Chapter 20 Autoimmunity and Autoimmune Disease
• Autoimmunity is associated with disease. Autoimmune mechanisms underlie many diseases, some organ-specific, others systemic in distribution, and autoimmune disorders can overlap – an individual may have more than one organ-specific disorder, or more than one systemic disease.
• Genetic factors play a role in the development of autoimmune diseases. Twin studies show that there is a heritable component to autoimmunity. The vast majority of autoimmune diseases are polygenic but HLA genes are particularly important.
• Self-reactive B and T cells persist even in normal subjects. Autoreactive B and T cells persist in normal subjects, but in disease are selected by autoantigen in the production of autoimmune responses.
• Controls on the development of autoimmunity can be bypassed. Microbial cross-reacting antigens and cytokine dysregulation can lead to autoimmunity.
• In most diseases associated with autoimmunity, the autoimmune process produces the lesions. The pathogenic role of autoimmunity can be demonstrated in experimental models. Human autoantibodies can be directly pathogenic. Immune complexes are often associated with systemic autoimmune disease. Autoantibody tests are valuable for diagnosis and sometimes for prognosis.
• Treatment of autoimmune disease has a variety of aims. Treatment of organ-specific diseases usually involves metabolic control. Treatment of systemic diseases includes the use of anti-inflammatory and immunosuppressive drugs. Biological therapies using monoclonal antibodies against pro-inflammatory cytokines have revolutionized the treatment of the autoimmune rheumatic diseases. B cell-directed therapies have proved highly effective in many autoimmune diseases.
Autoimmunity and autoimmune disease
Because the repertoire of specificities expressed by the B cells and T cells is generated randomly, it includes many that are specific for self components. The body therefore requires self-tolerance mechanisms to distinguish between self and non-self determinants to avoid autoreactivity (see Chapter 19). However, such mechanisms may fail and a number of diseases have been identified in which there is autoimmunity, with copious production of autoantibodies and autoreactive T cells. The targets of the autoimmune ‘attacks’ vary widely as we shall see.
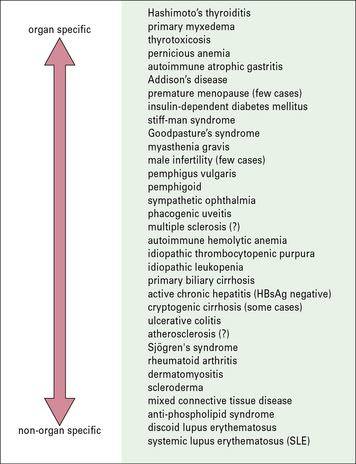
Autoimmune conditions present a spectrum between organ-specific and systemic disease
Hashimoto’s thyroiditis is highly organ-specific
One of the earliest examples in which the production of autoantibodies was associated with disease in a given organ is Hashimoto’s thyroiditis. (Thyroiditis is a condition that is most common in middle-aged women and often leads to the formation of a goiter or hypothyroidism.) The gland is infiltrated, sometimes to an extraordinary extent, with inflammatory lymphoid cells. These are predominantly mononuclear phagocytes, lymphocytes and plasma cells, and secondary lymphoid follicles are common (Fig. 20.1). The gland often also has regenerating thyroid follicles.
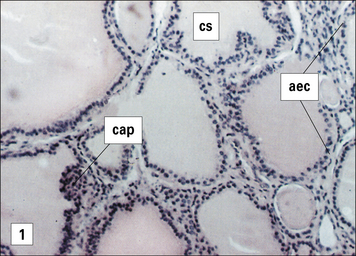
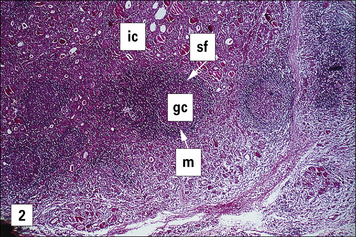
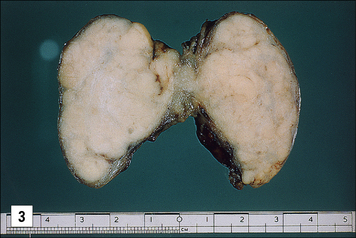
Fig. 20.1 Pathological changes in Hashimoto’s thyroiditis
((2) Reproduced from Woolf N. Pathology: basic and systemic. London: WB Saunders; 1998.)
The serum of patients with Hashimoto’s disease usually contains antibodies to thyroglobulin. These antibodies are demonstrable by agglutination and by precipitin reactions when present in high titer. Most patients also have antibodies directed against a cytoplasmic or microsomal antigen, also present on the apical surface of the follicular epithelial cells (Fig. 20.2), and now known to be thyroid peroxidase, the enzyme that iodinates thyroglobulin. The antibodies associated with Hashimoto’s thyroiditis react only with the thyroid, so the resulting lesion is highly localized.
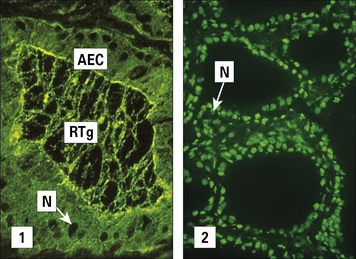
SLE is a systemic autoimmune disease
By contrast, the serum from patients with diseases such as systemic lupus erythematosus (SLE) reacts with many, if not all, of the tissues in the body. In SLE, one of the dominant antibodies is directed against the cell nucleus (see Fig. 20.2).
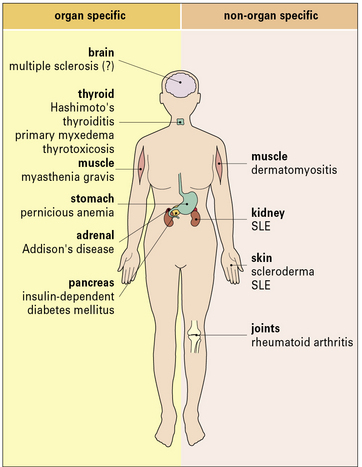
Hashimoto’s thyroiditis and SLE represent the extremes of the autoimmune spectrum (Fig. 20.3):
• the common target organs in organ-specific disease include the thyroid, adrenal, stomach, and pancreas;
• the non-organ-specific diseases, often termed systemic autoimmune diseases, which include rheumatological disorders, characteristically involve the skin, kidney, joints, and muscle (Fig. 20.4).
