CHAPTER 105 Barriers to Delivery of Therapeutics to Brain Tumors
Malignant gliomas are the most common type of primary brain tumor in adults. According to the Central Brain Tumor Registry of the United States, it is estimated that approximately 20,000 new cases develop each year.1 Treatment of malignant glioma represents one of the most formidable challenges in oncology. Despite treatment with surgery, radiation therapy, and chemotherapy, the prognosis remains poor, particularly for patients with glioblastoma multiforme (GBM), who have a median survival of 12 to 15 months.2 After recurrence, rapid tumor progression results in a median progression-free survival and overall survival of 9 and 25 weeks, respectively. The invasiveness of these tumors eliminates the possibility of curative surgical resection. Even at initial evaluation, infiltration by tumor cells extends at least 2 cm away from the radiographic contrast–enhancing mass.3,4 One of the main difficulties associated with treating malignant gliomas is the presence of the blood-brain barrier (BBB) and the partially functional blood-tumor barrier, which serves to prevent effective delivery of potentially active chemotherapeutic compounds. There is vast interest in and consequently multiple research efforts focused on the design of new approaches that will improve drug delivery to brain tumor cells with limited systemic toxicity. This chapter summarizes the methods that have been developed for overcoming the ability of brain tumor cells to remain safe from therapy behind multiple physical and physiologic barriers.
Blood-Brain Barrier
The BBB consists of a layer of endothelial cells that line the blood vasculature throughout the brain.5 These endothelial cells are held together by tight junctions produced in response to signals from astrocytes. Certain molecules, particularly large water-soluble molecules, are prohibited from diffusing through the gaps between these cells. Small, lipophilic molecules can, however, diffuse across the membranes, but they too are limited in distribution by diffusion. The BBB limits central nervous system (CNS) delivery of many common chemotherapeutic agents, and its permeability to most molecules can be predicted on the basis of each agent’s octanol-water partition coefficient and the unidirectional transfer coefficient (Kin).6–8 The octanol-water distribution coefficient measures solute lipophilicity, whereas Kin is a quantitative measure of the ability of a drug to pass from plasma into the brain. Kin is largely determined by lipid solubility because agents must first dissolve in the lipid membranes of the BBB to cross by lipid-mediated diffusion.9 Kin can be plotted against the partition coefficient by using 20 reference permeability markers that bind minimally to plasma proteins and cross the BBB by passive diffusion (Fig. 105-1).10 Many of the chemotherapeutic agents fall below the line predicted for BBB passive diffusion.
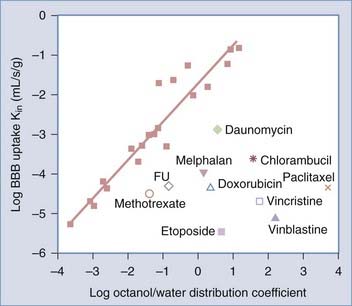
Active or facilitated transport is used for substances with low partition coefficients. This type of transport is dependent on ion channels, specific transporters, energy-dependent pumps, and receptor-mediated endocytosis. Glucose, amino acids, and small intermediate metabolites are carried into the brain via facilitated transport, whereas larger molecules such as insulin and transferrin are carried across the endothelial layer via receptor-mediated endocytosis.11
Certain factors contribute to poor uptake of chemotherapeutic agents across the BBB, including plasma protein binding, solute molecular weight, and active efflux transport. Most chemotherapeutic agents are bound to plasma proteins, which reduces the free fraction of the drug in plasma that is available to cross the BBB. The molecular weight of most of these agents exceeds 400 daltons, significantly larger than what would normally passively leak through the BBB.12,13 Another component of the BBB that makes it difficult for chemotherapeutic agents to function is its expression of high levels of drug efflux pumps, such as P-glycoprotein.14 These pumps actively remove chemotherapeutic drugs from the brain. A number of newer chemotherapeutic agents, such as gemcitabine, docetaxel, pemetrexed, irinotecan, and topotecan, which show promising antitumor activity against systemic tumors, have limited delivery across the BBB because of active efflux transport and plasma protein binding.15 The thymidine kinase inhibitor imatinib binds heavily to plasma proteins and is a substrate for active efflux pumps, and this is also suspected to be the case for erlotinib and gefinitib.16 The organic anion transporters and glutathione-dependent multidrug resistance–associated proteins also contribute to the efflux of organic anions from the brain and cerebrospinal fluid (CSF).17 There has been research focusing on blocking these types of transporters to improve therapeutic drug delivery across the BBB.
