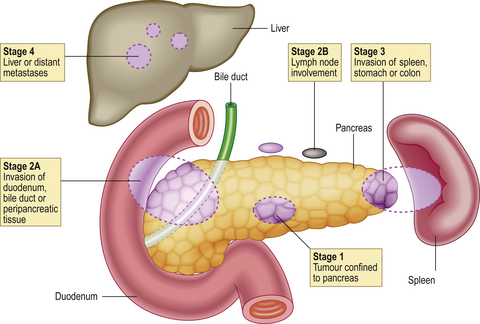
Cysts and cystic tumours
Pancreatic cysts are of two types:
True cystic tumours also occur—the benign cystadenoma and its malignant variant, cystadenocarcinoma.
Commonly confused conditions and entities relating to liver, biliary and pancreatic pathology
| Commonly confused | Distinction and explanation |
|---|---|
| Cholestasis and jaundice | Cholestasis is stagnation of bile, usually within the liver where, histologically, it is seen in liver cells or in the canaliculi between them or in bile ducts. Jaundice is yellowness of the tissues due to abnormally high levels of bilirubin. |
| Cholestasis and cholangitis | Cholestasis is stagnation of bile, usually within the liver where, histologically, it is seen in liver cells or in the canaliculi between them or in bile ducts. Cholangitis is inflammation of the bile ducts, usually precipitated by biliary obstruction with secondary bacterial infection. However, in sclerosing cholangitis the inflammatory activity may be minor. |
| Lupoid hepatitis and systemic lupus erythematosus (SLE) | Lupoid hepatitis, better known as autoimmune hepatitis, is ‘lupoid’ only because it shares with SLE a tendency to affect young adult women and the presence of anti-DNA auto-antibodies. |
| Haemosiderosis and haemochromatosis | Haemosiderosis means only the presence of excess iron, as haemosiderin, in the liver. This is a feature of haemochromatosis (genetic or acquired) characterised by excessive iron deposition accompanied by fibrosis and, eventually, cirrhosis. |
| Gallbladder empyema and emphysema | Empyema is the presence of pus in a hollow organ or body cavity. Emphysema (pulmonary or interstitial) is the presence of abnormal gas-filled spaces. |
| Cholecystitis and cholesterosis | Cholecystitis (acute or chronic) is inflammation of the gallbladder. Cholesterosis is the presence of lipid-filled histiocytes in the lamina propria of the gallbladder mucosa; it is clinically insignificant. |
| Pancreatic cysts and pseudocysts | Cysts have an epithelial lining and are developmental or neoplastic, in contrast to pseudocysts which are devoid of an epithelial lining and commonly develop in the pancreas after pancreatitis. |
Ala A., Walker A.P., Ashkan K., et al. Wilson’s disease. Lancet. 2007;369:397-408.
Burt A.D., Portmann B.C., Ferrell L.D.. MacSween’s pathology of the liver. Edinburgh: Churchill Livingstone; 2006.
Cruickshank A.H., Benbow E.W.. Pathology of the pancreas. Berlin: Springer Verlag; 1995.
Davis B.H., Kresina T.E.. Hepatic fibrogenesis. Clinics in Laboratory Medicine. 1996;16:261-275.
Desmet V.J., Rosai J.. Liver. In: Rosai J., editor. Rosai and Ackerman’s surgical pathology. St Louis: Mosby; 2004:917-1033.
Di Bisceglie A.M.. Hepatitis C. Lancet. 1998;351:351-355.
Goodman Z.D.. Drug hepatotoxicity. Clinics in Liver Disease. 2002;6:381-397.
Haugk B., Burt A.D. 2005 Non-alcoholic fatty liver disease. Pignatelli M., Underwood J.C.E., editors 2005. Recent advances in histopathology. Vol 21. London: RSM Press, pp 1-17
Herzer K., Sprinzl M.F., Galle P.R.. Hepatitis viruses: live and let die. Liver International. 2007;27:293-301.
Ishak K., Baptista A., Bianchi L., et al. Histological grading and staging of chronic hepatitis. Journal of Hepatology. 1995;22:696-699.
Kaplan M.M., Gershwin M.E.. Primary biliary cirrhosis. New England Journal of Medicine. 2005;353:1261-1273.
