CHAPTER 39 Basic Science of Central Nervous System Infections
Role of the Blood-Brain Barrier in Central Nervous System Infections
The BBB is composed of a specialized layer of microvascular endothelial cells, pericytes, and astrocyte foot processes (or ependymal cells in the case of the blood-ependymal barrier). Brain microvascular endothelial cells (BMECs) form monolayers with high transendothelial electrical resistance and highly selective macromolecular permeability, properties largely attributable to two features: (1) the formation of highly organized intercellular tight junctions and (2) a low rate of transcytosis relative to other endothelial subtypes.1 These features restrict the movement of pathogens from the intravascular space across the BBB into the brain parenchyma or CSF. However, many pathogens have developed strategies to cross the BBB despite these elaborate defenses. Three major pathways are used by pathogens to gain entry to the CNS across the BBB: (1) transcellular passage (e.g., Escherichia coli, group B streptococci [GBS]), (2) paracellular passage (e.g., protozoa), and (3) carriage within a transmigrating leukocyte, known as the “Trojan horse” mechanism (e.g., Listeria monocytogenes, Streptococcus suis, Mycobacterium tuberculosis, human immunodeficiency virus [HIV]). Most of these pathways across the BBB are poorly characterized for the vast majority of pathogens associated with CNS infection. Among the best characterized CNS-invasive pathogens with respect to passage across the BBB is E. coli. The following section discusses mechanisms involved in E. coli traversal of the BBB to highlight the complex interactions occurring between host and pathogen during the early stages of CNS infection. Additionally, established features of GBS passage across the BBB are highlighted to broaden the scope of mechanisms reviewed here.
Escherichia coli at the Blood-Brain Barrier Interface
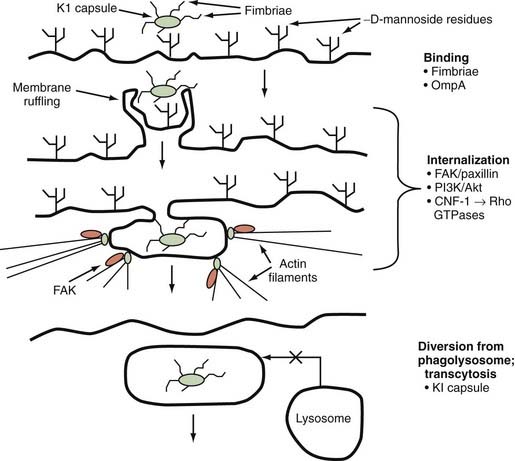
E. coli is a gram-negative bacterium implicated in the majority of cases of neonatal meningitis. In vitro studies of infection of human brain microvascular endothelial cells (HBMECs) by E. coli K1, an encapsulated strain responsible for most cases of neonatal meningitis, have shown that E. coli K1 interacts with these cells in a unique manner involving both host- and pathogen-specific structures and signaling pathways. These interactions lead to alterations in the host actin cytoskeleton, membrane protrusion and ruffling around bacteria, and endocytosis of bacteria into membrane-bound vacuoles, where bacterial determinants act to prevent lysosome fusion and influence intracellular vacuole trafficking to achieve transcytotic passage.2 Several bacterial determinants have been identified as part of the initial binding and invasion of HBMECs, including type 1 fimbriae, outer membrane protein A (OmpA), Ibe proteins, and cytotoxic necrotizing factor-1 (CNF-1).2 Type 1 fimbriae are adhesins that bind to α-D-mannosides on the surface of host cells, thereby allowing binding and interaction of the bacterium with the host cell. Type 1 fimbriae have been shown to play an important role in the binding of E. coli K1 to HBMECs; deletion of fimH, the gene for the major adhesin protein of type 1 fimbriae, significantly decreases binding of E. coli K1 to HBMECs, a finding that is reversed by genetic complementation of fimH in deletion mutants.3 OmpA also facilitates binding of E. coli K1 to HBMECs via interaction with surface glycoproteins containing N-acetylglucosamine residues.4 In vivo studies using an experimental neonatal rat model of meningitis have shown that deletion mutants of ompA are impaired in their ability to enter the CNS in comparison to the parent K1 strain, and N-acetylglucosamine oligosaccharides are able to block penetration of the CNS by wild-type E. coli K1 in the same animal model.5
Once binding has occurred, the process of cellular invasion must take place for E. coli K1 to ultimately infect the CNS. Invasion of HBMECs is dependent on the host actin cytoskeleton; E. coli K1 invasion can be completely inhibited in vitro by using inhibitors of the actin cytoskeleton such as cytochalasin D.6 Entry of E. coli K1 into membrane-bound vacuoles in HBMECs involves the formation of membrane projections, described as zipper-like structures, and subsequent membrane ruffling before internalization (Fig. 39-1).7 These events are related to several important signal transduction pathways in the host cell known to be involved in the regulation of endocytosis, cell membrane interactions, and the actin cytoskeleton. In particular, E. coli K1 leads to activation of focal adhesion kinase (FAK) and phosphorylation of paxillin, a cytoskeletal protein that interacts with and regulates the actin cytoskeleton.8 The exact mechanisms underlying FAK activation by E. coli K1 are unknown but appear to play a role in HBMEC invasion because overexpression of a dominant-negative form of FAK significantly inhibits HBMEC invasion by E. coli K1.8
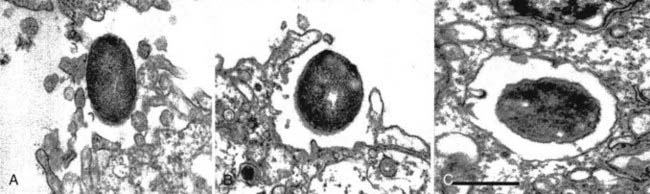
Another important signal pathway involved in E. coli K1 invasion of HBMECs is the family of phosphatidylinositol-3′-kinases (PI3Ks), which lie downstream of FAK activation. Pharmacologic inhibition of PI3Ks with LY-294002 significantly limits HBMEC invasion by E. coli K1.9 Akt/protein kinase B, a downstream effector of PI3K, is increased during E. coli K1 invasion of HBMECs.9 These observations indicate a role for the PI3K/Akt pathway in E. coli invasion of HBMECs, although the exact roles that these kinases play in the process have yet to be described.
Rho family guanosine triphosphatases (GTPases) have been shown to regulate a diverse array of processes that affect the actin cytoskeleton, cell motility, and cell-cell/cell-matrix interactions.10 E. coli strains associated with urinary tract infections and meningitis have been shown to produce CNF-1, an AB-type toxin with deaminase activity that targets this family of GTPases.11 CNF-1 activates Rho GTPases and increases the in vitro uptake of bacteria by nonprofessional phagocytes such as epithelial and endothelial cells.11 Deletion of the cnf1 gene from E. coli K1 significantly decreases the invasion of HBMECs, and this deletion is associated with decreased activation of RhoA and Cdc42, GTPases that are activated during E. coli K1 invasion of these cells.11
Once E. coli K1 bacteria have been internalized in membrane-bound vacuoles within HBMECs, these organisms must survive the internal hostile milieu and avoid destruction in the lysosome to gain entry into the CNS. The K1 capsule appears to play a very important role in preventing the normal maturation of endosomes and fusion of vacuoles with the lysosome. K1 isogenic deletion mutants (K1−) have been shown to traffic through the endosomal system and colocalize with cathepsin D, thus confirming fusion of the lysosome with the vacuoles containing these bacteria.12 The exact mechanisms underlying the K1 capsular effect on endosomal trafficking have yet to be elucidated, however.
In summary, a neuroinvasive strain of E. coli uses specific tools to gain entry into the CNS by binding/uptake into HBMECs and subsequent diversion of the normal protective trafficking of endosomal compartments to the lysosome (Fig. 39-2). These events allow E. coli K1 strains to cross the highly selective BBB and cause meningitis. The next section reviews the most recent information pertaining to crossing the BBB by GBS, gram-positive bacteria that remain a prominent cause of sepsis and meningitis in neonates and infants.
Group B Streptococci: If You Can’t Beat Them, Join Them
Like E. coli K1 strains, GBS express a polysaccharide capsule that serves many functions in the process of host invasion, including protection from opsonization, complement-mediated lysis, antibody-mediated clearance, and phagocytosis.13 Nine distinct GBS capsular serotypes have been identified, with serotype III strains dominating the clinical isolates associated with meningitis.13 Virulent strains of GBS have terminal sialic acid residues coating the surface of the polysaccharide capsule; GBS strains lacking these residues are less virulent than isogenic counterparts carrying these sialic acid groups.14 Many host glycoproteins have the same terminal sialic acid residues (α2 → α3 N-acetylneuraminic acid), so the presence of these residues on the GBS capsule may provide the bacterium with some protection from immune surveillance via molecular mimicry.13 These residues also inhibit activation of the alternative complement pathway, thus preventing opsonophagocytosis of GBS.13
Like E. coli K1, GBS must bind to the luminal surface of BMECs to initiate invasion of the CNS. Importantly, GBS are able to bind to several components of the extracellular matrix, including fibrinogen, laminin, and immobilized but insoluble fibronectin. ScpB, a C5 peptidase anchored in the GBS membrane, has been identified as a selective fibronectin-binding protein that differentially associates with bound fibronectin.13 FbsA is a surface-anchored protein that binds fibrinogen, and the gene fbsA is regulated by RogB, a transcriptional regulator that positively regulates several genes involved in binding to the extracellular matrix.13
Recently, GBS pili have been shown to play a role in GBS interactions with HBMECs, and targeted deletion of the gene for the pilus accessory surface protein PilA significantly reduces the ability of GBS to adhere to HBMECs, whereas deletion of pilB, the gene encoding the major pilus structural protein, does not affect adherence but significantly reduces HBMEC invasion by GBS.15
The dissociation of adherence and invasion observed with the pili mutants just described supports the hypothesis that these events, adherence and invasion, are mediated by distinct virulence factors expressed by GBS. Other invasion mediators that have been identified for GBS and HBMECs include a gene involved in modification of lipoteichoic acid (LTA), expression of a β-hemolysin/cytolysin, and factors that alter host cell signaling pathways. LTA is a major structural component of the surface of gram-positive bacteria and may mediate amphiphilic interactions with host cell membrane phospholipids. Allelic exchange of a glycosyltransferase homologue in GBS, iagA, reduces LTA anchoring in the GBS cell wall and significantly reduces HBMEC invasion in vitro and the development of meningitis in a murine in vivo model.16 β-Hemolysin/cytolysin is a pore-forming exotoxin expressed by GBS that is known to promote GBS invasion of HBMECs at sublytic concentrations and cause HBMEC cytolysis at higher concentrations.17 β-Hemolysin/cytolysin also induces HBMEC synthesis of interleukin-8 (IL-8), an extremely potent neutrophil chemoattractant, and promotes neutrophil transmigration across HBMEC monolayers.17 Mice hematogenously infected with a GBS mutant lacking this exotoxin had lower brain bacterial counts and lower mortality than did mice infected with the parent wild-type strain expressing this toxin.17
As with E. coli K1, GBS have been shown to modulate a number of host signaling pathways to facilitate passage across the BBB. In fact, the FAK/paxillin/PI3K pathway is a common target involved in both E. coli K1 and GBS invasion of HBMECs. Inhibition of FAK signaling via a dominant-negative form of FAK and pharmacologic inhibition of PI3K with LY-294002 both significantly inhibit GBS invasion of HBMECs.18 Another shared pathway involved in E. coli K1 and GBS invasion of HBMECs involves RhoA GTPase; RhoA levels are increased during GBS invasion of HBMECs, as are levels of Rac1.19 Inhibition of RhoA and Rac1 with a geranylgeranyl transferase I inhibitor, GGTI-288, and expression of dominant-negative forms of these GTPases both result in significantly reduced HBMEC invasion by GBS.19
Innate Immunity in the Central Nervous System
Microglia: Ramón y Cajal’s “Third Element”
Santiago Ramón y Cajal (1852-1934) divided the histology of the brain into “elements,” including neurons (first element), astrocytes (second element), and a third category of non-neuronal, nonastrocytic cells that he termed the third element. This third element contained cells later identified as oligodendrocytes, as well as a group of small, highly branched cells that were morphologically distinct from other cells in the CNS. These cells were eventually termed microglia by Pio del Rio Hortega (1882-1945), who went on to further characterize these cells as a distinct entity in the brain parenchyma.20 Like monocytes and macrophages, these cells are derived from the bone marrow and share many features of monocytes/macrophages with respect to immune modulation.20
Microglia can exist in several different states, as defined by surface markers, morphology, migration status, and function. Resting microglia are small cells with few surface markers and prominent thin branches that are constantly reorganizing and sampling the microenvironment of the brain parenchyma.21 A large number of stimuli activate resting microglia, with the nature of the stimulus influencing the structural and functional changes that occur during activation. Cellular debris from CNS damage, particularly free adenosine triphosphate, activates microglia and induces a morphologic change from a small, ramified cell to an ameboid cell capable of phagocytosing such debris in a manner similar to macrophages.21 Microglia, like macrophages, express a large number of receptors associated with various mediators of the inflammatory response to tissue damage, pathogens, and immune stimuli. A major family of receptors expressed by microglia includes pathogen-associated molecular pattern (PAMP) receptors known as the Toll-like receptors (TLRs). Eleven TLRs (TLR1 to TLR11) have been identified to date in humans. Microglia express TLR1 to TLR9, which allows them to detect and respond to a huge array of PAMPs; LTA (TLR2), double-stranded RNA (TLR3), lipopolysaccharide (LPS; TLR4), flagellin (TLR5), single-stranded RNA (TLR7), and unmethylated CpG DNA (TLR9) are examples.22 Engagement of these receptors by cognate ligands triggers signal transduction of multiple intracellular pathways responsible for the inflammatory response. LPS-induced activation of TLR4 has been well characterized in microglia and leads to activation of nuclear factor κB (NF-κB), cytokine production (interferon-β [IFN-β]), tumor necrosis factor-α (TNF-α)], signal transducer and activator of transcription-1α (STAT-1α), production of reactive oxygen species (ROS), and production of nitric oxide (·NO, or NO).23 Flagellin-mediated activation of TLR5 on microglia has been shown to upregulate expression of TLRs 1, 2, 4, and 5, as well as IL-6.24 TLR3 activation in microglia has been demonstrated in response to HIV, and TLR9 activation in microglia leads to the production of multiple cytokines and chemokines (TNF-α, IL-1β, IL-6, IL-12, macrophage inflammatory protein-1α [MIP-1α], MIP-1β).25,26
