449 Parkinson’s Disease and Other Movement Disorders
PARKINSON’S DISEASE AND RELATED DISORDERS
Parkinson’s disease (PD) is the second commonest neurodegenerative disease, exceeded only by Alzheimer’s disease (AD). Its cardinal clinical features were first described by the English physician James Parkinson in 1817. It is noteworthy that James Parkinson was a general physician who captured the essence of this condition based on a visual inspection of a mere handful of patients. It is estimated that approximately 1 million persons in the United States, 1 million in Western Europe, and 5 million worldwide suffer from this disorder. PD affects men and women of all races, all occupations, and all countries. The mean age of onset is about 60 years. The frequency of PD increases with aging, but cases can be seen in patients in their 20s and even younger. Based on the aging of the population and projected demographics, it is estimated that the prevalence of the disease will dramatically increase in the next several decades.
Clinically, PD is characterized by rest tremor, rigidity, bradykinesia (slowing), and gait impairment, known as the “cardinal features” of the disease. Additional features can include freezing of gait, postural instability, speech difficulty, autonomic disturbances, sensory alterations, mood disorders, sleep dysfunction, cognitive impairment, and dementia (Table 449-1).
|
CLINICAL FEATURES OF PARKINSON’S DISEASE |
Pathologically, the hallmark features of PD are degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc), reduced striatal dopamine, and intracytoplasmic proteinaceous inclusions known as Lewy bodies that primarily contain the protein alpha synuclein (Fig. 449-1). While interest has primarily focused on the dopamine system, neuronal degeneration with inclusion body formation can also affect cholinergic neurons of the nucleus basalis of Meynert (NBM), norepinephrine neurons of the locus coeruleus (LC), serotonin neurons in the raphe nuclei of the brainstem, and neurons of the olfactory system, cerebral hemispheres, spinal cord, and peripheral autonomic nervous system. This “nondopaminergic” pathology is likely responsible for the development of nondopaminergic clinical features listed in Table 449-1 characterized by their lack of satisfactory response to dopaminergic replacement therapy. There is evidence that Lewy body pathology first begins in the peripheral autonomic nervous system, olfactory system, and dorsal motor nucleus of the vagus nerve in the lower brainstem, and then spreads in a predictable and sequential manner to affect the upper brainstem and cerebral hemispheres (Braak staging). These studies suggest that degeneration of dopamine neurons develops in a mid-stage of the disease. Indeed, epidemiologic studies suggest that clinical symptoms reflecting this nondopaminergic degeneration, such as constipation, anosmia, rapid eye movement (REM) behavior sleep disorder, and cardiac denervation, can precede the onset of the classic motor features of PD.
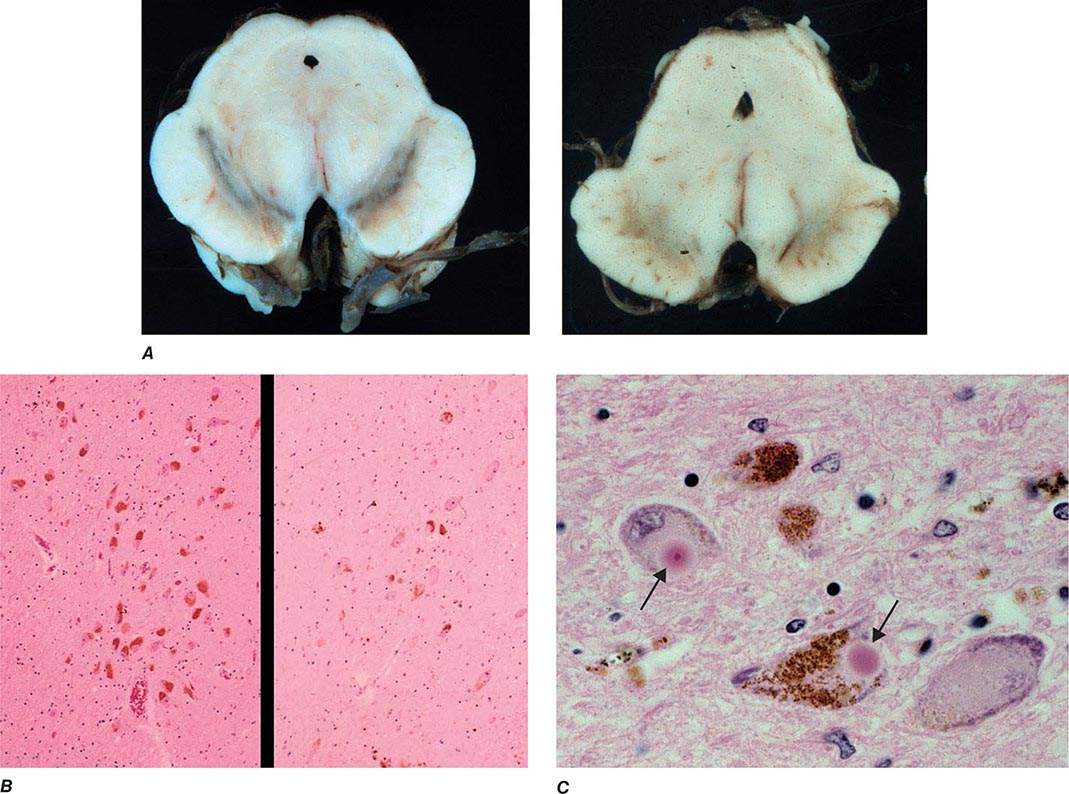
FIGURE 449-1 Pathologic specimens from a patient with Parkinson’s disease (PD) compared to a normal control demonstrating (A) reduction of pigment in SNc in PD (right) versus control (left), (B) reduced numbers of cells in SNc in PD (right) compared to control (left), and (C) Lewy bodies (arrows) within melanized dopamine neurons in PD. SNc, substantia nigra pars compacta.
DIFFERENTIAL DIAGNOSIS
Parkinsonism is a generic term that is used to define a syndrome manifest as bradykinesia with rigidity and/or tremor. It has a differential diagnosis (Table 449-2) that reflects damage to different components of the basal ganglia. The basal ganglia are comprised of a group of subcortical nuclei that include the striatum (putamen and caudate nucleus), subthalamic nucleus (STN), globus pallidus pars externa (GPe), globus pallidus pars interna (GPi), and the SNc (Fig. 449-2). Among the different forms of parkinsonism, PD is the most common (approximately 75% of cases). Historically, PD was diagnosed based on the presence of two of three parkinsonian features (tremor, rigidity, bradykinesia). However, postmortem studies found a 24% error rate when diagnosis was based on these criteria. Clinicopathologic correlation studies subsequently determined that parkinsonism associated with rest tremor, asymmetry, and a good response to levodopa was more likely to predict the correct pathologic diagnosis. With these revised criteria (known as the U.K. Brain Bank Criteria), a clinical diagnosis of PD is confirmed pathologically in as many as 99% of cases. A more complete definition of PD is now needed to incorporate the fact that there is widespread pathology beyond the dopaminergic system, nondopamine and nonmotor clinical features, and a premotor stage of the disease.
|
DIFFERENTIAL DIAGNOSIS OF PARKINSONISM |

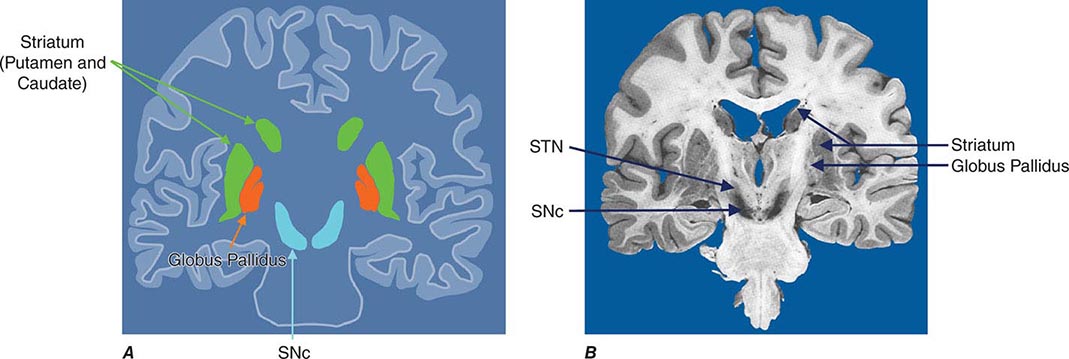
FIGURE 449-2 Basal ganglia nuclei. Schematic (A) and postmortem (B) coronal sections illustrating the various components of the basal ganglia. SNc, substantia nigra pars compacta; STN, subthalamic nucleus.
Imaging of the brain dopamine system in PD with positron emission tomography (PET) or single-photon emission computed tomography (SPECT) shows reduced uptake of striatal dopaminergic markers, particularly in the posterior putamen with relative sparing of the caudate nucleus (Fig. 449-3), reflecting the degeneration of nigrostriatal dopamine neurons. Imaging can be useful in patients where there is diagnostic uncertainty (e.g., dystonic tremor, essential tremor) or in research studies, but is rarely necessary in routine practice because the diagnosis can usually be established on clinical criteria alone. This may change in the future when there is a disease-modifying therapy and it is important to make the diagnosis as early as possible. Genetic testing is not routinely used at present, but can be helpful for identifying at-risk individuals in a research setting. Mutations of the LRRK2 gene (see below) have attracted particular interest because they are the commonest cause of familial PD and are responsible for approximately 1% of typical sporadic cases of the disease. Mutations in LRRK2 are a particularly common cause of PD in Ashkenazi Jews and North African Berber Arabs. The penetrance of the most common LRRK2 mutation ranges from 28 to 74% and is strongly correlated to the age of the carrier, with 50% affected by age 60 years. Mutations in the parkin gene should be considered in patients with onset prior to 40 years of age.
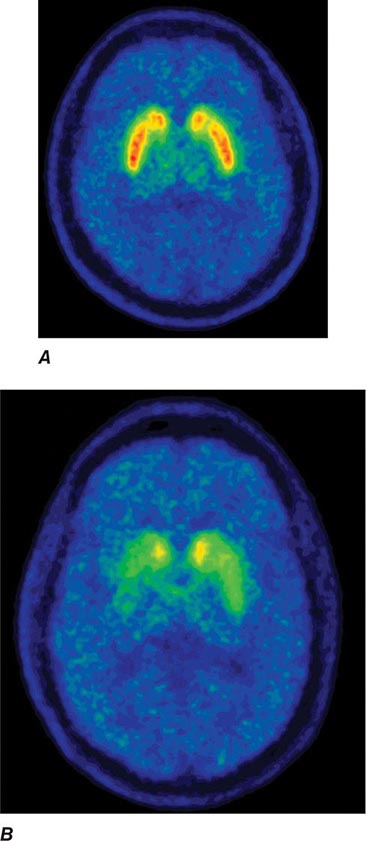
FIGURE 449-3 [11C]Dihydrotetrabenazine positron emission tomography (a marker of VMAT2) in healthy control (A) and Parkinson’s disease (B) patient. Note the reduced striatal uptake of tracer, which is most pronounced in the posterior putamen and tends to be asymmetric. (Courtesy of Dr. Jon Stoessl.)
Atypical and Secondary Parkinsonism Atypical parkinsonism refers to a group of neurodegenerative conditions that usually are associated with more widespread neurodegeneration than is found in PD (often involvement of striatum and/or globus pallidus as well as the SNc). As a group, they present with parkinsonism (rigidity and bradykinesia) but have a slightly different clinical picture than PD, reflecting differences in the underlying pathology. In these conditions, parkinsonism is typically characterized by early speech and gait impairment, absence of rest tremor, no motor asymmetry, poor or no response to levodopa, and an aggressive clinical course. In the early stages, they may show some modest benefit from levodopa and be difficult to distinguish from PD. Pathologically, neurodegeneration typically involves degeneration of the SNc but occurs without Lewy bodies (see below for individual conditions). Neuroimaging of the dopamine system is usually not helpful, because dopamine depletion can be seen in both PD and atypical parkinsonism. By contrast, metabolic imaging of the basal ganglia/thalamus network (using 2-F-deoxiglucose PET) may be helpful, showing a pattern of decreased activity in the GPi with increased activity in the thalamus, the reverse of what is seen in PD.
Multiple-system atrophy (MSA) manifests as a combination of parkinsonian, cerebellar, and autonomic features and can be divided into a predominant parkinsonian (MSA-p) or cerebellar (MSA-c) form. Clinically, MSA is suspected when a patient presents with atypical parkinsonism in conjunction with cerebellar signs and/or early and prominent autonomic dysfunction, usually orthostatic hypotension (Chap. 454). Pathologically, MSA is characterized by degeneration of the SNc, striatum, cerebellum, and inferior olivary nuclei coupled with characteristic glial cytoplasmic inclusions (GCIs) that stain for α-synuclein. Magnetic resonance imaging (MRI) can show pathologic iron accumulation in the striatum on T2-weighted scans, high signal change in the region of the external surface of the putamen (putaminal rim) in MSA-p, or cerebellar and brainstem atrophy (the pontine “hot cross buns” sign [Fig. 454-2]) in MSA-c. Mutations in the CoQ2 gene encoding parahydroxybenzoate-polyprenyl transferase, an enzyme involved in the biosynthesis of coenzyme Q10 (CoQ10), a cofactor of the mitochondrial respiratory chain, have been identified in familial and sporadic forms of MSA.
Progressive supranuclear palsy (PSP) is a form of atypical parkinsonism that is characterized by slow ocular saccades, eyelid apraxia, and restricted eye movements with particular impairment of downward gaze. Patients frequently experience hyperextension of the neck with early gait disturbance and falls. In later stages, speech and swallowing difficulty and cognitive impairment become evident. MRI may reveal a characteristic atrophy of the midbrain with relative preservation of the pons, the “hummingbird sign” on midsagittal images. Pathologically, PSP is characterized by degeneration of the SNc, striatum, subthalamic nucleus, midline thalamic nuclei, and pallidum along with neurofibrillary tangles and inclusions that stain for the tau protein.
Corticobasal ganglionic degeneration is less common and is usually manifest by asymmetric dystonic contractions and clumsiness of one hand coupled with cortical sensory disturbances manifest as apraxia, agnosia, focal limb myoclonus, or alien limb phenomenon (where the limb assumes a position in space without the patient being aware of it). Dementia may occur at any stage of the disease. Both cortical and basal ganglia features are required to make this diagnosis. MRI frequently shows asymmetric cortical atrophy. Pathologic findings include achromatic neuronal degeneration with tau deposits. Because other disorders such as PSP can present with a similar clinical picture, the term corticobasal ganglia syndrome should be used until a precise diagnosis can be confirmed pathologically.
Secondary parkinsonism can occur as a result of drugs, stroke, tumor, infection, or exposure to toxins such as carbon monoxide or manganese. Dopamine-blocking agents such as the neuroleptics are the commonest cause of secondary parkinsonism. These drugs are most widely used in psychiatry, but physicians should be aware that drugs such as metoclopramide and chlorpromazine, which are primarily used to treat gastrointestinal problems, are also neuroleptic agents and common causes of secondary parkinsonism (as well as acute and tardive dyskinesias; see below). Other drugs that can cause secondary parkinsonism include tetrabenazine, calcium channel blockers (flunarizine, cinnarizine), amiodarone, and lithium.
Finally, parkinsonism can be seen as a feature of other degenerative disorders such as Wilson’s disease, Huntington’s disease (especially the juvenile form known as the Westphal variant), dopa-responsive dystonia, and neurodegenerative disorders with brain iron accumulation such as pantothenate kinase (PANK)–associated neurodegeneration (formerly known as Hallervorden-Spatz disease).
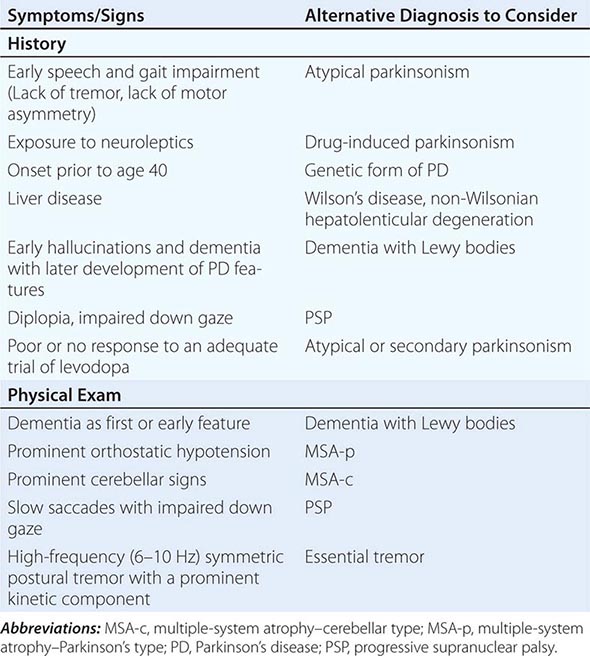
Some features that suggest parkinsonism might be due to a condition other than PD are shown in Table 449-3.
|
FEATURES SUGGESTING AN ATYPICAL OR SECONDARY CAUSE OF PARKINSONISM |
ETIOLOGY AND PATHOGENESIS
Most PD cases occur sporadically (~85–90%) and are of unknown cause. Twin studies suggest that environmental factors likely play an important role in patients older than 50 years, with genetic factors being more important in younger patients. Epidemiologic studies also suggest increased risk with exposure to pesticides, rural living, and drinking well water and reduced risk with cigarette smoking and caffeine. However, no environmental factor has yet been proven to cause typical PD. The environmental hypothesis received support with the demonstration in the 1980s that MPTP (1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine), a byproduct of the illicit manufacture of a heroin-like drug, caused a PD-like syndrome in addicts in northern California. MPTP is transported to the central nervous system, where it is oxidized to form MPP+, a mitochondrial toxin that is selectively taken up by, and damages, dopamine neurons. However, MPTP or MPTP-like compounds have not been linked to sporadic PD.