Blood-Tumor Barrier
Although the endothelial tight junction component of the BBB can be compromised significantly in the enhancing part of a glioma and hence result in the hallmark of contrast enhancement on computed tomography or magnetic resonance imaging, other remaining physiologic barriers and new physical barriers can restrict delivery of drug to brain tumors. When compared with the normal, ordered vasculature of healthy tissues, blood vessels in tumors are often highly abnormal, such as distended capillaries with leaky walls and sluggish flow, and thereby lead to inconsistent drug delivery. “Leakage” from the tumor vasculature results in accumulation of interstitial fluid that subsequently increases intratumoral interstitial pressure, thus limiting the diffusion of drugs into brain tumors.18 It has been shown that tumors can consist of areas of normal interstitial pressure, in which the pressure is 1 to 2 mm Hg, interposed with peritumoral areas in which interstitial fluid pressure can be 50 mm Hg or greater.19 Recent studies have shown that targeting vascular endothelial growth factor with the drug bevacizumab may decrease interstitial pressure to allow greater entry of drug into the tumor.20 Highly irregular blood flow results in localized hypoxia, which subsequently leads to resistance of tumor to certain anticancer agents and to radiotherapy as well. Increased bioreductive enzyme expression is an adaptive strategy for solid tumors to detoxify anticancer drugs, and the hypoxic environment further contributes to the increased bioreductive activity of tumors.21
Blood–Cerebrospinal Fluid Barrier
The specialized tight junctions of epithelial cells in the choroid plexus (the blood-CSF barrier) that separate CSF from blood are also responsible for barrier function.22 The tight junctions of the blood-CSF barrier, in comparison to the BBB, are between the epithelial cells of the choroid plexus rather than at the endothelial layer. By sealing neighboring epithelial choroidal cells continuously together, the tight junctions strongly restrict the paracellular movement of solutes and thus considerably limit the diffusion of polar drugs into the CNS via the choroid plexus. The fenestrated choroid plexus capillaries provide little resistance to the movement of small molecules. However, the blood-CSF barrier does not contribute significantly to entry of drug into the brain parenchyma because the surface area of the blood-CSF barrier is approximately 1000-fold less than the surface area of the BBB.23 Similar to the BBB, however, the blood-CSF barrier also contains drug-metabolizing enzymes located within the choroid plexus and in some circumventricular organs, and these enzymes primarily serve a protective role against exogenously administered molecules.24
Drug Modifications for Enhanced Drug Delivery to Brain Tumors
Lipophilic Analogues
As stated previously, the BBB allows the diffusion of small, nonionic, lipid-soluble molecules, whereas larger, more water-soluble ionic molecules do not readily cross (Table 105-1). There are endogenous transporters that allow some large and polar molecules to be transported into the brain. To improve drug delivery to brain tumors, certain strategies have been formulated that involve passive drug uptake into brain by means of lipophilic analogues. Carmustine is an alkylating agent used to treat brain tumors and other well-known malignancies. Multiple carmustine analogues have been studied in clinical trials and have demonstrated decreased alkylating activity and increased dose-limiting toxicity when compared with carmustine.25 This is probably due to factors affecting drug-receptor interactions or increased binding to plasma proteins resulting in lower drug concentrations available for diffusion into the brain. Moreover, lipophilic analogues are less soluble in brain interstitial fluid, which limits their activity against tumor cells.26
| ADVANTAGES | DISADVANTAGES | |
|---|---|---|
| Drug Modifications | ||
| Lipophilic analogues | Allows passage of large, polar molecules | Limited activity against tumor cells |
| Lipophilic prodrugs | Better penetration than with parent drug | Require chemical transformation to achieve active form |
| ADEPT/GDEPT | ||
ADEPT, antibody-directed enzyme prodrug therapy; BBB, blood-brain barrier; BBBD, blood-brain barrier disruption; CED, convection-enhanced delivery; GDEPT, gene-directed enzyme prodrug therapy.
Lipophilic Prodrugs
As an alternative to analogues, prodrugs require chemical or biochemical transformation to achieve the active form within the body.27,28 Prodrugs are designed to overcome the pharmaceutical or pharmokinetic limitations, or both, of the parent molecule to better penetrate the BBB. Lipophilic ester prodrugs of the anticancer agent chlorambucil have been developed to increase efficacy in the treatment of brain tumors.29 After equimolar doses of chlorambucil and chlorambucil–tertiary butyl ester, brain delivery of the ester was 35-fold greater than that of chlorambucil. However, despite an enhanced CNS delivery ratio, none of the prodrugs demonstrated anticancer activity superior to the equimolar administration of chlorambucil against a brain-sequestered carcinosarcoma in the rat.30
Antibody- and Gene-Directed Enzyme Prodrug Therapy
Further strategies have been used to enhance prodrug activity at the site of action with minimal effect on the rest of the body. If the prodrug can be selectively cleaved to the active drug by specific enzymes near the site of action, function would be enhanced. Prodrug-activating enzymes were initially targeted to tumor cells by using antibodies and more specifically genes. In antibody-directed enzyme prodrug therapy, enzymes that activate prodrugs are directed to human tumor xenografts by conjugating them to tumor-selective monoclonal antibodies.23 Once the enzyme is conjugated with an antitumor antibody, it is delivered to the tumor by intravenous (IV) infusion. When the conjugate is cleared from blood, a prodrug is then delivered. The prodrug is now able to be activated by the tumor-associated enzyme. Martin and associates were able to use this strategy in phase I clinical trial in patients with nonresectable metastatic colon carcinoma.31 Unfortunately, there have been limitations to this strategy in that some tumor-specific antigens inactivate the antibody-enzyme complexes delivered. These limitations are what led to the development of gene-directed enzyme prodrug therapy (GDEPT).
GDEPT was the first type of gene therapy used for treating human brain tumors. In GDEPT, an inactive prodrug can be activated to release a cytotoxic drug by an enzyme that has been delivered to the tumor for expression. Specific enzymes used include thymidine kinase (TK), nitroreductase, and cytochrome P-450. The most studied approach uses the TK gene inserted into herpes simplex virus, followed by combination treatment with the prodrug ganciclovir. In one trial using this strategy in patients with malignant glioma, 12 of 15 patients showed moderate response to the treatment, but no complete responses were observed.32 As with other approaches to gene therapy, this method is limited by the difficulty of achieving selective gene delivery to a sufficient number of tumor cells.
Chemical Drug Delivery System and Carrier-Mediated Drug Transport
By using specific properties of the BBB, this system locks drugs in the brain on arrival and prevents them from recrossing the BBB.33 Linking an active drug molecule to a bioremovable lipophilic targeting moiety (targeter) creates a complex that can then be oxidized to form a charge that has the effect of capturing the ionized drug–targeter inside the brain. Subsequently, slow release of the drug from the targeter results in sustained and brain-specific release of the free active drug. This system has been studied consistently with the drug lomustine.34
The carrier-mediated drug delivery approach uses the facilitative endogenous transport systems that are present in brain endothelial cells. Specific transport systems that exist for the brain include glucose, amino acids, choline, vitamins, low-density lipoprotein, and nucleosides.35–40 Glucose and the large neutral amino acids have high transport capacity, which makes them the most used agents for delivery of drug to the brain.41 Several amino acid mimetic agents such as L-dopa, α-methyldopa, and baclofen are drugs in clinical use that are readily taken up into the brain by these transport systems.
Receptor/Vector-Mediated Drug Targeting
This approach makes use of the endogenous BBB transport system by aiming at improving brain uptake by coupling nontransportable therapeutic molecules to a drug transport vector.23 These vectors may include endogenous peptides such as insulin or transferrin, modified proteins, or antireceptor-specific monoclonal antibodies. A significant increase in radioactivity in rat brain tumor versus normal brain was achieved after IV administration of the radiolabeled epidermal growth factor peptide conjugated to the antitransferrin monoclonal antibody transport vector.42
Barrier Disruption Strategies for Enhancing Drug Delivery
Although advances in chemical modification of drugs to make them lipophilic and the use of carriers to circumvent the BBB have been explored, transient BBB disruption (BBBD) became a favorable option in the recent past. Techniques that transiently disrupt the BBB were investigated, including techniques that create a paracellular route of transport through the endothelium by opening tight junctions and those that create a transcellular pathway through the endothelium. Several endogenous compounds, including neurotransmitters, hormones, and inflammatory mediators, can readily open the tight junctions of the BBB.43 BBBD has also been seen in physiologic states such as hypertension, hypoxia, and ischemia. Hypertonic substances, including mannitol and biologically active agents such as bradykinin and angiotensin peptides, have also been shown to disrupt the BBB.
