[level-membership-for-anesthesiology-category]
Chapter 2 Basic Pharmacologic Principles
Pharmacokinetics
3. What factors govern drug absorption?
4. How is absorption via buccal mucosa significantly different from drug absorption via the stomach?
5. What aspects of absorption make transdermal drug delivery distinct from other modes of drug delivery? Name some examples of drugs for which a transdermal application is clinically important.
6. What is the mechanism for the offset of local anesthetic effects following nerve block?
7. What is “first order” transfer? How does doubling the dose of a drug affect the shape of a plot of drug absorbed versus time?
8. How does absorption rate from its delivery site affect peak plasma concentration of a drug? What does absorption rate mean regarding the relative safety of intercostal nerve blocks?
9. Define distribution. Define volume of distribution.
10. Distinguish central volume of distribution from peripheral volume of distribution.
11. What factors increase a peripheral volume of distribution for a drug?
12. What are two empiric models of peripheral volumes of distribution that are clinically useful?
13. Generally speaking, what is clearance of a drug? What is the difference between systemic clearance and “intercompartmental” clearance?
14. How are most anesthetic drugs removed from the body?
15. What processes are used in the liver to metabolize drugs?
16. What drugs are metabolized by cytochrome CYP 3A4?
17. What drugs or substances induce CYP 3A4? What drugs or substances inhibit CYP 3A4?
18. What function important to anesthesia does CYP 2D6 have? What drugs inhibit CYP 2D6, and what clinical implication does this have?
19. Why do remifentanil, succinylcholine, and esmolol generally vanish from the plasma so quickly after intravenous administration?
20. Why is the pharmacokinetics of succinylcholine less predictable than other drugs cleared by ester hydrolysis?
21. Define “linear” pharmacokinetics.
22. Describe the formula for rate of drug metabolism in terms of liver blood flow.
23. What is an extraction ratio? What is the formula for clearance in terms of hepatic blood flow? What are the units for clearance?
24. In the case of a drug exhibiting “linear” pharmacokinetics, what is significant about the constant relationship between metabolic rate and drug concentration?
25. What does it mean to say that a drug’s metabolism is “flow limited Name a drug whose metabolism is “flow limited.”
26. What does it mean to say that a drug’s metabolism is “capacity limited What effect does an alteration of blood flow to the liver have on drugs whose metabolism is “capacity limited
27. How is the maximum metabolic rate (Vm) of the liver defined? What is Km?
28. What factors may alter the maximum metabolic rate for a drug in the liver? How do changes in maximum metabolic rate for a drug alter the clearance of “flow limited” and “capacity limited” drugs?
29. What does it mean for a drug to have saturable pharmacokinetics?
30. What class of drugs significant to anesthesia practiceare eliminated by the kidneys?
31. Why is a normal serum creatinine value in an elderly person not a reliable indicator of the individual’s ability to clear drugs in the kidney?
32. Describe the clearance of propofol from the bloodstream following intravenous injection.
33. What is distribution clearance? What is the clinical significance of this phenomenon?
34. What is the capacity for plasma proteins to bind most anesthetic drugs? How does the number of protein binding sites for a drug in plasma influence the amount of a drug in the plasma bound to proteins?
35. What effect does a change in plasma protein concentrations have on the apparent potency of a drug?
36. Sketch a graph plotting time versus the amount of drug for a first order pharmacokinetic process. What does it mean to say that a first order pharmacokinetic process demonstrates exponential decay?
37. What do the rate constants between pharmacokinetic compartments relate?
38. For anesthetic drugs, which compartment model best reflects their pharmacokinetic behavior? What about anesthetic drug pharmacokinetics makes this model appropriate? What do the “compartments” correspond to?
39. Why is it impossible to achieve a steady state drug concentration with a bolus of drug followed by a simple infusion when a drug is best described by a multiple compartment model?
40. What is the time course of drug effect? Why does this exist for anesthesia drugs, and what pharmacokinetic properties does this process exhibit?
41. Define context-sensitive half-time. What are its limitations? Why is context-sensitive half-time a more meaningful concept with regards to the offset of anesthetic drug effects than drug half-life?