Activation of microglia by invading pathogens has many consequences, some beneficial to the host and some detrimental. Microglia, like other members of the monocyte lineage, are capable of NO synthesis and a respiratory burst, processes directed at producing oxidative damage to the offending pathogen. Microglia produce NO via inducible nitric oxide synthase (iNOS, NOS-2), which leads to the formation of peroxynitrite (ONOO−), a highly toxic product capable of damaging both host and pathogen.27
Microglia also possess the metabolic machinery (reduced nicotinamide adenine dinucleotide phosphate oxidase) necessary for the generation of superoxide anion (·O2−), an extremely reactive oxygen species that can damage nucleic acids, lipids, and proteins.28 Pathogens expressing superoxide dismutase are able to neutralize this effective defense. Microglia also function as phagocytes in the CNS under both physiologic and pathophysiologic conditions. Microglia are the major scavengers of cell debris in the CNS and interact, in part, with apoptotic bodies expressing externalized phosphatidylserine.29 The orphan receptor TREM-2 (triggering receptor expressed on myeloid cells-2) is important in transforming microglia into phagocytes, and activation of TREM-2 enhances phagocytosis while suppressing the production of proinflammatory cytokines, events that may be important for prevention of the autoimmune responses to the autoantigens present in apoptotic bodies.30
Microglia also regulate the response of other CNS cells to injury or infection and recruit cells from outside the CNS via the production of a wide variety of cytokines, chemokines, and lipid mediators. Table 39-1 lists some of the cytokines and chemokines known to be generated by microglia in response to a number of activating stimuli.31 Microglia also produce factors that support glial and neuronal cells in their microenvironment, including nerve growth factor, NT-3, brain-derived neurotropic factor, glial-derived neurotropic factor, and basic fibroblast growth factor.31 Many potent lipid mediators are synthesized by microglia, including prostaglandins (D2, E2, F2α, thromboxane B2), leukotriene B4, and platelet-activating factor.32,33 Several of these lipid mediators serve an autocrine role; the EP2 receptor on microglia participates in the activation of microglia, and antagonism of this receptor may have a neuroprotective effect by preventing excessive microglial neurotoxicity.34 The critical role of microglia in orchestrating the innate immune response has been established, and experimental evidence for this role is expanding rapidly; an extensive review of this evidence can be found elsewhere.31,35,36
TABLE 39-1 Cytokines and Chemokines Produced by Microglial Cells
| CYTOKINES | CHEMOKINES (CHEMOATTRACTANT CYTOKINES) |
|---|---|
| IL-1α/IL-1β | CXCL1 (growth-regulated oncogene-α) |
| IL-1 receptor antagonist | CXCL2/3 (MIP-2) |
| IL-3 | CXCL8 (IL-8) |
| IL-4 | CXCL10 (IP-10) |
| IL-6 | CCL2 (MCP-1) |
| IL-10 | CCL3 (MIP-1α) |
| IL-12 | CCL4 (MIP-1β) |
| IL-13 | CCL5 (RANTES) |
| IL-15 | CCL22 (macrophage-derived chemokine) |
| IL-18 | |
| TNF-α | |
| TGF-β | |
| M-CSF |
IL, interleukin; IP-10, interferon-γ–inducible protein-10; MCP-1, monocyte chemoattractant protein-1; M-CSF, macrophage colony-stimulating factor; MIP-1α, macrophage inflammatory protein-1α; RANTES, regulated on activation, normal T cell expressed and secreted; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α.
Data from Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140-155.
In addition to initiating the innate immune response to a variety of pathogens in the CNS, microglia also function as antigen-presenting cells and are able to prime CD4+ T cells to initiate the adaptive immune response. Various TLR ligands (LPS, peptidoglycan, polyinosinic-polycytidylic acid [poly-I:C], CpG DNA) and infection with Theiler’s murine encephalomyelitis virus result in increased expression of major histocompatibility class II complexes and costimulatory molecules on microglia, events that favor efficient antigen presentation and development of an adaptive immune response.26 Cytomegalovirus (CMV), a gamma herpes virus capable of invading the human CNS, elicits CXCL10 (IFN-γ–inducible protein-10 [IP-10]) production from primary microglia but not from astrocytes (see later).37 CXCL10 is an important chemokine involved in IFN-γ–induced T-cell recruitment, a process critical for control of CMV infection.37 Remarkably, astrocytes infected with CMV produce the viral homologue of IL-10, UL111a, which suppresses the production of CXCL10 from activated microglia.37 Thus, CMV is able to suppress T-cell recruitment in the CNS by subverting the production of an important antiviral chemokine by microglia.
Microglia, as potent regulators of the proinflammatory response to CNS injury and infection, are also able to downregulate these proinflammatory responses. Microglia express anti-inflammatory cytokines (IL-4, IL-10, IL-13, transforming growth factor-β [TGF-β]) and actively phagocytose apoptotic T cells after stimulation by IFN-β and IFN-γ.31,38,39 Production of IL-4 and IL-13 ultimately triggers microglial apoptosis via autocrine receptor engagement, thereby providing a means of balancing inflammation in response to a specific CNS insult.40
Astrocytes: Stellar Actors in Central Nervous System Immunopathogenesis
Like microglia, astrocytes express several TLRs, but to a more limited degree, which allows them to participate in the initial innate immune response to CNS invasion. The close proximity of astrocyte foot processes to BBB tight junctions and the huge surface area presented by the brain endothelium also highly favor early interaction between astrocytes and invading pathogens. TLRs expressed by astrocytes include 1, 3, 4, 5, and 9, with little or no TLRs 2, 6, 7, 8, or 10.22,41 Among the expressed TLRs, TLR3 levels are the most prominent by quantitative real-time polymerase chain reaction (PCR).41 TLR3 ligation in astrocytes (by poly-I:C) triggers the production and release of IFN-β, CXCL10, and IL-6, as well as an increase in the expression of TLRs 1 to 5 and 9 mRNA by quantitative real-time PCR.41 Astrocytes are the main source of IL-6 production in the CNS; IL-6 is a multifunctional cytokine with diverse biologic activities, including neurotropism, protection of neurons from glutamate toxicity and ischemic injury, astrocyte proliferation, inflammation, and modulation of Fas/FasL expression in astrocytes.42 This latter effect on Fas/FasL expression may affect the general immunologic state of the CNS in that astrocytes normally express both elements of the Fas apoptosis apparatus yet do not succumb to this autocrine signaling.43 The lack of astrocyte apoptosis despite Fas/FasL expression has been attributed to low-level expression of procaspase-8, whereas FasL expression by astrocytes is thought to provide a molecular barrier to circulating lymphocytes entering the CNS, cells that are highly sensitive to Fas/FasL-induced apoptosis.44,45
In addition to the production of important immunologically active cytokines and chemokines, astrocytes also respond to chemokines through expression of chemokine receptors. Multiple chemokine receptors have been identified in astrocytes, and ligation of these receptors has many downstream effects on astrocyte function, including regulation of chemokine production and receptor expression (Table 39-2).46 Of note, astrocytes express CXCR4, the receptor for stromal cell–derived factor-1α (SDF-1α), a chemokine with autocrine activity leading to influx of Ca2+ and chemostasis of astrocytes. Both CCR5 and CXCR4 serve as CD4-associated coreceptors for HIV. Although astrocytes lack CD4 receptors, CXCR4 binds to the gp120 glycoprotein of HIV, an event that leads to activation of the mitogen-activated protein kinases (MAPKs) extracellular signal–regulated kinase-1 (ERK-1) and ERK-2.46 HIV also infects astrocytes via a CD4-independent mechanism, which results in a restricted (i.e., nonlytic, nonproductive) infection with incorporation of the HIV provirion into genomic DNA. Nonetheless, this restrictive infection alters astrocyte functions: increased expression of immunomodulatory molecules such as monocyte chemotactic protein-1 (MCP-1), complement factor 3 (C3), and iNOS and decreased glutamate uptake (see later).47 The decreased glutamate uptake and increased iNOS expression by HIV-infected astrocytes probably contribute to the neurotoxicity observed with HIV infection of the CNS.
TABLE 39-2 Select Astrocyte Chemokine Receptors and Ligands and Astrocyte Responses to Receptor Activation
| CHEMOKINE RECEPTOR | LIGAND | ASTROCYTE RESPONSE |
|---|---|---|
| CCR1 | CCL3 | Chemotaxis, chemokine synthesis |
| CCL4 | Ca2+ mobilization, chemokine synthesis, chemostasis | |
| CCR2 | CCL2 | Chemotaxis |
| CCL11 | Inhibition of forskolin-induced cAMP production, chemotaxis | |
| CCR3 | CCL3 | Chemotaxis, chemokine synthesis |
| CCR5 | CCL5 | Ca2+ mobilization |
| CXCR2 | CXCL2/3 | Chemokine synthesis |
| CXCL8 | Complement protein 3 synthesis | |
| CXCR4 | CXCL12 | Ca2+ mobilization, inhibition of forskolin-induced cAMP production, phosphorylation of ERK-1/2 |
cAMP, cyclic adenosine monophosphate; ERK, extracellular signal–regulated kinase.
Data from Dorf ME, Berman MA, Tanabe S, et al. Astrocytes express functional chemokine receptors. J Neuroimmunol. 2000;111:109-121.
One of the major functions of astrocytes is uptake of glutamate at synaptic junctions in the CNS. Glutamate is an excitatory neurotransmitter that is highly neurotoxic, mainly via two mechanisms: (1) hyperactivation of neurons and (2) inhibition of cysteine uptake leading to oxidative damage to neurons through glutathione depletion. Astrocytes “soak up” glutamate in the CNS via excitatory amino acid transporters and rapidly convert glutamate to nontoxic glutamine by expression of the enzyme glutamine synthase.48 This glutamine is then exported from astrocytes, taken back into neurons, and converted back to glutamate via a mitochondrial-based glutaminase for use as a neurotransmitter.48 Pathogen-associated factors or inflammatory mediators released in the CNS during infection, or both, can alter the ability of astrocytes to control the CNS glutamate concentration and result in glutamate-induced neurotoxicity as a by-product of infection. For example, HIV gp120 has been shown to reduce astrocyte expression of glutamine synthase, and patients with HIV-associated dementia have higher brain glutamate levels than do nonaffected controls.49,50 Glucocorticoid-induced expression of glutamine synthase is inhibited by IL-1β and TNF-α.51
Matrix Metalloproteinases, “You Can’t Have Your Cake and Eat It Too!”
MMPs are a subgroup of the metzincin family of proteases that share a common Zn2+ binding site in their catalytic domain (…HExxHxxGxxH…, where x is any amino acid) and an associated methionine within a β turn, biochemical elements important for proteolytic activity.52 There are 24 members of the MMP group in mammals, and they can be further subdivided according to their domain structure (additional details can be found in the review by Yong52). Most MMPs are secreted from cells, although some may be associated with cell surface molecules such as integrins (e.g., pro-MMP-2) or the hyaluronan receptor (active MMP-9) or are linked to the cell membrane via a glycophosphatidylinositol link.52 MMPs degrade a wide variety of substrates, including extracellular matrix components, receptors, growth factors, and adhesion molecules.52 As a consequence, MMPs must be tightly regulated to prevent extensive tissue destruction or unintended biologic sequelae. The first level of control involves the transcriptional regulation of MMP genes based on specific activation signals. MMPs may also undergo posttranslational modification and are highly compartmentalized within cells, thereby allowing further intracellular control of MMP activity.52 The second level of MMP regulation is based on secretion of these enzymes as zymogens, or proenzymes, which require additional cleavage to become active proteases. Finally, specific inhibitors, known as tissue inhibitors of metalloproteinases (TIMP-1 to TIMP-4), are expressed in tissues to counteract MMPs.52
MMPs have been implicated as playing a role, either beneficial or detrimental, in many different diseases affecting the CNS, including infections, ischemic or traumatic injury, and autoimmune disorders.52 The cellular sources of these MMPs vary depending on the specific pathologic condition but generally include cellular elements of the CNS (microglia, astrocytes, neurons), endothelial cells, and infiltrating leukocytes, especially neutrophils and macrophages.52 Neutrophils, in particular, represent an important early source of MMP-9 because these cells are among the “first responders” of the innate immune response to injury or infection and contain preformed stores of this MMP.52 In general, MMPs can have complex effects on the inflammatory state of the CNS. The BBB is rich in potential substrates for MMPs, including type IV collagen, fibronectin, and laminin; MMP degradation of the BBB may favor transmigration of leukocytes, as well as the movement of macromolecules and water, thus contributing to brain edema. Injection of heat-killed Neisseria meningitidis results in BBB disruption and increased intracranial pressure (ICP), phenomena that are inhibited by the administration of batimastat, an MMP inhibitor.53 MMPs also modify or degrade cytokines and chemokines relevant to this inflammatory state. For example, MMP-9 cleaves six amino acids from the N-terminal of IL-8 to produce a truncated form of IL-8 that is more potent with respect to neutrophil chemoattraction.54 In contrast, MMPs degrade IL-1β, thus demonstrating anti-inflammatory activity.55
CNS infections in which MMPs have specifically been shown to play a role include pneumococcal, gram-negative bacterial, and tuberculous meningitis, as well as viral infections such as HIV, human T-cell lymphotropic virus type I, and mumps and parasitic infections such as cerebral malaria and Angiostrongylus-associated meningoencephalitis.56–61 We focus here on the MMPs and pneumococcal meningitis as an example of the complex roles played by MMPs in CNS infection.