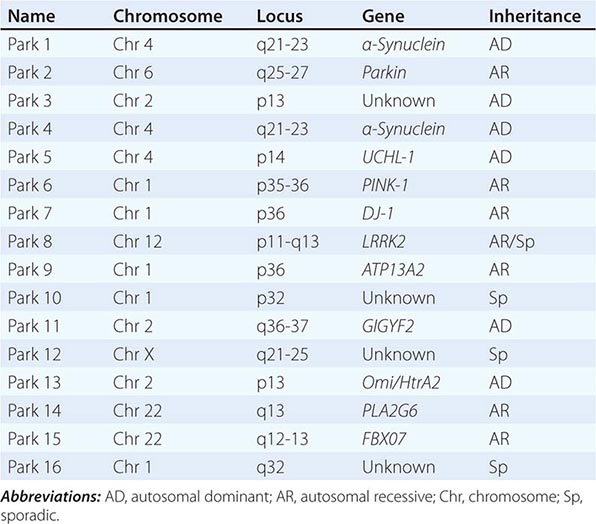
About 10–15% of cases are familial in origin, and multiple specific mutations and gene associations have been identified (Table 449-4). Genetic factors have also been linked to sporadic cases, with several typical PD cases found to carry the LRRK2 mutation, and genome-wide association studies (GWAS) implicating alpha synuclein, tau, and HLA as risk factors. It has been proposed that most cases of PD may be due to a “double hit” involving an interaction between a gene mutation that induces susceptibility coupled with exposure to a toxic environmental factor that may induce epigenetic or somatic DNA alterations. In this scenario, both factors are required for PD to ensue, while the presence of either one alone is not sufficient to cause the disease.
|
GENETIC CAUSES OF PARKINSON’S DISEASE |
Several factors have been implicated in the pathogenesis of cell death in PD, including oxidative stress, inflammation, mitochondrial dysfunction, and proteolytic stress. Recent studies have demonstrated that with aging dopamine neurons switch from sodium to calcium pacing through calcium channels, potentially making these high-energy neurons vulnerable to calcium-mediated neurotoxicity. Whatever the pathogenic mechanism, cell death appears to occur, at least in part, by way of a signal-mediated apoptotic or “suicidal” process. Each of these mechanisms offers a potential target for neuroprotective drugs. However, it is not clear which of these factors is primary, if the mechanism is the same in each individual case, if they act by way of a network such that a cocktail of agents might be required to provide neuroprotection, or if the findings to date merely represent epiphenomena unrelated to the true cause of cell death that remains undiscovered (Fig. 449-4).
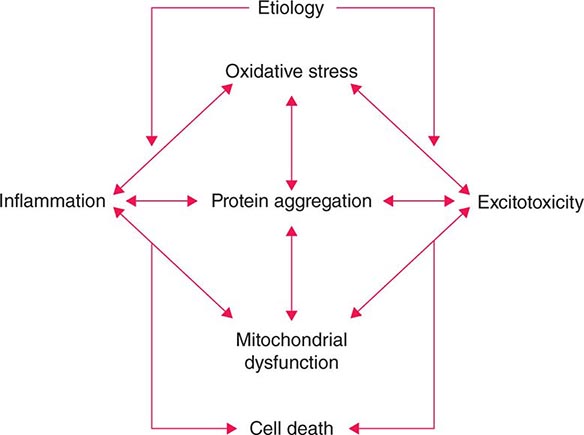
FIGURE 449-4 Schematic representation of how pathogenetic factors implicated in Parkinson’s disease interact in a network manner, ultimately leading to cell death. This figure illustrates how interference with any one of these factors may not necessarily stop the cell death cascade. (Adapted from CW Olanow: Movement Disorders 22:S-335, 2007.)
Gene mutations may not cause all cases of PD, but may be helpful in pointing to specific pathogenic pathways and mechanisms that are central to a neurodegenerative process that might be relevant to all forms of the disease. To date, most interest has focused on pathways implicated by mutations in α-synuclein, LRRK2, and PINK1/Parkin.
Most interest has focused on α-synuclein. Mutations in α-synuclein cause rare familial forms of PD, and α-synuclein constitutes the major component of Lewy bodies in patients with sporadic PD (Fig. 449-1). Furthermore, duplication or triplication of the wild-type α-synuclein can also cause a form of PD, indicating that increased production of the normal protein alone can cause the disease. More recently, Lewy pathology was discovered to have developed in healthy embryonic dopamine neurons that had been implanted into the striatum of PD patients, suggesting that the abnormal protein had transferred from affected cells to healthy unaffected dopamine neurons. Based on these findings, it has been proposed that α-synuclein is a prion and PD is a prion disorder. Here it is proposed that, like the prion protein PrPC, α-synuclein can misfold to form β-rich sheets, generate toxic oligomers and aggregates, polymerize to form amyloid plaques (i.e., Lewy bodies), cause neurodegeneration, and spread to involve unaffected neurons. Indeed, injection of α-synuclein fibrils into the striatum promotes the development of Lewy pathology in host neurons, neurodegeneration, behavioral abnormalities, and the spread of α-synuclein pathology to anatomically connected sites. Further support for this hypothesis comes from the demonstration that inoculation of α-synuclein derived from human Lewy bodies induces widespread Lewy pathology in mice and primates. Collectively, this evidence supports the possibility that neuroprotective therapies for PD might be developed based on inhibiting accumulation or accelerating removal of α-synuclein aggregates.
Mutations in the glucocerebrosidase (GBA) gene associated with Gaucher’s disease numerically represent the most important risk factor for the development of PD. While the responsible mechanism is not precisely known, it is noteworthy that GBA mutations are associated with altered autophagy and lysosomal function and could impair the clearance of α-synuclein.
Six different LRRK2 mutations have been linked to PD, with Gly2019Ser being the most common. The mechanism responsible for cell death with this mutation is not known but is thought to involve changes in kinase activity with altered phosphorylation of target proteins (including autophosphorylation) and possibly lysosomal dysfunction. Kinase inhibitors can block toxicity associated with LRRK2 mutations in laboratory models, and there has been much interest in developing drugs directed at this target. However, kinase inhibitors are likely to be toxic, the physiologic role of LRRK2 is not known, and the large majority of PD patients do not carry a LRRK2 mutation.
Mutations in PINK1 and parkin have implicated mitochondrial dysfunction as a possible cause of PD. Recent studies suggest a role for parkin and PINK1 proteins in the turnover and clearance of damaged mitochondria (mitophagy), and mutations in parkin and PINK1 cause mitochondrial dysfunction in transgenic animals that can be corrected with overexpression of parkin. This is a particularly attractive target because postmortem studies in PD patients show a defect in complex I of the respiratory chain in SNc neurons.
Thus, evidence is accumulating that genetics plays an important role in both familial and “sporadic” forms of PD. It is anticipated that better understanding of the pathways responsible for cell death caused by these mutations will permit the development of more relevant animal models of PD and targets for the development of neuroprotective drugs.
PATHOPHYSIOLOGY OF PD
The classic model of the organization of the basal ganglia in the normal and PD states is provided in Fig. 449-5. With respect to motor function, a series of neuronal circuits or loops link the basal ganglia nuclei with corresponding cortical motor regions in a somatotopic manner. The striatum is the major input region of the basal ganglia, while the GPi and SNr are the major output regions. The input and output regions are connected via direct and indirect pathways that have reciprocal effects on the activity of the output pathway. The output of the basal ganglia provides inhibitory (GABAergic) tone to thalamic and brainstem neurons that in turn connect to motor systems in the cerebral cortex and spinal cord that control motor function. Physiologically, decreased neuronal activity in the GPi/SNr is associated with movement facilitation and vice versa. Dopaminergic projections from SNc neurons serve to modulate neuronal firing and to stabilize the basal ganglia network. The basal ganglia and similar cortical loops are now thought to also play an important role in regulating normal behavioral, emotional, and cognitive functions.
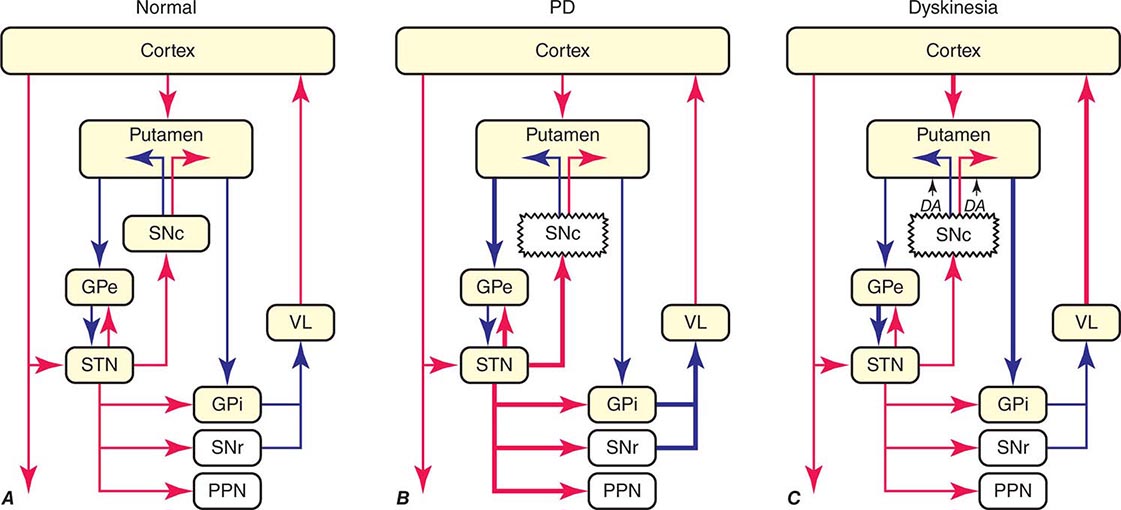
FIGURE 449-5 Basal ganglia organization. Classic model of the organization of the basal ganglia in the normal (A), Parkinson’s disease (PD) (B), and levodopa-induced dyskinesia (C) state. Inhibitory connections are shown as blue arrows and excitatory connections as red arrows. The striatum is the major input region and receives its major input from the cortex. The GPi and SNr are the major output regions, and they project to the thalamocortical and brainstem motor regions. The striatum and GPi/SNr are connected by direct and indirect pathways. This model predicts that parkinsonism results from increased neuronal firing in the STN and GPi and that lesions or DBS of these targets might provide benefit. This concept led to the rationale for surgical therapies for PD. The model also predicts that dyskinesia results from decreased firing of the output regions, resulting in excessive cortical activation by the thalamus. This component of the model is not completely correct because lesions of the GPi ameliorate rather than increase dyskinesia in PD, suggesting that firing frequency is just one of the components that lead to the development of dyskinesia. DBS, deep brain stimulation; GPe, external segment of the globus pallidus; GPi, internal segment of the globus pallidus; PPN, pedunculopontine nucleus; SNc, substantia nigra, pars compacta; SNr, substantia nigra, pars reticulata; STN, subthalamic nucleus; VL, ventrolateral thalamus. (Derived from JA Obeso et al: Trends Neurosci 23:S8, 2000.)
In PD, dopamine denervation with loss of dopaminergic tone leads to increased firing of neurons in the STN and GPi, excessive inhibition of the thalamus, reduced activation of cortical motor systems, and the development of parkinsonian features (Fig. 449-5). The current role of surgery in the treatment of PD is based on this model, which predicted that lesions or high-frequency stimulation of the STN or GPi might reduce this neuronal overactivity and improve PD features.
|
TREATMENT |
PARKINSON’S DISEASE |
LEVODOPA
Since its introduction in the late 1960s, levodopa has been the mainstay of therapy for PD. Experiments in the late 1950s by Carlsson demonstrated that blocking dopamine uptake with reserpine caused rabbits to become parkinsonian; this could be reversed with the dopamine precursor, levodopa. Subsequently, Hornykiewicz demonstrated a dopamine deficiency in the striatum of PD patients and suggested the potential benefit of dopaminergic replacement therapy. Dopamine does not cross the blood-brain barrier (BBB), so clinical trials were initiated with levodopa, a precursor of dopamine. Studies over the course of the next decade confirmed the value of levodopa and revolutionized the treatment of PD.
Levodopa is routinely administered in combination with a peripheral decarboxylase inhibitor to prevent its peripheral metabolism to dopamine and the development of nausea and vomiting due to activation of dopamine receptors in the area postrema that are not protected by the BBB. In the United States, levodopa is combined with the decarboxylase inhibitor carbidopa (Sinemet), whereas in many other countries, it is combined with benserazide (Madopar). Levodopa is also available in controlled-release formulations as well as in combination with a catechol-O-methyltransferase (COMT) inhibitor (see below). Levodopa remains the most effective symptomatic treatment for PD and the gold standard against which new therapies are compared. No current medical or surgical treatment provides antiparkinsonian benefits superior to what can be achieved with levodopa. Levodopa benefits the classic motor features of PD, prolongs independence and employability, improves quality of life, and increases life span. Almost all PD patients experience improvement, and failure to respond to an adequate trial should cause the diagnosis to be questioned.
There are, however, important limitations of levodopa therapy. Acute dopaminergic side effects include nausea, vomiting, and orthostatic hypotension. These are usually transient and can generally be avoided by gradual titration. If they persist, they can be treated with additional doses of a peripheral decarboxylase inhibitor (e.g., carbidopa) or a peripheral dopamine-blocking agent such as domperidone (not available in the United States). More important are motor complications (see below) that develop in the majority of patients treated long-term with levodopa. In addition, the disease continues to progress, and features such as falling, freezing, autonomic dysfunction, sleep disorders, and dementia may emerge with disease progression that are not adequately controlled by levodopa. Indeed, these nondopaminergic features (especially falling and dementia) are the primary source of disability and the main reason for nursing home placement for patients with advanced PD
Levodopa-induced motor complications consist of fluctuations in motor response (“on” episodes when the drug is working and “off” episodes when parkinsonian features return) and involuntary movements known as dyskinesias (Fig. 449-6). When patients initially take levodopa, benefits are long-lasting (many hours) even though the drug has a relatively short half-life (60–90 min). With continued treatment, however, the duration of benefit following an individual dose becomes progressively shorter until it approaches the half-life of the drug. This loss of benefit is known as the wearing-off effect. In more severe cases, patients may experience a delay in turning on (delayed-on) or no response at all to a given dose (no-on). Dyskinesias tend to occur at the time of levodopa peak plasma concentration and maximal clinical benefit (peak-dose dyskinesia). They are usually choreiform in nature but can manifest as dystonic movements, myoclonus, or other movement disorders. They are not troublesome when mild, but can be disabling when severe, and can limit the ability to fully use levodopa to control PD features. In more advanced states, patients may cycle between “on” periods complicated by disabling dyskinesias and “off” periods in which they suffer from severe parkinsonism and painful dystonic postures. Patients may also experience “diphasic dyskinesias,” which occur as the levodopa dose begins to take effect and again as it wears off. These dyskinesias typically consist of transient, stereotypic, rhythmic movements that predominantly involve the lower extremities and are frequently associated with parkinsonism in other body regions. They can be relieved by increasing the dose of levodopa, although higher doses may induce more severe peak-dose dyskinesia.
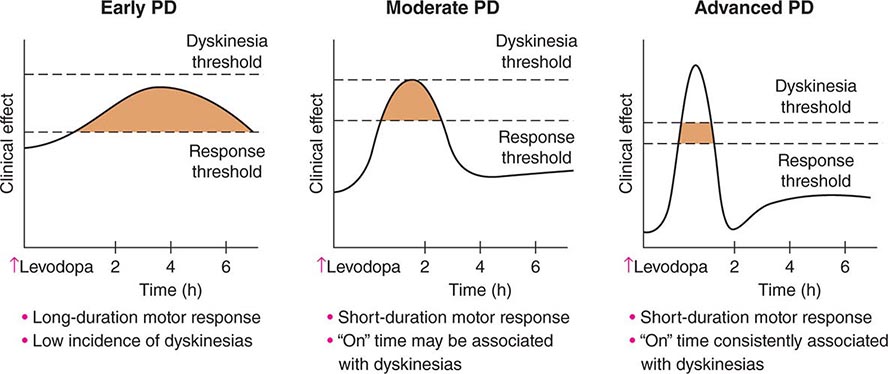
FIGURE 449-6 Changes in motor response associated with chronic levodopa treatment. Levodopa-induced motor complications. Schematic illustration of the gradual shortening of the duration of a beneficial motor response to levodopa (wearing off) and the appearance of dyskinesias complicating “on” time. PD, Parkinson’s disease.
The cause of levodopa-induced motor complications is not precisely known. They are more likely to occur in females, younger individuals with more severe disease, and with the use of higher doses (mg/kg) of levodopa. The classic model of the basal ganglia has been useful for understanding the origin of motor features in PD, but has proved less valuable for understanding levodopa-induced dyskinesias (Fig. 449-5). The model predicts that dopamine replacement might excessively inhibit the pallidal output system, thereby leading to increased thalamocortical activity, enhanced stimulation of cortical motor regions, and the development of dyskinesia. However, lesions of the pallidum that completely destroy its output are associated with amelioration rather than induction of dyskinesia as suggested by the classic model. It is now thought that dyskinesia results from levodopa-induced alterations in the GPi neuronal firing pattern (pauses, bursts, synchrony, etc.) and oscillatory activity, and not simply the firing frequency alone. This in turn leads to the transmission of misinformation from pallidum to thalamus/cortex, resulting in dyskinesia. Surgical lesions or high-frequency stimulation might ameliorate dyskinesia by interfering with (blocking or masking) this abnormal neuronal activity and preventing the transfer of misinformation to motor systems.