42. Why is morphine not an appropriate choice for continuous infusion during anesthesia? Define context-sensitive effect site decrement time.
Pharmacodynamics
43. When a drug’s concentration is equal to its dissociation constant for the binding of a certain receptor, what may be said regarding occupation of those receptors? If a dissociation constant is relatively high, what does this mean regarding the nature of the binding between a receptor and a drug?
44. Define receptor full agonist. What is a partial agonist?
45. Define receptor antagonist. What is an inverse agonist?
46. Distinguish efficacy from affinity.
47. What is the difference between competitive and noncompetitive antagonism?
48. How does binding of a receptor by a drug result in drug effect?
49. What three types of receptors are of most significance to anesthesia? For each type of receptor, name several drugs important in anesthesia whose effects are mediated by that receptor type.
50. Sketch a graph plotting the dose of a drug (or measure of exposure) versus its response (drug effect). If a dose-response curve is “shifted to the left,” what does this mean regarding drug (or exposure) potency?
51. Define ED50, LD50, and therapeutic index. Why is a drug with a higher therapeutic index safer?
52. Describe the interaction of hypnotics and opioids, specifically with regard to fentanyl and isoflurane MAC.
53. What is a response surface?
54. Define additive drug interactions, supraadditive interactions, and infraadditive interactions.
Answers
1. Pharmacokinetics is the process by which the body “disposes” of drugs via absorption, distribution, metabolism, and elimination. It can be thought of as “what the body does to the drug.” (35)
2. Pharmacodynamics is the process by which drugs interact with specific receptors in the body to produce pharmacologic effects. It can be thought of as “what the drug does to the body.” (35)
Pharmacokinetics
3. Drug absorption is governed by route of delivery, bioavailability of the drug, and possibly first-pass metabolism. (36)
4. Drug absorption via buccal mucosa differs from absorption from lower in the gastrointestinal tract because presence of food does not hinder delivery of drug to the mucosa. Also, venous outflow from the buccal mucosa returns directly to the systemic circulation, thereby avoiding the potential for the first-pass hepatic effect that is present for drugs absorbed in the stomach, which first enter the portal venous system. (36)
5. Transdermal drug delivery is distinct in that skin is designed to be a significant barrier to absorption. This means that drugs delivered transdermally will have a markedly delayed onset of action following administration. Also, the skin serves as a depot of the drug, resulting in prolonged drug effect following removal of the skin application. Examples of drugs delivered transdermally include clonidine, scopolamine, nitroglycerin, and fentanyl. (36)
6. Local anesthetics applied in nerve blocks have their pharmacologic effects ended by movement of the drug away from the site of action. The process by which the body absorbs this locally applied bolus of drug, thus ending its local effects, is the same by which the body absorbs drugs injected into tissues for the purpose of eliciting systemic drug effects that follow absorption. (36)
7. “First order” transfer is when the rate of drug absorption is proportional to the concentration gradient. Doubling the dose of a drug does not affect the shape of the curve (absorption over time) when a “first order” transfer is occurring. Concentrations will be exactly twice as high at all times, but peak absorption will occur at the same time and the shape of the curve will be identical. (36)
8. Absorption rate from drug delivery sites significantly affects peak plasma concentrations. The higher the absorption rate, the higher the peak plasma concentration that will result. Nerve blocks at sites with rapid absorption result in higher peak plasma concentrations of the local anesthetic injected, providing a risk of toxicity relatively greater than nerve blocks at sites with slower absorption. Intercostal blocks are performed in areas of relatively high absorption. (36)
9. Distribution is the process by which an injected drug mixes with blood and body tissues after its administration. Measuring plasma concentration of a drug allows calculation of a mixing volume, or volume of distribution. Volume of distribution is thus a calculated number (dose of drug administered intravenously divided by plasma concentration) that reflects the apparent volume of body tissues that the drug is distributed across, assuming all the tissues it is distributed across are in equilibrium with plasma concentration. Higher levels of drug remaining in the plasma after drug administration lead to a smaller calculated volume of distribution. (36)
