[level-membership-for-pediatrics-category]
Chapter 45 Asthma
The term status asthmaticus has been used to denote a more severe form of asthma attack, but its definition varies widely among different authors. To some authors, status asthmaticus is an asthma attack that does not respond to initial treatments with bronchodilators,1,2 whereas to others it indicates severe asthma that leads to respiratory failure and requires mechanical ventilatory support.3 For the purposes of this text, status asthmaticus is defined as an asthma attack that fails to respond to initial doses of nebulized β2-adrenergic and anticholinergic agents and systemic corticosteroid drugs and that requires admission to the hospital for continuation of treatment. Patients who experience relentless progression of respiratory signs and symptoms and require admission to the intensive care unit (ICU) are reported as having near-fatal asthma.4
Epidemiology and Risk Factors
Asthma is the most common chronic illness in childhood, affecting approximately 6.7 million children and adolescents in the United States, or 9.1% of persons younger than 18 years.5 Asthma is also a very common discharge diagnosis in children’s hospitals, accounting for approximately 5.6% of all hospital admissions and more than 150,000 admissions each year in the United States alone.5 The prevalence of asthma worldwide is highly variable, with greater than twentyfold differences in prevalence of symptoms encountered among centers located in various parts of the world.6 The highest prevalence rates for asthma are found in the United Kingdom, Australia, New Zealand, and the Republic of Ireland; very low prevalence rates occur in eastern Europe, the Indian subcontinent, and China.6 Race is a significant factor in determining prevalence and severity of asthma in children and young adults. African Americans are 4.1 times more likely to require treatment for asthma in the emergency department, two times more likely to be hospitalized for asthma, and 7.6 times more likely to die compared with white persons.5 Socioeconomic status also has been shown to negatively correlate with asthma prevalence, morbidity, and mortality in the United States.7
Asthma prevalence has increased steadily during the past 27 years in the United States.5 However, while the rate of nonurgent asthma-related outpatient visits continues to increase, emergency department visits and death rates have steadily declined during the past decade.5
The incidence of asthma-related respiratory failure requiring mechanical ventilation is difficult to determine because of variability in diagnostic criteria and reporting practices. Nonetheless, up to 36% of adult patients admitted to an inner-city medical ICU with near-fatal asthma require invasive mechanical ventilation.8 This figure appears to be significantly lower for children, considering that only 22 (10.2%) of 237 patients treated for near-fatal asthma underwent mechanical ventilation during a 2-year period in the pediatric ICU (PICU) of a tertiary children’s hospital (A.T. Rotta, unpublished data), and that only 14 (8.6%) of 163 patients required intubation in another study.9
The majority of patients with asthma who experience respiratory failure or arrest do so during the first stages of therapy or prior to arrival in the emergency department.9 Therefore early identification and close monitoring of patients at high risk for near-fatal asthma could be advantageous. High-risk patients often have a history of ICU admissions,10 mechanical ventilation,2,10 seizures or syncope during an attack,11 PaCO2 greater than 45 torr,2,10 attacks precipitated by food,10 or a history of rapidly progressive and sudden respiratory deterioration.1 These patients are likely to use more than two canisters of β-agonist metered-dose inhalers per month12 and often are poorly compliant or are receiving insufficient steroid therapy.13,14 Denial or failure to perceive the severity of an attack are factors frequently associated with near-fatal asthma.15,16 Although unquestionably some patients at risk for near-fatal asthma simply ignore early warning signs and do not seek adequate therapy, a subgroup of patients actually lacks normal perception of disease severity. Some patients with near-fatal asthma exhibit reduced chemosensitivity to hypoxia and blunted perception of dyspnea.17 Other patients have a decreased perceptual sensitivity of inspiratory muscle loads and display abnormal respiratory-related evoked potentials.18
Although many of these high-risk factors are commonly present in patients with near-fatal asthma, they fail to identify a significant number of cases. In one study, 33% of patients who died of asthma were judged to have a history of trivial or mild asthma, whereas 32% had never been admitted to the hospital with an asthma exacerbation.1 Some of these patients may in fact have what likely represents a distinct clinical entity known as sudden asphyxial asthma, a condition marked by acute onset of severe airway obstruction and hypoxia that rapidly leads to cardiorespiratory arrest in patients known to have only mild asthma or no asthma history at all.19,20
Pathophysiology
Asthma is primarily an inflammatory disease and, as such, it is marked by highly redundant pathways and complex interactions among inflammatory cells, mediators, and the airway epithelium21 (Figure 45-1). Functionally, asthma is characterized by variable airflow obstruction and airway hyperresponsiveness associated with airway inflammation. Pathologically, it is marked by mast cell degranulation, accumulation of eosinophils and CD4 lymphocytes, hypersecretion of mucus, thickening of the subepithelial collagen layer, and smooth muscle hypertrophy and hyperplasia.22
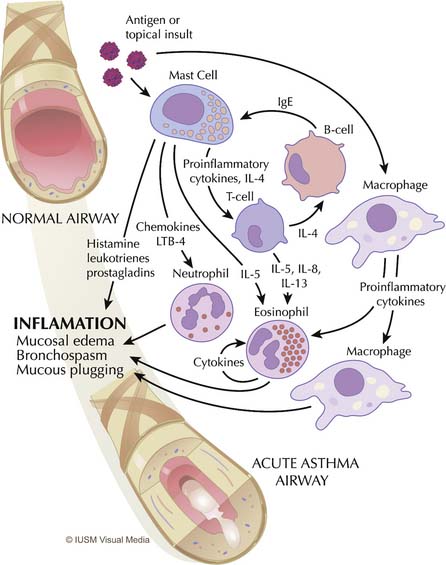
Mast cells, eosinophils, macrophages, and T lymphocytes are central to the derangements that occur during an acute attack (see Figure 45-1). The usual cascade begins with the activation and degranulation of mast cells in response to allergens or topical insults. The mast cells in turn promote activation of T lymphocytes by presenting these cells to the allergenic particles. The inflammatory process is then amplified by T-lymphocyte release of cytokines and chemokines. Increasing evidence exists that airway inflammation in asthma is the result of T-lymphocyte activation with the production of TH2 cytokines, such as interleukin (IL)-4, IL-5, IL-8, and IL-13.23 The presence of these TH2 cytokines leads to further augmentation of the inflammatory process through overexuberant production of immunoglobulin E (IgE) by B cells, stimulation of airway epithelial cells, and eosinophil chemotaxis. IgE stimulates mast cells to release leukotrienes, whereas interleukins (particularly IL-5) promote maturation and migration of activated eosinophils into the airway.24 This highly inflammatory milieu results in stimulation of airway epithelial cells and continued augmentation of the inflammatory process by further release of leukotrienes, prostaglandins, nitric oxide, adhesion molecules, and platelet-activating factor. This process results in overproduction of mucus and epithelial cell destruction that lead to airway plugging and denudation of the airway surface. Epithelial denudation is known to expose nerve endings, resulting in hyperirritable airways25 that become more susceptible to spasm and obstruction when challenged by subsequent exposure to allergens,26 inhaled irritants such as cigarette smoke and pollution,27 respiratory tract infections,28 psychological stress,29 and exercise,30 among other insults. Mucus itself is pathological in content,31,32 and mucus hypersecretion has been underappreciated as a cause of respiratory failure in persons with severe asthma, when in fact strong evidence exists that it may be a principal cause.31–33
Inflammation-mediated edema, mucus hypersecretion, airway plugging, and bronchospasm lead to the severe airway obstruction seen in patients with status asthmaticus and near-fatal asthma. The resulting obstruction and increased airway resistance create an impediment for inspiratory and expiratory gas flow, which leads to deranged pulmonary mechanics and increased lung volumes.32
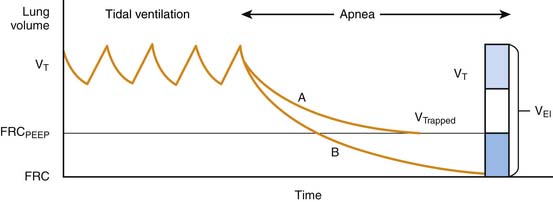
Airway plugging can result in ventilation/perfusion mismatching and increased oxygen requirements. Hypoxemia is common in patients with a severe asthma attack, but it is generally easily corrected with supplemental oxygen34 and is only weakly correlated with pulmonary function abnormalities.35 More frequently, airway plugging and obstruction lead to regional alveolar hyperinflation associated with reduced perfusion, resulting in a significantly increased pulmonary dead space. Most patients with this condition exhibit an increased respiratory rate in attempt to achieve a higher minute volume and compensate for the ventilation abnormality. Unfortunately, in patients with more severe disease, airway obstruction also results in significant prolongation of expiratory time, which, coupled with initiation of inspiration prior to completion of the previous exhalation, leads to dynamic hyperinflation, gas trapping, and the development of abnormally high lung volumes36 (Figure 45-2).
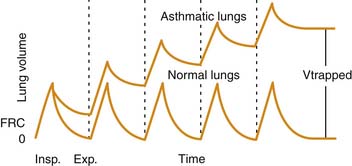
The higher lung volumes that result from incomplete alveolar emptying and dynamic hyperinflation serve as an adaptation mechanism to allow for higher expiratory flows than would have been possible at lower, more physiologic lung volumes. This higher expiratory flow is accomplished, however, at a high energy cost. Expiration becomes an active process, and the use of accessory muscles is required to overcome the high resistances to airflow both during inspiration and exhalation.37 During a severe attack, inspiratory transpulmonary pressures in excess of 50 cm H2O may be generated, compared with approximately 5 cm H2O during normal breathing.38 The increased muscle work is accompanied by an increase in blood flow to the diaphragm, but this flow often is insufficient to meet the much greater metabolic demands.39 Failure to promptly relieve the airway obstruction and reduce the work of breathing eventually leads to respiratory muscle fatigue, inadequate ventilation, and respiratory failure.
States of advanced airway obstruction and dynamic hyperinflation typical of severe asthma attacks have a significant impact on the circulatory system. The highly negative intrapleural pressures generated by spontaneously breathing patients during inspiration favor transcapillary edema fluid movement into the air spaces.37 They also cause a phasic increase in left ventricular afterload and a decrease in cardiac output40 that is clinically manifested as pulsus paradoxus.41 Right ventricular afterload may be increased during severe asthma as a result of pulmonary vasoconstriction related to hypoxia and acidosis. A state of increased pulmonary vascular resistance resulting from dynamic hyperinflation also can increase right ventricular afterload, further affecting cardiac output.41–43
Clinical Assessment
History
The child with an asthma exacerbation usually presents with complaints of difficulty breathing and shortness of breath. The presence of these complaints in a child known to have had previous asthma exacerbations is highly suggestive of the diagnosis. A significant percentage of children have a history of a coexisting viral upper respiratory infection, whereas some describe exposure to known allergic triggers. Circumstances permitting, time should be taken to inquire about the presence of high-risk factors (Box 45-1) for near-fatal asthma and the adequacy of maintenance intercrisis therapy.
Physical Examination
Wheezing, which is a common clinical finding in patients with acute asthma exacerbations, is the audible manifestation of the transmitted turbulence to airflow in the intrathoracic intrapulmonary airways. Wheezing may be predominantly expiratory as a result of the dynamic phasic compression of conducting airways, but it also can be biphasic. Wheezing in persons with severe asthma usually is symmetrical. An asymmetrical distribution suggests regional mucous plugging, atelectasis, pneumothorax, or the presence of a foreign body. The degree of wheezing correlates poorly with disease severity,44 because wheezes are heard only in the presence of airflow. As such, a patient with severe airway obstruction and very limited airflow may have a silent chest upon arrival at the emergency department, but loud wheezes may develop after effective therapy is instituted. Likewise, in a patient with loud wheezes that continue to worsen, a silent chest may develop as a prelude to respiratory failure.
