Chapter 46 Bacterial Cell Wall Synthesis Inhibitors
| Abbreviations | |
|---|---|
| CSF | Cerebrospinal fluid |
| ESBLs | Extended-spectrum β-lactamases |
| GI | Gastrointestinal |
| IM | Intramuscular |
| IV | Intravenous |
| MIC | Minimal inhibitory concentration |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| NAG | N-acetylglucosamine |
| NAM | N-acetylmuramate |
| PBPs | Penicillin-binding proteins |
Therapeutic Overview
Since its introduction, however, many strains of bacteria, particularly Staphylococcus aureus, have become resistant to penicillin through several mechanisms including the production of metabolic enzymes called β-lactamases, altered penicillin binding proteins (PBPs), and reduced drug permeability by resistant bacteria (see Table 45-6). Resistance to penicillin by the induction of β-lactamases led to the development of semisynthetic penicillins resistant to hydrolysis by these enzymes as well as numerous compounds with greater activity against gram-negative organisms. The development of resistance to β-lactams is an ongoing clinical problem and is increasing at a dramatic rate.
β-lactams. Resistance to vancomycin, however, is an increasing problem in enterococcus species. Even more alarming has been the discovery of clinical isolates of S. aureus with reduced vancomycin susceptibility, or even full resistance mediated by transfer of resistance genes from enterococcus species.
| Therapeutic Overview |
|---|
| β-Lactams |
| Penicillins, Cephalosporins, Carbapenems, and Monobactams |
| Bactericidal: inhibit many gram-positive and gram-negative organisms |
| Agents differ by: |
| Organism inhibited |
| Pharmacokinetics |
| Bacterial resistance |
| Glycopeptides and Polypeptides |
| Vancomycin—Bactericidal; inhibits many methicillin-resistant staphylococci |
| Bacitracin—Bactericidal; topical use only for gram-positive bacteria |
Mechanisms of Action
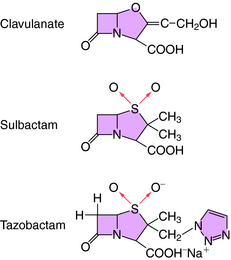
All β-lactam antibiotics have a four-membered ring structure containing a cyclic amide (the lactam); the β indicates that the amine is located on the second carbon relative to the carbonyl group (Fig. 46-1, A). This small ring is structurally strained with low inherent stability, which explains why some penicillins are not effective when administered orally, because they readily undergo hydrolysis, particularly in the presence of high stomach acidity. In addition, this ring is subject to hydrolysis by the β-lactamases, representing a major mechanism of resistance to these compounds. This is why some penicillin derivatives are marketed in combination with β-lactamase inhibitors such as clavulanate, sulbactam, and tazobactam, all of which contain the β-lactam ring structure (Fig. 46-2).
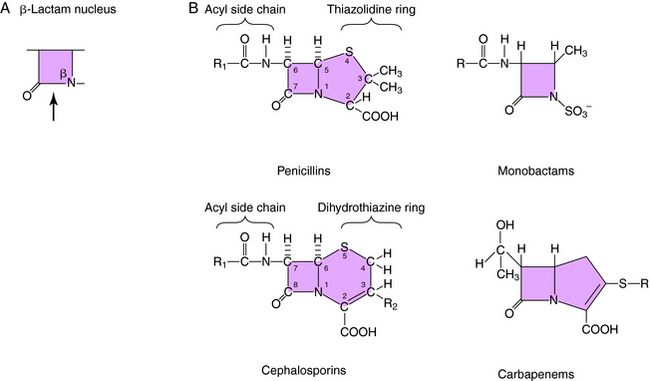
All of the β-lactam antibiotics, except the monobactams, have a second ring fused to the β-lactam ring (see Fig. 46-1, B). For penicillins the second ring is a thiazolidine, whereas for cephalosporins it is a dihydrothiazine. Carbapenems have an unsaturated ring with an external sulfur. Different structural groups positioned at the side chains (R) give rise to compounds with differing antibiotic properties.