Current information suggests that altered neuronal firing patterns and motor complications relate to nonphysiologic levodopa replacement. Striatal dopamine levels are normally maintained at a relatively constant level. In PD, dopamine neurons degenerate and striatal dopamine is dependent on the peripheral availability of levodopa. Intermittent doses of short-acting levodopa result in fluctuating plasma levels because of variability in transit of the drug from the stomach to the duodenum where it is absorbed and the short half-life of the drug. This variability results in exposure of dopamine receptors to pathologically high and low concentrations of dopamine. It has been hypothesized that more continuous delivery of levodopa might prevent the development of motor complications. Indeed, a recent controlled study demonstrated that continuous intraintestinal infusion of levodopa/carbidopa intestinal gel (Duodopa) is associated with significant improvement in “off” time and in “on” time without dyskinesia in advanced PD patients compared with optimized standard oral levodopa.
Behavioral alterations can also be encountered in levodopa-treated patients. A dopamine dysregulation syndrome has been described where patients have a craving for levodopa and take frequent and unnecessary doses of the drug in an addictive manner. PD patients taking high doses of levodopa can develop purposeless, stereotyped behaviors such as the meaningless assembly and disassembly or collection and sorting of objects. This is known as punding, a term taken from the Swedish description of the meaningless behaviors seen in chronic amphetamine users. Hypersexuality and other impulse-control disorders are occasionally encountered with levodopa, although these are more commonly seen with dopamine agonists.
DOPAMINE AGONISTS
Dopamine agonists are a diverse group of drugs that act directly on dopamine receptors. Unlike levodopa, they do not require metabolism to an active product and do not undergo oxidative metabolism. Initial dopamine agonists were ergot derivatives (e.g., bromocriptine, pergolide, cabergoline) and were associated with ergot-related side effects, including cardiac valvular damage. They have largely been replaced by a second generation of nonergot dopamine agonists (e.g., pramipexole, ropinirole, rotigotine). In general, dopamine agonists do not have comparable efficacy to levodopa. They were initially introduced as adjuncts to levodopa to enhance motor function and reduce “off” time in fluctuating patients. Subsequently, it was shown that dopamine agonists, possibly because they are relatively long-acting, are less prone than levodopa to induce dyskinesia. For this reason, many physicians initiate therapy with a dopamine agonist, although supplemental levodopa is eventually required in virtually all patients. Both ropinirole and pramipexole are available as orally administered immediate (tid) and extended-release (qd) formulations. Rotigotine is administered as a once-daily transdermal patch. Apomorphine is a dopamine agonist with efficacy comparable to levodopa, but it must be administered parenterally and has a very short half-life and duration of activity (45 min). It is generally administered by injection as a rescue agent for the treatment of severe “off” episodes. Apomorphine can also be administered by continuous subcutaneous infusion and has been demonstrated to reduce both “off” time and dyskinesia in advanced patients. However, this approach has not been approved in the United States.
Dopamine agonist use is associated with a variety of side effects. Acute side effects are primarily dopaminergic and include nausea, vomiting, and orthostatic hypotension. As with levodopa, these can usually be avoided by slow titration. Side effects associated with chronic use include hallucinations and cognitive impairment. Sedation with sudden unintended episodes of falling asleep while driving a motor vehicle have been reported. Patients should be informed about this potential problem and should not drive when tired. Dopamine agonists can also be associated with impulse-control disorders, including pathologic gambling, hypersexuality, and compulsive eating and shopping. The precise cause of these problems, and why they appear to occur more frequently with dopamine agonists than levodopa, remains to be resolved, but reward systems associated with dopamine and alterations in the ventral striatum and orbitofrontal regions have been implicated. In general, chronic side effects are dose-related and can be avoided or minimized with lower doses. Injections of apomorphine and patch delivery of rotigotine can be complicated by development of skin lesions at sites of administration.
MAO-B INHIBITORS
Inhibitors of monoamine oxidase type B (MAO-B) block central dopamine metabolism and increase synaptic concentrations of the neurotransmitter. Selegiline and rasagiline are relatively selective suicide inhibitors of the MAO-B enzyme. Clinically, MAO-B inhibitors provide antiparkinsonian benefits when used as monotherapy in early disease and reduced “off” time when used as an adjunct to levodopa in patients with motor fluctuations. MAO-B inhibitors are generally safe and well tolerated. They may increase dyskinesia in levodopa-treated patients, but this can usually be controlled by down-titrating the dose of levodopa. Inhibition of the MAO-A isoform prevents metabolism of tyramine in the gut, leading to a potentially fatal hypertensive reaction known as a “cheese effect” because it can be precipitated by foods rich in tyramine such as some cheeses, aged meats, and red wine. Selegiline and rasagiline do not functionally inhibit MAO-A and are not associated with a cheese effect with doses typically used in clinical practice. There are theoretical risks of a serotonin reaction in patients receiving concomitant selective serotonin reuptake inhibitor (SSRI) antidepressants, but these are rarely encountered.
Interest in MAO-B inhibitors has also focused on their potential to have disease-modifying effects. MPTP toxicity can be prevented experimentally by coadministration of an MAO-B inhibitor that blocks its conversion to the toxic pyridinium ion MPP+. MAO-B inhibitors also have the potential to block the oxidative metabolism of dopamine and prevent oxidative stress. In addition, both selegiline and rasagiline incorporate a propargyl ring within their molecular structure that provides antiapoptotic effects in laboratory models. The DATATOP study showed that selegiline significantly delayed the time until the emergence of disability, necessitating the introduction of levodopa, in untreated PD patients. However, it could not be determined whether this was due to a neuroprotective effect that slowed disease progression or a symptomatic effect that merely masked ongoing neurodegeneration. More recently, the ADAGIO study demonstrated that early treatment with rasagiline 1 mg/d, but not 2 mg/d, provided benefits that could not be achieved when treatment with the same drug was initiated at a later time point, consistent with a disease-modifying effect; however, the long-term significance of these findings is uncertain.
COMT INHIBITORS
When levodopa is administered with a decarboxylase inhibitor, it is primarily metabolized in the periphery by COMT. Inhibitors of COMT increase the elimination half-life of levodopa and enhance its brain availability. Combining levodopa with a COMT inhibitor reduces “off” time and prolongs “on” time in fluctuating patients while enhancing motor scores. Two COMT inhibitors have been approved, tolcapone and entacapone. There is also a combination tablet of levodopa, carbidopa, and entacapone (Stalevo).
Side effects of COMT inhibitors are primarily dopaminergic (nausea, vomiting, increased dyskinesia) and can usually be controlled by down-titrating the dose of levodopa by 20–30%. Severe diarrhea has been described with tolcapone, and to a lesser degree with entacapone, and necessitates stopping the medication in 5–10% of individuals. Cases of fatal hepatic toxicity have been reported with tolcapone, and periodic monitoring of liver function is required. This problem has not been encountered with entacapone. Discoloration of urine can be seen with both COMT inhibitors due to accumulation of a metabolite, but it is of no clinical concern.
It has been proposed that initiating levodopa in combination with a COMT inhibitor to enhance its elimination half-life could provide more continuous levodopa delivery if administered at frequent intervals and reduce the risk of motor complications. While this result has been demonstrated in a preclinical MPTP model, and continuous infusion reduces both “off” time and dyskinesia in advanced PD patients, no benefit of initiating levodopa with a COMT inhibitor compared to levodopa alone was detected in early PD patients in the STRIDE-PD study. This may have been because the combination was not administered at frequent enough intervals to provide continuous levodopa availability. For now, the main value of COMT inhibitors continues to be in patients who experience motor fluctuations.
OTHER MEDICAL THERAPIES
Centrally acting anticholinergic drugs such as trihexyphenidyl and benztropine were used historically for the treatment of PD, but they lost favor with the introduction of dopaminergic agents. Their major clinical effect is on tremor, although it is not certain that this benefit is superior to what can be obtained with agents such as levodopa and dopamine agonists. Still, they can be helpful in individual patients with severe tremor. Their use is limited particularly in the elderly, due to their propensity to induce a variety of side effects including urinary dysfunction, glaucoma, and particularly cognitive impairment.
Amantadine also has historical importance. Originally introduced as an antiviral agent, it was appreciated to also have antiparkinsonian effects that are now thought to be due to N-methyl-D-aspartate (NMDA) receptor antagonism. While some physicians use amantadine in patients with early disease for its mild symptomatic effects, it is most widely used as an antidyskinesia agent in patients with advanced PD. Indeed, it is the only oral agent that has been demonstrated in controlled studies to reduce dyskinesia without worsening parkinsonian features, although benefits may be relatively transient. Cognitive impairment is a major concern. Other side effects include livido reticularis and weight gain. Amantadine should always be discontinued gradually because patients can experience withdrawal-like symptoms.
Several new classes of drug are currently being investigated in an attempt to enhance antiparkinsonian effects, reduce off time, and treat or prevent dyskinesia. These include adenosine A2A antagonists, nicotinic agonists, glutamate antagonists, and 5-HT1A agonists.
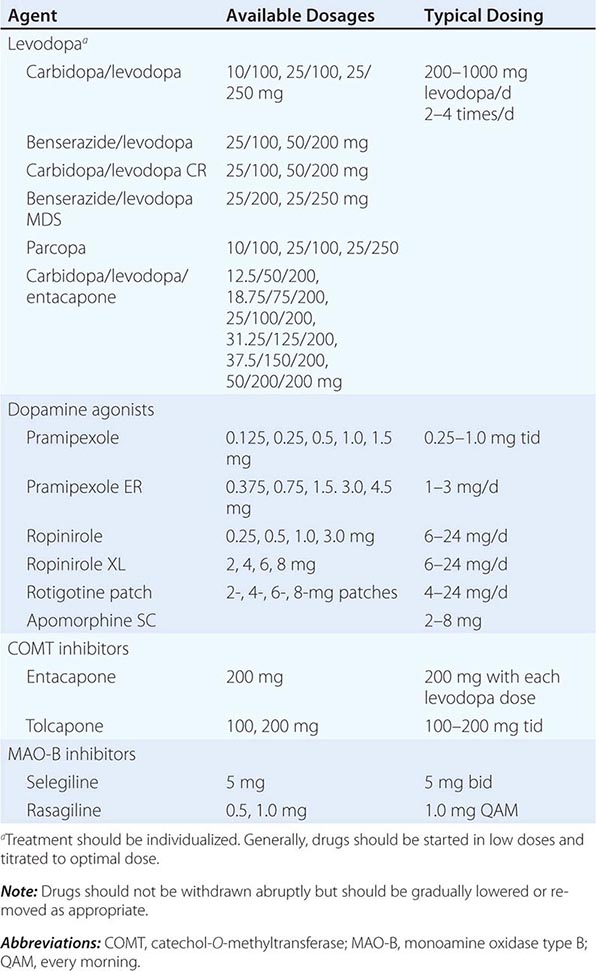
A list of the major drugs and available dosage strengths is provided in Table 449-5.
|
DRUGS COMMONLY USED FOR TREATMENT OF PARKINSON’S DISEASEa |
NEUROPROTECTION
Despite the many therapeutic agents available for the treatment of PD, patients continue to experience disease progression with intolerable disability. A neuroprotective therapy that slows or stops disease progression remains the major unmet therapeutic need in PD. As noted above, trials of certain drugs (e.g., selegiline and rasagiline) have provided positive results consistent with a disease-modifying effect. However, it is not possible to determine if the positive results were due to neuroprotection with slowing of disease progression or confounding symptomatic effects that mask ongoing progression. CoQ10, a mitochondrial bioenhancer and antioxidant, attracted attention with a positive preliminary trial, but this was not replicated in larger double-blind studies.
SURGICAL TREATMENT
Surgical treatments for PD have been used for more than a century. Lesions placed in the motor cortex improved tremor but were associated with motor deficits, and this approach was abandoned. Subsequently, it was appreciated that lesions placed into the ventral intermediate (VIM) nucleus of the thalamus reduced contralateral tremor without inducing hemiparesis, but these lesions did not meaningfully help other more disabling features of PD. In the 1990s, it was shown that lesions placed in the posteroventral portion of the GPi (motor territory) improved rigidity and bradykinesia as well as tremor. Importantly, pallidotomy was also associated with marked improvement in contralateral dyskinesia. This procedure gained favor with greater understanding of the pathophysiology of PD (see above). However, this procedure is not optimal for patients with bilateral disease, because bilateral lesions are associated with side effects such as dysphagia, dysarthria, and impaired cognition, and has largely been replaced by deep brain stimulation (DBS). Unilateral lesions of the STN are associated with a larger antiparkinsonian benefit and reduced levodopa requirement, but there is a concern about the risk of hemiballismus, and this procedure is not commonly performed.
Most surgical procedures for PD performed today use DBS. Here, an electrode is placed into the target area and connected to a stimulator inserted SC over the chest wall. DBS simulates the effects of a lesion without necessitating making a brain lesion. The precise mechanism whereby DBS works is not fully resolved but may act by disrupting the abnormal signal associated with PD and motor complications. The stimulation variables can be adjusted with respect to electrode configuration, voltage, frequency, and pulse duration in order to maximize benefit and minimize adverse side effects. In cases with intolerable side effects, stimulation can be stopped and the system removed. The procedure does not require making a lesion in the brain and is thus suitable for performing bilateral procedures with relative safety.
DBS for PD primarily targets the STN or the GPi. It provides dramatic results, particularly with respect to reducing “off” time and dyskinesias, but does not improve or prevent the development of features that fail to respond to levodopa such as freezing, falling, and dementia. The procedure is thus primarily indicated for patients who suffer disability resulting from severe tremor, or levodopa-induced motor complications that cannot be satisfactorily controlled with drug manipulation. In such patients, DBS has been shown to improve quality of life in comparison to best medical therapy. Side effects can be seen with respect to the surgical procedure (hemorrhage, infarction, infection), the DBS system (infection, lead break, lead displacement, skin ulceration), or the stimulation itself (ocular and speech abnormalities, muscle twitches, paresthesias, depression, and rarely suicide). Recent studies indicate that benefits following DBS of the STN and GPi are comparable, but that GPi stimulation may be associated with a reduced frequency of depression. Although not all PD patients are candidates, the procedure is profoundly beneficial for many. Studies of DBS in early PD patients show benefits in comparison to medical therapy, but this must be weighed against the cost of the procedure and the risk of side effects. Long-term studies demonstrate continued benefits with respect to the classical motor features of PD, but DBS does not prevent the development of nondopaminergic features, which continue to be a source of disability. Studies continue to evaluate the optimal way to use DBS (low- vs high-frequency stimulation, closed systems, etc.). Comparison of DBS to other therapies aimed at improving motor function without causing dyskinesia, such as Duodopa and apomorphine infusions, remains to be performed. Studies are examining additional DBS targets that might benefit gait dysfunction, depression, and cognitive impairment in PD patients.
EXPERIMENTAL THERAPIES FOR PD
There has been considerable scientific and public interest in a number of novel interventions that are being investigated as possible treatments for PD. These include cell-based therapies (such as transplantation of fetal nigral dopamine cells or dopamine neurons derived from stem cells), gene therapies, and trophic factors. Transplant strategies are based on the concept of implanting dopaminergic cells into the striatum to replace degenerating SNc dopamine neurons. Fetal nigral mesencephalic cells have been demonstrated to survive implantation, re-innervate the striatum in an organotypic manner, and restore motor function in PD models. However, two double-blind studies failed to show significant benefit of fetal nigral transplantation in comparison to a sham operation with respect to their primary endpoints. Additionally, grafting of fetal nigral cells is associated with a previously unrecognized form of dyskinesia that persists after lowering or even stopping levodopa. This has been postulated to be related to unregulated release of dopamine from serotonin neurons. In addition, there is evidence that after many years, transplanted healthy embryonic dopamine neurons from unrelated donors develop PD pathology and become dysfunctional, suggesting transfer of α-synuclein from affected to unaffected neurons in a prion-like manner (see discussion above). Perhaps most importantly, it is not clear how replacing dopamine cells alone will improve nondopaminergic features such as falling and dementia, which are the major sources of disability for patients with advanced disease. These same concerns apply to dopamine neurons derived from stem cells, which have not yet been properly tested in PD patients and bear the additional concern of tumors and unanticipated side effects. The short-term future for this technology as a treatment for PD, at least in its current state, is therefore not promising, and there is no scientific basis to warrant routine treatment with stem cells as is being marketed in some countries.
Trophic factors are a series of proteins that enhance neuronal growth and restore function to damaged neurons. There are several different trophic factors that have been demonstrated to have beneficial effects on dopamine neurons in laboratory studies. Glial-derived neurotrophic factor (GDNF) and neurturin have attracted particular attention as possible therapies for PD. However, double-blind trials of intraventricular and intraputaminal infusions of GDNF failed to show benefits compared to placebo in PD patients, possibly because of inadequate delivery of the trophic molecule throughout the target region.