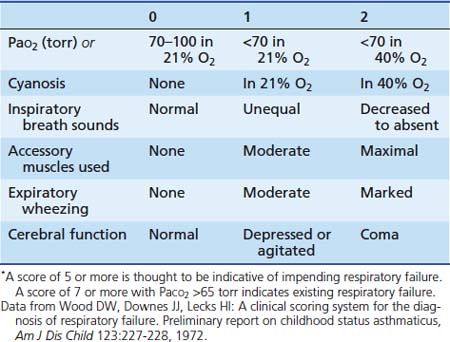
An objective assessment of disease severity is important in evaluating a patient’s response to therapy. Wood and colleagues45 developed a practical clinical asthma score composed of five variables with three different grades that allows for semiquantitative assessment of disease severity (Table 45-1). This clinical asthma score has been shown to correlate well with the need for prolonged bronchodilator therapy and hospitalization.46 However, although clinical asthma scores seem to be useful for assessing the severity of an attack, they are not as effective in prospectively identifying patients who require prolonged hospitalization or in whom complications and subsequent disability develop.47,48
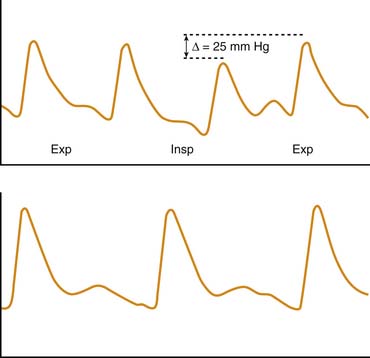
A less frequently used but more objective method of assessing disease severity and progression in patients with severe asthma is measurement of the pulsus paradoxus. Originally described by Adolf Kussmaul49 in a patient with constrictive pericarditis, pulsus paradoxus also is observed in conditions in which pleural pressure swings are exaggerated, such as status asthmaticus and near-fatal asthma. The simplest definition of pulsus paradoxus is an exaggeration of the physiologic inspiratory decrease in systolic blood pressure50 (Figure 45-3). It has been suggested that the term pulsus paradoxus is inappropriate to describe this phenomenon,51 because an accentuated inspiratory decrease in systolic pressure in the same direction as the normally occurring change cannot be described as a paradox. However, the true paradox described by Kussmaul49 was “the presence of a pulse slight and irregular, disappearing during inspiration and returning upon expiration despite the continued presence of the cardiac impulse during both respiratory phases.”50 Several mechanisms have been implicated in the occurrence of pulsus paradoxus in persons with asthma, and it is likely that various mechanisms contribute differently depending on the adequacy of intravascular volume, the magnitude of pleural pressure swings, the degree of pulmonary hyperinflation, and the state of cardiac contractility. These mechanisms include increased left ventricular afterload from highly negative intrapleural pressure52; decreased left ventricular preload as a result of inspiratory blood pooling in the pulmonary vasculature53; impaired left ventricular diastolic filling caused by a leftward shift of the interventricular septum resulting from increased venous return to the right heart54; constraint of cardiac filling because of longitudinal inspiratory deformation of the pericardium41; and increased right ventricular afterload with decreased filling of the left ventricle as a result of hyperinflation, acidosis, and hypoxia.55 The pulsus paradoxus can be measured easily in a patient who is spontaneously breathing by transducing pressure signals from an indwelling arterial catheter or by using a manual sphygmomanometer. In the latter technique, the cuff is inflated 20 mm Hg above the systolic pressure and then deflated until the first Korotkoff sounds are heard (systolic blood pressure). Initially, Korotkoff sounds are heard only during expiration. The cuff is then carefully deflated until the point where the sounds are heard equally during both inspiration and expiration. The difference between the highest systolic pressure and the pressure at which all Korotkoff sounds are heard is the magnitude of the pulsus paradoxus. During normal breathing, this difference is less than 5 mm Hg, but it is generally greater than 10 mm Hg during acute asthma exacerbations and greater than 20 mm Hg in patients with more severe disease.56 Changes in the magnitude of pulsus paradoxus during the course of therapy are good indicators of disease severity and clinical response to treatment.41,56
Laboratory Data
Arterial Blood Gas Analysis
Arterial blood gas measurements provide objective information on the adequacy of ventilation and oxygenation of the patient with asthma. The typical blood gas abnormality encountered in the early phase of asthma is relative hypoxemia with hypocapnia (PaCO2 <35 torr), reflecting hyperventilation.57 With worsening of airway obstruction, PaCO2 measurements return to the normal range of approximately 40 torr. However, this “normal” PaCO2 should not be viewed as reassuring when taken in the context of prolonged expiratory time, tachypnea, and accessory muscle use.58 In fact, PaCO2 greater than 40 torr in a patient with status asthmaticus should be interpreted as a sign of evolving respiratory muscle fatigue and warrants close clinical observation. Sicker patients often exhibit a mixed respiratory and metabolic acidosis.59 Lactic acidosis is frequently encountered in these patients and is thought to represent exaggerated lactate production by the respiratory muscles and tissue hypoxia.60
Muscle Enzymes
At least one third of patients with acute severe asthma exhibit an elevated plasma creatine kinase (CK) level.61 Although such elevations seem to be more pronounced in patients with marked acidemia or in those presenting with more severe respiratory insufficiency, a convincing correlation between disease severity and CK elevation has not been established.61 Myoglobin, a heme protein present in skeletal and cardiac muscle, is often also elevated in patients with near–fatal asthma.61 Elevations of CK–myocardial bound (CK–MB) isoenzyme develop in some patients with near–fatal asthma, suggesting a possible myocardial injury. This scenario certainly is plausible, considering that many patients are hypoxemic, acidotic, have high myocardial energy demand, and are receiving medications with adverse cardiac effects. However, plasma myoglobin and CK–MB elevations cannot be solely attributed to myocardial injury because the lungs and respiratory muscles are also known sources of these substances.62 Cardiospecific troponin T is a very sensitive and specific marker of myocardial cell damage63 and should be used preferentially to address the question of cardiomyocyte involvement in patients with severe asthma.
Electrocardiography
Patients with status asthmaticus or near-fatal asthma with significant airway obstruction and hyperinflation may exhibit a change in the mean frontal P–wave vector. A P–wave axis greater than 60 degrees has been associated with hyperinflation in both pediatric and adult patients with airway obstruction and is thought to represent positional atrial changes caused by inferior displacement of the diaphragm.64
Twelve–lead electrocardiography and continuous cardiac monitoring are valuable tools in the care of patients with near-fatal asthma in the ICU environment. These patients usually receive high doses of β-agonist drugs and may show evidence of hypokalemia (low–voltage T waves) or cardiac arrhythmias.65,66 The already increased myocardial energy demand resulting from airway obstruction is compounded by the chronotropic and vasodilatory effects inherent to β–agonist drugs and may lead to myocardial ischemia, particularly in adult patients with restricted coronary perfusion. Pediatric patients also may exhibit electrocardiographic (ECG) and enzymatic evidence of myocardial ischemia, particularly during treatment with intravenously administered isoproterenol.67 However, despite the fact that a study reported that a high percentage (66%) of patients exhibited nonspecific ST segment changes or other criteria suggestive of ischemia, these changes were not well correlated with initiation of terbutaline therapy or elevations in cardiac troponin T.68
Treatment
Initial Management in the Emergency Department
Patients with moderate or severe acute asthma also should receive a dose of systemic corticosteroid in the emergency department, which usually is administered prior to the second dose of albuterol. Prednisone (2 mg/kg) can be administered orally and is generally well tolerated. Oral prednisone is superior to inhaled fluticasone in children with severe asthma as evidenced by greater improvement in pulmonary function and lower hospitalization rates.69 The role of corticosteroid drugs in reversing an acute asthma attack in the emergency department has been the subject of debate, considering that these drugs require at least 4 to 6 hours for peak effects to be manifested.70 However, regardless of considerations about onset of action, acute suppression of inflammation is a cornerstone of acute asthma treatment and should be initiated as early as possible. Sicker patients with severe asthma exacerbations, those unable to tolerate oral medication because of respiratory distress or emesis, or those with a history of nausea during intensive β–agonist therapy should be given parenteral corticosteroid drugs such as methylprednisolone (2 mg/kg administered intravenously, followed by 0.5 to 1 mg/kg/dose administered intravenously every 6 hours).
Inhaled or nebulized anticholinergic agents such as ipratropium bromide are now considered an important adjunct in the treatment of persons with moderate and severe asthma exacerbations in the emergency department. In patients treated with one dose of a corticosteroid, use of ipratropium bromide (500 μg/2.5 mL) in conjunction with the second and third albuterol (salbutamol) doses has been associated with greater clinical improvement71 and reduced hospitalization rates compared with corticosteroid and albuterol (salbutamol) alone.72
Admission Criteria
The majority of patients with an acute asthma exacerbation respond to treatment in the emergency department and are discharged home. Among patients whose symptoms persist despite initial treatment, most can be safely managed in the general pediatric inpatient ward. Indications for hospitalization after treatment in the emergency department are loosely defined but may include (1) an inadequate response to three or four aerosol treatments; (2) relapse within 1 hour of receiving treatment with aerosols and steroids; (3) persistent SpO2 measurements of less than 91% in room air; (4) the need for oxygen therapy; (5) a significant reduction in peak expiratory flow rate; (6) having unreliable family support or being unable to comply with outpatient treatment; and (7) multiple visits for the same episode.73,74 Patients who require higher levels of monitoring or more invasive and aggressive treatment or who deteriorate during hospitalization in the general pediatric ward should be admitted to the PICU.
Management in the Intensive Care Unit
Oxygen
Sick patients with asthma are likely to exhibit hypoxemia as a result of intrapulmonary shunts caused by mucus plugging and atelectasis. Treatment with β–agonist agents also can contribute to hypoxemia by abolishing regional pulmonary hypoxic vasoconstriction and increasing intrapulmonary shunt.75,76 Therefore humidified oxygen should be offered both as a carrier gas for nebulizations and continuously between treatments.77 Supplemental oxygen can be safely incorporated into the treatment algorithm because, unlike in some adult patients with severe chronic obstructive pulmonary disease78 or asthma,79 no evidence exists to suggest that supplemental oxygen suppresses the respiratory drive in children with near–fatal asthma.
Corticosteroids
Corticosteroid drugs play a central role in the treatment of patients with status asthmaticus and near-fatal asthma, considering that these conditions are predominantly inflammatory in nature. Glucocorticosteroid agents modulate airway inflammation by a number of mechanisms, including direct interaction with cytosolic receptors and glucocorticosteroid response elements in gene promoters and indirect effects on binding of transcription factors, such as nuclear factor–κB, and on other cell signaling processes, such as posttranscriptional events.80 Gene products suppressed by glucocorticosteroid agents include a wide range of cytokines (IL–1, IL–2, IL–3, IL–4, IL–5, IL–6, IL–7, IL–8, IL–11, IL–12, IL–13, tumor necrosis factor–α, and granulocyte-macrophage colony–stimulating factor), adhesion molecules (intracellular adhesion molecule–1 and vascular cell adhesion molecule-1), and inducible enzymes, including NO synthase and cyclooxygenase–2.81 Transcription of other genes, such as lipocortin–1 and the β2–adrenergic receptor, may be enhanced.81 Glucocorticosteroid agents also decrease airway mucus production, reduce inflammatory cell infiltration and activation, and attenuate capillary permeability.82–85
In children with status asthmaticus or near–fatal asthma, glucocorticosteroid drugs should be administered by the IV route. The oral route can be used in selected cases, but inhaled glucocorticosteroid drugs play no role in the treatment of the sick hospitalized patient.21,69 The most common agent used in the United States is methylprednisolone because of its wide availability as an IV preparation and lack of mineralocorticoid effects. The usual dose of methylprednisolone is 0.5 to 1 mg/kg/dose, administered intravenously every 6 hours. Hydrocortisone, an agent with both glucocorticoid and mineralocorticoid activity, can be used as an alternative at doses of 2 to 4 mg/kg/dose, administered intravenously every 6 hours. Short courses of steroids usually are well tolerated without significant adverse effects.84 However, hypertension, hyperglycemia, mood disorders, and serious viral infections, such as fatal varicella, have been reported in previously well patients with asthma who have received glucocorticosteroid drugs.84,86,87 Duration of corticosteroid therapy is dictated by the severity of illness and the clinical response. Once initiated, treatment in patients with status asthmaticus or near-fatal asthma is generally continued for 5 to 7 days. Longer treatment courses necessitate gradual weaning of the drug to decrease the chances of symptomatic adrenal insufficiency or relapse. Prophylaxis with an H2 blocker should be considered because of the possibility of steroid-associated gastritis and gastric perforation.88
β–Agonists
The β–agonist properties of the sympathomimetic agents cause bronchial smooth muscle relaxation and hence bronchodilatation. These agents also can increase diaphragmatic contractility, enhance mucociliary clearance, and inhibit bronchospastic mediators from mast cells.89 Therefore β–agonists, along with systemic corticosteroids, are the mainstay of pharmacotherapy in persons with near-fatal asthma. β2–receptor selectivity is desirable to avoid adverse effects of nonselective α– and β1–adrenergic receptor stimulation. However, despite β2 selectivity, cardiovascular adverse effects remain a dose–limiting factor. The relative potency of various agents for the β2 receptor is as follows: isoproterenol > fenoterol > albuterol > terbutaline > isoetharine > metaproterenol.90 Of these agents, only albuterol and terbutaline are widely used in clinical practice, with some centers still using isoproterenol in selected occasions.
Once bound to the β–adrenergic receptor, β–agonists activate adenyl cyclase, resulting in increased intracellular cyclic adenosine monophosphate (cAMP) levels, which leads to bronchial and vascular smooth muscle relaxation. Dose-response curves demonstrate that large dose increases fail to enhance bronchodilation significantly; however, as the degree of bronchial constriction increases, the bronchodilation dose–response curve shifts to the right, indicating the need for a higher dose to achieve the desired response.90
The most frequent untoward adverse effects of β–agonist agents are skeletal muscle tremor, nausea, and tachycardia. These adverse effects are common to both nonselective and selective β–agonist drugs administered by IV and inhalational routes. Other cardiovascular adverse effects include blood pressure instability (predominantly diastolic hypotension) and cardiac dysrhythmias.91,92 Myocardial ischemia has been well documented as a complication of IV isoproterenol administration to children with near-fatal asthma.67,93 However, continuous IV infusions of terbutaline appear to be safe and are not associated with significant cardiotoxicity.68 Prolongations of the QTc interval and hypokalemia have been observed during IV infusions of β–agonist drugs.94 Hypokalemia occurs in the setting of relatively stable total body potassium and is the result of intracellular potassium shifting that results, at least in part, from an increased number of sodium-potassium pumps and not from augmented potassium elimination.95 Therefore supraphysiologic potassium supplementation is rarely necessary. A less frequently recognized adverse effect of β–agonist agents is hypoxemia, which is likely related to decreased regional hypoxic pulmonary vasoconstriction in areas of atelectasis and the resultant increased intrapulmonary shunt.75,76
Albuterol (Salbutamol)
Albuterol is the most β2–specific aerosol agent available in the United States. It usually is administered every 20 minutes during the initial phase of treatment at a dose of 0.05 to 0.15 mg/kg. The optimal dose and frequency of albuterol are controversial because less than 1% of the nebulized drug is deposited in the lung.96 Moreover, spontaneous tidal volume, breathing pattern, and technique are unpredictable yet major determinants of drug delivery. After the initial series of three albuterol treatments, continuous albuterol nebulization should be started for patients who require nebulization treatments more frequently than every 1 hour.
Continuous albuterol nebulization appears to be superior to repeated intermittent dosing and has not been shown to cause significant cardiotoxicity.97–99 A small prospective randomized study in children with near–fatal asthma and impending respiratory failure indicated that children treated with continuous albuterol nebulization had more rapid clinical improvement and shorter hospitalizations compared with children treated with intermittent albuterol doses.98 Continuous administration of albuterol also was associated with more efficient allocation of respiratory therapists’ time98 and could offer the added advantage of more hours of uninterrupted sleep to patients who often are already exhausted.100 The usual dose of continuously administered albuterol ranges between 0.15 and 0.45 mg/kg/h, with a maximum dose of 20 mg/h. Higher doses of albuterol have been used in patients who are unresponsive to standard treatment.51 However, we do not support this practice, because the intensification of adverse effects can outweigh any small incremental gain in bronchodilatation.
The availability of levalbuterol has generated some controversy. Albuterol is a 50:50 mixture of R–albuterol (levalbuterol), the active enantiomer that causes bronchodilation, and S–albuterol, which was thought to be inactive in humans. The U.S. Food and Drug Administration has approved levalbuterol, the pure R–isomer, as a preservative–free nebulizer solution.101 The purported advantage of using levalbuterol over albuterol stems from the fact that S–albuterol may not be completely inert and has a longer elimination half–life than R–albuterol.102,103 However, the notion that S–albuterol is not inert and that it is capable of clinically significant adverse effects is not universally accepted.104–106 A large randomized controlled trial of levalbuterol versus racemic albuterol in children with asthma demonstrated a decreased rate of hospitalization in patients treated with levalbuterol. However, this study had methodological problems, as the primary outcome variable (rate of hospital admission) was left to the discretion of the treating physicians and none of the secondary outcome variables were significantly different between treatment groups once the patients had been admitted to the hospital.107 More recent randomized clinical studies in children with asthma failed to show definitive evidence that levalbuterol is superior to a regular racemic albuterol.108,109 Furthermore, although the cost of levalbuterol has decreased significantly in the past few years, this drug continues to be more expensive than albuterol (C. A. Thomas, PharmD, Riley Hospital for Children, personal communication, 2011). Considering the lower cost of albuterol and the paucity of clinical evidence supporting the superiority of levalbuterol, we continue to favor albuterol as the routine bronchodilator of choice in children with near–fatal asthma.