An individual may have more than one autoimmune disease
• Thyroid antibodies occur with a high frequency in patients with pernicious anemia who have gastric autoimmunity, and these patients have a higher incidence of thyroid autoimmune disease than the normal population. Similarly, patients with thyroid autoimmunity have a high incidence of stomach autoantibodies and to a lesser extent the clinical disease itself (pernicious anemia).
• Around 15% of patients with primary Sjögren’s syndrome have concomitant autoimmune hypothyroidism, and up to 40% have thyroid antibodies. Around 10% of SLE patients have thyroid antibodies in their serum.
• 30% of patients with SLE and Sjögren’s syndrome have a second, third or even fourth autoimmune disease.
• where the antigen is localized in a particular organ, type II hypersensitivity (e.g. autoimmune hemolytic anemia) and type IV cell-mediated reactions, as in type 1 insulin-dependent diabetes, are most important (see Chapters 24–26);
• in systemic disorders such as SLE, type III immune complex deposition leads to inflammation through a variety of mechanisms, including complement activation and phagocyte recruitment. In rheumatoid arthritis immune complexes directly stimulate production of cytokines such as TNFα.
Genetic factors in autoimmunity
Within the families of patients with organ-specific autoimmunity, not only is there a general predisposition to develop organ-specific antibodies, other genetically controlled factors tend to select the organ that is mainly affected. Thus, although relatives of patients with Hashimoto’s thyroiditis and families of patients with pernicious anemia both have a higher than normal incidence and titer of thyroid autoantibodies, the relatives of patients with pernicious anemia have a far higher frequency of gastric autoantibodies (Fig. 20.5), indicating that there are genetic factors that differentially select the stomach as the target within these families.
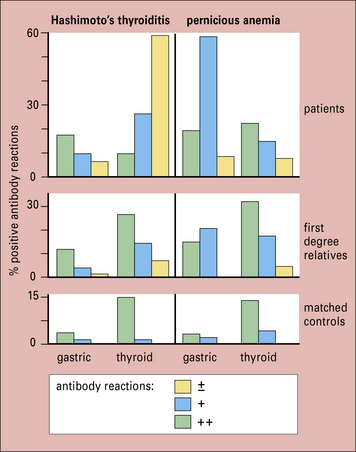
Fig. 20.5 Overlap between thyroid and gastric autoimmunity
(Data adapted from Doniach D, Roitt IM, Taylor KB. Ann NY Acad Sci 1965;124:605.)
Certain HLA haplotypes predispose to autoimmunity
Further evidence for the operation of genetic factors in autoimmune disease comes from their tendency to be associated with particular HLA specificities (Fig. 20.6). For most autoimmune diseases, the MHC region which is located on the short arm of chromosome 6, provides the strongest genetic component to disease susceptibility. Recent high-density genome mapping in multiple autoimmune diseases has demonstrated complex, multilocus effects that span the entire region, with both shared and unique loci across diseases.
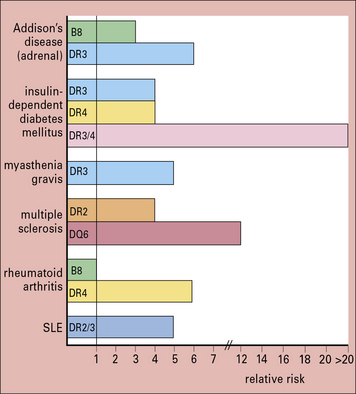
The haplotype B8, DR3 is common in both the organ-specific diseases and systemic autoimmune diseases like SLE and myositis, though Hashimoto’s thyroiditis tends to be associated more with DR5. Interestingly, for type 1 diabetes mellitus, DQ2/8 heterozygotes have a greatly increased risk of developing the disease (see Fig. 20.6).
Genes outside the HLA region also confer susceptibility to autoimmunity
Although HLA risk factors tend to dominate, autoimmune disorders are genetically complex and genome-wide searches for mapping the genetic intervals containing genes for predisposition to disease also reveal a plethora of non-HLA genes (Fig. 20.7) affecting:
• lymphocyte activation (receptor signaling pathways and co-stimulation);
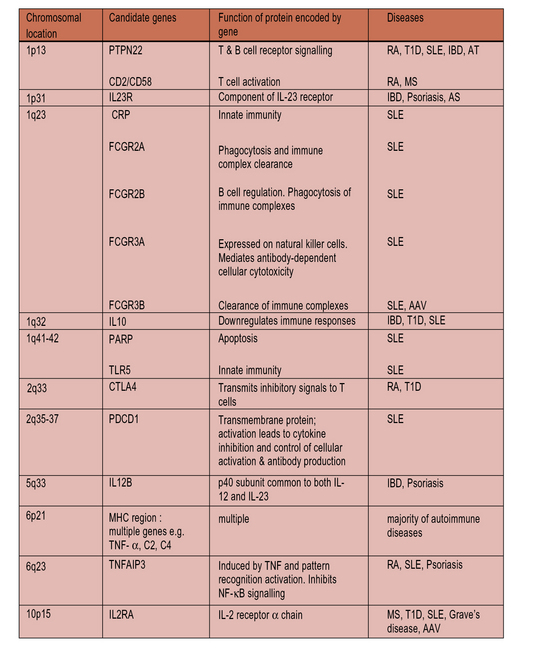

Autoimmunity is associated with genes that control lymphocyte activation
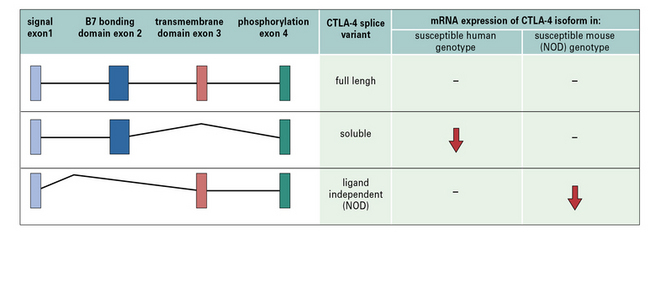
 Autoimmunity is associated with a multitude of genes encoding molecules expressed by lymphocytes which modulate co-stimulation signals. These include CD2/CD58, TNFSF15, and ISOSLG. A notable example is a single nucleotide polymorphism (SNP) linked to CTLA-4 (Fig. 20.w1), a molecule present on Tregs (see Chapter 19).
