[level-membership-for-surgery-category]Chapter 26
Atherosclerotic Risk Factors
General Considerations
Christos Liapis, John Kakisis
The word atherosclerosis is derived from two Greek roots: αθηρι (athere, gruel) and σκληρóς (skleros, hard). In 1755, von Haller first applied the term atheroma to a common type of plaque that, on sectioning, exuded yellow pultaceous content from its core.1 In 1904, Marchand introduced the term atherosclerosis in acknowledgment of the consistent association of fatty degeneration and arterial stiffening.2
Atherosclerosis is a systemic disease of large and medium-sized arteries in which lipid and fibrous material accumulate within the intimal layer. It should not be confused with the more general term arteriosclerosis, which refers to generalized thickening and loss of elasticity of arteries as a result of an increased amount of basement material and plasma protein deposition.3
Advances in our understanding of the pathogenesis of atherosclerotic vascular disease gave birth to the concept of cardiovascular risk factors, initially referred to in the Framingham Heart Study.4 Risk factor assessment is important to accurately guide primary and secondary prevention, whereas compliance with risk factor modification is still an area of concern.
Some risk factors are hereditary (and therefore not controllable), but others are acquired or related to behavioral and environmental factors and thus potentially amenable to manipulation. More recently, new risk factors have emerged.
This chapter summarizes the interaction of risk factors with the arterial wall during the atherosclerotic process and briefly reports the currently considered major risk factors. The emerging role and potential management of minor risk factors and markers of atherosclerosis are reviewed. Finally, the importance of compliance with these strategies, including knowledge that appeared in the literature since the publication of the seventh edition of the textbook, is discussed.
Risk Factors and Atherogenesis
The interaction of risk factors with the arterial wall initiates the atherosclerotic process (Fig. 26-1). The starting point for atheroma formation is endothelial dysfunction (reported also as “activation” because cells frequently respond normally to a noxious stimulus).5,6 Chapter 5 covers the mechanisms of atherogenesis in detail; this chapter provides a summary of the key mechanisms related to individual risk factors.
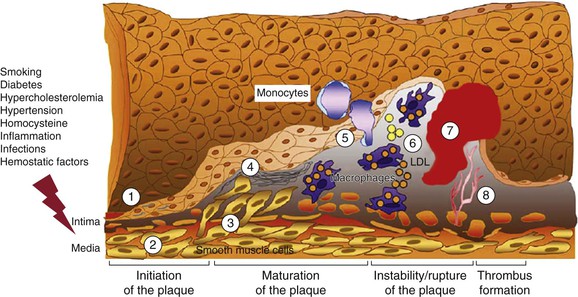
Figure 26-1 Evolution of arterial wall changes and plaque formation in the response-to-injury hypothesis: 1, endothelial dysfunction; 2, vascular smooth muscle cell hypertrophy; 3, migration and proliferation of vascular smooth muscle cells; 4, matrix elaboration; 5, expression of adhesion molecules and migration of monocytes; 6, uptake of low-density lipoprotein (LDL) and formation of foam cells; 7, thrombus formation; 8, angiogenesis and neovascularization. (Modified from Defraigne JO: Development of atherosclerosis for the vascular surgeon. In Liapis CD, Balzer K, Fernandes e Fernandes J, Benedetti-Valentini F, editors: Vascular surgery, New York, 2007, Springer, p 24.)
Interaction with Endothelial Cells: Endothelial Dysfunction
Currently, the most important contributors to endothelial dysfunction are hemodynamic disturbances, hypercholesterolemia, and inflammation. Etiologic culprits also include cigarette toxins, homocysteine, and a wide spectrum of infectious agents. Inflammatory cytokines (e.g., tumor necrosis factor [TNF]) can also stimulate the expression of proatherogenic genes in endothelial cells.7,8
The response of the endothelium to these stimuli can be rapid. For instance, forearm vascular reactivity increases substantially in the 4-hour period after ingestion of a fatty meal.9 Conversely, low-density lipoprotein (LDL) lowering reduces vascular reactivity.9
Experimental models of tobacco exposure, diabetes mellitus, hypercholesterolemia, and hypertension are characterized by common endothelial abnormalities, such as increased generation of oxidative stress and reductions in bioactivity or synthesis (or both) of endothelium-derived nitric oxide, which results in attenuation of vascular tone.10–14
Chronic endothelial injury eventually leads to endothelial dysfunction and increased permeability (Fig. 26-2). Concomitant events include LDL oxidation and accumulation in the subendothelial space of the intima.15 Oxidized LDL has a proinflammatory and proatherogenic effect, and it has recently been suggested that several of the neoepitopes generated during oxidation are highly immunogenic and result in the generation of autoantibodies.16
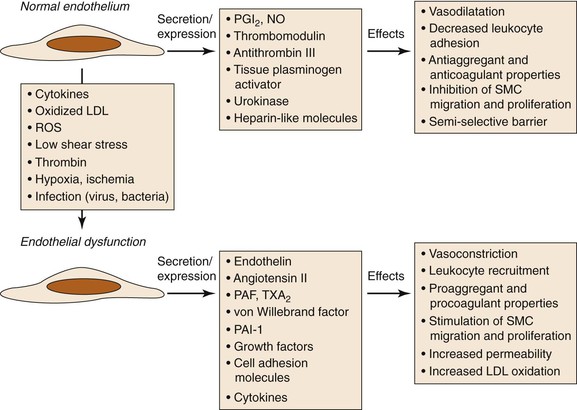
Figure 26-2 Consequences of endothelial dysfunction. Normal endothelium displays antiaggregant, anticoagulant, and vasodilatative properties along with inhibition of cell proliferation. After exposure to various agents causing endothelial dysfunction, these functions are modified toward procoagulant and vasoconstrictive activities together with stimulation of cell recruitment and proliferation. LDL, Low-density lipoprotein; NO, nitric oxide; PAF, platelet-activating factor; PAI-1, plasminogen activator inhibitor-1; PGI2, prostacyclin; ROS, reactive oxygen species; SMC, smooth muscle cell; TXA2, thromboxane A2. (Modified from Defraigne JO: Development of atherosclerosis for the vascular surgeon. In Liapis CD, Balzer K, Fernandes e Fernandes J, Benedetti-Valentini F, editors: Vascular surgery, New York, 2007, Springer, p 25.)
The greater endothelial elaboration of oxygen-derived free radicals activates oxidant-sensitive transcriptional proteins such as nuclear factor κB (NF-κB), which induces the expression of adhesion molecules and thus initiates the inflammatory process.17–22
Both cell recruitment into the endothelial surface and oxidized LDL stimulate the production of growth factors and chemotactic agents by endothelial cells.23 These substances attract and stimulate the proliferation of both macrophages and vascular smooth muscle cells (VSMCs).
Macrophages
As monocytes are attracted to the endothelium and migrate to the subendothelial space, they mature into macrophages and upregulate pattern recognition receptors, including scavenger receptors and Toll-like receptors. Scavenger receptors mediate internalization of oxidized LDL, which results in foam cell formation (the earliest event in the formation of fatty streaks). Toll-like receptors transmit activating signals that lead to the release of cytokines, proteases, and vasoactive molecules. Activated macrophages attract additional monocytes and stimulate VSMCs in a positive feedback loop. As foam cells accumulate in the subendothelial space, they distort the overlying endothelium and may eventually even rupture through the endothelial surface.23
Accumulating evidence indicates that macrophages are responsible for some key features of vulnerable plaque. An intense infiltration of macrophages is invariably observed at the site of plaque rupture, where the fibrous cap appears to be undermined. These macrophages are synthesizing cathepsins and abundant amounts of matrix metalloproteinases (MMP-1, MMP-3, and MMP-9). MMPs degrade the extracellular matrix, which weakens the fibrous cap.24–26
T Lymphocytes
T lymphocytes recruited to the intima interact with macrophages and can generate a chronic immune inflammatory state. The T lymphocytes found in atherosclerotic lesions are polyclonal, which indicates that these cells do not develop in response to a single antigen. It is not clear whether the T lymphocytes respond to specific antigens (e.g., bacterial or viral antigens or modified arterial wall constituents and lipoproteins) or are nonspecifically activated by the local inflammatory milieu.7
Nevertheless, T lymphocytes in atherosclerotic lesions recognize antigens and mount helper T cell type 1 responses with secretion of proinflammatory cytokines, which in turn can stimulate macrophages as well as endothelial cells and VSMCs.27
Subtypes of T lymphocytes called regulatory T cells, previously shown to maintain immunologic tolerance, control the development and progression of atherosclerosis. The function of regulatory T lymphocytes is modulated by chemokines and by costimulatory pathways.28,29
Complement
Activation of complement seems to play a role in both initiation of atherosclerosis and acceleration of the disease.30 Complement activation can occur by the classical (antibody dependent), the alternative (antibody independent), the lectin, or the coagulation pathway.31 Whereas complement activation by the classical and lectin pathways may be protective by removal of apoptotic cells and cell debris from atherosclerotic plaques, activation of the complement cascade by the alternative pathway may be proatherogenic and may play a role in plaque destabilization, leading to its rupture and the onset of acute cardiovascular events. In this context, various complement components, including C3, C3a, C4, C5a, and mannose-binding lectin, have served as biomarkers in prospective clinical studies, associated with an increased risk of death, myocardial infarction (MI), or restenosis after percutaneous transluminal angioplasty.31
Platelets
Platelets play an important role in stimulating the progression of atherosclerotic lesions by secreting growth factors and vasoactive substances (e.g., platelet-derived growth factor, transforming growth factor-α, transforming growth factor-β, epidermal growth factor, and insulin-like growth factor-1) after their adherence to the vessel wall in sites of endothelial ulceration.32,33 These substances recruit and stimulate proliferation of VSMCs.
Recently, platelets have been suggested as initial role players in the development of atherosclerotic lesions by recruiting and binding to leukocytes, endothelial cells, and circulating progenitor cells and initiating transformation of monocytes into macrophages. Platelets internalize oxidized phospholipids, express various scavenger receptors that are able to regulate LDL uptake, and promote foam cell formation.34,35 Moreover, platelets have been identified as key effectors of inflammation throughout plaque development through the secretion of several inflammatory molecules, such as CD40L, P-selectin, RANTES, and Toll-like receptors.36 These molecules exacerbate the inflammation and induce the transition from chronic to acute disease, featuring increased instability of the atherosclerotic lesion that results in plaque rupture and thrombosis.36
Vascular Smooth Muscle Cells
The mediators (e.g., platelet-derived growth factor, fibroblast growth factor, transforming growth factor-β, interleukin [IL]–1, and MMPs) released by endothelial cells, macrophages, lymphocytes, and platelets induce a change in the phenotype of VSMCs from the quiescent “contractile” phenotype to the active “synthetic” state. VSMCs in the synthetic state can migrate and proliferate from the media to the intima, where they produce excessive amounts of extracellular matrix (e.g., collagen, elastin, and proteoglycans) that transforms the lesion into a fibrous plaque. VSMCs are also capable of functions typically attributed to other cell types. Like macrophages, VSMCs can express a variety of receptors for lipid uptake and can form foam-like cells, thereby participating in the early accumulation of plaque lipid.
Like endothelial cells, VSMCs can also express a variety of adhesion molecules (e.g., vascular cell adhesion molecule-1 [VCAM-1] and intercellular adhesion molecule-1 [ICAM-1]) to which monocytes and lymphocytes can adhere and migrate into the vessel wall.37 The lesion grows until it is transformed from a fibrous plaque to a complex plaque. Apoptosis, proliferation, and migration of VSMCs are vital to the pathogenesis of atherosclerosis and plaque rupture. VSMCs are the only cells within plaque capable of synthesizing structurally important collagen isoforms.
Currently, the development of therapeutic approaches targeting VSMC migration and proliferation represents an active field of research, beyond drug-eluting stents and balloons that are already standard procedures for the prevention of restenosis.38–40
Vulnerable Plaque
Vulnerable atherosclerotic plaque (high-risk or unstable plaque) is associated with an increased risk of disruption, distal embolization, and vascular events. Vulnerable plaque is an advanced histologic lesion with a large lipid core (filled with lipid and cell debris), thin fibrous cap, ulceration, intraluminal thrombosis, and intraplaque hemorrhage as well as intense infiltration by macrophages and other inflammatory cells.41–44
The most widely used histologic classification of atherosclerotic plaque is the American Heart Association–recommended Stary classification.44 Inflammation plays a key role in the pathogenesis of atherosclerosis. In this process, the immune system and oxidative stress seem to be involved in the initiation, propagation, and activation of lesions in the arterial wall.41–44 Unstable plaque is dominated by inflammatory cells that destroy the fibrous cap and are responsible for endothelial denudation and thrombogenicity of the plaque contents (Fig. 26-3). Rupture depends on the balance between inflammatory cell activity and the VSMC-driven repair process.45 Activated macrophages, T lymphocytes, and mast cells produce a variety of molecules—inflammatory cytokines, proteases, coagulation factors, radicals, and vasoactive molecules—that are expressed in plaque and may modulate remodeling of the extracellular matrix, cell proliferation, and cell death (apoptosis) and thereby ultimately destabilize these lesions.41–44,46
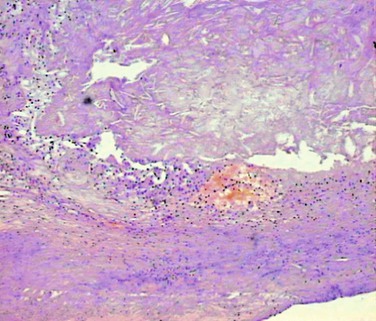
Figure 26-3 Unstable atherosclerotic plaque on histologic examination. Type VI plaque according to Stary’s classification,44 in which one or more of the following are present: surface defects (ulceration), hematoma, and thrombosis.
The likelihood of plaque rupture is a balance between the tensile strength of the plaque and the hemodynamic stress exerted on it.47 In ruptured plaque, the fibrous cap appears to be eroded at the shoulder of the lesion (where the fibrous cap meets the intima of the normal segment of the vessel wall).
Furthermore, local infection (e.g., induced by cytomegalovirus, herpes simplex virus, or Chlamydophila pneumoniae) may influence plaque stability.48
Several invasive and noninvasive imaging methods have been used to identify vulnerable plaque, but all have their limitations (Table 26-1).43 Thus far there are no widely available modalities to adequately provide both anatomic and functional information about vulnerable atherosclerotic plaque. New modalities are on the way, and the future looks promising.49–51
Table 26-1
Advantages and Disadvantages of the Different Imaging Techniques Used for Identification of Vulnerable Plaque
| Imaging Technique | Advantages | Disadvantages |
| High-resolution duplex ultrasound scanning | High resolution; noninvasive; no x-ray exposure; information on arterial wall thickness; qualitative and quantitative analysis of plaque | Limited information on plaque activity, possible improvement with the use of contrast-enhanced ultrasound |
| Digital subtraction angiography | Information on diameter stenosis and luminal surface | Invasive; may underestimate the degree of stenosis; no vessel wall imaging; no information on plaque composition and activity; contrast agent; x-ray exposure |
| Magnetic resonance angiography | Noninvasive; no x-ray exposure; no contrast agent; information on plaque composition | Less accurate in severe stenosis (unless contrast is used) |
| Helical computed tomography angiography | Noninvasive; differentiates between soft, intermediate, and calcified plaque | Contrast agent; x-ray exposure; overestimation of stenosis; no information on plaque activity |
| Intravascular ultrasound | Characterizes vessel wall and plaque morphology early in the disease process; differentiates stable and unstable plaque | Invasive; no information on plaque activity |
| Positron emission tomography | High sensitivity; functional information (plaque activity); molecular imaging | Lack of anatomic information unless combined with computed tomography; radiation exposure |
| Single-photon emission computed tomography | High sensitivity; functional information (plaque activity); molecular imaging | Lack of anatomic information unless combined with computed tomography; radiation exposure |
| Intravascular thermography | Functional information (plaque activity) | Invasive; risk of endothelial damage; difficult interpretation |
| Optical coherence tomography | High resolution (fibrous, lipid, and calcified components of the plaque can be distinguished); small probe size | Invasive; needs blood displacement; limited time window for imaging (2 seconds); limited length of penetration (1-2 mm) |
| Elastography | Differentiates lipid-rich and fibrous tissues; high sensitivity and specificity | Invasive |
| Near-infrared spectroscopy | Information on tissue chemical composition; detects the lipid core, the fibrous cap, and inflammation | Influenced by flowing blood; lack of anatomic information; limited independent role |
Assessment and Management of Risk Factors
The prevalence plus severity of atherosclerosis and peripheral arterial disease (PAD) or coronary artery disease (CAD) in individuals and groups is related to several “old” and “new” risk factors. Thus far there has been no universal agreement on the exact classification of the various cardiovascular risk factors and risk markers.
Classification of Risk Factors
An American Heart Association prevention conference statement in 199952,53 classified risk factors into three categories: “traditional”/conventional risk factors, which appear to have a direct causal role in atherogenesis; predisposing factors, which mediate some risk through the causal factors but may also have independent effects; and conditional risk factors, which have “an association with an increased risk for CAD, although their causative, independent, and quantitative contributions to CAD are not well documented.” The authors suggested that these factors may enhance risk in the presence of causative risk factors, hence the term “conditional.”
Since then, extensive research in all clinical fields of atherosclerosis has identified several novel and emerging risk factors or “markers” of atherosclerosis, which, however, need further confirmatory studies. As soon as sufficient evidence is available, some of them will move to the category of conditional risk factors, but it remains to be elucidated which ones will ever prove to be predisposing or even conventional.
A contemporary classification of risk factors is presented in Box 26-1.5,52–58
Conventional Risk Factors
Conventional risk factors for atherosclerosis are similar in the limbs and in the carotid, coronary, and other vascular beds. The four major risk factors for vascular disease59–65 are discussed in detail in other chapters of this section—smoking (see Chapter 27), diabetes mellitus (see Chapter 28), hyperlipidemia (see Chapter 29), and hypertension (see Chapter 30)—and are not discussed here.