10. Central volume of distribution is the apparent volume immediately (within a minute) following intravenous drug injection. It anatomically consists of the heart, the great vessels, and the lungs. Peripheral volumes of distribution are those volumes of distribution that are calculated after the injected drug has had time to distribute to tissues to which distribution of drug is slower. These peripheral tissues include muscle, fat, and bone. While there is an anatomic correlation to central and peripheral volumes of distribution, volume of distribution is a calculated number that does not necessarily equate to an actual physical volume. (36)
11. The solubility of the drug in the tissue relative to the solubility in the blood or plasma determines the peripheral volume of distribution. If the drug is highly soluble in the tissue, then less of it will stay in the plasma. Sampling the plasma concentration of the drug will result in calculation of a higher volume of distribution than if plasma levels remained higher. Drug properties that lower free plasma levels include low levels of binding to plasma proteins, a lower degree of ionization, and higher lipid solubility. (36)
12. One clinically useful model to describe peripheral volumes of distribution divides the body into tissue beds: “vessel rich group” (brain, most organs), muscle group, fat group, and “vessel poor group” (skin, cartilage, ligaments). Another is to identify the number of compartments in the body needed to explain the pharmacokinetics of the drug in question. The pharmacokinetics of most anesthetic drugs can be explained by a three compartment model (one central volume of distribution, and two peripheral volumes of distribution). In spite of names given to different compartments in different models, such compartments are empiric, and do not necessarily correlate directly to underlying anatomic structures or physiologic processes. (36-37)
13. Clearance is the removal of drug from tissue. Systemic clearance is when the drug is permanently removed from the body. “Intercompartmental” clearance is when the drug leaves the body tissue in question but moves into a different body tissue. (37)
14. Most anesthetic drugs are removed from the body by hepatic metabolism. (37)
15. In the liver, drugs are metabolized through the processes of oxidation, reduction, conjugation, and hydrolysis. Oxidation and reduction occurs via the cytochrome P-450 system. (37)
16. Drugs important to anesthesia that are metabolized by CYP 3A4 include acetaminophen, alfentanil, dexamethasone, fentanyl, lidocaine, methadone, midazolam, and sufentanil. Also, propofol is partly oxidized by CYP 3A4. (37)
17. Rifampin, rifabutin, tamoxifen, glucocorticoids, carbamazepine, barbiturates, and St. John’s wort induce CYP 3A4, increasing the metabolism of substrates of CYP 3A4 (hastening clearance). Inhibitors of CYP 3A4 include midazolam, propofol, grapefruit juice, antifungal drugs, protease inhibitors, “mycin” antibiotics, and selective serotonin reuptake inhibitors (SSRIs). In the case of midazolam, this has been shown to prolong the effects of other drugs metabolized by CYP 3A4, such as alfentanil and fentanyl. (37)
18. CYP 2D6 is the cytochrome in the liver responsible for the conversion of codeine to morphine (the active metabolite of codeine). CYP 2D6 is inhibited by quinidine and SSRIs. The clinical implication of this is that codeine, oxycodone, and hydrocodone, which all rely on activity of CYP 2D6 for production of the active metabolite from which their clinically relevant pharmacologic effects are derived, are poor analgesic choices for patients receiving SSRIs. (37)
19. Remifentanil, succinylcholine, and esmolol are cleared in the plasma and tissue by ester hydrolysis. This occurs very quickly because these esterases are so abundant. (37)
20. The pharmacokinetics of succinylcholine are less reliable than that of other drugs cleared by plasma and tissue esterases because it is metabolized specifically by butylcholinesterase (formerly known as “pseudocholinesterase”). Defects in the gene for butylcholinesterase lead to a potentially significant slowing in the metabolism of succinylcholine. (37)
21. “Linear” pharmacokinetics are said to exist for a drug when the rate of the drug’s metabolism is directly proportional to its concentration. This is a general characteristic of anesthetic drugs. (37)
22. Rate of metabolism equals liver blood flow times the difference in drug concentration between blood flowing into the liver and blood flowing out. (38)
23. The extraction ratio of a drug is the fraction of the drug that is removed from the plasma during passage through the liver. Clearance by the liver is equal to hepatic blood flow multiplied by the extraction ratio. (Therefore, units of clearance are liters per minute.) Hepatic extraction ratios are unchanging properties of specific drugs. More of a drug is metabolized by the liver when the drug is being delivered to the liver in increasing concentrations. This must be true for the extraction ratio to remain constant. (37-38)
24. For most anesthetic drugs, metabolic rate is proportional to drug concentration (“linear” pharmacokinetics). The proportionality constant that relates the drug concentration to the metabolic rate is another definition of clearance.