Autoimmunity is associated with a multitude of genes encoding molecules expressed by lymphocytes which modulate co-stimulation signals. These include CD2/CD58, TNFSF15, and ISOSLG. A notable example is a single nucleotide polymorphism (SNP) linked to CTLA-4 (Fig. 20.w1), a molecule present on Tregs (see Chapter 19).
SLE is associated with multiple gene loci
Genome wide-association studies have identified multiple genetic loci associated with SLE (see Fig. 20.7). Null alleles which cause a deficiency of one of the early complement components (C1q, C2, or C4) strongly predispose individuals to a lupus-like disease, although there is a lower incidence compared to ‘classic’ SLE of certain clinical features, notably glomerulonephritis and serological characteristics (ANA and anti-dsDNA antibodies) in these individuals.
Autoimmunity and autoimmune disease
Autoimmunity results from antigen-driven self-reactive lymphocytes
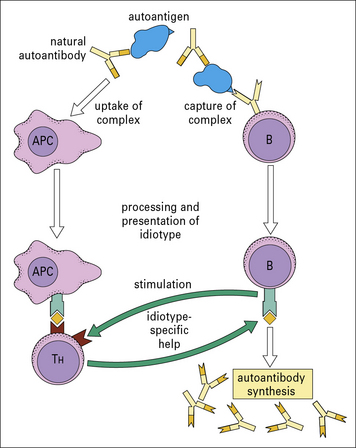
Given that autoreactive B cells exist, the question remains whether they are stimulated to proliferate and produce autoantibodies by interaction with autoantigens or by some other means, such as non-specific polyclonal activators or idiotypic interactions (see below and Fig. 20.8).
The most direct evidence for autoimmunity being antigen driven comes from studies of the Obese strain chicken, which spontaneously develops thyroid autoimmunity. If the thyroid gland (the source of antigen) is removed at birth, the chickens mature without developing thyroid autoantibodies (Fig. 20.9). Furthermore, once thyroid autoimmunity has developed, later removal of the thyroid leads to a gross decline of thyroid autoantibodies, usually to undetectable levels.
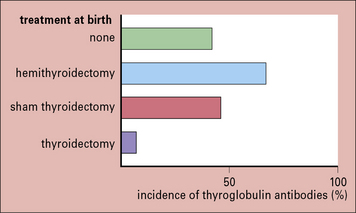
Fig. 20.9 Effect of neonatal thyroidectomy on Obese chickens
(Based on data from de Carvalho et al. J Exp Med 1982;155:1255.)
In organ-specific disorders, there is ample evidence for T cells responding to antigens present in the organs under attack. But in non-organ-specific autoimmunity, identification of the antigens recognized by T cells is often inadequate. However, histone-specific T cells are generated in patients with SLE and histone could play a ‘piggyback’ role in the formation of anti-DNA antibodies by substituting for natural antibody in the mechanism outlined in Figure 20.8.
In this view SLE, for example, might sometimes be initiated as an ‘idiotypic disease’, following the model shown in Figure 20.8. In this scheme, autoantibodies are produced normally at low levels by B cells using germline genes. If these then form complexes with the autoantigen, the complexes can be taken up by APCs (including B cells) and components of the complex, including the antibody idiotype, presented to T cells. Idiotype-specific T cells would then provide help to the autoantibody-producing B cells.
The ‘waste disposal’ hypothesis of SLE
Antibodies to nuclear components are the serological hallmark of SLE. The question arising from this observation is how nuclear components normally hidden are detected by the immune system as antigen. The answer appears to lie with apoptosis. There is strong evidence that SLE (and possibly other autoimmune diseases) are diseases of failure of clearance of apoptotic cells, i.e. due to decreased macrophage ‘scavenger’ function. When a cell undergoes apoptosis (programmed cell death), blebs of cellular material are formed on the cell surface. Antigens normally buried deep within the cell (and so not detected by the immune system) are exposed on the cell surface. In healthy individuals these apoptotic cells are efficiently cleared. However, in SLE apoptosis is defective; it has been demonstrated that scavenging of apoptotic debris in vitro by macrophages from lupus patients is less efficient than by macrophages from healthy controls. Thus the antigens contained within the apoptotic blebs may trigger an autoimmune response. Apoptotic blebs vary in size; larger blebs contain antigens including Sm, Mi-2, Ro-60, and La, whilst smaller blebs contain fodrin, Jo-1, Ro-52, and ribosomal P. Antibodies against all of these targets can be found in systemic autoimmune diseases such as SLE, Sjögren’s syndrome, and myositis (Fig. 20.10).
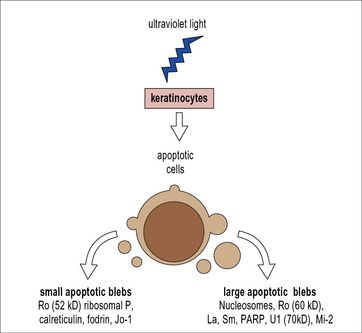
Fig. 20.10 Induction of surface blebs during apoptosis
(Based on Rahman A, Isenberg DA: Systemic lupus erythematosus. N Engl J Med, 2008; 358:929–939.)