Osmotic Blood-Brain Barrier Disruption
Transient osmotic disruption of the BBB and blood-CSF and blood-tumor barriers can be achieved within a localized vascular distribution by intra-arterial (IA) infusion of a hyperosmotic agent, usually mannitol.44 By injecting hypertonic solution, rapid diffusion of fluid out of the cells takes place and causes shrinkage of endothelial cells and subsequent opening of the tight junctions of the BBB for several hours (Fig. 105-2).45 This infusion of hypertonic solution is usually followed by the IA administration of chemotherapy. Animal studies suggest that this method is able to increase concentrations of chemotherapeutic agents in the brain parenchyma up to 90-fold.46 Delivery of methotrexate to the CNS is enhanced 4- to 7-fold when administered IA after osmotic BBBD as compared with IA administration without BBBD.47 The ability of BBBD to improve the therapeutic efficacy of chemotherapy has been variable in humans. In chemoresponsive tumors, BBBD-associated chemotherapy compared favorably with conventional chemotherapy.48–50 In primary CNS lymphoma phase II studies, a significant difference in survival was found when comparing patients treated with BBBD chemotherapy with or without prior whole-brain radiotherapy.51 A concern with the use of BBBD is the potential for neurotoxicity from the high concentrations of chemotherapy delivered to the normal brain. Chemotherapeutic agents such as doxorubicin, cisplatin, and taxanes cause neurotoxicity with BBBD, even though they are well tolerated systemically.52 The efficacy of BBBD-mediated chemotherapy for malignant gliomas is still being explored.
Biochemical Disruption of the Blood-Brain Barrier
This technique for BBBD is less invasive than the aforementioned, and it may be more reliable because it gives a more precise time window for delivery of drug to the brain. Mediators of the inflammatory response, including leukotrienes, histamine, and vasoactive peptides, have been found to cause transient vascular leakage and increased permeability of blood vessels.53 Bradykinin, a peripheral vasodilator, increases tight junction permeability by activating the B2 receptors of endothelial cells.54 Previous studies have shown that the bradykinin agonist RMP-7 can safely permeabilize the BBB. Animal models were used to compare the effects of RMP-7 within tumor, brain tissue proximal to tumor, and brain tissue distal from tumor. In one study, the effect of RMP-7 on enhancing BBB permeation was evaluated for three compounds with different physical characteristics: carboplatin, dextran, and carmustine. RMP-7 increased permeability of the vascular barriers to the hydrophilic compounds carboplatin and dextran. Greater effect was seen on the blood-brain-tumor barrier than on the nontumor BBB. No effect of RMP-7 was seen in permeabilizing the BBB for the lipophilic drug carmustine. In a multinational clinical trial, IA RMP-7 was evaluated in combination with the anticancer agent carboplatin for the treatment of malignant brain tumors.55 Because of high levels of toxicity, however, these clinical trials were discontinued.
Ultrasound-Mediated Blood-Brain Barrier Disruption
BBBD over a large volume, as seen with osmotic and biochemical disruption, may prove a disadvantage for many applications because the agents may have undesired side effects as a result of their spread within the CNS. To prevent this problem, localized and reversible image-guided BBBD would provide anatomically or functionally targeted drug delivery while preserving an intact BBB to protect the nontargeted regions. A new approach to focal CNS delivery is BBBD induced by magnetic resonance image–guided focused ultrasound.56 Consistent vascular leakage without tissue damage was achieved by localizing cavitation-generated mechanical stress to blood vessel walls by IV injection of preformed gas bubbles just before pulsed ultrasound treatment.57 Ultrasound caused reversible focal opening of the BBB that healed completely within 24 hours. A rat brain study demonstrated that the locations in the brain that were exposed to ultrasound showed significantly higher concentrations of liposomal doxorubicin and that clinically relevant levels were reached.58 Because of attenuation of ultrasound by bone, a bone window is created with a craniotomy. Recent studies have shown, however, that focal, trans-skull ultrasound exposure of brain tissue may be accomplished by using a large–surface-area phased array.59
Methods for Delivery of Drugs Directly to the Brain Parenchyma
Implanted Polymers
Implanted, biodegradable polymers have been used for the treatment of brain tumors in a number of clinical trials. The underlying concept is to provide continuous drug delivery to the tumor with a drug-impregnated wafer that has a controlled sustained-release rate. Brem and colleagues studied the treatment of gliomas with carmustine-loaded biodegradable polymers as a drug delivery wafer. This matrix releases the drug by a combination of diffusion and hydrolytic polymer degradation. In their initial randomized clinical trial they were able to show an increase in mean survival time for patients with recurrent glioma.60 This result led to the first Food and Drug Administration (FDA)-approved polymer wafer, Gliadel. A subsequent randomized trial in patients with newly diagnosed malignant glioma led to an expanded indication from the FDA to include patients with either newly diagnosed or recurrent glioma who are undergoing gross total tumor resection.61 These studies showed evidence of efficacy, and thus this drug was approved by the FDA for use. There have, however, been some criticisms related to the modest efficacy and association with toxicity. Additional small, early-stage clinical trials using polymers impregnated with chemotherapeutic agents such as paclitaxel, doxorubicin, 5-fluorouracil, mitomycin, nimustine hydrochloride, and mitoxantrone have also been conducted.62–66 None of these combinations have been developed commercially at this time. Disadvantages of these local delivery strategies include an increased risk for local neurotoxicity with higher polymer/drug concentrations, poor wound healing, and limited drug penetration beyond the resection cavity, which fails to provide therapeutic drug concentrations to tumor cells that are just centimeters away.
