CHAPTER 88 Autoimmune Hepatitis
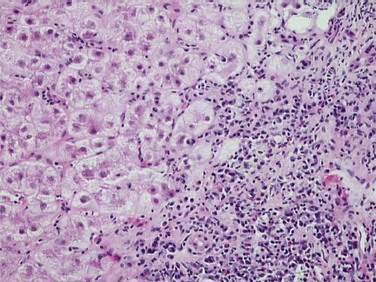
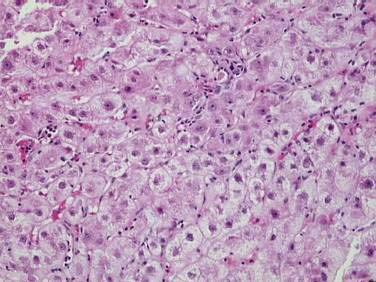
Autoimmune hepatitis (AIH) is a disorder of unknown cause characterized by unresolving inflammation of the liver and by the presence of interface hepatitis on histologic examination (Fig. 88-1), hypergammaglobulinemia, and autoantibodies.1 Diagnosis requires the exclusion of other chronic liver diseases that have similar features, including Wilson disease, chronic viral hepatitis, drug-induced liver disease, nonalcoholic fatty liver disease, and the immune cholangiopathies of primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC). Panacinar or lobular hepatitis is within the histologic spectrum of AIH (Fig. 88-2), and centrilobular (Rappaport zone 3) necrosis has been described in AIH and may indicate an early stage of the disease before the development of interface hepatitis.2
DIAGNOSTIC CRITERIA
An international panel codified the diagnostic criteria of AIH in 1992, and an expanded panel updated them in 1999.3 The propensity for an acute, rarely fulminant, presentation has been recognized,2 and the requirement for six months of disease activity to establish chronicity has been waived.3 Cholestatic histologic changes, including bile duct injury and ductopenia, are incompatible findings, but trivial biliary changes within the background of classic histologic features do not preclude the diagnosis.4,5
The serologic tests essential for diagnosis are assays for antinuclear antibodies (ANA), smooth muscle antibodies (SMA), and antibodies to liver-kidney microsome type 1 (anti-LKM1).1,6,7 These assays are based on the indirect immunofluorescence of rodent tissues or Hep-2 cell lines or on enzyme immunoassays using microtiter plates with adsorbed recombinant or highly purified antigens. Atypical perinuclear anti-neutrophil cytoplasmic antibodies (atypical pANCA) are common in type 1 AIH, PSC, and chronic ulcerative colitis.6,7 They are directed against antigens within the nucleus, rather than the cytoplasm, of granulocytes, and the reactivities localize to the proteins within the lamina of the nucleus. Because these antibodies are directed against nuclear envelope antigens rather than cytoplasmic antigens, a better term for them is anti-neutrophil nuclear antigens or “ANNA.”8 Atypical perinuclear anti-neutrophil cytoplasmic antibodies have been useful in evaluating patients who lack the conventional autoantibodies.6,7 Celiac disease can be associated with liver disease that resembles AIH and should be excluded in patients with cryptogenic chronic hepatitis by screening for immunoglobulin (Ig) A antibodies to tissue transglutaminase or endomysium.6,7,9 IgA endomysial antibodies are more predictive of celiac disease in patients with AIH than are IgA antibodies to tissue transglutaminase, which can be stimulated by hepatic inflammation and fibrogenesis.6,7
New autoantibodies continue to be characterized in the hope of improving diagnostic specificity and prognostic value, but none has been incorporated into conventional diagnostic algorithms.6,7,10 Antibodies to soluble liver antigen (anti-SLA), actin (anti-actin), chromatin (anti-chromatin), asialoglycoprotein receptor (ASGPR), and liver cytosol type 1 (anti-LC1) have been associated with severe AIH, poor treatment response, and relapse after drug withdrawal.10–13 Their major clinical limitation has been their infrequent individual occurrence in AIH.
CLINICAL CRITERIA
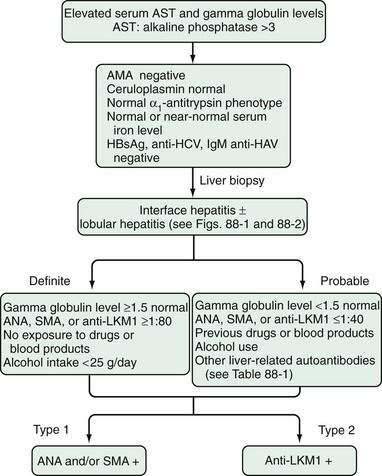
The definite diagnosis of AIH requires exclusion of other similar diseases; laboratory findings that indicate substantial immunoreactivity; and histologic features of interface hepatitis (Fig. 88-3).3 A probable diagnosis is justified when findings are compatible with AIH but insufficient for a definite diagnosis (see Fig. 88-3).3 Patients who lack conventional autoantibodies but who are seropositive for investigational markers, such as antibodies to ASGPR, SLA, actin, or LC1, are classified as having probable disease.3
SCORING CRITERIA
The original scoring system proposed by the International Autoimmune Hepatitis Group accommodates the diverse manifestations of AIH and renders an aggregate score that reflects the net strength of the diagnosis before and after glucocorticoid treatment (Table 88-1).3 By weighing each component of the syndrome, discrepant features can be accommodated and biases associated with isolated inconsistencies can be avoided. The original scoring system ensures the comparability of study populations in clinical trials, provides a comprehensive template for systematically assessing all features of the disease, and can render a diagnosis of AIH in patients with few or atypical features.3 The original scoring system is not a discriminative diagnostic index, and it should not be used to distinguish AIH from other liver diseases.
Table 88-1 Revised Original Scoring System for the Diagnosis of Autoimmune Hepatitis
| CATEGORY | VARIABLE | SCORE |
|---|---|---|
| Gender | Female | +2 |
| AP/AST | >3 | −2 |
| <1.5 | +2 | |
| Gamma globulin or IgG level above normal | >2.0* | +3 |
| 1.5-2.0* | +2 | |
| 1.0-1.5* | +1 | |
| <1.0 | 0 | |
| ANA, SMA, or anti-LKM1 titer | >1 : 80 | +3 |
| 1 : 80 | +2 | |
| 1 : 40 | +1 | |
| <1 : 40 | 0 | |
| AMA | Positive | −4 |
| Viral markers | Positive | −3 |
| Negative | +3 | |
| Drug history | Yes | −4 |
| No | +1 | |
| Alcohol | <25 g/day | +2 |
| >60 g/day | −2 | |
| HLA | DR3 or DR4 | +1 |
| Immune disease | Thyroiditis, ulcerative colitis, synovitis, others | +2 |
| Other liver-defined autoantibodies | Anti-SLA, anti-actin, anti-LC1, pANCA | +2 |
| Histologic features | Interface hepatitis | +3 |
| Plasmacytic infiltrate | +1 | |
| Rosettes | +1 | |
| None of above | −5 | |
| Biliary changes | −3 | |
| Other features | −3 | |
| Treatment response | Complete | +2 |
| Relapse | +3 | |
| Pretreatment Score | ||
| Definite diagnosis | >15 | |
| Probable diagnosis | 10-15 | |
| Post-treatment Score | ||
| Definite diagnosis | >17 | |
| Probable diagnosis | 12-17 |
AMA, antimitochondrial antibodies; ANA, antinuclear antibodies; anti-LC1, antibodies to liver cytosol type 1; anti-LKM1, antibodies to liver-kidney microsome type 1; anti-SLA, antibodies to soluble liver antigen; AP/AST (or AP/ALT), ratio of serum alkaline phosphatase level to serum aspartate aminotransferase (or serum alanine aminotransferase) level; HLA, human leukocyte antigen; IgG, immunoglobulin G; pANCA, perinuclear anti-neutrophil cytoplasmic antibodies; SMA, smooth muscle antibodies.
* Times upper limit of normal.
Adapted from Alvarez F, Berg PA, Bianchi FB, et al. International Autoimmune Hepatitis Group report: Review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 1999; 31:929-38.
A simplified scoring system has been developed to ease clinical application and is based on four clinical components that include the presence and level of autoantibody expression by indirect immunofluorescence, serum IgG concentration, histologic features, and viral markers (Table 88-2).14 The original scoring system has greater sensitivity for the diagnosis than the simplified system (100% vs. 95%), but the simplified system has greater specificity (90% vs. 73%) and predictability (92% vs. 82%).15 Whereas the original scoring system is useful for evaluating patients in whom every component must be assessed because of few or atypical findings, the simplified scoring system is useful for excluding AIH in patients with other conditions and concurrent immune features.
Table 88-2 Simplified Scoring System for Diagnosis of Autoimmune Hepatitis*
| CATEGORY | VARIABLE | SCORE |
|---|---|---|
| Autoantibodies† | ||
| Antinuclear antibodies or smooth muscle antibodies | 1 : 40 | +1 |
 1 : 80 1 : 80 |
+2 | |
| Antibodies to liver-kidney microsome type 1 |  1 : 40 1 : 40 |
+2 |
| Antibodies to soluble liver antigen | Positive | +2 |
| Immunoglobulin Level | ||
| Immunoglobulin G | >Upper limit of normal | +1 |
| >1.1 times upper limit of normal | +2 | |
| Histologic Findings | ||
| Morphologic features | Compatible with autoimmune hepatitis | +1 |
| Typical of autoimmune hepatitis | +2 | |
| Viral Disease | ||
| Absence of viral hepatitis | No viral markers | +2 |
| Pretreatment Aggregate Score | ||
| Definite diagnosis | ≥7 | |
| Probable diagnosis | 6 |
* Adapted from Hennes EM, Zeniya M, Czaja AJ, et al. Simplified diagnostic criteria for autoimmune hepatitis. Hepatology 2008; 48:169-76.
† Autoantibody titers as determined by indirect immunofluorescence.
PATHOGENESIS
The pathogenic mechanisms of AIH are unknown. The most popular hypotheses invoke a constellation of interactive factors that include a triggering agent, genetic predisposition, and various determinants of autoantigen display, immunocyte activation, and effector cell expansion (Fig. 88-4).16–18 Proposed triggering factors include infectious agents, drugs, and toxins. The lag time between exposure to the trigger and onset of the disease can be long, and the triggering factor may not be needed for perpetuation of the disorder. The CD4+ helper T cell is the principal effector cell, and its activation is the initial step in the pathogenic pathway.