Predisposing Risk Factors
Advanced Age
Although atherosclerosis may be present in younger individuals, age has a dominant influence. All forms of cardiovascular disease become more prevalent in the elderly. In several studies, the risk for PAD increased 1.5- to 2.0-fold for every 10-year rise in age,59,66–68 whereas the prevalence of intracranial atherosclerosis increases by 5% for every 1-year rise in age.69 A recent systematic review of 40 studies showed that the prevalence of carotid artery disease among people younger than 70 years was 4.8% in men and 2.2% in women, rising to 12.5% and 6.9%, respectively, among those 70 years of age and older.70 Similarly, death rates from CAD increase with each decade of life up to the age of 85 years. The death rate from CAD in white men aged 25 to 34 years is about 10 per 100,000; by the age of 55 to 64 years, it has increased 10-fold to nearly 1000 per 100,000.71
Obesity
Obesity predisposes patients to the development of hypertension, diabetes mellitus, and hyperlipidemia. Excess adiposity has recently been shown to play a direct role in initiating atherosclerosis in that fat cells are capable of affecting the systemic vasculature through a variety of mechanisms, among which altered arterial homeostasis, endothelial dysfunction, and inflammation are the most prominent.72–74 Weight loss interventions modify not only the risk factors but also arterial function.
In the Cardiovascular Health Study, each 5-unit increase in body mass index in midlife and in older age was associated with about a 30% increase in PAD prevalence and incidence.75 The European Prospective Investigation into Cancer and Nutrition (EPIC) study showed that general (as indicated by the body mass index) and abdominal (as indicated by the waist circumference or the waist-to-hip ratio) adiposity are both associated with the risk of death.76 Abdominal adiposity is a better predictor of death in patients with a low body mass index. The underlying mechanism is that adipose tissue, especially adipose tissue from visceral fat deposits, secretes mediators that are important in the development of chronic diseases, such as respiratory ailments, cancer, and atherosclerosis. In support of this theory, the risk of death from cardiovascular disease or cancer had been found to be higher among patients with a higher body mass index.77
Physical Inactivity
It is well established that the absence of even moderate levels of physical activity increases risk for cardiovascular morbidity and mortality in the whole population. It is hypothesized that physical activity consisting of either structured training or self-controlled body motivation results in “pleiotropic” actions on the cardiovascular system, which is explained, at least in part, by the modification of traditional and novel cardiovascular risk factors (e.g., hypertension, dyslipidemia, obesity, insulin resistance, endothelial dysfunction, hyperglycemia, inflammation, hypercoagulability, and oxidative stress).78–80 However, only 60% of the beneficial effect of physical activity on cardiovascular disease can be attributed to favorable changes in known risk factors, indicating that physical inactivity may be an independent risk factor for cardiovascular disease.80,81 Moreover, a substantial and expanding body of evidence has demonstrated that low exercise capacity is an independent predictor of cardiovascular and overall mortality.82–85 Therefore, lifelong exercise training should be incorporated in all strategies of primary or secondary prevention against cardiovascular diseases.86–89
Gender
Gender influences the risk for atherosclerosis and its preceding risk factors (e.g., hyperlipidemia, hypertension, and insulin resistance). Epidemiology, symptoms, and progression of cardiovascular disease are different between men and women. Indeed, cardiovascular disease develops in women about 10 years later than in men and typically after menopause. Premenopausal (estrogenic) women are relatively protected against atherosclerosis and its consequences compared with age-matched men. Estrogen alters serum lipoprotein levels through estrogen receptor–mediated effects on hepatic expression of apoproteins. With the onset of menopause, LDL levels rise, whereas high-density lipoprotein (HDL) levels fall, thereby changing a previously antiatherogenic lipid profile to one similar to that of men. However, several clinical trials have failed to demonstrate any utility of postmenopausal hormonal therapy for prevention of vascular disease in women.90–92
Family History and Genetics
Most cardiovascular events result from the interactions of genetic and environmental factors, none of which can cause disease by itself.
Familial predisposition to atherosclerosis is multifactorial and related to familial clustering of other risk factors (e.g., hypertension, hyperlipidemia, and diabetes), direct genetic linkage to atherosclerosis, or family habits associated with environmental risk factors.93 After adjustment for known risk factors, familial history of CAD remains an independent predictor of the disease, associated with a twofold to threefold increased risk of CAD.65 Particularly, individuals with a familial history of CAD of early onset (age at onset before 55 years in men and before 65 years in women) should be regarded as a high-risk group.65 Similarly, three family studies have assessed the heritability of the ankle-brachial index and demonstrated that about 20% to 50% of the interindividual variability in the ankle-brachial index is attributable to genetic determinants.94 Although aneurysm development is a combination of atherosclerosis and degeneration, the significant association with family history indicates genetic predisposition.95
Approximately 40 quantitative trait loci for atherosclerotic disease have been found in humans. Linkage analysis has identified specific genes that may contribute to cardiovascular risk, for example, myocyte enhancer factor-2 (MEF2A) with risk for MI96 and arachidonate 5-lipoxygenase–activating protein (ALOX5AP) with risk for MI and stroke.97 In femoral artery samples from patients with PAD, 366 genes were differentially regulated in intermediate lesions and 447 in advanced lesions.98 Among the upregulated genes, several genes involved in immune and inflammatory responses were identified.
In the future, a complete identification of genetic markers of atherosclerosis may be used in clinical prediction algorithms and even in therapeutic strategies. Resources such as the Human Genome Project and the international HapMap project are expected to change the practice of cardiovascular medicine in the 21st century.99
Behavioral and Socioeconomic Factors
Poor community socioeconomic conditions, depression, social isolation, work- or family-related stress, and type A behavioral patterns have been correlated with CAD and the severity and progression of atherosclerosis. These psychosocial risk factors are conveyed by behavioral pathways, such as inappropriate diet, smoking, sedentary lifestyle, and inadequate use of medical resources, and psychobiologic mechanisms, such as disturbed autonomic and hormonal regulation, which is directly involved in the pathogenesis of cardiovascular diseases.100–104
The risk for angina and ischemic stroke is two to four times higher in blacks than in whites, whereas the risk for PAD is highest in non-Hispanic blacks,105,106 not explained and not related to diabetes, hypertension, or body mass index.107
Conditional Risk Factors
Conditional risk factors had initially been proposed to facilitate classification of factors that may enhance the risk for cardiovascular disease in the presence of causative risk factors. Even though evidence for these factors has improved toward “independency,” additional prospective studies and randomized trials are needed to provide level A evidence.5,52–58
Homocysteine
Homocysteine is an intermediate metabolic product in the transmethylation and transsulfuration reactions that are essential for normal cell growth, differentiation, and function. It is involved in the transformation of methionine to cysteine.108 Plasma levels of homocysteine are regulated by environmental factors, such as vitamin B levels, as well as by genetically determined factors, such as the rare cystathionine β-synthase deficiency and enzyme activity of 5,10-methylenetetrahydrofolate reductase.109
Reports suggest that elevated homocysteine levels are an independent risk factor for vascular disease.110 Elevated homocysteine is associated with a mildly increased risk for CAD, stroke, PAD, and venous thromboembolism with a risk ranging between 0.8 and 3.4.111–113 In a meta-analysis of studies that adjusted for six or seven Framingham risk factors, each 5-µmol/L increase in homocysteine level conferred an approximately 9% increase in the risk for cardiovascular events.114
Homocysteine exerts its atherothrombotic effect through several potential mechanisms. It enhances thrombosis by forming a less porous fibrin network; by activating platelet aggregation and procoagulant factors V and VII, thrombomodulin, tissue factor pathway inhibitor, and plasminogen activator inhibitor (PAI); and by inhibiting activation of protein C. It also triggers proinflammatory activity by activating NF-κB and by promoting monocyte differentiation through NAD(P)H oxidase–mediated oxidant stress, endothelial dysfunction by inhibition of nitric oxide availability by endothelial cells and formation of reactive oxygen species, the LDL oxidation usually detected in cases of atherosclerotic lesions, proliferation of smooth muscle cells, and expression of tissue factor in endothelial cells and macrophages.115–119
Plasma levels of homocysteine are usually regulated by supplementation with B vitamins, either dietary or prescribed as folate.117,120 However, several clinical trials have demonstrated that lowering of homocysteinemia with vitamin supplementation is not effective in reducing cardiovascular risk,121–123 and even a harmful effect from combined B vitamin treatment has been suggested.124
C-Reactive Protein
C-reactive protein (CRP) is an acute phase protein. It is primarily synthesized by hepatocytes, driven by IL-6 with synergistic enhancement of IL-1 or TNF.125 A rise in CRP levels by as much as 1000-fold is not uncommon in both infectious and noninfectious disorders, including MI.126 Until recently, it has been the primary inflammatory marker used in clinical practice, and its predictive value can be estimated only through high-precision assays (high-sensitivity CRP [hsCRP]), with acceptable precision down to or below 0.3 mg/L (2.86 nmol/L). It is within these lower, previously “normal” ranges that hsCRP levels seem to have predictive ability for cardiovascular events. An hsCRP level higher than 10 mg/L (95.24 nmol/L), for example, should be discarded and measurement repeated in 2 weeks to allow the acute inflammation to subside before retesting.
Observational as well as prospective studies have consistently reported that elevated CRP serum levels definitely have prognostic value for cardiovascular events and mortality. CRP has been suggested to induce a prothrombotic state by induction of tissue factor expression in human monocytes.127 It can activate or inhibit the complement system, driving the inflammation in atherosclerotic lesions.128 CRP has also been reported to decrease the expression and bioactivity of endothelial nitric oxide synthase,129 with a subsequent effect on vasodilatation. CRP inhibits both basal and vascular endothelial growth factor–stimulated angiogenesis, whereas it promotes endothelial apoptosis in a nitric oxide–dependent fashion.130 CRP has been found to synergistically enhance angiotensin II–induced proinflammatory effects, such as cellular migration and proliferation, and lesion collagen and elastin content.131 CRP induces the release of monocyte chemotactic protein-1 (MCP-1) and endothelin-1 and thereby upregulates adhesion molecules and chemoattractant chemokines in endothelial cells and VSMCs.132 Finally, CRP has been suggested to mediate the proliferation and activation of VSMCs through activation of NF-κB,133 which leads to accumulation of these cells in the vascular intima and initiates atherogenesis.
Several therapeutic strategies orientated toward a reduction in CRP have been proposed, including cyclooxygenase inhibitors (aspirin and cyclooxygenase 2 inhibitors), angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and especially statins.134 In a randomized clinical trial comprising 17,802 apparently healthy women and men with LDL cholesterol levels of 130 mg/dL or lower and hsCRP levels of 2.0 mg/dL or higher, LDL cholesterol levels were reduced by 50% and hsCRP levels by 37% in patients randomized to 20 mg daily rosuvastatin instead of placebo.135 After a median follow-up of 1.9 years, the rates of most endpoints, including MI, stroke, arterial revascularization, and unstable angina, were reduced by about 50%, whereas all-cause mortality was reduced by 20%. Given the short follow-up time, the beneficial outcome of rosuvastatin may reflect the plaque stabilization effect of lowering CRP levels rather than the inhibition of bulky progression of existing plaque.
However, the causal association between CRP and vascular disease has been challenged by other studies showing that polymorphisms in the gene encoding CRP are associated with marked increases in CRP levels but are not in themselves associated with an increased risk of ischemic vascular disease.136 Similarly, morphometric analyses of atherosclerotic plaques in CRP-deficient animals revealed equivalent or increased atherosclerotic lesions compared with controls, an experimental result that does not support a proatherogenic role of CRP.137
Fibrinogen
Fibrinogen is a glycoprotein that circulates at high concentration in blood. Fibrinogen initially mediates platelet aggregation. Later in clot formation it is converted to fibrin. The fibrin matrix gives the clot shape, strength, flexibility, and stability.
Levels of fibrinogen have been demonstrated to be elevated in patients with acute thrombosis. Fibrinogen increases plasma viscosity and induces VSMC proliferation. The Gothenburg Heart Study was the first prospective trial to show an association between fibrinogen levels and subsequent risk for cardiovascular disease.138 In the Northwick Park Heart Study,139 fibrinogen level appeared to be as effective as total cholesterol concentration in predicting future risk for CAD. Higher levels of fibrinogen predict subsequent acute coronary syndromes, whereas lower levels, despite elevated cholesterol levels, are associated with a lower risk for acute coronary syndromes.140 It remains unclear, however, whether elevated fibrinogen levels are a cause or a consequence of atherosclerosis. In the Copenhagen City Heart Study, the relative risk of stroke development was almost double in patients with higher fibrinogen levels. Nevertheless, elevated levels were not associated with echolucent unstable carotid plaque.141 On the contrary, the CARDIA (Coronary Artery Risk Development in Young Adults) study demonstrated that an elevated fibrinogen concentration in persons aged 25 to 37 years is independently associated with subclinical cardiovascular disease (as indicated by the prevalence of coronary artery calcification and increased mean carotid intima-media thickness) in the subsequent decade.142
Modification of fibrinogen levels has failed to reduce the overall risk for MI or sudden cardiac death.143
Emerging Factors and Markers of Atherosclerosis
Inflammatory Markers
Apart from CRP and fibrinogen, several other inflammatory markers have been associated with atherosclerosis and cardiovascular disease (Table 26-2).
Table 26-2
Inflammatory Biomarkers and Different Stages of Evolution of Atherosclerotic Plaque
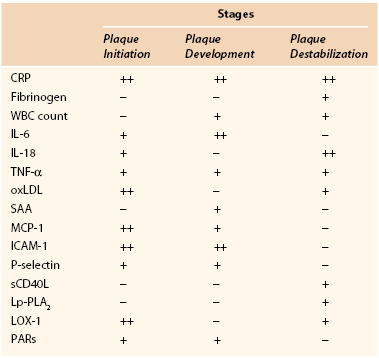
CRP, C-reactive protein; ICAM-1, intercellular adhesion molecule-1; IL, interleukin; LOX-1, lectin-like oxidized LDL receptor-1; Lp-PLA2, lipoprotein-associated phospholipase A2; MCP-1, monocyte chemotactic protein-1; oxLDL, oxidized low-density lipoprotein; PARs, protease-activated receptors; SAA, serum amyloid A; sCD40L, soluble CD40 ligand; TNF-α, tumor necrosis factor-α; WBC, white blood cell.
Serum Amyloid A.
Serum amyloid A is an acute-phase reactant, an apolipoprotein associated with HDL, that is predominantly produced by the liver. Nevertheless, it is also implicated in chronic inflammatory diseases (rheumatoid arthritis, atherosclerosis). Serum amyloid A has been shown to have a number of potentially proatherogenic effects that can lead to the destabilization of an atherosclerotic plaque: promotion of chemotaxis for monocytes and neutrophils; stimulation of the production of other proinflammatory cytokines, such as IL-1β and TNF-α; reduction of reverse cholesterol transport; and induction of the MMPs.144 Serum amyloid A has also been shown to promote thrombosis by increasing tissue factor, leading again to the destabilization of atherosclerotic plaques.144 Serum amyloid A was found to be marginally associated with cardiovascular disease,145 especially with extension of CAD in women.146
White Blood Cell Count.
In peripheral blood, the white blood cell count is usually increased in inflammatory and infectious conditions and could be affected in plaque inflammation as well. A higher leukocyte count has been associated with a greater cardiovascular risk. In a meta-analysis of seven prospective studies comparing the top with the bottom third of the value distribution, the relative risk of coronary disease was 1.4 (95% CI, 1.3 to 1.5),147 thus rendering leukocytes a valuable marker. Consistent with this, the decrease in the leukocyte count, achieved by pravastatin in patients with CAD, has been found to be an independent predictor of inhibition of the progression of coronary atherosclerosis.148
Cytokines.
Cytokines are key regulatory glycoproteins allied to inflammatory and immunologic processes that modulate all aspects of vascular inflammation. They are intimately associated with atherogenesis and modulate plaque morphology and stabilization. Many cytokines have been implicated in atheroma formation and complication. Several studies have established the role of IL-6 as an independent predictor of PAD in community screening, irrespective of ethnicity.149–151 IL-6 enhances cell adhesion molecule expression and the production of acute-phase reactants such as CRP and TNF-α by hepatocytes. Exogenous administration of IL-18 to mice enhances atherosclerotic lesions,152 whereas inhibition of IL-18 results in a reduction in atherosclerosis.153 IL-18 has a bearing on the progression and stability of human atherosclerotic plaque.154 TNF-α is involved in the progression of atherosclerosis from the initial stages of intimal thickening to the subsequent vessel occlusion. It stimulates expression of selectin and adhesion molecules and production of MMP-1, MMP-9, MMP-11, and MMP-13 in the endothelium, VSMCs, and macrophages.155 Locally within the atheroma, it increases expression of tissue factor, a potent thrombogenic protein.156 Finally, MCP-1 is the most widely studied chemokine in PAD and CAD,157 and it is consistently expressed in both animal and human atherosclerotic plaque.
Endothelial Adhesion Molecules.
Induction of endothelial adhesion molecules (ICAM-1, P-selectin) is considered one of the earliest steps in atherogenesis. Adhesion of leukocytes to the vascular endothelium and subsequent migration into the intima are key events in the atherosclerotic process. Several cytokines induce expression of ICAM-1, VCAM-1, and selectins by endothelial and smooth muscle cells and promote leukocyte adhesion at the site of the vascular lesion.158
Circulating Soluble CD40 Ligand.
Largely derived from activated platelets, circulating soluble CD40 ligand (sCD40L) can trigger an inflammatory reaction in vascular endothelial cells by the secretion of cytokines and chemokines. Interaction of membrane-bound CD40L and sCD40L with the CD40 receptor leads to the release of matrix MMPs and subsequent destabilization of the plaque.159 Elevated plasma concentrations of sCD40L at baseline predict a subsequent increased risk for future cardiovascular events in apparently healthy women and stable angina patients.160
Protease-Activated Receptors.
Members of the G protein–coupled receptors, protease-activated receptors mediate the cellular effects of thrombin. Protease-activated receptors contribute to the proinflammatory phenotype observed in endothelial dysfunction, and their upregulation in VSMCs seems to be a key element in the pathogenesis of atherosclerosis and restenosis.161
Lipoprotein-Associated Phospholipase A2.
Lipoprotein-associated phospholipase A2 is a proinflammatory enzyme secreted by macrophages and leukocytes. Its activity is associated with a risk for CAD and ischemic stroke.162
Because of their involvement in the atherosclerotic process, some of the inflammatory markers became important therapeutic targets. However, trials using TNF-α antagonists have failed to show any improvement in cardiac function.163,164 Statins have been found to have an independent effect on reducing MCP-1 levels.165 Nevertheless, no marked differences in TNF-α levels have been demonstrated in PAD patients receiving statin therapy.166 Peroxisome proliferator–activated receptor-γ agonists may inhibit production of cytokines and expression of adhesion molecules in endothelial cells167 and reduce production and entry of MMPs into plaque.168 Overall, the clinical value of anticytokine therapies remains to be proved.