Convection-Enhanced Delivery
A more recently described approach for achieving both local and regional drug delivery was proposed by Bobo and coworkers in 1994.67 CED involves the continuous injection under positive pressure of a solute containing a therapeutic agent. CED relies on pressure-driven bulk flow of infusate as a means to deliver therapeutic agents to the CNS. The bulk flow mechanism is created by a small pressure gradient from a pump that pushes solute through a catheter targeted within the CNS. There are several advantages of CED over traditional delivery methods: (1) CED bypasses the BBB and can be used to infuse therapeutic agents with large or small molecular weights and (2) it provides targeted delivery into the region or structure into which the catheter is placed. This limits the potential for neurotoxicity because the therapeutic agent can be delivered to an entire targeted region while minimizing systemic toxicity or widespread neurotoxicity. Histologic studies have shown that inflammation adjacent to the catheter tract and at the catheter tip is typically limited to a 50-µm radius.68 Furthermore, CED within the defined infusion parameters does not produce cerebral edema or measurable increases in intracranial pressure.69
In CED, one or more catheters are stereotactically implanted through a bur hole into or adjacent to either the enhancing portion of a tumor or the nonenhancing infiltrative surrounding tissue, or into both. Pressure-driven flow of drug is achieved via an infusion pump, and the agent is directly infused into the target tissue at a predetermined concentration, rate, and duration. The volume of distribution in brain tumors achieved with CED differs markedly from that in normal brain. This difference may be due to a number of factors, including increased interstitial fluid pressure within and adjacent to brain tumors as compared with normal brain and an altered BBB. The increased interstitial fluid pressure observed in brain tumors creates a pressure gradient that can potentially drive infusate out of high-pressure areas within the tumor into relatively low-pressure areas in surrounding normal tissue. This pressure gradient and the marked heterogeneity of drug distribution within the tumor itself are potential limiting factors in drug delivery by this method.70
The use of CED for a variety of agents to treat malignant gliomas has been widely investigated in preclinical models and clinical trials. Hadaczek and colleagues examined the distribution of adeno-associated virus type 2–thymidine kinase (AAV2-TK) and AAV2–aromatic L-amino acid decarboxylase (AAV2-AADC) in monkey brains after CED and demonstrated that this is an efficient method for delivery of the AAV2 vector.71 The safety and efficacy of intratumoral CED of paclitaxel in patients harboring recurrent GBMs was examined in a phase I/II clinical study.72 Of the 15 patients who received 20 cycles of intratumoral CED of paclitaxel, 5 complete responses and 6 partial responses were observed, for a response rate of 73%. CED of paclitaxel was found to have a high antitumor response rate, although it was also associated with a significant incidence of treatment-associated complications.72 Initial studies of chronic topotecan infusion, a topoisomerase I inhibitor, into rats bearing gliomas showed a more than fivefold increase in survival time without significant neurological toxicity.73
Another class of agents that has been investigated for use in CED is the so-called targeted immunotoxins. Protein toxins produced by bacteria represent novel and potent cytotoxic agents that may be coupled to specific carrier-ligands used for cellular targeting. The carrier-ligand provides tumor-selective properties by recognition of a cell surface receptor on the tumor and promotes binding of the toxin-carrier complex before entry into the cell. These toxins tend to be far more potent than traditional chemotherapeutic agents, and direct delivery into the brain is probably the only means by which they can be administered safely. A phase I clinical trial using a transferrin conjugated to a diphtheria toxin and delivered via CED showed good tumor response in malignant brain tumors without significant neurotoxicity.74 A prospective, randomized phase III study was opened to study the efficacy of this toxin, delivered via CED into unresectable tumors, versus best medical therapy for the treatment of recurrent GBM. Unfortunately, the study was halted before completion of its accrual because of an intermediate futility analysis indicating a less than 20% chance of a positive outcome for the study. Clinical studies of CED using interleukin 4 (IL-4)- and IL-13–linked cytotoxins have also showed promising results in early-stage clinical trials,75,76 but a phase III trial of an IL-13–linked cytotoxin delivered via CED failed to meet its goal of improving survival more than 50% over carmustine (Gliadel).77
CED of therapeutic agents remains a promising strategy for treating malignant gliomas. Although initial clinical trials have failed to show survival benefit for new agents delivered by this approach, multiple earlier stage trials have addressed only a fraction of the myriad technical and technologic issues that surround this novel approach. The development of CED has been limited by the fact that both new technologies and novel therapeutic agents are being developed simultaneously. New trials are being planned to investigate agents that can be co-infused with radiographic tracers (Fig. 105-3), as well as novel catheters that avoid problems with backflow and will potentially provide more reliable drug distribution.
Other Approaches for Enhancing Delivery of Drugs to the Brain
Intraventricular/Intrathecal Drug Delivery
Using the CSF space for delivery of chemotherapeutic agents has been a valuable method of treatment for patients with CNS malignancies. However, there is little bulk flow between the intercellular space of the brain and the CSF space. Hence, little drug is able to diffuse from the CSF spaces into the parenchyma, which has prohibited this delivery system from being an optimal one for the treatment of intraparenchymal brain tumors.78 Intrathecal delivery is hence a technique that is typically reserved for leptomeningeal spread of malignant cells, as is often seen in primary CNS lymphoma and various types of metastatic carcinomas.79
Intra-arterial Therapy
Delivery of a higher concentration of drug increases transition across the BBB. One study showed that IA infusion delivered a greater amount of chloroethylnitrosourea into the brain tumor than the IV route did.80,81 For the drug etoposide, its concentration was found to be four times higher after IA than IV administration.82 The route of delivery had an impact on brain delivery of cisplatin, with IA administration increasing delivery to glioma twofold in comparison to IV administration.83 None of the studies, however, have shown improved survival with IA versus IV delivery. This delivery technique has also been limited by the high degree of drug toxicity and lack of treatment of the contralateral hemisphere.
Liposomal Drug Encapsulation
The use of liposomes for targeted drug delivery is somewhat controversial because liposomes do not specifically target tumor cells. Furthermore, systemically administered liposomes are selectively removed from the circulation by phagocytic cells of the reticuloendothelial system, which makes liposomes good delivery systems for tumors of the liver and spleen. To make them better targeting agents for CNS tumors, their surface can be modified by the attachment of antibodies to tumor-associated antigens (immunoliposomes). In one study, 80-nm liposomes were pegylated on the surface with a specific polymer to allow successful brain delivery.84 Subsequent brain delivery of immunoliposomes containing 3H-daunomycin was successful. These liposomes can carry up to 10,000 drug molecules, thus making this immunoliposome delivery system highly efficient for delivery of drug to the brain. Future clinical investigation is focused on optimizing the delivery of these agents by both the systemic circulation and CED.