Kasai Y., Takeda S., Takagi H.. Pathogenesis of hepatocellular carcinoma: a review from the viewpoint of molecular analysis. Seminars in Surgical Oncology. 1996;12:155-159.
Krawitt E.L.. Autoimmune hepatitis. New England Journal of Medicine. 2006;354:54-66.
Neoptolemos J., Lemoine N.. Pancreatic cancer: molecular and clinical advances. Oxford: Blackwell Science; 1995.
Pietrangelo A.. Molecular insights into the pathogenesis of hereditary haemochromatosis. Gut. 2006;55:564-568.
Rosai J.. Gallbladder and extrahepatic bile ducts. In: Rosai J., editor. Rosai and Ackerman’s surgical pathology. St Louis,: Mosby; 2004:1035-1060.
Rosai J.. Pancreas and ampullary region. In: Rosai J., editor. Rosai and Ackerman’s surgical pathology. St Louis: Mosby; 2004:1061-1114.
Sanders G., Kingsnorth A.N.. Gallstones. British Medical Journal. 2007;335:295-299.
Scheuer P.J.. Assessment of liver biopsies in chronic hepatitis: how is it best done? Journal of Hepatology. 2003;38:240-242.
Scheuer P.J., Lefkowitch J.H.. Liver biopsy interpretation. London: W B Saunders; 2006.
Teckman J., Perlmutter D.H.. Conceptual advances in pathogenesis and treatment of childhood metabolic liver disease. Gastroenterology. 1995;108:1263-1279.
Tsukamoto H., Lu S.C.. Current concepts in the pathogenesis of liver injury. FASEB Journal. 2001;15:1335-1349.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Chapter 16 Liver, biliary system and exocrine pancreas
COMMON CLINICAL PROBLEMS FROM LIVER AND BILIARY SYSTEM DISEASE
Pathological basis of hepatic signs and symptoms
| Sign or symptom | Pathological basis |
|---|---|
| Jaundice | Haemolysis (increased formation of bilirubin), liver disease (impaired conjugation and/or excretion) or biliary obstruction |
| Dark urine | Conjugated hyperbilirubinaemia (water-soluble) |
| Pale faeces | Biliary obstruction causing lack of bile pigments |
| Spider naevi | Secondary to hyperoestrogenism |
| Gynaecomastia | |
| Oedema | Reduced plasma oncotic pressure due to hypoalbuminaemia |
| Xanthelasma | Cutaneous lipid deposits due to hypercholesterolaemia in chronic biliary obstruction |
| Steatorrhoea | Malabsorption of fat due to lack of bile (e.g. biliary obstruction) |
| Pruritus | Biliary obstruction resulting in bile salt accumulation |
| Ascites | Combination of hypoalbuminaemia, portal hypertension and secondary hyperaldosteronism |
| Bruising or bleeding | Impaired hepatic synthesis of clotting factors |
| Hepatomegaly | Increased size of liver due to inflammation (e.g. hepatitis), infiltration (e.g. amyloid, fat) or tumour (primary or secondary) |
| Haematemesis | Ruptured oesophageal varices due to portal hypertension |
| Encephalopathy | Failure of liver to remove exogenous or endogenous substances mimicking or altering balance of neurotransmitters |
LIVER
NORMAL STRUCTURE AND FUNCTION
Forming the interface between the gastrointestinal tract and the rest of the body, the liver is of crucial importance in metabolising, storing or excreting the absorbed products of digestion. The liver has numerous other vital functions; therefore, the clinical consequences of liver disease are often wide-ranging and, if severe, life-threatening. However, considerable functional reserve and reparative capacity enables many patients to tolerate cellular injury or losses that, in other organs, would imperil their survival.
This wedge-shaped organ, weighing approximately 1.5 kg in the adult, is situated in the right hypochondrial region of the abdominal cavity. It has four lobes: the right is larger than the left; the smaller caudate lobe is situated posteriorly and the quadrate lobe is more anterior. The liver receives blood from two sources:
Blood leaves the liver through the hepatic veins, which drain into the inferior vena cava.