Intravenously administered albuterol is not available in the United States. However, the efficacy of albuterol infusions in patients with severe asthma has been well established in countries where the IV preparation is available.110–112
Terbutaline
Terbutaline is a relatively selective β2–agonist with a mechanism of action that is similar to that of albuterol. It is the most commonly used parenteral β–agonist in the United States and is available for nebulization, subcutaneous injection, and IV use. Because of its lower β1–receptor affinity, subcutaneous administration of terbutaline has largely supplanted the use of epinephrine in persons with severe acute asthma. The use of subcutaneous terbutaline is limited in the PICU environment; it is reserved for patients with acute worsening of the respiratory status who do not have vascular access and in whom access cannot be easily obtained. Subcutaneous terbutaline is more commonly used in the acute management of sick patients in the emergency department and in the prehospital setting. The usual subcutaneous terbutaline dose is 0.01 mg/kg/dose (maximum 0.25 mg) subcutaneously every 20 minutes for three doses, as necessary.
Terbutaline is more commonly used in the ICU environment through the IV route. This therapy is indicated for patients with near-fatal asthma who fail to improve or show signs of deterioration during treatment with nebulized β2–agonists, ipratropium bromide, and steroids. The usual IV terbutaline doses are 0.1 to 10 μg/kg/min as a continuous infusion,91 prepared in 0.9% normal saline solution or D5W. In our clinical experience, however, most patients are started on a dose of 1 μg/kg/min and the dose is titrated to effect, with doses higher than 4 μg/kg/min rarely necessary. Patients receiving doses lower than 1 μg/kg/min can be given a loading dose of 10 μg/kg over 10 minutes to accelerate the onset of action.
Anticholinergic Agents
Anticholinergic agents have become an important part of the treatment of children with severe acute asthma. The prototypical anticholinergic agent used in treating patients with asthma is ipratropium bromide, a quaternary ammonium compound formed by the introduction of an isopropyl group to the N atom of atropine. Unlike atropine (a tertiary ammonium compound), ipratropium bromide does not cross the blood-brain barrier, thus preventing the occurrence of central anticholinergic adverse effects. Considering that bronchial smooth muscle tone is influenced by the parasympathetic tone, ipratropium bromide can produce bronchodilation by inhibition of cholinergic-mediated bronchospasm.113 An unexpected but important property of ipratropium bromide is the lack of negative effect on ciliary bronchial epithelium, unlike the marked inhibition of ciliary beating and mucociliary clearance produced by atropine.113
Nebulized ipratropium bromide (250- to 500-μg doses) can be used every 20 minutes during the first hour in the emergency department. The recommended dose for continuation therapy is 250 to 500 μg, given every 6 hours. After inhalation, peak responses usually develop over 30 to 90 minutes, and clinical effects may persist for more than 4 hours.113 Systemic effects are minimal because less than 1% of an inhaled dose of ipratropium bromide is absorbed into the circulation. However, extrapulmonary effects such as mydriasis and blurred vision have been reported as a result of topical ocular absorption of the drug.114,115
The addition of ipratropium bromide to nebulized albuterol in the treatment of bronchospasm makes pharmacological sense, because albuterol causes bronchodilatation by increasing cAMP levels, while the effect of ipratropium bromide is mediated by a decrease in cyclic guanosine monophosphate. The combined use of ipratropium bromide and nebulized albuterol in treating children with asthma who present to the emergency department has proved to be cost effective and reduces the rate of admission to the hospital.71,72 However, the routine addition of repeated doses of nebulized ipratropium bromide to a standard regimen of β2–agonist agents and systemic steroid drugs in hospitalized children with status asthmaticus does not appear to confer a significant benefit.116,117 Considering the high safety profile of inhaled ipratropium bromide treatments, the benefits of its use in the emergency department, and the lack of data specific to the PICU population, we find it reasonable to administer ipratropium bromide along with standard therapy for critically ill patients with status asthmaticus until definitive evidence becomes available.
Magnesium Sulfate
Magnesium is a physiologic calcium antagonist that causes smooth muscle relaxation as a result of inhibition of calcium uptake. It has been known for more than 60 years that magnesium causes bronchorelaxation in patients with asthma,118 but its incorporation as an adjunct in the treatment of patients with severe asthma has occurred only recently. Numerous reports, case series, and randomized controlled trials have suggested clinical improvement when asthmatic patients with severe airway obstruction receive IV magnesium sulfate infusions in the emergency department or ICU.119,120 Magnesium appears to be as effective as albuterol when delivered by nebulization121 and has been used successfully by some practitioners as a liquid vehicle for albuterol nebulization.122
The indication for IV magnesium sulfate in children with status asthmaticus or near–fatal asthma is still controversial because of the paucity of randomized controlled trials. Some studies suggest that magnesium sulfate infusions are associated with significant improvements in short-term pulmonary function,123–126 whereas another study failed to show improvement in disease severity or a reduction in hospitalization rates.127 The usual dose of magnesium sulfate in children with status asthmaticus or near-fatal asthma is 25 to 40 mg/kg/dose, intravenously, infused over 20 to 30 minutes.126 The onset of clinical response is rapid (occurring in minutes) and is generally observed during the initial infusion. During the infusion patients should be carefully monitored for adverse effects, which include hypotension, nausea, and flushing. Serious toxicity involving cardiac arrhythmias, muscle weakness, areflexia, and respiratory depression has not been reported with the use of magnesium sulfate in persons with acute asthma, when used as directed. The IV infusion of magnesium sulfate under controlled conditions appears to be safe, and a subset of patients with status asthmaticus and near-fatal asthma clearly responds to this mode of therapy.123–126 A systematic review of the published randomized controlled trials supports the use of magnesium sulfate in addition to β2–agonist agents and systemic steroid drugs in the treatment of persons with severe acute asthma.128
Methylxanthine Agents
The exact molecular mechanism of theophylline–mediated bronchodilation is unclear but is thought to involve, at least in part, its action as a phosphodiesterase-4 inhibitor, reducing the degradation of cAMP, which in turn mediates cellular responses that result in bronchial smooth muscle relaxation.129 Other mechanisms of action have been proposed, including inhibition of phosphoinositide 3–kinase activity,130 adenosine receptor antagonism,131 increasing histone deacetylase activity,132 stimulation of endogenous catecholamine release,133 prostaglandin antagonism,134 and alterations in intracellular calcium mobilization.135 Theophylline is also known to cause inhibition of afferent neuronal activity,136 thereby leading to inhibition of bronchospasm mediated by reflex activation of cholinergic pathways. Theophylline has antiinflammatory and immunomodulatory actions137 and is known to augment diaphragmatic contractility and increase respiratory drive.138
The bronchodilator effects of theophylline in isolated human bronchial preparations in vitro occur at concentrations greater than 70 μmol/L, which is capable of inducing a 50% reversal of bronchoconstriction.139 Such high local concentrations presumably would be achieved with plasma levels greater than 10 to 20 μg/mL.140 In clinical practice, however, this range poses a difficult problem because of the narrow window between therapeutic levels and toxicity, which often overlap. The half–life of theophylline ranges from 3 to 7 hours.141 Therefore theophylline is generally administered as a continuous IV infusion to avoid significant fluctuations in serum concentrations. Aminophylline is equivalent to 80% theophylline and also is administered by continuous IV infusion. When a decision is made to initiate therapy with theophylline or aminophylline, a loading dose is given to achieve serum levels between 10 and 20 μg/mL. Assuming a normal average volume of distribution, a 1 mg/kg dose of theophylline (1.25 mg/kg of aminophylline) raises the serum concentration by 2 μg/mL. The loading dose should be administered over 20 minutes and should be followed immediately by the continuous infusion of the drug. Empiric doses can be started for patients with normal hepatic and cardiac function as follows: infants younger than 6 months: 0.5 mg/kg/h; infants aged 6 months to 1 year: 0.85 to 1 mg/kg/h; children aged 1 to 9 years: 1 mg/kg/h; and children older than 9 years: 0.75 mg/kg/h. Patients with compromised hepatic and cardiovascular function should be started at a dose of 0.25 mg/kg/h. Obese patients should have doses calculated by ideal body weight to prevent toxicity. Serum drug levels should be monitored 30 to 60 minutes after the loading dose and frequently during the continuous infusion, considering that steady–state concentrations are not achieved until approximately five half–lives, which corresponds to 24 to 36 hours of infusion.
A number of studies in adults and children with acute asthma indicate that therapy with theophylline or aminophylline is of no clinical benefit.142–144 More recently, randomized, placebo–controlled trials tested the efficacy of aminophylline9 and theophylline145 in children with near–fatal asthma in the ICU environment. Aminophylline treatment resulted in significantly improved physiologic outcomes, such as oxygenation and pulmonary function testing, but did not decrease ICU length of stay and was associated with adverse effects such as nausea and vomiting.9 Theophylline was associated with faster clinical improvement, but it had no effect on PICU length of stay and led to a significantly higher frequency of vomiting compared with control subjects.145
Helium-Oxygen Mixtures
Helium is a biologically inert gas that is less dense than any other known gas except hydrogen and is about one seventh as dense as air. The medicinal application of helium and oxygen mixtures (heliox) in the treatment of asthma and extrathoracic airway obstruction has been known for approximately 7 decades.146 Because of its low density, heliox reduces the Reynolds number. This effect is associated with a reduced likelihood of turbulent gas flow while facilitating laminar gas flow in the airways, thus decreasing the work of breathing in situations associated with high airway resistance.147 Heliox provides a theoretical benefit to patients with obstructive lesions of the extrathoracic and intrathoracic airways. Several reports advocate the benefit of heliox in the management of children with extrathoracic airway obstruction.147,148 The role of heliox in patients with asthma is less clear.
Research using heliox mixtures has demonstrated a greater percentage of lung particle retention and a greater delivery of albuterol from both metered–dose inhalers and nebulizers,149,150 suggesting that one of the beneficial effects of heliox use in patients with asthma is improved deposition of aerosolized drugs. A recent study in children with moderate to severe asthma exacerbations showed that 70%/30% heliox–driven continuous nebulized albuterol treatments were associated with a greater degree of clinical improvement compared with oxygen–driven continuous nebulized albuterol.149
Heliox has been recommended by some persons as a useful adjunct in adult patients with severe asthma, both during spontaneous breathing and during mechanical ventilation.151–154 Anecdotal reports suggest that heliox is associated with improvement in pulmonary function in children with acute asthma.155,156 However, a small randomized crossover trial of heliox in spontaneously breathing patients with severe asthma failed to show improvement in pulmonary function or dyspnea scores.157 Additionally, a systematic review of seven prospective, controlled trials in children and adults failed to provide support for the use of heliox in patients with moderate or severe acute asthma.158 The paucity of well–executed, randomized, controlled studies makes it impossible to assess the therapeutic effect of heliox in children with asthma at this time. In addition, should heliox be beneficial in some patients, the duration of administration and optimal helium–oxygen mixture remain undetermined. Until more sound information emerges, heliox remains an unproved therapy for pediatric asthma, and its use should be restricted to individual attempts in selected patients with severe refractory near–fatal asthma who did not respond to more conventional treatments.159 The need to use 80:20 or 70:30 helium–oxygen mixtures to take full advantage of the lower gas density properties may further thwart the use of heliox in sicker patients who exhibit significant hypoxemia.
Ketamine
Ketamine hydrochloride is a dissociative anesthetic agent available in a solution for IV or intramuscular administration. The term dissociative anesthetic is derived from the strong feeling of dissociation from the environment that is experienced by the subject to whom it is administered. After IV administration, a sensation of dissociation is generally experienced within 15 seconds, and unconsciousness becomes apparent after another 30 seconds. This reaction is followed by intense analgesia that lasts approximately 40 to 60 minutes and amnesia that may persist for up to 2 hours. Some patients, particularly older children, may experience a postanesthesia emergence reaction with confusion, agitation, and hallucinations. Usual ketamine doses do not significantly affect hypoxic or hypercarbic respiratory drive.160 Pharyngeal and laryngeal reflexes are maintained, and although the cough reflex is somewhat depressed, airway obstruction does not normally occur. Aside from its anesthetic properties, ketamine exerts a number of other effects, including sialorrhea. It increases airway secretions, cardiac output, heart rate, blood pressure, metabolic rate, cerebral blood flow, and intracranial pressure.161 Pulmonary vascular resistance is not altered, and hypoxic pulmonary vasoconstriction is preserved. Ketamine inhibits bronchospasm and lowers airway resistance, presumably through blockage of N-methyl-D-aspartate receptors in airway smooth muscle.162 The bronchodilatory effect of ketamine makes it an attractive agent in patients with asthma who require sedation and anesthesia for intubation or mechanical ventilation.163,164 However, the bronchodilatory effects of ketamine are often obliterated by the significant observed increase in airway secretions and sialorrhea.
Some controversy exists regarding the use of ketamine in nonintubated patients with near-fatal asthma, with the goal of avoiding the need for mechanical ventilation. Limited evidence suggests that this strategy may be viable in selected patients.165 In our experience, the administration of ketamine to nonintubated children with severe refractory asthma frequently precedes the need to intubate and is rarely associated with significant and noticeable clinical improvement. For this reason, attempts at administering ketamine to nonintubated children with severe refractory asthma should always take place in the ICU under strictly monitored conditions and with personnel capable of rapidly establishing an airway for initiation of ventilatory support.
Mechanical Ventilation
Indications
Some patients may benefit from attempts to attenuate respiratory muscle fatigue with a trial of noninvasive ventilation.132 However, the use of bilevel positive airway pressure requires patient cooperation and a well–fitted and sealed mask, which may prove difficult, if not impossible, to achieve in an anxious and agitated child with impending respiratory failure.
