[level-membership-for-pediatrics-category]
Chapter 525 Bartter and Gitelman Syndromes and Other Inherited Tubular Transport Abnormalities
525.1 Bartter Syndrome
Rajasree Sreedharan and Ellis D. Avner
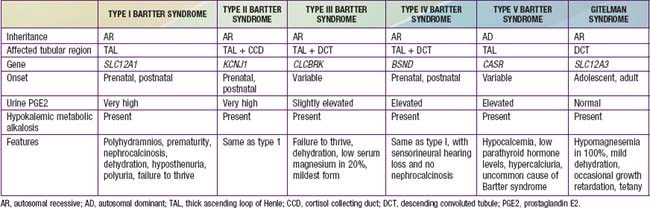
Bartter syndrome is a group of disorders characterized by hypokalemic metabolic alkalosis with hypercalciuria and salt wasting (Chapter 52) (Table 525-1). Antenatal Bartter syndrome (types I, II, IV) (also called hyperprostaglandin E syndrome) typically manifests in infancy and has a more-severe phenotype than classic Bartter syndrome (type III), including maternal polyhydramnios, neonatal salt wasting, and severe episodes of recurrent dehydration. The milder phenotype, classic Bartter syndrome, manifests in childhood with failure to thrive and a history of recurrent episodes of dehydration. A phenotypically related disease, Gitelman syndrome has a distinct genetic defect and is discussed in Chapter 525.2 (see Table 525-1). One distinct variant of antenatal Bartter syndrome is associated with sensorineural deafness (type IV).
Pathogenesis
The biochemical features of Bartter syndrome, including hypokalemic metabolic alkalosis with hypercalciuria, resemble those seen with chronic use of loop diuretics and reflect a defect in sodium, chloride, and potassium transport in the ascending loop of Henle. The loss of sodium and chloride, with resultant volume contraction, stimulates the renin-angiotensin II-aldosterone (RAA) axis. Aldosterone promotes sodium uptake and potassium secretion, exacerbating the hypokalemia. It also stimulates hydrogen ion secretion distally, worsening the metabolic alkalosis. Hypokalemia stimulates prostaglandin synthesis, which further activates the RAA axis. Bartter syndrome has been associated with 5 distinct genetic defects in loop of Henle transporters (see Table 525-1). Each contributes, in some manner, to sodium and chloride transport. Mutations in the genes that encode the Na+/K+/2Cl− transporter (NKCC2, the site of action of furosemide), the luminal potassium channel (ROMK), combined chloride channel (CLC-Ka, CLC-Kb), or subunit of chloride channels (barttin) cause neonatal Bartter syndrome. Isolated defects in the genes that produce the basolateral chloride channel ClC-Kb cause classic Bartter syndrome.
525.2 Gitelman Syndrome
Rajasree Sreedharan and Ellis D. Avner
Gitelman syndrome (often called a “Bartter syndrome variant”) is a rare autosomal recessive cause of hypokalemic metabolic alkalosis, with distinct features of hypocalciuria and hypomagnesemia. Patients with Gitelman syndrome typically present in late childhood or early adulthood (see Table 525-1).
525.3 Other Inherited Tubular Transport Abnormalities
Rajasree Sreedharan and Ellis D. Avner
Inherited abnormalities in distinct transporters in each segment of the nephron have now been identified and the molecular defects have been characterized. Renal tubular acidosis and nephrogenic diabetes insipidus are discussed in detail in Chapters 523 and 524, respectively. Cystinuria is an autosomal recessive disorder seen primarily in patients of Middle Eastern descent and is characterized by recurrent stone formation. The disease is caused by a defective high-affinity transporter for L-cystine and dibasic amino acids present in the proximal tubule.
Bökenkamp A, Böckenhauer D, Cheonh HI, et al. Dent-2 disease: a mild variant of Lowe syndrome. J Pediatr. 2009;155:94-99.
Chadha V, Alon US. Hereditary renal tubular disorders. Sem Nephrol. 2009;29:399-411.
Cho HY, Lee BH, Choi HJ, et al. Renal manifestations of Dent disease and Lowe syndrome. Pediatr Nephrol. 2008;23:243-249.
Devuyst O, Konrad M, Jeunemaitre X, et al. Tubular disorders of electrolyte regulation. In: Avner ED, Harmon WE, Niaudet P, et al, editors. Pediatric nephrology. ed 6. Heidelberg, Germany: Springer-Verlag; 2009:929-978.
Garnier A, Dreux S, Vargas-Poussou R, et al. Bartter syndrome prenatal diagnosis based on amniotic fluid biochemical analysis. Pediatr Res. 2010;67(3):300-303.
Kleta R, Bockenhauer D. Bartter syndrome and other salt-losing tubulopathies. Nephron Physiol. 2006;104:73-80.
Seyberth HW. An improved terminology and classification of Bartter-like syndromes. Nat Clin Pract Nephrol. 2008;4:560-570.
Sung CC, Chen YS, Lin SH. Family paralysis. Lancet. 2011;377:352.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 525 Bartter and Gitelman Syndromes and Other Inherited Tubular Transport Abnormalities
525.1 Bartter Syndrome
Rajasree Sreedharan and Ellis D. Avner
Bartter syndrome is a group of disorders characterized by hypokalemic metabolic alkalosis with hypercalciuria and salt wasting (Chapter 52) (Table 525-1). Antenatal Bartter syndrome (types I, II, IV) (also called hyperprostaglandin E syndrome) typically manifests in infancy and has a more-severe phenotype than classic Bartter syndrome (type III), including maternal polyhydramnios, neonatal salt wasting, and severe episodes of recurrent dehydration. The milder phenotype, classic Bartter syndrome, manifests in childhood with failure to thrive and a history of recurrent episodes of dehydration. A phenotypically related disease, Gitelman syndrome has a distinct genetic defect and is discussed in Chapter 525.2 (see Table 525-1). One distinct variant of antenatal Bartter syndrome is associated with sensorineural deafness (type IV).
Pathogenesis
The biochemical features of Bartter syndrome, including hypokalemic metabolic alkalosis with hypercalciuria, resemble those seen with chronic use of loop diuretics and reflect a defect in sodium, chloride, and potassium transport in the ascending loop of Henle. The loss of sodium and chloride, with resultant volume contraction, stimulates the renin-angiotensin II-aldosterone (RAA) axis. Aldosterone promotes sodium uptake and potassium secretion, exacerbating the hypokalemia. It also stimulates hydrogen ion secretion distally, worsening the metabolic alkalosis. Hypokalemia stimulates prostaglandin synthesis, which further activates the RAA axis. Bartter syndrome has been associated with 5 distinct genetic defects in loop of Henle transporters (see Table 525-1). Each contributes, in some manner, to sodium and chloride transport. Mutations in the genes that encode the Na+/K+/2Cl− transporter (NKCC2, the site of action of furosemide), the luminal potassium channel (ROMK), combined chloride channel (CLC-Ka, CLC-Kb), or subunit of chloride channels (barttin) cause neonatal Bartter syndrome. Isolated defects in the genes that produce the basolateral chloride channel ClC-Kb cause classic Bartter syndrome.