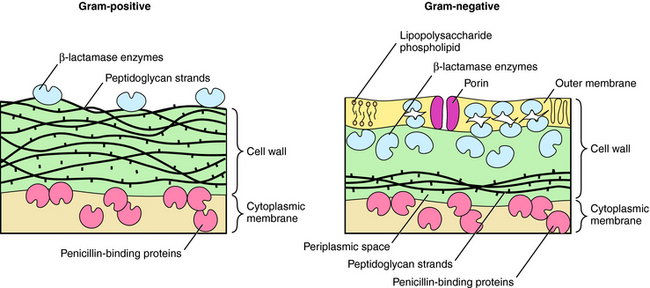
β-lactams interfere with bacterial cell wall synthesis. Although the outer cellular coverings of gram-positive and negative bacteria differ, both have a rigid cell wall composed of a highly cross-linked peptidoglycan matrix (Fig. 46-3). Gram-negative bacteria contain an outer lipopolysaccharide membrane exterior to several peptidoglycan layers. Gram-positive bacteria lack the lipopolysaccharide layer but contain many more (15 to 30) layers of peptidoglycan. The cell wall is assembled in a series of steps, originating within the cytoplasm of the bacteria and terminating outside the cytoplasmic membrane.
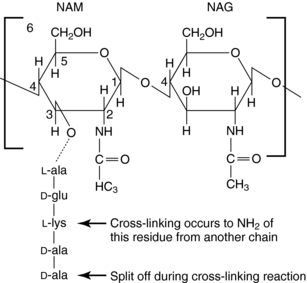
The glycan part of the peptidoglycan is composed of repeating disaccharide units of N-acetylmuramate (NAM) attached to a pentapeptide and N-acetylglucosamine (NAG), connected through β-1,4-linkages (Fig. 46-4). This initial stage of peptidoglycan synthesis occurs in the cytoplasm. The NAG-NAM-pentapeptide is then transferred by a carrier across the cytoplasmic membrane, and the saccharide units are linked in sequence via the pentapeptides to form long chains of alternating disaccharides. This final stage involves a cross-linking reaction to form continuous two-dimensional sheets, a process that occurs outside the cytoplasmic membrane in the periplasm but is catalyzed by membrane-bound transpeptidase enzymes (Fig. 46-5). During the cross-linking reaction, the D-ala-D-ala terminus of the pentapeptide reacts with the transpeptidase to displace the final D-ala, forming an acylenzyme intermediate. This intermediate is reactive and readily couples to the free amino group of the third residue (L-lys) of the pentapeptide of an adjacent chain, thus completing the cross-linking and regenerating the enzyme. It is at the final cross-linking step during synthesis of the rigid peptidoglycan matrix that β-lactam and glycopeptide antibiotics exert their actions, albeit by different mechanisms.
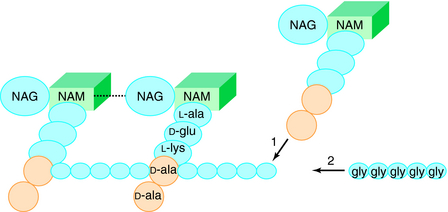
Mechanisms of Resistance to β-Lactams
As discussed in Chapter 45, bacterial resistance to antibiotics is a major therapeutic concern. Resistance to the β-lactams is common and occurs by three major mechanisms:
These mechanisms may coexist, and in some cases it takes a combination of mechanisms, such as decreased permeability and poor binding within a single organism, to confer resistance.
In gram-negative bacteria, β-lactams must pass an outer lipid membrane to reach the PBPs on the cytoplasmic membrane (see Fig. 46-3). Channels in the outer membrane, referred to as porins, allow β-lactams to pass through. Alterations in porin proteins that reduce the amount of drug reaching the PBPs have been observed. For example, P. aeruginosa can delete the porin protein through which imipenem passes and develop resistance. Some β-lactams are extremely resistant to β-lactamases but do not readily pass through porins of gram-negative outer membranes and thus fail to inhibit these bacteria.