Gene delivery offers the potential of providing widespread delivery throughout a target region and long-term expression of a therapeutic protein with a single procedure. Gene therapy involves placing the DNA of a therapeutic protein into a viral vector that can then be delivered to specific target regions. The DNA of the therapeutic protein is then incorporated into the genome of the host cells and released on a continual basis. The AAV2 virus has been most often used as the viral vector because it does not promote an inflammatory response, is not incorporated into the host genome, and is associated with long-lasting transgene expression. Clinical trials of AAV2 delivery of the trophic factor neurturin showed promising results in open label trials but failed in double-blind trials, possibly because axonal damage in PD prevented retrograde transport of the protein to dopamine neurons in the SNc where it is required to induce upregulation of repair genes required for the trophic response. However, a subsequent double-blind trial of AAV2-neurturin delivered into both the putamen and SNc also failed.
Gene delivery is also being explored as a means of delivering aromatic amino acid decarboxylase with or without tyrosine hydroxylase to the striatum to facilitate dopamine production and glutamic acid decarboxylase to the STN to inhibit overactive neuronal firing. None of these procedures has been established to be effective in PD patients. Furthermore, although gene delivery technology has great potential, this approach also carries the risk of unanticipated side effects, and current approaches directed at the nigrostriatal system do not address the nondopaminergic features of the illness.
MANAGEMENT OF THE NONMOTOR AND NONDOPAMINERGIC FEATURES OF PD
Although PD management has primarily focused on the dopaminergic features of the disease, management of the nondopaminergic features should not be ignored. Some nonmotor features, although not thought to reflect dopaminergic pathology, nonetheless benefit from dopaminergic drugs. For example, problems such as anxiety, panic attacks, depression, sweating, sensory problems, freezing, and constipation all tend to be worse during “off” periods and may improve with better dopaminergic control. Approximately 50% of PD patients suffer depression during the course of the disease, and depression is frequently underdiagnosed and undertreated. Antidepressants should not be withheld, particularly for patients with major depression. Serotonin syndromes have been a theoretical concern with the combined use of SSRIs and MAO-B inhibitors but are rarely encountered. Anxiety can be treated with short-acting benzodiazepines.
Psychosis can be a problem for some PD patients. In contrast to AD, hallucinations are typically visual, formed, and nonthreatening. Importantly, they can limit the use of dopaminergic agents to obtain satisfactory motor control. Psychosis in PD often responds to low doses of atypical neuroleptics and permits higher doses of levodopa to be tolerated. Clozapine is the most effective drug, but it can be associated with agranulocytosis, and regular monitoring is required. For this reason, many physicians start with quetiapine even though it has not been established to be effective in placebo-controlled trials. Hallucinations in PD patients are often a harbinger of a developing dementia.
Dementia in PD (PDD) is common, ultimately affecting as many as 80% of patients. Its frequency increases with aging and, in contrast to AD, primarily affects executive functions and attention, with relative sparing of language, memory, and calculations. When dementia precedes, or develops within 1 year after, the onset of motor dysfunction, it is by convention referred to as dementia with Lewy bodies (DLB; Chap. 448). These patients are particularly prone to have hallucinations and diurnal fluctuations. Pathologically, DLB is characterized by Lewy bodies distributed throughout the cerebral cortex (especially the hippocampus and amygdala) and is often also associated with AD pathology. It is likely that DLB and PDD represent a PD spectrum rather than separate disease entities. Mild cognitive impairment (MCI) frequently precedes the onset of dementia and is a more reliable index of impending dementia in PD than in the general population. Dopaminergic drugs can worsen cognitive function in demented patients and should be stopped or reduced to try and provide a compromise between antiparkinsonian benefit and preserved cognitive function. Drugs are usually discontinued in the following sequence: anticholinergics, amantadine, dopamine agonists, COMT inhibitors, and MAO-B inhibitors. Eventually, patients with cognitive impairment should be managed with the lowest dose of standard levodopa that provides meaningful antiparkinsonian effects and does not worsen mental function. Anticholinesterase agents such as rivastigmine and donepezil reduce the rate of deterioration of measures of cognitive function and can improve attention, but do not typically improve cognitive function in any meaningful way.
Autonomic disturbances are common and frequently require attention. Orthostatic hypotension can be problematic and contribute to falling. Initial treatment should include adding salt to the diet and elevating the head of the bed to prevent overnight sodium natriuresis. Low doses of fludrocortisone (Florinef) or midodrine provide control for most cases. Vasopressin, erythropoietin, and the norepinephrine precursor 3-0-methylDOPS can be used in more severe or refractory cases. If orthostatic hypotension is prominent in early disease, MSA should be considered. Sexual dysfunction can be helped with sildenafil or tadalafil. Urinary problems, especially in males, should be treated in consultation with a urologist to exclude prostate problems. Anticholinergic agents, such as oxybutynin (Ditropan), may be helpful. Constipation can be a very important problem for PD patients. Mild laxatives or enemas can be useful, but physicians should first ensure that patients are drinking adequate amounts of fluid and consuming a diet rich in bulk with green leafy vegetables and bran. Agents that promote gastrointestinal (GI) motility can also be helpful.
Sleep disturbances are common in PD patients, with many experiencing fragmented sleep with excess daytime sleepiness. Restless leg syndrome, sleep apnea, and other sleep disorders should be treated as appropriate. REM behavior disorder (RBD) is a syndrome comprised of violent movements and vocalizations during REM sleep, possibly representing acting out of dreams due to a failure of the normal inhibition of motor movements that typically accompanies REM sleep. Many PD patients have a history of antecedent RBD preceding the onset of the classic motor features of PD, and most cases of RBD go on to develop an α-synucleinopathy (PD or MSA). Low doses of clonazepam (0.5–1 mg at bedtime) are usually effective in controlling this problem. Consultation with a sleep specialist and polysomnography may be necessary to identify and optimally treat sleep problems.
NONPHARMACOLOGIC THERAPY
Gait dysfunction with falling is an important cause of disability in PD. Dopaminergic therapies can help patients whose gait is worse in “off” time, but there are currently no specific therapies available. Canes and walkers may become necessary to increase stability and reduce the risk of falling.
Freezing, where patients suddenly become stuck in place for seconds to minutes as if their feet were glued to the ground, is a major cause of falling. Freezing may occur during “on” or “off” periods. Freezing during “off” periods may respond to dopaminergic therapies, but there are no specific treatments for “on” period freezing. Some patients will respond to sensory cues such as marching in place, singing a song, or stepping over an imaginary line.
Exercise, with a full range of active and passive movements, has been shown to maintain and even improve function for PD patients, and active and passive exercises with full range of motion reduce the risk of arthritis and frozen joints. Some laboratory studies suggest the possibility that exercise might also have neuroprotective effects, but this has not been confirmed in PD. Exercise is generally recommended for all PD patients. It is less clear that physical therapy or specific exercises such as tai chi are required. It is important for patients to maintain social and intellectual activities to the extent possible. Education, assistance with financial planning, social services, and attention to home safety are important elements of the overall care plan. Information is available through numerous PD foundations and on the web, but should be reviewed with physicians to ensure accuracy. The needs of the caregiver should not be neglected. Caring for a person with PD involves a substantial work effort and there is an increased incidence of depression among caregivers. Support groups for patients and caregivers may be useful.
CURRENT MANAGEMENT OF PD
The management of PD should be tailored to the needs of the individual patient, and there is no single treatment approach that is universally accepted and applicable to all individuals. Clearly, if an agent could be demonstrated to have disease-modifying effects, it should be initiated at the time of diagnosis. Indeed, constipation, RBD, and anosmia may represent premotor features of PD and could permit the initiation of a disease-modifying therapy prior to the onset of the classical motor features of the disease. However, no therapy has yet been proved to be disease modifying. For now, physicians must use their judgment in deciding whether or not to introduce rasagiline (see above) or other drugs for their possible disease-modifying effects.
The next important issue to address is when to initiate symptomatic therapy. Several studies now suggest that it may be best to start therapy at the time of diagnosis (or soon after) in order to preserve beneficial compensatory mechanisms and possibly provide functional benefits even in the early stage of the disease. Levodopa remains the most effective symptomatic therapy for PD, and some recommend starting it immediately using low doses (≤400 mg/d), but others prefer to delay levodopa treatment, particularly in younger patients, in order to reduce the risk of inducing motor complications. An alternate approach is to begin with an MAO-B inhibitor and/or a dopamine agonist, and reserve levodopa for later stages when these drugs can no longer provide satisfactory control. In making this decision, the age, degree of disability, and side effect profile of the drug must all be considered. In patients with more severe disability, the elderly, those with cognitive impairment, or those in whom the diagnosis is uncertain, most physicians would initiate therapy with levodopa. Regardless of initial choice, it is important not to deny patients levodopa when they cannot be adequately controlled with alternative medications.
If motor complications develop, patients can initially be treated by manipulating the frequency and dose of levodopa or by combining lower doses of levodopa with a dopamine agonist, a COMT inhibitor, or an MAO-B inhibitor. Amantadine is the only drug that has been demonstrated to treat dyskinesia without worsening parkinsonism, but benefits may be short-lasting, and there are important side effects related to cognitive function. In advanced cases, it may be necessary to consider a surgical therapy such as DBS if the patient is a suitable candidate, but as described above, these procedures have their own set of complications. Continuous intraintestinal infusion of levodopa/carbidopa intestinal gel (Duodopa) appears to offer similar benefits to DBS, but also requires a surgical intervention with potentially serious complications. Continuous infusion of apomorphine is another treatment option and does not require surgery but is associated with potentially troublesome skin nodules. Comparative studies of these approaches in more advanced patients are awaited. There are ongoing efforts aimed at developing a long-acting oral or transdermal formulation of levodopa that mirrors the pharmacokinetic properties of a levodopa infusion. Such a formulation might provide all of the benefits of levodopa without motor complications and avoid the need for polypharmacy and surgical intervention.
A decision tree that considers the various treatment options and decision points for the management of PD is provided in Fig. 449-7.
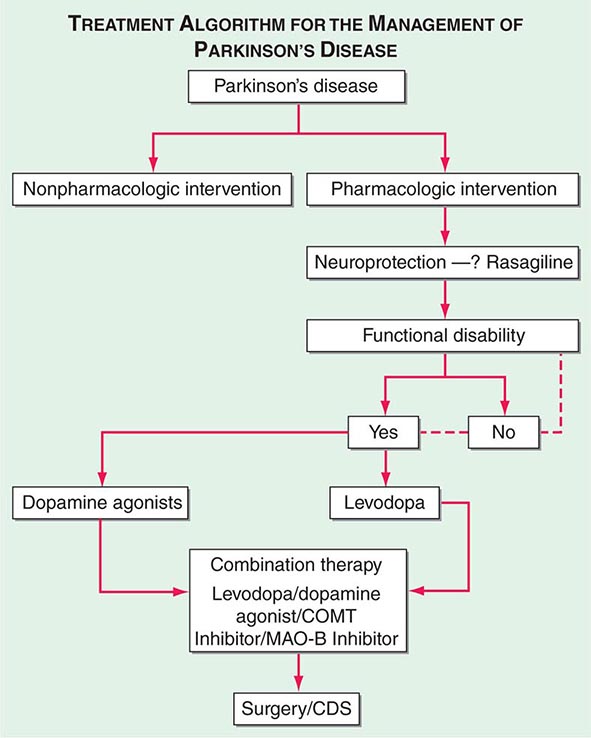
FIGURE 449-7 Treatment options for the management of Parkinson’s disease (PD). Decision points include: (1) Introduction of a neuroprotective therapy: No drug has been established to have or is currently approved for neuroprotection or disease modification, but there are several agents that have this potential based on laboratory and preliminary clinical studies (e.g., rasagiline 1 mg/d, coenzyme Q10 1200 mg/d, the dopamine agonists ropinirole, and pramipexole). (2) When to initiate symptomatic therapy: There is a trend toward initiating therapy at the time of diagnosis or early in the course of the disease because patients may have some disability even at an early stage, and there is the possibility that early treatment may preserve beneficial compensatory mechanisms; however, some experts recommend waiting until there is functional disability before initiating therapy. (3) What therapy to initiate: Many experts favor starting with a monoamine oxidase type B (MAO-B) inhibitor in mildly affected patients because of the good safety profile of the drug and the potential for a disease-modifying effect; dopamine agonists for younger patients with functionally significant disability to reduce the risk of motor complications; and levodopa for patients with more advanced disease, the elderly, or those with cognitive impairment. Recent studies suggest the early employment of polypharmacy using low doses of multiple drugs to avoid side effects associated with high doses of any one agent. (4) Management of motor complications: Motor complications are typically approached with combination therapy to try and reduce dyskinesia and enhance the “on” time. When medical therapies cannot provide satisfactory control, surgical therapies such as DBS or continuous infusion of levodopa/carbidopa intestinal gel can be considered. (5) Nonpharmacologic approaches: Interventions such as exercise, education, and support should be considered throughout the course of the disease. CDS, continuous dopaminergic stimulation; COMT, catechol-O-methyltransferase. (Adapted from CW Olanow et al: Neurology 72:S1, 2009.)
HYPERKINETIC MOVEMENT DISORDERS
Hyperkinetic movement disorders are characterized by involuntary movements unaccompanied by weakness and occurring in isolation or in combination (Table 449-6). The major hyperkinetic movement disorders and the diseases with which they are associated are considered in this section.
|
HYPERKINETIC MOVEMENT DISORDERS |

TREMOR
CLINICAL FEATURES
Tremor consists of alternating contractions of agonist and antagonist muscles in an oscillating, rhythmic manner. It can be most prominent at rest (rest tremor), on assuming a posture (postural tremor), or on actively reaching for a target (kinetic tremor). Tremor is also assessed based on distribution, frequency, and related neurologic dysfunction.
PD is characterized by a resting tremor, essential tremor (ET) by a postural tremor (trying to sustain a posture), and cerebellar disease by an intention or kinetic tremor (on reaching to touch a target). Normal individuals can have a physiologic tremor that typically manifests as a mild, high-frequency (10–12 Hz), postural or action tremor that is usually of no clinical consequence and often is only appreciated with an accelerometer. An enhanced physiologic tremor (EPT) can be seen in up to 10% of the population, often in association with anxiety, fatigue, a metabolic disturbance (e.g., hyperthyroidism, electrolyte abnormalities), drugs (e.g., valproate, lithium), or toxins (e.g., alcohol). Treatment is initially directed to the control of any underlying disorder and, if necessary, can often be improved with a beta blocker.
ESSENTIAL TREMOR
ET is the commonest movement disorder, affecting approximately 5–10 million persons in the United States. It can present in childhood but dramatically increases in prevalence over the age of 70 years. ET is characterized by a high-frequency tremor (6–10 Hz) that predominantly affects the upper extremities. The tremor is most often manifest as a postural or action (kinetic) tremor and, in severe cases, can interfere with functions such as eating and drinking. It is typically bilateral and symmetric but may begin on one side and remain asymmetric. Patients with severe ET can have an intention tremor with overshoot and slowness of movement. Tremor involves the head in ~30% of cases, voice in ~20%, tongue in ~20%, face/jaw in ~10%, and lower limbs in ~10%. The tremor is characteristically improved by alcohol and worsened by stress. Subtle impairment of coordination or tandem walking may be present, and disturbances of hearing, cognition, personality, mood, and olfaction have also been described, but usually the neurologic examination is normal aside from tremor. The major differential is a dystonic tremor (see below) or PD. PD can usually be differentiated from ET based on the presence of bradykinesia, rigidity, micrographia, and other parkinsonian features. However, the examiner should be aware that PD patients may have a postural tremor and ET patients may develop a rest tremor. These typically begin after a latency of a few seconds (emergent tremor). The examiner must take care to differentiate the effect of tremor on measurement of tone in ET from the cogwheel rigidity found in PD.
ETIOLOGY AND PATHOPHYSIOLOGY
The etiology and pathophysiology of ET are not known. Approximately 50% of cases have a positive family history with an autosomal dominant pattern of inheritance. Linkage studies have detected loci at chromosomes 3q13 (ETM-1), 2p22-25 (ETM-2), and 6p23 (ETM-3), but no causative genes have been identified to date. GWAS demonstrated an association with the LINGO1 gene, which is involved in oligodendrocyte differentiation and myelination, particularly in patients with young-onset ET. Recently, a nonsense mutation in the fused in sarcoma (FUS) gene was implicated as a cause of ET in a multigenerational family from Canada; this finding is of particular interest because different mutations in FUS are a known cause of familial amyotrophic lateral sclerosis (Chap. 452). It is likely that there are many other undiscovered genes for ET. The cerebellum and inferior olives have been implicated as possible sites of a “tremor pacemaker” based on the presence of cerebellar signs and increased metabolic activity and blood flow in these regions in some patients. Some pathologic studies have described cerebellar pathology with a loss of Purkinje cells and axonal torpedoes, but these findings are controversial and the precise pathologic correlate of ET remains to be defined.
TREATMENT
Many cases are mild and require no treatment other than reassurance. Occasionally, tremor can be severe and interfere with eating, writing, and activities of daily living. This is more likely to occur as the patient ages and is often associated with a reduction in tremor frequency. Beta blockers and primidone are the standard drug therapies for ET and help in about 50% of cases. Propranolol (20–120 mg daily, given in divided doses) is usually effective at relatively low doses, but higher doses may be effective in some patients. The drug is contraindicated in patients with bradycardia or asthma. Hand tremor tends to be most improved, while head tremor is often refractory. Primidone can be helpful but should be started at low doses (12.5 mg) and gradually increased (125–250 mg tid) to avoid sedation. Benefits have also been reported with gabapentin and topiramate. Botulinum toxin injections may be helpful for limb or voice tremor, but treatment can be associated with secondary muscle weakness. Surgical therapies targeting the VIM nucleus of the thalamus can be very effective for severe and drug-resistant cases.