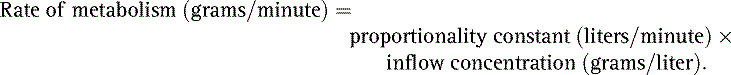
25. The clearance of a “flow limited” drug is limited only by the rate of blood flowing to the liver. Changes in hepatic blood flow result in a proportional change in drug clearance. Such drugs have extraction ratios near or equal to 1. The liver has a seemingly boundless ability to metabolize such drugs. One drug exhibiting “flow limited” clearance is propofol. (38)
26. The clearance of a “capacity limited” drug is limited by the liver’s ability to metabolize the drug. Such drugs have low extraction ratios (much less than 1). Alterations of liver blood flow have no effect on clearance of “capacity limited” drugs because liver blood flow has no effect on clearance of such drugs. One drug exhibiting “capacity limited” clearance is alfentanil. (38)
27. The maximum metabolic rate of the liver for a given drug (Vm) is the theoretical rate of drug metabolism if every possible enzyme in the liver were being used for that function. Km is the concentration of drug in the plasma associated with half of the maximum metabolic rate. (38)
28. Factors altering the maximum metabolic rate for a drug in the liver include enzyme inhibition, enzyme induction, and liver disease. Clearance of “flow limited” drugs is relatively insensitive to changes in maximum metabolic rate because there is such a reserve of metabolic capacity for these drugs. For clearance of such drugs to be affected by an alteration in metabolic rate requires such alteration to be massive. Clearance of “capacity limited” drugs is very sensitive to changes in the maximum metabolic rate, because the ability of the liver to metabolize the drug is so small. (38)
29. For a drug to have saturable pharmacokinetics means that the concentration of the drug in the plasma exceeds the concentration at which the metabolic rate of the drug is half its maximum (Km). Clearance of such drugs is a function of drug concentration (at clinical plasma concentrations). On the other hand, drugs that do not have saturable pharmacokinetics (i.e., their concentration is well below Km) are metabolized at a rate proportional to their concentration (most anesthetic drugs). Clearance of these drugs is a constant. (40)
30. Steroidal muscle relaxants, including pancuronium, vecuronium, and rocuronium are at least partially eliminated by the kidneys; 85% of pancuronium is eliminated by the kidneys, while 20% to 30% of vecuronium, and 10% to 20% of rocuronium are eliminated. (40)
31. Clearance of drugs by the kidney is achieved by filtration of drug from the plasma at the glomerulus and direct transport to the tubules. Creatinine clearance is generally a good indicator of renal ability to clear drugs because it is a good measure of glomerular filtration. Increasing age is an independent factor in the establishment of a patient’s creatinine clearance. Leaving serum creatinine unchanged and increasing a patient’s age results in shrinking of the numerator in the formula for creatinine clearance. In this, it can be seen that, creatinine clearance decreases with age even if serum creatinine never changes. Therefore, creatinine clearance may be decreased despite “normal” serum creatinine levels. In fact, this is inevitably true the older a person gets. Serum creatinine must eventually fall to below “normal” values for creatinine clearance to remain “normal” as very advanced age is reached. (40)
32. Propofol is primarily cleared by metabolism in the liver. Every bit of propofol that flows to the liver clears there, at all clinically significant doses and even in the presence of all but the most massive insults to the cellular processes responsible for its metabolism. Propofol’s clearance is actually greater than hepatic blood flow. This is only possible if, in addition to the robust ability of the liver to metabolize this drug, there are extrahepatic sites of drug metabolism. About a quarter of administered propofol is eliminated by the kidneys. Propofol is eliminated virtually completely by metabolism (metabolism occurring in both liver and kidney), with only a minute fraction (less than 1%) excreted in the urine unchanged. Renal elimination of propofol is not a function of filtration, but is a function of renal blood flow since the kidneys remove every molecule of the drug that enters it. (37-38, 40)
33. Distribution clearance is the transfer of a drug out of the plasma into peripheral tissues. The drug remains in active form and may be sequestered in tissues for an extended period, serving as a reservoir for recurrent or prolonged pharmacologic effects of drug administration. (40)
34. The capacity of plasma proteins to bind most anesthetic drugs is very large. That is, the number of sites on proteins available to bind drug is far greater than the number of molecules of an anesthetic drug administered at clinical levels. This does not necessarily mean that a lot of the drug will become bound to plasma proteins upon arrival in the bloodstream. Such binding is dependent on the rate constants for binding and dissociation each specific drug has for plasma proteins. The effect of an excess of protein binding sites is that the amount of drug bound to plasma proteins is purely a function of the concentration of the plasma protein. Whether this function allocates a large or small amount of drug to a protein-bound form is governed by the rate constants for binding and dissociation. (40)