 In mice deletion of some of the molecules involved in the clearance of apoptotic cells leads to autoantibody production and lupus-like disease. However, not all deletions resulting in defective clearance of apoptotic cells result in autoimmunity. Furthermore, immunization of mice with apoptotic cells does not produce autoimmune disease. This may be accounted for by the immunosuppressive capabilities of apoptotic cells, especially through release of anti-inflammatory cytokines from phagocytic cells. SLE-related autoantibodies and glomerulonephritis resembling human lupus nephritis can however be induced in mice by co-immunization with an apoptotic cell binding protein (human beta2 glycoprotein1) and the powerful adjuvant, LPS. Not only did the mice produce high titres of IgG anti-beta2 glycoprotein1 antibodies, but also antibodies to Ro, La, dsDNA, Sm, and nRNP in sequential order, recapitulating human disease. Knockout of CD28 abrogated these effects, indicating that CD28-mediated co-stimulation of T cells is required. This mouse model provides evidence of intermolecular epitope spread, and suggests that long-lived memory T cells responding to a single protein (beta2 glycoprotein1) are capable of providing help to other auto-antigen specific B cells if the protein interacts with a ‘scaffold’ containing the other autoantigens. Of note, immunization with either LPS or beta2 glycoprotein1 alone did not result in lupus autoantibody production.
In mice deletion of some of the molecules involved in the clearance of apoptotic cells leads to autoantibody production and lupus-like disease. However, not all deletions resulting in defective clearance of apoptotic cells result in autoimmunity. Furthermore, immunization of mice with apoptotic cells does not produce autoimmune disease. This may be accounted for by the immunosuppressive capabilities of apoptotic cells, especially through release of anti-inflammatory cytokines from phagocytic cells. SLE-related autoantibodies and glomerulonephritis resembling human lupus nephritis can however be induced in mice by co-immunization with an apoptotic cell binding protein (human beta2 glycoprotein1) and the powerful adjuvant, LPS. Not only did the mice produce high titres of IgG anti-beta2 glycoprotein1 antibodies, but also antibodies to Ro, La, dsDNA, Sm, and nRNP in sequential order, recapitulating human disease. Knockout of CD28 abrogated these effects, indicating that CD28-mediated co-stimulation of T cells is required. This mouse model provides evidence of intermolecular epitope spread, and suggests that long-lived memory T cells responding to a single protein (beta2 glycoprotein1) are capable of providing help to other auto-antigen specific B cells if the protein interacts with a ‘scaffold’ containing the other autoantigens. Of note, immunization with either LPS or beta2 glycoprotein1 alone did not result in lupus autoantibody production.Induction of autoimmunity
Molecular mimicry by cross-reactive microbial antigens can stimulate autoreactive lymphocytes
• no longer require a co-stimulatory signal; and
• have a higher avidity for the target, due to upregulation of accessory adhesion molecules (Fig. 20.11).
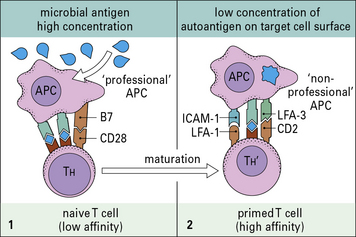
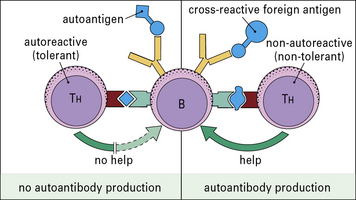
• these TH cells are tolerized at lower concentrations of autoantigens than the B cells; or
• because they recognize only cryptic epitopes. However, these ‘helpless’ B cells can be stimulated if the cross-reacting antigen bears a ‘foreign’ carrier epitope to which the T cells have not been tolerized (Fig. 20.12). The autoimmune process may persist after clearance of the foreign antigen if the activated B cells now focus the autoantigen on their surface receptors and present it to normally resting autoreactive T cells, which will then proliferate and act as helpers for fresh B cell stimulation.
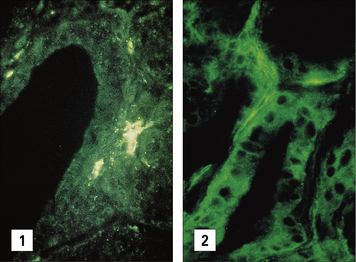
Cytokine dysregulation, inappropriate MHC expression, and failure of suppression may induce autoimmunity
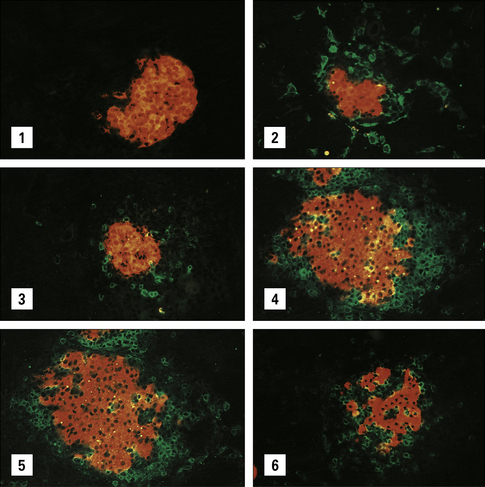
The surface expression of MHC class II in itself is not sufficient to activate the naive autoreactive T cells, but it may be necessary to allow a cell to act as a target for the primed autoreactive TH cells. It was therefore most exciting when cells taken from the glands of patients with Graves’ disease were found to be actively synthesizing class II MHC molecules (Fig. 20.13) and so were able to be recognized by CD4+ T cells.
Deficiencies in any of these factors may increase susceptibility to autoimmunity.
The feedback loop on TH cells and macrophages through the pituitary–adrenal axis is particularly interesting because defects at different stages in the loop turn up in a variety of autoimmune disorders (Fig. 20.14).
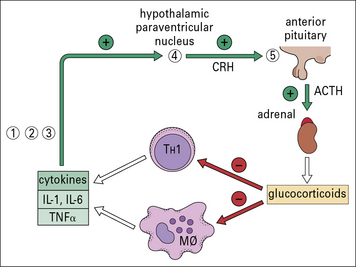
There is currently intense interest focused on the role of Tregs. Patients with rheumatoid arthritis, for example, reveal a deficiency of Treg function (see below and Fig. 20.w5).
Pre-existing defects in the target organ may increase susceptibility to autoimmunity
In the Obese strain chicken model of spontaneous thyroid autoimmunity, not only is there a low threshold of IFNγ induction of MHC class II expression by thymocytes, but when endogenous TSH is suppressed by thyroxine treatment, the uptake of iodine into the thyroid glands is far higher in the Obese strain than in a variety of normal strains. Furthermore, this is not due to any stimulating effect of the autoimmunity because immunosuppressed animals show even higher uptakes of iodine (Fig. 20.15).