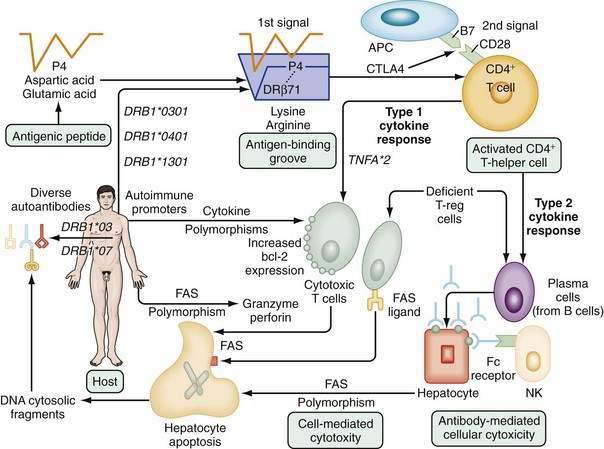
Molecular mimicry of a foreign antigen and a self-antigen is the most common explanation for the loss of self-tolerance, but this mechanism has not been established for any autoimmune disease.16–18 Genetic factors influence autoantigen presentation and CD4+ helper T cell recognition. The antigen-binding groove of the class II molecule of the major histocompatibility complex (MHC) is encoded by alleles that determine the groove’s configuration and ability to activate immunocytes. The susceptibility alleles of AIH in white North Americans and northern Europeans reside on the DRB1 gene and are DRB1*0301 and DRB1*0401 (see Fig. 88-4).19–21
Different ethnic groups have different susceptibility alleles, a finding that supports a “shared motif hypothesis” of pathogenesis.19–21 According to this hypothesis, the risk of disease relates to amino acid sequences in the antigen-binding groove of the class II MHC molecule, and multiple alleles encode the same or similar sequence (“shared motif”). The critical shared motif in white North Americans and northern Europeans with AIH is a six-amino-acid sequence represented by the code LLEQKR.19–21 This sequence is located between positions 67 and 72 of the DRβ polypeptide chain of the class II MHC molecule, and lysine (K) in position 71 is the critical determinant of susceptibility. DRB1*0301 and DRB1*0401 encode identical amino acid sequences in the DRβ67-72 region and affect susceptibility similarly.
DRB1*0404 and DRB1*0405 are the susceptibility alleles in Mexican, Japanese, mainland Chinese, and Argentine adults and encode a similar sequence, except for arginine (R) instead of lysine (K) at the DRβ71 position.20,21 Arginine is a positively charged amino acid that is structurally similar to lysine, and its substitution for lysine should not greatly alter the antigen-binding properties of the class II MHC molecule. By contrast, DRB1*1501 protects against AIH in white North Americans and northern Europeans, and this allele encodes isoleucine (I) instead of leucine (L) at position DRβ67 and alanine (A) instead of lysine (K) at position DRβ71. Alanine is a neutral, nonpolar amino acid that, when substituted for lysine, should greatly affect antigen presentation and immunocyte activation.
Antigenic peptides are selected for display by the nature of the amino acids that interact with residues within the antigen-binding groove (see Fig. 88-4).21,22 The critical six-amino-acid motif in AIH restricts the range of peptides that can be accommodated. Multiple self-antigens or foreign antigens may satisfy the minimal structural requirements and serve as immunogenic peptides. The ideal triggering epitope must have a negatively charged amino acid residue (aspartic acid or glutamic acid) at peptide position P4 to form a salt bridge with the positively charged lysine or arginine at DRβ71.21 Molecular modeling indicates that a negatively charged P4 residue in the antigenic peptide and the positively charged lysine or arginine at DRβ71 can form a P4-DRβ71 immunoreactive unit that is independent of the other residues within the antigen and antigen-binding groove. This minimal immunoreactive unit can be created by multiple antigenic peptides and class II MHC molecules, and the number of these units may affect susceptibility by a “dose effect.”
DRB1*1301 is associated with AIH in Argentine children22 and Brazilian patients23,24 and encodes ILEDER at positions DRβ67-72. Glutamic acid (E), aspartic acid (D), and glutamic acid (E) are at positions DRβ69, DRβ70, and DRβ71, respectively, in the class II MHC molecule, and the presence of these critically located but negatively charged amino acid residues argues against the “shared motif” hypothesis of pathogenesis. The “molecular footprint” hypothesis of pathogenesis holds that susceptibility to AIH in different regions and ethnic groups relates to indigenous factors or agents favored by certain genetic phenotypes.20,21 In South America, DRB1*1301 is associated with protracted hepatitis A virus infection, and persons with this allele may be “selected” from their environment to have prolonged exposure to viral and hepatic antigens that favor the development of AIH.25 An understanding of the individual susceptibility allele in different geographic regions may allow use of this “footprint” to track the cause of the disease.
The “autoimmune promoter hypothesis” of pathogenesis complements the “shared motif” and “molecular footprint” hypotheses by proposing that genetic promoters inside and outside the MHC can affect disease occurrence, either in synergy (epistasis) with the principal susceptibility factors or in lieu of them.18,20,21 Polymorphisms of the tumor necrosis factor (TNF)-α gene (TNFA*2),26 the cytotoxic T lymphocyte antigen 4 gene (CTLA4),27 and Fas gene promoter at position −670 (TNFRSF6)28 have been associated with increased immunoreactivity, disease severity, and early progression to cirrhosis in white North American and northern European patients. Constellations of autoimmune promoters, as yet undefined, may individualize the disease by affecting its occurrence, clinical phenotype, and outcome.
Liver cell destruction is accomplished by either cell-mediated cytotoxicity, antibody-dependent cell-mediated cytotoxicity, or a combination of both mechanisms (see Fig. 88-4).16–18 Cell-mediated cytotoxicity depends on the clonal expansion of CD8+ cytotoxic T cells that accomplish liver cell injury through the release of lymphokines. This mechanism is regulated by type 1 cytokines, and the −308 polymorphism of TNFA*2 may facilitate this pathway.26 Antibody-dependent cell-mediated cytotoxicity is regulated by type 2 cytokines, and the natural killer cell accomplishes liver cell destruction by binding of its Fc receptor with an antigen-antibody complex on the hepatocyte surface.16–18 The predominant mechanism depends on the phenotypic differentiation of the CD4+ helper T cell, which in turn reflects the cytokine milieu. The cytokine milieu may reflect polymorphisms of the cytokine genes that favor excessive production of some modulators, such as TNF-α, or deficient production of others.
Defects in the counter-regulatory cytokine milieu may also reflect reduced numbers of intrahepatic natural killer T (NKT) cells and the failure of T-regulatory (T-reg) cells (CD4+CD25+ cells) to modulate CD8+ T cell proliferation and cytokine production.18,29 Increased numbers of γδ T cells may also contribute to the cytodestructive process by recognizing antigens presented by the non-classic MHC molecules, and the recruitment and intrahepatic trafficking of cytotoxic T lymphocytes may be enhanced by the up-regulation of chemokines, such as CXCL16.18 Fibrogenesis, in turn, is fueled by the resulting inflammatory activity as perivascular hepatic stellate cells transform into myofibroblasts. The matrix proteins accumulate as tissue inhibitors retard the counteractive degradative actions of matrix metalloproteinases, and stellate cells continue to be activated in an autocrine fashion by transforming growth factor-β (TGF-β).30 Glucocorticoid therapy can favorably alter the cytokine milieu, improve the number and function of the T-reg cells, impair activation of TGF-β, promote disappearance of the metalloproteinase inhibitors, enhance degradation of the fibrotic liver matrix, and foster apoptosis of the hepatic stellate cells (see also Chapter 2).31
CLASSIFICATION
Two types of AIH have distinctive serologic profiles. Neither has been ascribed a unique cause, specific treatment strategy, or special type of behavior (Table 88-3). The terms are useful as clinical descriptors and as research designations to ensure homogeneous study populations.
Table 88-3 Classification of Autoimmune Hepatitis Based on Autoantibodies
| CLINICAL FEATURE | TYPE 1 | TYPE 2 |
|---|---|---|
| Signature autoantibodies | Smooth muscle | Liver-kidney microsome type 1 |
| Nuclear | ||
| Associated autoantibodies | Atypical pANCA | Liver cytosol type 1* |
| Actin* | Soluble liver antigen* | |
| Asialoglycoprotein receptor* | ||
| Chromatin* | ||
| Soluble liver antigen* | ||
| Putative autoantigen | Unknown | CYP2D6 |
| Age | Infants to elderly | Children (2-14) |
| Female | 78% | 89% |
| Concurrent immune diseases | 38% | 34% |
| Typical concurrent autoimmune diseases | Autoimmune thyroiditis | Autoimmune thyroiditis |
| Graves’ disease | Vitiligo | |
| Ulcerative colitis | Type 1 diabetes mellitus | |
| APECED | ||
| Organ-specific antibodies | 4% | 30% |
| Serum gamma globulin elevation | +++ | + |
| HLA associations | B8, DR3, DR4, DR13 | B14, DR3, C4A-Q0, DR7 |
| Allelic risk factors | DRB1*0301 and *0401 | DQB1*0201, DRB1*07, DRB1*03 |
| (white North Americans and northern Europeans) | (Germans and Brazilians) | |
| DRB1*1301 | ||
| (South Americans, especially children) | ||
| Glucocorticoid responsive | +++ | ++ |
APECED, autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy; CYP2D6, cytochrome P450 2D6; pANCA, perinuclear anti-neutrophil cytoplasmic antibodies.
* Autoantibodies with an asterisk are investigational only and not available for routine clinical use.
TYPE 1 AUTOIMMUNE HEPATITIS
Type 1 AIH is characterized by SMA, ANA, or both (see Table 88-3).1,7 Antibodies to actin have greater specificity, but less sensitivity, for the diagnosis of AIH than SMA.6 Atypical pANCA are found in as many as 90% of patients with type 1 AIH and typically are absent in type 2 AIH.6,7
Type 1 AIH can occur at any age and in either gender (see Table 88-3).7 Initial studies that suggested a bimodal age distribution probably reflected referral biases to tertiary medical centers. The disease has been described in infants and probably is underdiagnosed in the elderly.32 Seventy-eight percent of patients are women (female-to-male ratio 3.6 : 1), and 38% have concurrent extrahepatic immunologic diseases.7 Autoimmune thyroiditis (occurring in 12% of the cases), Graves’ disease (6%), and ulcerative colitis (6%) are the most common associated immune disorders. Rheumatoid arthritis, pernicious anemia, scleroderma, Coombs-positive hemolytic anemia, autoimmune thrombocytopenic purpura, symptomatic cryoglobulinemia, leukocytoclastic vasculitis, nephritis, erythema nodosum, systemic lupus erythematosus, and fibrosing alveolitis also may occur (less than 1% each). Cholangiography is warranted to exclude PSC in all patients who have concurrent ulcerative colitis (see Chapter 68).33
Type 1 AIH is associated with an abrupt onset of symptoms in 40% of cases and may manifest in a fulminant fashion.2 The acute presentation frequently reflects preexisting subclinical disease that is unmasked by progression or represents a spontaneous exacerbation of inflammatory activity. Features of chronicity are lacking in 8% of patients, in whom the presentation of the disorder is indistinguishable from that of acute viral or toxic hepatitis.
The target autoantigen of type 1 AIH is unknown. Human leukocyte antigen (HLA)-DR3 (DRB1*0301) and HLA-DR4 (DRB1*0401) are independent risk factors for the disease in white North Americans and northern Europeans.19–21 More than 80% of white patients in Great Britain and the United States possess either DRB1*0301, DRB1*0401, or both, compared with 42% of the unaffected white population. In South America, especially in children, DRB1*1301 is the principal susceptibility allele. These findings indicate that type 1 AIH is a complex polygenic disorder.