35. If plasma protein levels decrease due to aging or some disease process, then the free fraction of drug will increase. This will increase the apparent potency of the drug—with a couple of caveats. First, the increase in potency will be greater for drugs that are highly protein bound. A decrease in plasma protein levels has much more effect on drugs highly bound to plasma proteins. Potency of the drug will only appear to increase if the drug in question is more than 90% protein bound (free fraction <10%). Secondly, any noted increase in potency will only exist directly after intravenous bolus injection, while the volume of distribution is that of the central compartment, and protein binding of a drug is only to proteins in the plasma. Once drug concentration in the plasma equilibrates with peripheral tissues, then tissue proteins also participate in drug binding. The amount of drug binding by plasma proteins once the total body concentration of the drug is in equilibrium is trivial when compared to the amount bound by proteins in the tissue. For this reason, variations in plasma protein levels are significant to drug potency only immediately after administration, but have no significant effect on drug potency once drug concentration has equilibrated with the peripheral tissues. (40)
36. First order pharmacokinetic processes demonstrate exponential decay, which is to say that the rate of decrease in drug amount slows as the amount of the drug decreases. (Conversely, rate of decrease in the drug amount speeds up if the amount of drug increases.) (41, Figure 5-4)
37. Rate constants between compartments in pharmacokinetic compartment models relate flow of drug between compartments to the amount of drug in the driving compartment. (41)
38. For most anesthetic drugs, three distinct phases of pharmacokinetics following bolus injection can be distinguished, which is the defining characteristic of a three compartment pharmacokinetic model. These three phases correspond to rapid distribution, slow distribution, and the terminal phase. The distribution phases reflect the peripheral volumes initially filling with the drug, and the terminal phase reflects the drug being discharged back into the plasma. These three processes are definitive of a pharmacokinetic three compartment model, but do not discreetly correspond to the “three compartments” themselves. The three compartments themselves represent one central volume of distribution (central compartment) and two peripheral volumes of distribution (peripheral compartments). (41)
39. To achieve a steady state drug concentration for a drug best described by a multiple compartment model, the initial bolus of the drug must be followed by an infusion of drug that changes over time. The initial rate of infusion must be high enough to compensate for the drug leaving the central compartment for the peripheral compartment(s). After the peripheral compartment(s) have come to equilibrium with the plasma concentration, then the infusion must be decreased to a rate that exactly matches the rate of clearance of the drug. (42-43)
40. Time course of the drug effect is the delay between introduction of the drug into the plasma and the onset of measurable drug effect. This exists for anesthetic drugs because most of them produce their desired clinical effects via actions occurring outside the plasma. The drug must be delivered and diffuse into its target tissue before it produces effects, and this takes a certain amount of time. The site of the drug effect is connected to the plasma by a first order pharmacokinetic process, which means that the higher the drug concentration in the plasma, the faster the drug levels will rise in the target tissue site. The factor that relates the plasma concentration to the target tissue concentration is the equilibration rate constant. (43-44)
41. Context-sensitive half-time is the time necessary for a drug’s plasma concentration to decrease 50% after discontinuation of a continuous intravenous infusion of specific duration. The limitation of this value is that it fails to take into account equilibration delay, which is to say it does not factor in the time lag between changes in plasma drug concentration and concentration at target tissue sites. Context-sensitive half-time has much more clinical utility for anesthesia practice than drug half-life, though. Half-life is indicative of the time it takes to eliminate a drug from the body. Context-sensitive half-time is indicative of the time it takes plasma levels of drug to fall. Termination of the clinical effect of an anesthetic drug (i.e., awakening) is dependent on its plasma level (and by extension, corresponding target tissue concentration) of the drug and not the drug’s overall presence in the body. (44)