Infection
Several infectious agents have been associated with the formation and complications of atherosclerotic plaque. Of bacteria, C. pneumoniae, a human respiratory pathogen and an obligate intracellular parasite, has most strongly been associated with cardiovascular disease. Helicobacter pylori, the causative agent of peptic ulcer, has also been correlated with CAD. Cytomegalovirus, herpes simplex virus types 1 and 2, and hepatitis A virus are the viruses most often associated with atherosclerosis, at both the experimental and clinical levels.
Patients with acute MI were found to have higher antibody titers against C. pneumoniae than matched normal individuals do.169 Recent or chronic active infections could be associated with an increased risk for acute MI.170 C. pneumoniae was also detected by immunocytochemistry and polymerase chain reaction in carotid plaque171 and has been associated with cerebrovascular symptoms (Fig. 26-4).172 Serologic evidence enhances the association between C. pneumoniae DNA in peripheral blood and symptomatic carotid atherosclerotic disease.173 The severity of PAD has also been correlated with femoral plaque infection.174 Early reports suggested a link between C. pneumoniae infection and expansion and risk of rupture of abdominal aortic aneurysms.175 However, the lack of standardized methods of testing for C. pneumoniae infection led to conflicting findings. Overall, the presence of C. pneumoniae in atherosclerotic vascular tissues merely suggests an association; it does not establish an etiologic relationship.
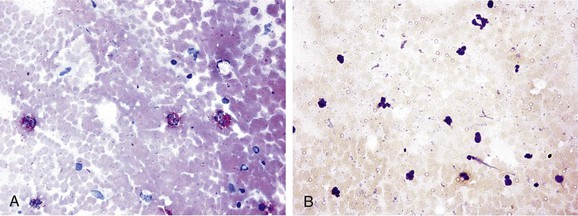
Figure 26-4 Immunohistochemistry of carotid plaque specimens. A, Characteristically (red fast stain) stained plaque for detection of Chlamydophila pneumoniae (formerly Chlamydia pneumoniae) with the use of a specific monoclonal antibody for Chlamydia, an avidin-biotin complex, and alkaline phosphatase (magnification ×400). B, Atheroma negative for C. pneumoniae (magnification ×200). (From Kaperonis EA, et al: The association of carotid plaque inflammation and Chlamydia pneumoniae infection with cerebrovascular symptomatology. J Vasc Surg 44:1198, 2006.)
Cytomegalovirus was demonstrated to be a risk factor for secondary coronary events or stroke in patients with elevated CRP.176,177 A number of potential mechanisms linking infection to atherosclerosis have been proposed. Modulation of lipid metabolism may be one mechanism by which pathogens may contribute to atherogenesis. Infectious agents may also damage the vascular endothelium and make it thrombogenic.178 Cytomegalovirus has been shown in in vitro models to enhance smooth muscle cell migration as well as to increase smooth muscle cell proliferation.179 Infection produces a wide spectrum of systemic manifestations, including the production of acute-phase proteins, circulating cytokines, and alterations in leukocytes, which may collectively influence the course of atherosclerosis. More likely, instead of a single pathogen, multiple pathogens are involved and the risk for CAD is related to the aggregate pathogen load—the so-called pathogen burden.180
Treatment trials of antibiotics for the prevention of cardiovascular events in animal models of atherosclerosis have produced results that were sufficiently encouraging to inspire multiple small-scale secondary prevention trials in patients. Numerous clinical trials have examined whether treatment of C. pneumoniae infection is beneficial in the secondary prevention of events in patients with stable and unstable CAD, but the results have been inconsistent. A large meta-analysis of randomized antibiotic trials181 has concluded that antichlamydial antibiotic therapy does not significantly improve major clinical outcomes in patients with CAD.
Vascular Calcification Markers
Atherosclerotic plaque calcification enhances plaque stability and decreases the likelihood of clinical events.182 The growing number of stimulatory and inhibitory molecules suggests that vascular calcification is an actively regulated process. Among these molecules, osteopontin, an acidic phosphoprotein, and osteoprotegerin, a member of the TNF-α receptor superfamily, have been demonstrated to inhibit mineral deposition as well as osteoclastogenesis, and they are constitutively expressed by a wide range of cell types in the vasculature.182–185
These bone matrix proteins, which attenuate vascular calcification, have emerged as novel markers of atherosclerotic plaque composition and cardiovascular disease prognosis. Data derived from clinical studies support the notion that increased serum levels of these markers are positively associated with acute cardiovascular events, coronary disease severity, and poor long-term cardiovascular outcomes.186–190 Studies have shown a strong relationship of serum osteopontin and osteoprotegerin levels with low carotid plaque echogenicity, and enhanced immunodetection of osteopontin and osteoprotegerin in human carotid plaque indicates their contribution to plaque instability (Fig. 26-5).191 Statins attenuate serum osteopontin and osteoprotegerin levels and enhance carotid plaque echogenicity, and thereafter stability, in patients with carotid stenosis.192
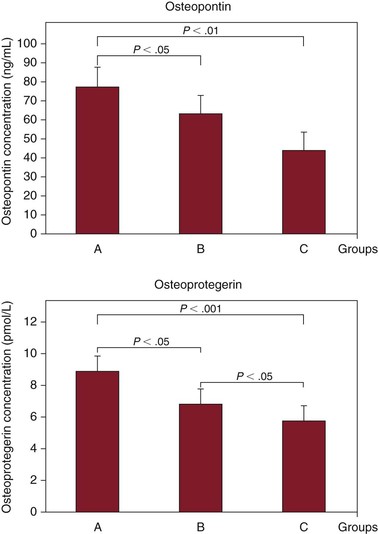
Figure 26-5 Relationship between serum levels of vascular calcification inhibitors and carotid plaque vulnerability. Serum osteopontin and osteoprotegerin concentrations are shown in the symptomatic (A), asymptomatic (B), and healthy control (C) groups. (From Kadoglou NP, et al: The relationship between serum levels of vascular calcification inhibitors and carotid plaque vulnerability. J Vasc Surg 47:55, 2008.)
Hemostatic Factors and Hypercoagulable States
Coagulation abnormalities have commonly been associated with the development of venous and arterial thrombosis. However, there is now sufficient evidence that apart from homocysteine, other hemostatic factors and hypercoagulable states are associated with atherosclerosis. Several circulating biomarkers that reflect inflammation, coagulation, fibrinolysis, and increased blood viscosity have been proposed as potential risk factors for the development of cardiovascular disease.193
The Caerphilly study, which monitored 2398 men for 4 years to establish which hemostatic markers add to the predictive value of conventional risk factors for CAD and stroke, suggests that fibrinogen, D-dimer, PAI activity, and procoagulant factor VII are independent risk factors that increase the potential for development of CAD and stroke in middle-aged men.194
The Edinburgh Artery Study, which observed 1592 individuals for 17 years to evaluate the relative contribution of inflammatory, hemostatic, and rheologic factors to incident MI and stroke, suggests that CRP, IL-6, fibrinogen, fibrin D-dimer, tissue plasminogen activator, leukocyte elastase, lipoprotein (a), and von Willebrand factor are strong predictors for the development of CAD.195
Among coagulation factors, high fibrinogen levels are associated with increased blood viscosity and risk for stroke, MI, and PAD.196 Increased levels depend on either environmental factors, such as smoking, or genetic polymorphisms, such as the −455G-A transition in the β chain of the fibrinogen gene.197 D-dimers that reflect plasmin and thrombin activity are also associated with increased risk for CAD and PAD.198 Other studies have demonstrated a higher prevalence of thrombophilic polymorphisms (prothrombin variant and factor V Leiden mutation) in patients with PAD, but their role as an independent risk factor has not been clearly stated.199,200
Case-control studies have indicated that proteins involved in fibrinolysis are related to the development of atherosclerosis; in particular, suppression of fibrinolysis by high plasma concentrations of PAI-1 has been associated with the development of CAD.201 Data associating PAI-1 levels and PAD are also available.202–204
Lipoprotein (a), a circulating lipoprotein resembling LDL cholesterol in core lipid composition, has structural similarity to plasminogen. Several clinical studies have correlated elevated lipoprotein (a) with CAD,205–207 cerebrovascular disease,208 and PAD.204,209,210
Antiphospholipid antibodies, such as lupus anticoagulant and anticardiolipin antibodies, are a heterogeneous family of autoantibodies associated with vascular damage. Antiphospholipid antibodies have been associated with PAD in various cohorts of patients,211 thus suggesting that they are an independent risk factor.204
Further prospective studies and meta-analyses are required to elucidate the underlying mechanisms, genetic or acquired, that link hemostatic factors and hypercoagulable states to atherosclerosis.
Long-term treatment with the direct thrombin inhibitor dabigatran etexilate has recently shown favorable effects in atherosclerosis progression and plaque stabilization in apolipoprotein E–deficient (ApoE−/−) mice.212 The modification of inflammatory mediators in plaque may provide a possible mechanism for this plaque-stabilizing effect.212
Matrix Metalloproteinases
MMPs are an ever-expanding family of zinc-dependent endopeptidases with proteolytic activity on one or more components of the extracellular matrix. On the basis of substrate specificity, MMPs are categorized into six groups. MMPs are predominantly found at several stages of atherosclerotic plaque development, and their activity is tightly regulated at three levels: control of gene transcription, secretion as latent enzymes and activation by proteases, and inhibition by tissue inhibitors (TIMPs). Almost all types of vascular cells secrete MMPs in normal conditions, whereas macrophages and lymphocytes constitute additional sources in atherosclerosis.213–217
Clinical and experimental studies provide a wealth of information on the crucial role of MMPs and TIMPs in intimal thickening and plaque destabilization. Atherosclerotic plaque is formed by net matrix deposition, in part mediated by migration and proliferation of VSMCs. Evidence of MMP-2 activation and MMP-9 upregulation during neointima formation is abundant. Regarding the underlying mechanisms, MMPs have been shown to catalyze the removal of basement membrane around VSMCs, to facilitate contact with the interstitial matrix, and to enhance the transformation of these cells to a more synthetic phenotype. All these effects further promote the migration and proliferation of VSMCs. At the initial stages of atherogenesis, MMPs have been also hypothesized to mediate subintimal infiltration of inflammatory cells. In addition, degenerative proteases seem to promote lipid–necrotic core formation in atherosclerotic plaque.216–218
Growing evidence supports the strong relationship of MMPs with plaque instability and subsequent cardiovascular events, revealing overproduction of MMPs by inflammatory cells in the rupture-prone regions of atherosclerotic plaque. This excessive proteolytic activity facilitates cleavage of the extracellular matrix and degradation of the fibrous cap with eventual acute plaque rupture. In clinical studies, increased serum levels of MMPs or MMP genotypes (e.g., MMP-3 polymorphism) were found to be closely associated with atherosclerotic manifestations such as CAD and ischemic stroke. Moreover, biomarker studies have shown greater levels of MMPs (e.g., MMP-9) in acute coronary syndromes, such as unstable angina and MI, than in stable CAD. Histologic analysis of specimens obtained from patients with unstable angina has shown a remarkable increase in intracellular MMP-9 levels in comparison to those with stable angina. Similarly, plaque extracted from symptomatic patients within 1 month before carotid endarterectomy contains fourfold higher concentrations of MMP-9. Other members of the MMP family, including excessive MMP-7, MMP-8, and MMP-12, have been reported in carotid plaque with the morphologic characteristics of vulnerability.218–221
Apart from arterial occlusive disease, MMPs have also been implicated in the development of abdominal aortic aneurysms. Several studies demonstrated increased expression and activity of MMPs (mostly MMP-2 and MMP-9) in the wall of aortic aneurysms as well as increased blood plasma concentration of MMPs in patients with abdominal aortic aneurysms.222–224 These studies have emphasized the importance of MMPs in the formation and expansion of abdominal aortic aneurysms as well as their potential use as therapeutic targets.225 For the pharmaceutical management of MMP activity, broad-spectrum inhibitors of MMPs and gene therapy with TIMPs have shown poor results. On the other hand, in vitro and in vivo models have proved that statins, apart from lipid modification, decrease MMP expression and activity and increase TIMP and collagen content in human atherosclerotic plaque. It seems that these lipid-lowering agents may exert their protective pleiotropic actions on the evolution of atherosclerotic plaque in part by modifying MMP activity.226,227 In parallel, peroxisome proliferator–activated receptors have been postulated to participate in atherogenesis inasmuch as they modulate proliferation and migration of VSMCs and regulate monocyte recruitment and foam cell formation in atherosclerotic plaque. Limited data have shown that a novel group of insulin-sensitizing drugs, thiazolidinediones, counterbalance the atherogenic actions of MMPs through activation of peroxisome proliferator–activated receptor-γ. These promising results remain to be proved in the long term.224–227
Adipokines
Obesity-associated atherosclerosis has recently been correlated with adipocyte-secreted factors called adipokines. Among these substances, leptin is upregulated in obesity and contributes to the development of vascular disease, whereas adiponectin is an antiatherogenic protein, concentrations of which are decreased in obesity-associated vascular disorders.
Leptin.
Leptin, a 167–amino acid protein, has a central role in fat metabolism; the increase in fat cells in number and size is coupled with an increase in leptin secretion. Leptin has been implicated in vascular processes as well, such as atherogenesis. It possesses angiogenic activity in endothelial cells and may elicit an angiogenic effect comparable to that of vascular endothelial growth factor. In addition, leptin receptors are expressed in atherosclerotic lesions. Through these receptors, leptin promotes vasodilatation. Besides that, leptin may induce vascular wall smooth muscle cell hypertrophy through induction of phosphorylation and activation of p38 mitogen–activated protein kinase and signal transducers and activators of transcription in a concentration- and time-dependent manner.228–231
Another effect of leptin is platelet aggregation. Through tyrosine phosphorylation, leptin (synergistically acting with adenosine diphosphate) induces platelet activation in a dose-dependent manner. Leptin also exerts many other potentially atherogenic effects, such as induction of endothelial dysfunction, stimulation of inflammatory reactions, oxidative stress, and decrease in paraoxonase activity. It may be of relevance that leptin-deficient and leptin receptor–deficient mice are protected from arterial thrombosis and neointimal hyperplasia in response to arterial wall injury.228–231 Inhibition of leptin signaling may thus be a promising strategy to slow the progression of atherosclerosis.
Adiponectin.
Adiponectin, a 247–amino acid protein, regulates the metabolism of lipids and glucose. Adiponectin may ameliorate insulin sensitivity and elevate HDL cholesterol, especially in obese persons; it may also serve as an anti-inflammatory molecule at the vascular wall, inhibit VSMC proliferation, and protect endothelium from macrophage adhesion and macrophage-induced injury.232–234
Adiponectin seems to have an important protective function against cardiovascular disease. Circulating levels lower than those in healthy control subjects have been found to be associated with conditions such as obesity, diabetes, and cardiovascular disease,235–239 whereas elevated plasma adiponectin in women confers protection from CAD.240 On the contrary, in studies of subjects with documented atherosclerotic cardiovascular disease, high adiponectin levels have been associated with increased mortality, which is a paradox to be resolved.241–243
Novel adipokines have also been shown to correlate with carotid plaque vulnerability, beneficially influenced by the use of statins.244
Endothelial Progenitor Cells
Endothelial progenitor cells are bone marrow–derived cells with an ability to repair damaged vascular endothelium (the initial atherogenic event), thus limiting the formation of atherosclerotic lesions. Alteration of the number of endothelial progenitor cells (e.g., endogenous mobilization or injection of ex vivo–generated endothelial progenitor cells) is a novel concept for modulating the balance of injury and repair mechanisms. In humans with coronary atherosclerotic disease, endothelial progenitor cells are a novel predictor of risk for cardiovascular mortality and morbidity. The number of endothelial progenitor cells correlates with endothelial function and is a better predictor than the patient’s combined Framingham risk factor score. Circulating endothelial progenitor cells inversely correlate with risk factors for atherosclerosis, whereas vasculoprotection, such as by statins, estrogens, and physical activity, may be partly mediated by progenitor cells.245–250
Creatinine
Renal impairment is associated with an increased risk for mortality in advanced PAD, irrespective of the presence of hypertension and diabetes.251 Recent evidence strongly suggests that renal disease and vascular disease progress in parallel and that treatment of vascular disease may also preserve renal function.252,253 Future studies will determine whether strategies that maintain renal function will result in a decreased risk for vascular events.
Urate
Uric acid levels have been found to be an independent risk factor for PAD and a predictor of CAD.254 Hyperuricemia is associated with worse functional status of the peripheral circulation in hypertensive patients.253,254
Microalbuminuria
Microalbuminuria is gaining recognition as a marker of an atherogenic milieu and cardiovascular disease. It has been found that the presence but not the magnitude of albuminuria is an important risk factor for PAD in diabetic but not in nondiabetic subjects.255 One hypothesis is that microalbuminuria may be a marker of risk for cardiovascular disease because it reflects subclinical vascular damage in the kidneys and other vascular beds.
Periodic screening for microalbuminuria could allow early identification of vascular disease and help stratify overall cardiovascular risk, especially in patients with risk factors such as hypertension and diabetes. Data from interventional studies suggest that treatment with angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers, statins, or strict glycemic control (in diabetics) offers significant reductions in cardiovascular or renal morbidity, or both, in patients with albuminuria.252,256
Oxidative Stress
Oxidative stress is the unifying mechanism for many cardiovascular risk factors, which additionally supports its central role in cardiovascular diseases. Oxidative biomarkers providing diagnostic or prognostic information (or both) have emerged.6
Oxidized LDL Cholesterol.
Oxidized LDL cholesterol is derived from the biochemical modification of LDL cholesterol and is proatherogenic. It is present in atherosclerotic lesions in humans.257
Lectin-like Oxidized LDL Receptor-1.
Lectin-like oxidized LDL receptor-1 is a scavenger receptor (membrane glycoprotein) that mediates the recognition and internalization of oxidized LDL. Lectin-like oxidized LDL receptor-1 overexpression contributes to atherosclerotic plaque formation and progression. Activation of lectin-like oxidized LDL receptor-1 by binding oxidized LDL stimulates intracellular signaling, gene expression, and production of superoxide radicals.258
Myeloperoxidase.
Myeloperoxidase, which generates the strong oxidizing agent hypochlorous acid, has been found to be correlated with risk for MI, endothelial dysfunction, and PAD.259–261
Dietary Antioxidants.
Many epidemiologic studies have indicated that a diet rich in fruits and vegetables is protective against the development and progression of cardiovascular disease. A large number of studies provide data suggesting that consumption of dietary antioxidants is associated with reduced risk for cardiovascular disease.6 Future studies are expected to clarify the role of oxidative biomarkers in the diagnosis and prognosis of cardiovascular disease and to determine their value in specific clinical populations.