However, more than 25% of nosocomial enterococcal isolates, primarily E. faecium, are now vancomycin resistant. Resistance is caused by production of a new pentapeptide ending in a terminal D-ala-D-lac instead of D-ala-D-ala, which does not bind vancomycin. There are also other types of resistance to vancomycin (Table 46-1); the vanA cluster of genes, transferred by a transposable genetic element, is the best characterized. This is an elegant resistance mechanism that contains at least eight genes. One gene product, vanS, functions like a transmembrane receptor, senses the presence of vancomycin, and activates the gene vanR to up regulate expression of three additional genes. The products of these three genes cleave the terminal D-ala-D-ala and insert D-ala-D-lac. Similar resistance genes are present in streptomyces that produce glycopeptide antibiotics and probably evolved as a self-preservation strategy for the organism. Vancomycin-resistant strains of S. aureus are emerging, mediated by increased numbers of vancomycin-binding sites in the cell wall. In 2002, the first two clinical strains of S. aureus isolates with high levels of vancomycin resistance appeared. These strains acquired the vanA gene cluster from vancomycin-resistant enterococci. Bacitracin is a polypeptide bactericidal antibiotic. It inhibits bacterial cell wall synthesis by interfering with dephosphorylation of the lipid carrier that moves the early cell wall components through the membrane.

Pharmacokinetics
All β-lactam agents exhibit time-dependent killing of bacteria and have little postantibiotic effect, unlike fluoroquinolones and aminoglycosides, which function by concentration-dependent mechanisms. Consequently, efficacy of the β-lactams depends on the amount of time the concentration of drug is above the minimal inhibitory concentration (MIC) for the organism at the site of infection. The pharmacokinetic parameters for penicillins of clinical interest are summarized in Table 46-2.
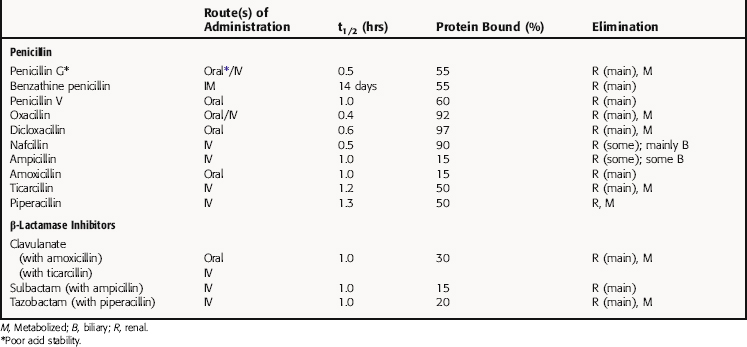
β-Lactamase-Resistant Penicillins
The β-lactamase-resistant penicillins oxacillin, cloxacillin, and dicloxacillin are acid stable and orally absorbed, but absorption is decreased in the presence of food. Peak plasma concentrations are achieved approximately 1 hour after ingestion, and they are all highly protein bound. Elimination is primarily via the kidney, with some biliary excretion and some liver metabolism. These drugs are minimally removed from the body by hemodialysis. Oxacillin is less effective orally; however, adequate plasma and CSF concentrations are achieved when it is administered by the intravenous (IV) route. Nafcillin is erratically absorbed when ingested orally, and the preferred route is IV. Elimination is primarily by biliary excretion. It enters the CSF in concentrations adequate to treat staphylococcal meningitis or brain abscesses.
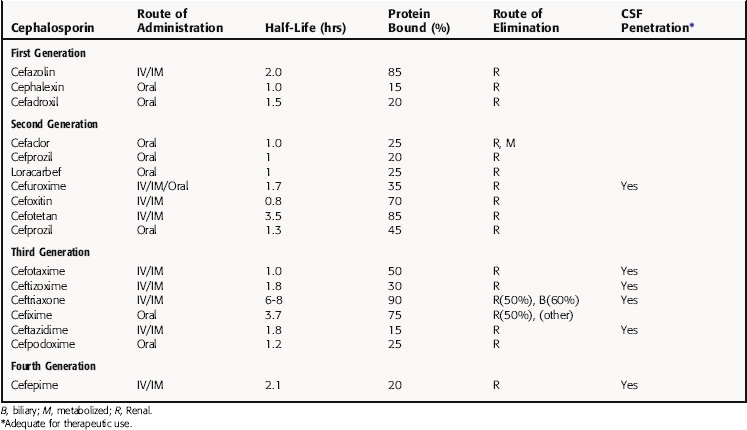
Many cephalosporins can be administered only parenterally. Although these compounds are distributed widely throughout the body, only a few of them enter the CSF in sufficient concentrations for the treatment of meningitis. Cephalosporins are generally eliminated by renal excretion, and their accumulation depends on renal status. Ceftriaxone is an exception, with significant biliary excretion. All cephalosporins reach high enough urinary concentrations to treat urinary tract infections. In the absence of common duct obstruction, biliary concentrations of all cephalosporins exceed plasma concentrations. Pharmacokinetic parameters for cephalosporins of clinical interest are listed in Table 46-3.