Intubation
A cuffed endotracheal tube should be introduced and its placement confirmed by a colorimetric method or capnography, auscultation, and chest radiograph. Special attention to the manual ventilation technique is needed to avoid fast rates that often are inadvertently applied immediately following intubation. Rapid respiratory rates applied to intubated children with severe airway obstruction lead to a state of high lung volume, significant dynamic hyperinflation, hypoxemia, and hemodynamic instability (hypotension). These patients require slow respiratory rates with very prolonged expiratory times to allow for adequate gas exchange and lung volumes. A helpful maneuver is to establish the timing of the next inspiration by using a stethoscope to auscultate for the disappearance of expiratory wheezes, thus marking the end of the previous exhalation. The occurrence of desaturation and hypotension following intubation should prompt an equipment check and confirmation of tube placement. A tension pneumothorax must be considered in patients with hypoxemia and hypotension who fail to improve rapidly after administration of fluids and optimization of ventilation (or brief endotracheal tube disconnection), particularly when unequal breath sounds are present.
Ventilator Settings
The goal of mechanical ventilation in patients with acute asthma should be to reverse hypoxemia (if present), relieve respiratory muscle fatigue, and maintain a level of alveolar ventilation compatible with an acceptable pH, while avoiding iatrogenic hyperinflation and levels of intrathoracic pressure that could adversely affect cardiac output. Therefore the choice of mechanical ventilator settings must take into consideration the significant derangements of lung mechanics and function that are inherent to persons with severe acute asthma. Ill–advised attempts to achieve a normal PaCO2 would require fast respiratory rates, high minute volumes, and very high airway pressures, which are associated with the development of barotrauma (pneumothorax and pneumomediastinum) and high mortality rates.166–168
A paradigm shift in the ventilatory management of patients with asthma occurred with the introduction of a strategy of controlled hypoventilation reported by Darioli and Perret.169 Their strategy resulted in no mortality in 34 episodes of mechanical ventilation in 26 patients and significantly lower complication rates in comparison with historical controls.169 This approach used tidal volumes between 8 and 12 mL/kg and targeted peak airway pressures up to 50 cm H2O. Tidal volumes were further reduced if the peak pressure limit could not be respected and higher Paco2 measurements were tolerated.169 A similar approach using respiratory rates lower than 12 breaths/min, tidal volumes between 8 and 12 mL/kg, peak inspiratory pressures of 40 to 45 cm H2O, and permissive hypercapnia also resulted in very few complications and no mortality or long–term morbidity in 19 mechanically ventilated children with near-fatal asthma.170
From a simplified perspective, the modes of ventilatory support for patients with severe acute asthma can be divided between pressure and volume preset. No definitive evidence exists to suggest that one particular mode of ventilation is superior to the other. However, to safely ventilate a patient with asthma, the characteristics of each mode must be understood. Pressure control modes use a decelerating gas flow and have the advantage of ensuring that a particular inspiratory pressure limit is respected. The main disadvantage of pressure control modes is that tidal volumes can vary greatly with changes in airway resistance and the state of hyperinflation. Volume control modes deliver a constant tidal volume, provided there is no significant air leak. An added advantage of volume control is that it allows for comparison of peak inspiratory pressure and plateau pressure measurements (peak-to-plateau pressure), which can serve as a longitudinal indicator of airway resistance and response to therapy. For these measurements, the plateau pressure is obtained by performing an inspiratory hold (a feature ubiquitous to most ventilators) and is then compared with the peak inspiratory pressure (Figure 45–4). An increasing peak–to–plateau pressure indicates increasing airway resistance, whereas a decreasing peak–to–plateau pressure suggests response to therapy. A disadvantage of volume control ventilation is that very high lung volumes can develop if exhalation is incomplete, because tidal volumes remain constant breath to breath. The option of using pressure–regulated volume control, a mode available in some ventilators, offers some of the advantages of pressure control and of volume control, including optimal inspiratory gas flow, assured tidal volumes, and minimized airway pressures.
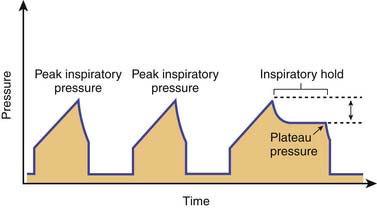
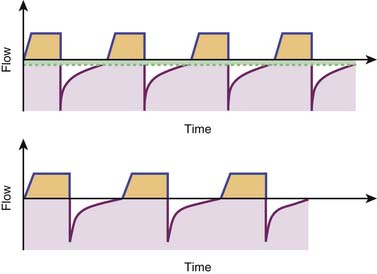
Use of positive end–expiratory pressure (PEEP) in intubated patients with asthma has been the focus of controversy. Externally applied PEEP may benefit patients with expiratory flow limitation resulting from dynamic compression of small airways by moving the equal pressure point, stenting collapsed or severely narrowed airways, and enabling decompression of upstream alveoli.171 The application of low levels of PEEP that are, by definition, lower than the level of auto–PEEP also may relieve dyspnea by facilitating ventilator triggering and synchronization for intubated patients capable of drawing spontaneous breaths.171,172 However, as elegantly demonstrated by Tuxen,173 use of PEEP in chemically paralyzed patients with severe airflow obstruction was uniformly associated with higher lung volumes, increased airway and intrathoracic pressures, and circulatory compromise (Figure 45-5).
Our personal preference is to use the volume control synchronized mandatory ventilation mode or the pressure regulated volume control mode, with tidal volumes of 8 to 12 mL/kg, which can be reduced as needed to generate peak inspiratory pressures of 45 cm H2O or less and plateau pressures 30 cm H2O or less. The initial tidal volume target of 8 to 12 mL/kg might seem high, particularly in the era of lung protective ventilation with reduced tidal volumes for patients with acute respiratory distress syndrome. However, it is important to note that the prototypical patient with near-fatal asthma does not have significant parenchymal lung injury or the heterogeneously decreased lung compliance typical of patients with acute respiratory distress syndrome and that tidal volumes are often reduced as needed to target conservative peak and plateau pressure goals, as previously discussed. Respiratory rate is initially set between 6 and 12 breaths/min, and inspiratory time is set between 1 and 1.5 seconds, allowing for expiratory times between 4 and 9 seconds. PEEP is set at zero for the patient under neuromuscular blockade. With intensification of therapy and clinical improvement, neuromuscular blockade is stopped and trigger sensitivity for spontaneous breaths is optimized. A low level of PEEP (lower than the measured auto-PEEP and never in excess of 8 cm H2O) is applied to facilitate synchronization between patient and machine, and spontaneous breaths are aided by the application of pressure support.
Use of high-level pressure support in the management of spontaneously breathing intubated patients with asthma with the goal of reducing inspiratory work while allowing the patient to actively assist with exhalation is an intriguing strategy that warrants further study.174
Ventilatory Monitoring
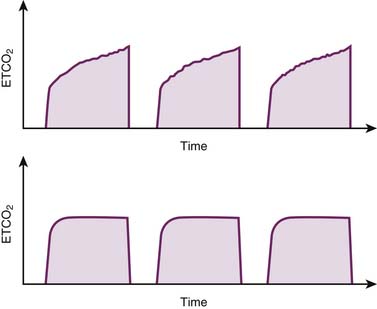
Regardless of the chosen mode of ventilation, patients with near-fatal asthma undergoing mechanical ventilation require very close monitoring. Frequent auscultation can provide valuable information regarding symmetry of breath sounds (e.g., pneumothorax or mucus plugging) and optimal length of exhalation. Monitoring modules capable of analyzing and displaying permutations of important variables, such as pressure, volume, flow, and time, can provide important information that assists in the optimization of ventilatory settings (Figure 45-6). Monitoring peak-to-plateau pressure differences allows for inferences regarding airway resistance and response to treatment. The shape of the capnography curve also may provide insights regarding adequacy of lung emptying (Figure 45-7), while integrated volumetric capnography can track changes in alveolar dead space over time.
Analgesia, Sedation, and Muscle Relaxation
Muscle relaxation with neuromuscular blockers should be maintained following initiation of mechanical ventilation until satisfactory gas exchange and clinical stability are achieved. Patients who exhibit hypercapnia during mechanical ventilation require continuation of neuromuscular blockers to abolish spontaneous respiratory movements that could worsen dynamic hyperinflation. However, use of neuromuscular blockers should be discontinued as soon as feasible to reduce the likelihood of serious neurologic complications, such as prolonged muscle weakness or paralysis, from the association of these agents and corticosteroid drugs.175,176 Reports of prolonged paralysis and myopathy after the concomitant use of corticosteroid drugs and aminosteroid-based agents, such as vecuronium and pancuronium, led to the preferential use of benzylisoquinolinium compounds, such as cisatracurium, in patients with asthma. However, this combination may not be completely safe, because prolonged muscle weakness has been observed in a patient treated with cisatracurium and corticosteroids.177
Inhalational Anesthetic Agents
Inhalational anesthetic agents have been used for their bronchodilatory effects in the treatment of mechanically ventilated patients with near-fatal asthma that is refractory to more conventional treatment modalities.178 The exact mechanism responsible for bronchodilatation during inhalational anesthesia is unknown but may involve direct inhibition of vagal tone.179 Various agents have been used successfully in both adult and pediatric patients with refractory near-fatal asthma, including halothane,180 isoflurane,181 enflurane,182 and sevoflurane.182,183 Although halothane is often recommended as an agent of choice, little evidence exists in humans to suggest that it is more effective than other agents.184 Sevoflurane compares favorably with halothane and appears to be less noxious to human airways than isoflurane or enflurane.185
Therapy for persons with refractory near-fatal asthma with an inhaled anesthetic agent should be performed only in a well-monitored ICU environment, under the direction of personnel experienced in the administration of these agents and their adverse effects. Patients treated with halothane can experience significant hypotension as a result of myocardial depression and require rapid fluid expansion and inotropic support.186,187 Halothane is associated with cardiac arrhythmias, particularly during concurrent administration of epinephrine,188 which explains why many physicians prefer a nonarrhythmogenic alternative such as isoflurane. Isoflurane does not have negative inotropic effects but may still cause hypotension because of vasodilatation.159 Considering that halothane and isoflurane result in equivalent bronchodilation, isoflurane is preferred for use in children because of its less significant adverse effects. The use of subanesthetic doses of inhalational anesthetic agents in attempts to avoid mechanical ventilation in spontaneously breathing patients with severe asthma during maximal medical treatment is an intriguing strategy that warrants further study.189
Bronchoscopy
Increased bronchial secretions and mucus plugging play a major role in the continued deterioration observed in some patients with severe acute asthma who fail to respond to maximal therapy.31–33 Mucous plugging and casts can cause atelectasis of large segments and worsen the heterogeneity of ventilation and dynamic hyperinflation (Figure 45-8). Thus a small percentage of mechanically ventilated patients with severe near-fatal asthma may require selective suction of mucus plugs, casts, or thick secretions by bronchoscopy.190 The combination of bronchial lavage with mucolytic agents such as N-acetylcysteine191 or recombinant human deoxyribonuclease192 and aggressive selective suction through a bronchoscope may be beneficial in patients with clinically significant mucous plugging who fail to respond to maximal therapy and traditional tracheal suction.
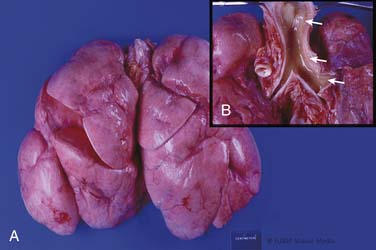
Extracorporeal Life Support
The use of extracorporeal life support (ECLS) has been reported in the management of the very few patients with near-fatal asthma who continue to exhibit a profound degree of clinical instability despite maximal therapy.193,194 Such cases are most unusual, as indicated by the very small number of patients with acute asthma as the primary diagnosis in the Extracorporeal Life Support Organization registry (1.36%, i.e., 173 of 12,763 pediatric and adult ECLS runs).195 Interestingly, the survival rate for persons with near-fatal asthma supported by ECLS is approximately 78%,195 which is remarkable considering that the vast majority of these patients were extraordinarily sick and had not responded to all forms of aggressive treatment.
Prognosis
The prognosis of patients with status asthmaticus or near-fatal asthma who receive proper medical therapy is excellent. Better understanding of the pathophysiology of airway obstruction and dynamic hyperinflation, coupled with improved mechanical ventilation strategies and aggressive pharmacologic treatment, has reduced the ICU mortality rate to nearly zero in these patients.196,197 Asthma fatalities still occur in patients with sudden onset of severe airway obstruction who do not come to medical attention prior to the development of respiratory failure or cardiorespiratory arrest.198,199 The treatment plan for patients admitted to the hospital with status asthmaticus or near-fatal asthma should be carefully reviewed prior to discharge to ensure adequate outpatient therapy, education, and follow-up in an attempt to reduce the likelihood of a preventable recurrence.
References are available online at http://www.expertconsult.com.
1. Robertson C.F., Rubinfeld A.R., Bowes G. Pediatric asthma deaths in Victoria: the mild are at risk. Pediatr Pulmonol. 1992;13:95-100.
2. Mountain R.D., Sahn S.A. Clinical features and outcome in patients with acute asthma presenting with hypercapnia. Am Rev Respir Dis. 1988;138:535-539.
3. Afzal M., Tharratt R.S. Mechanical ventilation in severe asthma. Clin Rev Allergy Immunol. 2001;20:385-397.
4. Restrepo R.D., Peters J. Near-fatal asthma: recognition and management. Curr Opin Pulm Med. 2008;14:13-23.
5. Akinbami L.J., Moorman J.E., Garbe P.L., et al. Status of childhood asthma in the United States, 1980-2007. Pediatrics. 2009;123(Suppl 3):S131-S145.
6. Warner J.O. Worldwide variations in the prevalence of atopic symptoms: what does it all mean? Thorax. 1999;54(Suppl 2):S46-S51.
7. Lwebuga-Mukasa J.S., Dunn-Georgiou E. The prevalence of asthma in children of elementary school age in western New York. J Urban Health. 2000;77:745-761.
8. Afessa B., Morales I., Cury J.D. Clinical course and outcome of patients admitted to an ICU for status asthmaticus. Chest. 2001;120:1616-1621.
9. Yung M., South M. Randomised controlled trial of aminophylline for severe acute asthma. Arch Dis Child. 1998;79:405-410.
10. Turner M.O., Noertjojo K., Vedal S., et al. Risk factors for near-fatal asthma. A case-control study in hospitalized patients with asthma. Am J Respir Crit Care Med. 1998;157:1804-1809.
11. Strunk R.C., Mrazek D.A., Fuhrmann G.S., et al. Physiologic and psychological characteristics associated with deaths due to asthma in childhood. A case-controlled study. JAMA. 1985;254:1193-1198.
