Chapter 49 Asymptomatic Electrocardiogram Abnormalities
Introduction
Asymptomatic Wolff-Parkinson-White Pattern
Case 1
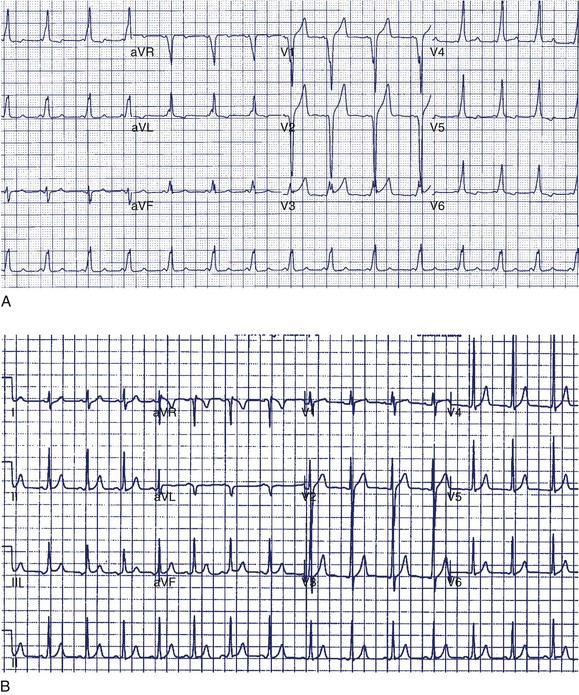
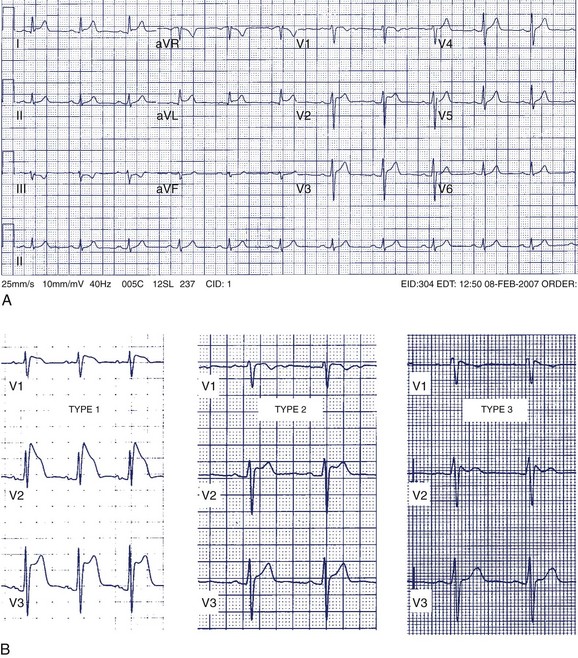
A 14-year-old boy was seen in the emergency department because of noncardiac chest pain following chest trauma during a basketball game. The electrocardiogram (ECG) seen in Figure 49-1 was recorded. He had never been aware of palpitations or syncope. Electrocardiographic pre-excitation was noted.
The most appropriate next step would be to:
The ECG findings now known as ventricular pre-excitation (δ-wave and short P-R interval) were first described in 1930.1 Initial interest was in the arrhythmias associated with the ECG pattern, although it was later observed that many individuals with identical ECG patterns remained asymptomatic and had a benign course. Radiofrequency (RF) ablation of accessory pathways has become the preferred treatment option for patients with Wolff-Parkinson-White (WPW) syndrome because it is curative and has a low rate of associated complications. The question that remains is whether this treatment should be extended as a prophylactic measure to asymptomatic individuals with the WPW pattern on ECG to prevent the small risk of sudden cardiac death (SCD).
Several studies have provided an estimate of the prevalence of the WPW pattern. It is estimated that 0.1% to 0.15% of the population will have ECG manifestations of WPW, of which approximately 50% will never have had symptoms.2 New cases arise at a rate of approximately 0.004% per year.2 However, the true incidence is likely higher, given that the WPW pattern is frequently intermittent and may be missed on ECG and that many individuals never receive an ECG in their lifetime.
Although estimates vary, the incidence of SCD among patients with WPW appears to be at most 0.1% per year.2,3 Munger et al did not report any SCDs in their large (113 patients) retrospective review. No deaths occurred among 293 patients studied in a recent prospective study that examined the usefulness of the EPS and ablation in asymptomatic individuals.4 Autopsy studies delving into the issue are generally limited by the difficulty in finding accessory pathways on routine tissue sections, but most cases of unexpected SCD are related to coronary or other unrecognized structural heart diseases. Nonetheless, SCD in an asymptomatic young individual with WPW pattern is a tragic event that cannot be ignored.
The most common cause of SCD in patients with WPW is ventricular fibrillation (VF) triggered by atrial fibrillation (AF). In these cases, AF is associated with a rapid ventricular response because of the presence of at least one accessory pathway with a short anterograde refractory period. It follows that only those patients who have accessory pathways capable of mediating a rapid ventricular rate are at risk for developing SCD. It has been found that VF rarely occurs when the shortest R-R (SRR) interval during induced AF is greater than 250 ms.5 AF is usually preceded by AV re-entrant tachycardia in these patients, presumably because of the electrophysiological, hemodynamic, and metabolic consequences of supraventricular tachycardia (SVT). Hence, the failure to induce tachycardia would independently predict a good prognosis.6 Interestingly, the presence of multiple accessory pathways in a given individual increases the risk of SCD by an odds ratio of 3.1, presumably because of increased ventricular desynchronization related to multiple pre-excitation wavefronts.7
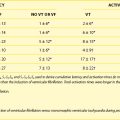
Because of the rarity of the outcome in question (<1/1000 patients/year) it is evident that predicting outcomes with any test is problematic. As one would expect from such a low incidence of SCD, the negative predictive value of the EPS is excellent. Klein et al have categorized the risk of SCD on the basis of the SRR interval during spontaneous or induced AF as follows8:
It must be emphasized that “definite” risk in this stratification is not equivalent to “high” risk. The use of the SRR less than 250 ms as a marker of risk in clinical practice is not as helpful as may be believed; 17% of asymptomatic individuals with WPW will have SRRs in this range, a far greater proportion than that expected to have an event.8 Using an SRR of less than 250 ms, sensitivity of 77.8%, specificity of 48.3%, and predictive accuracy of 18.9% can be obtained.9 Combining the findings of multiple accessory pathways and SRR less than 250 ms lowered the sensitivity (from 88% to 29%) but increased the specificity (from 36% to 92%) and the positive predictive value (from 9% to 22%).7 Pappone et al reported that the combination of a short pathway effective refractory period (ERP) (<250 ms) and inducible tachycardia had sensitivity of 93.7%, specificity of 67.6%, and positive and negative predictive values of 46.9% and 97.3%, respectively, for subsequent arrhythmia.6 Isoproterenol, which is often used in an attempt to induce tachycardia, AF, or both, will cause an even greater percentage of individuals to have an SRR less than 250 ms. Therefore the already poor positive predictive value of an SRR less than 250 ms can be expected to worsen. Despite its limitations, the EPS remains the gold standard for the determination of risk of SCD in asymptomatic patients with WPW.
