Autoimmune and Chronic Cholestatic Disorders of the Liver
Kenneth P. Batts
Introduction
The three most common liver syndromes with a putative autoimmune cause are autoimmune hepatitis (AIH), primary biliary cirrhosis (PBC), and primary sclerosing cholangitis (PSC). Overlap syndromes involve various combinations of these disorders. Other chronic cholestatic disorders discussed in this chapter include hepatobiliary involvement by immunoglobulin G4 (IgG4) disease. Some uncommon noncongenital chronic biliary diseases that can mimic PBC and PSC are reviewed. Pediatric cholestatic disorders (e.g., biliary atresia) are discussed in Chapter 54, and transplantation-related aspects of autoimmune liver disorders are covered in Chapter 52.
Autoimmune Hepatitis
Historically, AIH was called autoimmune-type chronic active hepatitis, but it later evolved to be called autoimmune hepatitis because it is an a priori chronic disease.1 AIH is an autoimmune-mediated syndrome characterized by severe chronic hepatitis that can progress to cirrhosis. It predominantly affects females. It is associated with polyclonal hypergammaglobulinemia, a variety of circulating autoantibodies, an immunogenetic predisposition (i.e., human leukocyte antigen [HLA]-B8, HLA-DR3, or HLA-DR4), absence of viral infection, and a favorable response to immunosuppressive therapy. AIH has been traditionally divided into type 1 (adult) and type 2 (pediatric) (Table 47.1).2
Table 47.1
Subtypes of Autoimmune Hepatitis
| Characteristic | Type 1 | Type 2 |
| Main autoantibody specificity | ANA, ASMA, antiactin | Anti-LKM1, anti-P450 IID6 |
| Genetic predisposition (HLA) | A1, B8, DR3, DR4 | B14, DR3, C4AQ0 |
| Age at onset (usual) | Adult | Pediatric |
| Special features | Lower serum IgG levels Onset more often fulminant May progress rapidly |
|
| Treatment | Corticosteroids | Corticosteroids |
ANA, Antinuclear antibody; anti-LKM1, anti–liver-kidney microsomal antibody 1; anti-P450 IID6, anti–cytochrome 450 IID6; ASMA, anti–smooth muscle antibody; HLA, human leukocyte antigen; IgG, immunoglobulin G.
The major symptoms and findings of AIH were initially incorporated into a scoring system by the International Autoimmune Hepatitis Group in 1992; the total score describes the probability of a patient having AIH.1 Subsequent updated3 and simplified4 versions exist. In these systems, a combination of clinical and laboratory features is used to predict the likelihood that a patient with chronic hepatitis has AIH. The original system had a high sensitivity for AIH but a low specificity for distinguishing AIH from PBC, which prompted the development of a revised system, which is summarized in Table 47.2.3,4
Table 47.2
Diagnostic Criteria for Autoimmune Hepatitis: Minimum Required Parameters
| Parameters | Score |
| History | |
| Sex | |
| Female | +2 |
| Male | 0 |
| Alcohol average consumption | |
| <25 g/day | +2 |
| >60 g/day | −2 |
| History of hepatotoxic drug use | |
| Yes | −4 |
| No | +1 |
| Other autoimmune diseases | |
| Present | +2 |
| Absent | 0 |
| Serum Biochemical Studies | |
| Serum ALP/ALT ratio | |
| <1.5 | +2 |
| >3.0 | −2 |
| Serum total globulin, γ-globulin, or IgG | |
| >2 × normal | +3 |
| 1.5-2 × normal | +2 |
| 1.0-1.5 × normal | +1 |
| Normal | 0 |
| Serology Studies | |
| ANA, ASMA, or LKM1 | |
| >1 : 80 | +3 |
| 1 : 80 | +2 |
| 1 : 40 | +1 |
| <1 : 40 | 0 |
| AMA | |
| Negative | 0 |
| Positive | −4 |
| Viral hepatitis markers | |
| Negative | +3 |
| Positive | −4 |
| Other autoantibodies | |
| Present | +2 |
| Absent | 0 |
| HLA-DR3 or -DR4 | |
| Present | +1 |
| Absent | 0 |
| Treatment Response | |
| Complete remission | +2 |
| Remission with subsequent relapse | +3 |
| Histology | |
| Interface hepatitis | +3 |
| Lymphoplasmacytic infiltrate | +1 |
| Hepatocyte rosetting | +1 |
| None of the above | −5 |
| Biliary changes | −3 |
| Other changes | −3 |
| Interpretation of Aggregate Scores | |
| Before therapy | |
| >15 | Definite AIH |
| 10-15 | Probable AIH |
| After therapy | |
| >17 | Definite AIH |
| 12-17 | Probable AIH |
AIH, Autoimmune hepatitis; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AMA, antimitochondrial antibody; ANA, antinuclear antibodies; ASMA, anti–smooth muscle antibody; HLA, human leukocyte antigen; LKM1, liver-kidney microsomal antibody 1.
From Alvarez F, Berg PA, Bianchi FB, et al. International Autoimmune Hepatitis Group report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929-938.
Knowledge of the pertinent clinical and laboratory parameters during interpretation of a liver biopsy sample is extremely helpful. Factors that favor AIH are female sex, a hepatitic rather than a cholestatic liver enzyme profile (i.e., serum aminotransferases are elevated more prominently than alkaline phosphatase), hypergammaglobulinemia, serum autoantibodies (see Pathogenesis), and absence of evidence of virus-, drug-, or alcohol-related liver disease. A clinical and biochemical response to immunosuppressive therapy is also an important criterion.3 However, independent of a biopsy, this scoring system should be interpreted with caution. Although useful in excluding AIH (because few patients with diseases other than AIH have scores that fall into the probable AIH category5), exceptions may occur for patients with nonalcoholic fatty liver disease.6
Clinical Features and Epidemiology
The incidence of classic (type 1) AIH is 1.9 cases per 100,000 individuals per year, which is a higher rate than for PBC or PSC. Patients of any age may be affected. Concurrent autoimmune diseases of other types are common (34% of women and 17% of men), and 78% of AIH patients are women.7 A subtype of AIH, characterized by the presence of anti–liver-kidney microsomal antibody (anti-LKM), is sometimes referred to as AIH type 2.8 It is more common among children and females than males.8
Individuals with certain human leukocyte antigen (HLA) types are at increased risk for AIH, although testing for the various phenotypes is not necessary in routine clinical practice. The HLA-A1, HLA-B8, HLA-DR3, and HLA-DR4 phenotypes are associated with type 1 AIH.1,8
In patients with AIH, serum biochemical values vary considerably, which is a reflection of the grade and stage of disease. During periods of inactive (quiescent) disease, serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) values may be normal. However, during disease flare-ups, the serum aminotransferase values may range from the low hundreds to an excess of 1000 IU/mL. Serum alkaline phosphatase values usually are normal or only minimally elevated. Bilirubin values are often normal but may be elevated during bouts of clinical activity. Hypergammaglobulinemia, which is usually evident even in early-stage disease, is a defining feature of AIH.1 Cases of AIH that progress to cirrhosis have elevations of bilirubin levels and biochemical evidence of cirrhosis (e.g., thrombocytopenia, decreased serum albumin, prolonged prothrombin time).
Pathogenesis
Similar to other types of autoimmune diseases, AIH is caused by a defect in suppressor T cells that leads to disordered immunoregulation and production of a variety of autoantibodies, some of which probably act against hepatocyte surface antigens. The defect in suppressor T cells may occur de novo (most common) or after a triggering episode. Hepatitis A10 and a variety of drugs11 are among the best described inciting factors. Although most of the autoantibodies in AIH patients are not specific for AIH and may not act directly in the pathogenesis of the disease, their identification is often helpful diagnostically.
Antinuclear antibodies (ANAs) are identified in approximately 80% of patients with AIH and typically occur in a titer greater than 1 : 40. Anti–smooth muscle antibody (ASMA) is positive in about 70% of AIH patients, also occurring in a titer greater than 1 : 40. The finding of high-titer ANA or ASMA, or both, in a patient with clinical suspicion of AIH defines the type 1 form of this disorder, which is the most common and represents the prototypical type. However, ANA and ASMA may be identified in the serum of patients without AIH and in patients with other types of liver diseases, such as hepatitis C, alcoholic liver disease, or nonalcoholic steatohepatitis, but in these cases, the titer is usually low.
The presence of anti-LKM is a characteristic feature of type 2 AIH, and it is usually detected in the absence of high-titer ANA or ASMA. High-titer anti-LKM in the absence of anti–hepatitis C virus antibody (anti-HCV) is designated type 2 AIH.12 This uncommon form of AIH occurs most often in children. Low-titer anti-LKM with anti-HCV is designated type 2b AIH by some, although it probably does not reflect an important pathologic entity. This type of AIH is more appropriately viewed as a variant of hepatitis C, which also has autoimmune features. Antibodies against soluble liver antigen (SLA), liver-pancreas (LP) antigen, and asialoglycoprotein receptor (ASGPR) may also be identified in type 1 AIH, but they are less diagnostically useful.13,14
Although the precise mechanism of immune attack on the liver remains unknown, based on the histopathology, it is postulated that hepatocytes, rather than biliary epithelial cells, are the primary target. Necrosis of hepatocytes is usually most prominent at the level of the limiting plate (i.e., zone 1, periportal hepatocytes). This process is also referred to as piecemeal necrosis, interface hepatitis,15 or troxis hepatitis.16 A predominant attack on hepatocytes in zone 3 (pericentral) occurs in some cases.17 Necrosis of random hepatocytes throughout the hepatic lobules is also common in AIH and is recognizable as acidophil bodies or their remnant, lipofuscin-laden macrophages.
Pathology
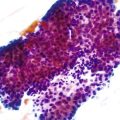
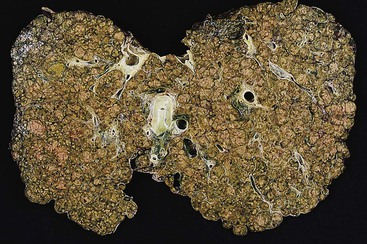
Gross examination of the liver in AIH contributes little diagnostically. In early-stage disease, the liver usually appears normal. If submassive or massive hepatocyte necrosis occurs during episodes of severe activity, the external surface of the liver may appear shriveled, and cross sections may show areas of confluent parenchymal collapse. While the disease progresses, cirrhosis eventually develops, and it is typically composed of tan to brown micronodules and macronodules (Fig. 47.1), similar to the pattern that occurs in chronic viral hepatitis. Occasionally, a greenish discoloration may be seen, similar to cirrhosis resulting from chronic biliary disorders.

Although the pathologic lesions in AIH vary considerably according to the grade and stage of disease (Table 47.3), chronic inflammation composed primarily of lymphocytes and a considerable number of plasma cells in portal tracts is a near-constant microscopic feature. However, during periods of quiescent disease (i.e., remission), portal inflammation may be the only histologic abnormality. In some cases, little or no inflammation is detected (Fig. 47.2). A small number of intraepithelial lymphocytes may be found within bile duct epithelium. However, bile duct destruction and loss are not features of AIH and should alert the pathologist to a possible diagnosis of PBC or an overlap syndrome.