12. Spitzer W.O., Suissa S., Ernst P., et al. The use of beta-agonists and the risk of death and near death from asthma. N Engl J Med. 1992;326:501-506.
13. Kolbe J., Vamos M., Fergusson W., et al. Determinants of management errors in acute severe asthma. Thorax. 1998;53:14-20.
14. Ordonez G.A., Phelan P.D., Olinsky A., et al. Preventable factors in hospital admissions for asthma. Arch Dis Child. 1998;78:143-147.
15. Birkhead G., Attaway N.J., Strunk R.C., et al. Investigation of a cluster of deaths of adolescents from asthma: evidence implicating inadequate treatment and poor patient adherence with medications. J Allergy Clin Immunol. 1989;84:484-491.
16. Martin A.J., Campbell D.A., Gluyas P.A., et al. Characteristics of near-fatal asthma in childhood. Pediatr Pulmonol. 1995;20:1-8.
17. Kikuchi Y., Okabe S., Tamura G., et al. Chemosensitivity and perception of dyspnea in patients with a history of near-fatal asthma. N Engl J Med. 1994;330:1329-1334.
18. Kifle Y., Seng V., Davenport P.W. Magnitude estimation of inspiratory resistive loads in children with life-threatening asthma. Am J Respir Crit Care Med. 1997;156:1530-1535.
19. Saetta M., Thiene G., Crescioli S., et al. Fatal asthma in a young patient with severe bronchial hyperresponsiveness but stable peak flow records. Eur Respir J. 1989;2:1008-1012.
20. Sur S., Crotty T.B., Kephart G.M., et al. Sudden-onset fatal asthma. A distinct entity with few eosinophils and relatively more neutrophils in the airway submucosa? Am Rev Respir Dis. 1993;148:713-719.
21. Kercsmar C.M. Current trends in management of pediatric asthma. Respir Care. 2003;48:194-205.
22. Wardlaw A.J., Brightling C., Green R., et al. Eosinophils in asthma and other allergic diseases. Br Med Bull. 2000;56:985-1003.
23. Brightling C.E., Symon F.A., Birring S.S., et al. TH2 cytokine expression in bronchoalveolar lavage fluid T lymphocytes and bronchial submucosa is a feature of asthma and eosinophilic bronchitis. J Allergy Clin Immunol. 2002;110:899-905.
24. Panzer S.E., Dodge A.M., Kelly E.A., et al. Circadian variation of sputum inflammatory cells in mild asthma. J Allergy Clin Immunol. 2003;111:308-312.
25. Laitinen L.A., Heino M., Laitinen A., et al. Damage of the airway epithelium and bronchial reactivity in patients with asthma. Am Rev Respir Dis. 1985;131:599-606.
26. Platts-Mills T.A., Rakes G., Heymann P.W. The relevance of allergen exposure to the development of asthma in childhood. J Allergy Clin Immunol. 2000;105:S503-S508.
27. Morkjaroenpong V., Rand C.S., Butz A.M., et al. Environmental tobacco smoke exposure and nocturnal symptoms among inner-city children with asthma. J Allergy Clin Immunol. 2002;110:147-153.
28. Papadopoulos N.G., Psarras S., Manoussakis E., et al. The role of respiratory viruses in the origin and exacerbations of asthma. Curr Opin Allergy Clin Immunol. 2003;3:39-44.
29. Schmaling K.B., McKnight P.E., Afari N. A prospective study of the relationship of mood and stress to pulmonary function among patients with asthma,. J Asthma. 2002;39:501-510.
30. Melo R.E., Sole D., Naspitz C.K. Exercise-induced bronchoconstriction in children: montelukast attenuates the immediate-phase and late-phase responses. J Allergy Clin Immunol. 2003;111:301-307.
31. Rogers D.F. Airway mucus hypersecretion in asthma: an undervalued pathology? Curr Opin Pharmacol. 2004;4:241-250.
32. Kuyper L.M., Pare P.D., Hogg J.C., et al. Characterization of airway plugging in fatal asthma. Am J Med. 2003;115:6-11.
33. Hays S.R., Fahy J.V. The role of mucus in fatal asthma. Am J Med. 2003;115:68-69.
34. Rodriguez-Roisin R., Ballester E., Roca J., et al. Mechanisms of hypoxemia in patients with status asthmaticus requiring mechanical ventilation. Am Rev Respir Dis. 1989;139:732-739.
35. Roca J., Ramis L., Rodriguez-Roisin R., et al. Serial relationships between ventilation-perfusion inequality and spirometry in acute severe asthma requiring hospitalization. Am Rev Respir Dis. 1988;137:1055-1061.
36. Levy B.D., Kitch B., Fanta C.H. Medical and ventilatory management of status asthmaticus. Intensive Care Med. 1998;24:105-117.
37. Stalcup S.A., Mellins R.B. Mechanical forces producing pulmonary edema in acute asthma. N Engl J Med. 1977;297:592-596.
38. Pride N.B., Permutt S., Riley R.L., et al. Determinants of maximal expiratory flow from the lungs. J Appl Physiol. 1967;23:646-662.
39. Martin J.G., Shore S.A., Engel L.A. Mechanical load and inspiratory muscle action during induced asthma. Am Rev Respir Dis. 1983;128:455-460.
40. Buda A.J., Pinsky M.R., Ingels N.B.Jr., et al. Effect of intrathoracic pressure on left ventricular performance. N Engl J Med. 1979;301:453-459.
41. Jardin F., Farcot J.C., Boisante L., et al. Mechanism of paradoxic pulse in bronchial asthma. Circulation. 1982;66:887-894.
42. Simmons D.H., Linde L.M., Miller J.H., et al. Relation between lung volume and pulmonary vascular resistance. Circ Res. 1961;9:465-471.
43. Williams M.H., Zohman L.R. Cardiopulmonary function in bronchial asthma. A comparison with chronic pulmonary emphysema. Am Rev Respir Dis. 1960;81:173-177.
44. McFadden E.R.Jr., Kiser R., DeGroot W.J. Acute bronchial asthma. Relations between clinical and physiologic manifestations. N Engl J Med. 1973;288:221-225.
45. Wood D.W., Downes J.J., Lecks H.I. A clinical scoring system for the diagnosis of respiratory failure. Preliminary report on childhood status asthmaticus. Am J Dis Child. 1972;123:227-228.
46. Keogh K.A., Macarthur C., Parkin P.C., et al. Predictors of hospitalization in children with acute asthma. J Pediatr. 2001;139:273-277.
47. van der Windt D.A., Nagelkerke A.F., Bouter L.M., et al. Clinical scores for acute asthma in pre-school children. A review of the literature. J Clin Epidemiol. 1994;47:635-646.
48. Baker M.D. Pitfalls in the use of clinical asthma scoring. Am J Dis Child. 1988;142:183-185.
49. Kussmaul A. Ueber schwielige mediastino-pericarditis und den paradoxen puls. Berl Klin Wschr. 1873;38:433-435.
50. Bilchick K.C., Wise R.A. Paradoxical physical findings described by Kussmaul: pulsus paradoxus and Kussmaul’s sign. Lancet. 2002;359:1940-1942.
51. Werner H.A. Status asthmaticus in children: a review. Chest. 2001;119:1913-1929.
52. Summer W.R., Permutt S., Sagawa K., et al. Effects of spontaneous respiration on canine left ventricular function. Circ Res. 1979;45:719-728.
53. Hitzig W.M. On mechanisms of inspiratory filling of the cervical veins and pulsus paradoxus in venous hypertension. J Mt Sinai Hosp. 1942;8:625-644.
54. Dohrnhorst A.C., Howard P., Leathart G.L. Pulsus paradoxus. Lancet. 1952;1:746-748.
55. Edmunds A.T., Godfrey S. Cardiovascular response during severe acute asthma and its treatment in children. Thorax. 1981;36:534-540.
56. Knowles G.K., Clark T.J. Pulsus paradoxus as a valuable sign indicating severity of asthma. Lancet. 1973;2:1356-1359.
57. McFadden E.R.Jr., Lyons H.A. Arterial-blood gas tension in asthma. N Engl J Med. 1968;278:1027-1032.
58. Weiss E.B., Faling L.J. Clinical significance of PaCO2 during status asthma: the cross-over point. Ann Allergy. 1968;26:545-551.
59. Mountain R.D., Heffner J.E., Brackett N.C.Jr., et al. Acid-base disturbances in acute asthma. Chest. 1990;98:651-655.
60. Appel D., Rubenstein R., Schrager K., et al. Lactic acidosis in severe asthma. Am J Med. 1983;75:580-584.
61. Lovis C., Mach F., Unger P.F., et al. Elevation of creatine kinase in acute severe asthma is not of cardiac origin. Intensive Care Med. 2001;27:528-533.
62. Tsung S.H. Several conditions causing elevation of serum CK-MB and CK-BB. Am J Clin Pathol. 1981;75:711-715.
63. Gerhardt W., Katus H., Ravkilde J., et al. S-troponin T in suspected ischemic myocardial injury compared with mass and catalytic concentrations of S-creatine kinase isoenzyme MB. Clin Chem. 1991;37:1405-1411.
64. Krishnan S.S., Stewart J., Amin N., et al. Electrocardiographic prediction of hyperinflation in children. Am J Respir Crit Care Med. 1997;156:2011-2014.
65. Udezue E., D’Souza L., Mahajan M. Hypokalemia after normal doses of nebulized albuterol (salbutamol). Am J Emerg Med. 1995;13:168-171.
66. Keller K.A., Bhisitkul D.M. Supraventricular tachycardia: a complication of nebulized albuterol. Pediatr Emerg Care. 1995;11:98-99.
67. Matson J.R., Loughlin G.M., Strunk R.C. Myocardial ischemia complicating the use of isoproterenol in asthmatic children. J Pediatr. 1978;92:776-778.
68. Chiang V.W., Burns J.P., Rifai N., et al. Cardiac toxicity of intravenous terbutaline for the treatment of severe asthma in children: a prospective assessment. J Pediatr. 2000;137:73-77.
69. Schuh S., Reisman J., Alshehri M., et al. A comparison of inhaled fluticasone and oral prednisone for children with severe acute asthma. N Engl J Med. 2000;343:689-694.
70. Rowe B.H., Spooner C., Ducharme F.M., et alEarly emergency department treatment of acute asthma with systemic corticosteroids Cochrane Database Syst 2000:Rev;CD002178
71. Qureshi F., Zaritsky A., Lakkis H. Efficacy of nebulized ipratropium in severely asthmatic children. Ann Emerg Med. 1997;29:205-211.
72. Qureshi F., Pestian J., Davis P., et al. Effect of nebulized ipratropium on the hospitalization rates of children with asthma. N Engl J Med. 1998;339:1030-1035.
73. Geelhoed G.C., Landau L.I., Le Souef P.N. Evaluation of SaO2 as a predictor of outcome in 280 children presenting with acute asthma. Ann Emerg Med. 1994;23:1236-1241.
74. Expert Panel Report 3. Guidelines for the diagnosis and management of asthma—summary report 2007. J Allergy Clin Immunol. 2007;120:S94-138.
75. Tal A., Pasterkamp H., Leahy F. Arterial oxygen desaturation following salbutamol inhalation in acute asthma. Chest. 1984;86:868-869.
76. Connett G., Lenney W. Prolonged hypoxaemia after nebulised salbutamol. Thorax. 1993;48:574-575.
77. Gleeson J.G., Green S., Price J.F. Air or oxygen as driving gas for nebulised salbutamol. Arch Dis Child. 1988;63:900-904.
78. Aubier M., Murciano D., Fournier M., et al. Central respiratory drive in acute respiratory failure of patients with chronic obstructive pulmonary disease. Am Rev Respir Dis. 1980;122:191-199.
79. Chien J.W., Ciufo R., Novak R., et al. Uncontrolled oxygen administration and respiratory failure in acute asthma. Chest. 2000;117:728-733.
80. Peters-Golden M., Sampson A.P. Cysteinyl leukotriene interactions with other mediators and with glucocorticosteroids during airway inflammation. J Allergy Clin Immunol. 2003;111:S37-42. discussion S43-38
81. Barnes P.J., Adcock I. Anti-inflammatory actions of steroids: molecular mechanisms. Trends Pharmacol Sci. 1993;14:436-441.
82. Dworski R., Fitzgerald G.A., Oates J.A., et al. Effect of oral prednisone on airway inflammatory mediators in atopic asthma. Am J Respir Crit Care Med. 1994;149:953-959.
83. Peters-Golden M., Thebert P. Inhibition by methylprednisolone of zymosan-induced leukotriene synthesis in alveolar macrophages. Am Rev Respir Dis. 1987;135:1020-1026.
84. Dunlap N.E., Fulmer J.D. Corticosteroid therapy in asthma. Clin Chest Med. 1984;5:669-683.
85. Fuller R.W., Kelsey C.R., Cole P.J., et al. Dexamethasone inhibits the production of thromboxane B2 and leukotriene B4 by human alveolar and peritoneal macrophages in culture. Clin Sci (Lond). 1984;67:653-656.
86. Welch M.J. Inhaled steroids and severe viral infections. J Asthma. 1994;31:43-50.
87. Koh Y.I., Choi I.S., Shin I.S., et al. Steroid-induced delirium in a patient with asthma: report of one case. Korean J Intern Med. 2002;17:150-152.
88. Dayton M.T., Kleckner S.C., Brown D.K. Peptic ulcer perforation associated with steroid use. Arch Surg. 1987;122:376-380.
89. Undem B.J., Lichtenstein L.M. Drugs used in the treatment of asthma. In: Hardman J.G., Limbird L.E., Gilman A.G., editors. Goodman & Gilman’s the pharmacological basis of therapeutics. New York: McGraw-Hill, 2001.
90. Kelly H.W. New beta 2-adrenergic agonist aerosols. Clin Pharm. 1985;4:393-403.
91. Stephanopoulos D.E., Monge R., Schell K.H., et al. Continuous intravenous terbutaline for pediatric status asthmaticus. Crit Care Med. 1998;26:1744-1748.
92. Katz R.W., Kelly H.W., Crowley M.R., et al. Safety of continuous nebulized albuterol for bronchospasm in infants and children. Pediatrics. 1993;92:666-669.
93. Maguire J.F., Geha R.S., Umetsu D.T. Myocardial specific creatine phosphokinase isoenzyme elevation in children with asthma treated with intravenous isoproterenol. J Allergy Clin Immunol. 1986;78:631-636.