Pharmacokinetic parameters of the carbapenem antibiotics imipenem, meropenem, and ertapenem, and the only monobactam, aztreonam, are summarized in Table 46-4. Imipenem enters the CSF only during inflammation and has high affinity for brain tissue. It is eliminated by glomerular filtration and tubular secretion and is inactivated by a dehydropeptidase in the renal tubules. To overcome this hydrolysis, imipenem is combined with a renal dehydropeptidase inhibitor, cilastatin. Cilastatin itself has no antibacterial activity and does not affect the properties of imipenem, except to prevent its hydrolysis. Minimal amounts of drug are excreted in bile, though biliary concentrations are adequate for treatment of biliary tract infections. Serum half-life increases as creatinine clearance falls and is increased in patients with renal insufficiency. Meropenem also penetrates well into most fluids and tissues as well as the CSF after IV administration. Because meropenem is excreted unchanged in the urine (is not hydrolyzed by renal dehydropeptidase), dosages must be adjusted in renal insufficiency. Ertapenem has the advantage over imipenem and meropenem of a relatively long half-life, enabling once-daily dosing. It is highly protein bound and mainly renally excreted, requiring dosage changes with severe renal impairment. It is also less susceptible to hydrolysis by renal dehydropeptidase and is not administered with cilastatin.
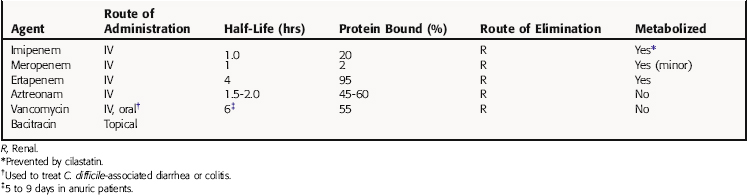
Pharmacokinetic parameters for vancomycin and bacitracin are summarized in Table 46-4. Vancomycin is administered IV, except for the treatment of C. difficile-associated diarrhea or colitis, when it is administered orally and acts locally. Vancomycin administered IV enters many body fluids, including bile and pleural, pericardial, peritoneal, and synovial fluids. It crosses the meninges during inflammation. Vancomycin is eliminated by glomerular filtration and is not metabolized; thus dosage should be adjusted on the basis of renal function. Vancomycin is not removed efficiently by hemodialysis or peritoneal dialysis. Monitoring plasma concentrations is necessary to ensure therapeutic concentrations are achieved and toxicity averted in patients with depressed renal function.
Bacitracin is applied topically and is combined in several preparations with neomycin or polymyxin.
Relation of Mechanisms of Action to Clinical Response
Penicillins and Cephalosporins
Penicillins are classified by their main antibacterial activities as follows:
β-Lactamase Inhibitor Combinations
The β-lactamase inhibitors inactivate the β-lactamases of S. aureus and of many gram-negative bacteria, including plasmid-mediated common β-lactamases in E. coli, Haemophilus, Neisseria, Salmonella, and Shigella species, and chromosomal β-lactamases in Klebsiella, Moraxella, and Bacteroides species. None of the β-lactamase inhibitors bind to the chromosomal ampC β-lactamases in Pseudomonas, Enterobacter, Citrobacter, and Serratia species. Beta-lactamase inhibitors differ in relative potency, and this difference is reflected in the ratio of inhibitor to the paired penicillin. Sulbactam, the weakest inhibitor, is available in a 1:2 ratio of sulbactam to ampicillin; the ratio of tazobactam to piperacillin is 1:8, whereas the ratio of clavulanate to ticarcillin is 1:30. The major differences between the penicillin β-lactamase inhibitor combinations lie in the different spectra of the penicillin component. All combinations have excellent activity against anaerobes.