42. Morphine is not an appropriate choice for continuous infusion during anesthesia, because it has a very slow plasma-effect site equilibration. Although morphine has rapid plasma pharmacokinetics (a short context-sensitive half-time), it has very slow blood-brain equilibration (a long context-sensitive effect site decrement time). Context-sensitive effect site decrement time relates the time course of effect site concentration with the duration of drug delivery. Drugs such as morphine, which do not equilibrate quickly between their plasma concentrations and target tissue concentrations, do not have their clinical effects well represented by context-sensitive half-time values. (44)
Pharmacodynamics
43. When a drug’s concentration is equal to its dissociation constant for the binding of a certain receptor, then then 50% of those receptors are bound by the drug. A high dissociation constant means that the receptor has low affinity for the drug (weak binding). A low dissociation constant means that the receptor has high affinity for the ligand drug (tight binding). (45)
44. A full agonist is a drug that activates a receptor to its maximum capacity. A partial agonist is a drug that binds a receptor and produces a less effective response than a full agonist, even at high concentrations. (45)
45. An antagonist is a drug that blocks access to a receptor by agonists, but itself produces no activation of the receptor. An inverse agonist is a drug that binds a receptor and produces a pharmacologic response that is below the baseline response measured in the absence of the drug. This is usually thought to result from the inverse agonist acting to block access to the receptor by endogenous agonist(s). (45)
46. Efficacy of a drug is the level of activation of receptors that results when the drug interacts with the receptors. Affinity refers to how much of a drug binds to receptors at any given drug concentration. These are completely independent properties; therefore, drugs may have identical affinities for a receptor type, but have very different efficacies. The difference between, for example, full agonists and antagonists is not a function of their relative affinities for the receptor in question. By definition, it is a function of their relative efficacies. (45)
47. Competitive antagonism is when an antagonist competes with an agonist for receptor binding, causing the total number of receptors bound by the agonist to decrease. The competitive antagonist displaces an agonist, without blocking it, and can therefore be displaced from the receptor by an increased dose of agonist. A noncompetitive antagonist alters the conformation of the receptor by irreversibly binding to the receptor complex. (45)
48. Binding of a receptor by a drug causes the receptor to favor, and spend more time in, one of its conformations more than it did before being bound by the drug. The particular conformation favored by receptor-drug interaction facilitates the particular biochemical cascade that results in the drug effect. (45)
49. G-protein-coupled receptors mediate the action of opioids, serotonin, all vasoactive amines, prostaglandins, and histamine. Ligand-gated ion channels mediate the action of propofol, midazolam, thiopental, ketamine, and muscle relaxants. Voltage-gated ion channels are the target of local anesthetic action. (46)
50. Shifting of a dose-response curve “to the left” means that a lower dose of drug is necessary to provide an effect. This means that a shift in the curve “to the left” represents an increase in drug potency. Conversely, shifting the curve “to the right” represents a decrease in drug potency. (46, Figure 5-11)
51. ED50 is the dose of drug required to produce a specific effect in 50% of individuals. LD50 is the dose required to produce death in 50% of individuals. Therapeutic index is the ratio between LD50 and ED50. The higher the therapeutic index, the further the lethal dose of the drug is from the therapeutic dose. This makes administering the drug safer because giving a dose greater than that needed for a therapeutic response is less likely to climb to the level that constitutes a lethal dose. (46)
52. Fentanyl administration in conjunction with isoflurane administration results in an effective decrease in the MAC of the volatile anesthetic. This decrease in MAC is proportional to fentanyl dose, but it levels off and never reaches zero because fentanyl alone cannot ensure nonresponsiveness. (46-47)
53. A response surface is a three-dimensional surface that shows the effect of the combination of any two drugs. (46-47)
54. If drugs have additive effects, their concomitant administration results in pharmacologic effects equivalent to the sum of the effects each would have produced if administered alone. If drugs have supraadditive (synergistic) effects, their concomitant administration produces pharmacologic effects in excess of the sum of each drug’s individual effects. If drugs have infraadditive effects, their concomitant administration produces pharmacologic effects more than what either would produce if administered alone, but less than if the individual effects of each drug were added to one another. (47-48)
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 2 Basic Pharmacologic Principles