Bile is formed in the liver and drains from it into the right and left hepatic ducts; these fuse to form the common bile duct to be joined by the cystic duct, which communicates with the gallbladder where the bile is stored and concentrated.
Most of the liver comprises liver cells (hepatocytes). These are arranged in plates one cell thick, bordering the vascular sinusoids through which flows hepatic arterial and portal venous blood. The blood flowing through the vascular sinusoids is separated from the liver cells by a thin fenestrated (porous) barrier of cells (endothelial cells and phagocytic Kupffer cells) and the space of Disse. Within the space of Disse the basement membrane is interrupted, thus allowing free exchange of molecules at the liver cell membrane. Blood flowing through the vascular sinusoids drains into hepatic vein branches (central veins or terminal hepatic venules). Bile formed by the liver cells is secreted from them into minute canaliculi which run along the centre of the liver cell plates to drain into the bile duct branches in the portal tracts. Close to the vascular sinusoids in the vicinity of the terminal hepatic venules are stellate cells called the perisinusoidal cells of Ito; these are involved in hepatic fibrosis by synthesising collagen.
The portal tracts each contain three tubular structures, which are branches of:
These constitute the portal triad and are supported by collagen-rich connective tissue.
The microanatomy of the liver can be regarded conceptually as either acinar or lobular (Fig. 16.1):
Fig. 16.1 Comparison of acinar and lobular concepts of the microanatomical units of the liver. The acinar concept explains better the pathophysiology of the liver. Cells in acinar zone 3, being the most remote from the vascular supply in the hilum of the acinus, are consequently the most vulnerable to injury. Death of liver cells in zone 3 results in the observed necrosis bridging between portal tracts and central veins in severe liver injury. Lobular units are, however, often easier to perceive in histological sections.
Of the two microanatomical concepts—acinar or lobular—the ‘acinar concept’ is now considered to be more useful because it explains better many of the pathophysiological disturbances in liver disease. The zone of liver cells most remote from the axial vessels in the centre of the acinus (acinar zone 3) is the most susceptible to injury resulting from vascular insufficiency, as in circulatory shock or cardiac failure. As adjacent zones 3 are contiguous, liver cell death in this zone is often confluent.
The portal tracts are circumscribed by a boundary of liver cells, known as the limiting plate, which is breached in some forms of chronic liver inflammation; disruption of the limiting plate, when seen in biopsies, denotes that progression to cirrhosis is likely. The liver cells at the portal tract boundary can, in response to bile duct injury or obstruction, undergo a metaplastic change and proliferate to form new bile ductules.
Liver cells are rich in organelles, including numerous mitochondria, lysosomes, peroxisomes (microbodies), and rough and smooth endoplasmic reticulum, reflecting their wide range of metabolic functions. The cytoplasm is also laden with glycogen; this glycogen can be excessive in diabetes and in congenital deficiencies of glycogen debranching enzymes (the glycogenoses).
Liver cells synthesise albumin, clotting factors including fibrinogen, some complement components, alpha-1 antitrypsin, etc., and remove from the body many waste products and potentially toxic substances. Through the expression of specific receptors on the liver cells, the liver—the site of action of statins, the cholesterol-lowering drugs—has a major role in the uptake and metabolism of the low-density lipoproteins involved in atheroma (Ch. 13). Liver cells also metabolise or activate many other drugs. Extensive disease of the liver therefore affects many vital functions and has profound effects on the body.