Noninvasive methods for evaluating the risk for SCD have been described: Holter monitoring to determine whether intermittent pre-excitation is present (therefore making it extremely unlikely for the SRR of the pathway to be <250 ms), sudden loss of pre-excitation on an exercise test, with infusion of ajmaline or procainamide (suggesting a long refractory period for the pathway), and trans-esophageal pacing.10–12 With the exception of trans-esophageal pacing, all of these methods rely on assessments of accessory pathway conduction in sinus rhythm and are imperfect substitutes for a test that, in itself, has limitations. Trans-esophageal pacing has its proponents, but it provides less information than catheter EPS and arguably involves similar risk and discomfort.13
What, then, is the best approach for the management of the asymptomatic patient, given that SCD as a first manifestation of the WPW syndrome is a very rare occurrence? Since serious complications from RF ablation in experienced centers occur infrequently (~1%), it becomes difficult to make a categorical recommendation for or against ablation. Clearly, studying all patients with asymptomatic WPW and ablating those with an SRR less than 250 ms will result in increased morbidity and mortality among patients who may never develop any symptoms. However, an approach that involves not studying any asymptomatic individuals may result in some preventable deaths. Many physicians may prefer to accept a small procedural risk with a finite risk interval over a longer term risk of potentially lethal arrhythmias.14,15
Finally, two other factors may influence the discussion:
Discussion of Case 1
In this case, the δ-wave was prominent and could be easily monitored during a treadmill test. This is the least-invasive and most appropriate next step. If loss of pre-excitation occurs with exercise, this classifies the accessory pathway as benign and eliminates concerns of SCD. An EPS is probably premature at this point but may be required if the treadmill test is not prognostically helpful. Treadmill testing is particularly problematic when minimal pre-excitation occurs (see Figure 49-1, B). In such cases, an EPS may be the only method of risk evaluation. An echocardiogram is not useful under the circumstances because most patients with WPW have structurally normal hearts.
Repolarization Abnormalities
Many primary repolarization changes on the 12-lead ECG are benign or normal variants such as early repolarization. Others may result from the presence of structural heart disease (e.g., myocardial infarction [MI], dilated cardiomyopathy, and hypertrophic cardiomyopathy) or the consequence of pharmacologic agents (e.g., antiarrhythmic drugs). The congenital long QT syndrome (LQTS), short QT syndrome (SQTS), and Brugada syndrome are relatively uncommon diseases associated with potentially life-threatening ventricular arrhythmias. In general, diagnoses of these conditions are complicated because of the significant overlap in the ECG manifestations of normal and disease conditions. The management of these conditions continues to evolve. The following sections discuss the typical ECG changes and clinical features that accompany the most common pathologic repolarization abnormalities. Readers are also directed to Chapters 62 to 64 for additional information.
Long QT Syndrome
Case 2
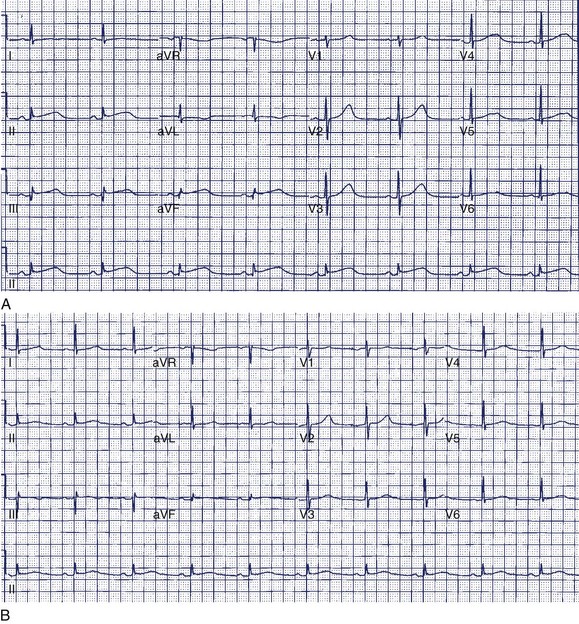
A 36-year-old accountant was referred for assessment after the unexpected death of his 24-year-old brother who was previously well. The postmortem examination revealed no obvious cause of death. A 27-year-old sister is alive and well. His mother, who was known to have epilepsy, had died at age 46 in a motor vehicle accident. The patient’s ECG shows a very long Q-T interval (QT = 600 ms, QTc = 545 ms) (Figure 49-2, A). He was not taking any medication and jogged regularly. An ECG done 2 years previously for an insurance physical was reported as normal (Figure 49-2, B).
The most appropriate course of action at this time would be to:
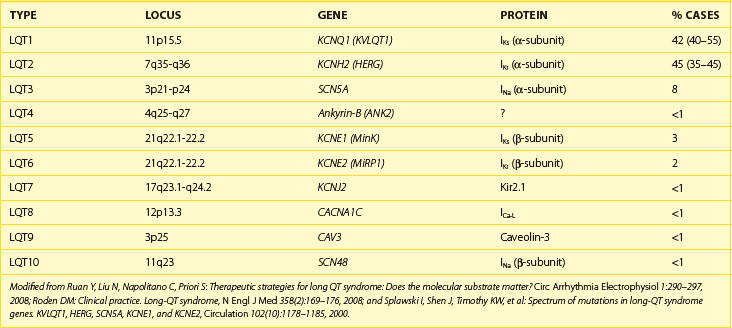
The congenital LQTS is a rare disorder (incidence 1 : 3000 to 1 : 10 000) characterized by prolongation of the Q-T interval, recurrent syncope, and SCD.16 Two major clinical variants were originally described: (1) Romano-Ward syndrome (autosomal dominant inheritance) and (2) Jervell Lange-Nielson syndrome (autosomal recessive inheritance), in which patients also have congenital deafness. Over the past 10 years, great advances have been made in understanding the genetic and cellular bases for LQTS. At least 10 distinct genotypic variants of LQTS encompassing hundreds of mutations have now been identified (Table 49-1), and a genetic diagnosis is now obtainable in 50% to 60% of patients.17 Three genotypes (LQT1, LQT2, and LQT3) account for more than 90% of cases.18–20 The principal abnormality in LQTS is prolongation of the action potential duration caused by a reduction in the outward potassium current (LQT1 and LQT2) or, less commonly, by a persistent inward sodium current during the plateau phase (LQT3). Action potential prolongation provokes the development of early afterdepolarizations, which may produce a triggered action potential resulting in a premature beat.21 Such impulses can initiate a polymorphic VT, known as torsades de pointes, which underlies the clinical symptoms of palpitations, syncope, or SCD caused by VF.