Table 47.3
Comparative Histopathology of Necroinflammatory Features in Autoimmune Hepatitis, Primary Biliary Cirrhosis, and Primary Sclerosing Cholangitis
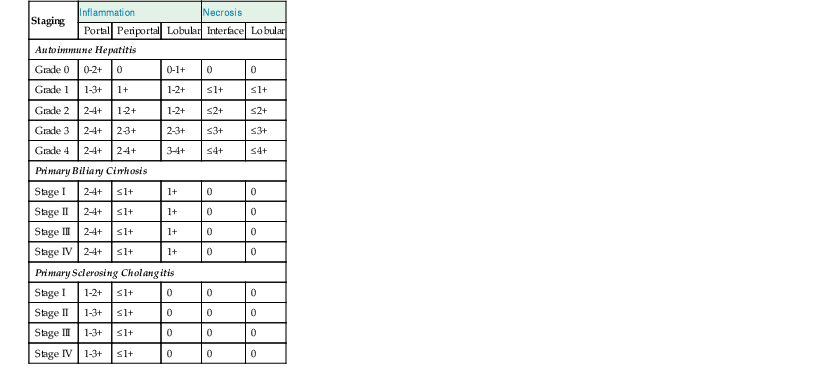
| Staging | Inflammation | Necrosis | |||
| Portal | Periportal | Lobular | Interface | Lobular | |
| Autoimmune Hepatitis | |||||
| Grade 0 | 0-2+ | 0 | 0-1+ | 0 | 0 |
| Grade 1 | 1-3+ | 1+ | 1-2+ | ≤1+ | ≤1+ |
| Grade 2 | 2-4+ | 1-2+ | 1-2+ | ≤2+ | ≤2+ |
| Grade 3 | 2-4+ | 2-3+ | 2-3+ | ≤3+ | ≤3+ |
| Grade 4 | 2-4+ | 2-4+ | 3-4+ | ≤4+ | ≤4+ |
| Primary Biliary Cirrhosis | |||||
| Stage I | 2-4+ | ≤1+ | 1+ | 0 | 0 |
| Stage II | 2-4+ | ≤1+ | 1+ | 0 | 0 |
| Stage III | 2-4+ | ≤1+ | 1+ | 0 | 0 |
| Stage IV | 2-4+ | ≤1+ | 1+ | 0 | 0 |
| Primary Sclerosing Cholangitis | |||||
| Stage I | 1-2+ | ≤1+ | 0 | 0 | 0 |
| Stage II | 1-3+ | ≤1+ | 0 | 0 | 0 |
| Stage III | 1-3+ | ≤1+ | 0 | 0 | 0 |
| Stage IV | 1-3+ | ≤1+ | 0 | 0 | 0 |
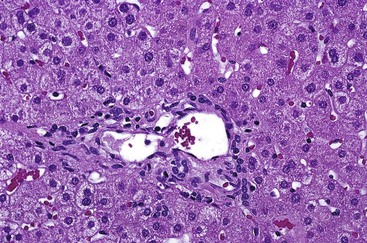
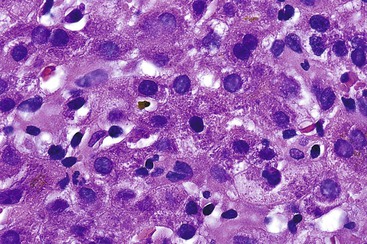
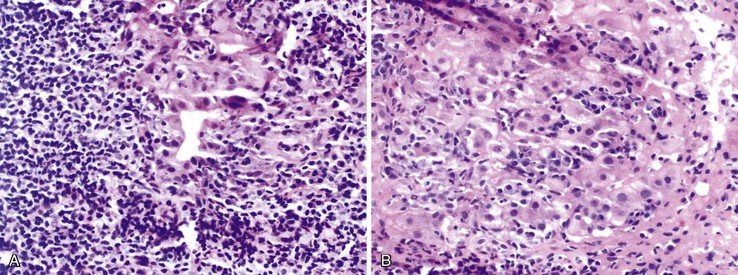
During flares of disease activity, interface hepatitis, which is the pathologic hallmark lesion of active AIH, is typically prominent. This lesion is characteristic of AIH, but it is not specific. Interface hepatitis is also common in viral- and drug-associated forms of chronic hepatitis, in PBC, and in other diseases. Interface hepatitis in AIH is characterized by a prominent lymphohistiocytic infiltrate at the portal tract mesenchymal-parenchymal junction with accompanying histologic evidence of liver cell damage (Fig. 47.3, A). CD8-positive T cells are a dominant subset of lymphocytes within areas of interface hepatitis, and CD4-positive T cells predominate within the portal tracts.18
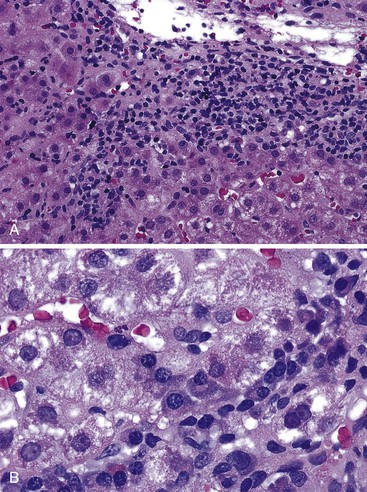
Damage caused by interface hepatitis may appear as overt necrosis of whole hepatocytes (i.e., acidophil bodies) or as slow destruction of hepatocytes caused by ingestion of small portions of cytoplasm by activated lymphocytes, described vividly by Wang and associates as troxis, which is the Greek word for “gnaw” or “chew.”16 In AIH, plasma cells are normally quite conspicuous, particularly compared with other disorders such as chronic viral hepatitis (see Fig. 47.3, B).
In addition to portal and periportal inflammation, lobular damage usually occurs in AIH. It manifests as some degree of anisonucleosis, ballooning degeneration, hepatocellular swelling, neocholangiole formation, cholestasis (rare), and acidophil bodies accompanied by a lymphocytic and plasmacytic infiltrate (Fig. 47.4). The degree of lobular damage contributes to the grade of disease. Although hepatocellular damage is usually panlobular, in some cases, zone 3 (centrilobular) damage may be prominent,16 resembling periportal interface hepatitis but occurring in the centrilobular zones of the liver (Fig. 47.5). This may represent an early form of AIH that precedes overt portal-dominant (classic) AIH.19
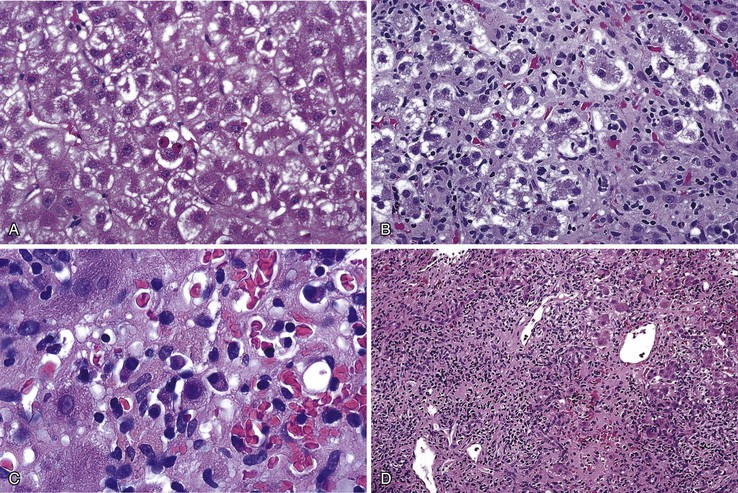
Lobular hepatitis is typically prominent during bouts of disease activity, but it may resolve spontaneously or as a result of immunosuppression. The activity grade can be assessed semiquantitatively, similar to hepatitis C. However, compared with chronic hepatitis C, AIH may have a much broader range of disease activity, from complete inactivity at one end of the spectrum to submassive or massive necrosis with confluent necrosis (see Fig. 47.4) at the other. Liver sampling is only rarely performed immediately after treatment, but rapid resolution of lobular necrosis with complete or incomplete resolution of portal inflammation and resolution of fibrosis is typical.
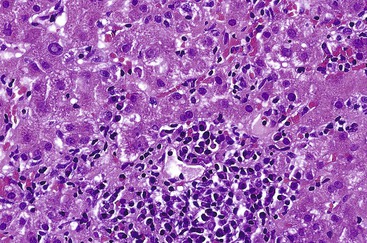
Cholestasis is usually not a feature of AIH, but a mild degree may be seen in more severe cases with marked lobular inflammation (Fig. 47.6). In contrast to hepatitis C, steatosis is not a typical feature of AIH. Lymphocytic venulitis involving portal or central veins may occur in high-grade disease, but it does not usually result in vein destruction or substantial endothelial damage, as in acute allograft rejection.
Similar to chronic viral hepatitis, progression of fibrosis (stage) from portal to initially periportal and then to bridging fibrosis and cirrhosis occurs in AIH. Even after cirrhosis has developed, interface hepatitis may persist within fibrous septa or regenerative nodules (i.e., active cirrhosis). Alternatively, the inflammation may subside completely, resulting in inactive cirrhosis.
Although AIH is a chronic disease, onset occurs at some point, and the disease may be regarded as acute. Relatively little is known regarding the pathology of acute-onset AIH. Lefkowitch and colleagues reported hepatocyte swelling, acidophil bodies, steatosis, cholestasis, piecemeal necrosis, and prominent portal plasma cells in initial liver biopsies from two patients with clinical features of AIH.20 Another study that examined liver samples from patients with clinical disease of less than 6 months’ duration found that with the exception of one case of lobular hepatitis with confluent necrosis (i.e., submassive hepatic necrosis), 25 of 26 of patients already had histologic changes of chronicity, such as portal inflammation and fibrosis (7 of 26) or bridging fibrosis (18 of 26).21 This suggests that most cases of clinically acute AIH represent a flare of previously occult chronic disease. This idea was further supported by observations in a related study that found that patients with AIH of less than 3 months’ duration were indistinguishable by clinical and laboratory tests from patients with disease of at least 12 months’ duration.22 These findings support the view that AIH is an a priori chronic disease.1
Differential Diagnosis
Acute-Onset Autoimmune Hepatitis
The histologic differential diagnosis of AIH is rather broad and depends on the clinical context of the affected patient. For patients who have severe, clinically acute hepatitis with prominent hepatocyte necrosis, the major differential diagnostic considerations include severe drug or toxic injury, fulminant A or B hepatitis, Epstein-Barr virus (EBV) (rare), and other types of systemic viral infection (see Table 47.3).
Distinguishing AIH from drug- or toxin-associated hepatitis can usually be accomplished by applying the principles of the scoring system of Alvarez and associates3 (see Table 47.2), which uses autoantibody status and hypergammaglobulinemia to establish a correct diagnosis. A thorough clinical history regarding use of drugs or toxins is essential.
In equivocal cases, a rapid response to immunosuppressive therapy helps support a diagnosis of AIH. Serologic testing for hepatitis A virus (HAV), hepatitis B virus (HBV), and EBV is usually quite reliable for distinguishing AIH and acute viral hepatitis. Acute hepatitis C is rarely a diagnostic concern, but when suspected, a positive serum assay result for hepatitis C virus (HCV) RNA is the best available diagnostic tool because anti-HCV antibodies may occur after the hepatitic episode. An initial absence of anti-HCV with subsequent development within weeks helps to support a diagnosis of acute hepatitis C infection.