Streptococcus pneumoniae is a gram-positive bacterium responsible for a number of infections in humans, including pneumonia, otitis media, sinusitis, sepsis, and meningitis. A rabbit model of experimental pneumococcal meningitis demonstrated a positive correlation between MMP-9 levels and CSF leukocyte counts and total protein levels.62 MMP-9 activity, as measured by gelatin zymography, localized predominantly to the intrathecal compartment and not to brain parenchyma, thus supporting the conclusion that the increase in MMP-9 activity during meningitis is derived from infiltrating leukocytes.62 MMP-9 may become activated during pneumococcal infection by ROS, as shown by the inhibition of MMP-9 activity in but not release from rat brain slices or neutrophils exposed to heat-inactivated pneumococci in the presence of ROS inhibitors.63 Pneumococci can directly activate MMP-9 via production of ZmpC, a pneumococcal zinc metalloproteinase.64 Interestingly, MMP-9 knockout mice (MMP-9−/−) do not develop different CNS pathology than wild-type controls when S. pneumoniae is injected directly into the brain parenchyma, but these mice are less able to clear systemic bacteremia, which suggests that MMP-9 expression may play a more important role in clearing systemic pneumococcal infection.65 Infant rat pups with experimental pneumococcal meningitis demonstrated a peak in brain parenchymal MMP-2 and MMP-9 levels 20 hours after infection and a peak in TIMP-1 levels 24 hours after infection.66 The MMP-9/TIMP-1 ratio was significantly elevated during the first 20 hours of infection, thus supporting an imbalance in proteinase and inhibitor levels.66 The elevation in MMP-9 was associated with proteolysis of collagen type IV in the meninges, perivascular spaces, and brain parenchyma, and parenchymal gelatinolytic activity correlated well with the degree of cortical damage.66 In a rat model of pneumococcal meningitis, brain levels of MMP-9 increased in infected rats treated with ceftriaxone but not in saline-injected rats, whereas treatment with ceftriaxone plus dexamethasone reduced MMP-9 levels in comparison to untreated controls or animals treated with just ceftriaxone.67 Glucocorticoids are known to inhibit MMP expression, and these findings are consistent with a potential protective effect of dexamethasone during pneumococcal meningitis. Tetracyclines inhibit the proteolytic activity of many MMPs, as well as the activity of TNF-α–converting enzyme (TACE), another proteolytic enzyme implicated in propagating CNS damage during meningitis.68 Infant rats with pneumococcal meningitis that were treated with ceftriaxone plus doxycycline had lower mortality, less cortical damage, and less BBB disruption than did rats treated with ceftriaxone alone.68 Both groups had sterile CSF by 40 hours after infection, with no differences in the time-kill curves between these groups during this time frame, thus suggesting that the effect of doxycycline was not due to enhanced bacterial clearance from CSF.68 These animal models provide evidence that MMPs, particularly MMP-9, play a pathophysiologic role in bacterial meningitis and that targeting MMPs with inhibitors may reduce mortality and cortical damage without significantly altering sterilization of the CNS.
Data regarding MMPs in human disease also exist and are consistent with the observations described in animal models. In 27 children with bacterial meningitis, 91% and 97% had elevated CSF levels of MMP-8 and MMP-9, respectively, when compared with uninfected control children.69 The majority of these children were infected with Haemophilus influenzae (n = 14) or N. meningitidis (n = 11), with only 2 infected with S. pneumoniae. However, elevated MMP-9 levels in CSF were associated (P < .05) with an increased risk for neurologic sequelae, including hearing loss and postinfection seizures.69 In 19 adults with bacterial meningitis (n = 7 with S. pneumoniae), all had elevated MMP-9 activity, as measured by gelatin zymography, in comparison to uninfected controls or patients with Guillain-Barré syndrome.53 Patients with bacterial meningitis also demonstrated increases in CSF TIMP-1 levels, and the MMP-9/TIMP-1 ratio was significantly elevated when compared with noninfected controls.53 MMP-9 levels in the infected patients correlated with CSF protein concentrations but not CSF leukocyte counts, whereas TIMP-1 levels correlated with CSF leukocyte counts but not protein levels.53 An investigation of 111 paired CSF and serum samples from patients with a range of neurological disorders, including both aseptic and bacterial meningitis, found that a CSF leukocyte count greater than 5/µL correlated well with elevated CSF MMP-9 activity by zymography (Spearman r = .755, P < .0001).70 A more recent study examined the correlation between serum MMP-2 levels and the α2-macroglobulin (α2M) index as a marker of increased BBB permeability in patients with infectious meningitis. The α2M index was defined as the ratio of α2M (CSF/serum) to albumin (CSF/serum). This study found that serum MMP-2 levels, as measured with an enzyme immunoassay, correlated well (r = .64, P < .0001) with the α2M index and were higher in patients with bacterial meningitis than in those with viral or fungal meningitis.70
Brain Edema and Neurotoxicity: Consequences of Central Nervous System Infection
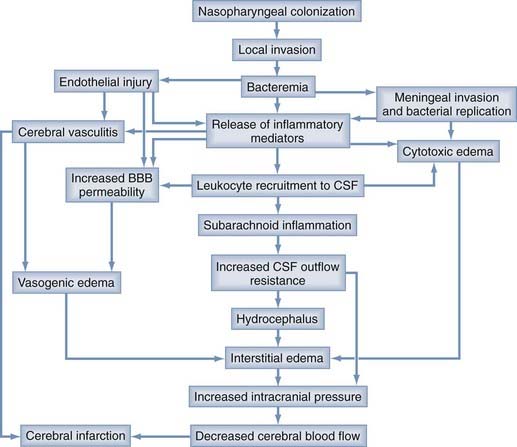
The severity of a CNS infection is dependent on many factors: the pathogenicity of the invading organism (i.e., the propensity of a pathogen to damage the host), the susceptibility of the host, the host immune response, and the timing and effectiveness of external interventions (e.g., antimicrobial agents, glucocorticoids, surgical therapies). Common mechanisms of injury to the CNS as a result of infection include brain edema and neurotoxicity. Brain edema can lead to increased ICP, decreased cerebral blood flow, ischemia, and herniation (Fig. 39-3). Specific infections can lead to increased ICP or herniation, or both, through a mass effect independent of the presence of brain edema, such as brain abscess or subdural empyema, or through effects on CSF drainage, such as cryptococcal or tuberculous meningitis. Neurotoxicity includes direct cytotoxic insults to neurons from pathogen-derived factors (e.g., pneumolysin from S. pneumoniae) or direct infection of neurons, as well as indirect cytotoxicity via immune system activation or altered neurophysiology (e.g., glutamate excitotoxicity). Earlier chapters discuss brain edema and the physiology of ICP in depth; this section addresses these issues as they relate to CNS infections.
Brain Edema and Central Nervous System Infections
Many factors contribute to the development of brain edema during the course of CNS infection, but most share a common final target, the BBB. The major cellular element of the BBB is the brain microvascular endothelium, a monolayer of overlapping endothelial cells connected by organized tight and adherens junctions and exhibiting little transcytosis, unlike the capillary endothelium in other tissues. BMECs behave much more like epithelial layers with respect to low paracellular permeability to macromolecules and cells, a feature that helps protect the brain parenchyma from unregulated fluid shifts based solely on Starling’s forces, such as oncotic or hydrostatic pressure. Alterations in BBB permeability occur during CNS infection when pathogen- or host-derived factors influence the complex machinery responsible for maintaining low BBB permeability. During infection of the CNS by viruses, bacteria, fungi, or parasites, the innate and adaptive immune responses induce influx of leukocytes into the CNS, a process associated with increased BBB permeability. This increase in BBB permeability is the sum of many individual, complex and interacting processes that involve leukocytic infiltration of the CNS, but changes in BBB permeability are not always dependent on leukocytes. Myriad proinflammatory and anti-inflammatory mediators are associated with this leukocyte infiltration and change in BBB permeability. Figure 39-3 provides a schematic view of the complex processes involved in the initiation and propagation of brain edema and neurotoxicity during bacterial meningitis; this scheme can also be applied to infection caused by nonbacterial meningitis. Some examples of specific pathogens and the mechanisms underlying alteration of the BBB during infection with these pathogens are discussed in the following sections.
Viruses Invading the Central Nervous System
A well-recognized feature of HIV-associated dementia is chronic breakdown of the BBB, a phenomenon attributed to immune dysregulation in the CNS via chronic activation of microglia, infiltration of the CNS by activated peripheral macrophages, and viral components such as gp120 and Tat.71 Both these proteins downregulate components of tight junctions, the major barrier to the paracellular pathway between BMECs.72,73 Tat possesses a cellular attachment domain that contains many positive charges, a feature that allows Tat to bind and cross cellular membranes. Exposure of BMECs to Tat leads to a number of important changes in BMEC physiology, including increased oxidative stress, expression of the early adhesion molecule E-selectin, and upregulation of inflammatory cytokines such as IL-6.74 Tat also decreases claudin-1 and claudin-5 localization to tight junctions; claudin-5, in particular, is an important component of tight junctions, and loss of this protein increases paracellular permeability across tight junctions significantly.73 The HIV envelope protein gp120 has been shown to decrease the expression of tight junction proteins.72 A recent study has demonstrated gp120-induced, proteasome-mediated degradation of zona occludens-1 (ZO-1) and ZO-2, important components of tight junctions in BMECs, but not degradation of other structural tight junction components such as occludin or claudin-1.75 This process is inhibited by lactacystin, a proteasome inhibitor, and enhanced by downregulation of the scaffold protein 14-3-3τ.75 Thus, HIV invasion of the CNS results in increased BBB permeability via several mechanisms, a process that allows continued viral invasion and progression of CNS disease.
West Nile virus (WNV), a single-stranded RNA flavivirus, causes severe, even fatal encephalitis in susceptible hosts. The mechanism by which WNV crosses the BBB is not fully elucidated, but inflammation appears to play a critical role. TLR3-deficient mice infected with WNV are significantly more resistant to WNV-related mortality (40% survival rate versus 0%) and demonstrate little or no infection of the CNS despite an increased viral load in the periphery.76 TLR3 recognizes double-stranded RNA and initiates an inflammatory response to cognate ligands. The lack of CNS involvement in WNV-infected, TLR3-deficient mice supports a role for TLR3-mediated inflammation in breakdown of the BBB and entry of WNV into the CNS. A recent study demonstrated that interference with the proinflammatory actions of macrophage migration inhibitory factor (MIF) in WNV-infected mice by antibodies, small molecule inhibitors, or MIF gene deletion results in a significant reduction in viral neuroinvasion, CNS leukocyte infiltration, and BBB breakdown.77 These data indicate that the innate immune response to WNV infection causes increased BBB permeability and facilitates entry of WNV into the CNS.
Herpes simplex virus (HSV) is the most common cause of sporadic viral meningoencephalitis in the United States. Brain edema is a common feature of this viral CNS infection, but the mechanisms underlying the edema are unclear. A recent study examined changes in aquaporin (AQP) expression in the brains of mice with experimental HSV encephalitis.78 AQPs are a family of protein water channels that are critical for water homeostasis in most organ systems, including the CNS. The predominant AQPs expressed in the CNS include AQP-1, which plays a role in CSF formation, and AQP-4, which is expressed heavily in astrocytes, especially at the BBB.79 AQP-4–deficient mice demonstrate less severe cerebral edema in models of bacterial meningitis, thus suggesting that AQP-4 is involved in the modulation of brain edema during CNS infection (see later).80 In mice with experimental HSV encephalitis, AQP-4 mRNA is significantly downregulated in the acute phase of disease (day 7 after inoculation), whereas AQP-1 and AQP-4 mRNA are both upregulated 6 months after inoculation.78 The initial downregulation of AQP-4 may be a protective response to prevent excessive brain edema, and therapies directed at AQP-4 regulation may prove beneficial in a wide variety of CNS infections associated with brain edema.
Bacterial Infections of the Central Nervous System
Increased permeability of the BBB most often occurs after bacteria have crossed this barrier and are already present in the CNS. Accordingly, a reduction in the BBB is not a prerequisite for initiation of a CNS infection but is a common consequence of infection and probably facilitates further invasion of the CNS by the offending organism. Bacterial products and the immunologic responses to bacteria both contribute to an increase in BBB permeability. Perhaps equally as important as the increased permeability of the BBB is the decreased clearance of CSF that occurs during meningitis. Scheld and colleagues monitored cerebrospinal hydrodynamics in rabbits during experimental meningitis by using a pressure device in direct continuity with the supracortical subarachnoid space.81 Rabbits were infected with either S. pneumoniae or E. coli (K1) to produce experimental meningitis, and a subgroup of animals were treated with penicillin G or methylprednisolone to assess the impact of antimicrobial or steroid therapy on CSF hydrodynamics. CSF outflow resistance increased 26-fold, from a baseline of 0.26 ± 0.04 mm Hg/µL per minute to 6.77 ± 3.52 mm Hg/µL per minute, during experimental meningitis and remained elevated up to 15 days later despite penicillin treatment. Administration of methylprednisolone early during pneumococcal meningitis reduced CSF outflow resistance 11.5-fold to 0.59 mm Hg/µL per minute. These observations highlight the extremely impaired drainage of CSF during bacterial meningitis, a process that itself promotes brain edema.