Cefoxitin is less active against gram-positive organisms than the first-generation agents, but it is more stable against β-lactamase degradation by Enterobacteriaceae (but not Enterobacter or Citrobacter species) and anaerobic bacteria. Also, it is not hydrolyzed by the plasmid-mediated ESBLs that destroy cefotaxime, ceftriaxone, and ceftazidime and has been used to treat aspiration pneumonia and intraabdominal and pelvic infections. Cefotetan inhibits many β-lactamase-producing Enterobacteriaceae and most Bacteroides species. It is also used to treat intraabdominal and pelvic infections. However, these two agents are not as active against B. fragilis as the penicillin β-lactamase inhibitor combinations discussed previously and are not as active against gram-negative bacilli as later cephalosporins. Consequently, use of second-generation cephalosporins has declined, although they are still used for perioperative prophylaxis.
Vancomycin should be reserved primarily for serious infections. It is also appropriate for therapy of staphylococcal infections in penicillin-allergic patients and is the drug of choice for treatment of MRSA infections. Pneumonia, endocarditis, osteomyelitis, and wound infections respond to vancomycin, and it also useful for treating infections of prosthetic valves and catheters caused by coagulase-negative staphylococci and Corynebacterium.
Orally administered vancomycin is prescribed to treat C. difficile-associated diarrhea or colitis that fails to respond to metronidazole (see Chapter 52) or that is severe and potentially life-threatening. Because the use of oral vancomycin is a risk factor for colonization and infection with vancomycin-resistant enterococci, however, it is not considered the agent of choice in most cases.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Problems associated with the use of bacterial cell wall synthesis inhibitors are summarized in the Clinical Problems Box; the frequencies of specific adverse reactions to β-lactam drugs are shown in Table 46-5.
| Reaction Type | Frequency (%) | Typical Drugs |
|---|---|---|
| Immunoglobulin E antibody allergy (anaphylaxis) | 0.004-0.4 | Penicillin G |
| Delayed-type hypersensitivity and contact dermatitis | 4-8 | Ampicillin |
| Idiopathic; rash | 4-8 | Ampicillin and cephalosporins |
| Gastrointestinal problems | 2-5 | Orally administered agents |
| Diarrhea | 25 | Ampicillin, cefixime, ceftriaxone, cefoxitin, β-lactamase inhibitor combinations |
| Enterocolitis | 1 | Any agent |
| Elevated hepatic aspartate aminotransferase | 1-4 | Oxacillin, Nafcillin |
| Interstitial nephritis | 1-2 | Nafcillin |
| Hemolytic anemia, serum sickness, cytotoxic antibody, hyperkalemia, neurological seizures, hemorrhagic cystitis | Rare | Any agent |
Although penicillins can cause a wide variety of adverse effects, serious adverse reactions are fortunately rare. Hypersensitivity, which can be life-threatening, is the most important and includes anaphylaxis, wheezing, angioedema, and urticaria. It occurs through immunoglobulin E-mediated antibody reactions, usually directed at the penicillin nucleus, which is common to all drugs of this class. Therefore a patient who develops anaphylaxis to a specific penicillin should be considered allergic to them all. Penicillins also can be partially degraded to compounds with varying allergenicity. The major determinants of penicillin allergy are penicilloyl acid derivatives (Fig. 46-6), but minor components of benzylpenicillin and benzylpenicilloate are important mediators of anaphylaxis. Anaphylactic reactions to penicillins are uncommon, occurring in 0.2% of 10,000 courses of treatment. In contrast, a morbilliform skin eruption-type of allergy occurs in 3% to 5% of patients receiving penicillin. Any of the β-lactams can cause Stevens-Johnson’s syndrome, a rare, life-threatening immune-complex-mediated hypersensitivity disorder of the skin and mucus membranes.
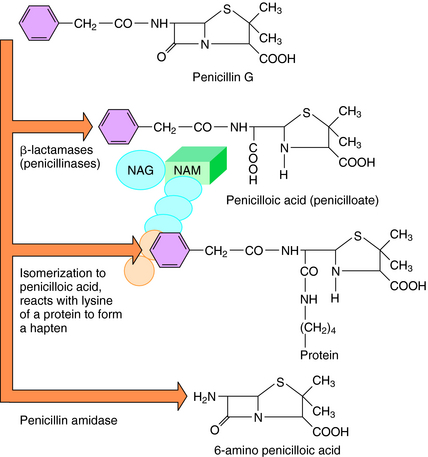
FIGURE 46–6 Some breakdown products of penicillin G in the presence of different enzymes and conditions.