Chronic Autoimmune Hepatitis
The pathology associated with typical AIH (i.e., classic pattern of chronic active hepatitis) may resemble chronic HBV infection with or without delta virus superinfection, hepatitis C, PBC, PSC, or chronic drug-related hepatitis (Table 47.4).
Table 47.4
Differential Diagnosis of Autoimmune Hepatitis
| Disease Pattern | Possible Diagnosis |
| Acute-onset pattern of predominant lobular hepatitis |
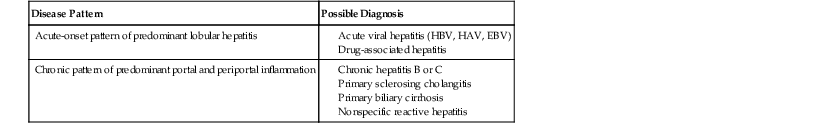
EBV, Epstein-Barr virus; HAV, hepatitis A virus; HBV, hepatitis B virus.
A diagnosis of chronic viral hepatitis is most often established serologically. However, the morphologic features may be helpful (Table 47.5). Brisk (grade 3 or 4) active chronic hepatitis usually excludes HCV, which almost always exhibits only minimal to mild disease, with transaminase levels that usually do not exceed 200 to 300 IU/mL. Occasionally, chronic HCV may exhibit fairly prominent plasma cell infiltrates. This has been designated by Czaja and associates as a form of chronic hepatitis C with autoimmune features.23 It likely accounts for examples of AIH type 2a (described earlier).
Table 47.5
Differentiation of Autoimmune Hepatitis from Chronic Viral Hepatitis
| Diseases | Helpful Hints |
| AIH vs. HCV |

AIH, Autoimmune hepatitis; HbcAg, hepatitis B core antigen HbeAg, hepatitis B e antigen; HbsAg, hepatitis B surface antigen; HBV, hepatitis B virus; HCV, hepatitis C virus.
Data from Alvarez F, Berg PA, Bianchi FB, et al. International Autoimmune Hepatitis Group report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929-938.
Chronic HBV infection may exhibit considerable disease activity that can mimic flares of AIH, but it is usually in the context of superimposed delta virus infection or emergence of a mutant strain of HBV. Prominent plasma cells help to distinguish AIH from chronic HBV infection, but the serologic profile is the most reliable means of ruling out chronic HBV infection.
Differentiating AIH from PBC and PSC is usually straightforward histologically, serologically, and biochemically (i.e., cholestatic enzyme pattern in PBC and PSC versus a hepatitic pattern in AIH) (Table 47.6; see also Table 47.3). The overlap syndromes of AIH and PBC and of AIH and PSC are discussed later in this chapter. Although PBC and PSC may demonstrate spillover of lymphocytes across the limiting plate into zone 1, neither disease demonstrates overt hepatocyte necrosis to any significant degree. In some cases, the number of portal plasma cells in PBC may be similar to that in AIH. However, PSC usually has fewer plasma cells. Although AIH, PBC, and PSC may have lymphocytes within bile duct epithelium (i.e., lymphocytic cholangitis), destruction of ducts and ductopenia are rare in AIH.24 Florid duct lesions characteristic of PBC and fibrous obliterative lesions characteristic of PSC are not seen in AIH, except in overlap syndromes.
Table 47.6
Differentiation of Autoimmune Liver Diseases
| Diseases | Helpful Hints |
| AIH vs. PBC |

AIH, Autoimmune hepatitis; PBC, primary biliary cirrhosis; PSC, primary sclerosing cholangitis.
Data from Alvarez F, Berg PA, Bianchi FB, et al. International Autoimmune Hepatitis Group report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929-938.
Grading and Staging
In liver pathology, the diagnosis should be rendered based on the cause whenever possible. For AIH, the grade of inflammatory activity should be reported. The simplest system for grading activity in AIH separates it into none, minimal, mild, moderate, and severe, which corresponds to a numerical grade of 0 through 4, respectively.12,25,26 This system is based primarily on the degree of piecemeal necrosis and lobular necrosis (see Table 47.3). If there is a discrepancy between these two patterns of necrosis, the more severe should prevail. The French METAVIR system devised for chronic viral hepatitis is similar, but it uses a scale of 0 to 3.27 The Knodell scoring system28 is useful for scientific studies, but its main drawback is that it does not distinguish necroinflammatory activity (i.e., grade) from fibrosis (i.e., stage). This problem was rectified in an updated version of this system published by Ishak and coworkers in 1995.29
An assessment of the degree of fibrosis (i.e., stage) should be reported. A variety of schemes exist, and the choice of a system is best left to the discretion of the pathologist based on input from the clinicians. The simplest system is based on a 0 to 4 scale that corresponds to absent, portal, periportal, bridging fibrosis, and cirrhosis, respectively (Table 47.7).12,25,26 The French METAVIR system is similar, but it uses a 0 to 3 scale.27 The updated Knodell system uses a 0 to 6 scheme.29

Natural History and Treatment
Because most cases of AIH respond to immunosuppressive therapy, the response to therapy can be included in the AIH scoring system.3 Initial treatment mainly uses corticosteroids with or without azathioprine. Eventually, many patients may be weaned from corticosteroids and maintained on azathioprine alone; 10% to 20% can eventually be weaned from all types of immunosuppression. Clinicians are often reluctant to discontinue immunosuppression if the serum transaminase levels remain elevated by more than two times normal or if histologic disease activity (grade 1 or greater in a four-grade system) persists.30 In cases that are steroid refractory or in which steroid side effects are an issue or azathioprine intolerance exists, cyclosporine, tacrolimus, or mycophenolate mofetil can be effective alternatives.31 For cases that progress to cirrhosis, liver transplantation is an acceptable form of therapy, although AIH may recur within the allograft.32
Primary Biliary Cirrhosis
PBC is a chronic bile duct–destructive disease that results in progressive cholestasis and cirrhosis if left untreated. Although the precise pathogenesis remains uncertain, considerable evidence indicates that it is an autoimmune disease that occurs in genetically predisposed individuals after stimulation by an environmental factor.33 PBC is associated with a variety of nonhepatic autoimmune diseases and elevated levels of serum autoantibodies.
The term PBC is commonly used even though it describes only the last stage of disease. A more appropriate term is the syndrome of PBC or chronic nonsuppurative destructive cholangitis.34,35 Although the latter term is fitting, it may also apply to other conditions, such as irreversible hepatic allograft rejection. Nevertheless, the term PBC is thoroughly entrenched in the literature and is likely to remain the term of choice by most clinicians. A PBC-like illness in patients who lack elevated levels of serum antimitochondrial antibodies (AMAs) (e.g., autoimmune cholangitis) is discussed later.
Clinical Features and Epidemiology
Approximately 90% of patients with PBC are female and between the ages of 40 and 60 years (range, 20 to 80 years). PBC occurs worldwide, and all races are susceptible. There is some familial clustering of PBC, with daughters of affected mothers at highest risk, but overall, less than 1% of first-degree relatives of patients with PBC develop the disorder.36 In a mostly white population in Olmsted County, Minnesota, the age-adjusted incidence of PBC is 4.5 and 0.7 per 100,000 women and men, respectively, with corresponding prevalence rates of 65.4 and 12.1 per 100,000.37
PBC coexists with other autoimmune diseases, with the strongest associations with connective tissue disorders such as rheumatoid arthritis, CREST syndrome (calcinosis, Raynaud disease, esophageal dysmotility, sclerodactyly, and telangiectasia), systemic lupus erythematosus, dermatomyositis, interstitial lung disease, and autoimmune thyroid disease.38 An association between celiac disease and PBC has been described.39 However, these associations most likely represent clustering of diseases in patients who are prone to autoimmune conditions rather than a direct pathogenetic link.
In early stages, PBC is usually asymptomatic and is detected only by identification of abnormal serum liver test results. In early disease, elevated serum levels of alkaline phosphatase and γ-glutamyltransferase (GGT) are typical. Serum cholesterol levels are usually elevated, and serum bilirubin levels are usually normal or only minimally elevated (<2 mg/dL). Serum ALT and AST levels are usually normal or only mildly elevated. Serum immunoglobulin M (IgM) levels may be mildly elevated.
In more advanced disease, the same general biochemical patterns persist. However, bilirubin levels tend to rise progressively, and the biochemical stigmata of cirrhosis (i.e., decreased serum albumin, prolonged prothrombin time, and thrombocytopenia) eventually become apparent. In later stages, the signs and symptoms of disease may be divided into those related to progressive cholestasis and those related to cirrhosis. Among the former are pruritus, xanthomas, jaundice, and osteoporosis. The signs and symptoms of cirrhosis in PBC are the same as for other causes of cirrhosis: esophagogastric varices, ascites, spider angiomas, and splenomegaly.
Pathogenesis
There is strong evidence that PBC represents an autoimmune attack directed toward autoantigens on biliary epithelium. PBC is thought to be initiated when tolerance to the dihydrolipoyl transacetylase (E2) subunit of the pyruvate dehydrogenase complex (PDC-E2) is lost, resulting in development of PDC-E2 specific–AMAs.33,40 The most conspicuous histologic features of PBC—lymphocytic and granulomatous destruction of intralobular and septal bile ducts—are tied to pathogenesis through the observation that intact immunoreactive PCD-E2 exists within apoptotic blebs of biliary epithelial cells.41 This protein has been demonstrated on allograft biliary epithelium in patients with recurrent PBC after liver transplantation.42
Although characteristic of PBC, AMAs are detected by using standard immunofluorescence techniques in only 92% to 95% of patients with clinical and histologic features of PBC, and they can be seen in patients without the typical clinical and histologic features of this disorder.43 ANAs are seen in approximately one third of patients, and the ANAs tend to be associated with a more rapid progression of disease.44 Approximately 5% to 8% of PBC patients lack AMAs (i.e., AMA-negative PBC), also known as autoimmune cholangitis (AC).
The type of inflammation, the lymphocytic and granulomatous duct destruction, the associated autoimmune diseases of other organs, and occurrence of overlap syndromes of PBC and AIH provide strong support of PBC as an autoimmune disorder. The precise pathogenesis remains unknown. However, T cell–mediated cytotoxicity and molecular mimicry after infection likely play a central role.33 Some have proposed a role for endothelin overproduction and biliary ischemia in the development of PBC.45
Pathology
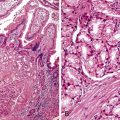
In early-stage PBC, the liver is often slightly enlarged and variably bile stained. In later stages, macronodular cirrhosis develops and is often associated with an intense green hue that reflects progressive cholestasis (Fig. 47.7). In contrast to PSC, cholangiectases and cholangitic abscesses are not seen.