The classic description of events in LQTS involves exercise-induced or emotion-induced syncope or cardiac arrest. Symptoms often begin in adolescence, though they may begin earlier in patients with LQT1.20 It is common for patients to be investigated and misdiagnosed as having a seizure disorder. Clinical events in LQTS are often seen in the context of heart rate acceleration precipitated by exercise, emotion, or sudden arousal.20,22,23 Patients with LQT1 are particularly prone to events during hyperadrenergic states, with swimming identified as a specific trigger in such patients.20,23 The history of arousal to an auditory stimulus such as a sudden loud noise (classically, a telephone call at night or an alarm clock going off) is strongly predictive of LQT2.20,22 Events that occur during rest or sleep are more suggestive of LQT3.20 Many of these circumstances represent the setting of sudden or marked changes in adrenergic tone and heart rate that may influence repolarization and subsequent vulnerability to arrhythmia.
The hallmark of LQTS is prolongation of the QTc (>440 ms in men and >460 ms in women), although considerable overlap occurs with QTc in normal populations. The QTc of genotype-positive patients overlaps with normal in up to one third because of variable penetrance.24–26 Also, 10% to 15% of QTc in normal adults are greater than 440 ms.27 Since repolarization is affected by factors such as sympathetic outflow, electrolyte balance, and pharmacologic agents, it is not surprising that considerable temporal variation may be present in QTc and that increased sampling improves diagnostic accuracy. In the absence of obvious reversible causes of QTc prolongation (Table 49-2), a diagnosis of LQTS can be made on the basis of ECG features and clinical presentation.28 The mean QTc does not differ among the LQT1, LQT2, and LQT3 types but is significantly longer in Jervell Lange-Neilson syndrome.20 In addition to QTc prolongation, qualitative abnormalities may also be found in LQTS, including ST-T wave changes, U waves, T-wave alternans, increased QT dispersion, and sinus bradycardia.21 Although not highly specific, characteristic ST-T wave morphologies may allow the prediction of LQT type: broad-based T waves typify LQT1; low amplitude, notched T waves occur in LQT2; and long isoelectric ST segment with late-onset T wave occur in LQT3.26
| FACTOR | MECHANISM |
|---|---|
| Bradycardia | ↑ APD |
| ↑ APD prolongation with class III agents | |
| Drugs* | Mainly IKr blockade |
| Electrolyte disorders (hypokalemia, hypomagnesemia, hypocalcemia) | Hypokalemia ↓ IKr and ↑ IKr sensitivity to pharmacologic blockers |
| Left ventricular hypertrophy/failure | ↓ K+ currents (Ito, IKr, IKs) |
| Changes to ICaL and intracellular calcium (Ca2+) | |
| Miscellaneous (e.g., anorexia, cerebrovascular disease, HIV infection, hypothyroidism, hypothermia, ionic contrast media) |
APD, Action potential duration.
* The major classes of drugs are antiarrhythmic drugs, antihistamines, macrolide antibiotics, antipsychotics, and antidepressants. An updated online list of specific drugs that prolong the Q-T interval is available at www.qtdrugs.org or www.torsades.org.
Modified from Walker BD, Krahn AD, Klein GJ, et al: Congenital and acquired long QT syndromes, Can J Cardiol 19(1):76–87, 2003.
It has been estimated that LQTS causes 3000 to 4000 SCDs per year in children and young adults in the United States.29 Long-term registry data suggest that annually the risk of syncope is 5% and the risk of LQTS-related death before the age of 50 years is approximately 1% in symptomatic patients, with the risk being significantly lower in asymptomatic patients.26,30 The most important predictor of risk is QTc duration, although age, gender, and genotype can be confounders. Using data from an international registry, three risk groups relating to the probability of experiencing a first cardiac event before the age of 40 years have been identified31:
Although a family history of cardiac events would intuitively appear to be prognostically important, this has not been borne out in clinical studies.32,33
β-Adrenoreceptor blockade is the mainstay of therapy (aiming for a reduction in peak exercise heart rate of >20%), and it is recommended for most patients with LQTS.34 Survival can be dramatically improved with aggressive treatment with β-blockers, left cervical sympathectomy, pacemakers, and an implantable cardioverter defibrillator (ICD).17,34 Lifestyle modifications, family screening, and genetic testing and counseling are also important considerations. In particular, genotyping may occasionally assist with diagnosis, family screening, and management.35 For example, β-blockers may be less effective in LQT3, and genotype-specific therapies such as potassium replacement for LQT2 and mexiletine for LQT3 are being evaluated.36,37
Discussion Case 2
The patient in question clearly has marked QTc prolongation in the absence of medication. The family history is a cause for concern and suggests that his brother and mother (LQTS is frequently misdiagnosed as epilepsy) both had LQTS. This man previously had a normal ECG, and it is not unusual for such patients to have variability of the Q-T interval at different times. In more borderline cases, scoring systems incorporating ECG, clinical features, and family history may be helpful.28 In this case, the best answer would be to recommend treatment with β-blocker. An argument can be made for an ICD in such a patient, although ICDs are usually reserved for high-risk patients such as those with recurrent symptoms who are on β-blockers or survivors of cardiac arrest (class IIa recommendation). Treadmill testing will not influence the decision to treat, although exercise may be diagnostically useful, especially if arrhythmias are observed. The EPS is generally of no value in LQTS. Where available, genetic counseling and family screening should also be offered.
Brugada Syndrome
Case 3
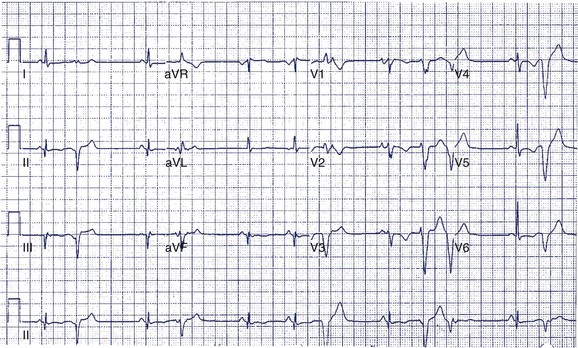
A 42-year-old man was referred for assessment of symptoms of atypical chest pain and ECG abnormalities (Figure 49-3). The patient was otherwise well. His father died suddenly in his mid-40s but no further details about his death are available.