LPS, a common structural element of gram-negative bacterial outer membranes, is recognized by TLR4 on innate immune cells, such as microglia in the CNS, and triggers a proinflammatory response. TLR4 knockout mice demonstrate impaired leukocyte recruitment to the CNS after intracerebral LPS injection.23 Intracisternal injection of LPS from H. influenzae type b (Hib) into rats produces increased BBB permeability, as measured by the accumulation of radiolabeled albumin in CSF, in a dose-dependent fashion (2 to 20 ng), although at higher doses (500 to 1000 ng) there is attenuation of the inflammatory response.82 The LPS-induced increase in BBB permeability does not occur in leukopenic rats, thus supporting the hypothesis that leukocytes are responsible for LPS-induced alterations in BBB permeability.82 However, an in vitro model of the rat BBB in which isolated primary rat cerebral microvascular endothelial monolayers were used demonstrated an increase in radiolabeled albumin flux across monolayers exposed to Hib LPS.83 The absence of leukocytes in this in vitro model indicates that LPS may increase BBB permeability independent of leukocytes through a direct effect on endothelial cells. Murine brain endothelial monolayers exposed to LPS secrete IL-1α, IL-6, IL-10, granulocyte-macrophage colony-stimulating factor, and TNF-α.84 These cytokines are released in a polarized fashion that favors secretion on the luminal side of the endothelial monolayer, and IL-6 production is further enhanced when the monolayer is exposed to LPS from the abluminal surface (akin to parenchymal exposure).84 LPS also triggers the expression of E-selectin and P-selectin on BMECs, as well as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule (ICAM).85 The selectins are critical to the initiation of leukocyte margination and rolling, which allows these cells to slow down and interact with adhesion molecules that facilitate leukocyte transmigration across the BBB, such as VCAM-1, ICAM, and integrins. When rats are injected intracisternally with encapsulated or unencapsulated isogenic strains of Hib, an increase in BBB permeability is noted in both groups, but differences are noted in clearance of Hib from CSF.86 Rats injected with unencapsulated bacteria are able to clear these bacteria from CSF more rapidly than are animals injected with encapsulated bacteria, a finding anticipated because of the known antiphagocytic properties of the capsule. Interestingly, when encapsulated or unencapsulated Hib is injected into leukopenic rats, an increase in BBB permeability is still observed despite reduced CSF pleocytosis in comparison to normal rats, and the degree of BBB permeability correlates with the CSF bacterial load, not the degree of CSF pleocytosis.86 These data suggest that leukocytes are not an absolute requirement for the increase in BBB permeability noted during Hib meningitis; additional host or bacterial factors are involved in reducing the barrier function of the BBB during Hib meningitis. Consistent with this hypothesis is the finding that Hib peptidoglycan (PGN) also induces meningeal inflammation and an increase in BBB permeability when injected intracisternally into rats, probably via TLR2 signaling and the associated inflammatory response.87
N. meningitidis is a gram-negative coccus responsible for epidemic outbreaks of bacterial meningitis, most commonly among human populations in developed countries sharing close living conditions, such as college students and military recruits. A murine brain endothelial cell line (MB114En) exposed to meningococcal whole cell lysates rapidly produces a large amount of NO via expression of iNOS (or NOS-2), a series of events that actually results in the death of endothelial cells and is prevented by an inhibitor of NOS-2 (L-NNA) or an inhibitor of poly(ADP-ribose) polymerase (3-aminobenzamide).88 This process is mediated by TLR2 and TLR4 and activation of the MAPK p38/NF-κB pathway.89
Although these studies have identified elements of gram-negative bacteria as modulators of BBB permeability, alterations in BBB permeability have also been shown with components of gram-positive bacteria. An in vitro model of the BBB was developed by using bovine BMECs cocultured with rat primary glial cells and subsequently exposed to the gram-positive cell wall components LTA and muramyl dipeptide (MDP, a subunit of PGN).90 Exposure of glial cells to purified LTA (10 µg/mL) from Staphylococcus aureus or S. pneumoniae led to an increase in barrier permeability to fluorescein isothiocyanate (FITC)-inulin after 72 hours as opposed to complete loss of the BBB after 24 hours of exposure to LPS (10 µg/mL).90 The increase in BBB permeability noted with LTA was augmented by the addition of MDP to LTA.90 The changes in BBB permeability associated with glial activation by LTA with or without MDP were associated with glial production of TNF-α, IL-1β, and NO. In this model, an increase in BBB permeability to FITC-albumin and a decrease in transendothelial electrical resistance could be produced by exposure of endothelial monolayers to TNF-α or IL-1β and blocked by the addition of antibodies against TNF-α and IL-1β to LTA-activated glial cells. Addition of an iNOS inhibitor to activated glial cells also partly reversed the reduction in transendothelial electrical resistance associated with LTA.90 These data indicate that glial cells contribute significantly to the increase in BBB permeability associated with LTA by the production of proinflammatory cytokines and NO.
The role of NO in BBB permeability has been examined further in vivo in a rat model of Hib meningitis. The CSF nitrite level, an indirect measure of NO production, increases significantly during the course of experimental Hib meningitis from basal levels to peak levels over the first 18 hours after intracisternal inoculation with live bacteria, but with no significant change in serum nitrite levels.91 The CSF nitrite level correlates well with increased BBB permeability, as measured by accumulation of radiolabeled albumin in the CSF from serum (r = .84, P = .018), and administration of a systemic NOS inhibitor, N-nitro-L-arginine methyl ester, reduces CSF nitrite levels and the degree of CSF pleocytosis associated with intracisternal Hib lipooligosaccharide.91
NO can also interact with other mediators to produce alterations in BBB permeability. Intrathecal administration of LPS to induce experimental meningitis in rats leads to a rapid increase in NO and prostaglandin E2 (PGE2) levels in the CSF that peaks about 6 to 8 hours after LPS administration.92 These increases in NO and PGE2 were temporally associated with an increase in BBB permeability, as measured by 14C-sucrose accumulation in brain parenchyma, thus supporting a role for these mediators in altering the BBB during meningitis.92
NO, in addition to myriad physiologic functions, can also react chemically with the ROS species ·O2− (superoxide) to form peroxynitrite (·NO + ·O2− → OONO−), an extremely reactive oxidant capable of damaging a wide variety of biomolecules. Peroxynitrite produces an increase in BBB permeability in response to experimental rodent viral CNS infections (Borna disease virus, rabies virus) via a TNF-α–independent mechanism, thus providing an additional mechanism for NO synthesis to modulate the BBB and immune response in the CNS.93,94
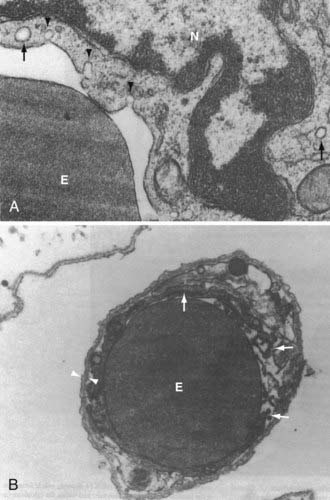
Alterations in BBB permeability as measured by leakage of macromolecules into the CSF during in vivo experimental meningitis correspond to morphologic changes in the cellular elements of the BBB. Experimental infection of rats with the encapsulated bacteria commonly responsible for human meningitis, S. pneumoniae, E. coli, or Hib, induces early changes in brain microvascular endothelium as examined by transmission electron microscopy.95 An early (4 hours after infection) and sustained increase in pinocytotic vesicles (10 and 18 hours after infection) was noted with all these bacterial species (Fig. 39-4A).95 The duration of infection is also associated with an increased observation of separation of intercellular junctions between BMECs (Fig. 39-4B).95
Functional changes in BBB permeability have micromolecular (changes in junctional protein levels, activation states, altered intracellular signaling pathways, and so on) and macromolecular (toxic injury to endothelial cells, endothelial apoptosis, altered endothelial migration, and so on) mechanisms that can and have been studied extensively. For example, an in vitro model of the human BBB using HBMECs examined changes in intercellular junctional regulation after exposure to E. coli K1.96 HBMEC monolayers exposed to E. coli K1 exhibit decreased transendothelial electrical resistance before traversal of these bacteria across the monolayer, but only when these bacteria are expressing OmpA. OmpA interacts with a 95-kD endothelial glycoprotein expressed only in brain microvascular endothelium and induces actin cytoskeletal rearrangements before bacterial internalization, a process dependent on protein kinase C-α (PKC-α).96 Activated PKC-α phosphorylates a specific protein at the adherens junction, vascular endothelial cadherin, which leads to dissociation of β-catenin from the adherens junction, decreased adherens junction intercellular adhesion, and increased paracellular permeability at the BBB.96 This entire process of PKC-α activation, β-catenin dissociation, and increased BBB permeability can be prevented by antibodies directed against E. coli K1 OmpA or its glycoprotein receptor.96
In addition to functional changes in brain endothelium, bacteria often produce toxins capable of directly damaging endothelial cells and thereby destroying the BBB via cellular loss. S. pneumoniae, the most common bacterial cause of meningitis, produces a pore-forming toxin called pneumolysin. Pneumolysin is a member of the family of cholesterol-dependent cytolysins (CDCs), a group of gram-positive bacterial toxins that share a carboxy-terminal undecapeptide sequence that binds to cholesterol in the membranes of eukaryotic cells and leads to oligomerization and pore formation.97 The CDC family includes proteins produced by many gram-positive human pathogens; some examples are streptolysin O (Streptococcus pyogenes), perfringolysin O (Clostridium perfringens), listeriolysin O (L. monocytogenes), and anthrolysin O (Bacillus anthracis). Pneumolysin lacks an amino-terminal signal sequence, which suggests that it is not secreted from pneumococci and requires autolysis for release.98,99 Pneumolysin is highly cytotoxic to HBMECs, and pneumococci unable to produce pneumolysin do not alter the permeability of HBMEC monolayers in vitro.100 Pneumolysin also plays a major role in neuronal damage during pneumococcal meningitis, as discussed in more detail later with respect to neurotoxicity.
Hemolytic-uremic syndrome, the leading cause of acute renal failure in children, is highly associated with infection by Shiga toxin (Stx1 or Stx2)-producing E. coli strains, and severe manifestations of this disease are complicated by CNS malfunction.101 Stx1 and Stx2 are AB-type protein toxins composed of five B subunits and one A subunit.102 The B subunits are responsible for binding to a cell surface glycolipid, globotriaosylceramide (Gb3), which results in endocytosis of Stx1/2 and retrograde transport of toxin to the cytosol of eukaryotic cells expressing Gb3.102 Once in the cytosol, Stx1/2 inactivates ribosomes by removal of the adenine at position 4342 of the 28S RNA of the 60S ribosomal subunit, a reaction that inactivates protein synthesis.102 Stx1 induces apoptosis of HBMECs, especially in the presence of TNF-α, and Stx2 induces apoptosis in HBMECs via upregulation of C/EBP homologue protein/growth arrest (CHOP) and caspase-3 activation.103,104
Neurotoxicity
Human Immunodeficiency Virus–Associated Neurotoxicity
Many investigations into the neurotoxic effects of HIV infection have been performed since the clinical recognition that patients with HIV/acquired immunodeficiency syndrome manifest a range of neurological maladies from mild cognitive impairment to full-blown dementia. Early investigations focused on target cells in the CNS, given the hypothesis that HIV was able to infect neurons directly. This hypothesis has been demonstrated in multiple studies to be incorrect; HIV does not infect human neurons directly, but invasion of the CNS clearly occurs, and this anatomic site can serve as a sequestration site for HIV latency.105 Our understanding of how HIV damages CNS neurons is currently incomplete, but many aspects have been elucidated and reveal a complex interplay among the virus, the immune response, and resident glial cells such as astrocytes and microglia. Once HIV has invaded the CNS, an event that occurs very early after primary infection of the host, infected monocytes, macrophages, microglia, and lymphocytes provide sources of lytic viral reproduction, accompanied by nonproductive infection of astrocytes. Several protein components of the HIV virion have been shown to influence neuronal survival, including Vpr, gp120, and Tat. HIV-1 Vpr, a viral regulatory protein, has been shown to form ion channels in the membranes of rat hippocampal neurons and induces caspase-8 activation with subsequent apoptosis of both undifferentiated and fully differentiated NT2 cells, a neuron-like cell line.106,107 Gp120, the HIV envelope glycoprotein, also induces apoptosis in NT2 cells and has been shown to significantly increase the production of ROS from glial cells in vitro by release of IL-1β, thereby leading to ROS-induced neurotoxicity.107,108 Tat, a regulatory protein that trans-activates genes located in the HIV long terminal repeats, is secreted from HIV-infected cells and taken up by a wide variety of cell types, including astrocytes and neurons.109 Exposure of neurons in vitro to Tat results in neuronal apoptosis via TNF-α and activation of a non–N-methyl-D-aspartate (NMDA) receptor.110 The direct neurotoxicity of Tat localizes to the carboxy-terminal portion of this protein, and a deletion mutant Tat protein lacking amino acids 31 to 61 (TatΔ31-61) does not induce neurotoxicity in cultured human fetal neurons.111 Although TatΔ31-61 does not produce direct neurotoxicity in vitro, this deletion mutant is still capable of generating indirect neurotoxicity via TNF-α production from macrophages, thus indicating that Tat neurotoxicity occurs by direct and indirect mechanisms.111 In contrast, both TNF-α and IL-1β upregulate expression of proteinase-activated receptor-2 (PAR-2) on neurons, and enhanced expression and activation of PAR-2 prevent the neurotoxicity associated with Tat.112 PAR-2 is a member of the family of PARs, or G protein–coupled receptors that require proteolytic cleavage to remove biochemical constraints on a contiguous receptor ligand and allow activation of PAR.113 In addition to activation by trypsin or mast cell tryptase, PAR-2 may also be activated by peptide sequences that mimic the intrinsic receptor ligand. Consistent with the protective role of activated PAR-2 on Tat-induced neurotoxicity, implantation of a PAR-2 peptide agonist in the mouse striatum prevented Tat-associated neurotoxicity in vivo.112
HIV also produces neuronal death via astrocytes and macrophages. Tat expression by astrocytes in vitro results in increased GFAP expression, a marker of astrocyte activation, and impairs glutamate uptake by astrocytes.114 The culture supernates from Tat-expressing astrocytes cause neuronal death.114 Additionally, HIV infection of human monocyte–derived macrophages increases macrophage glutamate synthesis via mitochondrial glutaminase, an enzyme present in macrophages that converts glutamine to glutamate.115 HIV-infected macrophage media induces neuronal apoptosis via glutamate-induced neurotoxicity, a process that is significantly inhibited when macrophages are treated with a mitochondrial glutaminase inhibitor.115
Neurotoxicity and Bacterial Meningitis
Despite advances in antimicrobial therapy for bacterial meningitis, neurological damage with resultant disabilities is a common outcome of bacterial meningitis, particularly when the causative organism is S. pneumoniae.116 The mechanisms underlying the neurotoxicity associated with bacterial meningitis can be shared by a group of invading pathogens or may be unique to a specific pathogen. Some examples of broad neurotoxic mechanisms are discussed, followed by some examples of pathogen-specific mechanisms.