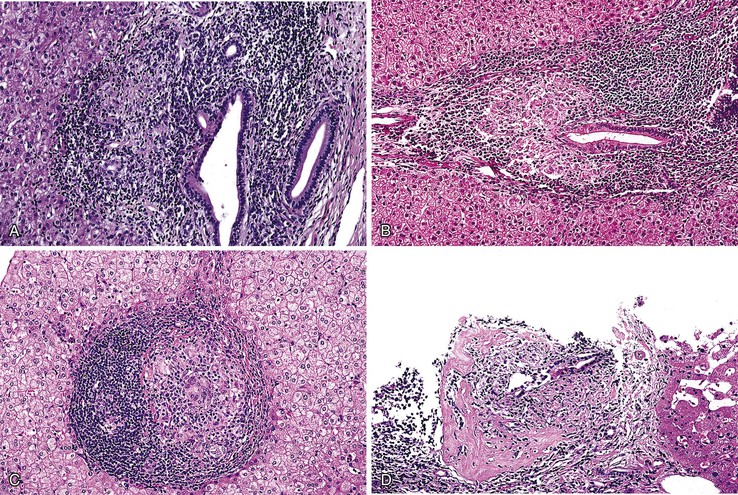
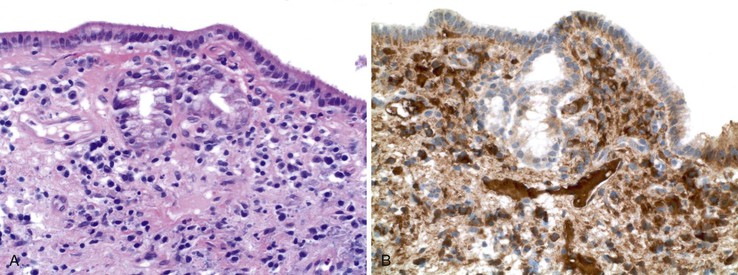
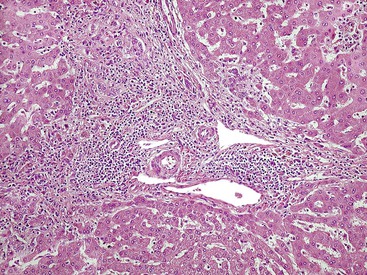
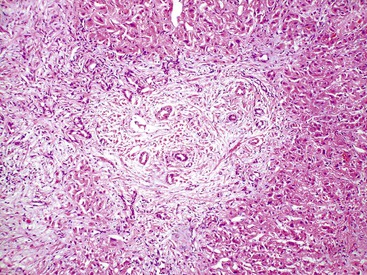
The hallmark lesion of PBC is destructive cholangitis that affects interlobular and septal bile ducts and results in duct loss and subsequent biliary cirrhosis. The term nonsuppurative destructive cholangitis accurately describes these lesions, but the more succinct term florid duct lesion is more commonly used.46 Because these lesions are usually focal, they may be missed on liver biopsy. The florid duct lesion is characterized by a portal lymphocytic infiltrate and epithelioid cells centered on septal and interlobular bile ducts, with histologic evidence of bile duct damage and destruction. Characteristic features include disruption of the bile duct basement membrane, intraepithelial lymphocytes and plasma cells, biliary epithelial cytoplasmic vacuolization, regenerative hyperplasia, and occasional mitotic figures. Granulomas are usually identified.
Florid duct lesions may reflect granulomatous duct destruction (Fig. 47.8) or more severe examples of lymphocytic cholangitis (Fig. 47.9). The bile duct damage in florid duct lesions is usually segmental in the longitudinal axis of the duct and in cross section, with only a portion of the duct affected at one time. Granulomas are often poorly defined and tend to be associated with the bile duct in an eccentric fashion, although concentric involvement may also be seen. Granulomas may also be found in portal lymphocytic infiltrates without an obvious connection to a bile duct. Less commonly, they may be found in hepatic lobules. With time, interlobular and septal bile ducts vanish, but small lymphohistiocytic aggregates may remain longer.
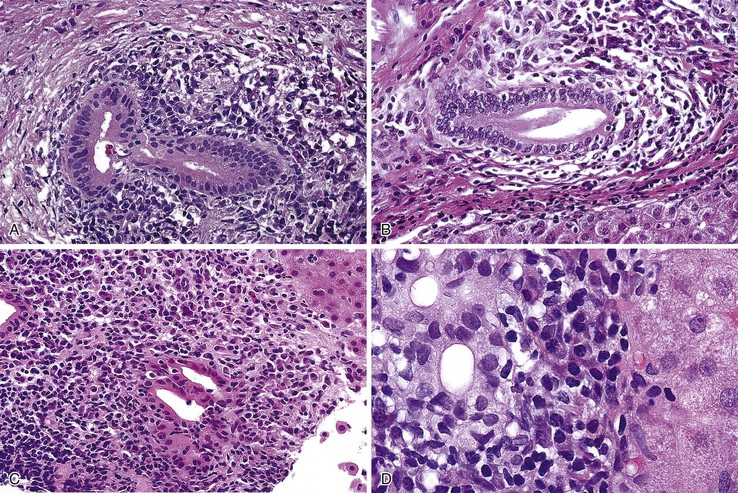
Bile duct changes are typically accompanied by a dense portal lymphoplasmacytic infiltrate (Fig. 47.10, A), which may be patchy in the early stages and may contain lymphoid aggregates and follicles, conspicuous plasma cells, and a few eosinophils.47 Biliary damage is likely primarily caused by the T cell infiltrates, but B cells appear to participate, primarily in early stages.48 When comparing AIH and PBC in terms of plasma cell subsets, IgG-positive cells strongly predominate over IgM-positive cells in AIH, but in PBC, IgM-positive plasma cells are much more conspicuous and may predominate.49 The portal lymphocytic infiltrate may spill over into the lobules, mimicking the appearance of interface hepatitis (see Fig. 47.10, B). In rare cases, mild interface hepatitis with hepatocyte necrosis may also develop.
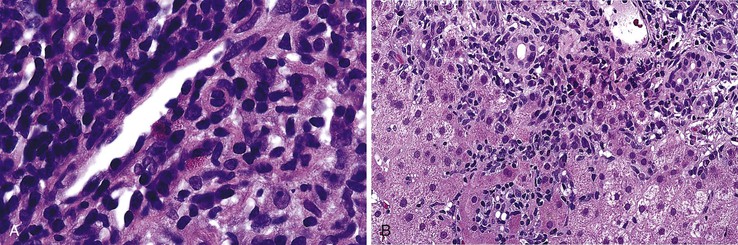
PBC lobules usually are almost normal. The normal-sized hepatocytes are not necrotic. In some cases, lymphocytes may aggregate within hepatic sinusoids in single-cell fashion, mimicking a lymphoproliferative disorder (Fig. 47.11, A). Scattered, usually small, noncaseating, lobular granulomas may be seen (see Fig. 47.11, B).
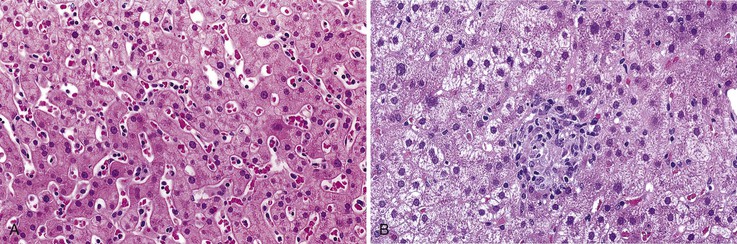
A subtle form of nodular regenerative hyperplasia has been observed in almost 50% of patients with early-stage disease. It may reflect portal venule damage by granulomas and contribute to the early development of portal hypertension.50
The progression of early PBC to cirrhosis is divided into four histologic stages.51 Although florid duct lesions may be seen at any stage, they are most common in stages I and II (Table 47.8). Stage I (i.e., portal stage) is characterized by a portal lymphocytic infiltrate with or without a florid duct lesion. In stage II (i.e., periportal stage), added features include piecemeal necrosis and delicate periportal fibrosis, which is often associated with ductular proliferation. Stage III (i.e., septal stage) is characterized by bridging necrosis or fibrous septa, and stage IV fibrosis (i.e., cirrhotic stage) is associated with nodular regeneration.
Table 47.8
Comparative Intrahepatic Bile Duct Findings in Autoimmune Hepatitis, Primary Biliary Cirrhosis, and Primary Sclerosing Cholangitis
| Staging | Lymphocytic Cholangitis | Florid Duct Lesion | Fibrous Cholangitis | Duct Loss (Ductopenia) |
| Autoimmune Hepatitis | ||||
| Grade 0 | 0-1+ | 0 | 0 | 0 |
| Grade 1 | 0-1+ | 0 | 0 | 0 |
| Grade 2 | 1-2+ | 0 | 0 | 0 |
| Grade 3 | 1-2+ | 0 | 0 | 0 |
| Grade 4 | 1-2+ | 0 | 0 | 0 |
| Primary Biliary Cirrhosis | ||||
| Stage I | 2-4+ | Common | 0 | 0 |
| Stage II | 2-4+ | Common | 0 | 1+ |
| Stage III | 2-4+ | Common | 0 | 1-3+ |
| Stage IV | 0-2+ | Few | 0 | 3-3+ |
| Primary Sclerosing Cholangitis | ||||
| Stage I | 1-2+ | 0 | Common | 0 |
| Stage II | 2-3+ | 0 | Common | 1+ |
| Stage III | 2-3+ | 0 | Common | 2-3+ |
| Stage IV | 2-3+ | 0 | Usual | 3-4+ |
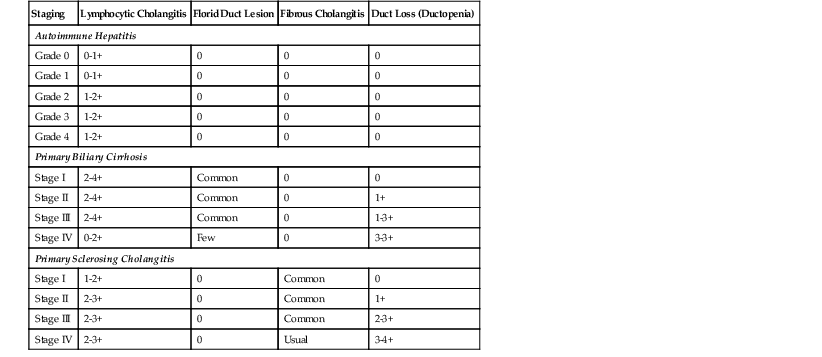
Regenerative nodules in PBC and in PSC are usually not as regular and round as in other forms of cirrhosis. The nodules often have a garland-shaped, irregular outline, not unlike pieces of a jigsaw puzzle. A progressive loss of ducts may occur early in the course of disease, with ductopenia usually being prominent in stage III and almost complete in stage IV. Florid duct lesions become less common with advancing stage,51 presumably reflecting loss of the target biliary epithelium.