DYSTONIA
CLINICAL FEATURES
Dystonia is a disorder characterized by sustained (>100 ms) or repetitive involuntary muscle contractions frequently associated with twisting and abnormal postures. Dystonia can range from minor contractions in an individual muscle group to severe and disabling involvement of multiple muscle groups. The frequency is estimated to be 300,000 cases in the United States but is likely to be much higher because many cases are not recognized. Dystonia is often brought out by voluntary movements (action dystonia) and can extend to involve muscle groups and body regions not required for a given action (overflow). It can be aggravated by stress and fatigue and attenuated by relaxation and sensory tricks such as touching the affected body part (geste antagoniste). Dystonia can be classified according to age of onset (childhood vs adult), distribution (focal, multifocal, segmental, or generalized), or etiology (primary or secondary).
PRIMARY DYSTONIAS
At least 16 gene mutations are associated with dystonia and classified as DYT1–DYT16. Idiopathic torsion dystonia (DYT1) or Oppenheim’s dystonia is predominantly a childhood-onset form of dystonia with an autosomal dominant pattern of inheritance that primarily affects Ashkenazi Jewish families. The majority of patients have an age of onset younger than 26 years (mean 14 years). In young-onset patients, dystonia typically begins in the foot or the arm and in 60–70% progresses to involve other limbs as well as the head and neck. In severe cases, patients can suffer disabling postural deformities that compromise mobility. Severity can vary within family members, with some affected relatives having severe disability and others a mild dystonia that may not even be appreciated. Most childhood-onset cases are linked to a mutation in the DYT1 gene located on chromosome 9q34, resulting in a trinucleotide GAG deletion with loss of one of a pair of glutamic acid residues in the protein torsin A. DYT1 mutations are found in 90% of Ashkenazi Jewish patients with DYT1 dystonia and probably relate to a founder effect that occurred about 350 years ago. There is variable penetrance, with only about 30% of gene carriers expressing a clinical phenotype. Why some gene carriers express dystonia and others do not is not known. The function of torsin A is unknown, but it is a member of the AAA+ (ATPase) family that resembles heat-shock proteins and may be related to protein processing and transport. The precise pathology responsible for DYT1 dystonia is not known.
Dopa-responsive dystonia (DRD) or the Segawa variant (DYT5) is a dominantly inherited form of childhood-onset dystonia caused by a mutation in the gene that encodes GTP cyclohydrolase-I, the rate-limiting enzyme for the synthesis of tetrahydrobiopterin. This mutation leads to a defect in the biochemical synthesis of tyrosine hydroxylase, the rate-limiting enzyme in the formation of dopamine. DRD typically presents in early childhood (1–12 years) and is characterized by foot dystonia that interferes with walking. Patients often experience diurnal fluctuations, with worsening of gait as the day progresses and improvement with sleep. DRD is typified by an excellent and sustained response to small doses of levodopa. Some patients may present with parkinsonian features, but can be differentiated from juvenile PD by normal striatal dopamine imaging and the absence of levodopa-induced dyskinesias. DRD may occasionally be confused with cerebral palsy because patients appear to have spasticity, increased reflexes, and Babinski responses (which likely reflect a dystonic contraction rather than an upper motor neuron lesion). Any patient suspected of having a childhood-onset dystonia should receive a trial of levodopa to exclude this treatable condition.
Mutations in the THAP1 gene (DYT6) on chromosome 8p21q22 have been identified in Amish families and are the cause of as many as 25% of cases of non-DYT1 young-onset primary torsion dystonia. These patients are more likely to have dystonia beginning in the brachial and cervical muscles, which later can become generalized and associated with speech impairment. Myoclonic dystonia (DYT11) results from a mutation in the epsilon-sarcoglycan gene on chromosome 7q21. It typically manifests as a combination of dystonia and myoclonic jerks, frequently accompanied by psychiatric disturbances.
FOCAL DYSTONIAS
These are the most common forms of dystonia. They typically present in the fourth to sixth decades and affect women more than men. The major types are as follows: (1) blepharospasm—dystonic contractions of the eyelids with increased blinking that can interfere with reading, watching television, and driving. This can sometimes be so severe as to cause functional blindness. (2) Oromandibular dystonia (OMD)—contractions of muscles of the lower face, lips, tongue, and jaw (opening or closing). Meige’s syndrome is a combination of OMD and blepharospasm that predominantly affects women older than age 60 years. (3) Spasmodic dysphonia—dystonic contractions of the vocal cords during phonation, causing impaired speech. Most cases affect the adductor muscles and cause speech to have a choking or strained quality. Less commonly, the abductors are affected, leading to speech with a breathy or whispering quality. (4) Cervical dystonia—dystonic contractions of neck muscles causing the head to deviate to one side (torticollis), in a forward direction (anterocollis), or in a backward direction (retrocollis). Muscle contractions can be painful and associated with a secondary cervical radiculopathy. (5) Limb dystonias—these can be present in either arms or legs and are often brought out by task-specific activities such as handwriting (writer’s cramp), playing a musical instrument (musician’s cramp), or putting (the yips). Focal dystonias can extend to involve other body regions (about 30% of cases) and are frequently misdiagnosed as psychiatric or orthopedic in origin. Their cause is not known, but genetic factors, autoimmunity, and trauma have been suggested. Focal dystonias are often associated with a high-frequency tremor that resembles ET. Dystonic tremor can usually be distinguished from ET because it tends to occur in conjunction with the dystonic contraction and disappears when the dystonia is relieved.
SECONDARY DYSTONIAS
These develop as a consequence of drugs or other neurologic disorders. Drug-induced dystonia is most commonly seen with neuroleptic drugs or after chronic levodopa treatment in PD patients and may be acute or chronic (see below). Secondary dystonia can also be observed following discrete lesions in the striatum and occasionally in the pallidum, thalamus, cortex, and brainstem due to infarction, anoxia, metabolic disorders, trauma, tumor, infection, or toxins such as manganese or carbon monoxide. In these cases, dystonia often assumes a segmental distribution, but it can be generalized when lesions are bilateral or widespread. More rarely, dystonia can develop following peripheral nerve injury and be associated with features of complex regional pain syndrome (Chap. 454). A psychogenic origin is responsible for some cases of dystonia presenting with fixed, immobile dystonic postures (see below).
DYSTONIA PLUS SYNDROMES
Dystonia may occur as a part of another neurodegenerative conditions such as Huntington’s disease, PD, Wilson’s disease, corticobasilar ganglionic degeneration, PSP, the Lubag form of dystonia-parkinsonism (DYT3), and mitochondrial encephalopathies. In contrast to the primary dystonias, dystonia is usually not the dominant neurologic feature in these conditions.
PATHOPHYSIOLOGY OF DYSTONIA
The pathophysiologic basis of dystonia is not completely known. The phenomenon is characterized by co-contracting synchronous bursts of agonist and antagonist muscle groups with recruitment of muscle groups that are not required for a given movement (overflow). Dystonia is characterized by derangement of the basic physiological principle of action-selection, leading to abnormal recruitment of inappropriate muscles for a given action with inadequate inhibition of this undesired motor activity. Physiologically, loss of inhibition is observed at multiple levels of the motor system (e.g., cortex, brainstem, spinal cord) accompanied by increased cortical excitability and reorganization. Attention has focused on the basal ganglia as the site of origin of at least some types of dystonia because there are alterations in blood flow and metabolism in these structures. Further, lesions of the GPi can induce dystonia, and surgical ablation or DBS of the globus pallidus can ameliorate dystonia. The dopamine system has also been implicated, because dopaminergic therapies can both induce and treat some forms of dystonia. Interestingly, no specific pathology has been consistently identified in primary dystonia.
CHOREAS
HUNTINGTON’S DISEASE (HD)
HD is a progressive, fatal, highly penetrant autosomal dominant disorder characterized by motor, behavioral, oculomotor, and cognitive dysfunction. The disease is named for George Huntington, a family physician who described cases on Long Island, New York, in the nineteenth century. Onset is typically between the ages of 25 and 45 years (range, 3–70 years) with a prevalence of 2–8 cases per 100,000 and an average age at death of 60 years. It is prevalent in Europe, North and South America, and Australia but is rare in African blacks and Asians. HD is characterized by rapid, nonpatterned, semipurposeful, involuntary choreiform movements, and for this reason was formerly referred to as Huntington’s chorea. In the early stages, the chorea tends to be focal or segmental, but progresses over time to involve multiple body regions. Dysarthria, gait disturbance, oculomotor abnormalities, behavioral disturbance, and cognitive impairment with dementia are also common features. With advancing disease, there tends to be a reduction in chorea and the emergence of dystonia, rigidity, bradykinesia, and myoclonus. Functional decline is often predicted by progressive weight loss despite adequate calorie intake. In younger patients (~10% of cases), HD can present as an akinetic-rigid or parkinsonian syndrome (Westphal variant). HD patients eventually develop behavioral and cognitive disturbances, and the majority progress to dementia. Depression with suicidal tendencies, aggressive behavior, and psychosis can be prominent features. HD patients may also develop non-insulin-dependent diabetes mellitus and neuroendocrine abnormalities (e.g., hypothalamic dysfunction). A clinical diagnosis of HD can be strongly suspected in cases of chorea with a positive family history, but genetic testing provides the ultimate confirmation of the diagnosis. The disease predominantly affects the striatum. Progressive atrophy of the heads of the caudate nuclei, which form the lateral margins of the lateral ventricles, can be visualized by MRI (Fig. 449-8), but the putamen can be equally or even more severely affected. More diffuse cortical atrophy is seen in the middle and late stages of the disease. Supportive studies include reduced metabolic activity in the caudate nucleus and putamen. Genetic testing can be used to confirm the diagnosis and to detect at-risk individuals in the family, but must be performed with caution and in conjunction with trained counselors, because positive results can worsen depression and generate suicidal reactions. The neuropathology of HD consists of prominent neuronal loss and gliosis in the caudate nucleus and putamen; similar changes are also widespread in the cerebral cortex. Intraneuronal inclusions containing aggregates of ubiquitin and the mutant protein huntingtin are found in the nuclei of affected neurons.
FIGURE 449-8 Huntington’s disease. A. Coronal fluid attenuated inversion recovery (FLAIR) magnetic resonance imaging shows enlargement of the lateral ventricles reflecting typical atrophy (arrows). B. Axial FLAIR image demonstrates abnormal high signal in the caudate and putamen (arrows).
In anticipation of developing neuroprotective therapies, there has been an intensive effort to define the premanifest stage of HD. Subtle motor impairment, cognitive alterations, and imaging changes can be detected in at-risk individuals who later go on to develop the manifest form of the disease. Defining the rate of progression of these features is paramount for future studies of putative disease-modifying therapies.
ETIOLOGY
HD is caused by an increase in the number of polyglutamine (CAG) repeats (>40) in the coding sequence of the huntingtin gene located on the short arm of chromosome 4. The larger the number of repeats, the earlier the disease is manifest. Intermediate forms of the disease with 36–39 repeats are described in some patients, typically with less severe clinical involvement. Acceleration of the process tends to occur, particularly in males, with subsequent generations having larger numbers of repeats and earlier age of disease onset, a phenomenon referred to as anticipation. The gene encodes the highly conserved cytoplasmic protein huntingtin, which is widely distributed in neurons throughout the central nervous system (CNS) but whose function is not known. Models of HD with striatal pathology can be induced by excitotoxic agents such as kainic acid and 3-nitropoprionic acid, which promote calcium entry into the cell and cytotoxicity. Mitochondrial dysfunction has been demonstrated in the striatum and skeletal muscle of symptomatic and presymptomatic individuals. Fragments of the mutant huntingtin protein can be toxic, possibly by translocating into the nucleus and interfering with transcriptional regulation of proteins. Neuronal inclusions found in affected regions in HD may represent a protective mechanism aimed at segregating and facilitating the clearance of these toxic proteins.
HUNTINGTON’S DISEASE–LIKE DISORDERS
A group of rare inherited conditions that can mimic HD, designated HD-like (HDL) disorders, have also been identified. HDL-1, -2, and -4 are autosomal dominant conditions that typically present in adulthood. HDL-1 is due to expansion of an octapeptide repeat in PRNP, the gene encoding the prion protein (Chap. 453e). Thus HDL-1 is properly considered a prion disease. Patients exhibit onset of personality change in the third or fourth decade, followed by chorea, rigidity, myoclonus, ataxia, and epilepsy. HDL-2 manifests in the third or fourth decade with a variety of movement disorders, including chorea, dystonia, or parkinsonism and dementia. Most patients are of African descent. Acanthocytosis can sometimes be seen in these patients, and this condition must be distinguished from neuroacanthocytosis. HDL-2 is caused by an abnormally expanded CTG/CAG trinucleotide repeat expansion in the junctophilin-3 (JPH3) gene. The pathology of HDL-2 consists of intranuclear inclusions immunoreactive for ubiquitin and expanded polyglutamine repeats. HDL-4, the most common condition in this group, is caused by expansion of trinucleotide repeats in TBP, the gene that encodes the TATA box binding protein involved in regulating transcription; this condition is identical to spinocerebellar ataxia (SCA) 17 (Chap. 451e), and most patients present primarily with ataxia rather than chorea. Mutations of the C9Orf gene associated with amyotrophic lateral sclerosis have also been reported in some individuals with an HDL phenotype.
OTHER CHOREAS
Chorea can be seen in a number of additional disorders. Sydenham’s chorea (originally called St. Vitus’s dance) is more common in females and is typically seen in childhood (5–15 years). It often develops in association with prior exposure to group A streptococcal infection and is thought to be autoimmune in nature. It is characterized by the acute onset of choreiform movements and behavioral disturbances. With the reduction in the incidence of rheumatic fever, the incidence of Sydenham’s chorea has fallen, but it can still be seen in developing countries. The chorea generally responds to dopamine-blocking agents, valproic acid, and carbamazepine, but is self-limited, and treatment is generally restricted to those with severe chorea. Chorea may recur in later life, particularly in association with pregnancy (chorea gravidarum) or treatment with sex hormones. Several reports have documented cases of chorea associated with NMDA receptor antibody–positive encephalitis following herpes simplex virus encephalitis.
Chorea-acanthocytosis (neuroacanthocytosis) is a progressive and typically fatal autosomal recessive disorder that is characterized by chorea coupled with red cell abnormalities on peripheral blood smear (acanthocytes). The chorea can be severe and associated with self-mutilating behavior, dystonia, tics, seizures, and a polyneuropathy. Mutations in the VPS13A gene encoding chorein have been described. A phenotypically similar X-linked form of the disorder has been described in older individuals who have reactivity with Kell blood group antigens (McLeod syndrome). A benign hereditary chorea of childhood (BHC1) due to mutations in the gene for thyroid transcription factor 1 and a late-onset benign senile chorea (BHC2) have also been described. It is important to ensure that patients with these types of choreas do not have HD.
Chorea may also occur in association with vascular diseases, hypo- and hyperglycemia, and a variety of infections and degenerative disorders. Systemic lupus erythematosus is the most common systemic disorder that causes chorea, which can last for days to years. Chorea can also be seen with hyperthyroidism, autoimmune disorders including Sjögren’s syndrome, infectious disorders including HIV disease, metabolic alterations, and polycythemia rubra vera; following open-heart surgery in the pediatric population; and in association with many medications (especially anticonvulsants, cocaine, CNS stimulants, estrogens, and lithium). Chorea is commonly seen in association with chronic levodopa treatment (discussed in the section on PD above). Chorea can also be seen in paraneoplastic syndromes associated with anti-CRMP-5 or anti-Hu antibodies (Chap. 122).
HEMIBALLISMUS
Hemiballismus is a violent form of chorea comprised of wild, flinging, large-amplitude movements on one side of the body. Proximal limb muscles tend to be predominantly affected. These movements may affect just one limb (monoballism) or, more exceptionally, both upper or lower limbs (paraballism). The movements may be so severe as to cause exhaustion, dehydration, local injury, and, in extreme cases, death. Fortunately, dopamine-blocking drugs can be very helpful, and importantly, hemiballismus is usually self-limiting and tends to resolve spontaneously after weeks or months. The most common cause is a partial lesion (infarct or hemorrhage) in the STN, but cases can also be seen with lesions in the putamen, thalamus, and parietal cortex. In extreme cases, pallidotomy can be very effective. Interestingly, surgically induced lesions and DBS of the STN in PD patients are usually not associated with hemiballismus.
TICS
A tic is a brief, rapid, recurrent, and seemingly purposeless stereotyped motor contraction. Motor tics can be simple, with movement only affecting an individual muscle group (e.g., blinking, twitching of the nose, jerking of the neck), or complex, with coordinated involvement of multiple muscle groups (e.g., jumping, sniffing, head banging, and echopraxia [mimicking movements]). Phonic (or vocal) tics can also be simple (e.g., grunting) or complex (e.g., echolalia [repeating other people’s words], palilalia [repeating one’s own words], and coprolalia [expression of obscene words]). Patients may also experience sensory tics, composed of unpleasant focal sensations in the face, head, or neck. These can be mild and of little clinical consequence or severe and disabling to the patient.
TOURETTE’S SYNDROME (TS)
TS is a neurobehavioral disorder named after the French neurologist Georges Gilles de la Tourette. It predominantly affects males, and the prevalence is estimated to be 0.03–1.6%, but it is likely that many mild cases do not come to medical attention. TS is characterized by multiple motor tics often accompanied by vocalizations (phonic tics). Patients characteristically can voluntarily suppress tics for short periods of time, but then experience an irresistible urge to express them. Tics vary in intensity and may be absent for days or weeks only to recur, occasionally in a different pattern. Tics tend to present between ages 2 and 15 years (mean 7 years) and often lessen or even disappear in adulthood. Associated behavioral disturbances include anxiety, depression, attention deficit hyperactivity disorder, and obsessive-compulsive disorder. Patients may experience personality disorders, self-destructive behaviors, difficulties in school, and impaired interpersonal relationships. Tics may present in adulthood and can also be seen in association with a variety of other disorders, including PD, HD, trauma, dystonia, drugs (e.g., levodopa, neuroleptics), and toxins.