The most appropriate next step to clarify the diagnosis of the ECG abnormality would be to:
Brugada syndrome is an inherited disorder (autosomal dominant with variable expression) characterized by syncope and SCD from polymorphic VT and VF.38 First described in 1992, it is now a recognized cause of SCD, particularly in southern Europe and in southeast Asia. Brugada syndrome is much more common in men and usually presents in the third and fourth decades of life. The primary abnormality is heterogeneous shortening of the action potential duration, particularly affecting the right ventricle, which results in phase 2 re-entry and polymorphic VT.39 In up to 20% of affected individuals, mutations may be demonstrated in the SCN5A gene that encodes the fast inward sodium (Na+) current (INa).40 In a canine right ventricular wedge preparation, it has been shown that the combination of a weaker INa in the presence of a large transient outward potassium (K+) current (Ito) results in a dramatic truncation of the epicardial action potential and marked transmural heterogeneity of repolarization.39
The Brugada ECG pattern is characterized by “pseudo–right bundle branch block (RBBB)” with ST-segment elevation in precordial leads V1 to V3 (reflecting predominant right ventricular abnormality). Three patterns of ST elevation have been described (see Figure 49-3, B):
Differentiation of the Brugada ECG pattern from prominent early repolarization changes in the right precordial leads of otherwise healthy patients may occasionally be difficult, and pharmacologic provocation may be particularly useful in such patients with nondiagnostic ECGs. Administration of the Na+ channel–blocking agents flecainide, ajmaline, and procainamide may convert a type 2 or 3 ECG pattern to the classic type 1 pattern, but reports of the sensitivity and specificity of this observation have been conflicting.41,42 Likewise, it has been noted that moving ECG leads V1 and V2 one or two interspaces higher (third and second interspaces, respectively) can increase the sensitivity of the ECG to diagnose a spontaneous type 1 pattern.43,44 Although none of the normal subjects had a Brugada pattern in the initial report, the specificity of this test requires a larger cohort for confirmation. The signal-averaged ECG will show late potentials in approximately 50% of patients and may be another marker of disease in borderline cases.45 Exercise stress testing does not usually provoke ventricular arrhythmias because most events occur at night or during episodes of high vagal tone.45
The diagnosis of Brugada syndrome is made on the basis of a spontaneous or induced type 1 Brugada pattern on ECG in concert with clinical symptoms (e.g., unexplained syncope or seizure, nocturnal agonal respiration), documented arrhythmia, or a family history of typical type 1 Brugada ECG pattern or SCD before the age of 45 years.46 A recent consensus conference on Brugada syndrome has recommended ICDs for those with aborted SCD as well as for those with syncope, seizures, and nocturnal agonal respiration without another obvious cause.45 This holds true whether the type 1 ECG is spontaneous or induced.
The risk of life-threatening events in asymptomatic patients is substantially lower, and the annual risk of sudden cardiac arrest has been variably reported to be 0.35% to 1.7%.47–49 Although controversial, for asymptomatic individuals with a spontaneous type 1 ECG, an EPS should be considered (class IIa indication). In asymptomatic patients with an induced type 1 ECG, an EPS is justified in those with a family history of SCD suspected to be caused by Brugada syndrome (class IIb indication). Induced type 1 ECG, in the absence of a family history of SCD, provides diagnostic data only, and close monitoring is recommended. However, it is noteworthy that a recent meta-analysis has raised questions about the usefulness of EPS for risk stratification in Brugada syndrome.50
Discussion Case 3
Without a family history of SCD, an ICD would not be recommended to this patient without evidence of spontaneous type 1 pattern, even in the setting of an inducible type 1 pattern with procainamide. This group of patients appears to remain at low risk for death caused by arrhythmia.43,50 In all circumstances, a clear discussion with patients, especially individuals who are highly risk averse, is required to delineate individual tolerances for small risks of arrhythmic events and ICD-related events.
Short QT Syndrome
The finding of a short Q-T interval on routine ECG should raise the possibility of congenital short QT syndrome (SQTS)—a recently described familial disorder characterized by marked shortening of the Q-T interval (QTc generally <340 ms) and narrow, high-amplitude T waves. SQTS is a rare condition, and its true prevalence is unknown. In several small series, the inheritance has been found to be autosomal dominant, and patients are at an increased risk of both atrial and ventricular arrhythmias, predominantly AF and SCD secondary to VF.51–53 The genetic abnormalities described for SQTS, so far, involve genes similar to those implicated in LQTS and Brugada syndrome, with gain-of-function mutations in K+ channel genes and loss-of-function mutations in L-type calcium (Ca2+) channels.54–57
The difficult clinical question is: Is it possible to distinguish a “pathologically short QTc” from an “abbreviated QTc at the low end of the normal range”? The reported QTc among patients diagnosed with SQTS in the published literature has been less than 340 ms, often less than 300 ms. In comparison, two community surveys have addressed the “normal range” of QTc. In a study of more than 12,000 healthy young individuals undergoing routine ECG, the shortest QTc was 335 ms, and no SCDs occurred in patients with QTc in the lowest 0.5% (335 to 360 ms). This suggests little overlap between SQTS and the normal range.58,59 However, in a separate community survey of more than 10,000 middle-aged Finnish participants, the shortest QTc was 305 ms, and the prevalence of a QTc less than 340 ms was 0.4% and less than 320 ms was 0.1%, suggesting some degree of overlap. Reassuringly, again, no SCDs occurred in individuals with a QTc less than 340 ms.
At present, few data are available to guide the risk stratification and management of patients with suspected SQTS. ICDs have been suggested as first-line therapy in patients with a personal or family history of SCD associated with SQTS.53,60
Early Repolarization Abnormalities
Early repolarization changes are present in 2% to 5% of the population, being more common in young athletic individuals. The ECG shows variable elevation of the J-point with or without associated slurring or notching. These changes are most commonly seen in the anterior precordial leads but may occur in other leads and show circadian variation.61 Although such changes are generally understood to be benign, a recent study from Haissaguerre et al described an increased incidence of early repolarization changes in the inferolateral leads in survivors of cardiac arrest caused by idiopathic VF compared with controls.62 Although, the findings of this study are provocative, the sensitivity, specificity, and predictive accuracy of this finding have yet to be established. Many asymptomatic individuals have early repolarization changes and yet are extremely unlikely to have increased risk. Therefore the abnormality may be an important diagnostic sign in high-risk patients with a history of unexplained syncope or a family history of SCD, but the same observation in an otherwise completely healthy patient should not cause alarm or warrant further investigation.61
Asymptomatic Arrhythmias
Ventricular Ectopy
Case 4
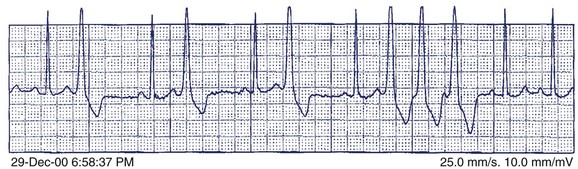
A 49-year-old man has Holter monitoring performed because of palpitations. No symptoms are noted in the patient’s diary, but asymptomatic ectopy similar to that seen on the 12-lead ECG in Figure 49-4 is recorded. He is referred for further assessment.