TYPE 2 AUTOIMMUNE HEPATITIS
Type 2 AIH is characterized by the expression of anti-LKM1 (see Table 88-3).1,7,34 Most affected persons are children (ages 2 to 14 years), but in Europe, especially in Germany and France, 20% of patients are adults.34 In the United States, type 2 AIH is rare, and only 4% of patients older than 18 years have anti-LKM1.35 The regional differences in prevalence may relate to ethnic differences in the genetic predisposition to the disease.36
Type 2 patients are younger than type 1 patients and may have different clinical and laboratory features (see Table 88-3).1,7,34 As with type 1 AIH, an acute or fulminant presentation is possible and important to recognize and treat early. Susceptibility to type 2 AIH has been associated with DQB1*0201, DRB1*07, and DRB1*03.37 DQB1*0201 is in strong linkage disequilibrium with DRB1*07 and DRB1*03 and has been proposed as the principal genetic determinant of the disease. The expression of anti-LKM1 has been associated with DRB1*07, and various aspects of type 2 AIH may have different genetic determinants.38
The target antigen of type 2 AIH is the cytochrome P450 2D6 mono-oxygenase (CYP2D6).39 This protein is a 50-kd microsomal drug-metabolizing enzyme, and its expression on the hepatocyte surface can be modulated by interleukins and TNF-α. Antibodies to LKM1 inhibit the activity of CYP2D6 in vitro but not in vivo, and lymphocytes extracted from the liver tissue of patients who have the disease exhibit immunoreactivity specific to the antigen.
CYP2D6 has been sequenced, cloned, and mapped, and five epitopes are recognized by anti-LKM1.40 The amino acid sequence spanning 193-212 of the CYP2D6 molecule is the target of anti-LKM1 in 93% of patients. Homologies exist between CYP2D6 and the genomes of the hepatitis C virus, cytomegalovirus, and herpes simplex type 1 virus. These molecular mimicries may result in cross-reacting antibodies and support the hypothesis that repeated viral infections may break self-tolerance and cause the disease.17
VARIANT FORMS
Patients who have atypical features of AIH currently lack an official designation and confident treatment strategy.41 They may have manifestations of AIH and another type of chronic liver disease or findings that are incompatible with AIH by current diagnostic criteria (Table 88-4).42,43
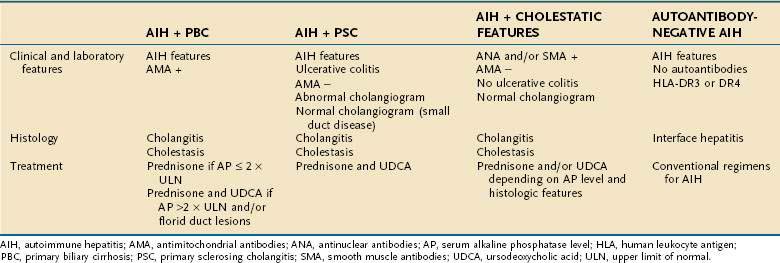
VARIANT WITH PRIMARY BILIARY CIRRHOSIS
AIH in patients who also have antimitochondrial antibodies (AMA) and histologic features of cholangitis constitutes an overlap syndrome with PBC (see Table 88-4).41–44 Typically, affected patients have low titers of AMA and concurrent features of bile duct injury or loss.45 Antibodies against the PBC-specific M2 mitochondrial antigens may be present46; histologic features of cholangitis, including destructive cholangitis, may be seen45; and copper staining of hepatic tissue indicative of cholestasis may be observed.45 Occurrence rates range from 5% of patients initially diagnosed as having AIH to 19% of patients initially diagnosed as having PBC (see also Chapter 89).42
The clinical course of the disease and response to treatment depend mainly on the predominant component of the disease. Patients who have high serum aspartate aminotransferase (AST) levels, serum alkaline phosphatase levels less than twice the upper limit of normal, moderate to severe interface hepatitis on histologic examination, and high diagnostic scores for AIH commonly respond to glucocorticoid therapy.42,45 By contrast, patients who have serum alkaline phosphatase levels greater than twice the upper limit of normal, serum gamma glutamyl transpeptidase levels at least five times the upper limit of normal, and florid bile duct lesions on histologic examination mainly have PBC and commonly respond to ursodeoxycholic acid in combination with glucocorticoids (prednisone, prednisolone, or budesonide).44
AMA can be detected in 18% of patients with AIH in the absence of cholestatic features and can appear and disappear during the course of the disease without evolution to a different syndrome or the need for different therapy.47,48 Histologic changes of bile duct injury are required in addition to AMA and other classic features of AIH to designate a variant form of AIH.
VARIANT WITH PRIMARY SCLEROSING CHOLANGITIS
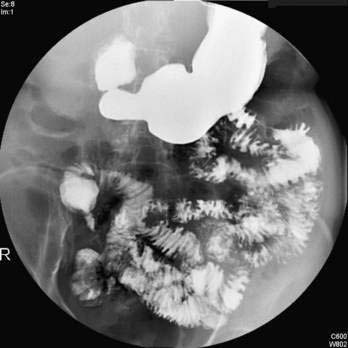
Histologic changes of lymphocytic, pleomorphic, or fibrous cholangitis; cholestatic laboratory findings; concurrent inflammatory bowel disease; or failure to respond to glucocorticoids constitute indications for cholangiography in patients who have AIH.41–43 As many as 41% of persons with one of these features have cholangiographic changes of PSC and are classifiable as having a variant syndrome of AIH (see Table 88-4).33 Furthermore, 54% of patients who have PSC have features that support a probable or definite diagnosis of AIH.42 The absence of characteristic cholangiographic changes does not preclude the diagnosis of PSC because small-duct disease may be present (see Table 88-4) (see Chapter 68).33,49 Magnetic resonance cholangiography has demonstrated unsuspected PSC of the large bile ducts in 8% of adults with AIH, and the possibility of PSC must be evaluated in all individuals with AIH and disease refractory to glucocorticoid therapy regardless of other features.50
Children with AIH may also have unsuspected bile duct changes. Autoimmune sclerosing cholangitis is a disorder described in children who have the clinical phenotype of AIH but abnormal findings on cholangiographic studies.51 Inflammatory bowel disease is frequently absent, and these children can respond to glucocorticoid therapy; in this respect, they may differ from adults with AIH and PSC.
Treatment is empirical and typically ineffective in adults.33,42,43 Glucocorticoids and ursodeoxycholic acid (13 to 15 mg/kg orally daily), alone or in combination, can be considered, depending on whether hepatitic or cholestatic features predominate. Some studies have suggested that high-dose ursodeoxycholic acid (20 to 25 mg/kg daily) has some value in typical PSC, and this treatment can be considered in conjunction with glucocorticoids.
VARIANT WITH CHOLESTATIC FEATURES
Eight percent of patients with AIH have histologic features of bile-duct injury and laboratory changes of cholestasis in the absence of AMA and cholangiographic changes of large-duct PSC.42,52 These individuals may have AMA-negative PBC, small-duct PSC, or a separate syndrome (“autoimmune cholangitis”) (see Table 88-4 and Chapter 89).42,52 This variant is probably a heterogeneous category that encompasses patients with atypical, early, or transitional features of classic disease.52 Persons who have this variant are inconsistently responsive to glucocorticoids and ursodeoxycholic acid.42,52 Preliminary experience suggests that these therapies can help to improve the clinical and laboratory abnormalities but not the histologic changes. Histologic findings of bile duct injury, including destructive cholangitis, may be seen in classic AIH in the absence of other cholestatic features.4,5 These changes are transient and probably represent collateral damage associated with severe inflammatory activity; they do not constitute a variant syndrome, nor do they change the diagnosis or affect the treatment strategy.
AUTOANTIBODY-NEGATIVE AUTOIMMUNE HEPATITIS
Thirteen percent of adults with chronic hepatitis of undetermined cause satisfy international criteria for the diagnosis of AIH but lack the characteristic autoantibodies (see Table 88-4).53 These patients commonly are designated as having cryptogenic chronic hepatitis and may be excluded inappropriately from therapies of potential benefit. Autoantibody-negative patients are similar in age, female predominance, frequency of concurrent immunologic diseases, histologic features, and laboratory findings to patients with classic AIH.53 Furthermore, they have similar frequencies of HLA-B8, HLA-DR3, and HLA-A1-B8-DR3,53 and they respond as well to glucocorticoid treatment as do their autoantibody-positive counterparts.53,54 These persons probably have a form of AIH that has escaped detection by conventional serologic assays, and they are candidates for a closely monitored treatment trial of glucocorticoids. Assays for atypical pANCA, anti-SLA, and IgA endomysial antibodies or antibodies to tissue transglutaminase occasionally yield positive results in these patients,6,7 and successive testing for conventional autoantibodies may demonstrate the late appearance of typical autoimmune markers in some cases (see Table 88-4).55
AUTOIMMUNE HEPATITIS AND CHRONIC HEPATITIS C
Eight percent of white North American adults with classic AIH have concurrent infection with hepatitis C virus (HCV), and 52% of patients with chronic hepatitis C have autoantibodies, concurrent immune diseases, or both.56 Identification of these patients as belonging to one or the other group is important because interferon therapy can enhance the immune manifestations of persons with AIH and concurrent HCV infection, and immunosuppressive treatment can increase serum viral levels in persons with chronic hepatitis C and background autoimmune features.
The nature and degree of the associated immune manifestations distinguish AIH with coincidental HCV infection from chronic hepatitis C with autoimmune features (Fig. 88-5).56,57 Concurrent immune diseases that reflect a cell-mediated response against autoantigens (autoimmune thyroiditis, Graves’ disease, inflammatory bowel disease) typify an autoimmune process and occur more commonly in AIH than in chronic hepatitis C.56,57 They also are more likely to have clinical consequences if the immune diseases are exacerbated during antiviral therapy. Serum autoantibody titers can increase during antiviral treatment, but this increase is neither pathogenic nor associated with clinical deterioration. Patients with chronic hepatitis C and concurrent autoimmune features have immune complex diseases (vasculitis, glomerulonephritis, and symptomatic cryoglobulinemia) more frequently than do patients with classic AIH, and these findings support a predominantly viral disease. The immune manifestations are driven mainly by viral antigen.56
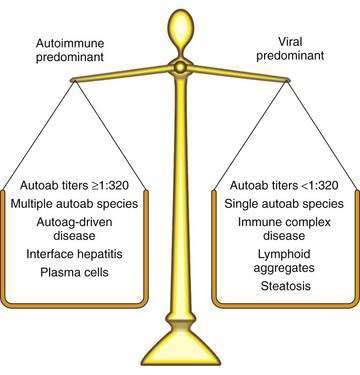
Patients with classic AIH typically express multiple antibodies in titers that exceed 1 : 320.56 By contrast, patients with chronic hepatitis C typically express one type of autoantibody (ANA or SMA) in titers that are usually less than 1 : 320. Furthermore, patients with AIH have higher serum levels of AST, gamma globulin, and IgG than do patients with chronic hepatitis C. High serum titers (greater than 1 : 320) of autoantibodies, marked hypergammaglobulinemia, multiple autoantibody types, or the presence of autoantigen-driven concurrent immune diseases indicates an autoimmune-predominant syndrome. Further differentiation requires liver biopsy evaluation.