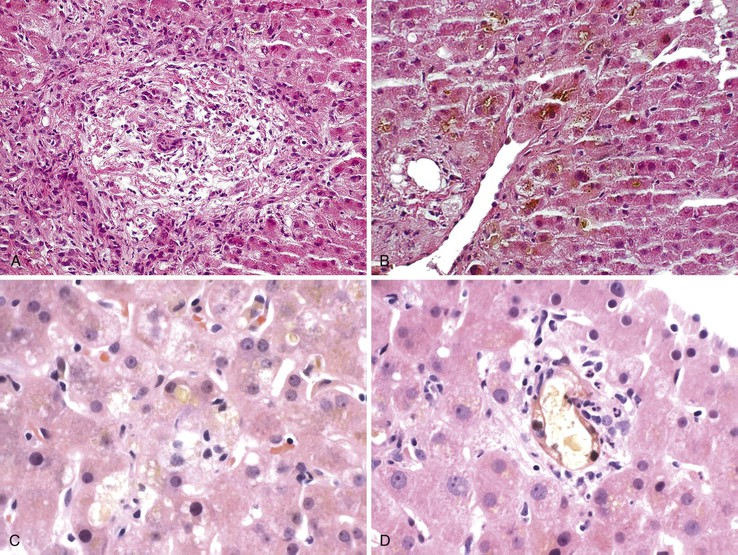
Periportal hepatocyte degeneration, which is thought to reflect bile acid (cholate) stasis, helps to distinguish PBC and PSC from chronic hepatitis resulting from autoimmune, viral, or drug causes.52 This feature becomes more prominent with advancing stages of disease and is similar in PBC and PSC. The finding of proliferating bile ductules at the limiting plate (Fig. 47.12, A and B) has been referred to as biliary piecemeal necrosis, in contrast to lymphocytic piecemeal necrosis that is typical of AIH.53 Biliary piecemeal necrosis is often accompanied by feathery degeneration of periportal or paraseptal hepatocytes, Mallory bodies, and deposition of orcein-positive copper-protein complexes (see Fig. 47.12, C and D). The combined, low-magnification effect of these features is a characteristic halo-like pattern surrounding regenerative nodules (see Fig. 47.12, E).
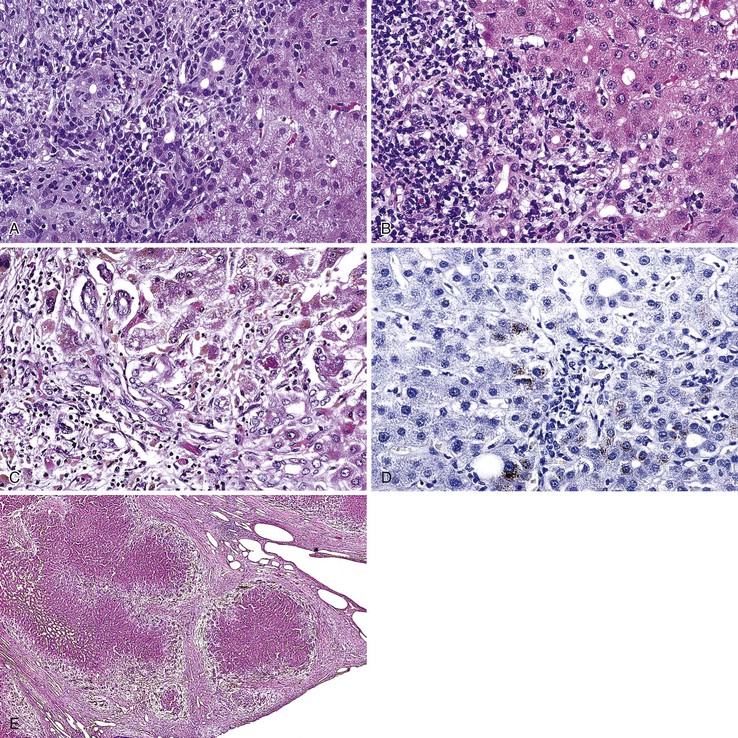
Differential Diagnosis
PBC shares many histologic features with PSC. In liver biopsy specimens, they may be indistinguishable. Unfortunately, the pathognomonic florid bile duct lesion of PBC is less prevalent in late-stage disease, and the pathognomonic fibrous obliterative cholangitis lesion of PSC is typically focal and may not be sampled in needle biopsies of the liver. Portal inflammation usually is more intense in PBC than in PSC. PBC commonly demonstrates mild sinusoidal lymphocytosis, a feature not normally seen in PSC (Table 47.9; see Tables 47.3, 47.6, and 47.8).
Table 47.9
Differential Diagnoses for Primary Biliary Cirrhosis or Autoimmune Cholangitis
| Component | Possible Diagnosis |
| Portal inflammation |
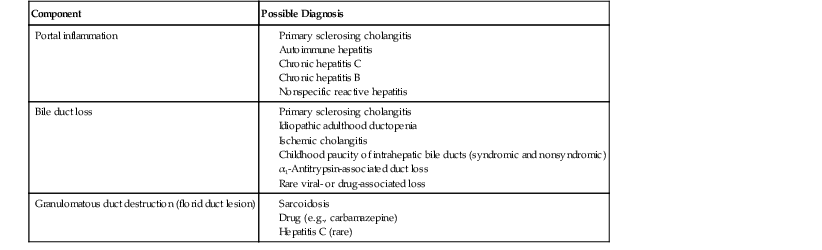
In the early stages, histologic manifestations of PBC and PSC may be indistinguishable from viral hepatitis (B or C) or AIH. All of these disorders may have a chronic active hepatitis pattern with portal and periportal inflammation (see Table 47.3). The presence of individual necrotic hepatocytes is not a hallmark feature of PBC or PSC, although acidophilic bodies may rarely occur. In contrast, hepatocyte necrosis is a common feature of active chronic viral hepatitis or AIH. Although PBC, PSC, and AIH may demonstrate spillover of lymphocytes into the periportal areas, this is not normally associated with visibly necrotic hepatocytes in PBC and PSC.
Granulomatous duct destruction is characteristic of PBC and AC (discussed later), but there are a variety of potential mimics (see Table 47.9). Florid duct lesions have rarely been described in hepatitis C.54 Uncommonly, drugs may cause lesions that mimic the type of florid duct lesion seen in PBC. Carbamazepine may cause a granulomatous type of hepatitis and acute cholangitis.55 Sarcoidosis can mimic PBC, particularly because of duct loss, cholestasis, and cirrhosis.56 The granulomas in sarcoidosis are usually better developed and more numerous than in PBC. Granulomas are only randomly associated with bile ducts and are commonly located within the hepatic lobules in sarcoidosis. The degree of portal inflammation usually is more intense in PBC compared with sarcoidosis. Lymphocytic duct destruction is also characteristic of graft-versus-host disease and acute cellular rejection. However, knowledge of the clinical setting allows distinction of these diseases from PBC.
Autoimmune Cholangitis
The term immunocholangitis was first used in Germany in 1987 to describe cases that resembled PBC histologically and clinically but had a serum autoantibody pattern similar to AIH.57 These patients constitute 5% to 8% of those with phenotypic evidence of PBC.58 In the mid-1990s, a number of publications addressed this issue, focusing primarily on the question of whether immunocholangitis should be considered AMA-negative PBC, an overlap syndrome with AIH, or a separate entity, such as AC59–61 or autoimmune cholangiopathy.62
It has become clear that AC is not equivalent to an AIH/PBC overlap syndrome. Comparison of the various clinical, histologic, and natural history aspects of PBC and AC indicates few differences between the two disorders.58 The similar response rates of patients with AC or PBC to ursodeoxycholic acid (UDCA) therapy and a similar natural history of AC and PBC after liver transplantation support the concept that AC is a form of PBC.63 The finding that most patients with AC show positive AMA levels when techniques more sensitive than standard indirect immunofluorescent microscopy are used further supports this theory.58
Natural History
Survival of patients with PBC depends greatly on the presence or absence of symptoms, stage of disease at the time of diagnosis, and whether UDCA therapy has been used because it delays progression of disease. Asymptomatic patients with early-stage disease have a longer time to transplantation or disease-related death. Patients with PBC are also at increased risk for hepatocellular carcinoma,64 although the risk is probably not as high as that seen in cirrhosis from other causes, such as viral hepatitis, hemochromatosis, or alcohol.
The Mayo model, a mathematical model for predicting the survival of patients with PBC (independent of a liver biopsy), has been developed65 and validated.66 It uses data (e.g., patient’s age, serum bilirubin and albumin levels, prothrombin time, presence or absence of edema) to estimate the likely survival time.
Treatment
Many therapies for PBC were initially attempted but had little success. Treatments included immunosuppressive drugs (e.g., azathioprine, d-penicillamine, corticosteroids, cyclosporine, methotrexate) and the antifibrotic agent colchicine.38 Since the late 1980s, several trials with UDCA, a synthetic bile acid, have been performed. UDCA is thought to reverse the potential hepatotoxicity of bile acids and inhibit eosinophil degranulation.47 It may also have immunosuppressive effects. Multiple trials at various institutions have shown that UDCA improves biochemical liver function.67–70 UDCA has little or no effect on hepatic inflammation or the prevalence of florid duct lesion development,71 but long-term (>5 years) UDCA therapy decreases the rate of progression to cirrhosis.72 In UDCA-treated patients, the rate of progression to cirrhosis is 4% and 17% at 5 and 10 years, respectively, for patients with stage I disease; 12% and 27%, respectively, for patients with stage II disease; and 59% and 76%, respectively, for patients with stage III disease.73
It appears that UDCA has a beneficial effect on overall and transplant-free survival in early-stage PBC and has become the mainstay of therapy.47,73,74 For patients with decompensated cirrhosis, orthotopic liver transplantation (OLT) has emerged as the most effective form of therapy. With the Mayo model, predicted versus actual survival rates for patients who have undergone liver transplantation were 55% and 79%, respectively, at 2 years after OLT and 22% and 68%, respectively, at 7 years after OLT.75 PBC may recur after liver transplantation76 at a rate of approximately 18% (see Chapter 52).32
Primary Sclerosing Cholangitis
PSC is a chronic biliary disorder characterized by involvement of the extrahepatic and, in most cases, intrahepatic biliary tree. Approximately 6% of cases show only intrahepatic biliary involvement (i.e., small duct PSC).77,78 By definition, the disease is idiopathic (i.e., primary) and does not result from other causes of cholangitis.79 The gallbladder may be involved.80 Polypoid gallbladder lesions in PSC may be malignant.81 It remains controversial whether IgG4-associated cholangitis should be regarded under the umbrella of PSC82 or as an independent entity; IgG4-associated sclerosing cholangitis is regarded as an independent entity in this chapter.
Clinical Features and Epidemiology
Most patients with PSC are adults. Most are 20 to 40 years of age at diagnosis. When children are affected, approximately 35% also have features of AIH.83,84 PSC occurs primarily in patients with idiopathic inflammatory bowel disease; 70% to 80% of patients with PSC also have chronic ulcerative colitis. Conversely, PSC develops in 2% to 7.5% of patients with ulcerative colitis and 1.4% to 3.4% of patients with Crohn’s disease.83 Patients with the haplotypes HLA-A1, HLA-B8, HLA-DR3, HLA-DR4, and HLA-DRW52A are at increased risk.85,86 In addition to its association with ulcerative colitis, other evidence of abnormal immune regulation in PSC includes the finding of perinuclear antinuclear cytoplasmic antibodies (pANCAs) in 80% of cases, antinuclear and smooth muscle antibodies in 20% to 50% of cases,83 and an association with other autoimmune diseases, such as thyroiditis, diabetes, and AIH.
Serum tests often reflect a chronic cholestatic process. In the early stages, elevation of alkaline phosphatase and GGT levels is almost universal. Elevation in the serum cholesterol level is also common, but serum ALT and AST levels are often only mildly (two to three times normal) elevated or almost normal. Serum bilirubin levels are usually normal. While the disease progresses, bilirubin levels may become slightly elevated, and biochemical evidence of cirrhosis (e.g., thrombocytopenia and decreased serum albumin) may become apparent. Markedly elevated serum bilirubin levels may be found in advanced (stage IV) disease. If these levels are found in early-stage disease, the physician should consider a dominant large duct stricture (benign or malignant) or choledocholithiasis.