Unmethylated CpG motifs in DNA released from pathogens can activate microglial TLR9 and thereby lead to the production of NO and the synthesis and release of TNF-α.117 Both NO and TNF-α are cytotoxic to neurons, and blockade of these mediators ameliorates the neurotoxicity associated with microglial activation via TLR9 ligation. Thus, this component of the innate immune response can result in neurotoxicity as a result of a wide array of invading pathogens whose DNA becomes available for TLR9 ligation. Similarly, components of the cell wall and outer membrane of bacteria are capable of eliciting an intense inflammatory response bearing all the hallmarks of the meningitis produced by the whole bacteria themselves. LTA activates both microglia and astrocytes via TLR2 ligation. In microglia, TLR2 activation by LTA results in the production and release of proinflammatory cytokines such as TNF-α, IL-6, and IL-1β, as well as upregulation of iNOS and increased production of NO.118,119 Coculture of neurons with glial cells (astrocytes or microglia) exposed to LTA leads to neuron apoptosis, and this outcome is dependent on the presence of glial cells. Furthermore, inhibition of NO or superoxide synthesis or scavenging of peroxynitrite all prevented, partially or completely, the induction of neuron apoptosis after glial LTA exposure.119 Nitrosative and oxidative damage to neurons secondary to activation of the innate immune effector cells in the CNS is a common mechanism involved in the neurotoxicity and neurodegeneration associated with CNS infections. In an infant rat model of GBS meningitis, treatment of the rats with a radical scavenger, α-phenyl-tert-butyl nitrone, at the onset of GBS infection abolished pathologic evidence of reactive oxygen intermediates, improved cerebral cortical blood flow, and prevented neuronal injury in both the cortex and dentate gyrus of the hippocampus.120 Although microglia and infiltrating phagocytic cells (neutrophils, macrophages) develop an oxidative burst on activation, thus providing a host-derived source for the generation of reactive oxygen intermediates, bacteria also contribute to this pool of intermediates via production of hydrogen peroxide (H2O2). The pneumococcus produces H2O2 as a virulence factor, and H2O2 induces apoptosis in neurons by mediating the release of apoptosis-inducing factor (AIF) from the damaged mitochondria.121 AIF is a flavoprotein with nicotinamide adenine dinucleotide oxidase activity, and its release from mitochondria and translocation to the nucleus result in chromatin condensation and DNA fragmentation in a caspase-independent manner.122 AIF also plays a major role in the mechanism of pneumolysin-induced neuronal death and is discussed further later.
We have already mentioned the role of glutamate excitotoxicity in the context of HIV neurodegeneration, and glutamate is also involved in the neurotoxicity induced by bacterial meningitis. A common finding of pneumococcal meningitis in animal models is the loss of neurons in the dentate gyrus of the hippocampus, predominantly via apoptosis.120 To investigate the contribution of glutamate to neuron apoptosis in the dentate gyrus, Tumani and associates examined glutamine synthetase activity in the brains of rabbits with pneumococcal meningitis.123 Significant increases in glutamine synthetase protein concentration and enzyme activity were found in the frontal cortex of infected rabbits when compared with uninfected controls, but no changes in this protein concentration or enzyme activity were noted in the dentate gyri of infected animals. Intravenous administration of a glutamine synthetase inhibitor (L-methionine sulfoximine) to infected rabbits undergoing treatment with ceftriaxone significantly increased the density of apoptotic neurons in the dentate gyri of these animals when compared with rabbits receiving ceftriaxone alone.123 These findings support an association between glutamate metabolism and apoptosis of neurons in the dentate gyrus during pneumococcal meningitis. Kynurenic acid, a by-product of L-tryptophan metabolism and an antagonist of excitatory neurotransmitters, inhibits necrosis and apoptosis of neurons in the cortex and hippocampus of rats with experimental GBS meningitis.124 In contrast to these data, blockade of the NMDA receptor subunit NR2B, the subunit most highly expressed in the developing hippocampus, with a selective and potent antagonist (RO 25-6981) in infant rats with pneumococcal meningitis did not alter hippocampal neuron apoptosis, although this intervention did significantly reduce the frequency of seizures.125
Pneumolysin, a CDC released from S. pneumoniae after cell lysis, provides an excellent example of a pathogen-specific factor involved in neurotoxicity. Live pneumococci induce neuronal death, a process associated with influx of Ca2+ into neurons, mitochondrial damage, and translocation of mitochondrial AIF to the cell nucleus.121 As mentioned previously, pneumococcal production of H2O2 accounts for some of these observations, but pneumococci lacking the ability to produce H2O2 are still able to kill primary neurons, whereas pneumococci lacking both H2O2 production and pneumolysin do not significantly affect neuronal survival.121 Pneumolysin colocalizes with apoptotic neurons in the dentate gyrus of rats with pneumococcal meningitis, and pneumolysin is sufficient to induce neuronal death in vitro.121 Pneumolysin has now been shown to act as a mitochondrial toxin that causes alterations in mitochondrial membrane potential and the release of AIF, thereby leading to neuronal death in a caspase-independent manner.126 Pneumococci deficient in pneumolysin (Δply) or autolysin (LytA−), the protein effector of pneumococcal autolysis, demonstrate significantly reduced virulence in a rat model of experimental meningitis.99 The contribution of autolysis to the pathogenesis of pneumococcal meningitis is relevant in the context of antibiotic-induced lysis of pneumococci in the CSF during meningitis. Grandgirard and coworkers examined differences in the inflammation associated with experimental pneumococcal meningitis treated with a lytic bactericidal antibiotic, ceftriaxone, and a nonlytic bactericidal antibiotic, daptomycin.127 Inflammation was quantified in this study by measuring MMP-9 and TNF-α concentrations in CSF, as well as by examining cortical damage and assessing CSF bacterial counts. Infected rabbits treated with daptomycin demonstrated significantly reduced MMP-9 concentrations in comparison to those treated with ceftriaxone, as well as reduced cortical damage. These observations support the concept that release of bacterial products, through either autolysis or antibiotic lysis, contributes directly to the inflammatory damage associated with bacterial meningitis.
Brain Abscess—Pus in the Parenchyma
Brain abscesses are space-occupying purulent infections within the substance of the brain. The cause of brain abscess is most often related to infection outside the CNS and can be subdivided according to the following associations: (1) related to infection of the paranasal sinuses or otologic structures, (2) related to odontogenic infection, (3) related to thoracopulmonary infections (e.g., lung abscess, empyema), (4) hematogenous spread from other sites (e.g., endocarditis), (5) direct inoculation (e.g., trauma, neurosurgical procedures), and (6) cryptogenic (≈20%). The microbiology of brain abscess is predictably related to the primary source of the abscess; Table 39-3 lists microbes associated with specific primary sources. There is a predominance of streptococcal species and anaerobes associated with primary sources in the sinuses, mouth, and lung, whereas staphylococci and Enterobacteriaceae are commonly found in brain abscesses associated with direct inoculation from trauma or neurosurgical procedures.
TABLE 39-3 Specific Pathogens Associated with Anatomic Sources for Brain Abscesses
| PREDISPOSING CONDITION | COMMON PATHOGENS |
|---|---|
| Otitis media or mastoiditis | Streptococci, Bacteroides spp., Prevotella spp., Enterobacteriaceae |
| Sinusitis | Streptococci, Haemophilus spp., Enterobacteriaceae, Bacteroides spp., Staphylococcus aureus |
| Dental infection | Fusobacterium spp., Prevotella spp., Bacteroides spp. streptococci |
| Direct inoculation (trauma, surgery) | Staphylococcus spp., Enterobacteriaceae, streptococci, Clostridium spp. |
| Lung infections (abscess, empyema), bronchiectasis | Actinomyces spp., streptococci, Fusobacterium spp., Bacteroides spp., Prevotella spp., Nocardia spp. |
| Endocarditis | Staphylococcus aureus, streptococci |
| Congenital heart disease | Streptococci, Haemophilus spp. |
| Unknown | Streptococci, Staphylococcus spp., Haemophilus spp., fastidious anaerobes |
Adapted from Tunkel AR. Brain abscess. In: Mandell GL, Bennet JE, Dolin R, eds. Principles and Practice of Infectious Diseases, 6th ed. Philadelphia: Elsevier; 2005:1150-1163.
The time course of brain abscess development in humans has been characterized by computed tomography and in animal models via necropsy and histology. Brain abscess begins as an early cerebritis (days 1 to 3), with edema formation, tissue necrosis, and neutrophil infiltration. This early phase is followed by an intermediate to late cerebritis with infiltration of macrophages and lymphocytes, and this process culminates in the formation of a capsule infiltrated with plasma cells and myofibroblasts.128,129 Encapsulation allows isolation of the infection and is thought to be responsible for the lack of significant clinical symptoms in patients with chronic brain abscess.
Experimental study of the pathogenesis of brain abscess was significantly advanced with the development of a rat model of brain abscess.130 Early studies involved stereotactic injection of a mixture of bacteria, including aerobes and anaerobes, to simulate the known microbiology of brain abscesses in humans.130–132 An important study by Costello and associates demonstrated that injecting microaerophilic or obligate anaerobic bacteria alone did not produce brain abscess, whereas the injection of facultative anaerobic organisms such as E. coli, S. aureus, or Streptococcus pyogenes produced abscesses, albeit with differences in potency (Table 39-4).132 The E. coli strains in this study were more virulent with respect to abscess formation than were the gram-positive organisms tested, and the K1-encapsulated strain was more virulent than the nonencapsulated E. coli strain. These important experiments demonstrate that microaerophilic and obligate anaerobes are probably not involved in the initiation of brain abscess despite the common isolation of these organisms from abscesses derived from different primary processes. The authors point out that this experimental model may not adequately reflect the initiation of brain abscesses in humans inasmuch as many of them are associated with mixed facultative aerobe/obligate anaerobe infections of the paranasal sinuses or dental structures, thereby leading to chronic exposure of the brain to mixed bacteria. Mixed facultative aerobe/obligate anaerobe infections are known to be synergistic in establishing infections at other sites in the body, and the clinical picture in human brain abscess probably reflects this synergistic advantage in establishing a brain abscess in normal human brain tissue.
TABLE 39-4 Relative Potency of Different Bacteria Producing Experimental Brain Abscesses after Intraparenchymal Injection
| BACTERIUM | ID50 (LOG10 COLONY-FORMING UNITS/mL) (95% CONFIDENCE INTERVAL) |
|---|---|
| Escherichia coli (K1 antigen positive) | 2.44 (1.03-2.77) |
| E. coli (K1 negative) | 3.64 (3.32-4.01) |
| Pseudomonas aeruginosa | 4.92 (4.63-5.35) |
| Staphylococcus aureus | 5.15 (4.45-5.67) |
| Streptococcus pyogenes | 5.88 (2.34-6.68) |
| Candida albicans | 6.24 (4.58-6.48) |
| Bacteroides fragilis | 7.66 (7.31-8.95) |
Data from Costello GT, Heppe R, Winn HR, et al. Susceptibility of brain to aerobic, anaerobic, and fungal organisms. Infect Immun. 1983;41:535-539.