Pharmacokinetics
3. What factors govern drug absorption?
4. How is absorption via buccal mucosa significantly different from drug absorption via the stomach?
5. What aspects of absorption make transdermal drug delivery distinct from other modes of drug delivery? Name some examples of drugs for which a transdermal application is clinically important.
6. What is the mechanism for the offset of local anesthetic effects following nerve block?
7. What is “first order” transfer? How does doubling the dose of a drug affect the shape of a plot of drug absorbed versus time?
8. How does absorption rate from its delivery site affect peak plasma concentration of a drug? What does absorption rate mean regarding the relative safety of intercostal nerve blocks?
9. Define distribution. Define volume of distribution.
10. Distinguish central volume of distribution from peripheral volume of distribution.
11. What factors increase a peripheral volume of distribution for a drug?
12. What are two empiric models of peripheral volumes of distribution that are clinically useful?
13. Generally speaking, what is clearance of a drug? What is the difference between systemic clearance and “intercompartmental” clearance?
14. How are most anesthetic drugs removed from the body?
15. What processes are used in the liver to metabolize drugs?
16. What drugs are metabolized by cytochrome CYP 3A4?
17. What drugs or substances induce CYP 3A4? What drugs or substances inhibit CYP 3A4?
18. What function important to anesthesia does CYP 2D6 have? What drugs inhibit CYP 2D6, and what clinical implication does this have?
19. Why do remifentanil, succinylcholine, and esmolol generally vanish from the plasma so quickly after intravenous administration?
20. Why is the pharmacokinetics of succinylcholine less predictable than other drugs cleared by ester hydrolysis?
21. Define “linear” pharmacokinetics.
22. Describe the formula for rate of drug metabolism in terms of liver blood flow.
23. What is an extraction ratio? What is the formula for clearance in terms of hepatic blood flow? What are the units for clearance?
24. In the case of a drug exhibiting “linear” pharmacokinetics, what is significant about the constant relationship between metabolic rate and drug concentration?
25. What does it mean to say that a drug’s metabolism is “flow limited Name a drug whose metabolism is “flow limited.”
26. What does it mean to say that a drug’s metabolism is “capacity limited What effect does an alteration of blood flow to the liver have on drugs whose metabolism is “capacity limited
27. How is the maximum metabolic rate (Vm) of the liver defined? What is Km?
28. What factors may alter the maximum metabolic rate for a drug in the liver? How do changes in maximum metabolic rate for a drug alter the clearance of “flow limited” and “capacity limited” drugs?
29. What does it mean for a drug to have saturable pharmacokinetics?
30. What class of drugs significant to anesthesia practiceare eliminated by the kidneys?
31. Why is a normal serum creatinine value in an elderly person not a reliable indicator of the individual’s ability to clear drugs in the kidney?
32. Describe the clearance of propofol from the bloodstream following intravenous injection.
33. What is distribution clearance? What is the clinical significance of this phenomenon?
34. What is the capacity for plasma proteins to bind most anesthetic drugs? How does the number of protein binding sites for a drug in plasma influence the amount of a drug in the plasma bound to proteins?