Miscellaneous
Alcohol in small amounts (<1 drink per day) is associated with higher HDL cholesterol and may have an atheroprotective effect. Conversely, larger amounts have been correlated with higher cardiovascular risk.262–264 Vitamins C and E could potentially slow the progression of PAD, whereas increased consumption of meat products has been associated with a lower ankle-brachial index.265–267
Several other minor risk factors and markers for atherosclerosis have been advocated, and many more are currently under investigation. However, their role in risk stratification beyond the conventional risk factors is yet to be proven.268
Factors Opposing Atherogenesis
Most of the research defining the pathophysiologic mechanism of atherosclerosis has focused on factors that precipitate vascular disease. However, a number of endogenous mechanisms oppose atherogenesis.
There is considerable epidemiologic, clinical, and experimental evidence supporting an antiatherogenic and antithrombotic role for HDL.269 HDL interrupts the process of atherogenesis at several key stages; it opposes atherosclerosis directly by removing cholesterol from foam cells, by inhibiting the oxidation of LDL, and by limiting the inflammatory processes that underlie atherosclerosis. HDL also has antithrombotic properties. HDL participates in reverse cholesterol transport (from the vessel to the liver) and also contains enzymes that metabolize platelet-activating factor (i.e., paraoxonase and platelet-activating factor acetylhydrolase).270 Catecholamines inhibit cholesterol biosynthesis in extrahepatic cells; they increase HDL binding activity, thereby enhancing efflux of cholesterol from cells. These effects are mediated by α2– and β2-adrenergic receptors.271 Tissue plasminogen activator is produced by endothelial cells272 and, by inducing fibrinolysis, may reduce the accretion of thrombus onto the vessel wall. Intracellular superoxide dismutase detoxifies oxygen-derived free radicals. Endothelium-derived nitric oxide and prostacyclin (prostaglandin I2) inhibit several processes that lead to the development of atherosclerotic plaque.273 These compounds are vasodilators, and they inhibit the adherence of platelets and leukocytes to the endothelium. In addition, both nitric oxide and prostacyclin inhibit the proliferation of VSMCs and macrophages and suppress the generation of oxygen-derived free radicals. The reduction in oxidative stress turns off the oxidant-responsive genes (e.g., MCP-1 and VCAM-1) that mediate monocyte binding and infiltration into the vessel wall.273,274
IL-10 (a potent anti-inflammatory cytokine) seems to inhibit monocyte–endothelial cell interactions, whereas the TIMPs counterbalance MMP activity.275 In addition, metallothioneins are antioxidant proteins that are expressed in response to injury; they protect cells from further damage and apoptosis.276 Furthermore, L-arginine can restore endothelial vasodilator function.277–280 It was found to enhance vascular nitric oxide production, to reduce superoxide anion generation, to suppress monocyte adherence, and to inhibit (and even reverse) the formation of intimal lesions in hypercholesterolemic rabbits.281–285 Adiponectin, the most abundantly secreted adipocyte-derived peptide hormone, has an overt antiatherosclerotic effect and protects against metabolic and cardiovascular diseases, as mentioned previously.286 Adiponectin increases hepatic and skeletal muscle sensitivity to insulin, enhances fatty acid oxidation, suppresses monocyte–endothelial cell interaction, supports endothelial cell growth, lowers blood pressure, and moderates adipose tissue growth. Finally, endothelial progenitor cells interact with mature endothelial cells through the expression of adhesion molecules and release of chemokines to repair damage to vascular endothelium.287–290
Factors Contributing to Acute Vascular Events
The role of trigger factors in acute vascular events, such as MI and stroke, has received increased attention in recent years. Heavy physical work, mental or emotional stress, recent heavy alcohol use, overeating, bursts of anger, and sexual activity as well as climatic factors are possible triggering factors for vascular events.291
A circadian pattern of vascular event rates (including MI, stroke, transient ischemic attack, angina, and sudden cardiac death) has consistently been shown during the early morning hours, immediately after getting up from sleep or shortly thereafter. Circadian variation has also been reported in certain physiologic parameters, such as blood pressure, heart rate, platelet aggregability, and refractoriness of the ventricle. The sympathetic nervous system has been implicated in the circadian variation in hemodynamic stress and blood coagulability, which can provoke vascular events in the morning hours. In addition, weekly variation has been noted, with more events occurring on Monday in workers but not in retired persons.292 Seasonal variation has also been noted in the rupture of abdominal aortic aneurysms, with more ruptures occurring in early winter.293
Mental stress may trigger vascular events by different mechanisms, such as impairing vasomotor response and heart rate variability and promoting thrombosis.294,295 Risk for vascular events has been shown to occur during stressful life events or special days. Thus, sporting events, earthquakes, threats of war, or Christmas, New Year’s Day, and birthdays may trigger vascular events by inducing autonomic dysfunction, such as ventricular arrhythmias and sudden death in patients with predisposing conditions.294,295 These triggers are most likely exerting their effects by the sympathetic nervous system, a probability that may in part explain the observation that β-adrenergic antagonists increase longevity in patients with atherosclerotic CAD. Furthermore, cocaine use confers an increased risk for vascular morbidity and mortality.296 The mechanisms responsible for this association are multifactorial; among them, platelet activation might play a substantial role.
Acute infections, such as lower respiratory tract and urinary tract infection, were also found to be associated with a transient (2 to 3 days) increase in risk for cardiovascular events, which lends strong support to the concept that in individuals with preexisting atherosclerosis, acute infection can trigger an acute inflammatory response that leads to a vascular event.297 The underlying mechanisms are not completely clear but may include, among others, endothelial dysfunction and higher adrenergic status during an acute infection. It is not known whether the transient increase in risk is due to a short-term alteration in endothelial function or to other mechanisms, such as changes in plaque composition, white blood cell activation, dehydration, or bed rest.
Furthermore, it has been suggested that ingestion of a fatty meal has an effect on the vascular endothelium and coagulation system and could be a trigger for acute coronary syndromes.298 In particular, after the ingestion of fatty meals, a constellation of changes in the vasculature can occur and result in both a hypercoagulable and a provasoconstrictor state. These acute changes in endothelial function, prothrombosis, and platelet activation in response to a fatty meal can potentially trigger, facilitate, and propagate the forces that drive acute coronary syndromes. In type 2 diabetes, adverse postprandial phenomena are exaggerated and prolonged and may therefore be expected to significantly contribute to the excess risk for vascular events.
Clinical Strategy
Assessment of risk factors is important to accurately predict cardiovascular risk and to guide primary and secondary prevention. Currently, several risk factors have been identified; however, level A clinical evidence and treatment guidelines exist for the traditional risk factors only: smoking, hyperlipidemia, hypertension, and diabetes. Nevertheless, assessment of conditional risk factors, such as homocysteine, hsCRP, fibrinogen, and lipoprotein (a), can be useful and necessary in many circumstances. Despite the fact that an established treatment is not yet available, evaluation for conditional risk factors may help more accurately assess the risk for PAD, CAD, or coronary heart disease and guide therapeutic decisions, especially when traditional risk factors are within borderline levels. Conditional risk factors may have greater potential as a means to augment risk assessment in the identification of persons who should be considered for lipid-lowering, antiplatelet, or other cardioprotective drug therapies as well as for increased emphasis on therapeutic lifestyle changes. A better potential use may be in patients at baseline risk, for whom an additional risk predictor may provide support for or against additional lifestyle or drug therapies. In addition, these factors might be considered for monitoring the effects of therapy (e.g., statins). Finally, novel and emerging risk factors are currently under intensive clinical evaluation but are not yet available for routine assessment and management. In the near future, some may be used to provide accurate individual-specific risk profiles and to facilitate clinical prediction algorithms for both chronic and acute events of atherosclerosis.
However, the major drawback for any clinical strategy, since it involves lifelong therapies, is the patient’s and physician’s compliance despite the various guidelines.299,300 Poor compliance with risk factor modification is a significant but often unrecognized risk factor for cardiovascular events. In a systematic review of implementation of established recommended secondary prevention measures in patients with PAD, antiplatelet, heart rate–lowering, blood pressure–lowering, and lipid-lowering agents were prescribed in 63%, 34%, 46%, and 45% of the patients, respectively.301 Glucose-lowering agents were prescribed in 81% and smoking cessation in 39% of the patients.301 In this context, the guideline goals for LDL cholesterol level are achieved by only 36% of patients, the body mass index goals by 33%, the exercise goals by 35%, and the HbA1c level goals by 24%.302
Not surprisingly, poor adherence to statin therapy has been found to be an independent risk factor for ischemic heart disease,303,304 whereas survival after MI has proved to be better for patients receiving adequate prevention.305,306 In a retrospective cohort study of 229,918 adults receiving statins, continuity of statin treatment (proportion of days covered ≥90%) conferred at least a 45% reduction in risk of death compared with patients with less than 10% proportion of days covered, in patients both with and without a known history of CAD.307 In another population-based sample of 137,277 patients younger than 65 years, patients with diabetes, hypertension, hypercholesterolemia, or congestive heart failure who maintained 80% to 100% medication adherence were significantly less likely to be hospitalized compared with patients with lower levels of adherence.308 As a result, higher medication costs were more than offset by medical cost reductions, producing a net reduction in overall health care costs.308
A meta-analysis has shown that as many as 6 of 10 patients may stop taking statins during the first 6 months after initiation of treatment.309 Whereas poor compliance has been associated with worse clinical outcome and increased cardiovascular morbidity and mortality, statin withdrawal may be even worse compared with not taking statins at all.310 In the PRISM (Platelet Receptor Inhibition in Ischemic Syndrome Management) study, if the statin therapy was withdrawn after admission for an acute coronary syndrome, cardiac risk increased compared with that of patients who continued to receive statins and tended to be higher compared with that of patients who never received statins.310 Similarly, statin discontinuation in the perioperative period in patients undergoing vascular surgery has been associated with a fivefold increased risk for postoperative troponin release and a sevenfold increased risk for the combination of MI and cardiovascular death.311 The pathophysiologic mechanism underlying these adverse clinical outcomes seems to extend beyond the cholesterol-lowering effects, including the pleiotropic statin activity. Indeed, several studies have documented the detrimental impact of statin withdrawal on endothelial function, inflammation, platelet activity, and angiotensin type 1 signaling.312
Determinants of poor patient adherence to treatment include the complexity of treatment, the number of daily pills, the number of chronic diseases, the development of adverse effects, the high cost of drug acquisition, and the lack of guideline compliance by physicians.313 Consequently, strategies to improve compliance with risk factor modification should include proper selection of agents, use of long-acting drugs to decrease the frequency of doses, and establishment of methods for monitoring of adherence, including educational programs, interdisciplinary approaches, and use of home-based technologies.314 In any case, increased awareness of the problem and active participation of a health care practitioner are of paramount importance to improve compliance with guidelines and to reduce cardiovascular morbidity and mortality.
Selected Key References
Andraws R, Berger JS, Brown DL. Effects of antibiotic therapy on outcomes of patients with coronary artery disease: a meta-analysis of randomized controlled trials. JAMA. 2005;293:2641–2647.
Fibrinogen Studies Collaboration. Associations of plasma fibrinogen levels with established cardiovascular disease risk factors, inflammatory markers, and other characteristics: individual participant meta-analysis of 154,211 adults in 31 prospective studies: the Fibrinogen Studies Collaboration. Am J Epidemiol. 2007;166:867–879.
Flu HC, Tamsma JT, Lindeman JH, Hamming JF, Lardenoye JH. A systematic review of implementation of established recommended secondary prevention measures in patients with PAOD. Eur J Vasc Endovasc Surg. 2010;39:70.
Grundy SM, Pasternak R, Greenland P, Smith S Jr, Fuster V. Assessment of cardiovascular risk by use of multiple-risk-factor assessment equations: a statement for healthcare professionals from the American Heart Association and the American College of Cardiology. Circulation. 1999;100:1481–1492.
Hackam DG, Anand SS. Emerging risk factors for atherosclerotic vascular disease: a critical review of the evidence. JAMA. 2003;290:932–940.
Ikonomidis I, Stamatelopoulos K, Lekakis J, Vamvakou GD, Kremastinos DT. Inflammatory and non-invasive vascular markers: the multimarker approach for risk stratification in coronary artery disease. Atherosclerosis. 2008;199:3–11.
Kadoglou NPE, Gerasimidis T, Moumtzouoglou A, Kapelouzou A, Sailer N, Fotiadis G, Vitta I, Katinios A, Bandios S, Voliotis K, Karayannacos PE, Liapis CD. Intensive lipid-lowering therapy ameliorates novel inflammatory and calcification markers and GSM score in patients with carotid stenosis. Eur J Vasc Endovasc Surg. 2008;35:661–668.
Murabito JM, D’Agostino RB, Silbershatz H, Wilson WF. Intermittent claudication. A risk profile from the Framingham Heart Study. Circulation. 1997;96:44–49.
Selvin E, Erlinger TP. Prevalence of and risk factors for peripheral arterial disease in the United States: results from the National Health and Nutrition Examination Survey, 1999-2000. Circulation. 2004;110:738–743.
Smith A, Patterson C, Yarnell J, Rumley A, Ben-Shlomo Y, Lowe G. Which hemostatic markers add to the predictive value of conventional risk factors for coronary heart disease and ischemic stroke? The Caerphilly study. Circulation. 2005;112:3080–3087.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Haimovici H, et al. Atherosclerosis. Biologic and surgical considerations. Haimovici H, et al. Vascular surgery principles and techniques. ed 4. Blackwell Science: Cambridge, Mass; 1996:127.
2. McMillan GC. Nature and definitions of atherosclerosis. Ann N Y Acad Sci. 1985;454:1.
3. Mackey RH, et al. Calcifications, arterial stiffness and atherosclerosis. Adv Cardiol. 2007;44:234–244.
4. Kannel WB, et al. Factors of risk in the development of coronary heart disease—six year follow-up experience: the Framingham study. Ann Intern Med. 1961;55:33.
5. Liapis CD, et al. Vascular surgery. Springer: New York; 2007.
6. Holtzman JL. Atherosclerosis and oxidant stress, a new perspective. Springer: New York; 2008.
7. Kumar V, et al. Robbins basic pathology. ed 8. Elsevier Saunders: Philadelphia; 2007.
8. Topper JN, et al. Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc Natl Acad Sci U S A. 1996;93:10417.
9. Vogel RA. Cholesterol lowering and endothelial function. Am J Med. 1999;107:479.
10. Rajagopalan S, et al. Angiotensin II–mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation: contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916.
11. Ohara Y, et al. Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest. 1993;91:2546.
12. Tesfamariam B, et al. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am J Physiol. 1992;263:H321.
13. Tsao PS, et al. Effects of diabetes on monocyte-endothelial interactions and endothelial superoxide production in fructose-induced insulin-resistant and hypertensive rats. Circulation. 1995;92:A2666.
14. Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol. 2000;20:2032.
15. Flavahan NA. Atherosclerosis or lipoprotein induced endothelial dysfunction: potential mechanisms underlying reduction in EDRF/nitric oxide activity. Circulation. 1992;85:1927.
16. Gounopoulos P, et al. Antibodies to oxidized low density lipoprotein: epidemiological studies and potential clinical applications in cardiovascular disease. Minerva Cardioangiol. 2007;55:821.
17. Cybulsky MI, et al. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788.
18. Berliner JA, et al. Atherosclerosis: basic mechanisms. Oxidation, inflammation, and genetics. Circulation. 1995;91:2488.
19. Tsao PS, et al. Fluid flow inhibits endothelial adhesiveness: nitric oxide and transcriptional regulation of VCAM-1. Circulation. 1996;94:1682.
20. Tsao PS, et al. Nitric oxide regulates monocyte chemotactic protein-1. Circulation. 1997;96:934.
21. Ross R. Cellular and molecular studies of atherosclerosis. Atherosclerosis. 1997;131:S3.
22. Berliner JA, et al. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J Clin Invest. 1990;85:1260.
23. Ross R. Cell biology of atherosclerosis. Annu Rev Physiol. 1995;57:791.
24. Elkington PT, et al. Analysis of matrix metalloproteinase secretion by macrophages. Methods Mol Biol. 2009;531:253.
25. Businaro R, et al. Cellular and molecular players in the atherosclerotic plaque progression. Ann N Y Acad Sci. 2012;1262:134.
26. Kong YZ, et al. Macrophage migration inhibitory factor induces MMP-9 expression: implications for destabilization of human atherosclerotic plaques. Atherosclerosis. 2005;178:207.
27. Hansson GK, et al. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;1:297.
28. Taleb S, et al. Regulatory T-cell immunity and its relevance to atherosclerosis. J Intern Med. 2008;263:489.
29. Kuiper J, et al. Immunomodulation of the inflammatory response in atherosclerosis. Curr Opin Lipidol. 2007;18:521.
30. Torzewski M, et al. Complement and atherosclerosis—united to the point of no return? Clin Biochem. 2013;46:20.
31. Speidl WS, et al. Complement in atherosclerosis: friend or foe? J Thromb Haemost. 2011;9:428.
32. Lievens D, et al. Platelets in atherosclerosis. Thromb Haemost. 2011;106:827.
33. Fuentes QE, et al. Role of platelets as mediators that link inflammation and thrombosis in atherosclerosis. Platelets. 2013;24:255.
34. Lindemann S, et al. Molecular pathways used by platelets to initiate and accelerate atherogenesis. Curr Opin Lipidol. 2007;18:566.
35. May AE, et al. Platelets: inflammatory firebugs of vascular walls. Arterioscler Thromb Vasc Biol. 2008;28:5.
36. Schulz C, et al. Platelets in atherosclerosis and thrombosis. Handb Exp Pharmacol. 2012;210:111.
37. Doran AC, et al. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812.
38. Azari BM, et al. Silencing of the F11R gene reveals a role for F11R/JAM-A in the migration of inflamed vascular smooth muscle cells and in atherosclerosis. Atherosclerosis. 2010;212:197.
39. Koon CM, et al. Salviae miltiorrhizae radix and Puerariae lobatae radix herbal formula mediates anti-atherosclerosis by modulating key atherogenic events both in vascular smooth muscle cells and endothelial cells. J Ethnopharmacol. 2011;138:175.
40. Natarajan R. Drugs targeting epigenetic histone acetylation in vascular smooth muscle cells for restenosis and atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:725.
41. Golledge J, et al. The symptomatic carotid plaque. Stroke. 2000;31:774.
42. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685.