94. Haalboom J.R., Deenstra M., Struyvenberg A. Hypokalaemia induced by inhalation of fenoterol. Lancet. 1985;1:1125-1127.
95. Tveskov C., Djurhuus M.S., Klitgaard N.A., et al. Potassium and magnesium distribution, ECG changes, and ventricular ectopic beats during beta 2-adrenergic stimulation with terbutaline in healthy subjects. Chest. 1994;106:1654-1659.
96. Fok T.F., Monkman S., Dolovich M., et al. Efficiency of aerosol medication delivery from a metered dose inhaler versus jet nebulizer in infants with bronchopulmonary dysplasia. Pediatr Pulmonol. 1996;21:301-309.
97. Montgomery V.L., Eid N.S. Low-dose beta-agonist continuous nebulization therapy for status asthmaticus in children. J Asthma. 1994;31:201-207.
98. Papo M.C., Frank J., Thompson A.E. A prospective, randomized study of continuous versus intermittent nebulized albuterol for severe status asthmaticus in children. Crit Care Med. 1993;21:1479-1486.
99. Craig V.L., Bigos D., Brilli R.J. Efficacy and safety of continuous albuterol nebulization in children with severe status asthmaticus. Pediatr Emerg Care. 1996;12:1-5.
100. Ackerman A.D. Continuous nebulization of inhaled beta-agonists for status asthmaticus in children: a cost-effective therapeutic advance? Crit Care Med. 1993;21:1422-1424.
101. Levalbuterol for asthma. Med Lett Drugs Ther. 1999;41:51-53.
102. Johansson F., Rydberg I., Aberg G., et al. Effects of albuterol enantiomers on in vitro bronchial reactivity. Clin Rev Allergy Immunol. 1996;14:57-64.
103. Walle T., Eaton E.A., Walle U.K., et al. Stereoselective metabolism of RS-albuterol in humans. Clin Rev Allergy Immunol. 1996;14:101-113.
104. Asmus M.J., Hendeles L., Weinberger M., et al. Levalbuterol has not been established to have therapeutic advantage over racemic albuterol. J Allergy Clin Immunol. 2002;110:325.
105. Ramsay C.M., Cowan J., Flannery E., et al. Bronchoprotective and bronchodilator effects of single doses of (S)-salbutamol, (R)-salbutamol and racemic salbutamol in patients with bronchial asthma. Eur J Clin Pharmacol. 1999;55:353-359.
106. Cockcroft D.W., Swystun V.A. Effect of single doses of S-salbutamol, R-salbutamol, racemic salbutamol, and placebo on the airway response to methacholine. Thorax. 1997;52:845-848.
107. Carl J.C., Myers T.R., Kirchner H.L., et al. Comparison of racemic albuterol and levalbuterol for treatment of acute asthma. J Pediatr. 2003;143:731-736.
108. Qureshi F., Zaritsky A., Welch C., et al. Clinical efficacy of racemic albuterol versus levalbuterol for the treatment of acute pediatric asthma. Ann Emerg Med. 2005;46:29-36.
109. Hardasmalani M.D., DeBari V., Bithoney W.G., et al. Levalbuterol versus racemic albuterol in the treatment of acute exacerbation of asthma in children. Pediatr Emerg Care. 2005;21:415-419.
110. Browne G.J., Penna A.S., Phung X., et al. Randomised trial of intravenous salbutamol in early management of acute severe asthma in children. Lancet. 1997;349:301-305.
111. Bohn D., Kalloghlian A., Jenkins J., et al. Intravenous salbutamol in the treatment of status asthmaticus in children. Crit Care Med. 1984;12:892-896.
112. Pirie J., Cox P., Johnson D., et al. Changes in treatment and outcomes of children receiving care in the intensive care unit for severe acute asthma. Pediatr Emerg Care. 1998;14:104-108.
113. Gross N.J. Ipratropium bromide. N Engl J Med. 1988;319:486-494.
114. Woelfle J., Zielen S., Lentze M.J. Unilateral fixed dilated pupil in an infant after inhalation of nebulized ipratropium bromide. J Pediatr. 2000;136:423-424.
115. Kizer K.M., Bess D.T., Bedford N.K. Blurred vision from ipratropium bromide inhalation. Am J Health Syst Pharm. 1999;56:914.
116. Craven D., Kercsmar C.M., Myers T.R., et al. Ipratropium bromide plus nebulized albuterol for the treatment of hospitalized children with acute asthma. J Pediatr. 2001;138:51-58.
117. Goggin N., Macarthur C., Parkin P.C. Randomized trial of the addition of ipratropium bromide to albuterol and corticosteroid therapy in children hospitalized because of an acute asthma exacerbation. Arch Pediatr Adolesc Med. 2001;155:1329-1334.
118. Haury V.G. Blood serum magnesium in bronchial asthma and its treatment by administration of magnesium sulfate. J Lab Clin Med. 1940;26:340-344.
119. Dib J.G., Engstrom F.M., Sisca T.S., et al. Intravenous magnesium sulfate treatment in a child with status asthmaticus. Am J Health Syst Pharm. 1999;56:997-1000.
120. Silverman R.A., Osborn H., Runge J., et al. IV magnesium sulfate in the treatment of acute severe asthma: a multicenter randomized controlled trial. Chest. 2002;122:489-497.
121. Mangat H.S., D’Souza G.A., Jacob M.S. Nebulized magnesium sulphate versus nebulized salbutamol in acute bronchial asthma: a clinical trial. Eur Respir J. 1998;12:341-344.
122. Hughes R., Goldkorn A., Masoli M., et al. Use of isotonic nebulised magnesium sulphate as an adjuvant to salbutamol in treatment of severe asthma in adults: randomised placebo-controlled trial. Lancet. 2003;361:2114-2117.
123. Ciarallo L., Sauer A.H., Shannon M.W. Intravenous magnesium therapy for moderate to severe pediatric asthma: results of a randomized, placebo-controlled trial. J Pediatr. 1996;129:809-814.
124. Devi P.R., Kumar L., Singhi S.C., et al. Intravenous magnesium sulfate in acute severe asthma not responding to conventional therapy. Indian Pediatr. 1997;34:389-397.
125. Gurkan F., Haspolat K., Bosnak M., et al. Intravenous magnesium sulphate in the management of moderate to severe acute asthmatic children nonresponding to conventional therapy. Eur J Emerg Med. 1999;6:201-205.
126. Ciarallo L., Brousseau D., Reinert S. Higher-dose intravenous magnesium therapy for children with moderate to severe acute asthma. Arch Pediatr Adolesc Med. 2000;154:979-983.
127. Scarfone R.J., Loiselle J.M., Joffe M.D., et al. A randomized trial of magnesium in the emergency department treatment of children with asthma. Ann Emerg Med. 2000;36:572-578.
128. Cheuk D.K., Chau T.C., Lee S.L. A meta-analysis on intravenous magnesium sulphate for treating acute asthma. Arch Dis Child. 2005;90:74-77.
129. Nicholson C.D., Shahid M. Inhibitors of cyclic nucleotide phosphodiesterase isoenzymes—their potential utility in the therapy of asthma. Pulm Pharmacol. 1994;7:1-17.
130. Foukas L.C., Daniele N., Ktori C., et al. Direct effects of caffeine and theophylline on p110 delta and other phosphoinositide 3-kinases. Differential effects on lipid kinase and protein kinase activities. J Biol Chem. 2002;277:37124-37130.
131. Feoktistov I., Biaggioni I. Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. An enprofylline-sensitive mechanism with implications for asthma. J Clin Invest. 1995;96:1979-1986.
132. Akingbola O.A., Simakajornboon N., Hadley E.F.Jr., et al. Noninvasive positive-pressure ventilation in pediatric status asthmaticus. Pediatr Crit Care Med. 2002;3:181-184.
133. Higbee M.D., Kumar M., Galant S.P. Stimulation of endogenous catecholamine release by theophylline: a proposed additional mechanism of action for theophylline effects. J Allergy Clin Immunol. 1982;70:377-382.
134. Horrobin D.F., Manku M.S., Franks D.J., et al. Methyl xanthine phosphodiesterase inhibitors behave as prostaglandin antagonists in a perfused rat mesenteric artery preparation. Prostaglandins. 1977;13:33-40.
135. Kolbeck R.C., Speir W.A.Jr., Carrier G.O., et al. Apparent irrelevance of cyclic nucleotides to the relaxation of tracheal smooth muscle induced by theophylline. Lung. 1979;156:173-183.
136. Barlinski J., Lockhart A., Frossard N. Modulation by theophylline and enprofylline of the excitatory non-cholinergic transmission in guinea-pig bronchi. Eur Respir J. 1992;5:1201-1205.
137. Kidney J., Dominguez M., Taylor P.M., et al. Immunomodulation by theophylline in asthma. Demonstration by withdrawal of therapy. Am J Respir Crit Care Med. 1995;151:1907-1914.
138. Aubier M., De Troyer A., Sampson M., et al. Aminophylline improves diaphragmatic contractility. N Engl J Med. 1981;305:249-252.
139. Rabe K.F., Tenor H., Dent G., et al. Phosphodiesterase isozymes modulating inherent tone in human airways: identification and characterization. Am J Physiol. 1993;264:L458-464.
140. Spina D. Theophylline and PDE4 inhibitors in asthma. Curr Opin Pulm Med. 2003;9:57-64.
141. Weinberger M., Hendeles L., Ahrens R. Clinical pharmacology of drugs used for asthma. Pediatr Clin North Am. 1981;28:47-75.
142. Strauss R.E., Wertheim D.L., Bonagura V.R., et al. Aminophylline therapy does not improve outcome and increases adverse effects in children hospitalized with acute asthmatic exacerbations. Pediatrics. 1994;93:205-210.
143. Rodrigo C., Rodrigo G. Treatment of acute asthma. Lack of therapeutic benefit and increase of the toxicity from aminophylline given in addition to high doses of salbutamol delivered by metered-dose inhaler with a spacer. Chest. 1994;106:1071-1076.
144. Goodman D.C., Littenberg B., O’Connor G.T., et al. Theophylline in acute childhood asthma: a meta-analysis of its efficacy. Pediatr Pulmonol. 1996;21:211-218.
145. Ream R.S., Loftis L.L., Albers G.M., et al. Efficacy of IV theophylline in children with severe status asthmaticus. Chest. 2001;119:1480-1488.
146. Barach A.L., Eckman M. The effects of inhalation of helium mixed with oxygen on the mechanics of respiration. J Clin Invest. 1936;15:47-61.
147. Gupta V.K., Cheifetz I.M. Heliox administration in the pediatric intensive care unit: an evidence-based review. Pediatr Crit Care Med. 2005;6:204-211.
148. Weber J.E., Chudnofsky C.R., Younger J.G., et al. A randomized comparison of helium-oxygen mixture (Heliox) and racemic epinephrine for the treatment of moderate to severe croup. Pediatrics. 2001;107:E96.
149. Kim I.K., Phrampus E., Venkataraman S., et al. Helium/oxygen-driven albuterol nebulization in the treatment of children with moderate to severe asthma exacerbations: a randomized, controlled trial. Pediatrics. 2005;116:1127-1133.
150. Hess D.R., Acosta F.L., Ritz R.H., et al. The effect of heliox on nebulizer function using a beta-agonist bronchodilator. Chest. 1999;115:184-189.
151. Gluck E.H., Onorato D.J., Castriotta R. Helium-oxygen mixtures in intubated patients with status asthmaticus and respiratory acidosis. Chest. 1990;98:693-698.
152. Shiue S.T., Gluck E.H. The use of helium-oxygen mixtures in the support of patients with status asthmaticus and respiratory acidosis. J Asthma. 1989;26:177-180.
153. Kass J.E., Castriotta R.J. Heliox therapy in acute severe asthma. Chest. 1995;107:757-760.
154. Manthous C.A., Hall J.B., Caputo M.A., et al. Heliox improves pulsus paradoxus and peak expiratory flow in nonintubated patients with severe asthma. Am J Respir Crit Care Med. 1995;151:310-314.
155. Kudukis T.M., Manthous C.A., Schmidt G.A., et al. Inhaled helium-oxygen revisited: effect of inhaled helium-oxygen during the treatment of status asthmaticus in children. J Pediatr. 1997;130:217-224.
156. Haynes J.M., Sargent R.J., Sweeney E.L. Use of heliox to avoid intubation in a child with acute severe asthma and hypercapnia. Am J Crit Care. 2003;12:28-30.
157. Carter E.R., Webb C.R., Moffitt D.R. Evaluation of heliox in children hospitalized with acute severe asthma. A randomized crossover trial. Chest. 1996;109:1256-1261.
158. Rodrigo G.J., Rodrigo C., Pollack C.V., et al. Use of helium-oxygen mixtures in the treatment of acute asthma: a systematic review. Chest. 2003;123:891-896.
159. Tobias J.D., Garrett J.S. Therapeutic options for severe, refractory status asthmaticus: inhalational anaesthetic agents, extracorporeal membrane oxygenation and helium/oxygen ventilation. Paediatr Anaesth. 1997;7:47-57.
160. Hirshman C.A., McCullough R.E., Cohen P.J., et al. Hypoxic ventilatory drive in dogs during thiopental, ketamine, or pentobarbital anesthesia. Anesthesiology. 1975;43:628-634.
161. Evers A.S., Crowder M. General anesthetics. In: Hardman J.G., Limbird L.E., Gilman A.G., editors. Goodman & Gilman’s the pharmacological basis of therapeutics. New York: McGraw-Hill, 2001.
162. Sato T., Hirota K., Matsuki A., et al. The role of the N-methyl-D-aspartic acid receptor in the relaxant effect of ketamine on tracheal smooth muscle. Anesth Analg. 1998;87:1383-1388.
163. L’Hommedieu C.S., Arens J.J. The use of ketamine for the emergency intubation of patients with status asthmaticus. Ann Emerg Med. 1987;16:568-571.
164. Nehama J., Pass R., Bechtler-Karsch A., et al. Continuous ketamine infusion for the treatment of refractory asthma in a mechanically ventilated infant: case report and review of the pediatric literature. Pediatr Emerg Care. 1996;12:294-297.
165. Sarma V.J. Use of ketamine in acute severe asthma. Acta Anaesthesiol Scand. 1992;36:106-107.
166. Scoggin C.H., Sahn S.A., Petty T.L. Status asthmaticus. A nine-year experience. JAMA. 1977;238:1158-1162.
167. Westerman D.E., Benatar S.R., Potgieter P.D., et al. Identification of the high-risk asthmatic patient. Experience with 39 patients undergoing ventilation for status asthmaticus. Am J Med. 1979;66:565-572.