Liver cells contain many enzymes, some of which are diagnostically important. Their release from damaged or dying liver cells into the blood, where their activity can be measured, indicates the presence and severity of liver disease (Table 16.1). These enzymes include:
Table 16.1 Diagnostic usefulness of serum analyses in liver disease
| Test | Deviation from normal | Interpretation |
|---|---|---|
| AlbuminNormal 35–50 g/l | ↓ | Liver failure |
| Prothrombin timeNormal < 15 s | ↑ | Liver failure |
| Alanine aminotransferase (ALT)Normal < 40 IU/l | ↑ | Hepatocellular injury |
| Aspartate aminotransferase (AST)Normal < 40 IU/l | ↑ | Hepatocellular injury |
| Gamma-glutamyltransferase (GGT)Normal < 50 IU/l | ↑ | Hepatocellular injury (centrilobular) |
| Alkaline phosphataseNormal < 100 IU/l | ↑ |
BilirubinNormal 5–12 μmol/l↑
IgM anti-HAV antibodyPresentHepatitis AHBsAgPresentHepatitis B or carrierHBeAgPresentActive hepatitis B infectionAnti-HCV antibodyPresentHepatitis C virus exposureHCV RNAPresentActive hepatitis C infectionCaeruloplasmin↓Wilson’s diseaseIgA↑Alcoholic cirrhosisIgG↑Autoimmune hepatitisIgM↑Primary biliary cirrhosisAnti-mitochondrial antibodyPresentPrimary biliary cirrhosisAnti-smooth muscle, antinuclear or anti-LKM antibodiesPresentAutoimmune hepatitisFerritin↑HaemochromatosisAlpha-1 antitrypsin↓Alpha-1 antitrypsin deficiencyAlpha-fetoprotein (AFP) (normally undetectable)↑Liver cell carcinoma
HAV, hepatitis A virus; HBsAg, hepatitis B surface antigen; HCV, hepatitis C virus; LKM, liver and kidney microsomal antigen.
All cells in the liver are capable of regeneration. The liver cells are classified as stable—that is, they are not normally replicating but will do so if the liver is injured. This regenerative capacity is vital in the recovery of patients with liver damage due to viruses, drugs or trauma, but if the damage is persistent or occurs repeatedly, it can result in loss of the normal acinar or lobular structure and its replacement by regenerative liver cell nodules which are functionally inefficient. This is the condition called cirrhosis.
Some changes occur naturally in the liver with age. In the fetus, the liver is a relatively larger organ compared to the rest of the body. It is a major site of haemopoiesis and the adult liver can revert to this activity in some haematological disorders. The fetal liver synthesises alpha-fetoprotein, a fetal serum protein, and this is replaced by albumin towards the end of gestation. Alpha-fetoprotein synthesis by the adult liver usually denotes the presence of a primary liver cell carcinoma. With advancing age, the liver shrinks and becomes dark brown due to an increased amount of lipofuscin pigment in the liver cells (‘brown atrophy’).
INVESTIGATION OF LIVER DISEASE
The investigation of a patient with liver disease commonly includes:
These investigations complement careful history-taking and a thorough clinical examination.
Biochemistry
Bilirubin
Bilirubin pigment is a breakdown product of the haem moiety of haemoglobin (Fig. 16.2). It is produced at sites of red cell destruction (e.g. spleen) and circulates in the blood in an unconjugated water-insoluble form bound to albumin. In the liver it is conjugated to glucuronic acid by the enzyme glucuronyl transferase. Conjugated bilirubin is water-soluble and can therefore appear in the urine if the outflow of bile from the liver is interrupted; the patient’s urine then becomes stained with conjugated bilirubin. Bilirubin is converted by bacteria in the intestine to faecal urobilinogen (stercobilinogen), some of which is absorbed and then excreted, mostly in the bile to complete its enterohepatic circulation or, in only trace amounts normally, by the kidneys to appear in the urine as urobilinogen. Stercobilinogen is oxidised to stercobilin (faecal urobilin), the principal faecal pigment.
Fig. 16.2 Simplified pathways of bilirubin metabolism. Excessive breakdown of haemoglobin, as in haemolytic anaemias, will lead to increased biliary excretion of bilirubin. Biliary obstruction will cause regurgitation of conjugated water-soluble bilirubin into the blood which is then excreted in the urine, causing it to darken. Liver cell damage in hepatitis will cause impaired biliary excretion of urobilinogen and conjugated bilirubin; these are excreted in the urine, causing it to darken. The enterohepatic circulation, which involves urobilinogen, also returns cholic acid and chenodeoxycholic acid to the liver; this enhances bile secretion.
In early or recovering viral hepatitis, impaired biliary excretion results in preformed stercobilinogen appearing in the urine in excess as urobilinogen; this is one sensitive marker of early liver injury. In well-established biliary obstruction, the urinary urobilinogen concentration falls, because the cessation of biliary excretion into the gut results in sustained absence of synthesis of faecal urobilinogen.