Ectopy in the Setting of a Structurally Normal Heart
Forms of benign VT found in patients with a structurally normal heart have been described to occur in both the left and right ventricles—called idiopathic VT. Some forms of idiopathic VT present with repetitive bursts of nonsustained monomorphic ectopy, especially from the right ventricular outflow tract (RVOT). The ectopy has a characteristic left bundle branch block (LBBB) morphology with large-amplitude R waves in the inferior leads with T-wave inversions. Figure 49-5 shows VPBs from the RVOT as recorded in lead II from the Holter monitor. Idiopathic forms of left ventricular tachycardia have also been recognized and can produce asymptomatic ectopy.63,64 Patients with asymptomatic idiopathic ectopy need no further investigations or therapy.
Arrhythmogenic Right Ventricular Dysplasia
The right ventricle can occasionally be the source of serious arrhythmia that presents as asymptomatic ectopy, and this needs to be distinguished from idiopathic forms of ectopy, including RVOT tachycardia. ARVD is characterized by palpitations, syncope, and SCD caused by ventricular arrhythmias in the setting of pathologic fibro-fatty infiltration of the right ventricle.65 It is most commonly diagnosed in young males, although cases at all ages and in both genders have been described. The disease is regarded as a progressive cardiomyopathy that may also affect the left ventricle, although the septum is typically spared. Both familial and sporadic forms of ARVD have been described, with the former usually displaying an autosomal dominant inheritance. ARVD may rarely be a component of the autosomal recessive Naxos disease, which is associated with hyperkeratosis of the palms and soles and woolly hair. ARVD is understood to be a disorder of cell adhesion and relates to mutations of genes that encode desmosomal proteins.66
In spite of considerable controversy regarding the sensitivity and specificity of testing, the diagnosis of ARVD is currently based on family history and the presence of ECG and structural abnormalities.65 The ECG in sinus rhythm may demonstrate abnormalities related to delayed right ventricular activation such as QRS prolongation, RBBB, a distinct low-frequency, a low-amplitude wave after the QRS (ε-wave), and T-wave inversion in the right precordial leads. Late potentials on the signal-averaged ECG are also seen in ARVD. Structural abnormalities can be found on echocardiography, right ventricular angiography, nuclear imaging, and MRI. Invasive EPS with electroanatomic mapping may identify areas of low-voltage electrograms, and programmed ventricular stimulation may induce one or more types of sustained monomorphic VT and less commonly VF. Finally, myocardial biopsy with immunohistochemical analysis and genetic testing may also be considered.67
ICDs are frequently prescribed for patients with ARVD and are generally recommended to patients with documented sustained VT or VF and to those otherwise deemed as “high risk,” including those with unexplained syncope, extensive or left ventricular involvement, or a malignant family history.34 EPS does not appear to be useful for risk stratification.68 Exercise restriction should also be discussed, and antiarrhythmic drug treatment and RF ablation are adjunctive measures for reducing the frequency of ventricular arrhythmias.69
When considering the evaluation of a patient with ventricular ectopy of LBBB morphology, historical features need to be carefully considered, especially when the ectopy does not arise from the RVOT. Major and minor Specific Task Force diagnostic criteria based on the arrhythmia, family history, ECG criteria, imaging, and histopathology of tissue have been created for ARVD.65 In patients with ARVD and a single risk factor for SCD, it is reasonable to consider ICD therapy (class IIa recommendation).68,70
Discussion Case 4
The baseline 12-lead ECG has features of ARVD with terminal low-amplitude voltage seen best in leads V1 to V3 (major criterion). Accompanying T-wave inversions are also seen (minor criterion). The ectopy has an LBBB morphology with left superior axis, which suggests a source from the right ventricular apex (minor criterion). It is clearly not from the RVOT and therefore cannot be immediately dismissed as benign. Imaging of the right ventricle is required. Echocardiography is highly variable for imaging the right ventricle and, in general, is not of sufficient quality to diagnose ARVD. In experienced hands, cardiac MRI is the best imaging modality for ARVD, and diagnostic criteria have been developed.65 Coronary angiography will not provide useful information. Any coronary lesions would have to be interpreted as incidental.
Atrial Fibrillation and Other Supraventricular Arrhythmia
Case 5
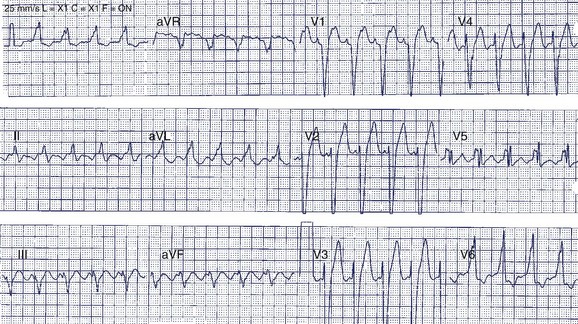
A 59-year-old asymptomatic man presented for preoperative assessment with the following ECG (Figure 49-6). An echocardiogram demonstrated reduced left ventricular function and an estimated ejection fraction of 35%.
The appropriate first management would be to schedule:
Not infrequently, patients are found to have paroxysmal or persistent AF of unknown duration and are unaware that any dysrhythmia is present.71 A similar presentation can occur in atrial flutter (see Figure 49-6). Several issues need to be addressed in such patients. These relate to decisions about occult tachycardia-induced cardiomyopathy, anticoagulation, and possible cardioversion.
Tachycardia-Induced Cardiomyopathy
The syndrome of tachycardia-induced cardiomyopathy has been well described in the setting of poorly controlled AF.72,73 Importantly, it is usually reversible if rate control can be achieved.72,73 Assessment of left ventricular function should be made in all such patients and aggressive rate control initiated in patients with impairment of left ventricular function. Repeated Holter monitoring can be performed to ensure adequate rate control, ideally below 100 beats/min except for periods of exercise. For those patients with persistent left ventricular dysfunction and failure of pharmacologic rate control, DC cardioversion, catheter ablation of atrial flutter or AF, or AV nodal ablation and permanent pacemaker insertion could be considered.
Anticoagulation
Decisions regarding the use of warfarin or aspirin for thromboembolic prophylaxis should be based on thromboembolic risk. The CHADS2 score (congestive heart failure, hypertension, age ≥75 years, and diabetes mellitus, each given 1 point; and a past history of transient ischemic attack or stroke given 2 points) is widely used for evaluating thromboembolic risk in those with nonvalvular AF.74 Warfarin is generally recommended to patients with a CHADS2 score of 2 or above. Importantly, the need for thromboembolic prophylaxis in asymptomatic patients with AF as well as in those with atrial flutter should be determined using the same guidelines.