35. What effect does a change in plasma protein concentrations have on the apparent potency of a drug?
36. Sketch a graph plotting time versus the amount of drug for a first order pharmacokinetic process. What does it mean to say that a first order pharmacokinetic process demonstrates exponential decay?
37. What do the rate constants between pharmacokinetic compartments relate?
38. For anesthetic drugs, which compartment model best reflects their pharmacokinetic behavior? What about anesthetic drug pharmacokinetics makes this model appropriate? What do the “compartments” correspond to?
39. Why is it impossible to achieve a steady state drug concentration with a bolus of drug followed by a simple infusion when a drug is best described by a multiple compartment model?
40. What is the time course of drug effect? Why does this exist for anesthesia drugs, and what pharmacokinetic properties does this process exhibit?
41. Define context-sensitive half-time. What are its limitations? Why is context-sensitive half-time a more meaningful concept with regards to the offset of anesthetic drug effects than drug half-life?
42. Why is morphine not an appropriate choice for continuous infusion during anesthesia? Define context-sensitive effect site decrement time.
Pharmacodynamics
43. When a drug’s concentration is equal to its dissociation constant for the binding of a certain receptor, what may be said regarding occupation of those receptors? If a dissociation constant is relatively high, what does this mean regarding the nature of the binding between a receptor and a drug?
44. Define receptor full agonist. What is a partial agonist?
45. Define receptor antagonist. What is an inverse agonist?
46. Distinguish efficacy from affinity.
47. What is the difference between competitive and noncompetitive antagonism?
48. How does binding of a receptor by a drug result in drug effect?
49. What three types of receptors are of most significance to anesthesia? For each type of receptor, name several drugs important in anesthesia whose effects are mediated by that receptor type.
50. Sketch a graph plotting the dose of a drug (or measure of exposure) versus its response (drug effect). If a dose-response curve is “shifted to the left,” what does this mean regarding drug (or exposure) potency?
51. Define ED50, LD50, and therapeutic index. Why is a drug with a higher therapeutic index safer?
52. Describe the interaction of hypnotics and opioids, specifically with regard to fentanyl and isoflurane MAC.
53. What is a response surface?
54. Define additive drug interactions, supraadditive interactions, and infraadditive interactions.
Answers
1. Pharmacokinetics is the process by which the body “disposes” of drugs via absorption, distribution, metabolism, and elimination. It can be thought of as “what the body does to the drug.” (35)
2. Pharmacodynamics is the process by which drugs interact with specific receptors in the body to produce pharmacologic effects. It can be thought of as “what the drug does to the body.” (35)
Pharmacokinetics
3. Drug absorption is governed by route of delivery, bioavailability of the drug, and possibly first-pass metabolism. (36)
4. Drug absorption via buccal mucosa differs from absorption from lower in the gastrointestinal tract because presence of food does not hinder delivery of drug to the mucosa. Also, venous outflow from the buccal mucosa returns directly to the systemic circulation, thereby avoiding the potential for the first-pass hepatic effect that is present for drugs absorbed in the stomach, which first enter the portal venous system. (36)
5. Transdermal drug delivery is distinct in that skin is designed to be a significant barrier to absorption. This means that drugs delivered transdermally will have a markedly delayed onset of action following administration. Also, the skin serves as a depot of the drug, resulting in prolonged drug effect following removal of the skin application. Examples of drugs delivered transdermally include clonidine, scopolamine, nitroglycerin, and fentanyl. (36)
6. Local anesthetics applied in nerve blocks have their pharmacologic effects ended by movement of the drug away from the site of action. The process by which the body absorbs this locally applied bolus of drug, thus ending its local effects, is the same by which the body absorbs drugs injected into tissues for the purpose of eliciting systemic drug effects that follow absorption. (36)
7. “First order” transfer is when the rate of drug absorption is proportional to the concentration gradient. Doubling the dose of a drug does not affect the shape of the curve (absorption over time) when a “first order” transfer is occurring. Concentrations will be exactly twice as high at all times, but peak absorption will occur at the same time and the shape of the curve will be identical. (36)
[/not-level-membership-for-anesthesiology-category]