Patients with classic AIH more commonly have severe interface hepatitis, moderate to severe portal plasma cell infiltration, and panacinar hepatitis in liver biopsy specimens than do patients with typical chronic hepatitis C.57 By contrast, patients with typical chronic hepatitis C have a higher frequency of portal lymphoid aggregates and steatosis than is seen in patients with classic AIH (see also Chapter 79). Histologic diagnoses based on these predominant patterns have high specificity (81% and 91%) and predictability (62% and 82%) for AIH and chronic hepatitis C, respectively.58 Their sensitivity for each clinical diagnosis, however, is low (40% and 57%, respectively). Treatment of patients with mixed features of AIH and chronic hepatitis C should be appropriate for the predominant disease and must be based on the nature of concurrent immune diseases, number and titers of associated autoantibodies, and histologic pattern. Combined therapies with glucocorticoid and antiviral drugs should be avoided.
EPIDEMIOLOGY
The incidence of AIH among white northern Europeans is 1.9 cases per 100,000 persons per year, and its point prevalence is 16.9 cases per 100,000 persons per year.59 In the United States, AIH affects 100,000 to 200,000 persons and accounts for 2.6% of the transplantations in the European Liver Transplant Registry and 5.9% in the National Institutes of Health Liver Transplantation Database. The frequency of AIH among patients with chronic liver disease in North America is between 11% and 23%.
The impact of genetic risk factors must be considered in assessing the occurrence of disease in different regions. The prevalence of AIH is greatest among northern European white persons who have a high frequency of HLA-DR3 and HLA-DR4, and AIH is found with similar frequency in the derivative populations of North America and Australia. The Japanese have a low frequency of HLA-DR3, and AIH in Japan is associated with HLA-DR4.20,21 All populations are susceptible to AIH, which has been described in African Americans, South Americans, Arabs, Japanese, mainland Chinese, and subcontinental Indians. The prevalence of AIH among Alaskan natives (43 per 100,000 population) is higher than that reported in a white Norwegian population (16.9 per 100,000).60
PROGNOSIS INDICES
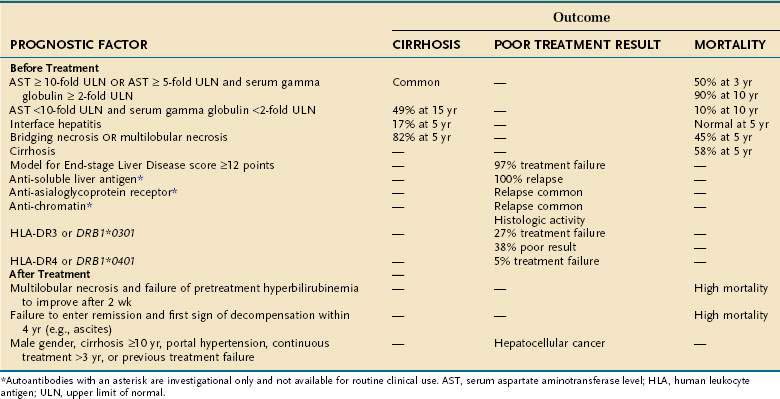
The prognosis for AIH relates mainly to the severity of liver inflammation at the initial medical consultation, as reflected in the laboratory indices and the histologic findings. HLA status and ethnicity influence disease occurrence, clinical phenotype, and treatment outcome (Table 88-5). Certain serologic markers have shown promise as prognostic instruments, and the Model for End-stage Liver Disease (MELD) score may be useful for identifying problematic patients early.
LABORATORY INDICES
Serum AST and gamma globulin levels reflect the severity of disease and immediate prognosis. Sustained severe elevations indicate a poor outcome unless therapy is started. Less severe laboratory abnormalities are associated with a better prognosis (see Table 88-5).1 Mild AIH may still progress, and the laboratory indices reflective of mild inflammatory activity are unable to predict disease behavior in individual patients.61
Spontaneous resolution is possible in 13% to 20% of patients regardless of disease activity. No features predict this outcome, and patients should not be managed with this expectation.61 Of persons who survive the early, most active stage of the disease, inactive cirrhosis develops in 41%. Patients who receive no treatment and who have initially severe disease and survive the first two years of illness typically survive long term.
HISTOLOGIC FINDINGS
The histologic findings at presentation also are indices of disease severity, and each pattern of liver cell injury has its own prognostic implication (see Table 88-5).1 Esophageal varices develop in 54% of patients with cirrhosis, and death from variceal hemorrhage occurs in 20% of those with varices if treatment of AIH is not instituted. Hepatocellular carcinoma (HCC) also can occur in patients with cirrhosis. The overall Kaplan-Meier 10-year probability of HCC is 2.9%, and the overall incidence of HCC is four cases per 1000 patient-years. The risk of HCC is small and mainly affects males and patients with longstanding cirrhosis (>10 years).62
HUMAN LEUKOCYTE ANTIGEN STATUS
White North American and northern European patients with HLA-DR3 are typically younger than patients with other HLA alleles and have more active disease (see Table 88-5).63,64 Whites with HLA-DR3 (DRB1*0301) respond less well to glucocorticoids than do patients with HLA-DR4 (DRB1*0401), whereas those with HLA-DR4 (DRB1*0401) have concurrent immune diseases more commonly and better outcomes than those with HLA-DR3 (DRB1*0301).63,64 By contrast, DRB1*1501 protects against AIH in white North Americans and northern Europeans. Other ethnic groups and individuals in other geographic regions may have different genetic predispositions that reflect diverse etiologic agents or antigenic exposures. At present, the clinical applications of HLA testing in AIH are uncertain, and determinations are not made routinely in practice.
ETHNICITY
Ethnicity may affect disease severity as well as presentation.65 Cirrhosis is present at accession more commonly, and hepatic synthetic function is decreased more frequently,66 in black North American patients with AIH than in white North American patients (85% vs. 38%). Both groups respond similarly to glucocorticoids, but black North American patients are younger at presentation than their white counterparts. These findings suggest that black North Americans have more aggressive disease than is seen in white North Americans and that their higher frequency of advanced disease reflects intrinsic disease behavior.
Studies in Alaskan natives, Turks, Japanese, South Americans, non-European, non-white patients, and Somalians also have emphasized clinical and prognostic differences among the racial groups. Alaskan natives have a higher frequency of acute icteric disease, asymptomatic illness, and advanced fibrosis at presentation than is characteristic of their white counterparts.60 Japanese patients lack the DRB1*0301 allele, which has been associated with disease occurrence, early age at onset, and poor treatment response in white populations.64 The clinical phenotype and treatment requirements in Japanese patients with AIH differ from those of their white counterparts. They typically have mild, late-onset disease that may respond to nonsteroidal medications such as ursodeoxycholic acid.
South American patients in Brazil and Argentina are younger than their North American white counterparts.23,24 They have more severe laboratory abnormalities, and DRB1*1301 is their principal susceptibility allele. African, Asian, and Arab patients also have an earlier age at onset than their white northern European counterparts and a higher frequency of cholestatic laboratory findings, greater occurrence of biliary changes on histologic examination, and poorer initial response to standard therapy than are found for white patients or other ethnic groups.67 Interwoven into the natural history of AIH in each ethnic population and geographic region are cultural and socioeconomic factors that may affect the time to diagnosis and access to treatment.
SEROLOGIC MARKERS
Emerging serologic tests for AIH reflect the goal of identifying markers that have prognostic as well as diagnostic value (see Table 88-5).6,7 Antibodies to SLA satisfy this role with a specificity of 99% for AIH and a strong association with severe disease and relapse after glucocorticoid withdrawal.11,12 The target autoantigen of anti-SLA is a 50-kd protein that may be a transfer ribonucleoprotein complex (tRNP(Ser)Sec) involved in the incorporation of selenocysteine into peptide chains.68 The expression of anti-SLA is closely associated with HLA-DR3, and antibodies to SLA may be surrogate markers of a genetic propensity for severe disease and relapse after drug withdrawal.11,12 Testing for anti-SLA may also be useful in reclassifying patients with cryptogenic chronic hepatitis.6,7
Antibodies to ASGPR are closely associated with histologic activity and also identify individuals who relapse after drug withdrawal possibly because of residual hepatic inflammation.6,7,13 Antibodies to chromatin and antibodies to actin are each being investigated as prognostic markers and can also identify patients who relapse after drug withdrawal (anti-chromatin) or die of liver failure or require liver transplantation (anti-actin).6,7,10
MODEL FOR END-STAGE LIVER DISEASE SCORE
The MELD score is an established method by which to predict early mortality associated with severe liver disease; it is based on the serum creatinine level, total serum bilirubin concentration, and international normalized ratio (INR).69 Survival predicted by the MELD score relates mainly to the degree of impaired liver function and not the presence of cirrhosis or the cause of liver disease. In AIH, a MELD score of ≥12 points at presentation identifies 97% of patients who fail to respond to glucocorticoid treatment. The specificity of this score for treatment failure is 68%, and its positive predictive value is 19% (see Table 88-5).69 Sensitivity for treatment failure is preferred over specificity in order to capture at presentation all patients at risk who can then be monitored more closely or considered for salvage therapies at the earliest sign of deterioration.
CLINICAL FEATURES
The clinical features of AIH frequently reflect the inflammatory activity of the liver disease or the complications of cirrhosis (Table 88-6).1,7 Cholestatic features may be present but do not dominate the clinical picture. Similarly, manifestations of liver decompensation, such as ascites, hepatic encephalopathy, and variceal bleeding, are uncommon findings at the initial medical consultation.
| OCCURRENCE (%) | |
|---|---|
| Symptoms | |
| Fatigue | 86 |
| Jaundice | 77 |
| Upper abdominal discomfort | 48 |
| Pruritus (mild) | 36 |
| None (at presentation) | 25-34 |
| Anorexia | 30 |
| Myalgias | 30 |
| Diarrhea | 28 |
| Cushingoid features | 19 |
| Fever (≤40°C) | 18 |
| Physical Findings | |
| Hepatomegaly | 78 |
| Jaundice | 69 |
| Spider angiomata | 58 |
| Concurrent immune disease | ≤38 |
| Splenomegaly | ≥32 |
| None | <25 |
| Ascites | 20 |
| Encephalopathy | 14 |
| Laboratory Features | |
| Elevated aspartate aminotransferase level | 100 |
| Hypergammaglobulinemia | 92 |
| Increased immunoglobulin G level | 91 |
| Hyperbilirubinemia | 83 |
| Alkaline phosphatase level ≥ 2-fold normal | 33 |
| Immunoserologic Markers* | |
| SMA, ANA, or anti-LKM1 | 100 |
| Atypical pANCA | 92 (type 1 AIH only) |
| Anti-asialoglycoprotein receptor* | 82 |
| Anti-actin* | 74 |
| Anti-chromatin* | 42 (ANA+ only) |
| Anti-liver cytosol 1* | 32 (type 2 AIH only) |
| Anti-soluble liver antigen* | 11-17 |
AIH, autoimmune hepatitis; ANA, antinuclear antibodies; anti-LKM1, antibodies to liver-kidney microsome type 1; pANCA, perinuclear anti-neutrophil cytoplasmic antibodies; SMA, smooth muscle antibodies.
* Autoantibodies with an asterisk are investigational or are not available for routine clinical use.