Etiology and Pathophysiology TS is thought to be a genetic disorder, but no specific gene mutation has been identified. Current evidence supports a complex inheritance pattern, with one or more major genes, multiple loci, low penetrance, and environmental influences. The risk of a family with one affected child having a second is about 25%. The pathophysiology of TS is not known, but alterations in dopamine neurotransmission, opioids, and second-messenger systems have been proposed. Some cases of TS may be the consequence of an autoimmune response to β-hemolytic streptococcal infection (pediatric autoimmune neuropsychiatric disorder associated with streptococcal infection [PANDAS]); however, this entity remains controversial.
MYOCLONUS
Myoclonus is a brief, rapid (<100 ms), shock-like, jerky movement consisting of single or repetitive muscle discharges. Myoclonic jerks can be focal, multifocal, segmental, or generalized and can occur spontaneously, in association with voluntary movement (action myoclonus) or in response to an external stimulus (reflex or startle myoclonus). Negative myoclonus consists of a brief loss of muscle activity (e.g., asterixis in hepatic failure). Myoclonic jerks can be severe and interfere with normal movement or benign and of no clinical consequence as is commonly observed in normal people when waking up or falling asleep (hypnogogic jerks).
Myoclonic jerks differ from tics in that they are not typically repetitive, can interfere with normal voluntary movement, and are not suppressible. They can arise in association with abnormal neuronal discharges in cortical, subcortical, brainstem, or spinal cord regions and can be associated with lesions in each of these regions, particularly in association with hypoxemia (especially following cardiac arrest), encephalopathy, and neurodegeneration. Reversible myoclonus can be seen with metabolic disturbances (renal failure, electrolyte imbalance, hypocalcemia), toxins, and many medications. Essential myoclonus is a relatively benign familial condition characterized by multifocal, very brief, lightning-like movements that are frequently alcohol sensitive. A mutation in the epsilon-sarcoglycan gene has been associated with a variety of myoclonus seen in association with dystonia (myoclonic dystonia).
DRUG-INDUCED MOVEMENT DISORDERS
This important group of movement disorders is primarily associated with drugs that block dopamine receptors (neuroleptics) or central dopaminergic transmission. These drugs are widely used in psychiatry, but it is important to appreciate that drugs used in the treatment of nausea or vomiting (e.g., prochlorperazine [Compazine]) or gastroesophageal disorders (e.g., metoclopramide) are neuroleptic agents. Hyperkinetic movement disorders secondary to neuroleptic drugs can be divided into those that present acutely, subacutely, or after prolonged exposure (tardive syndromes). Dopamine-blocking drugs can also be associated with a reversible parkinsonian syndrome for which anticholinergics are often concomitantly prescribed, but there is concern that this may increase the risk of developing a tardive syndrome.
ACUTE
Dystonia is the most common acute hyperkinetic drug reaction. It is typically generalized in children and focal in adults (e.g., blepharospasm, torticollis, or oromandibular dystonia). The reaction can develop within minutes of exposure and can be successfully treated in most cases with parenteral administration of anticholinergics (benztropine or diphenhydramine), benzodiazepines (lorazepam, clonazepam, or diazepam), or dopamine agonists. The abrupt onset of severe spasms may occasionally be confused with a seizure; however, there is no loss of consciousness, automatisms, or postictal features typical of epilepsy. The acute onset of chorea, stereotypic behavior, and tics may also be seen, particularly following exposure to CNS stimulants such as methylphenidate, cocaine, or amphetamines.
SUBACUTE
Akathisia is the commonest reaction in this category. It consists of motor restlessness with a need to move that is alleviated by movement. Therapy consists of removing the offending agent. When this is not possible, symptoms may be ameliorated with benzodiazepines, anticholinergics, beta blockers, or dopamine agonists.
TARDIVE SYNDROMES
These disorders develop months to years after initiation of neuroleptic treatment. Tardive dyskinesia (TD) is most common and typically presents with choreiform movements involving the mouth, lips, and tongue. In severe cases, the trunk, limbs, and respiratory muscles may also be affected. In approximately one-third of patients, TD remits within 3 months of stopping the drug, and most patients gradually improve over the course of several years. Abnormal movements may also develop or worsen after stopping the offending agent. The movements are often mild and more upsetting to the family than to the patient, but they can be severe and disabling, particularly in the context of an underlying psychiatric disorder. Atypical antipsychotics (e.g., clozapine, risperidone, olanzapine, quetiapine, ziprasidone, and aripiprazole) are thought to be associated with a lower risk of TD in comparison to traditional antipsychotics, although this remains to be established in controlled studies. Younger patients have a lower risk of developing neuroleptic-induced TD, whereas the elderly, females, and those with underlying organic cerebral dysfunction have been reported to be at greater risk. Chronic use is associated with increased risk, and specifically, the U.S. Food and Drug Administration has warned that use of metoclopramide for more than 12 weeks increases the risk of TD. Because TD can be permanent and resistant to treatment, antipsychotics should be used judiciously, atypical neuroleptics should be the preferred agent when possible, and the need for continued use should be regularly monitored.
Treatment primarily consists of stopping the offending agent. If the patient is receiving a traditional antipsychotic and withdrawal is not possible, replacement with an atypical antipsychotic should be tried. Abrupt cessation of a neuroleptic should be avoided because acute withdrawal can induce worsening. TD can persist after withdrawal of antipsychotics and can be difficult to treat. Benefits may occasionally be achieved with valproic acid, anticholinergics, or botulinum toxin injections. In refractory cases, catecholamine depleters such as tetrabenazine may be helpful, but this drug can be associated with dose-dependent sedation and orthostatic hypotension and may induce parkinsonism as a side effect. Other approaches include baclofen (40–80 mg/d), clonazepam (1–8 mg/d), or valproic acid (750–3000 mg/d). In some cases, the abnormal movement is refractory to therapy.
Chronic neuroleptic exposure can also be associated with tardive dystonia, with preferential involvement of axial muscles and characteristic rocking movements of the trunk and pelvis. Tardive dystonia can be more troublesome than tardive dyskinesia and frequently persists despite stopping medication. Valproic acid, anticholinergics, and botulinum toxin may occasionally be beneficial, but patients are frequently refractory to medical therapy. Tardive akathisia, tardive Tourette’s, and tardive tremor syndromes are rare but may also occur after chronic neuroleptic exposure.
Neuroleptic medications can also be associated with a neuroleptic malignant syndrome (NMS). NMS is characterized by the acute or subacute onset of muscle rigidity, elevated temperature, altered mental status, hyperthermia, tachycardia, labile blood pressure, renal failure, and markedly elevated creatine kinase levels. Symptoms typically evolve within days or weeks after initiating the drug. NMS can also be precipitated by the abrupt withdrawal of dopaminergic medications in PD patients. Treatment involves immediate cessation of the offending antipsychotic drug and the introduction of a dopaminergic agent (e.g., a dopamine agonist or levodopa), dantrolene, or a benzodiazepine. Treatment may need to be undertaken in an intensive care setting and include supportive measures such as control of body temperature (antipyretics and cooling blankets), hydration, electrolyte replacement, and control of renal function and blood pressure.
Drugs that have serotonin-like activity (tryptophan, MDMA or “ecstasy,” meperidine) or that block serotonin reuptake can induce a rare, but potentially fatal, serotonin syndrome that is characterized by confusion, hyperthermia, tachycardia, and coma as well as rigidity, ataxia, and tremor. Myoclonus is often a prominent feature, in contrast to NMS, which it resembles. Patients can be managed with propranolol, diazepam, diphenhydramine, chlorpromazine, or cyproheptadine as well as supportive measures.
A variety of drugs can also be associated with parkinsonism (see above) and hyperkinetic movement disorders. Some examples include phenytoin (chorea, dystonia, tremor, myoclonus), carbamazepine (tics and dystonia), tricyclic antidepressants (dyskinesias, tremor, myoclonus), fluoxetine (myoclonus, chorea, dystonia), oral contraceptives (dyskinesia), β-adrenergics (tremor), buspirone (akathisia, dyskinesias, myoclonus), and digoxin, cimetidine, diazoxide, lithium, methadone, and fentanyl (dyskinesias).
PAROXYSMAL DYSKINESIAS
Paroxysmal dyskinesias are a group of rare disorders characterized by episodic, brief involuntary movements that can manifest as various types of hyperkinetic movements, including chorea, dystonia, tremor, and myoclonus. There are two main categories: (1) paroxysmal kinesigenic dyskinesia, where the involuntary movements are triggered by sudden movement, and (2) paroxysmal nonkinesigenic dyskinesias, where the attacks are not induced by movement. There are rare cases of exercise-induced dyskinesia, where attacks are induced by prolonged exercise.
Paroxysmal kinesigenic dyskinesia (PKD) is characterized by brief, self-limited attacks induced by movement onset such as running but also occasionally by unexpected sound or photic stimulation. Attacks may affect one side of the body, last seconds to minutes at a time, and recur several times a day. They usually manifest as dystonic posturing of a limb but may also become generalized. PKD is most commonly familial with an autosomal dominant pattern of inheritance but may also occur secondary to various brain disorders such as multiple sclerosis or hyperglycemia. PKD is more frequent in males (4:1), and the onset is typically in the first or second decade of life. About 70% report sensory symptoms such as tingling or numbness of the affected limb preceding the attack by a few milliseconds. The evolution is relatively benign, and there is a trend toward resolution of the attacks over time. The cause is not known, but a mutation in the proline-rich transmembrane protein 2 (PRRT2) gene that may be involved in neurotransmitter release has now been identified. Treatment with low-dose anticonvulsant therapy such as carbamazepine or phenytoin is advised when the attacks are frequent and interfere with daily life activities and is effective in about 80% of patients. Some clinical features of PKD (abrupt and short-lasting attacks preceded by an “aura”) and its favorable response to anticonvulsant drugs have led to speculation that it is epileptic in origin, but this has not been established.
Paroxysmal nonkinesigenic dyskinesia (PNKD) involves attacks of generalized dyskinesias precipitated by alcohol, caffeine, stress, or fatigue. In comparison to PKD, the episodes have a relatively longer duration (minutes to hours) and are less frequent (one to three per day). PNKD is inherited as autosomal dominant with incomplete penetrance pattern in some 80% of cases. A missense mutation in the myofibrillogenesis regulator (MR-1) gene has been identified in several families. Recognition of the condition and elimination of the underlying precipitating factors, where possible, are the first priority. Tetrabenazine, neuroleptics, dopamine-blocking agents, propranolol, clonazepam, and baclofen may be helpful. Treatment may not be required if the condition is mild and self-limited. Most patients with PNKD do not benefit from anticonvulsant drugs, but some may respond to clonazepam or other benzodiazepines.
RESTLESS LEGS SYNDROME
Restless legs syndrome (RLS) is a neurologic disorder that affects approximately 10% of the adult population (it is rare in Asians) and can cause significant morbidity in some. It was first described in the seventeenth century by an English physician (Thomas Willis), but has only recently been recognized as being a bona fide movement disorder. The four core symptoms required for diagnosis are as follows: an urge to move the legs, usually caused or accompanied by an unpleasant sensation in the legs; symptoms that begin or worsen with rest; partial or complete relief by movement; and worsening during the evening or night.
Symptoms most commonly begin in the legs, but can spread to or even begin in the upper limbs. The unpleasant sensation is often described as a creepy-crawly feeling, paresthesia, or burning. In about 80% of patients, RLS is associated with periodic leg movements (PLMs) during sleep and occasionally while awake. These involuntary movements are usually brief, lasting no more than a few seconds, and recur every 5–90 s. The restlessness and PLMs are a major cause of sleep disturbance in patients, leading to poor-quality sleep and daytime sleepiness.
RLS is a heterogeneous condition. Primary RLS is genetic, and several loci have been found with an autosomal dominant pattern of inheritance, although penetrance may be variable. The mean age of onset in genetic forms is 27 years, although pediatric cases are recognized. The severity of symptoms is variable. Secondary RLS may be associated with pregnancy or a range of underlying disorders, including anemia, ferritin deficiency, renal failure, and peripheral neuropathy. The pathogenesis probably involves disordered dopamine function, which may be peripheral or central, in association with an abnormality of iron metabolism. Diagnosis is made on clinical grounds but can be supported by polysomnography and the demonstration of PLMs. The neurologic examination is normal. Secondary RLS should be excluded, and ferritin levels, glucose, and renal function should be measured.
Most RLS sufferers have mild symptoms that do not require specific treatment. General measures to improve sleep hygiene and quality should be attempted first. If symptoms remain intrusive, low doses of dopamine agonists, e.g., pramipexole (0.25–0.5 mg) or ropinirole (1–2 mg), are given 1–2 h before bedtime. Levodopa can be effective but is frequently associated with augmentation (spread and worsening of restlessness and its appearance earlier in the day) or rebound (reappearance sometimes with worsening of symptoms at a time compatible with the drug’s short half-life). Other drugs that can be effective include anticonvulsants, analgesics, and opiates. Management of secondary RLS should be directed to correcting the underlying disorder; for example, iron replacement for anemia. Iron infusion may also be helpful for severe primary RLS but requires expert supervision.
DISORDERS THAT MAY PRESENT WITH A COMBINATION OF PARKINSONISM AND HYPERKINETIC MOVEMENTS
WILSON’S DISEASE
Wilson’s disease (WD) is an autosomal recessive inherited disorder of copper metabolism that may manifest with neurologic, psychiatric, and liver disorders, alone or in combination. It is caused by mutations in the gene encoding a P-type ATPase. The disease was first comprehensively described by the English neurologist Kinnier Wilson at the beginning of the twentieth century, although at around the same time the German physicians Kayser and Fleischer separately noted the characteristic association of corneal pigmentation with hepatic and neurologic features. WD has a worldwide prevalence of approximately 1 in 30,000, with a gene carrier frequency of 1 in 90. About half of WD patients (especially younger patients) manifest with liver abnormalities. The remainder present with neurologic disease (with or without underlying liver abnormalities), and a small proportion have hematologic or psychiatric problems at disease onset.
Neurologic onset usually manifests in the second decade with tremor and rigidity. The tremor is usually in the upper limbs, bilateral, and asymmetric. Tremor can be on intention or occasionally resting and, in advanced disease, can take on a wing-beating characteristic. Other features include parkinsonism with bradykinesia, dystonia (particularly facial grimacing), dysarthria, and dysphagia. More than half of those with neurologic features have a history of psychiatric disturbances, including depression, mood swings, and overt psychosis. Kayser-Fleischer (KF) rings are seen in 80% of those with hepatic presentations and virtually all with neurologic features. KF rings represent the deposition of copper in Descemet’s membrane around the cornea. They consist of a characteristic grayish rim or circle at the limbus of the cornea and are best detected by slit-lamp examination. Neuropathologic examination is characterized by neurodegeneration and astrogliosis in the basal ganglia, particularly in the striatum.
WD should always be considered in the differential diagnosis of a movement disorder in the first decades of life. Low levels of blood copper and ceruloplasmin and high levels of urinary copper may be present, but normal levels do not exclude the diagnosis. A computed tomography (CT) scan usually reveals generalized brain atrophy in established cases, and ~50% have signal hypointensity in the caudate head, putamen, globus pallidum, substantia nigra, and red nucleus on T2-weighted MRI. However, correlation of imaging changes with clinical features is not good. It is very rare for WD patients with neurologic features not to have KF rings, and therefore when the diagnosis is considered, examination by slit-lamp is essential. Liver biopsy with demonstration of high copper levels remains the gold standard for the diagnosis.
In the absence of treatment, the course is progressive and leads to severe neurologic dysfunction and early death. Treatment is directed at reducing tissue copper levels and maintenance therapy to prevent reaccumulation. There is no clear consensus on treatment, and all patients should be managed in a unit with expertise in WD. Penicillamine is frequently used to increase copper excretion, but it may lead to a worsening of symptoms in the initial stages of therapy. Side effects are common and can to some degree be attenuated by coadministration of pyridoxine. Tetrathiomolybdate blocks the absorption of copper and can be used instead of penicillamine. Trientine and zinc are useful drugs for maintenance therapy. Effective treatment can reverse the neurologic features in most patients, particularly when started early. Some patients stabilize, and a few may still progress, especially those with hepatocerebral disease. KF rings tend to decrease after 3–6 months and disappear by 2 years. Adherence to maintenance therapy is a major challenge in long-term care.
NEURODEGENERATION WITH BRAIN IRON ACCUMULATION
Neurodegeneration with brain iron accumulation (NBIA) represents a group of inherited disorders characterized by iron accumulation in the basal ganglia. Clinically, they can manifest as a progressive neurologic disorder manifesting a variety of features including parkinsonism, dystonia, neuropsychiatric abnormalities, and retinal degeneration. Cognitive disorders and cerebellar dysfunction may also be seen. Presentation is usually in childhood, but adult cases have been described. Multiple genes have been identified to date. Pantothenate kinase–associated neurodegeneration (PKAN) formerly known as Hallervorden-Spatz disease and caused by a mutation in the PANK2 gene is the most common form of NBIA, accounting for about 50% of cases. Onset is usually in early childhood and is manifest as a combination of dystonia, parkinsonism, and spasticity. MRI shows a characteristic low signal abnormality in the center of the globus pallidus on T2-weighted scans known as the “eye of the tiger” sign caused by iron accumulation. Numerous other gene mutations have been described associated with iron accumulation including mutations in PLA2G6, C19orf12, FA2H, ATP13A2, WDR45, FTL, CP, and DCAF17. One must be cautious, however, not to assume that all cases with iron accumulation in the basal ganglia represent an NBIA, because iron accumulation in specific basal ganglia regions is normal, and excess iron accumulation may occur in the basal ganglia region as a consequence of neurodegeneration of multiple causes unrelated to a defect in iron metabolism.