Skin testing with benzylpenicilloyl polylysine, benzylpenicillin G, and Na+ benzylpenicilloate (see Fig. 46-6) is 95% successful in identifying people likely to have an anaphylactic reaction. However, a negative skin test does not exclude later development of a rash. Anaphylactic reactions to penicillins should be treated with epinephrine (see Chapter 11). There is no evidence that antihistamines or corticosteroids are beneficial. Whenever there is a history of an allergic reaction to penicillin or any other β-lactam antibiotic, the most practical approach is to use a different class of antibiotics.
Diarrhea is more common after oral ampicillin than amoxicillin, so the latter is generally preferred. Penicillins and almost all other antibiotics can cause Clostridium difficile enterocolitis. This organism can overgrow when the normal bowel flora is disrupted by antibiotic therapy, and it produces a cytotoxin and an enterotoxin that cause diarrhea and pseudomembrane formation. Distortion of normal intestinal flora by penicillins can also cause bowel function to be altered and cause colonization with resistant gram-negative bacilli or fungi such as Candida.
Imipenem and meropenem can cause allergic reactions similar to those produced by the penicillins and should not be administered to patients who have had anaphylactic reactions to penicillins or cephalosporins. Cutaneous eruptions and diarrhea can also occur. Rapid infusion of imipenem-cilastatin can produce nausea and emesis. Imipenem binds to brain tissue more avidly than does penicillin G and can cause seizures, which constitute its most
CLINICAL PROBLEMS OF BACTERIAL CELL WALL SYNTHESIS INHIBITORS
serious toxic reaction. Seizures have occurred in patients with decreased renal function and an underlying seizure focus; therefore imipenem should not be used to treat meningitis. In contrast, meropenem is unlikely to cause seizures and can be used safely to treat bacterial meningitis caused by susceptible organisms.
The most important side effect is ototoxicity, which can occur in patients with high plasma concentrations, but this is rarely seen at concentrations less than 30 µg/mL. The risk of ototoxicity is increased when vancomycin is given in combination with aminoglycosides. Nephrotoxicity is uncommon in patients receiving vancomycin alone but is noted in patients who also receive aminoglycosides (see Chapter 47). Phlebitis, which is common, can be avoided through the use of dilute solutions and slow infusion.
New Horizons
Since the discovery of penicillin in 1929 and its widespread availability in the 1940s, β-lactam antibiotics have been a mainstay of treatment of bacterial infections. However, their widespread use has resulted in increasing resistance through a variety of mechanisms. Inventive approaches to preventing resistance include the use of specific metabolic inhibitors, development of newer structures that are less susceptible to degradation, and the development of new classes of compounds. The main approaches used currently are to restrict their use unless absolutely necessary and to use the minimal durations of therapy needed. However, it is clear that new classes of compounds will be needed because most organisms eventually become resistant to these and other antibiotics. Fortunately, many new targets for antibiotic development are being identified by sequencing the genomes of specific bacterial species, which may shed new light on essential processes that can be targeted to specifically eradicate these infectious diseases. Such new targets are likely to include both metabolic and structural proteins, including those involved in synthesis and maintenance of bacterial cell walls.
Craig WA. Basic pharmacodynamics of antibacterials with clinical applications to the use of β-lactams, glycopeptides, and linezolid. Infect Dis Clin N Am. 2003;17:479-501.
Karchmer AW. Cephalosporins. In Mandell GL, Bennett JE, Dolin R, editors: Principles and Practice of Infectious Diseases, ed 5, New York: Churchill Livingstone, 2000.
Paterson DL, Bonomo RA. Extended spectrum beta-lactamases: A clinical update. Clin Microbiol Rev. 2005;18:657-686.
Zhanel GG, Wiebe R, Dilay L, et al. Comparative review of the carbapenems. Drugs. 2007;67:1027-1052.