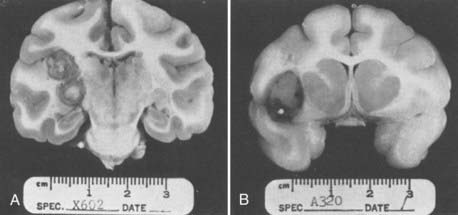
Before development of the rat brain abscess model, rhesus macaques (Macaca mulatta) were used to investigate the development and characteristics of brain abscesses in primates.131 S. epidermidis was injected either intracerebrally (n = five macaques) or intra-arterially via the carotid artery (on silicone cylinders, n = eight macaques); brain abscesses developed in five of five macaques injected intracerebrally as opposed to six of eight macaques injected with contaminated cylinders intra-arterially. Interestingly, abscesses in the animals injected intracerebrally developed thick capsules with an exuberant inflammatory response, beneficial prognostic features, whereas abscesses caused by “septic emboli” developed thin capsules, features historically associated with poorer clinical outcomes. Figure 39-5 displays the coronal sections of abscesses from intracerebral injection and septic embolization.131 Only recently have experiments begun to address the immunopathogenesis of brain abscesses, although in rodent models such as mice or rats as opposed to primates.
Kielian and Hickey published the first study that examined the host cytokine response to rat brain abscesses induced by direct inoculation of S. aureus.133 Using RNase protection assays and reverse transcriptase PCR, they found early induction of mRNA for IL-1α, IL-1β, IL-6, and TNF-α. Within 24 hours of S. aureus injection there was an increase in mRNA for KC, the murine homologue of IL-8, as well as increases in MCP-1 and MIP-1α. The increase in KC correlated histopathologically with the appearance of neutrophils at the injection site, and the increases in MCP-1 and MIP-1α heralded the influx of macrophages and lymphocytes. Additionally, the authors noted an increase in the expression of ICAM-1 and platelet endothelial adhesion molecule in microvessels at 24 and 48 hours after injection. These cytokine/chemokine/adhesion molecule changes agree well with the progression of brain abscess from an early, neutrophil-predominant cerebritis to an organizing lesion infiltrated with macrophages and lymphocytes.
The importance of chemokines and neutrophils in the early inflammatory response to brain abscess was highlighted in a study using an S. aureus mouse brain abscess model.134 Depletion of neutrophils from mice before the implantation of S. aureus–laden beads in the cerebral cortex led to higher bacterial counts and more severe abscesses than seen in control infected mice, thus supporting a role for neutrophils in the rapid containment of bacteria in brain parenchyma. Several chemokines were upregulated within 6 hours of S. aureus injection, including CCL1 to CCL4, CXCL1, and CXCL2; CXC1 and CXC2 are major neutrophil chemoattractants and share a common receptor, CXCR2. When CXCR2 knockout mice were injected with S. aureus–laden beads, the animals demonstrated poor neutrophil extravasation at the site of abscess formation, as well as an increased bacterial burden in these abscesses. As in the rat brain abscess model, IL-1 and TNF-α have been shown to play an important “containment” role in regulating the inflammatory response to S. aureus in the murine brain abscess model.135,136 TNF-α knockout mice injected intracerebrally with S. aureus have a higher mortality rate (54% versus 0%), higher bacterial loads, and higher inflammatory infiltrates, as well as delayed bacterial clearance rates, when compared with infected isogenic control mice.136
The expression of proinflammatory cytokines and chemokines is commonly triggered by engagement of TLRs, and this has been demonstrated in the murine brain abscess model. TLR2 and TLR4 are both important for the innate immune response to S. aureus brain abscess, as shown by the significantly delayed clearance of bacteria and increased mortality in TLR2 or TLR4 knockout mice injected intracerebrally with this bacterium.137 Interestingly, the TLR2 and TLR4 receptors share a common intracellular adaptor molecule, MyD88, and MyD88 knockout mice are also subject to more severe brain abscesses, with significantly increased tissue necrosis, than infected isogenic control mice are.138 However, unlike TLR2 or TLR4 knockout mice, MyD88 knockout mice do not have significantly increased bacterial loads, thus suggesting that MyD88 plays an important role in the containment of brain abscesses but not in clearance of bacteria from the initial cerebritis.
In addition to recruitment and activation of immune cells from outside the CNS, injection of S. aureus into cerebral cortex leads to the activation of astrocytes and microglial cells.130,133,139 The involvement of astrocytes in the pathogenesis of brain abscess is highlighted by the observation that mice lacking GFAP, an intermediary filament upregulated in activated astrocytes, demonstrate poor containment of primary S. aureus cerebritis and exhibit severe inflammation, increased bacterial loads, ventriculitis, vasculitis, and contralateral cerebral leukocyte infiltration.140 A major downregulator of microglia and astrocyte activation is the peroxisome proliferator–activated receptor-γ (PPAR-γ), and agonists of this nuclear regulator may modulate the inflammatory evolution of S. aureus brain abscesses in the murine model.141 In vitro studies of PPAR-γ agonists (15-deoxy-Δ12,14-prostaglandin J2, ciglitazone) on primary microglia or astrocytes do indeed demonstrate downregulation of proinflammatory cytokines, chemokines, and membrane inflammatory markers in these CNS cell types, although these effects in astrocytes were independent of PPAR-γ.142,143 Administration of ciglitazone to mice 3 days after injection of S. aureus intracerebrally was associated with reduced bacterial loads secondary to enhanced microglial phagocytosis; reduced expression of TNF-α, IL-1β, CXCL2, CCL3, and iNOS; and enhanced abscess encapsulation.143 These remarkable effects support the concept that effective control of brain abscesses requires a balance between proinflammatory and anti-inflammatory processes; factors favoring inflammation lead to excessive tissue damage, whereas impairment of specific inflammatory processes (e.g., CXCR2 knockout) leads to uncontrolled infection.
More evidence to support this “balance” hypothesis comes from observations on the effects of minocycline on experimental murine brain abscess. Minocycline has been shown to exert a number of neuroprotective effects, including modulation of microglial activation, inhibition of CNS cell apoptosis, and inhibition of proinflammatory signal transduction cascades.144 Kielian and colleagues injected mice intracerebrally with a minocycline-resistant strain of S. aureus to induce brain abscess and then administered minocycline to these mice at various time points.145 Minocycline administration, even as long as 3 days after injection, was associated with a reduction in brain abscess size, reduced mortality in the first 24 hours, transient reductions in IL-1β and CCL2 expression, and reduced astrocyte and microglial activation. Thus, the clinical effects of drugs such as ciglitazone and minocycline on human brain abscesses deserve further investigation as adjuncts to surgical and chemotherapeutic interventions.
Cerebrospinal Fluid Shunt Infections—The Role of Biofilms
Coagulase-negative staphylococci (CoNS) are common pathogens involved in the colonization and infection of CSF shunt devices, and these bacteria are notorious for their ability to form biofilm on prosthetic devices (Fig. 39-6). Several factors probably play a role in the initial adherence of CoNS to CSF shunt devices, including the surface materials of the shunt that interact with host tissues or fluids and bacterial virulence factors such as adhesins. Soon after placement, bioprosthetic materials are coated with host proteins, and these proteins may serve as receptors for adhesion molecules expressed by bacteria. These adhesion molecules are collectively known as “microbial surface components recognizing adhesive matrix molecules,” or MSCRAMMs.146 CoNS and S. aureus appear to adhere more readily to bioprosthetic material than do S. pneumoniae and E. coli, and the staphylococci adhere better to Teflon than to silicone.147 Live bacteria may not be necessary to initiate bacterial adherence to CSF shunt material; “dead” biofilm on sterile, unused silicone CSF shunt devices has been shown to facilitate the adherence of bacteria, with subsequent new biofilm formation.148 Once adherence has occurred, CoNS initiate the process of biofilm formation almost immediately. Biofilms are complex surface structures consisting of sessile bacteria embedded in an extracellular matrix that is permeated by water channels.149 Regulation of the transition state between planktonic (free-living) bacteria and sessile bacteria in biofilms is very often dependent on a phenomenon known as quorum sensing. Quorum sensing itself is a complex set of processes that allow bacteria in a community to regulate the gene expression and phenotypes of other bacteria in the same community. Biofilm formation by S. epidermidis is a process associated with phase variation and regulation of a DNA sequence known as the intercellular adhesion (ica) gene cluster.146 The ica gene cluster is organized as a four-gene operon (icaADBC) and encodes for the machinery necessary to synthesize a polysaccharide intercellular adhesin (PIA) that forms the extracellular matrix of S. epidermidis biofilms. PIA is composed of polymeric N-acetylglucosamine and accounts for the bulk of the extracellular matrix in CoNS biofilms.146 The icaADBC operon is under the transcriptional control of several systems in staphylococci, including repressors such as IcaR, TcaR, and the LuxS system, a quorum-sensing apparatus active in CoNS. Additionally, σB, a global stress regulator, acts as a positive regulator of biofilm formation in S. epidermidis, but not in S. aureus.146 More recent studies have also identified PIA-independent mechanisms of biofilm formation in staphylococci; an excellent and extensive review of staphylococcal biofilm regulation can be found in the review by O’Gara.146 Future advances in bioprosthetic device design, such as adhesion-resistant materials, antibiotic-impregnated devices, and devices designed to interfere with quorum sensing or transcriptional regulation of biofilm formation, may significantly reduce the infection rates associated with implanted bioprosthetic devices. Additionally, new chemotherapeutic interventions are needed; examples include bactericidal antimicrobials that penetrate biofilm and have a high threshold for the development of resistance, compounds that interfere with quorum sensing or transcriptional regulation of biofilm formation, or with both, and compounds that regulate phase variation among bacteria and drive sessile, biofilmed bacteria into the more susceptible planktonic form.
Albrecht J, Sonnewald U, Waagepetersen HS, et al. Glutamine in the central nervous system: function and dysfunction. Front Biosci. 2007;12:332-343.
Aravalli RN, Peterson PK, Lokensgard JR. Toll-like receptors in defense and damage of the central nervous system. J Neuroimmune Pharmacol. 2007;2:297-312.
Britt RH, Enzmann DR, Yeager AS. Neuropathological and computerized tomographic findings in experimental brain abscess. J Neurosurg. 1981;55:590-603.
Costello GT, Heppe R, Winn HR, et al. Susceptibility of brain to aerobic, anaerobic, and fungal organisms. Infect Immun. 1983;41:535-539.
Dorf ME, Berman MA, Tanabe S, et al. Astrocytes express functional chemokine receptors. J Neuroimmunol. 2000;111:109-121.
Fux CA, Quigley M, Worel AM, et al. Biofilm-related infections of cerebrospinal fluid shunts. Clin Microbiol Infect. 2006;12:331-337.
Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140-155.
Jack CS, Arbour N, Manusow J, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320-4330.
Kielian T, Hickey WF. Proinflammatory cytokine, chemokine, and cellular adhesion molecule expression during the acute phase of experimental brain abscess development. Am J Pathol. 2000;157:647-658.
Kim KS. Microbial translocation of the blood-brain barrier. Int J Parasitol. 2006;36:607-614.
Krantic S, Mechawar N, Reix S, et al. Apoptosis-inducing factor: a matter of neuron life and death. Prog Neurobiol. 2007;81:179-196.
Lehmann GL, Gradilone SA, Marinelli RA. Aquaporin water channels in central nervous system. Curr Neurovasc Res. 2004;1:293-303.
Meli DN, Christen S, Leib SL, et al. Current concepts in the pathogenesis of meningitis caused by Streptococcus pneumoniae. Curr Opin Infect Dis. 2002;15:253-257.
O’Gara JP. ica and beyond: biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol Lett. 2007;270:179-188.
Rock RB, Peterson PK. Microglia as a pharmacological target in infectious and inflammatory diseases of the brain. J Neuroimmune Pharmacol. 2006;1:117-126.
Rosado CJ, Kondos S, Bull TE, et al. The MACPF/CDC family of pore-forming toxins. Cell Microbiol. 2008;10:1765-1774.
Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annu Rev Neurosci. 1999;22:11-28.
Tunkel AR, Scheld WM. Pathogenesis and pathophysiology of bacterial meningitis. Clin Microbiol Rev. 1993;6:118-136.
Wood JH, Lightfoote WE, Ommaya AK. Cerebral abscesses produced by bacterial implantation and septic embolisation in primates. J Neurol Neurosurg Psychiatry. 1979;42:63-69.
Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6:931-944.
1 Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annu Rev Neurosci. 1999;22:11-28.
2 Kim BY, Kang J, Kim KS. Invasion processes of pathogenic Escherichia coli. Int J Med Microbiol. 2005;295:463-470.
3 Khan NA, Kim Y, Shin S, et al. FimH-mediated Escherichia coli K1 invasion of human brain microvascular endothelial cells. Cell Microbiol. 2007;9:169-178.
4 Wang Y, Kim KS. Role of OmpA and IbeB in Escherichia coli K1 invasion of brain microvascular endothelial cells in vitro and in vivo. Pediatr Res. 2002;51:559-563.
5 Prasadarao NV, Wass CA, Kim KS. Endothelial cell GlcNAc beta 1-4GlcNAc epitopes for outer membrane protein A enhance traversal of Escherichia coli across the blood-brain barrier. Infect Immun. 1996;64:154-160.
6 Prasadarao NV, Wass CA, Stins MF, et al. Outer membrane protein A–promoted actin condensation of brain microvascular endothelial cells is required for Escherichia coli invasion. Infect Immun. 1999;67:5775-5783.