OTHER DISORDERS
Acanthocytosis, some hereditary spinocerebellar atrophies and spastic parapareses, and HD can also present with parkinsonian features associated with involuntary movements. Diagnosis in these cases is best established with genetic testing.
PSYCHOGENIC DISORDERS
Virtually all movement disorders including tremor, tics, dystonia, myoclonus, chorea, ballism, and parkinsonism can be psychogenic in origin. Tremor affecting the upper limbs is the most common psychogenic movement disorder. Psychogenic movements can result from a somatoform or conversion disorder, malingering (e.g., seeking financial gain), or a factitious disorder (e.g., seeking psychological gain). Psychogenic movement disorders are common (estimated to be 2–3% of patients seen in a movement disorder clinic), more frequent in women, disabling for the patient and family, and expensive for society (estimated $20 billion annually). Clinical features suggesting a psychogenic movement disorder include an acute onset and a pattern of abnormal movement that is inconsistent with a known movement disorder. Diagnosis is based on the nonorganic quality of the movement, the absence of findings of an organic disease process, and positive features that specifically point to a psychogenic illness such as variability and distractibility. For example, the magnitude of a psychogenic tremor is increased with attention and diminishes or even disappears when the patient is distracted by being asked to perform a different task or is unaware that he or she is being observed. Other positive features suggesting a psychogenic problem include a tremor frequency that is variable or that entrains with the frequency of a designated movement in the contralateral limb, and a positive response to placebo medication. Associated features can include nonanatomic sensory findings, give-way weakness, astasia-abasia (an odd, gyrating gait; Chap. 32), and multiple somatic complaints with no underlying pathology (somatoform disorder). Comorbid psychiatric problems such as anxiety, depression, and emotional trauma may be present but are not necessary for the diagnosis of a psychogenic movement disorder to be made. Psychogenic movement disorders can occur as an isolated entity or in association with an underlying organic problem. The diagnosis can often be made based on clinical features alone, and unnecessary tests or medications can be avoided. Underlying psychiatric problems may be present and should be identified and treated, but many patients with psychogenic movement disorders have no obvious psychiatric pathology. Psychotherapy and hypnosis may be of value for patients with conversion reaction, and cognitive behavioral therapy may be helpful for patients with somatoform disorders. Patients with hypochondriasis, factitious disorders, and malingering have a poor prognosis.
450 |
Ataxic Disorders |
APPROACH TO THE PATIENT:
Ataxic Disorders
Symptoms and signs of ataxia consist of gait impairment, unclear (“scanning”) speech, visual blurring due to nystagmus, hand incoordination, and tremor with movement. These result from the involvement of the cerebellum and its afferent and efferent pathways, including the spinocerebellar pathways, and the frontopontocerebellar pathway originating in the rostral frontal lobe. True cerebellar ataxia must be distinguished from ataxia associated with vestibular nerve or labyrinthine disease, as the latter results in a disorder of gait associated with a significant degree of dizziness, light-headedness, or the perception of movement (Chap. 28). True cerebellar ataxia is devoid of these vertiginous complaints and is clearly an unsteady gait due to imbalance. Sensory disturbances can also on occasion simulate the imbalance of cerebellar disease; with sensory ataxia, imbalance dramatically worsens when visual input is removed (Romberg sign). Rarely, weakness of proximal leg muscles mimics cerebellar disease. In the patient who presents with ataxia, the rate and pattern of the development of cerebellar symptoms help to narrow the diagnostic possibilities (Table 450-1). A gradual and progressive increase in symptoms with bilateral and symmetric involvement suggests a genetic, metabolic, immune, or toxic etiology. Conversely, focal, unilateral symptoms with headache and impaired level of consciousness accompanied by ipsilateral cranial nerve palsies and contralateral weakness imply a space-occupying cerebellar lesion.
|
ETIOLOGY OF CEREBELLAR ATAXIA |
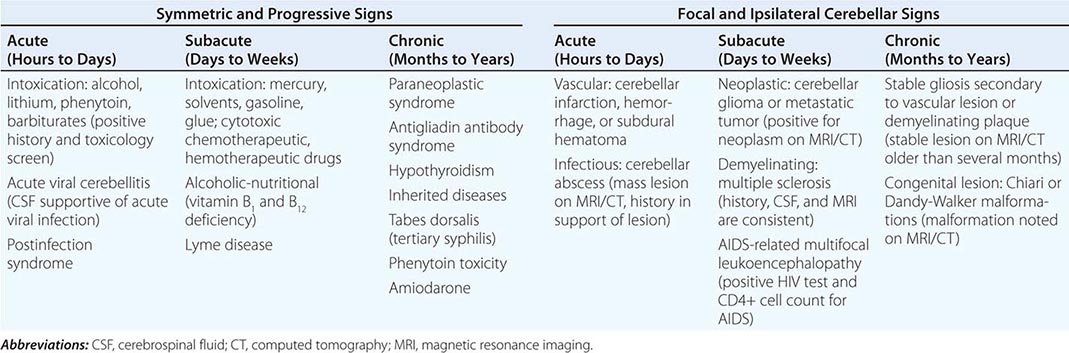
SYMMETRIC ATAXIA
Progressive and symmetric ataxia can be classified with respect to onset as acute (over hours or days), subacute (weeks or months), or chronic (months to years). Acute and reversible ataxias include those caused by intoxication with alcohol, phenytoin, lithium, barbiturates, and other drugs. Intoxication caused by toluene exposure, gasoline sniffing, glue sniffing, spray painting, or exposure to methyl mercury or bismuth are additional causes of acute or subacute ataxia, as is treatment with cytotoxic chemotherapeutic drugs such as fluorouracil and paclitaxel. Patients with a postinfectious syndrome (especially after varicella) may develop gait ataxia and mild dysarthria, both of which are reversible (Chap. 458). Rare infectious causes of acquired ataxia include poliovirus, coxsackievirus, echovirus, Epstein-Barr virus, toxoplasmosis, Legionella, and Lyme disease.
The subacute development of ataxia of gait over weeks to months (degeneration of the cerebellar vermis) may be due to the combined effects of alcoholism and malnutrition, particularly with deficiencies of vitamins B1 and B12. Hyponatremia has also been associated with ataxia. Paraneoplastic cerebellar ataxia is associated with a number of different tumors (and autoantibodies) such as breast and ovarian cancers (anti-Yo), small-cell lung cancer (anti-PQ-type voltage-gated calcium channel), and Hodgkin’s disease (anti-Tr) (Chap. 122). Another paraneoplastic syndrome associated with myoclonus and opsoclonus occurs with breast (anti-Ri) and lung cancers and neuroblastoma. Elevated serum anti-glutamic acid decarboxylase (GAD) antibodies have been associated with a progressive ataxic syndrome affecting speech and gait. For all of these paraneoplastic ataxias, the neurologic syndrome may be the presenting symptom of the cancer. Another immune-mediated progressive ataxia is associated with antigliadin (and antiendomysium) antibodies and the human leukocyte antigen (HLA) DQB1*0201 haplotype; in some affected patients, biopsy of the small intestine reveals villus atrophy consistent with gluten-sensitive enteropathy (Chap. 349). Finally, subacute progressive ataxia may be caused by a prion disorder, especially when an infectious etiology, such as transmission from contaminated human growth hormone, is responsible (Chap. 453e).
Chronic symmetric gait ataxia suggests an inherited ataxia (discussed below), a metabolic disorder, or a chronic infection. Hypothyroidism must always be considered as a readily treatable and reversible form of gait ataxia. Infectious diseases that can present with ataxia are meningovascular syphilis and tabes dorsalis due to degeneration of the posterior columns and spinocerebellar pathways in the spinal cord.
FOCAL ATAXIA
Acute focal ataxia commonly results from cerebrovascular disease, usually ischemic infarction or cerebellar hemorrhage. These lesions typically produce cerebellar symptoms ipsilateral to the injured cerebellum and may be associated with an impaired level of consciousness due to brainstem compression and increased intracranial pressure; ipsilateral pontine signs, including sixth and seventh nerve palsies, may be present. Focal and worsening signs of acute ataxia should also prompt consideration of a posterior fossa subdural hematoma, bacterial abscess, or primary or metastatic cerebellar tumor. Computed tomography (CT) or magnetic resonance imaging (MRI) studies will reveal clinically significant processes of this type. Many of these lesions represent true neurologic emergencies, as sudden herniation, either rostrally through the tentorium or caudal herniation of cerebellar tonsils through the foramen magnum, can occur and is usually devastating. Acute surgical decompression may be required (Chap. 330). Lymphoma or progressive multifocal leukoencephalopathy (PML) in a patient with AIDS may present with an acute or subacute focal cerebellar syndrome. Chronic etiologies of progressive ataxia include multiple sclerosis (Chap. 458) and congenital lesions such as a Chiari malformation (Chap. 456) or a congenital cyst of the posterior fossa (Dandy-Walker syndrome).
THE INHERITED ATAXIAS
These may show autosomal dominant, autosomal recessive, or maternal (mitochondrial) modes of inheritance. A genomic classification (Chap. 451e) has now largely superseded previous ones based on clinical expression alone.
Although the clinical manifestations and neuropathologic findings of cerebellar disease dominate the clinical picture, there may also be characteristic changes in the basal ganglia, brainstem, spinal cord, optic nerves, retina, and peripheral nerves. In large families with dominantly inherited ataxias, many gradations are observed from purely cerebellar manifestations to mixed cerebellar and brainstem disorders, cerebellar and basal ganglia syndromes, and spinal cord or peripheral nerve disease. Rarely, dementia is present as well. The clinical picture may be homogeneous within a family with dominantly inherited ataxia, but sometimes most affected family members show one characteristic syndrome, while one or several members have an entirely different phenotype.
AUTOSOMAL DOMINANT ATAXIAS
The autosomal spinocerebellar ataxias (SCAs) include SCA types 1 through 36, dentatorubropallidoluysian atrophy (DRPLA), and episodic ataxia (EA) types 1 to 7 (Chap. 451e). SCA1, SCA2, SCA3 (Machado-Joseph disease [MJD]), SCA6, SCA7, and SCA17 are caused by CAG triplet repeat expansions in different genes. SCA8 is due to an untranslated CTG repeat expansion, SCA12 is linked to an untranslated CAG repeat, and SCA10 is caused by an untranslated pentanucleotide repeat. The clinical phenotypes of these SCAs overlap. The genotype has become the gold standard for diagnosis and classification. CAG encodes glutamine, and these expanded CAG triplet repeat expansions result in expanded polyglutamine proteins, termed ataxins, that produce a toxic gain of function with autosomal dominant inheritance. Although the phenotype is variable for any given disease gene, a pattern of neuronal loss with gliosis is produced that is relatively unique for each ataxia. Immunohistochemical and biochemical studies have shown cytoplasmic (SCA2), neuronal (SCA1, MJD, SCA7), and nucleolar (SCA7) accumulation of the specific mutant polyglutamine-containing ataxin proteins. Expanded polyglutamine ataxins with more than ~40 glutamines are potentially toxic to neurons for a variety of reasons including: high levels of gene expression for the mutant polyglutamine ataxin in affected neurons; conformational change of the aggregated protein to a β-pleated structure; abnormal transport of the ataxin into the nucleus (SCA1, MJD, SCA7); binding to other polyglutamine proteins, including the TATA-binding transcription protein and the CREB-binding protein, impairing their functions; altering the efficiency of the ubiquitin-proteasome system of protein turnover; and inducing neuronal apoptosis. An earlier age of onset (anticipation) and more aggressive disease in subsequent generations are due to further expansion of the CAG triplet repeat and increased polyglutamine number in the mutant ataxin. The most common disorders are discussed below.
SCA1
SCA1 was previously referred to as olivopontocerebellar atrophy, but genomic data have shown that that entity represents several different genotypes with overlapping clinical features.
Symptoms and Signs SCA1 is characterized by the development in early or middle adult life of progressive cerebellar ataxia of the trunk and limbs, impairment of equilibrium and gait, slowness of voluntary movements, scanning speech, nystagmoid eye movements, and oscillatory tremor of the head and trunk. Dysarthria, dysphagia, and oculomotor and facial palsies may also occur. Extrapyramidal symptoms include rigidity, an immobile face, and parkinsonian tremor. The reflexes are usually normal, but knee and ankle jerks may be lost, and extensor plantar responses may occur. Dementia may be noted but is usually mild. Impairment of sphincter function is common, with urinary and sometimes fecal incontinence. Cerebellar and brainstem atrophy are evident on MRI (Fig. 450-1).
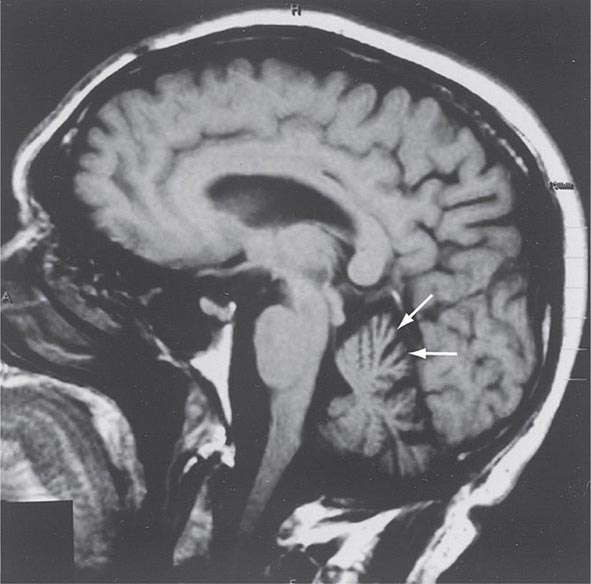
FIGURE 450-1 Sagittal magnetic resonance imaging (MRI) of the brain of a 60-year-old man with gait ataxia and dysarthria due to spinocerebellar ataxia type 1 (SCA1), illustrating cerebellar atrophy (arrows). (Reproduced with permission from RN Rosenberg, P Khemani, in RN Rosenberg, JM Pascual [eds]: Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 5th ed. London, Elsevier, 2015.)
Marked shrinkage of the ventral half of the pons, disappearance of the olivary eminence on the ventral surface of the medulla, and atrophy of the cerebellum are evident on gross postmortem inspection of the brain. Variable loss of Purkinje cells, reduced numbers of cells in the molecular and granular layer, demyelination of the middle cerebellar peduncle and the cerebellar hemispheres, and severe loss of cells in the pontine nuclei and olives are found on histologic examination. Degenerative changes in the striatum, especially the putamen, and loss of the pigmented cells of the substantia nigra may be found in cases with extrapyramidal features. More widespread degeneration in the central nervous system (CNS), including involvement of the posterior columns and the spinocerebellar fibers, is often present.
GENETIC CONSIDERATIONS
![]() SCA1 encodes a gene product, called ataxin-1, which is a novel protein of unknown function. The mutant allele has 40 CAG repeats located within the coding region, whereas alleles from unaffected individuals have ≤36 repeats. A few patients with 38–40 CAG repeats have been described. There is a direct correlation between a larger number of repeats and a younger age of onset for SCA1. Juvenile patients have higher numbers of repeats, and anticipation is present in subsequent generations. Transgenic mice carrying SCA1 developed ataxia and Purkinje cell pathology. Nuclear localization, but not aggregation, of ataxin-1 appears to be required for cell death initiated by the mutant protein.
SCA1 encodes a gene product, called ataxin-1, which is a novel protein of unknown function. The mutant allele has 40 CAG repeats located within the coding region, whereas alleles from unaffected individuals have ≤36 repeats. A few patients with 38–40 CAG repeats have been described. There is a direct correlation between a larger number of repeats and a younger age of onset for SCA1. Juvenile patients have higher numbers of repeats, and anticipation is present in subsequent generations. Transgenic mice carrying SCA1 developed ataxia and Purkinje cell pathology. Nuclear localization, but not aggregation, of ataxin-1 appears to be required for cell death initiated by the mutant protein.
SCA2
Symptoms and Signs Another clinical phenotype, SCA2, has been described in patients from Cuba and India. Cuban patients probably are descendants of a common ancestor, and the population may be the largest homogeneous group of patients with ataxia yet described. The age of onset ranges from 2–65 years, and there is considerable clinical variability within families. Although neuropathologic and clinical findings are compatible with a diagnosis of SCA1, including slow saccadic eye movements, ataxia, dysarthria, parkinsonian rigidity, optic disc pallor, mild spasticity, and retinal degeneration, SCA2 is a unique form of cerebellar degenerative disease.
GENETIC CONSIDERATIONS
![]() The gene in SCA2 families also contains CAG repeat expansions coding for a polyglutamine-containing protein, ataxin-2. Normal alleles contain 15–32 repeats; mutant alleles have 35–77 repeats.
The gene in SCA2 families also contains CAG repeat expansions coding for a polyglutamine-containing protein, ataxin-2. Normal alleles contain 15–32 repeats; mutant alleles have 35–77 repeats.
MACHADO-JOSEPH DISEASE/SCA3
MJD was first described among the Portuguese and their descendants in New England and California. Subsequently, MJD has been found in families from Portugal, Australia, Brazil, Canada, China, England, France, India, Israel, Italy, Japan, Spain, Taiwan, and the United States. In most populations, it is the most common autosomal dominant ataxia.