Nanoparticulate Systems
Because of the instability of liposomes, polymeric nanoparticles may be a better vehicle for controlled drug delivery methods for tumor treatment. They are made of natural or artificial polymers ranging in size between 10 and 1000 nm85 and are capable of being injected intravenously. These nanoparticles are coated with polysorbates to enhance the absorption of apolipoprotein E from blood plasma onto their surface. This allows them to interact with the low-density lipoprotein receptor and subsequently be taken up by endothelial cells so that they can be released within the cells and ultimately diffuse into the brain parenchyma. One study in rats using intravenously injected doxorubicin-loaded polysorbate 80–coated nanoparticles led to a 40% cure rate in rats bearing intracranial glioblastomas.86
Intranasal Applications
Intranasal delivery provides a practical, noninvasive method for delivering therapeutic agents to the brain because of the unique anatomic connection provided by the olfactory and trigeminal nerves.87 Intranasally administered drugs reach the parenchymal tissues of the brain and spinal cord or the CSF, or both, within minutes by using an extracellular route through perineural channels.88 In addition to bypassing the BBB, advantages of intranasal delivery include rapid delivery to the CNS, avoidance of hepatic first-pass drug metabolism, and elimination of the need for systemic delivery, thereby reducing systemic side effects. A recent preclinical study in rats used the telomerase inhibitor GRN163 and demonstrated uptake by intracerebral tumors and inhibition of tumor growth.89 This study was able to show a significant increased median survival time of the treated group. Further investigation of this approach is ongoing.
Magnetic Microspheres
A novel approach involving the use of magnetic particles potentially offers advantages over conventional drug administrations in terms of the ability to localize at the tumor and limiting systemic drug distribution. Magnetic anticancer agents encapsulated within a polymeric matrix are used to target tumors by retention of cationic particles within the tumor through the application of an external magnetic field.90 Bioadhesive cationic microspheres have been shown to increase particle retention at the brain-tumor barrier.91 Recently, small uncharged magnetic particles were evaluated for their ability to target intracerebral rat glioma-2 tumors in vivo.92 These studies demonstrated a significant increase in particle levels in brain tumor over particle levels in normal brain tissue.
Boron Neutron Capture Therapy
BNCT has been investigated in clinical trials since the 1950s.93 The rationale of this therapy is based on the nuclear reaction that occurs when boron 10 (10B) is irradiated with low-energy thermal neutrons to yield high–linear energy transfer alpha particles and recoiling lithium 7 nuclei.94 The high-energy, short-range alpha particles and 7Li are capable of destroying tumor cells within 10 µm of the site of the capture reaction. However, for BNCT to be successful, a sufficient amount of 10B must be selectively delivered to the tumor. 10B-enriched borocaptate sodium (BSH) is one of the most widely used boron compounds for BNCT because of its low systemic toxicity and selective localization in tumors.95 In small, early-phase clinical trials, BSH was used to treat grade III and grade IV gliomas and achieved mean survival rates significantly greater than those obtained with conventional radiation therapy.96 A number of other delivery agents for BNCT are currently under investigations for the treatment of brain tumors.
Abbott NJ, Romero IA. Transporting therapeutics across the blood-brain barrier. Mol Med Today. 1996;2(3):106-113.
Ali MJ, Navlitloha Y, Vavra MW, et al. Isolation of drug delivery from drug effect: problems of optimizing drug delivery parameters. Neuro Oncol. 2006;8:109-118.
Banks WA. Physiology and pathology of the blood-brain barrier: implication for microbial pathogenesis, drug delivery and neurodegenerative disorders. J Neurovirol. 1999;5:538-555.
Barth RF, Coderre JA, Vicente GH, et al. Boron neutron capture therapy of cancer: current status and future prospects. Clin Cancer Res. 2005;11:3987-4002.
Bartus RT, Elliott PJ, Dean RL, et al. Controlled modulation of BBB permeability using the bradykinin agonist, RMP-7. Exp Neurol. 1996;142:14-28.
Bobo RH, Laske DW, Akbasak A, et al. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci U S A. 1994;91:2076-2080.
Brem H, Mahaley MSJr, Vick NA, et al. Interstitial chemotherapy with drug polymer implants for the treatment of recurrent gliomas. J Neurosurg. 1991;74:441-446.
Chen MY, Lonser RR, Morrison PF, et al. Variables affecting convection-enhanced delivery to the striatum: a systematic examination of rate of infusion, cannula size, infusate concentration, and tissue-cannula sealing time. J Neurosurg. 1999;90:315-320.
Cloughesy TF, Black KL. Pharmacological blood-brain barrier modification for selective drug delivery. J Neurooncol. 1995;26:125-132.
Hashizume R, Ozawa T, Gryaznov SM, et al. New therapeutic approach for brain tumors: intranasal delivery of telomerase inhibitor GRN163. Neuro Oncol. 2008;10:112-120.
Huwyler J, Wu D, Pardridge WM. Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acad Sci U S A. 1996;93:14164-14169.
Hynynen K, Clement GT, McDonald N, et al. 500-Element ultrasound phased array system for noninvasive focal surgery of the brain: a preliminary rabbit study with ex vivo human skulls. Magn Reson Med. 2004;52:100-107.
Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1994;271:58-65.
Kurihara A, Pardridge WM. Imaging brain tumors by targeting peptide radiopharmaceuticals through the blood-brain barrier. Cancer Res. 1999;59:6159-6163.
Leggas M, Adachi A, Scheffer GL, et al. MrP4 confers resistance to topotecan and protects the brain from chemotherapy. Mol Cell Biol. 2004;24:7612-7621.
Muldoon LL, Soussain C, Jahnke K, et al. Chemotherapy delivery issues in central nervous system malignancy: a reality check. J Clin Oncol. 2007;25:2295-2305.
Neuwelt EA. Mechanisms of disease: the blood-brain barrier. Neurosurgery. 2004;54:131-142.
Pardridge WM. Recent advances in blood-brain barrier transport. Annu Rev Pharmacol Toxicol. 1988;28:25-39.
Rautio J, Chikhale PJ. Drug delivery systems for brain tumor therapy. Curr Pharm Des. 2004;10:1341-1353.