Ready fatigability is the most common symptom (seen in 86% of adult patients) (see Table 88-6). Weight loss is uncommon, and intense pruritus argues against the diagnosis. Hepatomegaly is the most common physical finding (78%), and jaundice is found in 69% of patients. Splenomegaly can be present in patients with and without cirrhosis (56% and 32%, respectively), as can spider angiomata. As many as 34% of patients are asymptomatic at initial consultation, and 25% of adults have a normal physical examination.7 The discordance between the severity of inflammatory activity and the presence of symptoms is most common in children, whose clinical status frequently does not accurately reflect the severity of the underlying liver disease.
Hyperbilirubinemia is present in 83% of patients, but the serum bilirubin level is greater than three times the upper limit of normal in only 46%.45 Similarly, the serum alkaline phosphatase level commonly is increased (81%), but elevations are two times the upper limit of normal in 33% and four times the upper limit of normal in only 10% of patients (see Table 88-6).45
Concurrent immunologic diseases are common and involve diverse organ systems, most frequently the thyroid.7 SMA, ANA, and anti-LKM1 are required for the diagnosis, and other autoantibodies may be present, as shown in Table 88-6,7 although these other autoantibodies do not have routine clinical applications.
SEVERE OR FULMINANT PRESENTATION
AIH can have an acute severe or fulminant presentation that can be mistaken for acute viral or toxic hepatitis, and misdiagnosis can delay or defer the institution of potentially life-saving therapy.2 Glucocorticoid therapy can be effective in suppressing the inflammatory activity in 36%-100% of patients, whereas delay in treatment can have a strong negative impact on outcome. Furthermore, unrecognized chronic disease can have a spontaneous exacerbation and appear acute. Affected patients invariably die if they are untreated or there is no response to glucocorticoid therapy within two weeks.70
ADVANCED AGE
Twenty percent of adults with AIH develop the disease after the age of 60 years, and they may be under-recognized because the liver disease is ascribed to the toxicity of medication or to concurrent illnesses that may also affect liver biochemical test levels.32,71 These elderly patients have a greater degree of hepatic fibrosis at presentation than young adults, and their higher frequencies of ascites and cirrhosis indicate that they have an aggressive disease that is commonly indolent and unsuspected.71 Enthusiasm for therapy is justifiably tempered by concerns regarding pre-existing osteopenia or other comorbid conditions. Elderly patients, however, typically respond well to glucocorticoid treatment, and preemptive efforts to maintain bone density with bisphosphonates, calcium and vitamin D supplements, and a regular exercise program can improve tolerance to the treatment.32,71 Elderly patients do have higher cumulative frequencies of drug-related complications and vertebral compression fractures than do young patients, but these consequences are associated with protracted durations of treatment or repeated relapses and re-treatment. Therapy in the elderly must be individualized to limit the duration of initial treatment, total cumulative dose of glucocorticoids, and re-treatment after relapse.32
ASYMPTOMATIC PATIENTS
AIH can be asymptomatic in 34% to 45% of patients.72,73 Typically, affected patients are men, and they have significantly lower serum alanine aminotransferase levels at presentation than do symptomatic patients. Histologic findings, including the frequency of cirrhosis, are similar in asymptomatic and symptomatic patients, and as many as 70% of asymptomatic patients become symptomatic during the course of their disease. The asymptomatic state does not preclude the need for glucocorticoid therapy if other objective manifestations of disease activity are present or emerge, and close surveillance of these patients for worsening inflammation is justified.
ASSOCIATED DISEASES
As many as 38% of patients with AIH have concurrent immune diseases, which may mask the underlying liver disease.1,7 Autoimmune thyroiditis, Graves’ disease, synovitis, and ulcerative colitis are the most common immune-mediated disorders associated with AIH in North American adults, whereas insulin-dependent diabetes mellitus, vitiligo, and autoimmune thyroiditis are the most common concurrent disorders in European children with AIH and anti-LKM1.34 Autoimmune sclerosing cholangitis can be present with or without inflammatory bowel disease in children with AIH,51 and PSC can be detected in 8% of adults with classic AIH50 and 44% of those with AIH and inflammatory bowel disease.33 Concurrent immune diseases typically do not affect the prognosis of AIH unless the underlying liver disease is unrecognized or associated with bile duct changes.33,50 Patients presenting with immune-mediated rheumatic diseases, celiac disease, or inflammatory bowel disease should be assessed for underlying AIH.7
HEREDITARY FORMS
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) is a syndrome caused by a single-gene mutation localized to chromosome 21q22.3 that affects the generation of an autoimmune regulator (AIRE) involved in the negative selection of autoreactive immunocytes in the thymus.74 The syndrome is characterized by multiple endocrine organ failure (parathyroids, adrenals, or ovaries), mucocutaneous candidiasis, and ectodermal dysplasia in various combinations that may include AIH. Unlike other autoimmune diseases, APECED has a recessive mendelian pattern of inheritance, complete penetrance of the gene, no HLA-DR associations, and no female predominance. Patients who have APECED and AIH may have particularly aggressive liver disease that does not respond well to standard immunosuppressive regimens.74
TREATMENT
INDICATIONS
The indications for treatment of AIH are shown in Table 88-7 and are based on manifestations of hepatic inflammation.1 The prognosis of untreated mild AIH is unclear, and the decision to treat patients with less than severe AIH is a highly individualized clinical judgment that reflects the perceived risk of the untreated disease, possible side effects of therapy, and patient’s physical and mental tolerance of the disease and its management. Untreated asymptomatic mild AIH may still progress, and there are no clinical indices that predict the course of disease. Most patients with AIH are treated regardless of disease activity at presentation, because of physicians’ uncertainty about the benign nature of mild or asymptomatic disease and the likelihood that the disease will remain mild long-term.61

REGIMENS
Prednisone, alone or at a lower dose in combination with azathioprine, is effective therapy (Table 88-8).1,75 No findings at presentation preclude a satisfactory response to therapy. The presence of ascites or hepatic encephalopathy identifies patients with a poor prognosis, but these findings do not preclude a full response to glucocorticoid therapy.70 Decompensated patients with multilobular necrosis on histologic examination in whom at least one laboratory parameter fails to normalize, or in whom hyperbilirubinemia does not improve, after two weeks of treatment have a high immediate mortality rate. These patients should be evaluated for liver transplantation (see Table 88-5).70,76 Patients in whom these parameters improve during the first two weeks of therapy have excellent immediate survival rates, and their drug treatment should be continued.70
Table 88-8 Preferred Treatment Regimens in Autoimmune Hepatitis
| Combination Therapy | Single-Drug Therapy | |
|---|---|---|
| Prednisone (mg/day) | Azathioprine (mg/day) | Prednisone (mg/day) |
| 30 mg × 1 wk | 50 mg until end point | 60 mg × 1 wk |
| 20 mg × 1 wk | 40 mg × 1 wk | |
| 15 mg × 2 wk | 30 mg × 2 wk | |
| 10 mg until end point | 20 mg until end point | |
DRUG ACTIONS
Glucocorticoids limit T cell activation by inhibiting cytokine production and the expression of adhesion molecules.31,77 Glucocorticoids are lipophilic and can diffuse into the cytosol of cells to bind the glucocorticoid receptor. The complex of drug and receptor then translocates to the nucleus where it inhibits cytokine gene expression and reduces the activity of nuclear factor-κB. Type 1 and type 2 cytokine pathways are affected, and both cellular and humoral immune responses are blunted. Repeated administration of glucocorticoids is required to achieve results in AIH because of a short biological half-life.Azathioprine is a purine antagonist that blocks the proliferation of lymphocytes.77 It is converted to 6-mercaptopurine in blood by a nonenzymatic, glutathione-based pathway, and this metabolite is, in turn, converted to 6-thioguanine by hypoxanthine guanine phosphoribosyl transferase. The 6-thioguanines are the active metabolites that interfere with purine nucleotide synthesis within the cell cycle, and they thereby impair proliferation of rapidly dividing T and B lymphocytes. Competing enzymatic pathways can convert 6-mercaptopurine to either 6-thiouric acid by xanthine oxidase or 6-methyl mercaptopurine by thiopurine methyltransferase (TPMT). Each end product is inactive, and the integrity of the enzymatic routes responsible for their production influences the erythrocyte concentrations of the active 6-thioguanine nucleotides. Drugs that inhibit xanthine oxidase activity, such as allopurinol, or deficiencies in TPMT activity can increase the therapeutic efficacy or the toxicity of the 6-thioguanine metabolites, respectively. Blood TPMT activity is also inducible by azathioprine, and this effect may further modulate the erythrocyte concentrations of the 6-thioguanine nucleosides. The immunosuppressive action of azathioprine is achieved slowly (three months or longer) because of the drug transformations and nuclear incorporations that are required to limit immunocyte proliferation. Azathioprine has weak or no immunosuppressive effect in AIH unless combined with prednisone
DRUG-RELATED SIDE EFFECTS
Cosmetic changes, such as facial rounding, dorsal hump formation, obesity, acne, striae, alopecia, and facial hirsutism, occur in 80% of patients after two years of treatment regardless of the regimen used.77 Severe side effects include osteopenia with vertebral compression, diabetes mellitus, cataracts, emotional instability, opportunistic infections, pancreatitis, and hypertension. Severe complications are uncommon and develop only after protracted therapy (more than 18 months) and on the regimen with the higher dose of prednisone (20 mg per day). Azathioprine with prednisone is preferred to a higher dose of prednisone alone because the combination produces fewer glucocorticoid-related side effects during comparable periods of treatment (10% vs. 44%). Treatment must be discontinued prematurely in 13% of patients, mainly because of intolerable obesity, cosmetic changes, brittle diabetes mellitus, or osteoporosis with vertebral compression (Table 88-9).
Table 88-9 Side Effects Associated with Therapy for Autoimmune Hepatitis*
| Prednisone | |
|---|---|
| TYPE | FREQUENCY |
| Azathioprine | |
|---|---|
| TYPE | FREQUENCY |
| 46% (especially with cirrhosis) | |
| 6% | |
| 5% | |
| 3% (after 10 yr) | |
| Rare | |
| Teratogenic during pregnancy | Rare (theoretical) |
* Adapted from Czaja AJ. Safety issues in the management of autoimmune hepatitis. Expert Opin Drug Saf 2008; 7:319-33.
Treatment with azathioprine can be complicated by cholestatic liver disease, nausea, emesis, rash, pancreatitis, arthralgias, opportunistic infections, and cytopenias.77 Five percent of patients treated with azathioprine develop early adverse reactions (nausea, vomiting, arthralgias, fever, skin rash, or pancreatitis), which warrant its discontinuation. The overall frequency of azathioprine-related side effects in patients treated with 50 mg daily is 10%, and the side effects typically improve after the dose is reduced or the therapy is discontinued.77 Cytopenia is the most common consequence of treatment; bone marrow failure is rare. Cytopenia occurs in 46% of patients, and severe hematologic abnormalities occur in 6%.77,78 These toxicities are not predictable by either genotyping or phenotyping for TPMT activity,78–80 and the most common association with cytopenia in these patients is cirrhosis and presumed hypersplenism associated with portal hypertension (see Table 88-9).78,80
Teratogenicity is a theoretical complication of therapy with azathioprine.77 Azathioprine has been administered successfully in pregnant women with AIH, pregnant mothers with inflammatory bowel disease, and women who have conceived while taking azathioprine after liver transplantation.77 Nevertheless, azathioprine has been associated with congenital malformations in pregnant mice, and these defects have included cleft palate, skeletal anomalies, hydrops fetalis, reduced thymic size, anemia, and hematopoietic depression. Furthermore, the placenta is only a partial barrier to the metabolites of azathioprine, and low levels of 6-thioguanine are detectable in the newborns of mothers treated for Crohn’s disease.77 The odds ratio of having a child with congenital malformations while taking azathioprine for inflammatory bowel disease is 3.4. Azathioprine is not an essential medication in the treatment of AIH and can be discontinued during pregnancy, in which case the liver disease can be managed successfully by adjustments in the dose of prednisone.