Symptoms and Signs MJD has been classified into three clinical types. In type I MJD (amyotrophic lateral sclerosis-parkinsonism-dystonia type), neurologic deficits appear in the first two decades and involve weakness and spasticity of extremities, especially the legs, often with dystonia of the face, neck, trunk, and extremities. Patellar and ankle clonus are common, as are extensor plantar responses. The gait is slow and stiff, with a slightly broadened base and lurching from side to side; this gait results from spasticity, not true ataxia. There is no truncal titubation. Pharyngeal weakness and spasticity cause difficulty with speech and swallowing. Of note is the prominence of horizontal and vertical nystagmus, loss of fast saccadic eye movements, hypermetric and hypometric saccades, and impairment of upward vertical gaze. Facial fasciculations, facial myokymia, lingual fasciculations without atrophy, ophthalmoparesis, and ocular prominence are common early manifestations.
In type II MJD (ataxic type), true cerebellar deficits of dysarthria and gait and extremity ataxia begin in the second to fourth decades along with corticospinal and extrapyramidal deficits of spasticity, rigidity, and dystonia. Type II is the most common form of MJD. Ophthalmoparesis, upward vertical gaze deficits, and facial and lingual fasciculations are also present. Type II MJD can be distinguished from the clinically similar disorders SCA1 and SCA2.
Type III MJD (ataxic-amyotrophic type) presents in the fifth to the seventh decades with a pancerebellar disorder that includes dysarthria and gait and extremity ataxia. Distal sensory loss involving pain, touch, vibration, and position senses and distal atrophy are prominent, indicating the presence of peripheral neuropathy. The deep tendon reflexes are depressed to absent, and there are no corticospinal or extrapyramidal findings.
The mean age of onset of symptoms in MJD is 25 years. Neurologic deficits invariably progress and lead to death from debilitation within 15 years of onset, especially in patients with types I and II disease. Usually, patients retain full intellectual function.
The major pathologic findings are variable loss of neurons and glial replacement in the corpus striatum and severe loss of neurons in the pars compacta of the substantia nigra. A moderate loss of neurons occurs in the dentate nucleus of the cerebellum and in the red nucleus. Purkinje cell loss and granule cell loss occur in the cerebellar cortex. Cell loss also occurs in the dentate nucleus and in the cranial nerve motor nuclei. Sparing of the inferior olives distinguishes MJD from other dominantly inherited ataxias.
GENETIC CONSIDERATIONS
![]() The gene for MJD maps to 14q24.3-q32. Unstable CAG repeat expansions are present in the MJD gene coding for a polyglutamine-containing protein named ataxin-3, or MJD-ataxin. An earlier age of onset is associated with longer repeats. Alleles from normal individuals have between 12 and 37 CAG repeats, whereas MJD alleles have 60–84 CAG repeats. Polyglutamine-containing aggregates of ataxin-3 (MJD-ataxin) have been described in neuronal nuclei undergoing degeneration. MJD-ataxin codes for a ubiquitin protease, which is inactive due to expanded polyglutamines. Proteosome function is impaired, resulting in altered clearance of proteins and cerebellar neuronal loss.
The gene for MJD maps to 14q24.3-q32. Unstable CAG repeat expansions are present in the MJD gene coding for a polyglutamine-containing protein named ataxin-3, or MJD-ataxin. An earlier age of onset is associated with longer repeats. Alleles from normal individuals have between 12 and 37 CAG repeats, whereas MJD alleles have 60–84 CAG repeats. Polyglutamine-containing aggregates of ataxin-3 (MJD-ataxin) have been described in neuronal nuclei undergoing degeneration. MJD-ataxin codes for a ubiquitin protease, which is inactive due to expanded polyglutamines. Proteosome function is impaired, resulting in altered clearance of proteins and cerebellar neuronal loss.
SCA6
Genomic screening for CAG repeats in other families with autosomal dominant ataxia and vibratory and proprioceptive sensory loss have yielded another locus. Of interest is that different mutations in the same gene for the α1A voltage-dependent calcium channel subunit (CACNLIA4; also referred to as the CACNA1A gene) at 19p13 result in different clinical disorders. CAG repeat expansions (21–27 in patients; 4–16 triplets in normal individuals) result in late-onset progressive ataxia with cerebellar degeneration. Missense mutations in this gene result in familial hemiplegic migraine. Nonsense mutations resulting in termination of protein synthesis of the gene product yield hereditary paroxysmal cerebellar ataxia or EA. Some patients with familial hemiplegic migraine develop progressive ataxia and also have cerebellar atrophy.
SCA7
This disorder is distinguished from all other SCAs by the presence of retinal pigmentary degeneration. The visual abnormalities first appear as blue-yellow color blindness and proceed to frank visual loss with macular degeneration. In almost all other respects, SCA7 resembles several other SCAs in which ataxia is accompanied by various noncerebellar findings, including ophthalmoparesis and extensor plantar responses. The genetic defect is an expanded CAG repeat in the SCA7 gene at 3p14-p21.1. The expanded repeat size in SCA7 is highly variable. Consistent with this, the severity of clinical findings varies from essentially asymptomatic to mild late-onset symptoms to severe, aggressive disease in childhood with rapid progression. Marked anticipation has been recorded, especially with paternal transmission. The disease protein, ataxin-7, forms aggregates in nuclei of affected neurons, as has also been described for SCA1 and SCA3/MJD.
SCA8
This form of ataxia is caused by a CTG repeat expansion in an untranslated region of a gene on chromosome 13q21. There is marked maternal bias in transmission, perhaps reflecting contractions of the repeat during spermatogenesis. The mutation is not fully penetrant. Symptoms include slowly progressive dysarthria and gait ataxia beginning at ~40 years of age with a range between 20 and 65 years. Other features include nystagmus, leg spasticity, and reduced vibratory sensation. Severely affected individuals are nonambulatory by the fourth to sixth decades. MRI shows cerebellar atrophy. The mechanism of disease may involve a dominant “toxic” effect occurring at the RNA level, as occurs in myotonic dystrophy.
DENTATORUBROPALLIDOLUYSIAN ATROPHY
DRPLA has a variable presentation that may include progressive ataxia, choreoathetosis, dystonia, seizures, myoclonus, and dementia. DRPLA is due to unstable CAG triplet repeats in the open reading frame of a gene named atrophin located on chromosome 12p12-ter. Larger expansions are found in patients with earlier onset. The number of repeats is 49 in patients with DRPLA and ≤26 in normal individuals. Anticipation occurs in successive generations, with earlier onset of disease in association with an increasing CAG repeat number in children who inherit the disease from their father. One well-characterized family in North Carolina has a phenotypic variant known as the Haw River syndrome, now recognized to be due to the DRPLA mutation.
EPISODIC ATAXIA
EA types 1 and 2 are two rare dominantly inherited disorders that have been mapped to chromosomes 12p (a potassium channel gene) for type 1 and 19p for type 2. Patients with EA-1 have brief episodes of ataxia with myokymia and nystagmus that last only minutes. Startle, sudden change in posture, and exercise can induce episodes. Acetazolamide or anticonvulsants may be therapeutic. Patients with EA-2 have episodes of ataxia with nystagmus that can last for hours or days. Stress, exercise, or excessive fatigue may be precipitants. Acetazolamide may be therapeutic and can reverse the relative intracellular alkalosis detected by magnetic resonance spectroscopy. Stop codon, nonsense mutations causing EA-2 have been found in the CACNA1A gene, encoding the α1A voltage-dependent calcium channel subunit (see “SCA6,” above).
AUTOSOMAL RECESSIVE ATAXIAS
Friedreich’s Ataxia This is the most common form of inherited ataxia, comprising one-half of all hereditary ataxias. It can occur in a classic form or in association with a genetically determined vitamin E deficiency syndrome; the two forms are clinically indistinguishable.
SYMPTOMS AND SIGNS Friedreich’s ataxia presents before 25 years of age with progressive staggering gait, frequent falling, and titubation. The lower extremities are more severely involved than the upper ones. Dysarthria occasionally is the presenting symptom; rarely, progressive scoliosis, foot deformity, nystagmus, or cardiopathy is the initial sign.
The neurologic examination reveals nystagmus, loss of fast saccadic eye movements, truncal titubation, dysarthria, dysmetria, and ataxia of trunk and limb movements. Extensor plantar responses (with normal tone in trunk and extremities), absence of deep tendon reflexes, and weakness (greater distally than proximally) are usually found. Loss of vibratory and proprioceptive sensation occurs. The median age of death is 35 years. Women have a significantly better prognosis than men.
Cardiac involvement occurs in 90% of patients. Cardiomegaly, symmetric hypertrophy, murmurs, and conduction defects are reported. Moderate mental retardation or psychiatric syndromes are present in a small percentage of patients. A high incidence of diabetes mellitus (20%) is found and is associated with insulin resistance and pancreatic β-cell dysfunction. Musculoskeletal deformities are common and include pes cavus, pes equinovarus, and scoliosis. MRI of the spinal cord shows atrophy (Fig. 450-2).
FIGURE 450-2 Sagittal magnetic resonance imaging (MRI) of the brain and spinal cord of a patient with Friedreich’s ataxia, demonstrating spinal cord atrophy. (Reproduced with permission from RN Rosenberg, P Khemani, in RN Rosenberg, JM Pascual [eds]: Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 5th ed. London, Elsevier, 2015.)
The primary sites of pathology are the spinal cord, dorsal root ganglion cells, and the peripheral nerves. Slight atrophy of the cerebellum and cerebral gyri may occur. Sclerosis and degeneration occur predominantly in the spinocerebellar tracts, lateral corticospinal tracts, and posterior columns. Degeneration of the glossopharyngeal, vagus, hypoglossal, and deep cerebellar nuclei is described. The cerebral cortex is histologically normal except for loss of Betz cells in the precentral gyri. The peripheral nerves are extensively involved, with a loss of large myelinated fibers. Cardiac pathology consists of myocytic hypertrophy and fibrosis, focal vascular fibromuscular dysplasia with subintimal or medial deposition of periodic acid-Schiff (PAS)-positive material, myocytopathy with unusual pleomorphic nuclei, and focal degeneration of nerves and cardiac ganglia.
GENETIC CONSIDERATIONS
![]() The classic form of Friedreich’s ataxia has been mapped to 9q13-q21.1, and the mutant gene, frataxin, contains expanded GAA triplet repeats in the first intron. There is homozygosity for expanded GAA repeats in >95% of patients. Normal persons have 7–22 GAA repeats, and patients have 200–900 GAA repeats. A more varied clinical syndrome has been described in compound heterozygotes who have one copy of the GAA expansion and the other copy a point mutation in the frataxin gene. When the point mutation is located in the region of the gene that encodes the amino-terminal half of frataxin, the phenotype is milder, often consisting of a spastic gait, retained or exaggerated reflexes, no dysarthria, and mild or absent ataxia.
The classic form of Friedreich’s ataxia has been mapped to 9q13-q21.1, and the mutant gene, frataxin, contains expanded GAA triplet repeats in the first intron. There is homozygosity for expanded GAA repeats in >95% of patients. Normal persons have 7–22 GAA repeats, and patients have 200–900 GAA repeats. A more varied clinical syndrome has been described in compound heterozygotes who have one copy of the GAA expansion and the other copy a point mutation in the frataxin gene. When the point mutation is located in the region of the gene that encodes the amino-terminal half of frataxin, the phenotype is milder, often consisting of a spastic gait, retained or exaggerated reflexes, no dysarthria, and mild or absent ataxia.
Patients with Friedreich’s ataxia have undetectable or extremely low levels of frataxin mRNA, as compared with carriers and unrelated individuals; thus, disease appears to be caused by a loss of expression of the frataxin protein. Frataxin is a mitochondrial protein involved in iron homeostasis. Mitochondrial iron accumulation due to loss of the iron transporter coded by the mutant frataxin gene results in oxidized intramitochondrial iron. Excess oxidized iron results in turn in the oxidation of cellular components and irreversible cell injury.
Two forms of hereditary ataxia associated with abnormalities in the interactions of vitamin E (α-tocopherol) with very-low-density lipoprotein (VLDL) have been delineated. These are abetalipoproteinemia (Bassen-Kornzweig syndrome) and ataxia with vitamin E deficiency (AVED). Abetalipoproteinemia is caused by mutations in the gene coding for the larger subunit of the microsomal triglyceride transfer protein (MTP). Defects in MTP result in impairment of formation and secretion of VLDL in liver. This defect results in a deficiency of delivery of vitamin E to tissues, including the central and peripheral nervous system, as VLDL is the transport molecule for vitamin E and other fat-soluble substitutes. AVED is due to mutations in the gene for α-tocopherol transfer protein (α-TTP). These patients have an impaired ability to bind vitamin E into the VLDL produced and secreted by the liver, resulting in a deficiency of vitamin E in peripheral tissues. Hence, either absence of VLDL (abetalipoproteinemia) or impaired binding of vitamin E to VLDL (AVED) causes an ataxic syndrome. Once again, a genotype classification has proved to be essential in sorting out the various forms of the Friedreich’s disease syndrome, which may be clinically indistinguishable.
Ataxia Telangiectasia • SYMPTOMS AND SIGNS Patients with ataxia telangiectasia (AT) present in the first decade of life with progressive telangiectatic lesions associated with deficits in cerebellar function and nystagmus. The neurologic manifestations correspond to those in Friedreich’s disease, which should be included in the differential diagnosis. Truncal and limb ataxia, dysarthria, extensor plantar responses, myoclonic jerks, areflexia, and distal sensory deficits may develop. There is a high incidence of recurrent pulmonary infections and neoplasms of the lymphatic and reticuloendothelial system in patients with AT. Thymic hypoplasia with cellular and humoral (IgA and IgG2) immunodeficiencies, premature aging, and endocrine disorders such as type 1 diabetes mellitus are described. There is an increased incidence of lymphomas, Hodgkin’s disease, acute T cell leukemias, and breast cancer.
The most striking neuropathologic changes include loss of Purkinje, granule, and basket cells in the cerebellar cortex as well as of neurons in the deep cerebellar nuclei. The inferior olives of the medulla may also have neuronal loss. There is a loss of anterior horn neurons in the spinal cord and of dorsal root ganglion cells associated with posterior column spinal cord demyelination. A poorly developed or absent thymus gland is the most consistent defect of the lymphoid system.
GENETIC CONSIDERATIONS
![]() The gene for AT (the ATM gene) encodes a protein that is similar to several yeast and mammalian phosphatidylinositol-3´-kinases involved in mitogenic signal transduction, meiotic recombination, and cell cycle control. Defective DNA repair in AT fibroblasts exposed to ultraviolet light has been demonstrated. The discovery of ATM permits early diagnosis and identification of heterozygotes who are at risk for cancer (e.g., breast cancer).
The gene for AT (the ATM gene) encodes a protein that is similar to several yeast and mammalian phosphatidylinositol-3´-kinases involved in mitogenic signal transduction, meiotic recombination, and cell cycle control. Defective DNA repair in AT fibroblasts exposed to ultraviolet light has been demonstrated. The discovery of ATM permits early diagnosis and identification of heterozygotes who are at risk for cancer (e.g., breast cancer).
MITOCHONDRIAL ATAXIAS
Spinocerebellar syndromes have been identified with mutations in mitochondrial DNA (mtDNA). Thirty pathogenic mtDNA point mutations and 60 different types of mtDNA deletions are known, several of which cause or are associated with ataxia (Chap. 462e).
GENETIC DIAGNOSTIC LABORATORIES
1. Baylor College of Medicine; Houston, Texas, 1-713-798-6522
http://www.bcm.edu/genetics/index.cfm?pmid=21387
2. GeneDx
3. Transgenomic, 1-877-274-9432
http://www.transgenomic.com/labs/neurology
GLOBAL FEATURES
![]() Ataxias with autosomal dominant, autosomal recessive, X-linked, or mitochondrial forms of inheritance are present on a worldwide basis. Machado-Joseph disease (SCA3) (autosomal dominant) and Friedreich’s ataxia (autosomal recessive) are the most common types in most populations. Genetic markers are now commercially available to precisely identify the genetic mutation for correct diagnosis and also for family planning. Early detection of asymptomatic preclinical disease can reduce or eliminate the inherited form of ataxia in some families on a global, worldwide basis.
Ataxias with autosomal dominant, autosomal recessive, X-linked, or mitochondrial forms of inheritance are present on a worldwide basis. Machado-Joseph disease (SCA3) (autosomal dominant) and Friedreich’s ataxia (autosomal recessive) are the most common types in most populations. Genetic markers are now commercially available to precisely identify the genetic mutation for correct diagnosis and also for family planning. Early detection of asymptomatic preclinical disease can reduce or eliminate the inherited form of ataxia in some families on a global, worldwide basis.
451e |
Classification of the Spinocerebellar Ataxias |
Ataxias with autosomal dominant, autosomal recessive, X-linked, or mitochondrial forms of inheritance are present on a worldwide basis. Machado-Joseph disease (SCA3) (autosomal dominant) and Friedreich’s ataxia (autosomal recessive) are the most common types in most populations. Mutation markers are now commercially available to identify carriers at risk in their families, which allows for precise identification of the genetic mutation for correct diagnosis and also for family planning. Identification of positive mutation carriers with family planning has allowed for early detection of asymptomatic preclinical disease to reduce or eliminate the inherited form of ataxia in specific families on a global, worldwide basis.
|
CLASSIFICATION OF THE SPINOCEREBELLAR ATAXIAS (SCA) |