Sawchuk RJ, Elmquist WF. Microdialysis in the study of drug transporters in the CNS. Adv Drug Deliv Rev. 2000;45:295-307.
Smith QR. A review of blood-brain barrier transport techniques. Methods Mol Med. 2003;89:193-208.
Vogelbaum MA. Convection enhanced delivery for treating brain tumors and selected neurological disorders: symposium review. J Neurooncol. 2007;83:97-109.
Vogelbaum MA. Convection enhanced delivery for the treatment of malignant gliomas: symposium review. J Neurooncol. 2005;73:57-69.
Westphal M, Ram Z, Riddle V, et alfor the Executive Committee of the Gliadel Study group. Gliadel wafer in initial surgery for malignant glioma: long-term follow-up of a multicenter controlled trial. Acta Neurochir (Wien). 2006;148:269-275.
1 Central Brain Tumor Registry of the United States (CBTRUS). Statistical report: primary brain tumors in the United States, 2000-2004. Chicago: Central Brain Tumor Registry of the United States; 2005.
2 Central Nervous System Cancers Practice Guidelines in Oncology, vol 2. Jenkintown, PA: National Comprehensive Cancer Network. 2005.
3 Watanabe M, Tanaka R, Takeda N. Magnetic resonance imaging and histopathology of cerebral gliomas. Neuroradiology. 1992;34:463-469.
4 Nicholas MK, Prados MD, Larson DA, et al. Malignant astrocytomas. In: Black P, Loeffler JS, editors. Cancer of the Nervous System. Cambridge: Blackwell Science; 1997:464-491.
5 Neuwelt EA. Mechanisms of disease: the blood-brain barrier. Neurosurgery. 2004;54:131-142.
6 Sawchuk RJ, Elmquist WF. Microdialysis in the study of drug transporters in the CNS. Adv Drug Deliv Rev. 2000;45:295-307.
7 Takasato Y, Rapoport SI, Smith QR. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol. 1984;247(Suppl 3):H484-H493.
8 Tofteng F, Larsen FS. Monitoring extracellular concentrations of lactate, glutamate, and glycerol by in vivo microdialysis in the brain during liver transplantation in acute liver failure. Liver Transplant. 2002;8:302-305.
9 Muldoon LL, Soussain C, Jahnke K, et al. Chemotherapy delivery issues in central nervous system malignancy: a reality check. J Clin Oncol. 2007;25:2295-2305.
10 Smith QR. A review of blood-brain barrier transport techniques. Methods Mol Med. 2003;89:193-208.
11 Pardridge WM. Introduction to the Blood-Brain Barrier: Methodology, Biology and Pathology. Cambridge: Cambridge University Press; 1998.
12 Banks WA. Physiology and pathology of the blood-brain barrier: implication for microbial pathogenesis, drug delivery and neurodegenerative disorders. J Neurovirol. 1999;5:538-555.
13 Wilson CB. Glioblastoma: the past, the present, and the future. Clin Neurosurg. 1992;38:32-48.
14 Schinkel AH, Wagenaar E, Mol C, et al. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. 1996;97:2517-2524.
15 Leggas M, Adachi A, Scheffer GL, et al. MrP4 confers resistance to topotecan and protects the brain from chemotherapy. Mol Cell Biol. 2004;24:7612-7621.
16 Breedveld P, Pluim D, Cipriani G, et al. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res. 2005;65:2577-2582.
17 Bradbury M. The Concept of a Blood-Brain Barrier. Chicester, UK: John Wiley & Sons; 1979.
18 Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1994;271:58-65.
19 Ali MJ, Navlitloha Y, Vavra MW, et al. Isolation of drug delivery from drug effect: problems of optimizing drug delivery parameters. Neuro Oncol. 2006;8:109-118.
20 Tong RT, Boucher Y, Kozin SV, et al. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731-3736.
21 Rockwell S. Use of hypoxia-directed drugs in the therapy of solid tumors. Semin Oncol. 1992;19:29-40.
22 Johanson CE, Jones HC. Promising vistas in hydrocephalus and cerebrospinal fluid research. Trends Neurosci. 2001;24:631-632.
23 Rautio J, Chikhale PJ. Drug delivery systems for brain tumor therapy. Curr Pharm Design. 2004;10:1341-1353.
24 Ghersi-Egea JF, Leininger-Muller B, Cecchelli R, et al. Blood-brain interfaces: relevance to cerebral drug metabolism. Toxicol Lett. 1995;82-83:645-653.
25 Pardridge WM. Recent advances in blood-brain barrier transport. Annu Rev Pharmacol Toxicol. 1988;28:25-39.
26 Brem H, Walter KA, Tamargo RJ, et al. Drug delivery to the brain. In: Domb AJ, ed. Polymeric Site-Specific Pharmacotherapy. New York: John Wiley and Sons, 1994;117-139.
27 Sinkula AA, Yalkowsky SH. Rationale for design of biologically reversible drug derivatives: prodrugs. J Pharm Sci. 1975;64:181-210.
28 Stella VJ, Charman WNA, Naringrekar VH. Prodrugs: do they have advantages in clinical practice? Drugs. 1985;29:455-473.
29 Greig NH, Genka S, Daly EM, et al. Physicochemical and pharmacokinetic parameters of seven lipophilic chlorambucil esters designed for brain penetration. Cancer Chemother Pharmacol. 1990;25:311-319.
30 Genka S, Deutsch J, Shetty UH, et al. Development of lipophilic anticancer agents for the treatment of brain tumors by the esterification of water-soluble chlorambucil. Clin Exp Metastasis. 1993;11:131-140.
31 Martin J, Stribbling SM, Poon GK, et al. Antibody-directed enzyme prodrug therapy: pharmacokinetics and plasma levels of prodrug and drug in a phase I clinical trial. Cancer Chemother Pharmacol. 1997;40:189-201.
32 Bodor N, Brewster ME. Problems of delivery of drugs to the brain. Pharmacol Ther. 1982;19:337-386.
33 Bodor N, Brewster ME. Chemical delivery systems. In: Juliano RL, editor. Targeted Drug Delivery. Berlin: Springer; 1991:231-284.
34 Oldenforf WH. Brain uptake of radiolabeled amino acids, amines and hexoses after arterial injection. Am J Physiol. 1971;221:1629-1639.