Cardioversion
Irrespective of the pressure from patients or referring physicians to perform DC cardioversion in asymptomatic patients, no evidence supports the claim that DC cardioversion ensures a better prognosis or reduced risk of stroke in sinus rhythm.75 Some patients with seemingly asymptomatic AF are unaware that they have had a reduction in functional status until sinus rhythm has been returned. For this reason, consideration is frequently given to perform at least a single cardioversion in minimally symptomatic patients. In general, maintenance of sinus rhythm is known to depend on the duration of AF as well as on left atrial dimension. The former is frequently difficult to assess in asymptomatic patients. Nonetheless, an echocardiographic assessment of left atrial dimension may give some insight into the likelihood of long-term maintenance of sinus rhythm. If a patient sees no improvement in functional status during sinus rhythm, repeated cardioversion is truly unwarranted.
Incessant Supraventricular Tachycardias
A small group of arrhythmogenic substrates allow for relatively slow but frequently incessant SVTs and tachycardia-induced cardiomyopathy.76 Patients may have a permanent form of junctional reciprocating tachycardia because of a slowly conducting accessory pathway or nodal pathway or may have an incessant atrial tachycardia.72,77 In the current era, RF ablation forms the first-line therapy in such patients. Alternatively, medication can be used, but maintenance of sinus rhythm is paramount for the improvement of left ventricular function. However, the demonstration of normal left ventricular function argues against any intervention in the truly asymptomatic individual with frequent paroxysmal SVT.
Asymptomatic Atrioventricular Nodal and His-Purkinje Disease
Case 6
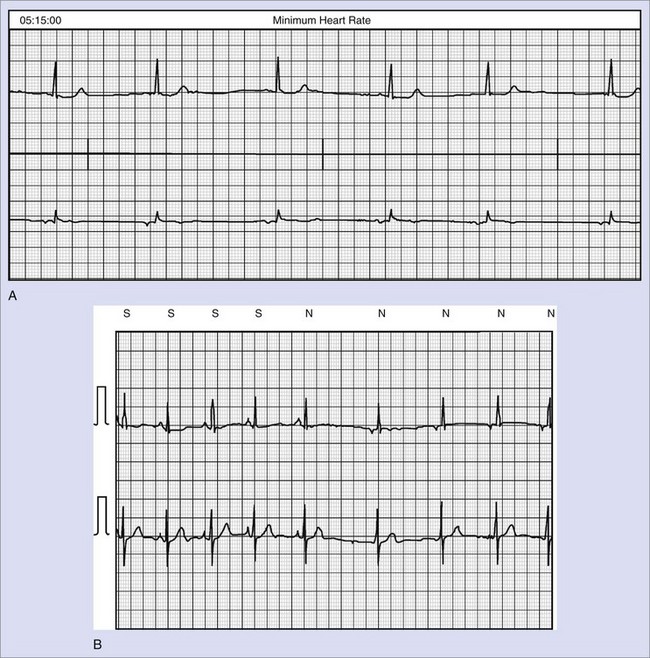
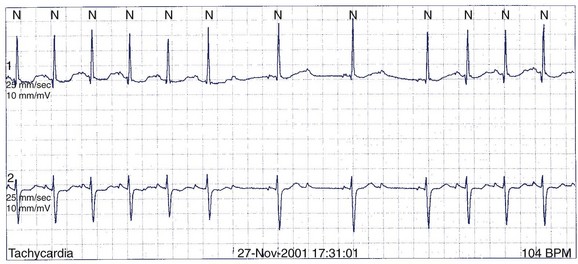
An otherwise healthy 54-year-old woman was referred for evaluation of an asymptomatic Holter monitor recording (Figure 49-7). Before referral, a diagnosis of Mobitz II second-degree AV block was made, and the patient was informed that she may require a permanent pacemaker. Is this correct?
Atrioventricular Block
Although unusual in the adult population, truly asymptomatic complete heart block is occasionally seen. Older patients with progressive His-Purkinje degeneration as the etiology tend to report symptoms of exercise intolerance or exertional dyspnea on directed questioning. The American College of Cardiology/American Heart Association/Heart Rhythm Society (ACC/AHA/HRS) Guidelines for Device Implantation state that awake, asymptomatic patients with third-degree AV block with documented asystole 3 seconds or more, an escape rate less than 40 beats/min, or an escape rhythm that is below the AV node have a class I indication for pacing.70 Asymptomatic third-degree AV block with faster average ventricular rates is considered a class IIa indication for pacing.
Asymptomatic type II second-degree AV block can occur at the level of the AV node or below the AV node within the His-Purkinje system. Block at the level of the AV node or within the His bundle (intra-His) produces a narrow QRS, whereas block below the His bundle most often produces a wide QRS. This is an important distinction because block at the level of the AV node does not require pacing. Associated bundle branch block or bifasicular block may be helpful to determine the level of block. Occasionally, invasive EPS may be required. The ACC/AHA/HRS Guidelines recognize that asymptomatic type II second-degree AV block with a wide QRS is classified as a class I indication for pacing. Type II second-degree AV block with a narrow QRS has been classified as a class IIa indication for pacing if block at the level of the node cannot be excluded, although the decision to pace should be highly individualized.70
Discussion Case 6
On close inspection, a 2 : 1 block is seen. It is important to determine the level of block in such cases—at the level of the AV node or in the His-Purkinje system. As noted in the earlier discussion, the importance is more than academic. In this case, the QRS complexes are narrow throughout. The P-R interval of the last conducted beat before the block is substantially longer than that of conducted beats following the dropped beats. This suggests that the 2 : 1 block is Mobitz I and likely at the level of the AV node. This is very common during sleep in healthy individuals. No pacemaker was inserted in this patient. The presence of distal conduction disease (bundle branch block, bifascicular or trifascicular block) suggests that the block is below the level of the AV node (Mobitz II) and that pacing may be indicated despite the asymptomatic nature of the abnormality.70
Among patients with asymptomatic bifascicular and trifascicular disease, a small incidence of progression to second- or third-degree AV block is seen.78 However, no single clinical or laboratory variable is predictive of the progression of AV block. Therefore asymptomatic bundle branch block alone and bifasicular or trifasicular block are not indications for pacing. Asymptomatic bundle branch block may be the first indication of insidious cardiac disease. As such, patients should be evaluated noninvasively with respect to cardiac size and function. In a similar vein, monitoring, especially at night, can record asymptomatic sinus bradycardia or pauses. Unless these are associated with symptoms, pacing is not required.70
Key References
Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: Report of the second consensus conference. Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111(5):659-670.
Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20(6):1391-1396.
Brugada P, Brugada R, Brugada J. Should patients with an asymptomatic Brugada electrocardiogram undergo pharmacological and electrophysiological testing? Circulation. 2005;112(2):279-292. discussion 279–292
Fisch GR, Zipes DP, Fisch C. Bundle branch block and sudden death. Prog Cardiovasc Dis. 1980;23(3):187-224.
Gage BF, Waterman AD, Shannon W, et al. Validation of clinical classification schemes for predicting stroke: Results from the National Registry of Atrial Fibrillation. JAMA. 2001;285(22):2864-2870.
Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: A familial cause of sudden death. Circulation. 2003;108(8):965-970.
Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: Clinical findings and diagnostic-therapeutic implications. Eur Heart J. 2006;27(20):2440-2447.
Klein GJ, Prystowsky EN, Yee R, et al. Asymptomatic Wolff-Parkinson-White. Should we intervene? Circulation. 1989;80(6):1902-1905.
Leitch JW, Klein GJ, Yee R, et al. Prognostic value of electrophysiology testing in asymptomatic patients with Wolff-Parkinson-White pattern. Circulation. 1990;82(5):1718-1723.
McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71(3):215-218.
Priori SG, Napolitano C. Should patients with an asymptomatic Brugada electrocardiogram undergo pharmacological and electrophysiological testing? Circulation. 2005;112(2):279-292. discussion 279–292
Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348(19):1866-1874.
Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358(2):169-176.
Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103(1):89-95.
Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102(10):1178-1185.
Wellens HJ. Should catheter ablation be performed in asymptomatic patients with Wolff-Parkinson-White syndrome? When to perform catheter ablation in asymptomatic patients with a Wolff-Parkinson-White electrocardiogram. Circulation. 2005;112(14):2201-2207. discussion 2216
Wyse DG, Waldo AL, DiMarco JP, et al. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med. 2002;347(23):1825-1833.
1 Wolff L, Parkinson J, White P. Bundle branch block with short P-R interval in healthy young people prone to paroxysmal tachycardia. Am Heart J. 1930;1930(5):685-704.
2 Munger TM, Packer DL, Hammill SC, et al. A population study of the natural history of Wolff-Parkinson-White syndrome in Olmsted County, Minnesota, 1953–1989. Circulation. 1993;87(3):866-873.
3 Klein GJ, Prystowsky EN, Yee R, et al. Asymptomatic Wolff-Parkinson-White. Should we intervene? Circulation. 1989;80(6):1902-1905.
4 Santinelli V, Radinovic A, Manguso F, et al. Asymptomatic ventricular preexcitation: A long-term prospective follow-up study of 293 adult patients. Circ Arrhythmia Electrophysiol. 2009;2:102-107.
5 Klein GJ, Bashore TM, Sellers TD, et al. Ventricular fibrillation in the Wolff-Parkinson-White syndrome. N Engl J Med. 1979;301(20):1080-1085.
6 Pappone C, Santinelli V, Rosanio S, et al. Usefulness of invasive electrophysiologic testing to stratify the risk of arrhythmic events in asymptomatic patients with Wolff-Parkinson-White pattern: Results from a large prospective long-term follow-up study. J Am Coll Cardiol. 2003;41(2):239-244.
7 Teo WS, Klein GJ, Guiraudon GM, et al. Multiple accessory pathways in the Wolff-Parkinson-White syndrome as a risk factor for ventricular fibrillation. Am J Cardiol. 1991;67(9):889-891.
8 Leitch JW, Klein GJ, Yee R, Murdock C. Prognostic value of electrophysiology testing in asymptomatic patients with Wolff-Parkinson-White pattern. Circulation. 1990;82(5):1718-1723.
9 Sharma AD, Yee R, Guiraudon G, Klein GJ. Sensitivity and specificity of invasive and noninvasive testing for risk of sudden death in Wolff-Parkinson-White syndrome. J Am Coll Cardiol. 1987;10(2):373-381.
10 Gaita F, Giustetto C, Riccardi R, et al. Stress and pharmacologic tests as methods to identify patients with Wolff-Parkinson-White syndrome at risk of sudden death. Am J Cardiol. 1989;64(8):487-490.
11 Eshchar Y, Belhassen B, Laniado S. Comparison of exercise and ajmaline tests with electrophysiologic study in the Wolff-Parkinson-White syndrome. Am J Cardiol. 1986;57(10):782-786.
12 Boahene KA, Klein GJ, Sharma AD, et al. Value of a revised procainamide test in the Wolff-Parkinson-White syndrome. Am J Cardiol. 1990;65(3):195-200.
13 Critelli G, Grassi G, Perticone F, et al. Transesophageal pacing for prognostic evaluation of preexcitation syndrome and assessment of protective therapy. Am J Cardiol. 1983;51(3):513-518.
14 Pappone C, Santinelli V. Should catheter ablation be performed in asymptomatic patients with Wolff-Parkinson-White syndrome? Catheter ablation should be performed in asymptomatic patients with Wolff-Parkinson-White syndrome. Circulation. 2005;112(14):2207-2215. discussion 2216
15 Wellens HJ. Should catheter ablation be performed in asymptomatic patients with Wolff-Parkinson-White syndrome? When to perform catheter ablation in asymptomatic patients with a Wolff-Parkinson-White electrocardiogram. Circulation. 2005;112(14):2201-2207. discussion 2216
16 Ruan Y, Liu N, Napolitano C, Priori S. Therapeutic strategies for long-QT syndrome: Does the molecular substrate matter? Circ Arrhythmia Electrophysiol. 2008;1:290-297.
17 Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358(2):169-176.
18 Walker BD, Krahn AD, Klein GJ, et al. Congenital and acquired long QT syndromes. Can J Cardiol. 2003;19(1):76-87.
19 Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102(10):1178-1185.
20 Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103(1):89-95.
21 Roden DM, Lazzara R, Rosen M, et al. Multiple mechanisms in the long-QT syndrome. Current knowledge, gaps, and future directions. The SADS Foundation Task Force on LQTS. Circulation. 1996;94(8):1996-2012.
22 Wilde AA, Jongbloed RJ, Doevendans PA, et al. Auditory stimuli as a trigger for arrhythmic events differentiate HERG-related (LQTS2) patients from KVLQT1-related patients (LQTS1). J Am Coll Cardiol. 1999;33(2):327-332.
23 Ackerman MJ, Tester DJ, Porter CJ, Edwards WD. Molecular diagnosis of the inherited long-QT syndrome in a woman who died after near-drowning. N Engl J Med. 1999;341(15):1121-1125.
24 Vincent GM. Role of DNA testing for diagnosis, management, and genetic screening in long QT syndrome, hypertrophic cardiomyopathy, and Marfan syndrome. Heart. 2001;86(1):12-14.