Enzymes
In liver cell injury, damage to the membranes of cells and their organelles allows intracellular enzymes to leak into the blood, where the elevated concentrations can be measured. Examples include ALT, AST and GGT. Their diagnostic usefulness is summarised in Table 16.1.
The enzyme alkaline phosphatase is normally present in bile. Obstruction to the flow of bile, by gallstones for example, causes regurgitation of alkaline phosphatase into the blood, resulting in increased serum concentrations.
Many of these enzymes are not exclusively specific to the liver; therefore the results of diagnostic serum assays need careful interpretation.
Albumin
Albumin is a major serum protein synthesised by the liver cells. It has a relatively long half-life, compared to that of clotting factors (see below), so liver damage has to persist before decreased serum levels are found. In chronic liver disease, such as cirrhosis, a low serum albumin concentration is an important manifestation of liver failure, which results in peripheral oedema and contributes to the presence of ascites due to a reduction in plasma oncotic pressure.
Clotting factors
Liver cells synthesise the vitamin K-dependent clotting factors, deficiency of which results in a bleeding tendency. This can be detected in the laboratory by measuring the prothrombin time. A prolonged bleeding and prothrombin time is a further manifestation of liver failure and, because these clotting factors have a relatively short half-life, deficiency may be found quite early in the course of the illness. The prothrombin time should be measured before performing a liver biopsy or undertaking surgery on a patient with liver disease to avoid the risk of unexpected haemorrhage. These clotting factor deficiencies can be corrected by administration of high doses of vitamin K or of the clotting factors themselves.
Immunology
Although insignificant amounts of immunoglobulins are synthesised in the liver, immunological abnormalities often accompany liver disease and are useful diagnostic markers. These abnormalities include the appearance in the patient’s blood of auto-antibodies to normal tissue antigens. The antibodies are not responsible for the tissue damage in the liver diseases with which they are associated. Examples include:
Polyclonal immunoglobulin elevations also occur:
Antibodies to hepatitis viruses are, in some instances (e.g. hepatitis A), clinically useful markers of these infections. Viral antigens or nucleic acids can also be tested for.
Imaging
Techniques used to visualise the liver and detect lesions within it include:
Biopsy
The two common types of liver biopsy are:
Both procedures carry a small but significant risk of haemorrhage and biliary leakage from the biopsy site. Bile duct obstruction is a contraindication to liver biopsy because of the increased risk of biliary peritonitis from bile leakage from the biopsy site. The risk must be outweighed by the likely therapeutic benefit to the patient resulting from an accurate assessment of their liver disease.
Most liver diseases produce diffuse abnormalities in the organ; a biopsy from any part of it will therefore be representative. Focal lesions such as tumours may be missed, particularly by percutaneous needle sampling, but the biopsy needle can be guided to them by using imaging techniques such as ultrasonography.
Liver biopsies are examined by light microscopy after sectioning and staining. Unlike renal biopsies, little additional clinically useful information is obtained by examining liver biopsies with the electron microscope.
JAUNDICE
Jaundice (or icterus) is the name given to yellowing of the skin and mucosal surfaces due to the presence of bilirubin. Usually jaundice is observable when the serum bilirubin concentration exceeds 40 micromol/l. Note, however, that:
The accumulation of bilirubin in the skin may cause some embarrassment to the patient and, often if due to biliary obstruction, discomfort due to pruritus.
Jaundice in infants
Physiological neonatal jaundice is relatively common, particularly in premature infants. Although it causes understandable parental anxiety, the jaundice is rarely severe and it fades as liver function matures. However, high bilirubin levels in infancy can be directly harmful. Because the neonatal blood–brain barrier is relatively permeable, unconjugated bilirubin can accumulate in the lipid-rich brain tissue, causing bilirubin encephalopathy or kernicterus; this can be avoided by phototherapy or, in severe cases, exchange transfusion.
Worsening jaundice may be one of the clinical features alerting to the presence of a congenital abnormality within the hepato-biliary system. Such abnormalities may be:
Structural congenital abnormalities include:
Functional abnormalities include congenital metabolic defects involving the liver (see Ch. 7) and congenital hyperbilirubinaemias.