35 Pardridge WM, Boado RJ. Molecular cloning and regulation of gene expression of blood-brain barrier glucose transporter. In: Pardridge WM, editor. The Blood-Brain Barrier: Cellular and Molecular Biology. New York: Raven Press; 1993:395-440.
36 Lefauconnier JM. Transport of amino acids. In: Bradbury MWB, editor. Physiology and Pharmacology of the Blood-Brain Barrier. Berlin: Springer-Verlag; 1992:117-150.
37 Segal MB, Zlokovic BV. The Blood-Brain Barrier, Amino Acids and Peptides. Dordrecht, The Netherlands: Kluwer; 1990.
38 Cornford EM, Baun LD, Oldendorf WH. Carrier mediated blood-brain barrier transport of choline analogs. J Neurochem. 1978;30:299-308.
39 Pratt OE. The movement of vitamins into the brain. In: Bradbury MWB, editor. Physiology and Pharmacology of the Blood-Brain Barrier. Berlin: Springer-Verlag; 1992:205-220.
40 Spector R, Eells J. Deoxynucleoside and vitamin transport into the central nervous system. Fed Proc. 1984;43:196-200.
41 Abbott NJ, Romero IA. Transporting therapeutics across the blood-brain barrier. Mol Med Today. 1996;2(3):106-113.
42 Kurihara A, Pardridge WM. Imaging brain tumors by targeting peptide radiopharmaceuticals through the blood-brain barrier. Cancer Res. 1999;59:6159-6163.
43 Greenwood J. Experimental manipulation of the blood-brain and blood-retinal barriers. In: Bradbury MWB, editor. Physiology and Pharmacology of the Blood-Brain Barrier. Germany: Springer-Verlag; 1992:459-486.
44 Neuwelt EA. Mechanisms of disease: the blood-brain barrier. Neurosurgery. 2004;54:131-142.
45 Rapoport SL, Robinson PJ. Tight-junctional modification as the basis of osmotic opening of the blood-brain barrier. Ann N Y Acad Sci. 1987;481:250-267.
46 Williams PC, Henner WD, Roman-Goldstein S, et al. Toxicity and efficacy of carboplatin and etoposide in conjunction with disruption of the blood-brain tumor barrier in the treatment of intracranial neoplasms. Neurosurgery. 1995;37:17-28.
47 Neuwelt EA, Frenkel EP, Rapoport SI, et al. Effect of osmotic blood-brain barrier disruption on methotrexate pharmacokinetics in the dog. Neurosurgery. 1980;7:36-43.
48 Dahlborg SA, Henner WD, Crossen JR, et al. Non-AIDS primary CNS lymphoma: the first example of a durable response in a primary brain tumor using enhanced chemotherapy delivery without cognitive loss and without radiotherapy. Cancer J Sci Am. 1996;2:168-174.
49 Kraemer DF, Fortin D, Doolittle ND, et al. Association of total dose intensity of chemotherapy in primary CNS lymphoma (human non-AIDS) and survival. Neurosurgery. 2001;48:1033-1041.
50 McAllister LD, Doolittle ND, Guastadisegni PE, et al. Cognitive outcomes and long-term follow-up after enhanced chemotherapy delivery for primary central nervous system lymphomas. Neurosurgery. 2000;46:51-61.
51 Neuwelt EA, Goldman DL, Dahlborg SA, et al. Primary CNS lymphoma treated with osmotic blood-brain barrier disruption: prolonged survival and preservation of cognitive function. J Clin Oncol. 1991;9:1580-1590.
52 Neuwelt EA, Barnett PA, Glasberg M, et al. Pharmacology and neurotoxicity of cis-diamminedichloroplatinum, bleomycin, 5-fluorouracil, and cyclophosphamide administration following osmotic blood-brain barrier modification. Cancer Res. 1983;43:5278-5285.
53 Cloughesy TF, Black KL. Pharmacological blood-brain barrier modification for selective drug delivery. J Neurooncol. 1995;26:125-132.
54 Bartus RT, Elliott PJ, Dean RL, et al. Controlled modulation of BBB permeability using the bradykinin agonist, RMP-7. Exp Neurol. 1996;142:14-28.
55 Ford J, Osborn C, Barton T, et al. A phase I study of intravenous RMP-7 with carboplatin in patients with progression of malignant glioma. Eur J Cancer. 1998;34:1807-1811.
56 Hynynen K, Clement GT, McDonald N, et al. 500-Element ultrasound phased array system for noninvasive focal surgery of the brain: a preliminary rabbit study with ex vivo human skulls. Magn Reson Med. 2004;52:100-107.
57 Hynynen K, McDannold N, Vykhodtseva N, et al. Noninvasive MR imaging–guided focal opening of the blood-brain barrier in rabbits. Radiology. 2001;220:640-646.
58 Treat LH, McDannold N, Hynynen K. Transcranial MRI-guided focused ultrasound-induced blood-brain barrier opening in rats. Ultrasonics Symposium IEEE.; 2004.
59 Clement GT, Hynynen K. A noninvasive method for focusing ultrasound through the human skull. Phys Med Biol. 2000;47:1219-1236.
60 Brem H, Mahaley MSJr, Vick NA, et al. Interstitial chemotherapy with drug polymer implants for the treatment of recurrent gliomas. J Neurosurg. 1991;74:441-446.
61 Westphal M, Ram Z, Riddle V, et al. for the Executive Committee of the Gliadel Study group. Gliadel wafer in initial surgery for malignant glioma: long-term follow-up of a multicenter controlled trial. Acta Neurochir (Wien). 2006;148:269-275.
62 Walter KA, Cahan MA, Gur A, et al. Interstitial Taxol delivered from a biodegradable polymer implant against experimental malignant glioma. Cancer Res. 1994;54:2207-2212.
63 Watts MC, Lesniak MS, Burke M, et al. Controlled release of Adriamycin in the treatment of malignant glioma (poster). Presented at American Association of Neurological Surgeons Annual Meeting; Denver, CO. 1997.
64 Menei P, Venier MC, Gamelin E, et al. Local and sustained delivery of 5-fluorouracil from biodegradable microspheres for the radiosensitization of glioblastoma: a pilot study. Cancer. 1999;86:325-330.