168. Picado C., Montserrat J.M., Roca J., et al. Mechanical ventilation in severe exacerbation of asthma. Study of 26 cases with six deaths. Eur J Respir Dis. 1983;64:102-107.
169. Darioli R., Perret C. Mechanical controlled hypoventilation in status asthmaticus. Am Rev Respir Dis. 1984;129:385-387.
170. Cox R.G., Barker G.A., Bohn D.J. Efficacy, results, and complications of mechanical ventilation in children with status asthmaticus. Pediatr Pulmonol. 1991;11:120-126.
171. Marini J.J. Should PEEP be used in airflow obstruction? Am Rev Respir Dis. 1989;140:1-3.
172. Stewart T.E., Slutsky A.S. Occult, occult auto-PEEP in status asthmaticus. Crit Care Med. 1996;24:379-380.
173. Tuxen D.V. Detrimental effects of positive end-expiratory pressure during controlled mechanical ventilation of patients with severe airflow obstruction. Am Rev Respir Dis. 1989;140:5-9.
174. Wetzel R.C. Pressure-support ventilation in children with severe asthma. Crit Care Med. 1996;24:1603-1605.
175. Polsonetti B.W., Joy S.D., Laos L.F. Steroid-induced myopathy in the ICU. Ann Pharmacother. 2002;36:1741-1744.
176. David W.S., Roehr C.L., Leatherman J.W. EMG findings in acute myopathy with status asthmaticus, steroids and paralytics. Clinical and electrophysiologic correlation. Electromyogr Clin Neurophysiol. 1998;38:371-376.
177. Davis N.A., Rodgers J.E., Gonzalez E.R., et al. Prolonged weakness after cisatracurium infusion: a case report. Crit Care Med. 1998;26:1290-1292.
178. Rooke G.A., Choi J.H., Bishop M.J. The effect of isoflurane, halothane, sevoflurane, and thiopental/nitrous oxide on respiratory system resistance after tracheal intubation. Anesthesiology. 1997;86:1294-1299.
179. Brown R.H., Mitzner W., Zerhouni E., et al. Direct in vivo visualization of bronchodilation induced by inhalational anesthesia using high-resolution computed tomography. Anesthesiology. 1993;78:295-300.
180. Restrepo R.D., Pettignano R., DeMeuse P. Halothane, an effective infrequently used drug, in the treatment of pediatric status asthmaticus: a case report. J Asthma. 2005;42:649-651.
181. Shankar V., Churchwell K.B., Deshpande J.K. Isoflurane therapy for severe refractory status asthmaticus in children. Intensive Care Med. 2006;32:927-933.
182. Burburan S.M., Xisto D.G., Rocco P.R. Anaesthetic management in asthma. Minerva Anesthesiol. 2007;73:357-365.
183. Schultz T.E. Sevoflurane administration in status asthmaticus: a case report. AANA J. 2005;73:35-36.
184. Lehane J.R., Jordan C., Jones J.G. Influence of halothane and enflurane on respiratory airflow resistance and specific conductance in anaesthetized man. Br J Anaesth. 1980;52:773-781.
185. Doi M., Ikeda K. Airway irritation produced by volatile anaesthetics during brief inhalation: comparison of halothane, enflurane, isoflurane and sevoflurane. Can J Anaesth. 1993;40:122-126.
186. Merin R.G., Kumazawa T., Luka N.L. Dose-dependent depression of cardiac function and metabolism by inhalation anesthetics in chronically instrumented dogs. Recent Adv Stud Cardiac Struct Metab. 1976;11:473-480.
187. Merin R.G., Kumazawa T., Luka N.L. Myocardial function and metabolism in the conscious dog and during halothane anesthesia. Anesthesiology. 1976;44:402-415.
188. Zink J., Sasyniuk B.I., Dresel P.E. Halothane-epinephrine-induced cardiac arrhythmias and the role of heart rate. Anesthesiology. 1975;43:548-555.
189. Baigel G. Volatile agents to avoid ventilating asthmatics. Anaesth Intensive Care. 2003;31:208-210.
190. Beach F.X., Williams N.E. Bronchial lavage in status asthmaticus. A long term review after treatment. Anaesthesia. 1970;25:378-381.
191. Henke C.A., Hertz M., Gustafson P. Combined bronchoscopy and mucolytic therapy for patients with severe refractory status asthmaticus on mechanical ventilation: a case report and review of the literature. Crit Care Med. 1994;22:1880-1883.
192. Durward A., Forte V., Shemie S.D. Resolution of mucus plugging and atelectasis after intratracheal rhDNase therapy in a mechanically ventilated child with refractory status asthmaticus. Crit Care Med. 2000;28:560-562.
193. Mikkelsen M.E., Woo Y.J., Sager J.S., et al. Outcomes using extracorporeal life support for adult respiratory failure due to status asthmaticus. ASAIO J. 2009;55:47-52.
194. Leiba A., Bar-Yosef S., Bar-Dayan Y., et al. Early administration of extracorporeal life support for near fatal asthma. Isr Med Assoc J. 2003;5:600-602.
195. Extracorporeal Life Support Organization Registry. International Summary. Ann Arbor: Extracorporeal Life Support Organization; MI, 2009.
196. Bellomo R., McLaughlin P., Tai E., et al. Asthma requiring mechanical ventilation. A low morbidity approach. Chest. 1994;105:891-896.
197. Kearney S.E., Graham D.R., Atherton S.T. Acute severe asthma treated by mechanical ventilation: a comparison of the changing characteristics over a 17 yr period. Respir Med. 1998;92:716-721.
198. Bohn D., Kissoon N. Acute asthma. Pediatr Crit Care Med. 2001;2:151-163.
199. Stein R., Canny G.J., Bohn D.J., et al. Severe acute asthma in a pediatric intensive care unit: six years’ experience. Pediatrics. 1989;83:1023-1028.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 45 Asthma
The term status asthmaticus has been used to denote a more severe form of asthma attack, but its definition varies widely among different authors. To some authors, status asthmaticus is an asthma attack that does not respond to initial treatments with bronchodilators,1,2 whereas to others it indicates severe asthma that leads to respiratory failure and requires mechanical ventilatory support.3 For the purposes of this text, status asthmaticus is defined as an asthma attack that fails to respond to initial doses of nebulized β2-adrenergic and anticholinergic agents and systemic corticosteroid drugs and that requires admission to the hospital for continuation of treatment. Patients who experience relentless progression of respiratory signs and symptoms and require admission to the intensive care unit (ICU) are reported as having near-fatal asthma.4
Epidemiology and Risk Factors
Asthma is the most common chronic illness in childhood, affecting approximately 6.7 million children and adolescents in the United States, or 9.1% of persons younger than 18 years.5 Asthma is also a very common discharge diagnosis in children’s hospitals, accounting for approximately 5.6% of all hospital admissions and more than 150,000 admissions each year in the United States alone.5 The prevalence of asthma worldwide is highly variable, with greater than twentyfold differences in prevalence of symptoms encountered among centers located in various parts of the world.6 The highest prevalence rates for asthma are found in the United Kingdom, Australia, New Zealand, and the Republic of Ireland; very low prevalence rates occur in eastern Europe, the Indian subcontinent, and China.6 Race is a significant factor in determining prevalence and severity of asthma in children and young adults. African Americans are 4.1 times more likely to require treatment for asthma in the emergency department, two times more likely to be hospitalized for asthma, and 7.6 times more likely to die compared with white persons.5 Socioeconomic status also has been shown to negatively correlate with asthma prevalence, morbidity, and mortality in the United States.7
Asthma prevalence has increased steadily during the past 27 years in the United States.5 However, while the rate of nonurgent asthma-related outpatient visits continues to increase, emergency department visits and death rates have steadily declined during the past decade.5
The incidence of asthma-related respiratory failure requiring mechanical ventilation is difficult to determine because of variability in diagnostic criteria and reporting practices. Nonetheless, up to 36% of adult patients admitted to an inner-city medical ICU with near-fatal asthma require invasive mechanical ventilation.8 This figure appears to be significantly lower for children, considering that only 22 (10.2%) of 237 patients treated for near-fatal asthma underwent mechanical ventilation during a 2-year period in the pediatric ICU (PICU) of a tertiary children’s hospital (A.T. Rotta, unpublished data), and that only 14 (8.6%) of 163 patients required intubation in another study.9
The majority of patients with asthma who experience respiratory failure or arrest do so during the first stages of therapy or prior to arrival in the emergency department.9 Therefore early identification and close monitoring of patients at high risk for near-fatal asthma could be advantageous. High-risk patients often have a history of ICU admissions,10 mechanical ventilation,2,10 seizures or syncope during an attack,11 PaCO2 greater than 45 torr,2,10 attacks precipitated by food,10 or a history of rapidly progressive and sudden respiratory deterioration.1 These patients are likely to use more than two canisters of β-agonist metered-dose inhalers per month12 and often are poorly compliant or are receiving insufficient steroid therapy.13,14 Denial or failure to perceive the severity of an attack are factors frequently associated with near-fatal asthma.15,16 Although unquestionably some patients at risk for near-fatal asthma simply ignore early warning signs and do not seek adequate therapy, a subgroup of patients actually lacks normal perception of disease severity. Some patients with near-fatal asthma exhibit reduced chemosensitivity to hypoxia and blunted perception of dyspnea.17 Other patients have a decreased perceptual sensitivity of inspiratory muscle loads and display abnormal respiratory-related evoked potentials.18
Although many of these high-risk factors are commonly present in patients with near-fatal asthma, they fail to identify a significant number of cases. In one study, 33% of patients who died of asthma were judged to have a history of trivial or mild asthma, whereas 32% had never been admitted to the hospital with an asthma exacerbation.1 Some of these patients may in fact have what likely represents a distinct clinical entity known as sudden asphyxial asthma, a condition marked by acute onset of severe airway obstruction and hypoxia that rapidly leads to cardiorespiratory arrest in patients known to have only mild asthma or no asthma history at all.19,20
Pathophysiology
Asthma is primarily an inflammatory disease and, as such, it is marked by highly redundant pathways and complex interactions among inflammatory cells, mediators, and the airway epithelium21 (Figure 45-1). Functionally, asthma is characterized by variable airflow obstruction and airway hyperresponsiveness associated with airway inflammation. Pathologically, it is marked by mast cell degranulation, accumulation of eosinophils and CD4 lymphocytes, hypersecretion of mucus, thickening of the subepithelial collagen layer, and smooth muscle hypertrophy and hyperplasia.22
Mast cells, eosinophils, macrophages, and T lymphocytes are central to the derangements that occur during an acute attack (see Figure 45-1). The usual cascade begins with the activation and degranulation of mast cells in response to allergens or topical insults. The mast cells in turn promote activation of T lymphocytes by presenting these cells to the allergenic particles. The inflammatory process is then amplified by T-lymphocyte release of cytokines and chemokines. Increasing evidence exists that airway inflammation in asthma is the result of T-lymphocyte activation with the production of TH2 cytokines, such as interleukin (IL)-4, IL-5, IL-8, and IL-13.23 The presence of these TH2 cytokines leads to further augmentation of the inflammatory process through overexuberant production of immunoglobulin E (IgE) by B cells, stimulation of airway epithelial cells, and eosinophil chemotaxis. IgE stimulates mast cells to release leukotrienes, whereas interleukins (particularly IL-5) promote maturation and migration of activated eosinophils into the airway.24 This highly inflammatory milieu results in stimulation of airway epithelial cells and continued augmentation of the inflammatory process by further release of leukotrienes, prostaglandins, nitric oxide, adhesion molecules, and platelet-activating factor. This process results in overproduction of mucus and epithelial cell destruction that lead to airway plugging and denudation of the airway surface. Epithelial denudation is known to expose nerve endings, resulting in hyperirritable airways25 that become more susceptible to spasm and obstruction when challenged by subsequent exposure to allergens,26 inhaled irritants such as cigarette smoke and pollution,27 respiratory tract infections,28 psychological stress,29 and exercise,30 among other insults. Mucus itself is pathological in content,31,32 and mucus hypersecretion has been underappreciated as a cause of respiratory failure in persons with severe asthma, when in fact strong evidence exists that it may be a principal cause.31–33
Inflammation-mediated edema, mucus hypersecretion, airway plugging, and bronchospasm lead to the severe airway obstruction seen in patients with status asthmaticus and near-fatal asthma. The resulting obstruction and increased airway resistance create an impediment for inspiratory and expiratory gas flow, which leads to deranged pulmonary mechanics and increased lung volumes.32
Airway plugging can result in ventilation/perfusion mismatching and increased oxygen requirements. Hypoxemia is common in patients with a severe asthma attack, but it is generally easily corrected with supplemental oxygen34 and is only weakly correlated with pulmonary function abnormalities.35 More frequently, airway plugging and obstruction lead to regional alveolar hyperinflation associated with reduced perfusion, resulting in a significantly increased pulmonary dead space. Most patients with this condition exhibit an increased respiratory rate in attempt to achieve a higher minute volume and compensate for the ventilation abnormality. Unfortunately, in patients with more severe disease, airway obstruction also results in significant prolongation of expiratory time, which, coupled with initiation of inspiration prior to completion of the previous exhalation, leads to dynamic hyperinflation, gas trapping, and the development of abnormally high lung volumes36 (Figure 45-2).
The higher lung volumes that result from incomplete alveolar emptying and dynamic hyperinflation serve as an adaptation mechanism to allow for higher expiratory flows than would have been possible at lower, more physiologic lung volumes. This higher expiratory flow is accomplished, however, at a high energy cost. Expiration becomes an active process, and the use of accessory muscles is required to overcome the high resistances to airflow both during inspiration and exhalation.37 During a severe attack, inspiratory transpulmonary pressures in excess of 50 cm H2O may be generated, compared with approximately 5 cm H2O during normal breathing.38 The increased muscle work is accompanied by an increase in blood flow to the diaphragm, but this flow often is insufficient to meet the much greater metabolic demands.39 Failure to promptly relieve the airway obstruction and reduce the work of breathing eventually leads to respiratory muscle fatigue, inadequate ventilation, and respiratory failure.