Oncogenicity is another possible complication of therapy with azathioprine.77 The incidence of extrahepatic neoplasms is 1 per 194 patient-years; the probability of tumor occurrence is 3% after 10 years; and the risk of malignancy is 1.4-fold greater than normal.81 The low but increased risk of malignancy does not contraindicate azathioprine therapy in AIH but emphasizes the importance of maintaining strict indications for treatment.
Blood TPMT activity is significantly lower in patients with AIH and intolerance to azathioprine than in patients with uncomplicated courses of treatment.79 Similar findings have been described in patients with inflammatory bowel disease and rheumatic conditions (see Chapter 111). These observations have suggested that routine screening of the blood TPMT level may identify persons with AIH at risk for azathioprine-related complications. The blood TPMT activity level, however, has not been predictive of toxicity in individual patients.79,80 Genotypic and phenotypic screening for blood TPMT activity has not reduced the frequency of azathioprine-induced side effects in patients with AIH compared with unscreened patients,78–80 nor has the occurrence of azathioprine-related side effects been associated with below-normal levels of TPMT activity.78 Near-zero enzyme activity occurs rarely in otherwise normal individuals (0.3% to 0.5%), and the value of screening to detect this unusually low enzyme deficiency remains uncertain, especially if some patients with low levels do not exhibit azathioprine toxicity.77,78 Testing for blood TPMT activity seems most appropriate in patients with pre-existing or progressive cytopenias and in those subjected to doses of azathioprine higher than the conventional schedule of 50 mg daily. Avoidance of azathioprine in patients with pre-existing or progressive cytopenias (leukocyte count below 2.5 × 109/L or platelet count below 50 × 109/L) and close monitoring (at three-month intervals) of the blood leukocyte and platelet counts in all patients taking the drug may be the best preventive strategy.77,78
END POINTS
Glucocorticoid therapy is continued until remission, treatment failure, incomplete response, or drug toxicity occurs (Fig. 88-6).1,75 Remission connotes absence of symptoms, resolution of all laboratory indices of active inflammation, and histologic improvement to normal liver tissue or inactive cirrhosis.82 Evaluation of liver tissue before drug withdrawal is essential to establish remission because histologic activity may be present in 55% of patients who satisfy other requirements for remission. Typically, histologic improvement lags behind clinical and laboratory resolution by three to eight months, and treatment should be extended for at least this period to compensate for the lag. Patients with normal serum AST and gamma globulin levels and normal liver biopsy findings immediately before drug withdrawal have a significantly lower frequency of relapse after drug withdrawal than patients with near-normal test levels and histology (60% vs. 90%, P < 0.001).82 Only 40% of treated patients, however, are able to improve fully, and complete resolution of the clinical and histologic manifestations of the disease does not preclude relapse after drug withdrawal.82
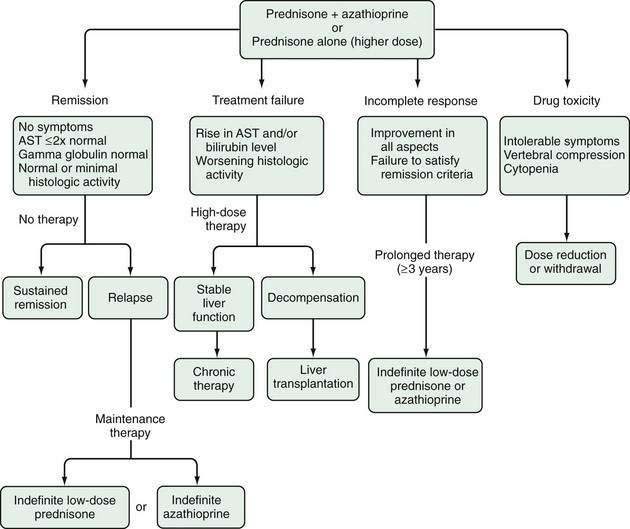
Figure 88-6. Treatment algorithm for autoimmune hepatitis. Patients who satisfy indications for glucocorticoid therapy are given prednisone in combination with azathioprine or a higher dose of prednisone alone (see Table 88-8). Treatment is continued until the criteria for a treatment end point are met. Possible end points are remission, treatment failure, incomplete response, and drug toxicity. Therapy can then be discontinued, increased in dose, or reduced in dose. Responses to the dose adjustments determine the need for other actions. The principal indices of inflammatory activity are serum aspartate aminotransferase (AST) and gamma globulin levels.
Treatment failure connotes deterioration during therapy (see Fig. 88-6).1,69,75 It is characterized by worsening of the serum AST or bilirubin levels by at least 67% of previous values, progressive histologic activity, or onset of ascites or encephalopathy. Conventional glucocorticoid therapy should be stopped, and a high-dose regimen should be instituted.
Incomplete response connotes improvement that is insufficient to satisfy remission criteria (see Fig. 88-6).1,75 Failure to achieve remission within three years indicates that remission is unlikely and warrants discontinuation of conventional treatment.
Drug toxicity justifies premature withdrawal of medication or a reduction in dose (see Fig. 88-6).1,75,77 Most side effects are reversible, and some consequences such as cataracts and osteopenia with vertebral compression have effective therapies. Weight gain, acne, edema, and diabetes may be consequences of the disease rather than the drugs.
RESULTS
Prednisone alone or in combination with azathioprine induces clinical, biochemical, and histologic remission in 65% of patients within three years.1,75 The average treatment interval until remission is 22 months. Therapy improves survival. The 10-year life expectancies following treatment for patients with and without cirrhosis at the time of the initial medical consultation are 89% and 90%, respectively. The overall 10-year survival rate is 93% and is comparable to that of an age- and sex-matched cohort from the population at large (94%). Patients who have histologic cirrhosis respond as well as do noncirrhotic patients and should receive similar treatment, with the same expectation of success. Twenty-one percent of patients managed with conventional regimens of prednisone alone or in combination with azathioprine sustain their remission for a median of 76 months after drug withdrawal, and an effort should be made to withdraw all patients from initial therapy after criteria for remission have been satisfied.83
Treatment with glucocorticoids also may reduce hepatic fibrosis.84 Fibrosis scores have improved in 56% of patients followed for 55 ± 9 months, and fibrosis did not progress in 33% of patients followed for 62 ± 14 months. Histologic activity indices decreased concurrently, and patients in whom the histologic activity indices improved had a higher frequency of improvement in the fibrosis scores (80% vs. 25%, P = .002). These findings suggest that improvement in hepatic fibrosis occurs in conjunction with reduction in liver inflammation and that glucocorticoids may facilitate the disappearance of fibrosis by suppressing inflammatory activity.30,84 Small case studies have also suggested that cirrhosis can disappear during treatment, but this possibility must await confirmation by assessments more reliably reflective of cirrhosis than conventional needle biopsy of the liver.
RELAPSE
Patients who enter remission commonly experience an exacerbation after drug withdrawal.1,75,83 Relapse is defined as the reappearance of histologic disease after discontinuation of drug therapy. An increase in the serum AST level to more than three-fold the upper limit of normal after discontinuation of drug therapy is invariably associated with interface hepatitis on histologic examination. This biochemical change is sufficient to diagnose relapse without requiring liver tissue assessment.
Relapse occurs in 50% of patients within six months of discontinuing therapy, and most patients (70% to 86%) experience an exacerbation within three years.82,83 Reinstitution of the original treatment induces another remission, but relapse commonly recurs after termination of therapy. Repeated relapse and re-treatment is associated with a cumulative morbidity and mortality. Cirrhosis develops more commonly (38% vs. 4%, P = 0.004); death from hepatic failure or need for liver transplantation occurs more often (20% vs. 0%, P = 0.008)85; and drug-induced side effects are more frequent (70% vs. 30%, P = 0.01) in persons who relapse than in those who sustain remission after drug withdrawal.77 The frequencies of each complication increase with each subsequent relapse and re-treatment. The optimal time to interrupt this sequence is after the first course of treatment and relapse, and the preferred treatment strategy is to implement maintenance therapy with azathioprine.86
After the initial relapse, therapy with prednisone and azathioprine is re-started and continued until clinical and laboratory resolution is achieved again (see Fig. 88-6).86 The dose of azathioprine is then increased to 2 mg/kg daily as the dose of prednisone is reduced. Azathioprine is then continued indefinitely as a chronic maintenance regimen. Eighty percent of patients are able to sustain remission in this fashion over a 10-year period of observation. Azathioprine-induced cytopenias compel dose reduction in 9% of patients; glucocorticoid-related side effects improve; and arthralgias associated with glucocorticoid withdrawal eventually resolve (but may be protracted). Malignancies of various types develop in 7% of patients, but an association with azathioprine is disputed.86
An alternative management strategy after the first relapse is to maintain suppression of inflammatory activity using daily, low-dose prednisone (see Fig. 88-6).87 Eighty-seven percent of patients can be managed long term on prednisone at less than 10 mg per day (median dose, 7.5 mg per day). The dose is titrated to the lowest level needed to prevent symptoms and to maintain serum AST levels below three times the upper limit of normal. Side effects attributable to previous glucocorticoid therapy resolve in 85% of patients; the immediate survival rate is comparable to that for persons who have relapsed and been treated with a conventional regimen of prednisone alone or a lower dose of prednisone and azathioprine (91% vs. 90%), and new complications do not occur. The azathioprine and low-dose prednisone maintenance regimens have not been compared directly, but the azathioprine schedule has intuitive appeal because it eliminates the need for prednisone.
Treatment after relapse does not need to be indefinite.83 The probability of ultimately achieving inactive disease that does not require continuous drug therapy is 28%. This possibility justifies an effort to withdraw treatment in all patients who have relapsed previously and who have stably maintained inactive disease on maintenance therapy for at least one year. This effort can be repeated if earlier withdrawal attempts have failed.83
TREATMENT FAILURE
Nine percent of patients deteriorate during glucocorticoid therapy (treatment failure) (see Fig. 88-6).1,69,75 High doses of prednisone alone (60 mg per day) or prednisone (30 mg per day) in conjunction with azathioprine (150 mg per day) constitute standard treatment in this group.1,75,88 Each schedule induces clinical and biochemical improvement in 70% of patients within two years. Histologic resolution, however, occurs in only 20%, and long-term therapy is frequently necessary. These patients are at risk of liver failure and serious drug toxicity. Liver transplantation must be considered at the first sign of hepatic decompensation. The development of ascites typically heralds the need for a transplant evaluation (see Table 88-5).