43. Daskalopoulou SS, et al. Carotid artery atherosclerosis: what is the evidence for drug action? Curr Pharm Des. 2007;13:1141.
44. Stary HC. Natural history and histological classification of atherosclerotic lesions: an update. Arterioscler Thromb Vasc Biol. 2000;20:1177.
45. Davies JR, et al. Targeting the vulnerable plaque: the evolving role of nuclear imaging. J Nucl Cardiol. 2005;12:234.
46. Rosner D, et al. Interferon-gamma induces Fas trafficking and sensitization to apoptosis in vascular smooth muscle cells via a PI3K- and Akt-dependent mechanism. Am J Pathol. 2006;168:2054.
47. Falk E. Why do plaques rupture? Circulation. 1992;86:SIII–30.
48. Libby P, et al. Roles of infectious agents in atherosclerosis and restenosis: an assessment of the evidence and need for future research. Circulation. 1997;96:4095.
49. Kusters DH, et al. Molecular imaging to identify the vulnerable plaque—from basic research to clinical practice. Mol Imaging Biol. 2012;14:523.
50. Faggioli GL, et al. Identification of carotid ‘vulnerable plaque’ by contrast-enhanced ultrasonography: correlation with plaque histology, symptoms and cerebral computed tomography. Eur J Vasc Endovasc Surg. 2011;41:238.
51. Timaran CH, et al. Atherosclerotic plaque composition assessed by virtual histology intravascular ultrasound and cerebral embolization after carotid stenting. J Vasc Surg. 2010;52:1188.
52. Grundy SM, et al. Assessment of cardiovascular risk by use of multiple-risk-factor assessment equations: a statement for healthcare professionals from the American Heart Association and the American College of Cardiology. Circulation. 1999;100:1481.
53. Smith SC Jr, et al. Prevention conference V: beyond secondary prevention: identifying the high-risk patient for primary prevention: executive summary. Circulation. 2000;101:111.
54. Kullo IJ, et al. Conditional risk factors for atherosclerosis. Mayo Clin Proc. 2005;80:219.
55. Vidula H, et al. Biomarkers of inflammation and thrombosis as predictors of near term mortality in patients with peripheral arterial disease: a cohort study. Ann Intern Med. 2008;148:85.
57. Hackam DG, et al. Emerging risk factors for atherosclerotic vascular disease: a critical review of the evidence. JAMA. 2003;290:932.
58. Pearson TA, et al. Markers of inflammation and cardiovascular disease. Application to clinical and public health practice. A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499.
59. Murabito JM, et al. Intermittent claudication. A risk profile from the Framingham Heart Study. Circulation. 1997;96:44.
60. Newman AB, et al. Ankle-arm index as a marker of atherosclerosis in the Cardiovascular Health Study. Cardiovascular Heart Study (CHS) Collaborative Research Group. Circulation. 1993;88:837.
61. Hirsch AT, et al. Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA. 2001;286:1317.
62. Selvin E, et al. Prevalence of and risk factors for peripheral arterial disease in the United States: results from the National Health and Nutrition Examination Survey, 1999-2000. Circulation. 2004;110:738.
63. Wattanakit K, et al. Risk factors for peripheral arterial disease incidence in persons with diabetes: the Atherosclerosis Risk in Communities (ARIC) Study. Atherosclerosis. 2005;180:389.
64. Gottsäter A. Managing risk factors for atherosclerosis in critical limb ischaemia. Eur J Vasc Endovasc Surg. 2006;32:478.
65. Teramoto T, et al. Risk factors of atherosclerotic diseases. Executive summary of Japan Atherosclerosis Society (JAS) guideline for diagnosis and prevention of atherosclerosis cardiovascular diseases for Japanese. J Atheroscler Thromb. 2007;14:267.
66. Hiatt WR, et al. Effect of diagnostic criteria on the prevalence of peripheral arterial disease: the San Luis Valley Diabetes Study. Circulation. 1995;91:1472.
67. Joensuu T, et al. Determinants of femoral and carotid artery atherosclerosis. J Intern Med. 1994;236:79.
68. Vogt MT, et al. Prevalence and correlates of lower extremity arterial disease in elderly women. Am J Epidemiol. 1993;137:559.
69. López-Cancio E, et al. The Barcelona–Asymptomatic Intracranial Atherosclerosis (AsIA) study: prevalence and risk factors. Atherosclerosis. 2012;221:221.
70. de Weerd M, et al. Prevalence of asymptomatic carotid artery stenosis according to age and sex: systematic review and metaregression analysis. Stroke. 2009;40:1105.
71. Hobson RW II, et al. Vascular surgery: principles and practice. Marcel Dekker: New York; 2004.
72. Meyers MR, et al. Endothelial dysfunction in obesity: etiological role in atherosclerosis. Curr Opin Endocrinol Diabetes Obes. 2007;14:365.
73. Brook RD. Obesity, weight loss and vascular function. Endocrine. 2006;29:21.
74. Mattu HS, et al. Role of adipokines in cardiovascular disease. J Endocrinol. 2013;216:T17.
75. Ix JH, et al. Association of body mass index with peripheral arterial disease in older adults: the Cardiovascular Health Study. Am J Epidemiol. 2011;174:1036.
76. Pischon T, et al. General and abdominal adiposity and risk of death in Europe. N Engl J Med. 2008;359:2105.
77. Jee SH, et al. Body-mass index and mortality in Korean men and women. N Engl J Med. 2006;355:779.
78. Kadoglou NP, et al. The impact of aerobic exercise training on novel adipokines, apelin and ghrelin, in patients with type 2 diabetes. Med Sci Monit. 2012;18:CR290.
79. Bowles DK, et al. Mechanism of beneficial effects of physical activity on atherosclerosis and coronary heart disease. J Appl Physiol. 2011;111:308.
80. Hamer M, et al. Physical activity and cardiovascular mortality risk: possible protective mechanisms? Med Sci Sports Exerc. 2012;44:84.
81. Mora S, et al. Physical activity and reduced risk of cardiovascular events: potential mediating mechanisms. Circulation. 2007;116:2110.
82. Savela S, et al. Leisure-time physical activity, cardiovascular risk factors and mortality during a 34-year follow-up in men. Eur J Epidemiol. 2010;25:619.
83. Gillum RF, et al. Physical activity, cognitive function, and mortality in a US national cohort. Ann Epidemiol. 2010;20:251.
84. Gulsvik AK, et al. Ageing, physical activity and mortality—a 42-year follow-up study. Int J Epidemiol. 2012;41:521.
85. Reddigan JI, et al. Relation of physical activity to cardiovascular disease mortality and the influence of cardiometabolic risk factors. Am J Cardiol. 2011;108:1426.
86. Berlin JA, et al. A meta-analysis of physical activity in the prevention of coronary heart disease. Am J Epidemiol. 1990;132:612.
87. Fletcher GF. The antiatherosclerotic effect of exercise and development of an exercise prescription. Cardiol Clin. 1996;14:85.
88. Kadoglou NPE, et al. Beneficial effects of combined treatment with rosiglitazone and exercise on cardiovascular risk factors in patients with type 2 diabetes. Diabetes Care. 2007;30:2242.
89. Kadoglou NP, et al. Exercise and carotid atherosclerosis. Eur J Vasc Endovasc Surg. 2008;35:264.
90. Vitale C, et al. Gender-specific characteristics of atherosclerosis in menopausal women: risk factors, clinical course and strategies for prevention. Climacteric. 2007;10:16.
91. Lisabeth L, et al. Stroke risk in women: the role of menopause and hormone therapy. Lancet Neurol. 2012;11:82.
92. Stramba-Badiale M. Postmenopausal hormone therapy and the risk of cardiovascular disease. J Cardiovasc Med (Hagerstown). 2009;10:303.
93. Chen Y, et al. Genetic and genomic insights into the molecular basis of atherosclerosis. Cell Metab. 2007;6:164.
94. McDermott MM, et al. The role of biomarkers and genetics in peripheral arterial disease. J Am Coll Cardiol. 2009;54:1228.
95. Krishna SM, et al. Genetic and epigenetic mechanisms and their possible role in abdominal aortic aneurysm. Atherosclerosis. 2010;212:16.
96. Topol EJ. The genomic basis of myocardial infarction. J Am Coll Cardiol. 2005;46:1456.
97. Helgadottir A, et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet. 2004;36:233.
98. Fu S, et al. Peripheral arterial occlusive disease: global gene expression analyses suggest a major role for immune and inflammatory responses. BMC Genomics. 2008;9:369.
99. Miller DT, et al. Atherosclerosis. The path from genomics to therapeutics. J Am Coll Cardiol. 2007;49:1589.
100. Rozanski A, et al. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation. 1999;99:2192.
101. Morland K, et al. The contextual effect of the local food environment on residents’ diets: the Atherosclerosis Risk in Communities study. Am J Public Health. 2002;92:1761.
102. Diez Roux AV, et al. Neighborhood characteristics and components of the insulin resistance syndrome in young adults: the Coronary Artery Risk Development in Young Adults (CARDIA) study. Diabetes Care. 2002;25:1976.
103. Wadsworth ME. Health inequalities in the life course perspective. Soc Sci Med. 1997;44:859.
104. Pollitt RA, et al. Evaluating the evidence for models of life course socioeconomic factors and cardiovascular outcomes: a systematic review. BMC Public Health. 2005;5:7.
105. Selvin E, et al. Prevalence of and risk factors for peripheral arterial disease in the United States: results from the National Health and Nutrition Examination Survey, 1999-2000. Circulation. 2004;110:738.
106. Saunders E, et al. Epidemiology of atherothrombotic disease and the effectiveness and risks of antiplatelet therapy: race and ethnicity considerations. Cardiol Rev. 2008;16:82.
107. Criqui MH, et al. Ethnicity and peripheral arterial disease: the San Diego Population Study. Circulation. 2005;112:2703.
108. Welch GN, et al. Homocysteine and atherothrombosis. N Engl J Med. 1998;338:1042.
110. Clarke R, et al. Hyperhomocysteinemia: an independent risk factor for vascular disease. N Engl J Med. 1991;324:1149.
111. Brattstrom L, et al. Impaired homocysteine metabolism in early onset cerebral and arterial occlusive disease. Atherosclerosis. 1990;81:51.
112. Darius H, et al. Are elevated homocysteine plasma levels related to peripheral arterial disease? Results from a cross-sectional study of 6880 primary care patients. Eur J Clin Invest. 2003;33:751.
113. Guilland JC, et al. Hyperhomocysteinemia: an independent risk factor or a simple marker of vascular disease? Pathol Biol. 2003;51:111.
114. Humphrey LL, et al. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clin Proc. 2008;83:1203.
115. Undas A, et al. Homocysteine and thrombosis: from basic science to clinical evidence. Thromb Haemost. 2005;94:907.
116. Eldibany MM, et al. Hyperhomocysteinemia and thrombosis: an overview. Arch Pathol Lab Med. 2007;131:872.
117. Carmody BJ, et al. Folic acid inhibits homocysteine-induced proliferation of human arterial smooth muscle cells. J Vasc Surg. 1999;30:1121.
118. Zhang D, et al. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine β-synthase–deficient mice. Circulation. 2009;120:1893.
119. Zhang D, et al. Severe hyperhomocysteinemia promotes bone marrow–derived and resident inflammatory monocyte differentiation and atherosclerosis in LDLr/CBS-deficient mice. Circ Res. 2012;111:37.
120. Malinow MR, et al. Reduction of plasma homocysteine levels by breakfast cereal fortified with folic acid in patients with coronary heart disease. N Engl J Med. 1998;338:1009.
121. Ciaccio M, et al. Hyperhomocysteinemia and cardiovascular risk: effect of vitamin supplementation in risk reduction. Curr Clin Pharmacol. 2010;5:30.
122. VITATOPS Trial Study Group. B vitamins in patients with recent transient ischaemic attack or stroke in the VITAmins TO Prevent Stroke (VITATOPS) trial: a randomised, double-blind, parallel, placebo-controlled trial. Lancet Neurol. 2010;9:855.
123. Holmes MV, et al. Effect modification by population dietary folate on the association between MTHFR genotype, homocysteine, and stroke risk: a meta-analysis of genetic studies and randomised trials. Lancet. 2011;378:584.
124. Bønaa KH, et al. Homocysteine lowering and cardiovascular events after acute myocardial infarction. [NORVIT Trial Investigators] N Engl J Med. 2006;354:1578.
125. Volanakis JE. Human C-reactive protein: expression, structure, and function. Mol Immunol. 2001;38:189.
126. Pietila K, et al. Serum C-reactive protein and infarct size in myocardial infarct patients with a closed versus an open infarct-related coronary artery after thrombolytic therapy. Eur Heart J. 1993;14:915.
127. Paffen E, et al. C-reactive protein does not directly induce tissue factor in human monocytes. Arterioscler Thromb Vasc Biol. 2004;24:975.
128. Giannakis E, et al. Multiple ligand binding sites on domain seven of human complement factor H. Int Immunopharmacol. 2001;1:433.
129. Venugopal SK, et al. Demonstration that C-reactive protein decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation. 2002;106:1439.
130. Verma S, et al. C-reactive protein attenuates endothelial progenitor cell survival, differentiation, and function: further evidence of a mechanistic link between C-reactive protein and cardiovascular disease. Circulation. 2004;109:2058.
131. Wang CH, et al. C-reactive protein upregulates angiotensin type 1 receptors in vascular smooth muscle. Circulation. 2003;107:1783.
132. Pasceri V, et al. Modulation of C-reactive protein–mediated monocyte chemoattractant protein-1 induction in human endothelial cells by anti-atherosclerosis drugs. Circulation. 2001;103:2531.
133. Hattori Y, et al. Vascular smooth muscle cell activation by C-reactive protein. Cardiovasc Res. 2003;58:186.
134. Ikonomidis I, et al. Inflammatory and non-invasive vascular markers: the multimarker approach for risk stratification in coronary artery disease. Atherosclerosis. 2008;199:3.
135. Ridker PM, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. [JUPITER Study Group] N Engl J Med. 2008;359:2195.
136. Zacho J, et al. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med. 2008;359:1897.
137. Teupser D, et al. No reduction of atherosclerosis in C-reactive protein (CRP)–deficient mice. J Biol Chem. 2011;286:6272.
138. Wilhelmsen L, et al. Fibrinogen as a risk factor for stroke and myocardial infarction. N Engl J Med. 1984;311:501.
139. Meade TO, et al. Haemostatic function and ischaemic heart disease: principal results of the Northwick Park Heart Study. Lancet. 1986;2:533.
140. Thompson SG, et al. Hemostatic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. [European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group] N Engl J Med. 1995;332:635.
141. Kofoed SC, et al. Fibrinogen predicts ischaemic stroke and advanced atherosclerosis, but not echolucent, rupture-prone carotid plaques. Eur Heart J. 2003;24:567.
142. Green D, et al. Elevated fibrinogen levels and subsequent subclinical atherosclerosis: the CARDIA Study. Atherosclerosis. 2009;202:623.
143. The Bezafibrate Infarction Prevention (BIP) Study Group. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation. 2000;102:21.
144. King VL, et al. Serum amyloid A in atherosclerosis. Curr Opin Lipidol. 2011;22:302.
145. Biasucci LM, et al. Elevated levels of C-reactive protein at discharge in patients with unstable angina predict recurrent instability. Circulation. 1999;99:855.
146. Johnson BD, et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: the National Heart, Lung, and Blood Institute–Sponsored Women’s Ischemia Syndrome Evaluation (WISE). Circulation. 2004;109:726.
147. Danesh J, et al. Association of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: meta-analyses of prospective studies. JAMA. 1998;279:1477.
148. Tani S, et al. Association of circulating leukocyte count with coronary atherosclerosis regression after pravastatin treatment. Atherosclerosis. 2008;198:360.
149. Tzoulaki I, et al. C-reactive protein, interleukin-6, and soluble adhesion molecules as predictors of progressive peripheral atherosclerosis in the general population: Edinburgh Artery Study. Circulation. 2005;112:976.
150. McDermott MM, et al. Patterns of inflammation associated with peripheral arterial disease: the InCHIANTI study. Am Heart J. 2005;150:276.
151. Allison MA, et al. The effect of novel cardiovascular risk factors on the ethnic-specific odds for peripheral arterial disease in the Multi-Ethnic Study of Atherosclerosis (MESA). J Am Coll Cardiol. 2006;48:1190.
152. Mallat Z, et al. Interleukin-18/interleukin-18 binding protein signalling modulates atherosclerotic lesion development and stability. Circ Res. 2001;89:E41.
153. Elhage R, et al. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E–knockout mice. Cardiovasc Res. 2003;59:234.
154. Mallat Z, et al. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 2001;104:1598.
155. Young JL, et al. Cytokines in the pathogenesis of atherosclerosis. Thromb Haemost. 2002;88:554.
156. Taubman MB, et al. Tissue factor in the pathogenesis of atherosclerosis. Thromb Haemost. 1997;78:200.
157. Hoogeveen RC, et al. Plasma MCP-1 level and risk for peripheral arterial disease and incident coronary heart disease: Atherosclerosis Risk in Communities study. Atherosclerosis. 2005;183:301.
159. André P, et al. Platelet derived CD40L. The switch hitting player of cardiovascular disease. Circulation. 2002;106:896.
160. Aukrust P, et al. Enhanced levels of soluble and membrane-bound CD40 ligand in patients with unstable angina. Circulation. 1999;100:614.
161. Hirano K. The roles of proteinase-activated receptors in the vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2007;27:27.
162. Oei HH, et al. Lipoprotein-associated phospholipase A2 activity is associated with risk of coronary heart disease and ischemic stroke: the Rotterdam Study. Circulation. 2005;111:570.
163. Chung ES, et al. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-α, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107:3133.
164. Muller-Ehmsen J, et al. TNF and congestive heart failure: therapeutic possibilities. Expert Opin Ther Targets. 2004;8:203.
165. Leu HB, et al. Fluvastatin reduces oxidative stress, decreases serum monocyte chemotactic protein-1 level and improves endothelial function in patients with hypercholesterolemia. J Formos Med Assoc. 2004;103:914.
166. DePalma RG, et al. Statins and biomarkers in claudicants with peripheral arterial disease: cross-sectional study. Vascular. 2006;14:193.
167. Jiang C, et al. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82.
168. Pasceri V, et al. Modulation of vascular inflammation in vitro and in vivo by peroxisome proliferator–activated receptor-γ activators. Circulation. 2000;101:235.