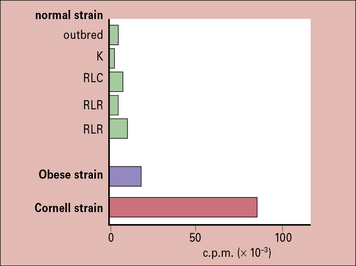
Other situations in which the production of autoantigen is affected are:
• diabetes mellitus, in which one of the genetic risk factors is linked to a transcription factor controlling the rate of insulin production; and
• rheumatoid arthritis, in which the agalacto IgG glycoform is abnormally abundant.
Autoimmune processes and pathology
• the autoimmunity is responsible for producing the lesions of the disease;
• there is a disease process that, through the production of tissue damage, leads to the development of autoantibodies;
• there is a factor that produces both the lesions and the autoimmunity.

The pathogenic role of autoimmunity can be demonstrated in experimental models
 The most direct test of whether autoimmunity is responsible for the lesions of disease is to induce autoimmunity deliberately in an experimental animal and see if this leads to the production of the pathological lesions.
The most direct test of whether autoimmunity is responsible for the lesions of disease is to induce autoimmunity deliberately in an experimental animal and see if this leads to the production of the pathological lesions.TH cells are essential for induction of autoimmunity in EAE
MBP-induced encephalomyelitis can be induced in susceptible strains by injecting T cells specific for MBP. These pathogenic T cells belong to the CD4/TH1 subset. It has been found that induction of disease can be prevented by treating the recipients with antibody to CD4 just before the expected time of disease onset, blocking the interaction of the TH cells’ CD4 with the class II MHC molecules of antigen-presenting target cells (see Chapter 8).
Examples of spontaneous autoimmunity
• removal of the thymus at birth appears to exacerbate the thyroiditis, suggesting that the thymus exerts a controlling effect on the disease through Tregs; but
• if the entire T cell population is abrogated by combining thymectomy with massive injections of anti-chick T cell serum, both autoantibody production and the attack on thyroid are completely inhibited.
T cells therefore play a variety of pivotal roles as mediators and regulators of this disease.
More directly, a diabetogenic CD4+ T cell clone can induce the chronic leukocytic infiltrate of T cells and macrophages, which damages the insulin-producing β cells of the pancreatic islets of Langerhans in the NOD murine model of type 1 diabetes mellitus (Fig. 20.w2).
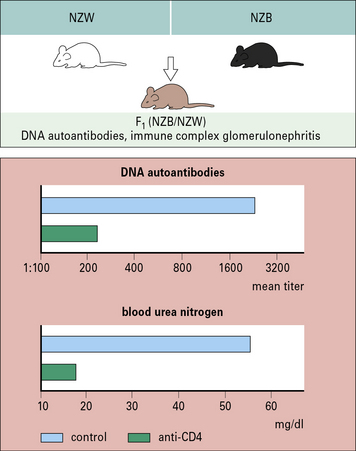
The dependence of yet another spontaneous model, the F1 hybrid of New Zealand Black and White strains, on the operation of immunological processes is aptly revealed by the suppression of the murine SLE (associated with immune complex glomerulonephritis and anti-DNA autoantibodies), by treatment with anti-CD4 antibody or azathioprine (Fig. 20.w3).
Human autoantibodies can be directly pathogenic
Autoantibodies can give rise to a wide spectrum of clinical thyroid dysfunction
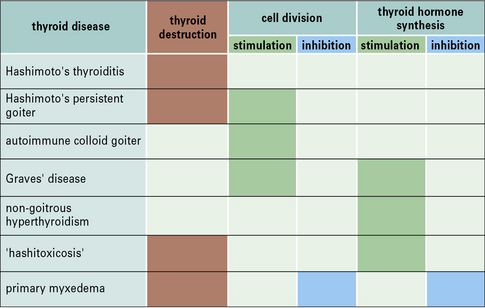
Different combinations of the various manifestations of thyroid autoimmune disease – chronic inflammatory cell destruction, and stimulation or inhibition of growth and thyroid hormone synthesis – can give rise to a wide spectrum of clinical thyroid dysfunction (Fig. 20.16).
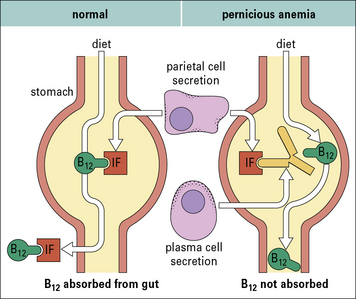
In pernicious anemia an autoantibody interferes with the normal uptake of vitamin B12
Subsequently, the factor in the serum that blocked vitamin uptake was identified as antibody against intrinsic factor. It is now known that plasma cells in the gastric mucosa of patients with PA secrete this antibody into the lumen of the stomach (Fig. 20.17).
Antibodies to the glomerular capillary basement membrane cause Goodpasture’s disease
Goodpasture’s disease is characterized clinically by rapidly progressive glomerulonephritis and pulmonary haemorrhage. Patients with Goodpasture’s disease have circulating antibodies to the glomerular capillary basement membrane which bind to the kidney and lung (see Fig. 25.3). Evidence for the direct pathogenity of these antibodies was demonstrated by the passive transfer of antibodies eluted from renal biopsy specimens into primates (whose renal antigens were similar to humans). The injected monkeys subsequently died from glomerulonephritis. Subsequent work has shown that these antibodies bind to the several non-collagenous-1 (NC-1) domains of type IV collagen in the GBM. Moreover, immunization of animals with NC-1 domains induces glomerulonephritis, providing a causal link between autoantigen and antibody.
Blood and vascular disorders caused by autoantibodies include AHA and ITP
The β2-glycoprotein is an abundant component of atherosclerotic plaques, and there is increasing attention to the idea that autoimmunity may initiate or exacerbate the process of lipid deposition and plaque formation in this disease, the two lead candidate antigens being heat-shock protein 60 (Fig. 20.18) and the low density lipoprotein, apoprotein B.