Alternative management strategies for treatment failure have included the administration of cyclosporine, tacrolimus, mycophenolate mofetil, ursodeoxycholic acid, budesonide, 6-mercaptopurine, methotrexate, and cyclophosphamide.75,89 In each instance, experience has been limited, and in most reports the preliminary results have been encouraging but uncorroborated. Among the new drugs used in treatment failure, only ursodeoxycholic acid has been evaluated by a randomized controlled clinical trial, and the findings did not support its use.75 Site-specific interventions will be possible as soon as the pathogenic mechanisms are clarified.75 These therapies may include peptides to block autoantigen display within the class II MHC molecules, agents such as CTLA4 to temper immunocyte response, T-cell vaccination, oral tolerance regimens, cytokine manipulations, mesenchymal stem cell transplantation, and gene therapies that can offset the over-expression of certain cytokines, limit fibrosis, and promote regeneration.
INCOMPLETE RESPONSE
In 13% of patients, clinical improvement is observed during therapy without achieving remission criteria (see Fig. 88-6).1,75 The diminishing benefit-to-risk ratio of protracted therapy justifies an alternative strategy. The administration of azathioprine (2 mg/kg daily) as the sole drug86 or a low-dose prednisone regimen87 is a reasonable approach. The goal of treatment is to reduce and stabilize disease activity on a drug schedule that is well tolerated.
DRUG TOXICITY
For patients with serious side effects from therapy, treatment usually can be continued with the single tolerated drug (prednisone or azathioprine) in an adjusted dose (see Fig. 88-6).1,75,77 Cyclosporine, 6-mercaptopurine, and cyclophosphamide also have been used successfully after drug toxicity in isolated cases.1,75
LIVER TRANSPLANTATION
Liver transplantation is effective in the treatment of decompensated AIH (see Chapter 95).1,75 Five-year survival rates for patients and grafts range from 83% to 92%, and the actuarial 10-year survival rate after transplantation is 75%. AIH recurs in at least 17% of patients, and AIH develops de novo in 3% to 5% of patients who undergo transplantation for nonautoimmune liver disease. Acute rejection, glucocorticoid-resistant rejection, and chronic rejection occur more commonly in patients undergoing transplantation for AIH than for other conditions, and patients with AIH are more difficult to withdraw from glucocorticoids.90
Recurrent AIH typically is mild and develops in patients who are inadequately immunosuppressed.89 Dose adjustments usually are sufficient to suppress the disease, but progression to cirrhosis and graft failure have been reported. De novo AIH is a clinical syndrome that affects both children and adults who undergo transplantation for nonautoimmune liver disease.90 Patients with de novo AIH in whom glucocorticoid therapy fails, experience worsening hepatic fibrosis with possible graft loss, and those who do not receive glucocorticoids progress to cirrhosis, require retransplantation, or die of liver failure. De novo AIH in some adults has been associated with severe centrilobular necrosis, and adult patients have been reported to express an atypical anti-liver-kidney cytosolic antibody of uncertain pathogenic significance.90 AIH should be included in the differential diagnosis of allograft dysfunction in all transplant recipients
Abdalian R, Dhar P, Jhaveri K, et al. Prevalence of sclerosing cholangitis in adults with autoimmune hepatitis: Evaluating the role of routine magnetic resonance imaging. Hepatology. 2008;47:949-57. (Ref 50.)
Alvarez F, Berg P, Bianchi F, et al. International Autoimmune Hepatitis Group report: Review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929-38. (Ref 3.)
Czaja AJ. Autoantibodies in autoimmune liver disease. Adv Clin Chem. 2005;40:127-64. (Ref 6.)
Czaja AJ. Autoimmune hepatitis—Part A: Pathogenesis. Expert Rev Gastroenterol Hepatol. 2007;1:113-28. (Ref 18.)
Czaja AJ. Autoimmune hepatitis—Part B: Diagnosis. Expert Rev Gastroenterol Hepatol. 2007;1:129-43. (Ref 7.)
Czaja AJ. Performance parameters of the diagnostic scoring systems for autoimmune hepatitis. Hepatology. 2008;48:1540-8. (Ref 15.)
Czaja AJ. Genetic factors associated with the occurrence, clinical phenotype and outcome of autoimmune hepatitis. Clin Gastroenterol Hepatol. 2008;6:379-88. (Ref 64.)
Czaja AJ. Safety issues in the management of autoimmune hepatitis. Expert Opin Drug Saf. 2008;7:319-33. (Ref 77.)
Czaja AJ, Carpenter HA. Empiric therapy of autoimmune hepatitis with mycophenolate mofetil: Comparison with conventional treatment for refractory disease. J Clin Gastroenterol. 2005;39:819-25. (Ref 88.)
Czaja AJ, Freese DK. Diagnosis and treatment of autoimmune hepatitis. Hepatology. 2002;36:479-97. (Ref 1.)
Hennes EM, Zeniya M, Czaja AJ, et al. Simplified diagnostic criteria for autoimmune hepatitis. Hepatology. 2008;48:169-76. (Ref 14.)
Montano-Loza AJ, Carpenter HA, Czaja AJ. Features associated with treatment failure in type 1 autoimmune hepatitis and predictive value of the model for end-stage liver disease. Hepatology. 2007;46:1138-45. (Ref 69.)
Montano-Loza A, Carpenter HA, Czaja AJ. Improving the end point of corticosteroid therapy in type 1 autoimmune hepatitis to reduce the frequency of relapse. Am J Gastroenterol. 2007;102:1005-12. (Ref 82.)
Montano-Loza A, Carpenter HA, Czaja AJ. Consequences of treatment withdrawal in type 1 autoimmune hepatitis. Liver Int. 2007;27:507-15. (Ref 85.)
Montano-Loza A, Czaja AJ. Current therapy for autoimmune hepatitis. Nature Clin Pract Gastroenterol Hepatol. 2007;4:202-14. (Ref 75.)
1. Czaja AJ, Freese DK. Diagnosis and treatment of autoimmune hepatitis. Hepatology. 2002;36:479-97.
2. Kessler WR, Cummings OW, Eckert G, et al. Fulminant hepatic failure as the initial presentation of acute autoimmune hepatitis. Clin Gastroenterol Hepatol. 2004;2:625-31.
3. Alvarez F, Berg P, Bianchi F, et al. International Autoimmune Hepatitis Group report: Review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929-38.
4. Czaja AJ, Carpenter HA. Autoimmune hepatitis with incidental histologic features of bile duct injury. Hepatology. 2001;34:659-65.
5. Czaja AJ, Muratori P, Muratori L, et al. Diagnostic and therapeutic implications of bile duct injury in autoimmune hepatitis. Liver Int. 2004;24:322-9.
6. Czaja AJ. Autoantibodies in autoimmune liver disease. Adv Clin Chem. 2005;40:127-64.
7. Czaja AJ. Autoimmune hepatitis—Part B: Diagnosis. Expert Rev Gastroenterol Hepatol. 2007;1:129-43.
8. Terjung B, Spengler U, Sauerbruch T, et al. “Atypical p-ANCA” in IBD and hepatobiliary disorders react with a 50-kilodalton nuclear envelope protein of neutrophils and myeloid cell lines. Gastroenterology. 2000;119:310-22.
9. Abdo A, Meddings J, Swain M. Liver abnormalities in celiac disease. Clin Gastroenterol Hepatol. 2004;2:107-12.
10. Czaja AJ, Shums Z, Norman GL. Nonstandard antibodies as prognostic markers in autoimmune hepatitis. Autoimmunity. 2004;37:195-201.
11. Czaja AJ, Donaldson PT, Lohse AW. Antibodies to soluble liver antigen/liver pancreas and HLA risk factors in type 1 autoimmune hepatitis. Am J Gastroenterol. 2002;97:413-19.
12. Ma Y, Okamoto M, Thomas MG, et al. Antibodies to conformational epitopes of soluble liver antigen define a severe form of autoimmune liver disease. Hepatology. 2002;35:658-64.
13. Czaja AJ, Pfeifer KD, Decker RH, et al. Frequency and significance of antibodies to asialoglycoprotein receptor in type 1 autoimmune hepatitis. Dig Dis Sci. 1996;41:1733-40.
14. Hennes EM, Zeniya M, Czaja AJ, et al. Simplified diagnostic criteria for autoimmune hepatitis. Hepatology. 2008;48:169-76.
15. Czaja AJ. Performance parameters of the diagnostic scoring systems for autoimmune hepatitis. Hepatology. 2008;48:1540-8.
16. Czaja AJ. Understanding the pathogenesis of autoimmune hepatitis. Am J Gastroenterol. 2001;96:1224-31.
17. Vergani D, Choudhuri K, Bogdanos DP, et al. Pathogenesis of autoimmune hepatitis. Clin Liver Dis. 2002;6:727-37.
18. Czaja AJ. Autoimmune hepatitis Part A: Pathogenesis. Expert Rev Gastroenterol Hepatol. 2007;1:113-28.
19. Strettell M, Donaldson P, Thomson L, et al. Allelic basis for HLA-encoded susceptibility to type 1 autoimmune hepatitis. Gastroenterology. 1997;112:2028-35.
20. Czaja A, Donaldson P. Genetic susceptibilities for immune expression and liver cell injury in autoimmune hepatitis. Immunol Rev. 2000;174:250-9.
21. Czaja AJ, Doherty DG, Donaldson PT. Genetic bases of autoimmune hepatitis. Dig Dis Sci. 2002;47:2139-50.
22. Pando M, Larriba J, Fernandez GC, et al. Pediatric and adult forms of type 1 autoimmune hepatitis in Argentina: Evidence for differential genetic predisposition. Hepatology. 1999;30:1374-80.
23. Goldberg AC, Bittencourt PL, Mougin B, et al. Analysis of HLA haplotypes in autoimmune hepatitis type 1: Identifying the major susceptibility locus. Hum Immunol. 2001;62:165-9.
24. Czaja AJ, Souto EO, Bittencourt PL, et al. Clinical distinctions and pathogenic implications of type 1 autoimmune hepatitis in Brazil and the United States. J Hepatol. 2002;37:302-8.
25. Fainboim L, Velasco VCC, Marcos CY, et al. Protracted, but not acute, hepatitis A virus infection is strongly associated with HLA-DRB1*1301, a marker for pediatric autoimmune hepatitis. Hepatology. 2001;33:1512-17.
26. Czaja A, Cookson S, Constantini P, et al. Cytokine polymorphisms associated with clinical features and treatment outcome in type 1 autoimmune hepatitis. Gastroenterology. 1999;117:645-52.
27. Agarwal K, Czaja AJ, Jones DEJ, et al. CTLA-4 gene polymorphism and susceptibility to type 1 autoimmune hepatitis. Hepatology. 2000;31:49-53.
28. Agarwal K, Czaja AJ, Donaldson PT. A functional Fas promoter polymorphism is associated with a severe phenotype in type 1 autoimmune hepatitis characterized by early development of cirrhosis. Tissue Antigens. 2007;69:227-35.
29. Longhi MS, Hussain MJ, Mitry RR, et al. Functional study of CD4+CD25+ regulatory T cells in health and autoimmune hepatitis. J Immunol. 2006;176:4484-91.
30. Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247-50.
31. Almawi WY. Molecular mechanisms of glucocorticoid effects. Mod Asp Immunobiol. 2001;2:78-82.
32. Czaja AJ. Clinical features, differential diagnosis and treatment of autoimmune hepatitis in the elderly. Drugs Aging. 2008;25:219-39.