Classification of jaundice
Jaundice may be classified as pre-hepatic, intrahepatic or post-hepatic, depending on the site of the lesion, or conjugated and unconjugated, based on chemical analysis of the bilirubin in the blood or by deduction from the colour of the patient’s urine. Only conjugated bilirubin is sufficiently water soluble to be excreted in the urine.
Pre-hepatic jaundice
The main cause of pre-hepatic jaundice is haemolysis, due for example to hereditary spherocytosis or autoimmune red cell destruction (see Ch. 23). In these conditions there is excessive production of bilirubin from the haemoglobin released from lysed red cells. Because the excess bilirubin is unconjugated, it is not excretable in the urine; the urine colour is normal (hence the synonym ‘acholuric jaundice’). The bile, however, may contain so much bilirubin that there is a risk of pigment gallstone formation.
Intrahepatic jaundice
Hepatic disorders in which jaundice may be a feature include:
In these conditions there is accumulation of bilirubin within the liver (intrahepatic cholestasis), often histologically evident in biopsies as plugs of bile pigment distending canaliculi. The excess bilirubin is predominantly conjugated, is therefore water soluble and is excreted in the urine, causing darkening; this is a simple but diagnostically useful observation.
Post-hepatic jaundice
Obstruction of the extrahepatic bile ducts is an important cause of jaundice necessitating urgent investigation and alleviation in order to prevent serious damage to the liver. Important causes are:
As with intrahepatic causes, some of which also directly interfere with biliary drainage (e.g. primary biliary cirrhosis, sclerosing cholangitis), the excess bilirubin is conjugated and darkens the urine. Conversely, the patient’s faeces are pale. Pruritus is a common and troublesome symptom, probably due to bile salt accumulation.
ACUTE LIVER INJURY
Liver injury is conveniently divided into acute and chronic for the purposes of description and clinical management. However, in practice, the same cause may produce either an acute or a chronic illness, in the latter event not necessarily with any preceding clinically evident acute phase. For example, viral hepatitis is considered here under the heading of acute liver injury, but it can lead to chronic liver damage.
Aetiology
The major causes of acute liver injury are:
Direct physical injury to the liver, such as laceration in a road traffic accident, is another important form of acute liver injury, but the focal nature of the injury contrasts with the diffuse injury produced by the agents listed above. Recovery from acute liver injury, focal or diffuse, is attributable to the capacity of the organ for cellular regeneration.
Clinicopathological features
The clinical and laboratory manifestations of acute liver injury are:
Most of the signs and symptoms of acute liver damage are predictable from the known functions of the liver. The best known is jaundice (or icterus) due to failure of the liver to secrete bile at the rate at which it is formed in the body from the destruction of red cells. Severe acute liver damage can lead to bruising and haemorrhage, due to clotting factor deficiency, and coma due to the accumulation of toxic metabolites that mimic neurotransmitters (‘false neurotransmitters’).
Laboratory investigations
Laboratory investigations will reveal evidence of liver cell damage, in that there will be elevated levels of serum enzymes, particularly the transaminases, and bilirubin. Liver cell damage results in some impairment of bilirubin conjugation, but also failure to excrete conjugated bilirubin and any stercobilinogen absorbed from the gut. Consequently, the urine is darkened by the presence of excess conjugated bilirubin and urobilin (derived by oxidation from urobilinogen) that cannot be excreted by the liver (Fig. 16.2). Eventually, as the liver damage persists, urobilinogen disappears from the urine because little or no bilirubin is being excreted by the liver. Jaundice due to bile duct obstruction—commonly by gallstones—also results in dark urine due to excess conjugated bilirubin that cannot be excreted by the liver; urobilinogen is usually absent, unless the obstruction is of very recent onset or intermittent, because no bilirubin reaches the intestine. Examination of urine and faeces (for colour) can therefore assist in the differential diagnosis of jaundice (Table 16.2).
Histology
In almost all cases of acute liver injury there will be liver cell degeneration or death and an inflammatory reaction. Superimposed on this uniform reaction to acute injury are, in many cases, diagnostic changes specific to the causative agent.