Immune complexes appear to be pathogenic in systemic autoimmunity
In the case of SLE, it can be shown that complement-fixing complexes of antibody with DNA and other nucleosome components such as histones are deposited in the kidney (see Fig. 25.3), skin, joints, and choroid plexus of patients, and must be presumed to produce type III hypersensitivity reactions as outlined in Chapter 25. A variety of different antibodies have been eluted from the kidney biopsies of patients with SLE. These include anti-dsDNA (nucleosomes), anti-Ro and anti-Sm/RNP. Whilst placing these antibodies at the scene of the crime, their mere presence does not prove they ‘pulled the trigger’. However, experiments using murine monoclonal anti-dsDNA antibodies in a rat kidney perfusion system, and other evidence from the use of human hybridoma derived anti-dsDNA antibodies in SCID mice, provides compelling evidence that some anti-dsDNA antibodies are genuinely pathogenic.
Autoantibodies to IgG provoke pathological damage in rheumatoid arthritis
The erosions of cartilage and bone in rheumatoid arthritis are mediated by macrophages and fibroblasts, which become stimulated by cytokines from activated T cells and immune complexes generated by a vigorous immunological reaction within the synovial tissue (Fig. 20.19).
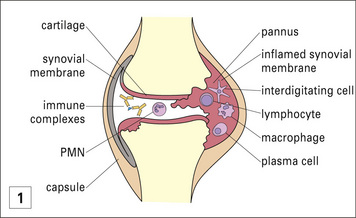
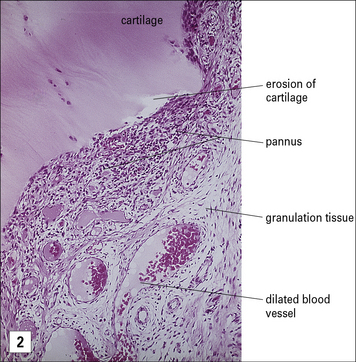
Fig. 20.19 Pathology of rheumatoid arthritis
(Histological section reproduced from Woolf N. Pathology: basic and systemic. London: W.B. Saunders; 1998.)
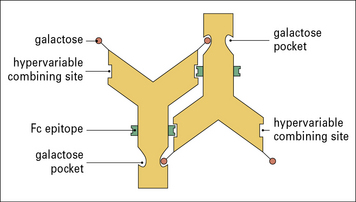
The complexes can arise through the self-association of IgG rheumatoid factors specific for the Fcγ domains – a process facilitated by the striking deficiency of terminal galactose on the biantennary N-linked Fc oligosaccharides (Fig. 20.20). This agalacto glycoform of IgG in complexes can exacerbate inflammation through reaction with mannose-binding lectin and production of TNFα.
Evidence for directly pathogenic T cells in human autoimmune disease is hard to obtain
In humans, evidence for a pivotal role of T cells in the development of autoimmune disease includes:
• the production of high-affinity, somatically mutated IgG autoantibodies characteristic of T-dependent responses;
• the isolation of thyroid-specific T cell clones from the glands of patients with Graves’ disease;
• the beneficial effect of ciclosporin in prediabetic individuals; and
Autoantibodies for diagnosis, prognosis, and monitoring
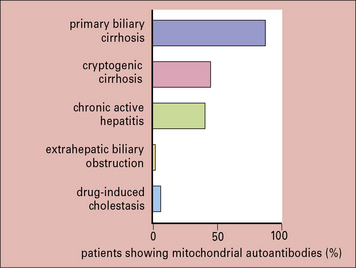
Whatever the relationship of autoantibodies to the disease process, they frequently provide valuable markers for diagnostic purposes. A particularly good example is the test for mitochondrial antibodies, used in diagnosing primary biliary cirrhosis (Fig. 20.21). Historically, exploratory laparotomy was needed to obtain this diagnosis, and was often hazardous because of the age and condition of the patients concerned. Some other diagnostically useful antibodies are listed below:
• antibodies to cyclic citrullinatated peptides (CCP) have a higher specificity than rheumatoid factor in the diagnosis of RA;
• the presence of anti-nuclear antibodies is one of the revised ACR criteria for SLE, but is non-specific. In contrast anti-dsDNA antibodies (also one of the ACR criteria) are highly specific for SLE, being present in 70% of SLE patients but in less than 0.5% of controls. Anti-Sm antibodies are found in 10% of Caucasian and 30% of Afro-Caribbean SLE patients; like dsDNA antibodies they have a high specificity for SLE;
• autoantibodies often have predictive value. For instance, individuals testing positively for antibodies to both insulin and glutamic acid decarboxylase have a high risk of developing type 1 diabetes mellitus.
Prognosis and disease subtype:
• in rheumatoid arthritis, CCP antibodies are associated with a poor prognosis and predict erosive disease;
• within the context of a patient with SLE the presence of specific autoantibodies are associated with specific disease manifestations. For example, anti-La antibodies are associated with features of Sjögren’s syndrome; anticardiolipin antibodies and anti-beta2 glycoprotein1 antibodies with thrombosis and miscarriage; anti-dsDNA antibodies with glomerulonephritis; anti-RNP with pulmonary hypertension, and anti-Ro with photosensitivity and the neonatal lupus syndrome.
• anti-dsDNA antibodies can be used as a measure of disease activity in SLE. A rising titer of anti-dsDNA antibodies often heralds a disease flare, especially if accompanied by a falling C3 level, and should prompt the clinician to monitor the patient more frequently;
• similarly, a rising ANCA titer may indicate impending relapse in AAV. However, the association between ANCA titer and disease activity in AAV is much less robust than that between anti-dsDNA antibody titer and lupus activity.
Treatment of autoimmune diseases
• hypothyroidism can be controlled by administration of thyroxin;
• type 1 diabetes mellitus can be controlled by administration of insulin;
• in pernicious anemia, metabolic correction is achieved by injection of vitamin B12;
• in myasthenia gravis metabolic correction is achieved by administration of cholinesterase inhibitors.