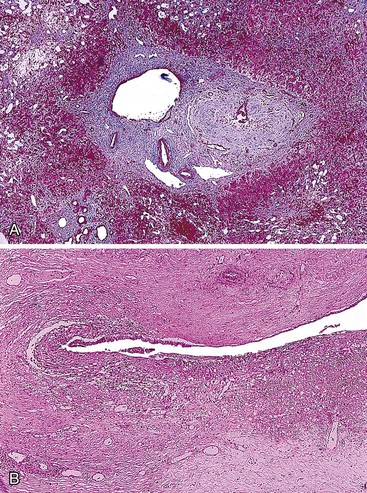
The hallmark of PSC is an abnormal cholangiogram. Endoscopic retrograde cholangiopancreatography (ERCP) and magnetic resonance cholangiopancreatography (MRCP) show comparable accuracy.83 With the exception of small duct PSC, cholangiography normally helps to establish the diagnosis by virtue of the characteristic, diffusely distributed, segmental, and multifocal strictures that lead to a beaded appearance with this radiologic test (Fig. 47.13).87 Rarely, other conditions may show similar cholangiographic findings (see Differential Diagnosis).
Pathogenesis
The pathogenesis of PSC is uncertain. Similar to PBC, a combination of clinical, biochemical, and morphologic features indicates that PSC represents an immune-mediated attack on biliary epithelium. However, in contrast to PBC, in which the direct immune target seems to be biliary epithelium, damage to biliary epithelium is not apparent in early PSC. Concentric fibrosis is the dominant histologic feature. The absence of a response to immunosuppressive therapy suggests that PSC is not a straightforward autoimmune process.
Some authorities have speculated that progressive periductal fibrosis may lead to an interruption of fluid and nutrient exchange between the peribiliary capillary plexus and the bile duct epithelium,87 which precipitates epithelial necrosis and eventually obliterates the lumen. The histologic changes in PSC are compatible with an immunologic attack on components of the subepithelial mesenchyme, leading to secondary fibrosis and possible ischemic epithelial damage.88 Further evidence of a possible role for microvascular damage in PSC includes the morphologic similarity between PSC and ischemic cholangitis, particularly after liver transplantation,89 and the observed displacement of peribiliary capillaries by connective tissue surrounding bile ducts in PSC.90
Natural History
Survival among untreated patients with PSC depends on the stage of disease at the time of diagnosis and the degree of cholangiographically evident bile duct abnormality. Typical survival from the time of diagnosis to death or liver transplantation is 12 to 18 years. The degree of cholangiographic disease is inversely correlated with survival.91,92
The course of progression has been assessed through examination of paired biopsy samples.93 Among patients with stage II disease, progression occurred in 42%, 66%, and 93% of patients at 1, 2, and 5 years, respectively. Among those with stage III disease, progression occurred in 14%, 25%, and 52%, respectively. Patients with PSC have a 4% to 14% risk for cholangiocarcinoma,94 which is associated with an extremely poor prognosis.95 Patients with small duct PSC, which is associated with a more favorable course than classic PSC, have a survival rate comparable to the general population and have a lower risk of hepatobiliary malignancy.96
Several mathematical models for predicting survival in PSC independent of biopsy have been developed. They are based on serum bilirubin, AST, and albumin levels and on the presence or absence of variceal bleeding,91 specific cholangiographic findings,92 and patient age at diagnosis.96
Pathology
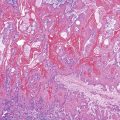
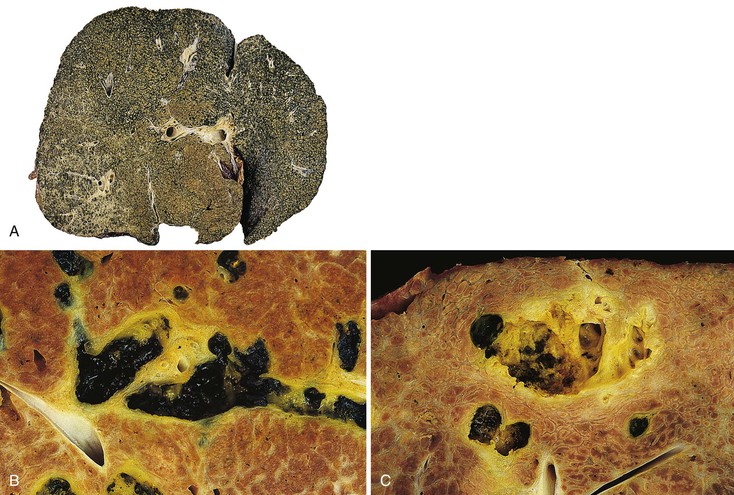
In early-stage disease, the liver is often slightly enlarged and bile stained to some degree. In later stages, similar to PBC, a macronodular type of cirrhosis develops that may be intensely green, reflecting progressive cholestasis (Fig. 47.14, A). In contrast to PBC, cholangiectases and cholangitic abscesses may develop. Cholangiectases are recognizable as cystic collections of bilious, sometimes calculous, dark green material that may be several centimeters in diameter, and they are usually located in the hilar region of the liver (see Fig. 47.14, B). Cholangitic abscesses, which presumably reflect superinfection of cholangiectases, have a more yellow appearance than cholangiectases (see Fig. 47.14, C). The large intrahepatic and extrahepatic ducts may develop areas of gross stricture and concentric fibrosis. Cholangiocarcinoma and bile duct carcinoma are recognizable as firm, white, sclerotic lesions or masses.
Large Ducts
Biopsies of the extrahepatic bile ducts in PSC usually reveal a thickened, fibrotic wall with a mixed inflammatory infiltrate. However, these alterations are nonspecific, because strictures unrelated to PSC (e.g., after bile duct surgery, after passage of gallstones with associated duct damage) may have similar features. Alterations of large intrahepatic ducts mimic those in the extrahepatic ducts. Both types of ducts may show segmental fibrosis with stricture formation. Areas of stricture frequently alternate with cholangiectases and occasional cholangitic abscesses.
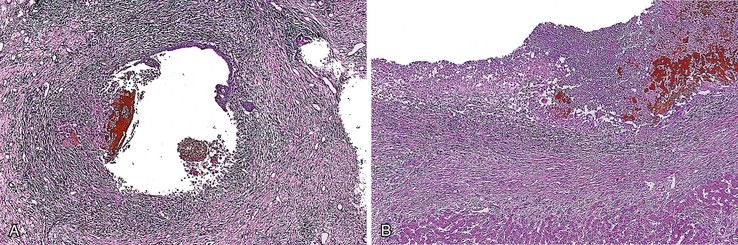
Histologic examination of cholangiectases reveals duct dilatation, epithelial atrophy with focal denudation, and mixed bilious material, neutrophils, and granulation tissue at the site of denudation (Fig. 47.15, A). Cholangitic abscesses are similar, but they tend to have a more exuberant neutrophilic infiltrate (see Fig. 47.15, B). Although these lesions strongly suggest PSC, they are rarely seen in liver biopsy samples.
Liver
The hallmark lesion of PSC is fibrous cholangitis, which may affect large ducts (perihilar intrahepatic or extrahepatic) and small intrahepatic bile ducts. When PSC affects small intrahepatic ducts without large duct involvement, small duct PSC may be diagnosed.97 Fibrous cholangitis is characterized by a mixed inflammatory infiltrate within or adjacent to damaged bile ducts associated with extensive concentric collagen deposition, which appears as a fibrous collar surrounding the duct combined with atrophy of the epithelium (Fig. 47.16, A and B). These lesions are frequently focal.
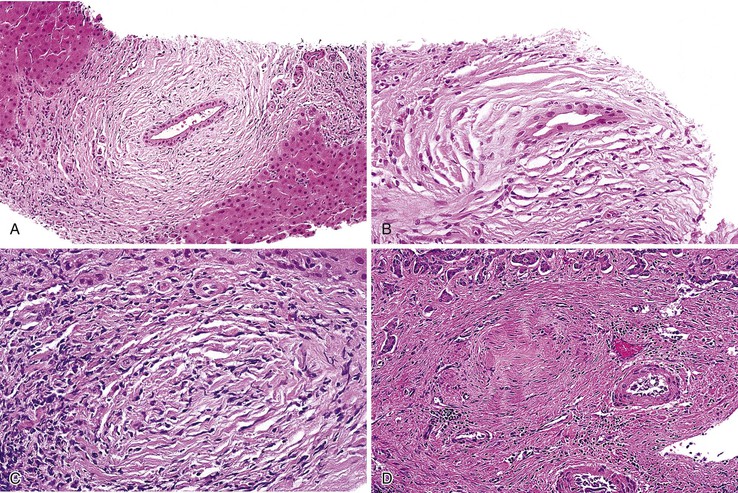
The end stage of fibrous cholangitis, sometimes referred to as fibrous-obliterative cholangitis, is characterized by complete loss of epithelium and the formation of fibrous scars that may be the only evidence of a preexisting bile duct (see Fig. 47.16, C and D). Unfortunately, fibrous-obliterative cholangitis is a rare finding among liver biopsy specimens. Ductopenia without ductal scars is the most common bile duct finding in needle biopsies from patients with advanced PSC.
With time, PSC proceeds through various histologic stages, similar to PBC (see Tables 47.3 and 47.8).98 Stage I disease is characterized by the presence of portal edema, inflammation, and ductular proliferation. At this stage, the changes may be indistinguishable from those of incomplete, secondary large duct obstruction, or they may resemble stage I PBC. Stage II is characterized by periportal fibrosis and inflammation with or without ductular proliferation. Scant lymphocytic piecemeal necrosis may be seen in some cases. In stage III disease, fibrous septa extend between adjacent portal tracts, and ductopenia becomes more prominent. Biliary piecemeal necrosis and cholate stasis, changes identical to those described in PBC, may become apparent (see Fig. 47.12, A through E). Similar to PBC, stage IV disease is the cirrhotic stage and is characterized by garland-shaped, regenerative nodules usually accompanied by ductular and biliary piecemeal necrosis that forms halos around the periphery of the nodules. Tissue samples often show near-complete loss of interlobular bile ducts at this stage.
Differential Diagnosis
The differential diagnosis of PSC includes histologic and cholangiographic categories, with some diagnoses spanning both classifications (Table 47.10; see Table 47.6). Histologic mimics have similarities in the pattern of inflammation or bile duct damage.
Table 47.10
Differential Diagnosis of Primary Sclerosing Cholangitis
| Component | Possible Diagnosis |
| Portal inflammation |

Inflammatory mimics include most of the major causes of chronic hepatitis (i.e., AIH, chronic HCV infection, and chronic HBV infection) and PBC or AC. PSC lacks the necrotic hepatocytes typical of AIH, hepatitis B, and hepatitis C in the lobules and periportal areas. The number of plasma cells in PSC usually is much lower than that seen in AIH and PBC. The pattern of sinusoidal lymphocytosis that is sometimes seen in PBC is much less conspicuous in PSC. Lymphocytic cholangitis can be seen in PSC, PBC, AIH, hepatitis C, and hepatitis B, although destructive cholangitis is seen only in PSC and PBC.