Also evident in liver biopsies will be the pattern of cell damage, from which the prognosis can be deduced (Fig. 16.3):
Fig. 16.3 Patterns of liver cell death and their clinicopathological significance. Death of the liver cells immediately surrounding central veins denotes cardiac failure, some other impediment to venous drainage, or some toxic cause (e.g. paracetamol overdose). Bridging necrosis (in acinar zone 3) is a feature of severe hepatitis. Interface hepatitis is death of liver cells at the margin of the portal tracts; this is a feature of chronic hepatitis due to a variety of causes. Apoptotic death of single cells is typical of acute viral hepatitis.
Viral hepatitis
Hepatitis viruses
The main hepatitis viruses (Table 16.3) are:
Table 16.3Hepatitis viruses: their characteristics and associated diseases (delta agent, a defective virus, is not included)
These hepatitis viruses are immunologically distinct. Infection usually confers life-long immunity to the infecting virus but not to the others.
The clinical features range from a trivial illness without jaundice (anicteric hepatitis) which may escape detection (this is a common result of HAV infection) to a more significant illness with jaundice and other clinical evidence of disturbed liver function. Sometimes the illness is dominated by jaundice, with little elevation of serum transaminases (cholestatic hepatitis). Severe infection leads to overt liver failure.
Yellow fever, caused by a group B arbovirus, shares many clinical and histological features with the illness usually designated viral hepatitis, but it is not normally included within this group for the purposes of description, mainly because its geographical distribution is very restricted.
The liver may also become infected by many other viruses, but these are not regarded as ‘hepatitis viruses’ because the infection is not confined to the liver. Examples include:
Hepatitis A virus
The main characteristics of hepatitis A are:
Infection by HAV used to be called ‘infectious hepatitis’ because of its common occurrence in epidemics, though it also occurs sporadically. In most countries, infection by the virus is common, usually in youth; the resulting illness is often very mild and jaundice absent or so slight that it escapes notice. Overt jaundice and clinical recognition of the infection is less common. Hepatitis sufficiently severe to warrant hospital admission is unusual, and long-term sequelae or death are exceptional rarities. It is therefore a relatively benign infection.
HAV passes from one individual to another by ‘faecal– oral’ transmission—usually indirectly, such as by the contamination of food and drinking water with sewage. Because the virus is excreted in the faeces before jaundice appears, thus leading to the recognition of the illness and isolation of the patient, many other individuals can be rapidly exposed to the hazard of infection. The incubation period is relatively short. HAV produces liver cell damage by a direct cytopathic effect.
Specific diagnosis is made by seeking an IgM-class antibody to HAV in the patient’s serum; this indicates recent infection. A carrier state does not exist.
Hepatitis B virus
The main characteristics of hepatitis B are:
Infection by HBV used to be called ‘serum hepatitis’ because it was known to be transmitted by blood and blood products. This is because infected, but apparently healthy, individuals can carry the virus in their blood and pass it on to others by the transfusion of blood or its products. This mode of transmission is much less common now that blood donors are screened by testing for the presence of the virus. However, the term ‘serum hepatitis’ has been abandoned because it misleadingly excludes transmission of the virus by other methods, notably venereally; the disease is often transmitted between homosexual males. HBV can also be transmitted by contaminated needles, such as may be used for tattooing or by drug addicts. There is a relatively high incidence of the carrier state in underdeveloped countries and the virus can be transmitted vertically from mother to child—in utero, during delivery or through intimate post-natal contact.
Specific diagnosis is made by seeking the hepatitis B surface antigen (HBsAg, formerly known as ‘Australia antigen’ because it was first detected in the serum of an Australian aborigine). The presence of the ‘e’ antigen (HBeAg) in the patient’s serum indicates active viral replication.
HBV produces liver cell damage not by a direct cytopathic effect but by causing viral antigens to appear on the cell surface (HBsAg); these are then recognised by the body’s immune system and the infected liver cells are destroyed (Fig. 16.4). Thus, if immunity is generally impaired or there is specific tolerance to the antigen, the virus can survive in the liver cells without causing damage; such patients become asymptomatic carriers of the virus and their body fluids are a hazard to other individuals. Liver biopsies of HBV-infected carriers show that the liver cells have a ground-glass texture to their cytoplasm due to the abundance of virus particles.
Fig. 16.4 Comparison of the pathogenesis of HAV and HBV hepatitis.
[/not-level-membership-for-pathology-category]