States of advanced airway obstruction and dynamic hyperinflation typical of severe asthma attacks have a significant impact on the circulatory system. The highly negative intrapleural pressures generated by spontaneously breathing patients during inspiration favor transcapillary edema fluid movement into the air spaces.37 They also cause a phasic increase in left ventricular afterload and a decrease in cardiac output40 that is clinically manifested as pulsus paradoxus.41 Right ventricular afterload may be increased during severe asthma as a result of pulmonary vasoconstriction related to hypoxia and acidosis. A state of increased pulmonary vascular resistance resulting from dynamic hyperinflation also can increase right ventricular afterload, further affecting cardiac output.41–43
Clinical Assessment
History
The child with an asthma exacerbation usually presents with complaints of difficulty breathing and shortness of breath. The presence of these complaints in a child known to have had previous asthma exacerbations is highly suggestive of the diagnosis. A significant percentage of children have a history of a coexisting viral upper respiratory infection, whereas some describe exposure to known allergic triggers. Circumstances permitting, time should be taken to inquire about the presence of high-risk factors (Box 45-1) for near-fatal asthma and the adequacy of maintenance intercrisis therapy.
Physical Examination
Wheezing, which is a common clinical finding in patients with acute asthma exacerbations, is the audible manifestation of the transmitted turbulence to airflow in the intrathoracic intrapulmonary airways. Wheezing may be predominantly expiratory as a result of the dynamic phasic compression of conducting airways, but it also can be biphasic. Wheezing in persons with severe asthma usually is symmetrical. An asymmetrical distribution suggests regional mucous plugging, atelectasis, pneumothorax, or the presence of a foreign body. The degree of wheezing correlates poorly with disease severity,44 because wheezes are heard only in the presence of airflow. As such, a patient with severe airway obstruction and very limited airflow may have a silent chest upon arrival at the emergency department, but loud wheezes may develop after effective therapy is instituted. Likewise, in a patient with loud wheezes that continue to worsen, a silent chest may develop as a prelude to respiratory failure.
An objective assessment of disease severity is important in evaluating a patient’s response to therapy. Wood and colleagues45 developed a practical clinical asthma score composed of five variables with three different grades that allows for semiquantitative assessment of disease severity (Table 45-1). This clinical asthma score has been shown to correlate well with the need for prolonged bronchodilator therapy and hospitalization.46 However, although clinical asthma scores seem to be useful for assessing the severity of an attack, they are not as effective in prospectively identifying patients who require prolonged hospitalization or in whom complications and subsequent disability develop.47,48
A less frequently used but more objective method of assessing disease severity and progression in patients with severe asthma is measurement of the pulsus paradoxus. Originally described by Adolf Kussmaul49 in a patient with constrictive pericarditis, pulsus paradoxus also is observed in conditions in which pleural pressure swings are exaggerated, such as status asthmaticus and near-fatal asthma. The simplest definition of pulsus paradoxus is an exaggeration of the physiologic inspiratory decrease in systolic blood pressure50 (Figure 45-3). It has been suggested that the term pulsus paradoxus is inappropriate to describe this phenomenon,51 because an accentuated inspiratory decrease in systolic pressure in the same direction as the normally occurring change cannot be described as a paradox. However, the true paradox described by Kussmaul49 was “the presence of a pulse slight and irregular, disappearing during inspiration and returning upon expiration despite the continued presence of the cardiac impulse during both respiratory phases.”50 Several mechanisms have been implicated in the occurrence of pulsus paradoxus in persons with asthma, and it is likely that various mechanisms contribute differently depending on the adequacy of intravascular volume, the magnitude of pleural pressure swings, the degree of pulmonary hyperinflation, and the state of cardiac contractility. These mechanisms include increased left ventricular afterload from highly negative intrapleural pressure52; decreased left ventricular preload as a result of inspiratory blood pooling in the pulmonary vasculature53; impaired left ventricular diastolic filling caused by a leftward shift of the interventricular septum resulting from increased venous return to the right heart54; constraint of cardiac filling because of longitudinal inspiratory deformation of the pericardium41; and increased right ventricular afterload with decreased filling of the left ventricle as a result of hyperinflation, acidosis, and hypoxia.55 The pulsus paradoxus can be measured easily in a patient who is spontaneously breathing by transducing pressure signals from an indwelling arterial catheter or by using a manual sphygmomanometer. In the latter technique, the cuff is inflated 20 mm Hg above the systolic pressure and then deflated until the first Korotkoff sounds are heard (systolic blood pressure). Initially, Korotkoff sounds are heard only during expiration. The cuff is then carefully deflated until the point where the sounds are heard equally during both inspiration and expiration. The difference between the highest systolic pressure and the pressure at which all Korotkoff sounds are heard is the magnitude of the pulsus paradoxus. During normal breathing, this difference is less than 5 mm Hg, but it is generally greater than 10 mm Hg during acute asthma exacerbations and greater than 20 mm Hg in patients with more severe disease.56 Changes in the magnitude of pulsus paradoxus during the course of therapy are good indicators of disease severity and clinical response to treatment.41,56
Laboratory Data
Arterial Blood Gas Analysis
Arterial blood gas measurements provide objective information on the adequacy of ventilation and oxygenation of the patient with asthma. The typical blood gas abnormality encountered in the early phase of asthma is relative hypoxemia with hypocapnia (PaCO2 <35 torr), reflecting hyperventilation.57 With worsening of airway obstruction, PaCO2 measurements return to the normal range of approximately 40 torr. However, this “normal” PaCO2 should not be viewed as reassuring when taken in the context of prolonged expiratory time, tachypnea, and accessory muscle use.58 In fact, PaCO2 greater than 40 torr in a patient with status asthmaticus should be interpreted as a sign of evolving respiratory muscle fatigue and warrants close clinical observation. Sicker patients often exhibit a mixed respiratory and metabolic acidosis.59 Lactic acidosis is frequently encountered in these patients and is thought to represent exaggerated lactate production by the respiratory muscles and tissue hypoxia.60
Muscle Enzymes
At least one third of patients with acute severe asthma exhibit an elevated plasma creatine kinase (CK) level.61 Although such elevations seem to be more pronounced in patients with marked acidemia or in those presenting with more severe respiratory insufficiency, a convincing correlation between disease severity and CK elevation has not been established.61 Myoglobin, a heme protein present in skeletal and cardiac muscle, is often also elevated in patients with near–fatal asthma.61 Elevations of CK–myocardial bound (CK–MB) isoenzyme develop in some patients with near–fatal asthma, suggesting a possible myocardial injury. This scenario certainly is plausible, considering that many patients are hypoxemic, acidotic, have high myocardial energy demand, and are receiving medications with adverse cardiac effects. However, plasma myoglobin and CK–MB elevations cannot be solely attributed to myocardial injury because the lungs and respiratory muscles are also known sources of these substances.62 Cardiospecific troponin T is a very sensitive and specific marker of myocardial cell damage63 and should be used preferentially to address the question of cardiomyocyte involvement in patients with severe asthma.
Electrocardiography
Patients with status asthmaticus or near-fatal asthma with significant airway obstruction and hyperinflation may exhibit a change in the mean frontal P–wave vector. A P–wave axis greater than 60 degrees has been associated with hyperinflation in both pediatric and adult patients with airway obstruction and is thought to represent positional atrial changes caused by inferior displacement of the diaphragm.64
Twelve–lead electrocardiography and continuous cardiac monitoring are valuable tools in the care of patients with near-fatal asthma in the ICU environment. These patients usually receive high doses of β-agonist drugs and may show evidence of hypokalemia (low–voltage T waves) or cardiac arrhythmias.65,66 The already increased myocardial energy demand resulting from airway obstruction is compounded by the chronotropic and vasodilatory effects inherent to β–agonist drugs and may lead to myocardial ischemia, particularly in adult patients with restricted coronary perfusion. Pediatric patients also may exhibit electrocardiographic (ECG) and enzymatic evidence of myocardial ischemia, particularly during treatment with intravenously administered isoproterenol.67 However, despite the fact that a study reported that a high percentage (66%) of patients exhibited nonspecific ST segment changes or other criteria suggestive of ischemia, these changes were not well correlated with initiation of terbutaline therapy or elevations in cardiac troponin T.68
Treatment
Initial Management in the Emergency Department
Patients with moderate or severe acute asthma also should receive a dose of systemic corticosteroid in the emergency department, which usually is administered prior to the second dose of albuterol. Prednisone (2 mg/kg) can be administered orally and is generally well tolerated. Oral prednisone is superior to inhaled fluticasone in children with severe asthma as evidenced by greater improvement in pulmonary function and lower hospitalization rates.69 The role of corticosteroid drugs in reversing an acute asthma attack in the emergency department has been the subject of debate, considering that these drugs require at least 4 to 6 hours for peak effects to be manifested.70 However, regardless of considerations about onset of action, acute suppression of inflammation is a cornerstone of acute asthma treatment and should be initiated as early as possible. Sicker patients with severe asthma exacerbations, those unable to tolerate oral medication because of respiratory distress or emesis, or those with a history of nausea during intensive β–agonist therapy should be given parenteral corticosteroid drugs such as methylprednisolone (2 mg/kg administered intravenously, followed by 0.5 to 1 mg/kg/dose administered intravenously every 6 hours).
Inhaled or nebulized anticholinergic agents such as ipratropium bromide are now considered an important adjunct in the treatment of persons with moderate and severe asthma exacerbations in the emergency department. In patients treated with one dose of a corticosteroid, use of ipratropium bromide (500 μg/2.5 mL) in conjunction with the second and third albuterol (salbutamol) doses has been associated with greater clinical improvement71 and reduced hospitalization rates compared with corticosteroid and albuterol (salbutamol) alone.72
Admission Criteria
The majority of patients with an acute asthma exacerbation respond to treatment in the emergency department and are discharged home. Among patients whose symptoms persist despite initial treatment, most can be safely managed in the general pediatric inpatient ward. Indications for hospitalization after treatment in the emergency department are loosely defined but may include (1) an inadequate response to three or four aerosol treatments; (2) relapse within 1 hour of receiving treatment with aerosols and steroids; (3) persistent SpO2 measurements of less than 91% in room air; (4) the need for oxygen therapy; (5) a significant reduction in peak expiratory flow rate; (6) having unreliable family support or being unable to comply with outpatient treatment; and (7) multiple visits for the same episode.73,74 Patients who require higher levels of monitoring or more invasive and aggressive treatment or who deteriorate during hospitalization in the general pediatric ward should be admitted to the PICU.
Management in the Intensive Care Unit
Oxygen
Sick patients with asthma are likely to exhibit hypoxemia as a result of intrapulmonary shunts caused by mucus plugging and atelectasis. Treatment with β–agonist agents also can contribute to hypoxemia by abolishing regional pulmonary hypoxic vasoconstriction and increasing intrapulmonary shunt.75,76 Therefore humidified oxygen should be offered both as a carrier gas for nebulizations and continuously between treatments.77 Supplemental oxygen can be safely incorporated into the treatment algorithm because, unlike in some adult patients with severe chronic obstructive pulmonary disease78 or asthma,79 no evidence exists to suggest that supplemental oxygen suppresses the respiratory drive in children with near–fatal asthma.
Corticosteroids
Corticosteroid drugs play a central role in the treatment of patients with status asthmaticus and near-fatal asthma, considering that these conditions are predominantly inflammatory in nature. Glucocorticosteroid agents modulate airway inflammation by a number of mechanisms, including direct interaction with cytosolic receptors and glucocorticosteroid response elements in gene promoters and indirect effects on binding of transcription factors, such as nuclear factor–κB, and on other cell signaling processes, such as posttranscriptional events.80 Gene products suppressed by glucocorticosteroid agents include a wide range of cytokines (IL–1, IL–2, IL–3, IL–4, IL–5, IL–6, IL–7, IL–8, IL–11, IL–12, IL–13, tumor necrosis factor–α, and granulocyte-macrophage colony–stimulating factor), adhesion molecules (intracellular adhesion molecule–1 and vascular cell adhesion molecule-1), and inducible enzymes, including NO synthase and cyclooxygenase–2.81 Transcription of other genes, such as lipocortin–1 and the β2–adrenergic receptor, may be enhanced.81 Glucocorticosteroid agents also decrease airway mucus production, reduce inflammatory cell infiltration and activation, and attenuate capillary permeability.82–85
In children with status asthmaticus or near–fatal asthma, glucocorticosteroid drugs should be administered by the IV route. The oral route can be used in selected cases, but inhaled glucocorticosteroid drugs play no role in the treatment of the sick hospitalized patient.21,69 The most common agent used in the United States is methylprednisolone because of its wide availability as an IV preparation and lack of mineralocorticoid effects. The usual dose of methylprednisolone is 0.5 to 1 mg/kg/dose, administered intravenously every 6 hours. Hydrocortisone, an agent with both glucocorticoid and mineralocorticoid activity, can be used as an alternative at doses of 2 to 4 mg/kg/dose, administered intravenously every 6 hours. Short courses of steroids usually are well tolerated without significant adverse effects.84 However, hypertension, hyperglycemia, mood disorders, and serious viral infections, such as fatal varicella, have been reported in previously well patients with asthma who have received glucocorticosteroid drugs.84,86,87 Duration of corticosteroid therapy is dictated by the severity of illness and the clinical response. Once initiated, treatment in patients with status asthmaticus or near-fatal asthma is generally continued for 5 to 7 days. Longer treatment courses necessitate gradual weaning of the drug to decrease the chances of symptomatic adrenal insufficiency or relapse. Prophylaxis with an H2 blocker should be considered because of the possibility of steroid-associated gastritis and gastric perforation.88
β–Agonists
The β–agonist properties of the sympathomimetic agents cause bronchial smooth muscle relaxation and hence bronchodilatation. These agents also can increase diaphragmatic contractility, enhance mucociliary clearance, and inhibit bronchospastic mediators from mast cells.89 Therefore β–agonists, along with systemic corticosteroids, are the mainstay of pharmacotherapy in persons with near-fatal asthma. β2–receptor selectivity is desirable to avoid adverse effects of nonselective α– and β1–adrenergic receptor stimulation. However, despite β2 selectivity, cardiovascular adverse effects remain a dose–limiting factor. The relative potency of various agents for the β2 receptor is as follows: isoproterenol > fenoterol > albuterol > terbutaline > isoetharine > metaproterenol.90 Of these agents, only albuterol and terbutaline are widely used in clinical practice, with some centers still using isoproterenol in selected occasions.
Once bound to the β–adrenergic receptor, β–agonists activate adenyl cyclase, resulting in increased intracellular cyclic adenosine monophosphate (cAMP) levels, which leads to bronchial and vascular smooth muscle relaxation. Dose-response curves demonstrate that large dose increases fail to enhance bronchodilation significantly; however, as the degree of bronchial constriction increases, the bronchodilation dose–response curve shifts to the right, indicating the need for a higher dose to achieve the desired response.90
[/not-level-membership-for-pediatrics-category]