• TNFα and IL-6 in rheumatoid arthritis;
• B cells in several autoimmune diseases (RA, SLE, AAV);

‘Biologics’ – a new paradigm in the treatment of autoimmune rheumatic disease
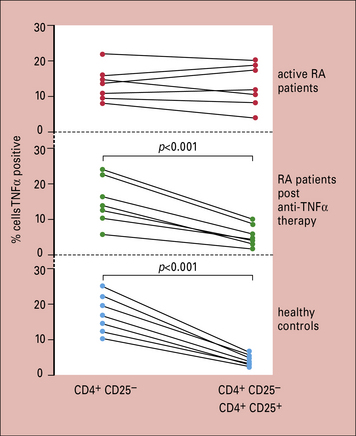
 The treatment of patients with rheumatoid arthritis has been revolutionized by the introduction of biological agents. Infliximab, a murine-human chimeric antibody against TNFα, not only markedly alleviates the symptoms of RA such as joint pain, stiffness and swelling, but also halts the progression of joint destruction (Fig. 20.w4). A number of other anti-TNF agents have subsequently become available including adalimumab (a fully humanized monoclonal antibody against TNF), etanercept (a soluble TNF receptor), and certolizumab (a pegylated anti-TNF antibody). It is fascinating to record that the compromised regulatory T cell function in the patients is reversed by such therapy (Fig. 20.w5). Similarly impressive results in RA are seen with B-cell depletion using rituximab, an anti-CD20 monoclonal antibody, and tocilizumab, a monoclonal antibody against IL-6. Other biologics licensed for use in RA include abatacept, which inhibits the co-stimulatory interaction between T cells and APCs, and anakinra, an IL-1 blocker.
The treatment of patients with rheumatoid arthritis has been revolutionized by the introduction of biological agents. Infliximab, a murine-human chimeric antibody against TNFα, not only markedly alleviates the symptoms of RA such as joint pain, stiffness and swelling, but also halts the progression of joint destruction (Fig. 20.w4). A number of other anti-TNF agents have subsequently become available including adalimumab (a fully humanized monoclonal antibody against TNF), etanercept (a soluble TNF receptor), and certolizumab (a pegylated anti-TNF antibody). It is fascinating to record that the compromised regulatory T cell function in the patients is reversed by such therapy (Fig. 20.w5). Similarly impressive results in RA are seen with B-cell depletion using rituximab, an anti-CD20 monoclonal antibody, and tocilizumab, a monoclonal antibody against IL-6. Other biologics licensed for use in RA include abatacept, which inhibits the co-stimulatory interaction between T cells and APCs, and anakinra, an IL-1 blocker.
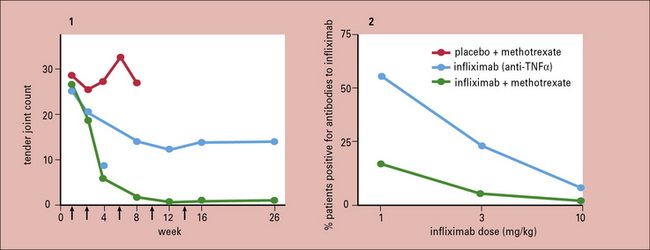
Fig. 20.w4 Synergy of anti-TNFα and methotrexate in the treatment of rheumatoid arthritis
(Data reproduced from Maini RN, et al. Arthritis Rheum 1998;41:1552, with permission of the authors and publishers.)
Less well-established approaches to treatment may become practicable
As we understand more about the molecular mechanisms underlying autoimmunity, targeted therapy is becoming increasingly possible (Fig. 20.22):
• several centers are trying out autologous stem cell transplantation following hematoimmunoablation with cytotoxic drugs for patients with severe SLE and vasculitis;
• repeated injection of Cop 1 (a random copolymer of alanine, glutamic acid, lysine, and tyrosine) reduces relapse rate in relapsing–remitting multiple sclerosis. Cop 1 was originally designed to simulate the postulated ‘guilty’ autoantigen, MBP, and induce experimental autoimmune encephalitis; paradoxically it had the opposite effect. This suggests it is possible to achieve antigen-specific immune suppression.
• eculizumab, a monoclonal antibody against complement component C5, has been used successfully in paroxysmal nocturnal hemoglobinuria. It may prove effective in SLE;
• TH cell subsets show considerable plasticity in vivo. Manipulation of the balance between pro-inflammatory TH17 cells and anti-inflammatory regulatory T cells may prove fruitful. Therapeutic interventions to increase the numbers of Tregs might increase self-tolerance and ‘put the brakes’ on inflammatory cascades. Adoptive transfer of polyclonal Tregs can prevent the onset of diabetes in NOD mice. However, it has become clear that successful treatment of established autoimmune disease by adoptive transfer of Tregs is antigen-specific; only Tregs specific for pancreatic antigens (and not polyclonal T regs) can reverse diabetes in NOD mice. This presents difficulties for treatment in human disease, where the inciting antigen is rarely known;
• inhibition of the activation or differentiation of TH17 cells, and/or blockade of linked cytokines such as IL17A and IL-23 may prove valuable. Trials of anti-IL17 monoclonal antibodies are already underway;
• small molecule therapies have the advantage of being cheaper to manufacture than monoclonal antibodies, of particular relevance in an era of spiraling healthcare costs. Kinases are intracellular enzymes that play a crucial role in signal transduction pathways controlling a number of cellular functions. Kinase inhibitors have been successfully used in oncology, but may also prove effective in autoimmune diseases. For example, a splenic tyrosine kinase inhibitor, reduces severity of antibody-mediated glomerulonephritis in rats, and is being trialled in RA.
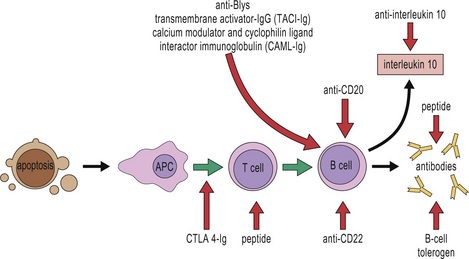
Fig. 20.22 Targeted therapeutic approaches in systemic lupus erythematosus
(Based on Rahman A, Isenberg DA: Systemic lupus erythematosus. N Engl J Med 2008; 358:929–939.)