7 Kim KS. Microbial translocation of the blood-brain barrier. Int J Parasitol. 2006;36:607-614.
8 Reddy MA, Wass CA, Kim KS, et al. Involvement of focal adhesion kinase in Escherichia coli invasion of human brain microvascular endothelial cells. Infect Immun. 2000;68:6423-6430.
9 Reddy MA, Prasadarao NV, Wass CA, et al. Phosphatidylinositol 3-kinase activation and interaction with focal adhesion kinase in Escherichia coli K1 invasion of human brain microvascular endothelial cells. J Biol Chem. 2000;275:36769-36774.
10 Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690-701.
11 Khan NA, Wang Y, Kim KJ, et al. Cytotoxic necrotizing factor-1 contributes to Escherichia coli K1 invasion of the central nervous system. J Biol Chem. 2002;277:15607-15612.
12 Hoffman JA, Wass C, Stins MF, et al. The capsule supports survival but not traversal of Escherichia coli K1 across the blood-brain barrier. Infect Immun. 1999;67:3566-3570.
13 Doran KS, Nizet V. Molecular pathogenesis of neonatal group B streptococcal infection: no longer in its infancy. Mol Microbiol. 2004;54:23-31.
14 Chaffin DO, Mentele LM, Rubens CE. Sialylation of group B streptococcal capsular polysaccharide is mediated by cpsK and is required for optimal capsule polymerization and expression. J Bacteriol. 2005;187:4615-4626.
15 Maisey HC, Hensler M, Nizet V, et al. Group B streptococcal pilus proteins contribute to adherence to and invasion of brain microvascular endothelial cells. J Bacteriol. 2007;189:1464-1467.
16 Doran KS, Engelson EJ, Khosravi A, et al. Blood-brain barrier invasion by group B Streptococcus depends upon proper cell-surface anchoring of lipoteichoic acid. J Clin Invest. 2005;115:2499-2507.
17 Doran KS, Liu GY, Nizet V. Group B streptococcal beta-hemolysin/cytolysin activates neutrophil signaling pathways in brain endothelium and contributes to development of meningitis. J Clin Invest. 2003;112:736-744.
18 Shin S, Paul-Satyaseela M, Lee JS, et al. Focal adhesion kinase is involved in type III group B streptococcal invasion of human brain microvascular endothelial cells. Microb Pathog. 2006;41:168-173.
19 Shin S, Kim KS. RhoA and Rac1 contribute to type III group B streptococcal invasion of human brain microvascular endothelial cells. Biochem Biophys Res Commun. 2006;345:538-542.
20 Barron KD. The microglial cell. A historical review. J Neurol Sci. 1995;134:57-68.
21 Raivich G, Banati R. Brain microglia and blood-derived macrophages: molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Res Brain Res Rev. 2004;46:261-281.
22 Aravalli RN, Peterson PK, Lokensgard JR. Toll-like receptors in defense and damage of the central nervous system. J Neuroimmune Pharmacol. 2007;2:297-312.
23 Zhou H, Lapointe BM, Clark SR, et al. A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharide. J Immunol. 2006;177:8103-8110.
24 Bernardino ALF, Myers TA, Alvarez X, et al. Toll-like receptors: insights into their possible role in the pathogenesis of Lyme neuroborreliosis. Infect Immun. 2008;76:4385-4395.
25 Suh HS, Zhao ML, Rivieccio M, et al. Astrocyte indoleamine 2,3-dioxygenase is induced by the TLR3 ligand poly(I:C): mechanism of induction and role in antiviral response. J Virol. 2007;81:9838-9850.
26 Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916-3924.
27 Li J, Baud O, Vartanian T, et al. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci U S A. 2005;102:9936-9941.
28 Wilkinson B, Landreth G. The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer’s disease. J Neuroinflammation. 2006;3:30.
29 Napoli I, Neumann H. Microglial clearance function in health and disease. Neuroscience. 2009;158:1030-1038.
30 Piccio L, Buonsanti C, Mariani M, et al. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. 2007;37:1290-1301.
31 Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140-155.
32 Shun-Fen T, Han-Yun H, Oi-Tong M. Prostaglandins and cyclooxygenases in glial cells during brain inflammation. Curr Drug Targets Inflamm Allergy. 2005;4:335-340.
33 Jaranowska A, Bussolino F, Sogos V, et al. Platelet-activating factor production by human fetal microglia. Effect of lipopolysaccharides and tumor necrosis factor-alpha. Mol Chem Neuropathol. 1995;24:95-106.
34 Caggiano AO, Kraig RP. Prostaglandin E receptor subtypes in cultured rat microglia and their role in reducing lipopolysaccharide-induced interleukin-1beta production. J Neurochem. 1999;72:565-575.
35 Rock RB, Peterson PK. Microglia as a pharmacological target in infectious and inflammatory diseases of the brain. J Neuroimmune Pharmacol. 2006;1:117-126.
36 Garden GA, Moller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1:127-137.
37 Cheeran MC, Hu S, Sheng WS, et al. CXCL10 production from cytomegalovirus-stimulated microglia is regulated by both human and viral interleukin-10. J Virol. 2003;77:4502-4515.
38 Hatalski CG, Hickey WF, Lipkin WI. Evolution of the immune response in the central nervous system following infection with Borna disease virus. J Neuroimmunol. 1998;90:137-142.
39 Chan A, Seguin R, Magnus T, et al. Phagocytosis of apoptotic inflammatory cells by microglia and its therapeutic implications: termination of CNS autoimmune inflammation and modulation by interferon-beta. Glia. 2003;43:231-242.
40 Yang MS, Park EJ, Sohn S, et al. Interleukin-13 and -4 induce death of activated microglia. Glia. 2002;38:273-280.
41 Jack CS, Arbour N, Manusow J, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320-4330.
42 Van Wagoner NJ, Benveniste EN. Interleukin-6 expression and regulation in astrocytes. J Neuroimmunol. 1999;100:124-139.
43 Becher B, D’Souza SD, Troutt AB, et al. Fas expression on human fetal astrocytes without susceptibility to fas-mediated cytotoxicity. Neuroscience. 1998;84:627-634.
44 Wosik K, Becher B, Ezman A, et al. Caspase 8 expression and signaling in Fas injury–resistant human fetal astrocytes. Glia. 2001;33:217-224.
45 Bechmann I, Steiner B, Gimsa U, et al. Astrocyte-induced T cell elimination is CD95 ligand dependent. J Neuroimmunol. 2002;132:60-65.
46 Dorf ME, Berman MA, Tanabe S, et al. Astrocytes express functional chemokine receptors. J Neuroimmunol. 2000;111:109-121.
47 Sabri F, Titanji K, De Milito A, et al. Astrocyte activation and apoptosis: their roles in the neuropathology of HIV infection. Brain Pathol. 2003;13:84-94.
48 Albrecht J, Sonnewald U, Waagepetersen HS, et al. Glutamine in the central nervous system: function and dysfunction. Front Biosci. 2007;12:332-343.
49 Visalli V, Muscoli C, Sacco I, et al. N-acetylcysteine prevents HIV gp 120–related damage of human cultured astrocytes: correlation with glutamine synthase dysfunction. BMC Neurosci. 2007;8:106.
50 Espey MG, Basile AS, Heaton RK, et al. Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology. 2002;58:1439-1440.
51 Huang TL, O’Banion MK. Interleukin-1 beta and tumor necrosis factor-alpha suppress dexamethasone induction of glutamine synthetase in primary mouse astrocytes. J Neurochem. 1998;71:1436-1442.
52 Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6:931-944.
53 Paul R, Lorenzl S, Koedel U, et al. Matrix metalloproteinases contribute to the blood-brain barrier disruption during bacterial meningitis. Ann Neurol. 1998;44:592-600.
54 Van den Steen PE, Proost P, Wuyts A, et al. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673-2681.
55 Ito A, Mukaiyama A, Itoh Y, et al. Degradation of Interleukin 1beta by matrix metalloproteinases. J Biol Chem. 1996;271:14657-14660.
56 Szklarczyk A, Stins M, Milward EA, et al. Glial activation and matrix metalloproteinase release in cerebral malaria. J Neurovirol. 2007;13:2-10.
57 Leppert D, Lindberg RL, Kappos L, et al. Matrix metalloproteinases: multifunctional effectors of inflammation in multiple sclerosis and bacterial meningitis. Brain Res Brain Res Rev. 2001;36:249-257.
58 Mastroianni CM, Liuzzi GM. Matrix metalloproteinase dysregulation in HIV infection: implications for therapeutic strategies. Trends Mol Med. 2007;13:449-459.
59 Umehara F, Okada Y, Fujimoto N, et al. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in HTLV-I–associated myelopathy. J Neuropathol Exp Neurol. 1998;57:839-849.
60 Sulik A, Wojtkowska M, Oldak E. Elevated levels of MMP-9 and TIMP-1 in the cerebrospinal fluid of children with echovirus type 30 and mumps meningitis. Scand J Immunol. 2008;68:323-327.
61 Chen KM, Liu JY, Lai SC, et al. Association of plasminogen activators and matrix metalloproteinase-9 proteolytic cascade with blood-CNS barrier damage of angiostrongyliasis. Int J Exp Pathol. 2006;87:113-119.
62 Azeh I, Mader M, Smirnov A, et al. Experimental pneumococcal meningitis in rabbits: the increase of matrix metalloproteinase-9 in cerebrospinal fluid correlates with leucocyte invasion. Neurosci Lett. 1998;256:127-130.
63 Meli DN, Christen S, Leib SL. Matrix metalloproteinase-9 in pneumococcal meningitis: activation via an oxidative pathway. J Infect Dis. 2003;187:1411-1415.
64 Oggioni MR, Memmi G, Maggi T, et al. Pneumococcal zinc metalloproteinase ZmpC cleaves human matrix metalloproteinase 9 and is a virulence factor in experimental pneumonia. Mol Microbiol. 2003;49:795-805.
65 Bottcher T, Spreer A, Azeh I, et al. Matrix metalloproteinase-9 deficiency impairs host defense mechanisms against Streptococcus pneumoniae in a mouse model of bacterial meningitis. Neurosci Lett. 2003;338:201-204.
66 Sellner J, Leib SL. In bacterial meningitis cortical brain damage is associated with changes in parenchymal MMP-9/TIMP-1 ratio and increased collagen type IV degradation. Neurobiol Dis. 2006;21:647-656.
67 Liu X, Han Q, Sun R, et al. Dexamethasone regulation of matrix metalloproteinase expression in experimental pneumococcal meningitis. Brain Res. 2008;1207:237-243.
68 Meli DN, Coimbra RS, Erhart DG, et al. Doxycycline reduces mortality and injury to the brain and cochlea in experimental pneumococcal meningitis. Infect Immun. 2006;74:3890-3896.
69 Leppert D, Leib SL, Grygar C, et al. Matrix metalloproteinase (MMP)-8 and MMP-9 in cerebrospinal fluid during bacterial meningitis: association with blood-brain barrier damage and neurological sequelae. Clin Infect Dis. 2000;31:80-84.
70 Yushchenko M, Weber F, Mader M, et al. Matrix metalloproteinase-9 (MMP-9) in human cerebrospinal fluid (CSF): elevated levels are primarily related to CSF cell count. J Neuroimmunol. 2000;110:244-251.
71 Buckner CM, Luers AJ, Calderon TM, et al. Neuroimmunity and the blood-brain barrier: molecular regulation of leukocyte transmigration and viral entry into the nervous system with a focus on neuroAIDS. J Neuroimmune Pharmacol. 2006;1:160-181.
72 Kanmogne GD, Primeaux C, Grammas P. HIV-1 gp120 proteins alter tight junction protein expression and brain endothelial cell permeability: implications for the pathogenesis of HIV-associated dementia. J Neuropathol Exp Neurol. 2005;64:498-505.
73 Andras IE, Pu H, Deli MA, et al. HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res. 2003;74:255-265.
74 Rusnati M, Presta M. HIV-1 Tat protein and endothelium: from protein/cell interaction to AIDS-associated pathologies. Angiogenesis. 2002;5:141-151.
75 Nakamuta S, Endo H, Higashi Y, et al. Human immunodeficiency virus type 1 gp120-mediated disruption of tight junction proteins by induction of proteasome-mediated degradation of zonula occludens-1 and -2 in human brain microvascular endothelial cells. J Neurovirol. 2008;14:186-195.
76 Wang T, Town T, Alexopoulou L, et al. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366-1373.
77 Arjona A, Foellmer HG, Town T, et al. Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J Clin Invest. 2007;117:3059-3066.
78 Martinez Torres FJ, Volcker D, Dorner N, et al. Aquaporin 4 regulation during acute and long-term experimental herpes simplex virus encephalitis. J Neurovirol. 2007;13:38-46.
79 Lehmann GL, Gradilone SA, Marinelli RA. Aquaporin water channels in central nervous system. Curr Neurovasc Res. 2004;1:293-303.
80 Papadopoulos MC, Verkman AS. Aquaporin-4 gene disruption in mice reduces brain swelling and mortality in pneumococcal meningitis. J Biol Chem. 2005;280:13906-13912.
81 Scheld WM, Dacey RG, Winn HR, et al. Cerebrospinal fluid outflow resistance in rabbits with experimental meningitis. Alterations with penicillin and methylprednisolone. J Clin Invest. 1980;66:243-253.
82 Wispelwey B, Lesse AJ, Hansen EJ, et al. Haemophilus influenzae lipopolysaccharide-induced blood brain barrier permeability during experimental meningitis in the rat. J Clin Invest. 1988;82:1339-1346.