Bile duct destruction with concentric periductal fibrosis, superficial epithelial necrosis, cholangiectases, and biliary strictures indistinguishable from PSC can be seen in ischemic cholangitis (discussed later; see Fig. 47.20).99,100 A chronic bile duct–destructive syndrome that may result in a biliary pattern of cirrhosis is associated with α1-antitrypsin deficiency. This syndrome occurs more commonly in the pediatric population and in those with homozygous disease. It is less common in adults and patients with heterozygous disease.101
Idiopathic adulthood ductopenia (IAD) is a syndrome characterized by idiopathic, progressive duct loss without the cholangiographic or clinical features of PSC or idiopathic inflammatory bowel disease.102 Ductopenia is defined as more than 50% loss of interlobular and septal bile ducts in 20 or more assessable portal tracts.102 Morphologically, IAD can mimic small duct PSC (see Miscellaneous Ductopenic Syndromes).
Cholangiographic mimics of PSC include any of the various causes of secondary sclerosing cholangitis (see Table 47.10),79 which are discussed in greater detail later in this chapter.
Treatment
No form of medical therapy has been shown to alter the natural history of PSC.103 UDCA does not cure or slow progression of PSC, although it may improve results of serum biochemical tests (e.g., alkaline phosphatase, GGT).104 Endoscopic therapy directed at relieving dominant biliary strictures can significantly improve survival and symptoms in patients with PSC.105,106
For patients with decompensated cirrhosis, liver transplantation has emerged as an effective therapy. With hepatic allografts, disease recurs at a rate of 5% to 20% at 4.5 years, but this has not been associated with a statistically significant decrease in patient survival.107 A diagnosis of recurrent PSC is often difficult because biliary tract ischemia and bile duct anastomotic strictures can mimic PSC in individual cases. Most of the data supporting recurrence come from studies that analyzed large series of patients in a multivariate fashion.107,108
Overlap Syndromes
All of the previously described types of autoimmune liver disease commonly coexist with a variety of nonhepatic autoimmune diseases. Predictably, all have been described in association with each other, in which case the disorder is called an overlap syndrome.109–112 A conceptual framework of overlap syndromes is presented in Figure 19.17, but several fundamental issues remain uncertain. For instance, it is unclear whether overlap syndromes represent distinct disease entities, but most authorities do not believe this theory. Diagnostic criteria for overlap syndromes considered to be distinct disease entities are lacking.111,112
Overlap syndromes have been estimated to affect 18% of patients with autoimmune liver disease by some authorities.110 However, the frequency depends on the stringency of the definition and the rigor with which diagnostic features are sought in weighing the numerous clinical, histologic, radiologic, and serologic overlaps that may occur.111,112
The most common type of overlap syndrome is combined AIH/PBC. Combined AIH/PSC is the next most common,109 and combined PBC/PSC is the least common113 and the most difficult to identify. AIH/PSC overlap syndrome may be more common in children with autoimmune liver disease; prospective cholangiography used to assess children with positive ANA, ASMA, or anti-LKM serum values showed that one half of the patients had abnormal cholangiographic results.114
Establishing a diagnosis of any overlap syndrome requires unequivocal histologic and clinical evidence of AIH, PBC, or PSC. For example, the mere presence of lymphocytic cholangitis in liver tissue from an otherwise typical case of AIH is not considered diagnostic of AIH/PBC or AIH/PSC overlap syndrome. Similarly, mild lymphocytic piecemeal necrosis identified in an otherwise typical example of PBC or PSC is not diagnostic of an overlap syndrome with AIH. An example of AIH/PSC overlap is shown in Figure 47.17. Application of the international AIH score is of limited value in addressing its putative context; the scoring system can effectively exclude a potential autoimmune component but lacks sensitivity in unmasking an AIH-associated overlap syndrome.115
The Venn diagram shown in Figure 47.18 represents a practical approach to overlap autoimmune liver diseases. For instance, key elements that would justify including AIH as part of an overlap syndrome include a hepatitic profile (e.g., elevated serum AST or ALT values, score of 2+ or more, lobular or interface hepatitis, lobular plasma cells) when there are no alternative explanations and, ideally, a positive response to corticosteroid therapy.
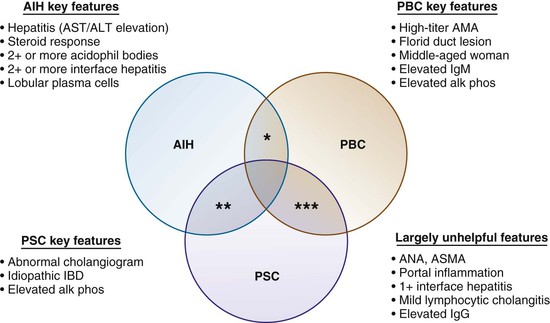
Features such as portal inflammation, mild interface hepatitis, mild lymphocytic cholangitis, elevations in ANA and ASMA values, and elevations in serum IgG levels are not useful in helping to establish an overlap syndrome such as AIH/PSC or AIH/PBD. Key features that would justify including PBC as part of an overlap syndrome are a strongly positive AMA value, elevated serum IgM levels, and florid duct lesions, although the latter are infrequently seen. An elevated serum alkaline phosphatase value and compatible age (typically, 45 to 60 years) and gender (90% female) are not considered useful inclusion criteria. Key features that would justify including PSC are a history of idiopathic inflammatory bowel disease and an abnormal, PSC-like cholangiogram. At resection, fibrous obliterative cholangitis and cholangiectases can be helpful, but they are not usually seen in needle biopsies of the liver.
Therapy is usually tailored to individual cases because there are no standard treatment regimens for overlap syndromes. Treatment of the dominant disease component is usually recommended (e.g., corticosteroids for AIH, UDCA for PBC). There are no known side effects of UDCA therapy, but corticosteroid therapy can exacerbate osteopenic bone disease, a common complication of biliary types of cirrhosis. Caution should be used in administering corticosteroid therapy for overlap syndromes.
Immunoglobulin G4–Associated Sclerosing Cholangitis
In 1998, a fibroinflammatory disorder of the biliary tree that mimicked PSC was reported in patients with autoimmune pancreatitis.116 However, in contrast to classic PSC, this disorder responded to corticosteroid therapy, suggesting that the cause was similar to autoimmune pancreatitis. It became clear that within the spectrum of IgG4-associated disease, which is characterized by hypergammaglobulinemia, high serum IgG4 levels, and a dense, IgG4-rich lymphoplasmacytic infiltrate, patients can develop secondary disease of the distal bile duct, which is often associated with autoimmune pancreatitis. IgG4 disease has been described in numerous anatomic locations other than the pancreatobiliary tree, including the retroperitoneum, salivary glands, esophagus, tonsils, mesentery, kidney, lungs, and lymph nodes. Tumefactive liver lesions that resemble inflammatory pseudotumor have been linked to IgG4 disease.117 Regardless of anatomic location of involvement, a response to steroid therapy is typical of IgG4 disease.
The criteria for diagnosing IgG4-associated sclerosing cholangitis (IgG4SC) vary. Tissue confirmation is not always necessary and depends on the strength of circumstantial evidence. The main differential diagnosis is between IgG4SC and PSC. Clinically, patients with IgG4SC tend to be older than those with PSC (mean age, 65 versus 41 years); are more likely to have obstructive jaundice, chronic pancreatitis, diabetes, and salivary gland swelling; and are less likely to have inflammatory bowel disease or biliary carcinoma.118 From a laboratory perspective, patients with IgG4SC are more likely than PSC patients to have elevated serum IgG4 levels (463 ± 355 versus 23 ± 10; normal < 135 mg/dL) and tend to have lower serum eosinophil counts (284 ± 219/mm3 versus 880 ± 1040/mm3). There were no significant differences in other common serum liver test values.118
Cholangiographically, IgG4SC patients are more likely than PSC patients to show segmental strictures and distal third common bile duct strictures, whereas PSC patients are more likely to have bandlike strictures, a beaded appearance, pruning, or a shaggy appearance. 118 Histologically, IgG4SC patients are less likely than PSC patients to have advanced (stage III or IV) liver fibrosis or fibrous obliterative cholangitis and tend to have more dense portal IgG4-positive plasma cell infiltrates. However, some periductal fibrosis can be seen in both conditions.118
IgG4SC is a clinicopathologic diagnosis, and a strict cutoff point for a significant increase in IgG4-positive cells that is diagnostic of IgG4SC is not known, but for practical purposes, 10 or more IgG4-positive plasma cells per high-power field (HPF) is regarded as abnormal. An example is shown in Figure 47.19. Obliterative phlebitis and storiform fibrosis are also features of IgG4SC, similar to those seen in IgG4 disease in other organs.
The distinction between IgG4SC and PSC can usually be established by using the information described earlier (Table 47.11), but reliance entirely on the histologic features is unwise. Examination of explanted livers from 98 patients with clinical features of PSC (not IgG4SC) showed that 23% of the putative PSC patients had moderate to marked IgG4-positive plasma cell infiltrates (moderate defined as 11 to –29 cells/HPF and marked defined as ≥30 cells/HPF), and this was associated with elevated serum IgG4 levels (mean, 200 mg/dL versus 72 mg/dL) and a more aggressive clinical course.119 It was unclear whether the study identified subtle or limited examples of IgG4SC or a subtype of PSC with increased numbers of IgG4-positive cells.
Table 47.11
Comparison of Immunoglobulin G4–Associated Sclerosing Cholangitis and Primary Sclerosing Cholangitis
| Characteristics | IgG4SC | PSC |
| Clinical Features | ||
| Mean age at diagnosis (yr) | 65 | 41 |
| Obstructive jaundice | More likely | Less likely |
| Associated coexisting conditions | Chronic pancreatitis Diabetes |
Idiopathic inflammatory bowel disease Biliary adenocarcinoma risk |
| Laboratory or Radiologic Features | ||
| Serum IgG4 level (mg/dL; normal, <135) | 463 ± 355 | 32 ± 10 |
| Serum eosinophil level (/mm3) | 284 ± 219 | 880 ± 1040 |
| Cholangiographic findings | Segmental, distal | Bandlike, beaded, shaggy pruning |
HLA, Human leukocyte antigen; IgG4SC, immunoglobulin G4–associated sclerosing cholangitis; PSC, primary sclerosing cholangitis.
Data from Nishino T, Oyama H, Hashimoto E, et al. Clinicopathologic distinction between sclerosing cholangitis with autoimmune pancreatitis and primary sclerosing cholangitis. J Gastroenterol. 2007;42:550-559. other liver disorders. J Autoimmune Dis. 2007;4:3-12.
Secondary Sclerosing Cholangitis
Secondary sclerosing cholangitis (SSC) is a syndrome characterized by intrahepatic or extrahepatic histologic and cholangiographic evidence of sclerosing cholangitis in a patient without PSC. This disorder is a consequence of other disease processes.79 SSC is not a distinct disease entity; it instead represents involvement of bile ducts by a variety of non-PSC diseases that result in a PSC-like syndrome (Box 47.1). Potential causes include immune disorders, ischemia, toxic injury, infiltrative disorders, infections, and idiopathic fibrosis, as reviewed by Abdalian and Heathcote.79
Immune disorders other than AIH, PBC, and PSC can cause intrahepatic duct loss and sclerosing cholangitis. IgG4SC is the dominant disorder. Rarely, connective tissue disorders, vasculitis, and idiopathic fibrosing disorders (e.g., retroperitoneal fibrosis), many of which may represent IgG4 disease, have been associated with sclerosing cholangitis. Intrahepatic duct loss also can occur in alloimmune injury (e.g., allograft rejection, graft-versus-host-disease).