65 Tajika Y, Muragaki Y, Hiyama H, et al. Local chemotherapy with slowly-releasing anticancer drug polymers for malignant brain tumors. J Control Rel. 1994;32:1-8.
66 DeMeco F, Li KW, Tyler BM, et al. Local delivery of mitoxantrone for the treatment of malignant brain tumors in rats. J Neurosurg. 2002;97:1173-1178.
67 Bobo RH, Laske DW, Akbasak A, et al. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci U S A. 1994;91:2076-2080.
68 Lonser RR, Walbridge S, Garmestani K, et al. Successful and safe perfusion of the primate brainstem: in vivo magnetic resonance imaging of macromolecular distribution during infusion. J Neurosurg. 2002;97:905-913.
69 Chen MY, Lonser RR, Morrison PF, et al. Variables affecting convection-enhanced delivery to the striatum: a systematic examination of rate of infusion, cannula size, infusate concentration, and tissue-cannula sealing time. J Neurosurg. 1999;90:315-320.
70 Vogelbaum MA. Convection enhanced delivery for the treatment of malignant gliomas: symposium review. J Neurooncol. 2005;73:57-69.
71 Hadaczek P, Kohutnicka M, Krauze MT, et al. Convection-enhanced delivery of adeno-associated virus type 2 (AAV2) into the striatum and transport of AAV2 within monkey brain. Hum Gene Ther. 2006;17:291-302.
72 Lidar Z, Mardor Y, Jonas T, et al. Convection-enhanced delivery of paclitaxel for the treatment of recurrent malignant glioma: a phase I/II clinical study. J Neurosurg. 2004;100:472-479.
73 Kaiser MG, Parsa AT, Fine RL, et al. Tissue distribution and antitumor activity of topotecan delivered by intracerebral clysis in a rat glioma model. Neurosurgery. 2000;47:1391-1398.
74 Weaver M, Laske DW. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J Neurooncol. 2003;65:3-13.
75 Rand RW, Kreitman RJ, Patronas N, et al. Intratumoral administration of recombinant circularly permuted interleukin-4–Pseudomonas exotoxin in patients with high-grade glioma. Clin Cancer Res. 2000;6:2157-2165.
76 Kunwar S, Chang SM, Prados MD, et al. Safety of intraparenchymal convection-enhanced delivery of cintredekin besudotox in early-phase studies. Neurosurg Focus. 2006;20(4):E15.
77 Vogelbaum MA. Convection enhanced delivery for treating brain tumors and selected neurological disorders: symposium review. J Neurooncol. 2007;83:97-109.
78 Blasberg RG. Methotrexate, cytosine arabinoside, and BCNU concentration in the brain after ventriculocisternal perfusion. Cancer Treat Rep. 1977;61:625-631.
79 Burch PA, Grossman SA, Reinhard CS. Spinal cord penetration of intrathecally administered cytarabine and methotrexate: a quantitative autoradiographic study. J Natl Cancer Inst. 1988;80:1211-1216.
80 Walter KA, Tamargo RJ, Olivi A, et al. Intratumoral chemotherapy. Neurosurgery. 1995;37:1128-1145.
81 Yamada K, Ushio Y, Hayakawa T, et al. Distribution of radiolabeled 1-(4-amino-2-methyl-5-pyrimidinyl)methyl-3-(2-chloroethyl)-3-nitrosourea hydrochloride in rat brain tumor: intraarterial versus intravenous administration. Cancer Res. 1987;47:2123-2128.
82 Savaraj N, Lu K, Feun LG, et al. Comparison of CNS penetration, tissue distribution, and pharmacology of VP16-213 by intracarotid and intravenous administration in dogs. Cancer Invest. 1987;5:11-16.
83 Nakagawa H, Fujita T, Izumoto S, et al. Cis-diamminedichloroplatinum (CDDP) therapy for brain metastases of lung cancer: distribution within the central nervous system after intravenous and intracarotid infusion. J Neurooncol. 1993;16:61-68.
84 Huwyler J, Wu D, Pardridge WM. Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acad Sci U S A. 1996;93:14164-14169.
85 Kreuter J. Nanoparticles. In: Swarbrick J, Boylan JC, editors. Encyclopedia of Pharmaceutical Technology, vol. 10. New York: Marcel Dekker; 1994:165-190.
86 Gelperina SE, Smirnova ZS, Khalanskiy AS, et al. Chemotherapy of brain tumors using doxorubicin bound to polysorbate 80–coated nanoparticles. Proceedings of the 3rd World Meeting APV/APGI, Berlin, April 3-6, 2000; 441-442.
87 Dhanda DA, Frey WH2nd, Leopold D, et al. Approaches for drug deposition in the human olfactory epithelium. Drug Deliv Technol. 2005;5:64-72.
88 Thorne RG, Pronk GJ, Padmanabhan V, et al. Delivery of insulin-like growth factor-1 to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience. 2004;127:481-496.
89 Hashizume R, Ozawa T, Gryaznov SM, et al. New therapeutic approach for brain tumors: intranasal delivery of telomerase inhibitor GRN163. Neuro Oncol. 2008:112-120.
90 Hassan EE, Gallo JM. Targeting anticancer drugs to the brain. I: Enhanced brain delivery of oxantrazole following administration in magnetic cationic microspheres. J Drug Target. 1993;1:7-14.
91 Gallo JM, Hassan EE. Receptor-mediated magnetic carriers: basis for targeting. Pharm Res. 1988;5:300-304.
92 Pulfer SK, Ciccotto SL, Gallo JM. Distribution of small magnetic particles in brain tumor–bearing rats. J Neurooncol. 1999;41:99-105.
93 Godwin JT, Farr LE, Sweet WH, et al. Pathological study of eight patients with glioblastoma multiforme treated by neutron capture therapy using boron 10. Cancer. 1955;8:601-615.
94 Barth RF, Coderre JA, Vicente GH, et al. Boron neutron capture therapy of cancer: current status and future prospects. Clin Cancer Res. 2005;11:3987-4002.
95 Soloway AH, Hatanaka H, Davis MA. Penetration of brain and brain tumor, VII. Tumor-binding sulfhydryl boron compounds. J Med Chem. 1967;10:714-717.
96 Hatanaka H, Nakagawa Y. Clinical results of long-surviving brain tumor patients who underwent boron neutron capture therapy. Int J Radiat Oncol Biol Phys. 1994;28:1061-1066.