83 Tunkel AR, Rosser SW, Hansen EJ, et al. Blood-brain barrier alterations in bacterial meningitis: development of an in vitro model and observations on the effects of lipopolysaccharide. In Vitro Cell Dev Biol. 1991;27A:113-120.
84 Verma S, Nakaoke R, Dohgu S, et al. Release of cytokines by brain endothelial cells: A polarized response to lipopolysaccharide. Brain Behav Immun. 2006;20:449-455.
85 Coisne C, Faveeuw C, Delplace Y, et al. Differential expression of selectins by mouse brain capillary endothelial cells in vitro in response to distinct inflammatory stimuli. Neurosci Lett. 2006;392:216-220.
86 Lesse AJ, Moxon ER, Zwahlen A, et al. Role of cerebrospinal fluid pleocytosis and Haemophilus influenzae type b capsule on blood brain barrier permeability during experimental meningitis in the rat. J Clin Invest. 1988;82:102-109.
87 Roord JJ, Apicella M, Scheld WM. The induction of meningeal inflammation and blood-brain barrier permeability by Haemophilus influenzae type b peptidoglycan. J Infect Dis. 1994;170:254-256.
88 Constantin D, Ala’Aldeent D, Murphy S. Transcriptional activation of nitric oxide synthase-2, and NO-induced cell death, in mouse cerebrovascular endothelium exposed to Neisseria meningitidis. J Neurochem. 2002;81:270-276.
89 Constantin D, Cordenier A, Robinson K, et al. Neisseria meningitidis–induced death of cerebrovascular endothelium: mechanisms triggering transcriptional activation of inducible nitric oxide synthase. J Neurochem. 2004;89:1166-1174.
90 Boveri M, Kinsner A, Berezowski V, et al. Highly purified lipoteichoic acid from gram-positive bacteria induces in vitro blood-brain barrier disruption through glia activation: role of pro-inflammatory cytokines and nitric oxide. Neuroscience. 2006;137:1193-1209.
91 Buster BL, Weintrob AC, Townsend GC, et al. Potential role of nitric oxide in the pathophysiology of experimental bacterial meningitis in rats. Infect Immun. 1995;63:3835-3839.
92 Jaworowicz DJJr, Korytko PJ, Singh LS, et al. Nitric oxide and prostaglandin E2 formation parallels blood-brain barrier disruption in an experimental rat model of bacterial meningitis. Brain Res Bull. 1998;46:541-546.
93 Hooper DC, Kean RB, Scott GS, et al. The central nervous system inflammatory response to neurotropic virus infection is peroxynitrite dependent. J Immunol. 2001;167:3470-3477.
94 Phares TW, Fabis MJ, Brimer CM, et al. A peroxynitrite-dependent pathway is responsible for blood-brain barrier permeability changes during a central nervous system inflammatory response: TNF-alpha is neither necessary nor sufficient. J Immunol. 2007;178:7334-7343.
95 Quagliarello VJ, Long WJ, Scheld WM. Morphologic alterations of the blood-brain barrier with experimental meningitis in the rat. Temporal sequence and role of encapsulation. J Clin Invest. 1986;77:1084-1095.
96 Sukumaran SK, Prasadarao NV. Escherichia coli K1 invasion increases human brain microvascular endothelial cell monolayer permeability by disassembling vascular-endothelial cadherins at tight junctions. J Infect Dis. 2003;188:1295-1309.
97 Rosado CJ, Kondos S, Bull TE, et al. The MACPF/CDC family of pore-forming toxins. Cell Microbiol. 2008;10:1765-1774.
98 Hirst RA, Kadioglu A, O’Callaghan C, et al. The role of pneumolysin in pneumococcal pneumonia and meningitis. Clin Exp Immunol. 2004;138:195-201.
99 Hirst RA, Gosai B, Rutman A, et al. Streptococcus pneumoniae deficient in pneumolysin or autolysin has reduced virulence in meningitis. J Infect Dis. 2008;197:744-751.
100 Zysk G, Schneider-Wald BK, Hwang JH, et al. Pneumolysin is the main inducer of cytotoxicity to brain microvascular endothelial cells caused by Streptococcus pneumoniae. Infect Immun. 2001;69:845-852.
101 Eriksson KJ, Boyd SG, Tasker RC. Acute neurology and neurophysiology of haemolytic-uraemic syndrome. Arch Dis Child. 2001;84:434-435.
102 Obrig TG. Shiga toxin mode of action in E. coli O157:H7 disease. Front Biosci. 1997;2:d635-d642.
103 Eisenhauer PB, Jacewicz MS, Conn KJ, et al. Escherichia coli Shiga toxin 1 and TNF-alpha induce cytokine release by human cerebral microvascular endothelial cells. Microb Pathog. 2004;36:189-196.
104 Fujii J, Wood K, Matsuda F, et al. Shiga toxin 2 causes apoptosis in human brain microvascular endothelial cells via C/EBP homologous protein. Infect Immun. 2008;76:3679-3689.
105 Kramer-Hammerle S, Rothenaigner I, Wolff H, et al. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111:194-213.
106 Piller SC, Ewart GD, Jans DA, et al. The amino-terminal region of Vpr from human immunodeficiency virus type 1 forms ion channels and kills neurons. J Virol. 1999;73:4230-4238.
107 Patel CA, Mukhtar M, Pomerantz RJ. Human immunodeficiency virus type 1 Vpr induces apoptosis in human neuronal cells. J Virol. 2000;74:9717-9726.
108 Walsh K, Megyesi J, Wilson J, et al. Antioxidant protection from HIV-1 gp120-induced neuroglial toxicity. J Neuroinflammation. 2004;1:8.
109 King JE, Eugenin EA, Buckner CM, et al. HIV tat and neurotoxicity. Microbes Infect. 2006;8:1347-1357.
110 New DR, Maggirwar SB, Epstein LG, et al. HIV-1 Tat induces neuronal death via tumor necrosis factor-alpha and activation of non–N-methyl-D-aspartate receptors by a NFκB-independent mechanism. J Biol Chem. 1998;273:17852-17858.
111 Nath A, Psooy K, Martin C, et al. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol. 1996;70:1475-1480.
112 Noorbakhsh F, Vergnolle N, McArthur JC, et al. Proteinase-activated receptor-2 induction by neuroinflammation prevents neuronal death during HIV infection. J Immunol. 2005;174:7320-7329.
113 Dale C, Vergnolle N. Protease signaling to G protein–coupled receptors: implications for inflammation and pain. J Recept Signal Transduct Res. 2008;28:29-37.
114 Zhou BY, Liu Y, Kim B, et al. Astrocyte activation and dysfunction and neuron death by HIV-1 Tat expression in astrocytes. Mol Cell Neurosci. 2004;27:296-305.
115 Tian C, Erdmann N, Zhao J, et al. HIV-infected macrophages mediate neuronal apoptosis through mitochondrial glutaminase. J Neurochem. 2008;105:994-1005.
116 Meli DN, Christen S, Leib SL, et al. Current concepts in the pathogenesis of meningitis caused by Streptococcus pneumoniae. Curr Opin Infect Dis. 2002;15:253-257.
117 Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916-3924.
118 Chien HF, Yeh KY, Jiang-Shieh YF, et al. Signal transduction pathways of nitric oxide release in primary microglial culture challenged with gram-positive bacterial constituent, lipoteichoic acid. Neuroscience. 2005;133:423-436.
119 Kinsner A, Pilotto V, Deininger S, et al. Inflammatory neurodegeneration induced by lipoteichoic acid from Staphylococcus aureus is mediated by glia activation, nitrosative and oxidative stress, and caspase activation. J Neurochem. 2005;95:1132-1143.
120 Leib SL, Kim YS, Chow LL, et al. Reactive oxygen intermediates contribute to necrotic and apoptotic neuronal injury in an infant rat model of bacterial meningitis due to group B streptococci. J Clin Invest. 1996;98:2632-2639.
121 Braun JS, Sublett JE, Freyer D, et al. Pneumococcal pneumolysin and H2O2 mediate brain cell apoptosis during meningitis. J Clin Invest. 2002;109:19-27.
122 Krantic S, Mechawar N, Reix S, et al. Apoptosis-inducing factor: a matter of neuron life and death. Prog Neurobiol. 2007;81:179-196.
123 Tumani H, Smirnov A, Barchfeld S, et al. Inhibition of glutamine synthetase in rabbit pneumococcal meningitis is associated with neuronal apoptosis in the dentate gyrus. Glia. 2000;30:11-18.
124 Leib SL, Kim YS, Ferriero DM, et al. Neuroprotective effect of excitatory amino acid antagonist kynurenic acid in experimental bacterial meningitis. J Infect Dis. 1996;173:166-171.
125 Kolarova A, Ringer R, Tauber MG, et al. Blockade of NMDA receptor subtype NR2B prevents seizures but not apoptosis of dentate gyrus neurons in bacterial meningitis in infant rats. BMC Neurosci. 2003;4:21.
126 Braun JS, Hoffmann O, Schickhaus M, et al. Pneumolysin causes neuronal cell death through mitochondrial damage. Infect Immun. 2007;75:4245-4254.
127 Grandgirard D, Schurch C, Cottagnoud P, et al. Prevention of brain injury by the nonbacteriolytic antibiotic daptomycin in experimental pneumococcal meningitis. Antimicrob Agents Chemother. 2007;51:2173-2178.
128 Enzmann DR, Britt RH, Yeager AS. Experimental brain abscess evolution: computed tomographic and neuropathologic correlation. Radiology. 1979;133:113-122.
129 Britt RH, Enzmann DR, Yeager AS. Neuropathological and computerized tomographic findings in experimental brain abscess. J Neurosurg. 1981;55:590-603.
130 Flaris NA, Hickey WF. Development and characterization of an experimental model of brain abscess in the rat. Am J Pathol. 1992;141:1299-1307.
131 Wood JH, Lightfoote WE, Ommaya AK. Cerebral abscesses produced by bacterial implantation and septic embolisation in primates. J Neurol Neurosurg Psychiatry. 1979;42:63-69.
132 Costello GT, Heppe R, Winn HR, et al. Susceptibility of brain to aerobic, anaerobic, and fungal organisms. Infect Immun. 1983;41:535-539.
133 Kielian T, Hickey WF. Proinflammatory cytokine, chemokine, and cellular adhesion molecule expression during the acute phase of experimental brain abscess development. Am J Pathol. 2000;157:647-658.
134 Kielian T, Barry B, Hickey WF. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J Immunol. 2001;166:4634-4643.
135 Kielian T, Bearden ED, Baldwin AC, et al. IL-1 and TNF-alpha play a pivotal role in the host immune response in a mouse model of Staphylococcus aureus–induced experimental brain abscess. J Neuropathol Exp Neurol. 2004;63:381-396.
136 Stenzel W, Soltek S, Miletic H, et al. An essential role for tumor necrosis factor in the formation of experimental murine Staphylococcus aureus–induced brain abscess and clearance. J Neuropathol Exp Neurol. 2005;64:27-36.
137 Stenzel W, Soltek S, Sanchez-Ruiz M, et al. Both TLR2 and TLR4 are required for the effective immune response in Staphylococcus aureus–induced experimental murine brain abscess. Am J Pathol. 2008;172:132-145.
138 Kielian T, Phulwani NK, Esen N, et al. MyD88-dependent signals are essential for the host immune response in experimental brain abscess. J Immunol. 2007;178:4528-4537.
139 Baldwin AC, Kielian T. Persistent immune activation associated with a mouse model of Staphylococcus aureus–induced experimental brain abscess. J Neuroimmunol. 2004;151:24-32.
140 Stenzel W, Soltek S, Schluter D, et al. The intermediate filament GFAP is important for the control of experimental murine Staphylococcus aureus–induced brain abscess and Toxoplasma encephalitis. J Neuropathol Exp Neurol. 2004;63:631-640.
141 Kielian T, Drew PD. Effects of peroxisome proliferator–activated receptor-gamma agonists on central nervous system inflammation. J Neurosci Res. 2003;71:315-325.
142 Phulwani NK, Feinstein DL, Gavrilyuk V, et al. 15-Deoxy-Delta12,14-prostaglandin J2 (15d-PGJ2) and ciglitazone modulate Staphylococcus aureus–dependent astrocyte activation primarily through a PPAR-gamma–independent pathway. J Neurochem. 2006;99:1389-1402.
143 Kielian T, Syed MM, Liu S, et al. The synthetic peroxisome proliferator–activated receptor-gamma agonist ciglitazone attenuates neuroinflammation and accelerates encapsulation in bacterial brain abscesses. J Immunol. 2008;180:5004-5016.
144 Blum D, Chtarto A, Tenenbaum L, et al. Clinical potential of minocycline for neurodegenerative disorders. Neurobiol Dis. 2004;17:359-366.
145 Kielian T, Esen N, Liu S, et al. Minocycline modulates neuroinflammation independently of its antimicrobial activity in Staphylococcus aureus–induced brain abscess. Am J Pathol. 2007;171:1199-1214.
146 O’Gara JP. ica and beyond: biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol Lett. 2007;270:179-188.
147 Livni G, Yuhas Y, Ashkenazi S, et al. In vitro bacterial adherence to ventriculoperitoneal shunts. Pediatr Neurosurg. 2004;40:64-69.
148 Fux CA, Quigley M, Worel AM, et al. Biofilm-related infections of cerebrospinal fluid shunts. Clin Microbiol Infect. 2006;12:331-337.
149 Braxton EEJr, Ehrlich GD, Hall-Stoodley L, et al. Role of biofilms in neurosurgical device-related infections. Neurosurg Rev. 2005;28:249-255.