25 Locati EH, Zareba W, Moss AJ, et al. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: Findings from the International LQTS Registry. Circulation. 1998;97(22):2237-2244.
26 Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation. 1991;84(3):1136-1144.
27 Taggart NW, Haglund CM, Tester DJ, Ackerman MJ. Diagnostic miscues in congenital long-QT syndrome. Circulation. 2007;115(20):2613-2620.
28 Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88(2):782-784.
29 Vincent GM. The molecular genetics of the long QT syndrome: Genes causing fainting and sudden death. Annu Rev Med. 1998;49:263-274.
30 Schwartz PJ. Idiopathic long QT syndrome: Progress and questions. Am Heart J. 1985;109(2):399-411.
31 Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348(19):1866-1874.
32 Kaufman ES, McNitt S, Moss AJ, et al. Risk of death in the long QT syndrome when a sibling has died. Heart Rhythm. 2008;5(6):831-836.
33 Kimbrough J, Moss AJ, Zareba W, et al. Clinical implications for affected parents and siblings of probands with long-QT syndrome. Circulation. 2001;104(5):557-562.
34 Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114(10):e385-e484.
35 Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: Clinical impact. Circulation. 1999;99(4):529-533.
36 Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92(12):3381-3386.
37 Ruan Y, Liu N, Bloise R, et al. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation. 2007;116(10):1137-1144.
38 Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20(6):1391-1396.
39 Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100(15):1660-1666.
40 Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392(6673):293-296.
41 Brugada R, Brugada J, Antzelevitch C, et al. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. 2000;101(5):510-515.
42 Gussak I, Antzelevitch C, Bjerregaard P, et al. The Brugada syndrome: Clinical, electrophysiologic and genetic aspects. J Am Coll Cardiol. 1999;33(1):5-15.
43 Shimizu W, Matsuo K, Takagi M, et al. Body surface distribution and response to drugs of ST segment elevation in Brugada syndrome: Clinical implication of eighty-seven-lead body surface potential mapping and its application to twelve-lead electrocardiograms. J Cardiovasc Electrophysiol. 2000;11(4):396-404.
44 Sangwatanaroj S, Prechawat S, Sunsaneewitayakul B, et al. New electrocardiographic leads and the procainamide test for the detection of the Brugada sign in sudden unexplained death syndrome survivors and their relatives. Eur Heart J. 2001;22(24):2290-2296.
45 Alings M, Wilde A. “Brugada” syndrome: Clinical data and suggested pathophysiological mechanism. Circulation. 1999;99(5):666-673.
46 Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: Report of the second consensus conference. Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111(5):659-670.
47 Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: Insights for risk stratification and management. Circulation. 2002;105(11):1342-1347.
48 Brugada P, Brugada R, Brugada J. Should patients with an asymptomatic Brugada electrocardiogram undergo pharmacological and electrophysiological testing? Circulation. 2005;112(2):279-292. discussion 279–292
49 Priori SG, Napolitano C. Should patients with an asymptomatic Brugada electrocardiogram undergo pharmacological and electrophysiological testing? Circulation. 2005;112(2):279-292. discussion 279–292
50 Gehi AK, Duong TD, Metz LD, et al. Risk stratification of individuals with the Brugada electrocardiogram: A meta-analysis. J Cardiovasc Electrophysiol. 2006;17(6):577-583.
51 Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: A new clinical syndrome? Cardiology. 2000;94(2):99-102.
52 Gaita F, Giustetto C, Bianchi FW, et al. Short QT Syndrome: A familial cause of sudden death. Circulation. 2003;108(8):965-970.
53 Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: Clinical findings and diagnostic-therapeutic implications. Eur Heart J. 2006;27(20):2440-2447.
54 Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109(1):30-35.
55 Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109(20):2394-2397.
56 Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96(7):800-807.
57 Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115(4):442-449.
58 Gallagher MM, Magliano G, Yap YG, et al. Distribution and prognostic significance of QT intervals in the lowest half centile in 12,012 apparently healthy persons. Am J Cardiol. 2006;98(7):933-935.
59 Anttonen O, Junttila MJ, Rissanen H, et al. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation. 2007;116(7):714-720.
60 Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: Pharmacological treatment. J Am Coll Cardiol. 2004;43(8):1494-1499.
61 Wellens HJ. Early repolarization revisited. N Engl J Med. 2008;358(19):2063-2065.
62 Haissaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358(19):2016-2023.
63 Lin FC, Finley CD, Rahimtoola SH, Wu D. Idiopathic paroxysmal ventricular tachycardia with a QRS pattern of right bundle branch block and left axis deviation: A unique clinical entity with specific properties. Am J Cardiol. 1983;52(1):95-100.
64 Belhassen B, Shapira I, Pelleg A, et al. Idiopathic recurrent sustained ventricular tachycardia responsive to verapamil: An ECG-electrophysiologic entity. Am Heart J. 1984;108(4 Pt 1):1034-1037.
65 McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71(3):215-218.
66 Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2005;16(8):927-935.
67 Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360(11):1075-1084.
68 Corrado D, Leoni L, Link MS, et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108(25):3084-3091.
69 Wichter T, Borggrefe M, Haverkamp W, et al. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation. 1992;86(1):29-37.
70 Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices): Developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. Circulation. 2008;117(21):e350-e408.
71 Humphries KH, Kerr CR, Connolly SJ, et al. New-onset atrial fibrillation: Sex differences in presentation, treatment, and outcome. Circulation. 2001;103(19):2365-2370.
72 Fenelon G, Wijns W, Andries E, Brugada P. Tachycardiomyopathy: Mechanisms and clinical implications. Pacing Clin Electrophysiol. 1996;19(1):95-106.
73 Packer DL, Bardy GH, Worley SJ, et al. Tachycardia-induced cardiomyopathy: A reversible form of left ventricular dysfunction. Am J Cardiol. 1986;57(8):563-570.
74 Gage BF, Waterman AD, Shannon W, et al. Validation of clinical classification schemes for predicting stroke: Results from the National Registry of Atrial Fibrillation. JAMA. 2001;285(22):2864-2870.
75 Wyse DG, Waldo AL, DiMarco JP, et al. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med. 2002;347(23):1825-1833.
76 Gillette PC, Smith RT, Garson AJr, et al. Chronic supraventricular tachycardia. A curable cause of congestive cardiomyopathy. JAMA. 1985;253(3):391-392.
77 Shinbane JS, Wood MA, Jensen DN, et al. Tachycardia-induced cardiomyopathy: A review of animal models and clinical studies. J Am Coll Cardiol. 1997;29(4):709-715.
78 Fisch GR, Zipes DP, Fisch C. Bundle branch block and sudden death. Prog Cardiovasc Dis. 1980;23(3):187-224.