169. Saikku P, et al. Serological evidence of an association of a novel chlamydia, TWAR, with chronic coronary heart disease and acute myocardial infarction. Lancet. 1988;2:983.
170. Arcari CM, et al. Association between Chlamydia pneumoniae and acute myocardial infarction in young men in the United States military: the importance of timing of exposure measurement. Clin Infect Dis. 2005;40:1123.
171. Grayston JT, et al. Chlamydia pneumoniae (TWAR) in atherosclerosis of the carotid artery. Circulation. 1995;92:3397.
172. Kaperonis EA, et al. The association of carotid plaque inflammation and Chlamydia pneumoniae infection with cerebrovascular symptomatology. J Vasc Surg. 2006;44:1198.
173. Sessa R, et al. Chlamydia pneumoniae DNA in patients with symptomatic carotid atherosclerotic disease. J Vasc Surg. 2003;37:1027.
174. Kaperonis EA, et al. Inflammation and Chlamydia pneumoniae infection correlate with the severity of peripheral arterial disease. Eur J Vasc Endovasc Surg. 2006;31:509.
175. Lindholt JS, et al. Indicators of infection with Chlamydia pneumoniae are associated with expansion of abdominal aortic aneurysms. J Vasc Surg. 2001;34:212.
176. Muhlestein JB, et al. Cytomegalovirus seropositivity and C-reactive protein have independent and combined predictive value for mortality in patients with angiographically demonstrated coronary artery disease. Circulation. 2000;102:1817.
177. Smieja M, et al. Multiple infections and subsequent cardiovascular events in the Heart Outcome Prevention Evaluation (HOPE) study. Circulation. 2003;107:251.
178. Vercellotti GM. Effect of viral activation of the vessel wall on inflammation and thrombosis. Blood Coagul Fibrinolysis. 1998;9:S3.
179. Zhou YF, et al. Cytomegalovirus infection of rats increases neointimal response to vascular injury without consistent evidence of direct infection of the vascular wall. Circulation. 1999;100:1569.
180. Epstein SE, et al. Infection and atherosclerosis: emerging mechanistic paradigms. Circulation. 1999;100:e20.
181. Andraws R, et al. Effects of antibiotic therapy on outcomes of patients with coronary artery disease: a meta-analysis of randomized controlled trials. JAMA. 2005;293:2641.
182. Abedin M, et al. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. 2004;24:1161.
183. Johnson RC, et al. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. 2006;99:1044.
184. O’Brien ER, et al. Osteopontin is synthesized by macrophage, smooth muscle, and endothelial cells in primary and restenotic human coronary atherosclerotic plaques. Arterioscler Thromb. 1994;14:1648.
185. Schoppet M, et al. RANK ligand and osteoprotegerin: paracrine regulators of bone metabolism and vascular function. Arterioscler Thromb Vasc Biol. 2002;22:549.
186. Kiechl S, et al. Osteoprotegerin is a risk factor for progressive atherosclerosis and cardiovascular disease. Circulation. 2004;109:2175.
187. Jono S, et al. Serum osteoprotegerin levels are associated with the presence and severity of coronary artery disease. Circulation. 2002;106:1192.
188. Ohmori R, et al. Plasma osteopontin levels are associated with the presence and extent of coronary artery disease. Atherosclerosis. 2003;170:333.
189. Kurata M, et al. Osteopontin and carotid atherosclerosis in patients with essential hypertension. Clin Sci (Lond). 2006;111:319.
190. Golledge J, et al. Osteoprotegerin and osteopontin are expressed at high concentrations within symptomatic carotid atherosclerosis. Stroke. 2004;35:1636.
191. Kadoglou NPE, et al. The relationship between serum levels of vascular calcification inhibitors and carotid plaque vulnerability. J Vasc Surg. 2008;47:55.
192. Kadoglou NPE, et al. Intensive lipid-lowering therapy ameliorates novel inflammatory and calcification markers and GSM score in patients with carotid stenosis. Eur J Vasc Endovasc Surg. 2008;35:661.
193. Lowe GD, et al. Classical and emerging risk factors for cardiovascular disease. Semin Vasc Med. 2002;2:353.
194. Smith A, et al. Which hemostatic markers add to the predictive value of conventional risk factors for coronary heart disease and ischemic stroke? The Caerphilly study. Circulation. 2005;112:3080.
195. Tzoulaki I, et al. Relative value of inflammatory, hemostatic and rheological factors for incident myocardial infarction and stroke. Circulation. 2007;115:2119.
196. Lee AJ, et al. Fibrin D-dimer, haemostatic factors and peripheral arterial disease. Thromb Haemost. 1995;3:828.
197. Martiskainen M, et al. Fibrinogen gene promoter −455A allele as a risk factor for macular stroke. Stroke. 2003;34:886.
198. Al Zahrani H, et al. Increased fibrin turnover in peripheral arterial disease: comparison with a population study. Clin Hemorheol. 1992;12:867.
199. Foley PWX, et al. Activated protein C resistance, factor V Leiden and peripheral arterial disease. Card Surg. 1997;5:157.
200. Arruda VR, et al. Prevalence of prothrombin gene variant (nt20210A) in venous thrombosis and arterial disease. Thromb Haemost. 1997;78:1430.
201. Kohler HP, et al. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. 2000;342:1792.
202. Strano A, et al. Plasma levels of the molecular markers of coagulation and fibrinolysis in patients with peripheral arterial disease. Semin Thromb Haemost. 1996;22:35.
203. McDermott MM, et al. Relation of levels of hemostatic factors and inflammatory markers to the ankle brachial index. Am J Cardiol. 2003;92:194.
204. Sofi F, et al. Thrombophilic risk factors for symptomatic peripheral arterial disease. J Vasc Surg. 2005;41:255.
205. Stein JH, et al. Lp(a) excess and coronary heart disease. Arch Intern Med. 1997;157:1170.
206. Clarke R, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. [PROCARDIS Consortium] N Engl J Med. 2009;361:2518.
208. Misirli H, et al. Relation of lipid and Lp(a) to ischemic stroke. J Clin Neurosci. 2002;2:127.
209. Sutton-Tyrrell K, et al. Lipoprotein (a) and peripheral atherosclerosis in older adults. Atherosclerosis. 1996;122:11.
210. Valentine JR, et al. Lp(a) lipoprotein is an independent discriminating risk factor for premature peripheral atherosclerosis among white men. Arch Intern Med. 1994;154:801.
211. Puisieux F, et al. Association between anticardiolipin antibodies and mortality in patients with peripheral arterial disease. Am J Med. 2000;109:635.
212. Kadoglou NP, et al. The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E–deficient mice: dabigatran etexilate and atherosclerosis. Cardiovasc Drugs Ther. 2012;26:367.
213. Visse R, et al. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827.
214. Nagase H, et al. Matrix metalloproteinases. J Biol Chem. 1999;274:21491.
215. Plutzky J. The vascular biology of atherosclerosis. Am J Med. 2003;115:55S.
216. Lijnen HR. Metalloproteinases in development and progression of vascular disease. Pathophysiol Haemost Thromb. 2003;33:275.
217. Galis ZS, et al. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251.
218. Alvarez B, et al. Serum values of metalloproteinase-2 and metalloproteinase-9 as related to unstable plaque and inflammatory cells in patients with greater than 70% carotid artery stenosis. J Vasc Surg. 2004;40:469.
219. Jiang X, et al. The expression of matrix metalloproteinases-9, transforming growth factor-β1 and transforming growth factor-β receptor I in human atherosclerotic plaque and their relationship with plaque stability. Chin Med J (Engl). 2004;117:1825.
220. Kadoglou NP, et al. Matrix metalloproteinases and diabetic vascular complications. Angiology. 2005;56:173.
221. Kadoglou NP, et al. Matrix metalloproteinases: contribution to pathogenesis, diagnosis, surveillance and treatment of abdominal aortic aneurysms. Curr Med Res Opin. 2004;20:419.
222. Takagi H, et al. Circulating matrix metalloproteinase-9 concentrations and abdominal aortic aneurysm presence: a meta-analysis. Interact Cardiovasc Thorac Surg. 2009;9:437.
223. Rizas KD, et al. Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiol Rev. 2009;17:201.
224. Saratzis A, et al. Abdominal aortic aneurysm: a review of the genetic basis. Angiology. 2011;62:18.
225. Lim CS, et al. Matrix metalloproteinases in vascular disease—a potential therapeutic target? Curr Vasc Pharmacol. 2010;8:75.
226. Furman C, et al. Rosuvastatin reduces MMP-7 secretion by human monocyte-derived macrophages: potential relevance to atherosclerotic plaque stability. Atherosclerosis. 2004;174:93.
227. Guo H, et al. Rosuvastatin inhibits MMP-2 expression and limits the progression of atherosclerosis in LDLR-deficient mice. Arch Med Res. 2009;40:345.
228. Paraskevas KI, et al. Leptin: a promising therapeutic target with pleiotropic action besides body weight regulation. Curr Drug Targets. 2006;7:761.
229. Beltowski J. Leptin and atherosclerosis. Atherosclerosis. 2006;189:47.
230. Fantuzzi G, et al. Adipose tissue and atherosclerosis: exploring the connection. Arterioscler Thromb Vasc Biol. 2007;27:996.
231. Considine RV. Human leptin: an adipocyte hormone with weight-regulatory and endocrine functions. Semin Vasc Med. 2005;5:15.
232. Stefan N, et al. Plasma adiponectin concentration is associated with skeletal muscle insulin receptor tyrosine phosphorylation, and low plasma concentration precedes a decrease in whole-body insulin sensitivity in humans. Diabetes. 2002;51:1884.
233. Kantartzis K, et al. The relationships of plasma adiponectin with a favorable lipid profile, decreased inflammation, and less ectopic fat accumulation depend on adiposity. Clin Chem. 2006;52:1934.
234. Ouchi N, et al. Adiponectin as an anti-inflammatory factor. Clin Chim Acta. 2007;380:24.
235. Gable DR, et al. Adiponectin and its gene variants as risk factors for insulin resistance, the metabolic syndrome and cardiovascular disease. Atherosclerosis. 2006;188:231.
236. Arita Y, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79.
237. Im JA, et al. Association between hypoadiponectinemia and cardiovascular risk factors in nonobese healthy adults. Metabolism. 2006;55:1546.
238. Rothenbacher D, et al. Adiponectin, risk of coronary heart disease and correlations with cardiovascular risk markers. Eur Heart J. 2005;26:1640.
239. Hasan-Ali H, et al. Serum adiponectin and leptin as predictors of the presence and degree of coronary atherosclerosis. Coron Artery Dis. 2011;22:264.
240. Zyriax BC, et al. Factors contributing to the risk of cardiovascular disease reflected by plasma adiponectin: data from the coronary risk factors for atherosclerosis in women (CORA) study. Atherosclerosis. 2008;200:403.
241. Persson J, et al. High plasma adiponectin concentration is associated with all-cause mortality in patients with carotid atherosclerosis. Atherosclerosis. 2012;225:491.
242. Pilz S, et al. Adiponectin and mortality in patients undergoing coronary angiography. J Clin Endocrinol Metab. 2006;91:4277.
243. Dekker JM, et al. Prognostic value of adiponectin for cardiovascular disease and mortality. J Clin Endocrinol Metab. 2008;93:1489.
244. Kadoglou NP, et al. Adipokines: a novel link between adiposity and carotid plaque vulnerability. Eur J Clin Invest. 2012;42:1278.
245. Shantsila E, et al. Endothelial progenitor cells in cardiovascular disorders. J Am Coll Surg. 2007;49:741.
246. Vasa M, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:E1.
247. Hill JM, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593.
248. Dimmeler S, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391.
249. Laufs U, et al. Physical training increases endothelial progenitor cells, inhibits neointima formation, and enhances angiogenesis. Circulation. 2004;109:220.
250. Strehlow K, et al. Estrogen increases bone marrow–derived endothelial progenitor cell production and diminishes neointima formation. Circulation. 2003;107:3059.
251. Mlekusch W, et al. Serum creatinine predicts mortality in patients with peripheral artery disease: influence of diabetes and hypertension. Atherosclerosis. 2004;175:361.
252. Daskalopoulou SS, et al. Peripheral arterial disease: a missed opportunity to administer statins so as to reduce cardiac morbidity and mortality. Curr Med Chem. 2005;12:763.
253. Daskalopoulou SS, et al. Uric acid levels and vascular disease. Curr Med Res Opin. 2004;20:951.
254. Langlois M, et al. Serum uric acid in hypertensive patients with and without peripheral arterial disease. Atherosclerosis. 2003;168:163.
255. Tseng CH, et al. Effect of angiotensin blockade on the association between albuminuria and peripheral arterial disease in elderly Taiwanese patients with type 2 diabetes mellitus. Circ J. 2005;69:965.
256. Wattanakit K, et al. Albuminuria and peripheral arterial disease: results from the Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis. 2008;201:212.
257. Ylä-Herttuala S, et al. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Invest. 1989;84:1086.
259. Tsimikas S. Oxidative biomarkers in the diagnosis and prognosis of cardiovascular disease. Am J Cardiol. 2006;98:9P.
260. Haslacher H, et al. Plasma myeloperoxidase level and peripheral arterial disease. Eur J Clin Invest. 2012;42:463.
261. Chen LQ, et al. Race-specific associations of myeloperoxidase with atherosclerosis in a population-based sample: the Dallas Heart Study. Atherosclerosis. 2011;219:833.
262. Steinberg D, et al. Alcohol and atherosclerosis. Ann Intern Med. 1991;114:967.
263. Notzon FC, et al. Causes of declining life expectancy in Russia. JAMA. 1998;279:793.
264. Mukamal KJ, et al. Roles of drinking pattern and type of alcohol consumed in coronary heart disease in men. N Engl J Med. 2003;348:109.
265. Donnan PT, et al. Diet as a risk factor for peripheral arterial disease in the general population: the Edinburgh Artery Study. Am J Clin Nutr. 1993;57:917.
266. Langlois M, et al. Serum vitamin C concentration is low in peripheral arterial disease and is associated with inflammation and severity of atherosclerosis. Circulation. 2001;103:1863.
267. Salonen RM, et al. Six-year effect of combined vitamin C and E supplementation on atherosclerotic progression: the Antioxidant Supplementation in Atherosclerosis Prevention (ASAP) Study. Circulation. 2003;107:947.
268. U.S. Preventive Services Task Force. Using nontraditional risk factors in coronary heart disease risk assessment: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2009;151:474.
269. Barter P. The role of HDL-cholesterol in preventing atherosclerotic disease. Eur Heart J Suppl. 2005;7:F4.
270. Zimmerman GA, et al. Adhesion and signaling in vascular cell-cell interactions. J Clin Invest. 1997;100:S3.
271. Krone W, et al. Effects of calcium antagonists and adrenergic antihypertensive drugs on plasma lipids and cellular cholesterol metabolism. J Cardiovasc Pharmacol. 1987;10:S199.
272. Vaughan DE. The renin-angiotensin system and fibrinolysis. Am J Cardiol. 1997;79:12.
273. Cooke JP, et al. Nitric oxide synthase: role in the genesis of vascular disease. Annu Rev Med. 1997;48:489.
274. Tsao PS, et al. Nitric oxide regulates monocyte chemotactic protein-1. Circulation. 1997;96:934.
275. Mostafa Mtairag E, et al. Effects of interleukin-10 on monocyte/endothelial cell adhesion and MMP-9/TIMP-1 secretion. Cardiovasc Res. 2001;49:882.
276. Daskalopoulou SS, et al. Metallothionein expression in the high-risk carotid atherosclerotic plaque. Curr Med Res Opin. 2007;23:659.
277. Drexler H, et al. Effect of L-arginine on coronary endothelial function in cardiac transplant recipients: relation to vessel wall morphology. Circulation. 1994;89:1615.
278. Clarkson P, et al. Oral L-arginine improves endothelium-dependent dilation in hypercholesterolemic young adults. J Clin Invest. 1996;97:1989.
279. Lerman A, et al. Long-term L-arginine supplementation improves small-vessel coronary endothelial function in humans. Circulation. 1998;97:2123.
280. Bode-Böger SM, et al. L-Arginine induces nitric oxide–dependent vasodilation in patients with critical limb ischemia. A randomized, controlled study. Circulation. 1996;93:85.
281. Cooke JP, et al. Antiatherogenic effects of L-arginine in the hypercholesterolemic rabbit. J Clin Invest. 1992;90:1168.
282. Wang BY, et al. Dietary arginine prevents atherogenesis in the coronary artery of the hypercholesterolemic rabbit. J Am Coll Cardiol. 1994;23:452.
283. Böger RH, et al. Supplementation of hypercholesterolaemic rabbits with L-arginine reduces the vascular release of superoxide anions and restores NO production. Atherosclerosis. 1995;117:273.
284. Tsao PS, et al. Enhanced endothelial adhesiveness in hypercholesterolemia is attenuated by L-arginine. Circulation. 1994;89:2176.
285. Candipan RC, et al. Regression or progression. Dependency on vascular nitric oxide. Arterioscler Thromb Vasc Biol. 1996;16:44.
286. Stern N, et al. Hypoadiponectinemia as a marker of adipocyte dysfunction—part I: the biology of adiponectin. J Cardiometab Syndr. 2007;2:174.
287. Fadini GP, et al. Endothelial progenitor cells in the natural history of atherosclerosis. Atherosclerosis. 2007;194:46.
288. Wu Y, et al. Essential role of ICAM-1/CD18 in mediating EPC recruitment, angiogenesis and repair to the infarcted myocardium. Circ Res. 2006;99:315.
289. Duan H, et al. LFA-1 and VLA-4 involved in human high proliferative potential–endothelial progenitor cells homing to ischemic tissue. Thromb Haemost. 2006;96:807.
290. Ceradini DJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858.
291. Willich SN. Circadian variation and triggering of cardiovascular events. Vasc Med. 1999;4:41.
292. Willich SN, et al. Weekly variation of acute myocardial infarction. Increased Monday risk in the working population. Circulation. 1994;90:87.
293. Liapis C, et al. Seasonal variation in the incidence of ruptured abdominal aortic aneurysm. Eur J Vasc Surg. 1992;6:416.
294. Saposnik G, et al. Does a birthday predispose to vascular events? Neurology. 2006;67:300.
295. Witte DR, et al. Cardiovascular mortality in Dutch men during 1996 European football championship: longitudinal population study. BMJ. 2000;321:1552.
296. Callahan KP, et al. Platelet activation as a universal trigger in the pathogenesis of acute coronary events after cocaine abuse. Swiss Med Wkly. 2001;131:487.