33. Perdigoto R, Carpenter H, Czaja A. Frequency and significance of chronic ulcerative colitis in severe corticosteroid-treated autoimmune hepatitis. J Hepatol. 1992;14:325-31.
34. Homberg J, Abuaf N, Bernard O, et al. Chronic active hepatitis associated with anti-liver-kidney microsome antibody type 1: A second type of “autoimmune” hepatitis. Hepatology. 1987;7:1333-9.
35. Czaja A, Manns M, Homburger H. Frequency and significance of antibodies to liver/kidney microsome type 1 in adults with chronic active hepatitis. Gastroenterology. 1992;103:1290-5.
36. Muratori P, Czaja AJ, Muratori L, et al. Genetic distinctions between autoimmune hepatitis in Italy and North America. World J Gastroenterol. 2005;11:1862-6.
37. Djilali-Saiah I, Renous R, Caillat-Zucman S, et al. Linkage disequilibrium between HLA class II region and autoimmune hepatitis in pediatric patients. J Hepatol. 2004;40:904-9.
38. Muratori P, Czaja AJ, Muratori L, et al. Evidence of a genetic basis for the different geographical occurrences of liver/kidney microsomal antibody type 1 in hepatitis C. Dig Dis Sci. 2007;52:179-84.
39. Manns M, Griffin K, Sullivan K, et al. LKM-1 autoantibodies recognize a short linear sequence in P450IID6, a cytochrome P-450 monooxygenase. J Clin Invest. 1991;88:1370-8.
40. Ma Y, Bogdanos DP, Hussain MJ, et al. Polyclonal T-cell responses to cytochrome P450IID6 are associated with disease activity in autoimmune hepatitis type 2. Gastroenterology. 2006;130:868-82.
41. Czaja A. The variant forms of autoimmune hepatitis. Ann Intern Med. 1996;125:588-98.
42. Czaja A. Frequency and nature of the variant syndromes of autoimmune liver disease. Hepatology. 1998;28:360-5.
43. Ben-Ari Z, Czaja AJ. Autoimmune hepatitis and its variant syndromes. Gut. 2001;49:589-94.
44. Chazouilleres O, Wendum D, Serfaty L, et al. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: Clinical features and response to therapy. Hepatology. 1998;28:296-301.
45. Kenny RP, Czaja AJ, Ludwig J, et al. Frequency and significance of antimitochondrial antibodies in severe chronic active hepatitis. Dig Dis Sci. 1986;31:705-11.
46. Czaja A, Carpenter H, Manns M. Antibodies to soluble liver antigen, P450IID6, and mitochondrial complexes in chronic hepatitis. Gastroenterology. 1993;105:1522-8.
47. O’Brien C, Joshi S, Feld JJ, et al. Long-term follow-up of antimitochondrial antibody-positive autoimmune hepatitis. Hepatology. 2008;48:550-6.
48. Montano-Loza AJ, et alCarpenter HA, Czaja AJ, et al. Frequency, behavior and prognostic implications of antimitochondrial antibodies in type 1 autoimmune hepatitis. J Clin Gastroenterol. 2008;42:1047-53.
49. Angulo P, Maor-Kendler Y, Lindor KD. Small-duct primary sclerosing cholangitis: A long-term follow-up study. Hepatology. 2002;35:1494-500.
50. Abdalian R, Dhar P, Jhaveri K, et al. Prevalence of sclerosing cholangitis in adults with autoimmune hepatitis: Evaluating the role of routine magnetic resonance imaging. Hepatology. 2008;47:949-57.
51. Gregorio GV, Portmann B, Karani J, et al. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: A 16-year prospective study. Hepatology. 2001;33:544-53.
52. Czaja A, Carpenter H, Santrach P, et al. Autoimmune cholangitis within the spectrum of autoimmune liver disease. Hepatology. 2000;31:1231-8.
53. Czaja A, Carpenter H, Santrach P, et al. The nature and prognosis of severe cryptogenic chronic active hepatitis. Gastroenterology. 1993;104:1755-61.
54. Gassert DJ, Garcia H, Tanaka K, et al. Corticosteroid-responsive cryptogenic chronic hepatitis: Evidence of seronegative autoimmune hepatitis. Dig Dis Sci. 2007;52:2433-7.
55. Czaja A. Behavior and significance of autoantibodies in type 1 autoimmune hepatitis. J Hepatol. 1999;30:394-401.
56. Czaja AJ. The autoimmune hepatitis/hepatitis C overlap syndrome: Does it exist? Falk Symposium No. 136. Leuschner U, Broome U, Stiehl A, editors. Cholestatic Liver Diseases: Therapeutic Options and Perspectives. 2004. Lancaster Publishing Services, Lancaster, UK: 132
57. Czaja AJ, Carpenter HA. Histological findings in chronic hepatitis C with autoimmune features. Hepatology. 1997;26:459-66.
58. Czaja AJ, Carpenter HA. Sensitivity, specificity and predictability of biopsy interpretations in chronic hepatitis. Gastroenterology. 1993;105:1824-32.
59. Boberg K, Aadland E, Jahnsen J, et al. Incidence and prevalence of primary biliary cirrhosis, primary sclerosing cholangitis, and autoimmune hepatitis in a Norwegian population. Scand J Gastroenterol. 1998;33:99-103.
60. Hurlburt KJ, McMahon BJ, Deubner H, et al. Prevalence of autoimmune hepatitis in Alaska natives. Am J Gastroenterol. 2002;97:2402-7.
61. Czaja AJ. Features and consequences of untreated type 1 autoimmune hepatitis. Liver Int. 2009;29:816-23.
62. Montano-Loza AJ, Carpenter HA, Czaja AJ. Predictive factors for hepatocellular carcinoma in type 1 autoimmune hepatitis. Am J Gastroenterol. 2008;103:1944-51.
63. Czaja A, Strettell M, Thomson L, et al. Associations between alleles of the major histocompatibility complex and type 1 autoimmune hepatitis. Hepatology. 1997;25:317-23.
64. Czaja AJ. Genetic factors associated with the occurrence, clinical phenotype and outcome of autoimmune hepatitis. Clin Gastroenterol Hepatol. 2008;6:379-88.
65. Verma S, Torbenson M, Thuluvath PJ. The impact of ethnicity on the natural history of autoimmune hepatitis. Hepatology. 2007;46:1828-35.
66. Lim KN, Casanova RL, Boyer TD, et al. Autoimmune hepatitis in African Americans: Presenting features and responses to therapy. Am J Gastroenterol. 2001;96:3390-4.
67. Zolfino T, Heneghan MA, Norris S, et al. Characteristics of autoimmune hepatitis in patients who are not of European caucasoid ethnic origin. Gut. 2002;50:713-17.
68. Costa M, Rodriques-Sanchez JL, Czaja AJ, et al. Isolation and characterization of cDNA encoding the antigenic protein of the human tRNA(Ser)Sec complex recognized by autoantibodies from patients with type 1 autoimmune hepatitis. Clin Exp Immunol. 2000;121:364-74.
69. Montano-Loza AJ, Carpenter HA, Czaja AJ. Features associated with treatment failure in type 1 autoimmune hepatitis and predictive value of the model for end-stage liver disease. Hepatology. 2007;46:1138-45.
70. Czaja A, Rakela J, Ludwig J. Features reflective of early prognosis in corticosteroid-treated severe autoimmune chronic active hepatitis. Gastroenterology. 1988;95:448-53.
71. Czaja AJ, Carpenter HA. Distinctive clinical phenotype and treatment outcome of type 1 autoimmune hepatitis in the elderly. Hepatology. 2006;43:532-8.
72. Kogan J, Safadi R, Ashur Y, et al. Prognosis of symptomatic versus asymptomatic autoimmune hepatitis. A study of 68 patients. J Clin Gastroenterol. 2002;35:75-81.
73. Feld JJ, Dinh H, Arenovich T, et al. Autoimmune hepatitis: Effect of symptoms and cirrhosis on natural history and outcome. Hepatology. 2005;42:53-62.
74. Clemente M, Obermayer-Straub P, Meloni A, et al. Cytochrome P450 1A2 is a hepatic autoantigen in autoimmune polyglandular syndrome type 1. J Clin Endocrinol Metab. 1997;82:1353-61.
75. Montano-Loza A, Czaja AJ. Current therapy for autoimmune hepatitis. Nature Clin Pract Gastroenterol Hepatol. 2007;4:202-14.
76. Czaja AJ. Corticosteroids or not in severe acute or fulminant autoimmune hepatitis: Therapeutic brinksmanship and the point beyond salvation. Liver Transpl. 2007;13:953-5.
77. Czaja AJ. Safety issues in the management of autoimmune hepatitis. Expert Opin Drug Saf. 2008;7:319-33.
78. Czaja AJ, Carpenter HA. Thiopurine methyltransferase deficiency and azathioprine intolerance in autoimmune hepatitis. Dig Dis Sci. 2006;51:968-75.
79. Langley PG, Underhill J, Tredger JM, et al. Thiopurine methyltransferase phenotype and genotype in relation to azathioprine therapy in autoimmune hepatitis. J Hepatol. 2002;37:441-7.
80. Heneghan MA, Allan ML, Bornstein JD, et al. Utility of thiopurine methyltransferase genotyping and phenotyping, and measurement of azathioprine metabolites in the management of patients with autoimmune hepatitis. J Hepatol. 2006;45:584-91.
81. Wang K, Czaja A, Beaver S, et al. Extrahepatic malignancy following long-term immunosuppressive therapy of severe hepatitis B surface antigen-negative chronic active hepatitis. Hepatology. 1989;10:39-43.
82. Montano-Loza A, Carpenter HA, Czaja AJ. Improving the end point of corticosteroid therapy in type 1 autoimmune hepatitis to reduce the frequency of relapse. Am J Gastroenterol. 2007;102:1005-12.
83. Czaja AJ, Menon KVN, Carpenter HA. Sustained remission after corticosteroid therapy for type 1 autoimmune hepatitis: A retrospective analysis. Hepatology. 2002;35:890-7.
84. Czaja AJ, Carpenter HA. Decreased fibrosis during corticosteroid therapy of autoimmune hepatitis. J Hepatol. 2004;40:644-50.
85. Montano-Loza A, Carpenter HA, Czaja AJ. Consequences of treatment withdrawal in type 1 autoimmune hepatitis. Liver Int. 2007;27:507-15.
86. Johnson P, McFarlane I, Williams R. Azathioprine for long-term maintenance of remission in autoimmune hepatitis. N Engl J Med. 1995;333:958-63.
87. Czaja AJ. Low dose corticosteroid therapy after multiple relapses of severe HBsAg-negative chronic active hepatitis. Hepatology. 1990;11:1044-9.
88. Czaja AJ, Carpenter HA. Empiric therapy of autoimmune hepatitis with mycophenolate mofetil: Comparison with conventional treatment for refractory disease. J Clin Gastroenterol. 2005;39:819-25.
89. Manns MP, Bahr MJ, Woynarowski M, et al. Budesonide 3 mg TID is superior to prednisolone in combination with azathioprine in the treatment of autoimmune hepatitis [abstract]. J Hepatol. 2008;48(Suppl 2):S369.
90. Czaja AJ. Autoimmune hepatitis after liver transplantation and other lessons of self-intolerance. Liver Transpl. 2002;8:505-13.