Ischemic cholangitis is another important cause of sclerosing cholangitis, occurring independently or as a component of other disorders that cause SSC. It is likely that at least some of the autoimmune, infectious, fibrosing, and infiltrative causes of sclerosing cholangitis have an element of ischemic injury as a result of microscopic vascular damage to the peribiliary vascular plexus (discussed later).
Infectious disorders (e.g., bacterial infection, recurrent pyogenic cholangitis) and immune deficiencies (e.g., AIDS cholangiopathy) occasionally complicated by superimposed viral or bacterial infections can cause SSC.79 The patient’s clinical features and culture results are usually considered more helpful for diagnostic purposes than liver biopsy, although Cryptosporidium, Mycobacterium avium-intracellulare complex, cytomegalovirus, or other opportunistic pathogens are rarely identified histologically.
Infiltrative processes can cause SSC through the tumefactive effect of tumor cells or secondary vascular damage. Eosinophilic cholangitis is characterized by a dense eosinophilic bile duct infiltrate. It may be isolated to the biliary system or represent a component of more generalized eosinophilic gastroenteritis. Peripheral blood eosinophilia, a clinical history of allergies or asthma, absence of evidence of parasitic infection, and a rapid response to corticosteroid therapy are clues to its diagnosis.120 When the main biliary tract is affected, a PSC-like appearance may develop initially, with resolution expected after therapy. Peripheral liver biopsies often show a mechanical obstruction–like picture but without bile duct loss. Periductal eosinophilia is observed in some cases. Mast cell disease, Langerhans cell histiocytosis, and metastases are rare infiltrative causes of SSC.79
Ischemic Cholangitis
Ischemic damage to the biliary tract (i.e., ischemic cholangitis99) is an underrecognized disorder. It may be one of the most common causes of SSC. Patients at risk for ischemic cholangitis include those with prior biliary or upper abdominal surgery or trauma, thrombotic or vasculitic disorders, ABO-incompatible liver allografts, and hepatic intraarterial chemotherapy (particularly floxuridine). Clinical manifestations depend on the severity of bile duct damage but usually mimic PSC with the formation of biliary strictures, secondary obstructive cholestasis, occasional intrahepatic bile duct loss, and superimposed infective cholangitis.
The condition is not considered reversible. Some cases are managed by stenting, whereas others require resection of the diseased portion of bile duct, which may necessitate liver transplantation.
An important distinction between hepatocytes and biliary epithelial cells (intrahepatic and extrahepatic) is that the former receive a dual blood supply (i.e., hepatic arterial and portal venous), whereas the latter depend only on hepatic arterial blood. Blood is supplied to the mid-extrahepatic bile duct through the 3- and 9-o’clock arteries, which are derived from the right hepatic artery superiorly and the retroduodenal artery inferiorly.121 However, there are a variety of anatomic variations. For instance, the extrahepatic blood supply may be derived from the hepatic artery in some individuals or from the retroduodenal arteries in others. At the ultrastructural level, a rich anastomosing plexus of arterioles and venules surrounds the bile ducts.122
Ischemic cholangitis may be caused by a primary insult to the hepatic arteries, other major arteries, or the microscopic arteriolar vessels (i.e., peribiliary vascular plexus). The hepatic arteries may be compromised by surgical or traumatic interruption of the arteries (e.g., during liver transplantation) and thrombosis (e.g., spontaneous, secondary tumor, intraarterial chemotherapy). Smaller arteries may be involved in systemic vasculitides, ABO-incompatible transplantation, autoimmune disorders, and microthrombotic disease, such as hemolytic uremic syndrome,123 which overlaps with immune-mediated SSC (see Table 47.10). One case of presumed ischemic cholangitis in a critically ill patient was thought to result from microthrombosis related to severe systemic illness with generalized hypotension and hypoxemia (i.e., shock cholangitis).124
Ischemic cholangitis initially manifests with atrophic-appearing biliary epithelium, followed in more severe cases by sloughing of the biliary epithelium (Fig. 47.20). This results in contact of bile with the underlying mesenchyme, which may cause an exuberant mixed neutrophilic, lymphocytic, and histiocytic reaction. This may result in bile duct dilation (i.e., cholangiectases), bile plugs, choledocholithiasis, and bacterial superinfection due to bile stasis (i.e., cholangitic abscesses).
Fibrosis eventually occurs, causing a beaded cholangiographic appearance reminiscent of PSC. In needle biopsy specimens of peripheral liver, the dominant histologic picture is typically that of large duct obstruction, with biliary atrophy or bile duct loss recognizable in more severe cases. In long-standing cases, biliary-type fibrosis and cirrhosis may develop. In liver explants or autopsy specimens, the full spectrum of biliary damage is often apparent, with cholangiectases, cholangitic abscesses, strictures, and biliary sludge or stones, all of which mimic PSC.
Miscellaneous Ductopenic Syndromes
The mechanisms of bile duct loss remain poorly understood.125 Ductopenia in the absence of an identifiable cause is called idiopathic adulthood ductopenia.102 IAD is a syndrome characterized by progressive duct loss and its complications. It has no apparent cause (i.e., no morphologic or serologic features of PBC/AC, no abnormal cholangiogram or history of inflammatory bowel disease to suggest PSC, and no other known bile duct–destructive condition).
Patients with IAD fall into two main categories: those with progressive disease that may ultimately lead to liver transplantation and those with a benign clinical course.102 Rather than a distinct syndrome, IAD may represent a mixture of late-onset paucity of intrahepatic bile ducts, small duct PSC in the absence of inflammatory bowel disease, AC in the absence of granulomas or typical serum autoantibodies, and postviral duct destruction.102
The histologic features in early-stage disease consist of focal bile duct dropout (>50% loss is the definition of ductopenia), with relatively minor nonspecific inflammation, absence of granulomas or other specific features, and perhaps mild ductular proliferation (Fig. 47.21). Late-stage disease shows progressive ductopenia, changes of cholestasis, and biliary-type fibrosis, as described earlier in PBC and PSC.
Ductopenia has rarely been associated with adverse drug reactions. Small intrahepatic ducts are affected most commonly. In these cases, duct regeneration may occur.126 An unusual association between Hodgkin disease and ductopenia (i.e., vanishing bile duct syndrome) exists,127 although the mechanism is unclear.
Obstructive Cholangitis
Clinical Features
In obstructive cholangitis (i.e., large duct obstruction), mechanical interruption of extrahepatic bile flow caused by biliary stones within large ducts or the bile duct, ampullary strictures, or masses may produce a clinical and histologic picture that depends on the time frame (acute or chronic) and degree of stenosis (partial or complete obstruction). Nausea and vomiting are common early symptoms. Jaundice typically develops in cases of severe obstruction. Common duct obstruction with secondary (ascending) cholangitis may elicit the Charcot triad of symptoms (i.e., right upper quadrant pain, fever, and jaundice), with additional hypotension and mental status changes resulting in the Raynaud pentad, a poor prognostic sign. In patients with chronic, low-grade obstruction, progressive hepatic fibrosis and chronic biliary-type cirrhosis may evolve with the development of progressive pruritus and other consequences of established cirrhosis (e.g., portal hypertension, coagulopathies). Biochemically, acute bile duct obstruction is usually associated with a significant elevation in serum aminotransferase (ALT and AST), alkaline phosphatase, and GGT levels, with hyperbilirubinemia in proportion to the degree of obstruction but not often exceeding 25 mg/dL. In patients with chronic bile duct obstruction, serum aminotransferase levels tend to decrease, and alkaline phosphatase and GGT levels persist or slowly increase.
Pathology
Obstructive cholangitis does not have specific gross pathologic features. Histologically, liver biopsies from patients with acute bile duct obstruction may show portal edema, ductular proliferation with neutrophils (i.e., cholangiolitis), tortuous bile ducts that may appear increased in number on histologic sectioning, and, in some cases, intraluminal or intraepithelial bile duct neutrophils (i.e., cholangitis). Bile pigment may accumulate within the bile duct lumens and be visible on histologic examination. Even in the acute stage of extrahepatic large duct obstruction, the pathologist often notices a temporal progression of histologic changes. For instance, within the first 1 to 2 weeks of obstruction, cholestasis begins in the zone 3 (perivenular) region, which is often associated with portal edema and portal inflammation. In this early acute period, the pattern of cholestasis may mimic a drug effect, infectious hepatitis, or sepsis or shock.
Cholestasis in the early phase is often intracytoplasmic and canalicular with occasional large bile plugs. Thereafter, a marked marginal bile ductular reaction associated with neutrophils (i.e., cholangiolitis) develops in more than 80% of cases. In the following weeks and with continued obstruction, the parenchymal and portal changes become more developed, and cholestasis becomes more pronounced. Cholestasis extends to zone 2 and zone 1 (periportal) regions, the deposits become larger in size and number, and cholestatic liver cell rosettes associated with dense bile concretions may develop. Cholestatic feathery degeneration is common: hepatocytes are enlarged, rarefied, and degenerated. Lyses of liver cells and release of bile leads to the formation of bile lakes and bile infarcts, which are defined as clusters of confluent liver cells with feathery degeneration, bile stasis, and necrosis with or without macrophages. These features are virtually diagnostic of obstructive cholangitis. Bile infarcts are caused by the toxic action of bile salts. With repair, foreign-body giant cells and fibrous tissue become apparent.
If the obstructive cause is not reversed in the acute period, chronic changes develop, including further cholate stasis, copper accumulation, Mallory bodies, fibrosis, and cirrhosis. The bile ductular reaction may become extremely prominent. Eventually, a process resembling SSC develops. The small and large interlobular bile ducts show proliferation, branching, and increased tortuosity. The epithelium may appear degenerated or ulcerated, resulting in periductal fibrosis. Ductular proliferation is usually prominent. Canalicular cholestasis, often pericentral (zone 3) predominant, may be seen in severe cases (Fig. 47.22, A). Eventually, persistent chronic obstruction (Fig. 47.23) may result in a biliary cirrhosis–like histologic appearance, except that the bile ducts usually persist.
Differential Diagnosis
The major differential diagnosis for acute bile duct obstruction is cholestatic drug injury. In some cases, knowledge of the clinical features is needed to establish a correct diagnosis. The histiologic features that favor bile duct obstruction include portal edema, zone 3 canalicular cholestasis, and acute cholangitis with bile accumulation within the lumens of the bile ducts. Drug reactions may also cause zonal cholestasis but not usually prominent edema. Cholestasis associated with systemic shock or sepsis (i.e., cholangitis lenta) tends to cause the formation of bile plugs in dilated zone 1 (periportal) cholangioles, which contrasts with drug reactions and acute bile duct obstruction. Nevertheless, all of these disorders may show brisk ductular proliferation with or without neutrophils.
For chronic biliary tract obstruction, the major differential diagnoses are chronic ductopenic syndromes (i.e., PBC, PSC, and SSC). All of these disorders show progressive biliary-type fibrosis and cholate stasis. However, in contrast to PBC, PSC, and SSC, bile ducts usually persist and remain viable in cases of chronic biliary tract obstruction. All of these disorders, including chronic bile duct destruction, may show some degree of ductular proliferation at the portal-parenchymal interface (i.e., limiting plate).