297. Smeeth L, et al. Risk of myocardial infarction and stroke after acute infection or vaccination. N Engl J Med. 2004;351:2611.
298. Anderson RA, et al. Is the fatty meal a trigger for acute coronary syndromes. Atherosclerosis. 2001;159:9.
299. Liapis CD, et al. ESVS Guidelines: Section A—prevention in patients with carotid stenosis. [ESVS Guidelines Collaborators; European Society for Vascular Surgery] Curr Vasc Pharmacol. 2010;8:673.
300. Smith SC Jr, et al. AHA/ACCF Secondary Prevention and Risk Reduction Therapy for Patients with Coronary and other Atherosclerotic Vascular Disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation. [World Heart Federation and the Preventive Cardiovascular Nurses Association] Circulation. 2011;124:2458.
301. Flu HC, et al. A systematic review of implementation of established recommended secondary prevention measures in patients with PAOD. Eur J Vasc Endovasc Surg. 2010;39:70.
302. Kinikini D, et al. Meeting AHA/ACC secondary prevention goals in a vascular surgery practice: an opportunity we cannot afford to miss. [American Heart Association; American College of Cardiology] J Vasc Surg. 2006;43:781.
303. Corrao G, et al. Results of a retrospective database analysis of adherence to statin therapy and risk of nonfatal ischemic heart disease in daily clinical practice in Italy. Clin Ther. 2010;32:300.
304. Perreault S, et al. Impact of better adherence to statin agents in the primary prevention of coronary artery disease. Eur J Clin Pharmacol. 2009;65:1013.
305. Bramlage P, et al. The effect of optimal medical therapy on 1-year mortality after acute myocardial infarction. Heart. 2010;96:604.
306. Danchin N, et al. Impact of combined secondary prevention therapy after myocardial infarction: data from a nationwide French registry. [USIC 2000 investigators] Am Heart J. 2005;150:1147.
307. Shalev V, et al. Continuation of statin treatment and all-cause mortality: a population-based cohort study. Arch Intern Med. 2009;169:260.
308. Sokol MC, et al. Impact of medication adherence on hospitalization risk and healthcare cost. Med Care. 2005;43:521.
310. Heeschen C, et al. Withdrawal of statins increases event rates in patients with acute coronary syndromes. [Platelet Receptor Inhibition in Ischemic Syndrome Management (PRISM) Investigators] Circulation. 2002;105:1446.
311. Schouten O, et al. Effect of statin withdrawal on frequency of cardiac events after vascular surgery. Am J Cardiol. 2007;100:316.
312. Jasińska-Stroschein M, et al. Novel mechanistic and clinical implications concerning the safety of statin discontinuation. Pharmacol Rep. 2011;63:867.
313. Sanz G, et al. Polypill and global cardiovascular health strategies. Semin Thorac Cardiovasc Surg. 2011;23:24.
314. Munger MA, et al. Medication nonadherence: an unrecognized cardiovascular risk factor. MedGenMed. 2007;9:58.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]Chapter 26
Atherosclerotic Risk Factors
General Considerations
Christos Liapis, John Kakisis
The word atherosclerosis is derived from two Greek roots: αθηρι (athere, gruel) and σκληρóς (skleros, hard). In 1755, von Haller first applied the term atheroma to a common type of plaque that, on sectioning, exuded yellow pultaceous content from its core.1 In 1904, Marchand introduced the term atherosclerosis in acknowledgment of the consistent association of fatty degeneration and arterial stiffening.2
Atherosclerosis is a systemic disease of large and medium-sized arteries in which lipid and fibrous material accumulate within the intimal layer. It should not be confused with the more general term arteriosclerosis, which refers to generalized thickening and loss of elasticity of arteries as a result of an increased amount of basement material and plasma protein deposition.3
Advances in our understanding of the pathogenesis of atherosclerotic vascular disease gave birth to the concept of cardiovascular risk factors, initially referred to in the Framingham Heart Study.4 Risk factor assessment is important to accurately guide primary and secondary prevention, whereas compliance with risk factor modification is still an area of concern.
Some risk factors are hereditary (and therefore not controllable), but others are acquired or related to behavioral and environmental factors and thus potentially amenable to manipulation. More recently, new risk factors have emerged.
This chapter summarizes the interaction of risk factors with the arterial wall during the atherosclerotic process and briefly reports the currently considered major risk factors. The emerging role and potential management of minor risk factors and markers of atherosclerosis are reviewed. Finally, the importance of compliance with these strategies, including knowledge that appeared in the literature since the publication of the seventh edition of the textbook, is discussed.
Risk Factors and Atherogenesis
The interaction of risk factors with the arterial wall initiates the atherosclerotic process (Fig. 26-1). The starting point for atheroma formation is endothelial dysfunction (reported also as “activation” because cells frequently respond normally to a noxious stimulus).5,6 Chapter 5 covers the mechanisms of atherogenesis in detail; this chapter provides a summary of the key mechanisms related to individual risk factors.
Figure 26-1 Evolution of arterial wall changes and plaque formation in the response-to-injury hypothesis: 1, endothelial dysfunction; 2, vascular smooth muscle cell hypertrophy; 3, migration and proliferation of vascular smooth muscle cells; 4, matrix elaboration; 5, expression of adhesion molecules and migration of monocytes; 6, uptake of low-density lipoprotein (LDL) and formation of foam cells; 7, thrombus formation; 8, angiogenesis and neovascularization. (Modified from Defraigne JO: Development of atherosclerosis for the vascular surgeon. In Liapis CD, Balzer K, Fernandes e Fernandes J, Benedetti-Valentini F, editors: Vascular surgery, New York, 2007, Springer, p 24.)
Interaction with Endothelial Cells: Endothelial Dysfunction
Currently, the most important contributors to endothelial dysfunction are hemodynamic disturbances, hypercholesterolemia, and inflammation. Etiologic culprits also include cigarette toxins, homocysteine, and a wide spectrum of infectious agents. Inflammatory cytokines (e.g., tumor necrosis factor [TNF]) can also stimulate the expression of proatherogenic genes in endothelial cells.7,8
The response of the endothelium to these stimuli can be rapid. For instance, forearm vascular reactivity increases substantially in the 4-hour period after ingestion of a fatty meal.9 Conversely, low-density lipoprotein (LDL) lowering reduces vascular reactivity.9
Experimental models of tobacco exposure, diabetes mellitus, hypercholesterolemia, and hypertension are characterized by common endothelial abnormalities, such as increased generation of oxidative stress and reductions in bioactivity or synthesis (or both) of endothelium-derived nitric oxide, which results in attenuation of vascular tone.10–14
Chronic endothelial injury eventually leads to endothelial dysfunction and increased permeability (Fig. 26-2). Concomitant events include LDL oxidation and accumulation in the subendothelial space of the intima.15 Oxidized LDL has a proinflammatory and proatherogenic effect, and it has recently been suggested that several of the neoepitopes generated during oxidation are highly immunogenic and result in the generation of autoantibodies.16
Figure 26-2 Consequences of endothelial dysfunction. Normal endothelium displays antiaggregant, anticoagulant, and vasodilatative properties along with inhibition of cell proliferation. After exposure to various agents causing endothelial dysfunction, these functions are modified toward procoagulant and vasoconstrictive activities together with stimulation of cell recruitment and proliferation. LDL, Low-density lipoprotein; NO, nitric oxide; PAF, platelet-activating factor; PAI-1, plasminogen activator inhibitor-1; PGI2, prostacyclin; ROS, reactive oxygen species; SMC, smooth muscle cell; TXA2, thromboxane A2. (Modified from Defraigne JO: Development of atherosclerosis for the vascular surgeon. In Liapis CD, Balzer K, Fernandes e Fernandes J, Benedetti-Valentini F, editors: Vascular surgery, New York, 2007, Springer, p 25.)
The greater endothelial elaboration of oxygen-derived free radicals activates oxidant-sensitive transcriptional proteins such as nuclear factor κB (NF-κB), which induces the expression of adhesion molecules and thus initiates the inflammatory process.17–22
Both cell recruitment into the endothelial surface and oxidized LDL stimulate the production of growth factors and chemotactic agents by endothelial cells.23 These substances attract and stimulate the proliferation of both macrophages and vascular smooth muscle cells (VSMCs).
Macrophages
As monocytes are attracted to the endothelium and migrate to the subendothelial space, they mature into macrophages and upregulate pattern recognition receptors, including scavenger receptors and Toll-like receptors. Scavenger receptors mediate internalization of oxidized LDL, which results in foam cell formation (the earliest event in the formation of fatty streaks). Toll-like receptors transmit activating signals that lead to the release of cytokines, proteases, and vasoactive molecules. Activated macrophages attract additional monocytes and stimulate VSMCs in a positive feedback loop. As foam cells accumulate in the subendothelial space, they distort the overlying endothelium and may eventually even rupture through the endothelial surface.23
Accumulating evidence indicates that macrophages are responsible for some key features of vulnerable plaque. An intense infiltration of macrophages is invariably observed at the site of plaque rupture, where the fibrous cap appears to be undermined. These macrophages are synthesizing cathepsins and abundant amounts of matrix metalloproteinases (MMP-1, MMP-3, and MMP-9). MMPs degrade the extracellular matrix, which weakens the fibrous cap.24–26
T Lymphocytes
T lymphocytes recruited to the intima interact with macrophages and can generate a chronic immune inflammatory state. The T lymphocytes found in atherosclerotic lesions are polyclonal, which indicates that these cells do not develop in response to a single antigen. It is not clear whether the T lymphocytes respond to specific antigens (e.g., bacterial or viral antigens or modified arterial wall constituents and lipoproteins) or are nonspecifically activated by the local inflammatory milieu.7
Nevertheless, T lymphocytes in atherosclerotic lesions recognize antigens and mount helper T cell type 1 responses with secretion of proinflammatory cytokines, which in turn can stimulate macrophages as well as endothelial cells and VSMCs.27
Subtypes of T lymphocytes called regulatory T cells, previously shown to maintain immunologic tolerance, control the development and progression of atherosclerosis. The function of regulatory T lymphocytes is modulated by chemokines and by costimulatory pathways.28,29
Complement
Activation of complement seems to play a role in both initiation of atherosclerosis and acceleration of the disease.30 Complement activation can occur by the classical (antibody dependent), the alternative (antibody independent), the lectin, or the coagulation pathway.31 Whereas complement activation by the classical and lectin pathways may be protective by removal of apoptotic cells and cell debris from atherosclerotic plaques, activation of the complement cascade by the alternative pathway may be proatherogenic and may play a role in plaque destabilization, leading to its rupture and the onset of acute cardiovascular events. In this context, various complement components, including C3, C3a, C4, C5a, and mannose-binding lectin, have served as biomarkers in prospective clinical studies, associated with an increased risk of death, myocardial infarction (MI), or restenosis after percutaneous transluminal angioplasty.31
Platelets
Platelets play an important role in stimulating the progression of atherosclerotic lesions by secreting growth factors and vasoactive substances (e.g., platelet-derived growth factor, transforming growth factor-α, transforming growth factor-β, epidermal growth factor, and insulin-like growth factor-1) after their adherence to the vessel wall in sites of endothelial ulceration.32,33 These substances recruit and stimulate proliferation of VSMCs.
Recently, platelets have been suggested as initial role players in the development of atherosclerotic lesions by recruiting and binding to leukocytes, endothelial cells, and circulating progenitor cells and initiating transformation of monocytes into macrophages. Platelets internalize oxidized phospholipids, express various scavenger receptors that are able to regulate LDL uptake, and promote foam cell formation.34,35 Moreover, platelets have been identified as key effectors of inflammation throughout plaque development through the secretion of several inflammatory molecules, such as CD40L, P-selectin, RANTES, and Toll-like receptors.36 These molecules exacerbate the inflammation and induce the transition from chronic to acute disease, featuring increased instability of the atherosclerotic lesion that results in plaque rupture and thrombosis.36
Vascular Smooth Muscle Cells
The mediators (e.g., platelet-derived growth factor, fibroblast growth factor, transforming growth factor-β, interleukin [IL]–1, and MMPs) released by endothelial cells, macrophages, lymphocytes, and platelets induce a change in the phenotype of VSMCs from the quiescent “contractile” phenotype to the active “synthetic” state. VSMCs in the synthetic state can migrate and proliferate from the media to the intima, where they produce excessive amounts of extracellular matrix (e.g., collagen, elastin, and proteoglycans) that transforms the lesion into a fibrous plaque. VSMCs are also capable of functions typically attributed to other cell types. Like macrophages, VSMCs can express a variety of receptors for lipid uptake and can form foam-like cells, thereby participating in the early accumulation of plaque lipid.
Like endothelial cells, VSMCs can also express a variety of adhesion molecules (e.g., vascular cell adhesion molecule-1 [VCAM-1] and intercellular adhesion molecule-1 [ICAM-1]) to which monocytes and lymphocytes can adhere and migrate into the vessel wall.37 The lesion grows until it is transformed from a fibrous plaque to a complex plaque. Apoptosis, proliferation, and migration of VSMCs are vital to the pathogenesis of atherosclerosis and plaque rupture. VSMCs are the only cells within plaque capable of synthesizing structurally important collagen isoforms.
Currently, the development of therapeutic approaches targeting VSMC migration and proliferation represents an active field of research, beyond drug-eluting stents and balloons that are already standard procedures for the prevention of restenosis.38–40
Vulnerable Plaque
Vulnerable atherosclerotic plaque (high-risk or unstable plaque) is associated with an increased risk of disruption, distal embolization, and vascular events. Vulnerable plaque is an advanced histologic lesion with a large lipid core (filled with lipid and cell debris), thin fibrous cap, ulceration, intraluminal thrombosis, and intraplaque hemorrhage as well as intense infiltration by macrophages and other inflammatory cells.41–44
The most widely used histologic classification of atherosclerotic plaque is the American Heart Association–recommended Stary classification.44 Inflammation plays a key role in the pathogenesis of atherosclerosis. In this process, the immune system and oxidative stress seem to be involved in the initiation, propagation, and activation of lesions in the arterial wall.41–44 Unstable plaque is dominated by inflammatory cells that destroy the fibrous cap and are responsible for endothelial denudation and thrombogenicity of the plaque contents (Fig. 26-3). Rupture depends on the balance between inflammatory cell activity and the VSMC-driven repair process.45 Activated macrophages, T lymphocytes, and mast cells produce a variety of molecules—inflammatory cytokines, proteases, coagulation factors, radicals, and vasoactive molecules—that are expressed in plaque and may modulate remodeling of the extracellular matrix, cell proliferation, and cell death (apoptosis) and thereby ultimately destabilize these lesions.41–44,46
Figure 26-3 Unstable atherosclerotic plaque on histologic examination. Type VI plaque according to Stary’s classification,44 in which one or more of the following are present: surface defects (ulceration), hematoma, and thrombosis.
The likelihood of plaque rupture is a balance between the tensile strength of the plaque and the hemodynamic stress exerted on it.47 In ruptured plaque, the fibrous cap appears to be eroded at the shoulder of the lesion (where the fibrous cap meets the intima of the normal segment of the vessel wall).
Furthermore, local infection (e.g., induced by cytomegalovirus, herpes simplex virus, or Chlamydophila pneumoniae) may influence plaque stability.48
Several invasive and noninvasive imaging methods have been used to identify vulnerable plaque, but all have their limitations (Table 26-1).43 Thus far there are no widely available modalities to adequately provide both anatomic and functional information about vulnerable atherosclerotic plaque. New modalities are on the way, and the future looks promising.49–51
Table 26-1
Advantages and Disadvantages of the Different Imaging Techniques Used for Identification of Vulnerable Plaque
| Imaging Technique | Advantages | Disadvantages |
| High-resolution duplex ultrasound scanning | High resolution; noninvasive; no x-ray exposure; information on arterial wall thickness; qualitative and quantitative analysis of plaque | Limited information on plaque activity, possible improvement with the use of contrast-enhanced ultrasound |
| Digital subtraction angiography | Information on diameter stenosis and luminal surface | Invasive; may underestimate the degree of stenosis; no vessel wall imaging; no information on plaque composition and activity; contrast agent; x-ray exposure |
| Magnetic resonance angiography | Noninvasive; no x-ray exposure; no contrast agent; information on plaque composition | Less accurate in severe stenosis (unless contrast is used) |
| Helical computed tomography angiography | Noninvasive; differentiates between soft, intermediate, and calcified plaque | Contrast agent; x-ray exposure; overestimation of stenosis; no information on plaque activity |
| Intravascular ultrasound | Characterizes vessel wall and plaque morphology early in the disease process; differentiates stable and unstable plaque | Invasive; no information on plaque activity |
| Positron emission tomography | High sensitivity; functional information (plaque activity); molecular imaging | Lack of anatomic information unless combined with computed tomography; radiation exposure |
| Single-photon emission computed tomography | High sensitivity; functional information (plaque activity); molecular imaging | Lack of anatomic information unless combined with computed tomography; radiation exposure |
| Intravascular thermography | Functional information (plaque activity) | Invasive; risk of endothelial damage; difficult interpretation |
| Optical coherence tomography | High resolution (fibrous, lipid, and calcified components of the plaque can be distinguished); small probe size | Invasive; needs blood displacement; limited time window for imaging (2 seconds); limited length of penetration (1-2 mm) |
| Elastography | Differentiates lipid-rich and fibrous tissues; high sensitivity and specificity | Invasive |
| Near-infrared spectroscopy | Information on tissue chemical composition; detects the lipid core, the fibrous cap, and inflammation | Influenced by flowing blood; lack of anatomic information; limited independent role |
Assessment and Management of Risk Factors
The prevalence plus severity of atherosclerosis and peripheral arterial disease (PAD) or coronary artery disease (CAD) in individuals and groups is related to several “old” and “new” risk factors. Thus far there has been no universal agreement on the exact classification of the various cardiovascular risk factors and risk markers.
Classification of Risk Factors
An American Heart Association prevention conference statement in 199952,53 classified risk factors into three categories: “traditional”/conventional risk factors, which appear to have a direct causal role in atherogenesis; predisposing factors, which mediate some risk through the causal factors but may also have independent effects; and conditional risk factors, which have “an association with an increased risk for CAD, although their causative, independent, and quantitative contributions to CAD are not well documented.” The authors suggested that these factors may enhance risk in the presence of causative risk factors, hence the term “conditional.”
Since then, extensive research in all clinical fields of atherosclerosis has identified several novel and emerging risk factors or “markers” of atherosclerosis, which, however, need further confirmatory studies. As soon as sufficient evidence is available, some of them will move to the category of conditional risk factors, but it remains to be elucidated which ones will ever prove to be predisposing or even conventional.