Critical thinking: Autoimmunity and autoimmune disease (see p. 440 for explanations)
| Investigation | Result |
|---|---|
| radiograph of hands | soft tissue swelling, but no bone erosions |
| chest radiograph | a small pleural effusion at the right lung base |
| full blood count | a mild normocytic, normochromic anemia and mild lymphocytopenia |
| C-reactive protein levels | normal |
| erythrocyte sedimentation rate | raised |
| rheumatoid factor | negative |
| serum IgG levels | raised |
| anti-nuclear antibodies (ANA) | positive by immunofluorescence |
| anti-double-stranded DNA, anti-RNA, and anti-histone | positive by ELISA antibodies |
| complement (C3 and C4) levels | low |
| skin biopsy from an area unaffected by the rash | deposition of IgG and complement components at the junction between dermis and epidermis (lupus ‘band’ test) |
Arbuckle M.R., McClain M.T., Rubertone M.V., et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533.
Betterle C., Greggio N.A., Volpato M. Clinical Review 93: Autoimmune polyglandular syndrome type 1. J Clin Endocrinol Metab. 1998;83:1049–1055.
Edwards J.C., Szczepanski L., Szechinski J., et al. Efficacy of the novel B cell targeted therapy, rituximab, in patients with active rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581.
Damsker J.M., Hansen A.M., Caspi R.R. TH1 and TH17 cells: adversaries and collaborators. Ann N Y Acad Sci. 2010;1183:211–221.
Rioux J.D., Goyette P., Vyse T.J., et alInternational MHC and Autoimmunity Genetics Network. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc Natl Acad Sci USA. 2009;106:18680–18685.
Keymeulen B., Vandemeulebroucke E., Ziegler A.G., et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352:2598–2608.
Levine J.S., Subang R., Nasr S.H., et al. Immunization with an apoptotic cell-binding protein recapitulates the nephritis and sequential autoantibody emergence of systemic lupus erythematosus. J Immunol. 2006;177:6504–6516.
McGaha T.L., Sorrentino B., Ravetch J.V. Restoration of tolerance in lupus targeted inhibiting receptor expression. Science. 2005;307:590–593.
Notley C.A., Ehrenstein M.R. The yin and yang of regulatory T cells and inflammation in RA. Nat Rev Rheumatol. 2010;6:572.
Park H., Li Z., Yang X.O., et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141.
Rahman A., Isenberg D.A. Systemic lupus erythematosus. N Engl J Med. 2008;358:929–939.
Roitt I.M., Doniach D., Campbell P.N., Vaughan-Hudson R. Auto-antibodies in Hashimoto’s disease (lymphadenoid goitre). Lancet. 1956(ii):820–821.
Suber T., Rosen A. Apoptotic cell blebs: repositories of autoantigens and contributors to immune context. Arthritis Rheum. 2009;60:2216–2219.
Wakeland E.K., Liu K., Graham R.R., Behrens T.W. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 2001;15:397–408.
Zenewicz L.A., Abraham C., Flavell R.A., Cho J.H. Unraveling the genetics of autoimmunity. Cell. 2010;140:791–797.
Botto M., Dell’Agnola C., Bygrave A.E., et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–59.
Carr E.J., Clatworthy M.R., Lowe C.E., et al. Contrasting genetic association of IL2RA with SLE and ANCA-associated vasculitis. BMC Med Genet. 2009;10:22.
Davies K.A., Erlendsson K., Beynon H.L., et al. Splenic uptake of immune complexes in man is complement dependent. J Immunol. 1993;151:3866–3873.
Ding B., Padyukov L., Lundström E., et al. Different patterns of associations with anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis in the extended major histocompatibility complex region. Arthritis Rheum. 2009;60:30–38.
Herrmann M., Voll R.E., Zoller O.M., et al. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:1241–1250.
Isenberg D.A., Manson J.J., Ehrenstein M.R., et al. Anti-dsDNA antibodies- at journey’s end? Rheumatology. 2007;46:1052–1056.
Kain R., Exner M., Brandes R., et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med. 2008;14:1088–1096.
Lerner R.A., Glassock R.J., Dixon F.J. The role of anti-glomerular basement membrane antibody in the pathogenesis of human glomerulonephritis. J Exp Med. 1967;126:989–1004.
Lin J.P., Cash J.M., Doyle S.Z., et al. Familial clustering of rheumatoid arthritis with other autoimmune diseases. Hum Genet. 1998;103:475–482.
Mattey D.L., Dawes P.T., Clarke S., et al. Relationship among the HLA-DRB1 shared epitope, smoking, and rheumatoid factor production in rheumatoid arthritis. Arthritis Rheum. 2002;47:403–407.
McKinney E., Lyons P.A., Carr E.J., et al. A CD8+ T cell transcription signature predicts prognosis in autoimmune disease. Nat Med. 2010;16:586–591.
Niewold T.B., Hua J., Lehman T.J., et al. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8:492–502.
Pinching A.J., Rees A.J., Pussell B.A., et al. Relapses in Wegener’s granulomatosis: the role of infection. Br Med J. 1980;281:836–838.
Rioux J.D., Goyette P., Vyse T.J., et al. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc Natl Acad Sci USA. 2009;106:18680–18685.
Silman A.J., Newman J., MacGregor A.J. Cigarette smoking increases the risk of rheumatoid arthritis: Results from a nationwide study of disease-discordant twins. Arthritis Rheum. 1996;39:732–735.
Stone J.H., Merkel P.A., Spiera R., Seo P., et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–232.
Tan E.M., Feltkamp T.E., Smolen J.S., et al. Range of antinuclear antibodies in “healthy” individuals. Arthritis Rheum. 1997;40:1601–1611.
Wakeland E.K., Liu K., Graham R.R., Behrens T.W. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 2001;15:397–408.
Weinblatt M., Kavanaugh A., Genovese M., et al. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med. 2010;363:1303–1312.
Yamada H., Nakashima Y., Okazaki K., et al. TH1 but not TH17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis. 2008;67:1299–1304.