A contemporary classification of risk factors is presented in Box 26-1.5,52–58
Conventional Risk Factors
Conventional risk factors for atherosclerosis are similar in the limbs and in the carotid, coronary, and other vascular beds. The four major risk factors for vascular disease59–65 are discussed in detail in other chapters of this section—smoking (see Chapter 27), diabetes mellitus (see Chapter 28), hyperlipidemia (see Chapter 29), and hypertension (see Chapter 30)—and are not discussed here.
Predisposing Risk Factors
Advanced Age
Although atherosclerosis may be present in younger individuals, age has a dominant influence. All forms of cardiovascular disease become more prevalent in the elderly. In several studies, the risk for PAD increased 1.5- to 2.0-fold for every 10-year rise in age,59,66–68 whereas the prevalence of intracranial atherosclerosis increases by 5% for every 1-year rise in age.69 A recent systematic review of 40 studies showed that the prevalence of carotid artery disease among people younger than 70 years was 4.8% in men and 2.2% in women, rising to 12.5% and 6.9%, respectively, among those 70 years of age and older.70 Similarly, death rates from CAD increase with each decade of life up to the age of 85 years. The death rate from CAD in white men aged 25 to 34 years is about 10 per 100,000; by the age of 55 to 64 years, it has increased 10-fold to nearly 1000 per 100,000.71
Obesity
Obesity predisposes patients to the development of hypertension, diabetes mellitus, and hyperlipidemia. Excess adiposity has recently been shown to play a direct role in initiating atherosclerosis in that fat cells are capable of affecting the systemic vasculature through a variety of mechanisms, among which altered arterial homeostasis, endothelial dysfunction, and inflammation are the most prominent.72–74 Weight loss interventions modify not only the risk factors but also arterial function.
In the Cardiovascular Health Study, each 5-unit increase in body mass index in midlife and in older age was associated with about a 30% increase in PAD prevalence and incidence.75 The European Prospective Investigation into Cancer and Nutrition (EPIC) study showed that general (as indicated by the body mass index) and abdominal (as indicated by the waist circumference or the waist-to-hip ratio) adiposity are both associated with the risk of death.76 Abdominal adiposity is a better predictor of death in patients with a low body mass index. The underlying mechanism is that adipose tissue, especially adipose tissue from visceral fat deposits, secretes mediators that are important in the development of chronic diseases, such as respiratory ailments, cancer, and atherosclerosis. In support of this theory, the risk of death from cardiovascular disease or cancer had been found to be higher among patients with a higher body mass index.77
Physical Inactivity
It is well established that the absence of even moderate levels of physical activity increases risk for cardiovascular morbidity and mortality in the whole population. It is hypothesized that physical activity consisting of either structured training or self-controlled body motivation results in “pleiotropic” actions on the cardiovascular system, which is explained, at least in part, by the modification of traditional and novel cardiovascular risk factors (e.g., hypertension, dyslipidemia, obesity, insulin resistance, endothelial dysfunction, hyperglycemia, inflammation, hypercoagulability, and oxidative stress).78–80 However, only 60% of the beneficial effect of physical activity on cardiovascular disease can be attributed to favorable changes in known risk factors, indicating that physical inactivity may be an independent risk factor for cardiovascular disease.80,81 Moreover, a substantial and expanding body of evidence has demonstrated that low exercise capacity is an independent predictor of cardiovascular and overall mortality.82–85 Therefore, lifelong exercise training should be incorporated in all strategies of primary or secondary prevention against cardiovascular diseases.86–89
Gender
Gender influences the risk for atherosclerosis and its preceding risk factors (e.g., hyperlipidemia, hypertension, and insulin resistance). Epidemiology, symptoms, and progression of cardiovascular disease are different between men and women. Indeed, cardiovascular disease develops in women about 10 years later than in men and typically after menopause. Premenopausal (estrogenic) women are relatively protected against atherosclerosis and its consequences compared with age-matched men. Estrogen alters serum lipoprotein levels through estrogen receptor–mediated effects on hepatic expression of apoproteins. With the onset of menopause, LDL levels rise, whereas high-density lipoprotein (HDL) levels fall, thereby changing a previously antiatherogenic lipid profile to one similar to that of men. However, several clinical trials have failed to demonstrate any utility of postmenopausal hormonal therapy for prevention of vascular disease in women.90–92
Family History and Genetics
Most cardiovascular events result from the interactions of genetic and environmental factors, none of which can cause disease by itself.
Familial predisposition to atherosclerosis is multifactorial and related to familial clustering of other risk factors (e.g., hypertension, hyperlipidemia, and diabetes), direct genetic linkage to atherosclerosis, or family habits associated with environmental risk factors.93 After adjustment for known risk factors, familial history of CAD remains an independent predictor of the disease, associated with a twofold to threefold increased risk of CAD.65 Particularly, individuals with a familial history of CAD of early onset (age at onset before 55 years in men and before 65 years in women) should be regarded as a high-risk group.65 Similarly, three family studies have assessed the heritability of the ankle-brachial index and demonstrated that about 20% to 50% of the interindividual variability in the ankle-brachial index is attributable to genetic determinants.94 Although aneurysm development is a combination of atherosclerosis and degeneration, the significant association with family history indicates genetic predisposition.95
Approximately 40 quantitative trait loci for atherosclerotic disease have been found in humans. Linkage analysis has identified specific genes that may contribute to cardiovascular risk, for example, myocyte enhancer factor-2 (MEF2A) with risk for MI96 and arachidonate 5-lipoxygenase–activating protein (ALOX5AP) with risk for MI and stroke.97 In femoral artery samples from patients with PAD, 366 genes were differentially regulated in intermediate lesions and 447 in advanced lesions.98 Among the upregulated genes, several genes involved in immune and inflammatory responses were identified.
In the future, a complete identification of genetic markers of atherosclerosis may be used in clinical prediction algorithms and even in therapeutic strategies. Resources such as the Human Genome Project and the international HapMap project are expected to change the practice of cardiovascular medicine in the 21st century.99
Behavioral and Socioeconomic Factors
Poor community socioeconomic conditions, depression, social isolation, work- or family-related stress, and type A behavioral patterns have been correlated with CAD and the severity and progression of atherosclerosis. These psychosocial risk factors are conveyed by behavioral pathways, such as inappropriate diet, smoking, sedentary lifestyle, and inadequate use of medical resources, and psychobiologic mechanisms, such as disturbed autonomic and hormonal regulation, which is directly involved in the pathogenesis of cardiovascular diseases.100–104
The risk for angina and ischemic stroke is two to four times higher in blacks than in whites, whereas the risk for PAD is highest in non-Hispanic blacks,105,106 not explained and not related to diabetes, hypertension, or body mass index.107
Conditional Risk Factors
Conditional risk factors had initially been proposed to facilitate classification of factors that may enhance the risk for cardiovascular disease in the presence of causative risk factors. Even though evidence for these factors has improved toward “independency,” additional prospective studies and randomized trials are needed to provide level A evidence.5,52–58
Homocysteine
Homocysteine is an intermediate metabolic product in the transmethylation and transsulfuration reactions that are essential for normal cell growth, differentiation, and function. It is involved in the transformation of methionine to cysteine.108 Plasma levels of homocysteine are regulated by environmental factors, such as vitamin B levels, as well as by genetically determined factors, such as the rare cystathionine β-synthase deficiency and enzyme activity of 5,10-methylenetetrahydrofolate reductase.109
Reports suggest that elevated homocysteine levels are an independent risk factor for vascular disease.110 Elevated homocysteine is associated with a mildly increased risk for CAD, stroke, PAD, and venous thromboembolism with a risk ranging between 0.8 and 3.4.111–113 In a meta-analysis of studies that adjusted for six or seven Framingham risk factors, each 5-µmol/L increase in homocysteine level conferred an approximately 9% increase in the risk for cardiovascular events.114
Homocysteine exerts its atherothrombotic effect through several potential mechanisms. It enhances thrombosis by forming a less porous fibrin network; by activating platelet aggregation and procoagulant factors V and VII, thrombomodulin, tissue factor pathway inhibitor, and plasminogen activator inhibitor (PAI); and by inhibiting activation of protein C. It also triggers proinflammatory activity by activating NF-κB and by promoting monocyte differentiation through NAD(P)H oxidase–mediated oxidant stress, endothelial dysfunction by inhibition of nitric oxide availability by endothelial cells and formation of reactive oxygen species, the LDL oxidation usually detected in cases of atherosclerotic lesions, proliferation of smooth muscle cells, and expression of tissue factor in endothelial cells and macrophages.115–119
Plasma levels of homocysteine are usually regulated by supplementation with B vitamins, either dietary or prescribed as folate.117,120 However, several clinical trials have demonstrated that lowering of homocysteinemia with vitamin supplementation is not effective in reducing cardiovascular risk,121–123 and even a harmful effect from combined B vitamin treatment has been suggested.124
C-Reactive Protein
C-reactive protein (CRP) is an acute phase protein. It is primarily synthesized by hepatocytes, driven by IL-6 with synergistic enhancement of IL-1 or TNF.125 A rise in CRP levels by as much as 1000-fold is not uncommon in both infectious and noninfectious disorders, including MI.126 Until recently, it has been the primary inflammatory marker used in clinical practice, and its predictive value can be estimated only through high-precision assays (high-sensitivity CRP [hsCRP]), with acceptable precision down to or below 0.3 mg/L (2.86 nmol/L). It is within these lower, previously “normal” ranges that hsCRP levels seem to have predictive ability for cardiovascular events. An hsCRP level higher than 10 mg/L (95.24 nmol/L), for example, should be discarded and measurement repeated in 2 weeks to allow the acute inflammation to subside before retesting.
Observational as well as prospective studies have consistently reported that elevated CRP serum levels definitely have prognostic value for cardiovascular events and mortality. CRP has been suggested to induce a prothrombotic state by induction of tissue factor expression in human monocytes.127 It can activate or inhibit the complement system, driving the inflammation in atherosclerotic lesions.128 CRP has also been reported to decrease the expression and bioactivity of endothelial nitric oxide synthase,129 with a subsequent effect on vasodilatation. CRP inhibits both basal and vascular endothelial growth factor–stimulated angiogenesis, whereas it promotes endothelial apoptosis in a nitric oxide–dependent fashion.130 CRP has been found to synergistically enhance angiotensin II–induced proinflammatory effects, such as cellular migration and proliferation, and lesion collagen and elastin content.131 CRP induces the release of monocyte chemotactic protein-1 (MCP-1) and endothelin-1 and thereby upregulates adhesion molecules and chemoattractant chemokines in endothelial cells and VSMCs.132 Finally, CRP has been suggested to mediate the proliferation and activation of VSMCs through activation of NF-κB,133 which leads to accumulation of these cells in the vascular intima and initiates atherogenesis.
Several therapeutic strategies orientated toward a reduction in CRP have been proposed, including cyclooxygenase inhibitors (aspirin and cyclooxygenase 2 inhibitors), angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and especially statins.134 In a randomized clinical trial comprising 17,802 apparently healthy women and men with LDL cholesterol levels of 130 mg/dL or lower and hsCRP levels of 2.0 mg/dL or higher, LDL cholesterol levels were reduced by 50% and hsCRP levels by 37% in patients randomized to 20 mg daily rosuvastatin instead of placebo.135 After a median follow-up of 1.9 years, the rates of most endpoints, including MI, stroke, arterial revascularization, and unstable angina, were reduced by about 50%, whereas all-cause mortality was reduced by 20%. Given the short follow-up time, the beneficial outcome of rosuvastatin may reflect the plaque stabilization effect of lowering CRP levels rather than the inhibition of bulky progression of existing plaque.
However, the causal association between CRP and vascular disease has been challenged by other studies showing that polymorphisms in the gene encoding CRP are associated with marked increases in CRP levels but are not in themselves associated with an increased risk of ischemic vascular disease.136 Similarly, morphometric analyses of atherosclerotic plaques in CRP-deficient animals revealed equivalent or increased atherosclerotic lesions compared with controls, an experimental result that does not support a proatherogenic role of CRP.137
Fibrinogen
Fibrinogen is a glycoprotein that circulates at high concentration in blood. Fibrinogen initially mediates platelet aggregation. Later in clot formation it is converted to fibrin. The fibrin matrix gives the clot shape, strength, flexibility, and stability.
Levels of fibrinogen have been demonstrated to be elevated in patients with acute thrombosis. Fibrinogen increases plasma viscosity and induces VSMC proliferation. The Gothenburg Heart Study was the first prospective trial to show an association between fibrinogen levels and subsequent risk for cardiovascular disease.138 In the Northwick Park Heart Study,139 fibrinogen level appeared to be as effective as total cholesterol concentration in predicting future risk for CAD. Higher levels of fibrinogen predict subsequent acute coronary syndromes, whereas lower levels, despite elevated cholesterol levels, are associated with a lower risk for acute coronary syndromes.140 It remains unclear, however, whether elevated fibrinogen levels are a cause or a consequence of atherosclerosis. In the Copenhagen City Heart Study, the relative risk of stroke development was almost double in patients with higher fibrinogen levels. Nevertheless, elevated levels were not associated with echolucent unstable carotid plaque.141 On the contrary, the CARDIA (Coronary Artery Risk Development in Young Adults) study demonstrated that an elevated fibrinogen concentration in persons aged 25 to 37 years is independently associated with subclinical cardiovascular disease (as indicated by the prevalence of coronary artery calcification and increased mean carotid intima-media thickness) in the subsequent decade.142
Modification of fibrinogen levels has failed to reduce the overall risk for MI or sudden cardiac death.143
Emerging Factors and Markers of Atherosclerosis
Inflammatory Markers
Apart from CRP and fibrinogen, several other inflammatory markers have been associated with atherosclerosis and cardiovascular disease (Table 26-2).
Table 26-2
Inflammatory Biomarkers and Different Stages of Evolution of Atherosclerotic Plaque
CRP, C-reactive protein; ICAM-1, intercellular adhesion molecule-1; IL, interleukin; LOX-1, lectin-like oxidized LDL receptor-1; Lp-PLA2, lipoprotein-associated phospholipase A2; MCP-1, monocyte chemotactic protein-1; oxLDL, oxidized low-density lipoprotein; PARs, protease-activated receptors; SAA, serum amyloid A; sCD40L, soluble CD40 ligand; TNF-α, tumor necrosis factor-α; WBC, white blood cell.
Serum Amyloid A.
Serum amyloid A is an acute-phase reactant, an apolipoprotein associated with HDL, that is predominantly produced by the liver. Nevertheless, it is also implicated in chronic inflammatory diseases (rheumatoid arthritis, atherosclerosis). Serum amyloid A has been shown to have a number of potentially proatherogenic effects that can lead to the destabilization of an atherosclerotic plaque: promotion of chemotaxis for monocytes and neutrophils; stimulation of the production of other proinflammatory cytokines, such as IL-1β and TNF-α; reduction of reverse cholesterol transport; and induction of the MMPs.144 Serum amyloid A has also been shown to promote thrombosis by increasing tissue factor, leading again to the destabilization of atherosclerotic plaques.144 Serum amyloid A was found to be marginally associated with cardiovascular disease,145 especially with extension of CAD in women.146
White Blood Cell Count.
In peripheral blood, the white blood cell count is usually increased in inflammatory and infectious conditions and could be affected in plaque inflammation as well. A higher leukocyte count has been associated with a greater cardiovascular risk. In a meta-analysis of seven prospective studies comparing the top with the bottom third of the value distribution, the relative risk of coronary disease was 1.4 (95% CI, 1.3 to 1.5),147 thus rendering leukocytes a valuable marker. Consistent with this, the decrease in the leukocyte count, achieved by pravastatin in patients with CAD, has been found to be an independent predictor of inhibition of the progression of coronary atherosclerosis.148
Cytokines.
Cytokines are key regulatory glycoproteins allied to inflammatory and immunologic processes that modulate all aspects of vascular inflammation. They are intimately associated with atherogenesis and modulate plaque morphology and stabilization. Many cytokines have been implicated in atheroma formation and complication. Several studies have established the role of IL-6 as an independent predictor of PAD in community screening, irrespective of ethnicity.149–151 IL-6 enhances cell adhesion molecule expression and the production of acute-phase reactants such as CRP and TNF-α by hepatocytes. Exogenous administration of IL-18 to mice enhances atherosclerotic lesions,152 whereas inhibition of IL-18 results in a reduction in atherosclerosis.153 IL-18 has a bearing on the progression and stability of human atherosclerotic plaque.154 TNF-α is involved in the progression of atherosclerosis from the initial stages of intimal thickening to the subsequent vessel occlusion. It stimulates expression of selectin and adhesion molecules and production of MMP-1, MMP-9, MMP-11, and MMP-13 in the endothelium, VSMCs, and macrophages.155 Locally within the atheroma, it increases expression of tissue factor, a potent thrombogenic protein.156 Finally, MCP-1 is the most widely studied chemokine in PAD and CAD,157 and it is consistently expressed in both animal and human atherosclerotic plaque.
Endothelial Adhesion Molecules.
Induction of endothelial adhesion molecules (ICAM-1, P-selectin) is considered one of the earliest steps in atherogenesis. Adhesion of leukocytes to the vascular endothelium and subsequent migration into the intima are key events in the atherosclerotic process. Several cytokines induce expression of ICAM-1, VCAM-1, and selectins by endothelial and smooth muscle cells and promote leukocyte adhesion at the site of the vascular lesion.158
Circulating Soluble CD40 Ligand.
Largely derived from activated platelets, circulating soluble CD40 ligand (sCD40L) can trigger an inflammatory reaction in vascular endothelial cells by the secretion of cytokines and chemokines. Interaction of membrane-bound CD40L and sCD40L with the CD40 receptor leads to the release of matrix MMPs and subsequent destabilization of the plaque.159 Elevated plasma concentrations of sCD40L at baseline predict a subsequent increased risk for future cardiovascular events in apparently healthy women and stable angina patients.160
Protease-Activated Receptors.
Members of the G protein–coupled receptors, protease-activated receptors mediate the cellular effects of thrombin. Protease-activated receptors contribute to the proinflammatory phenotype observed in endothelial dysfunction, and their upregulation in VSMCs seems to be a key element in the pathogenesis of atherosclerosis and restenosis.161
Lipoprotein-Associated Phospholipase A2.
Lipoprotein-associated phospholipase A2 is a proinflammatory enzyme secreted by macrophages and leukocytes. Its activity is associated with a risk for CAD and ischemic stroke.162
[/not-level-membership-for-surgery-category]
