[level-membership-for-critical-care-medicine-category]
97 Ascites
 Definition and Diagnosis
Definition and Diagnosis
Ascites is the abnormal accumulation of fluid in the peritoneal cavity.1 Patients with ascites generally present on clinical examination with abdominal distention and a fluid wave or shifting dullness on abdominal percussion, but the abdominal examination findings also may be normal if the amount of ascites is not massive.
Diagnostic imaging can confirm the diagnosis of ascites. Ultrasonography is the easiest and most sensitive technique for the detection of ascitic fluid, being capable of visualizing very small volumes (5-10 mL). Computed tomography (CT) is also very sensitive for detecting ascites (Figure 97-1). Small amounts of ascitic fluid localize in the perihepatic area and in Morrison’s pouch (the hepatorenal space).
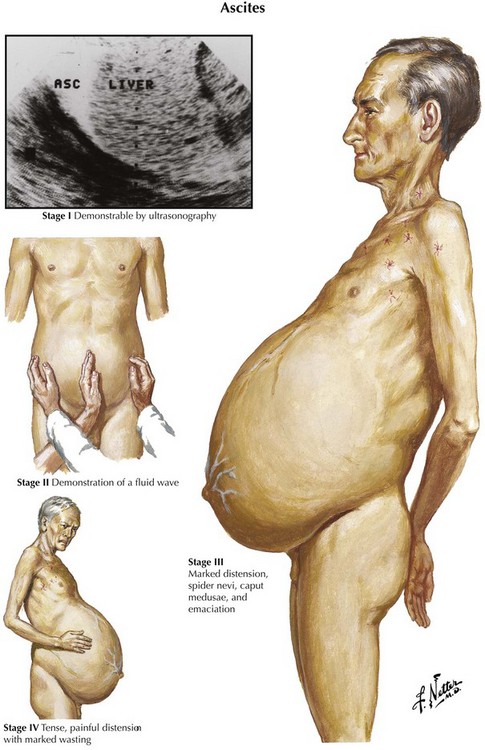
A diagnostic paracentesis (20 mL)2 is performed to determine the etiology of the ascites as well as to exclude or establish a diagnosis of spontaneous bacterial peritonitis (SBP). A diagnostic paracentesis should be performed in any person with new-onset ascites. Paracentesis to evaluate for SBP is also indicated for cirrhotic patients with known ascites who require hospitalization or sustain clinical deterioration, such as worsening encephalopathy or unexplained fever. A missed or delayed diagnosis of SBP can lead to sepsis and significant morbidity and mortality.
Peritoneal fluid from patients with new-onset ascites of unknown origin should be assayed for cell count, albumin level, culture, total protein concentration, Gram stain, and cytologic analysis.3 Serum albumin concentration should be measured as well.
The serum ascites albumin gradient (SAAG, serum albumin concentration—ascitic fluid albumin concentration) is the best diagnostic measure for classification of ascites (Table 97-1).4 The SAAG is very specific and sensitive for distinguishing ascites due to portal hypertension (SAAG > 1.1 g/dL) from that occurring as a result of other pathogenetic mechanisms such as inflammation or peritoneal malignancy (SAAG ≤ 1.1 g/dL). Ideally, specimens should be obtained simultaneously. In the past, ascites was classified as being an exudate (protein concentration ≥ 2.5 g/dL) or a transudate (protein concentration < 2.5 g/dL), but this classification scheme is no longer used because of its poor sensitivity and specificity.5 The total protein level may provide additional clues about diagnosis when used with the SAAG; that is, high SAAG and high protein concentration is seen in most cases of ascites due to hepatic congestion, whereas low serum ascites albumin gradient and high protein concentration characterizes malignant ascites. The terms high albumin gradient and low albumin gradient should replace the terms transudate and exudate in the description of ascites.
TABLE 97-1 Causes of Ascites Based on Normal or Diseased Peritoneum and Serum-to-Ascites Albumin Gradient (SAAG)
| Normal Peritoneum | |
| Portal Hypertension (SAAG > 1.1 g/dL) | Hypoalbuminemia (SAAG < 1.1 g/dL) |
| Hepatic congestion | Nephrotic syndrome |
| Congestive heart failure | Protein-losing enteropathy |
| Constrictive pericarditis | Severe malnutrition with anasarca |
| Tricuspid insufficiency | |
| Budd-Chiari syndrome | |
| Liver disease | Miscellaneous Conditions (SAAG < 1.1 g/dL) |
| Cirrhosis | Chylous ascites |
| Alcoholic hepatitis | Pancreatic ascites |
| Fulminant hepatic failure | Bile ascites |
| Massive hepatic metastases | Nephrogenic ascites |
| Urine ascites | |
| Ovarian disease | |
| Diseased Peritoneum (SAAG < 1.1 g/dL) | |
| Infections | Other Rare Conditions |
| Bacterial peritonitis | Familial Mediterranean fever |
| Tuberculous peritonitis | Vasculitis |
| Fungal peritonitis | Granulomatous peritonitis |
| HIV-associated peritonitis | Eosinophilic peritonitis |
| Malignant Conditions | |
| Peritoneal carcinomatosis | |
| Primary mesothelioma | |
| Pseudomyxoma peritonei | |
| Hepatocellular carcinoma | |
 Pathophysiology
Pathophysiology
Ascites is the most common complication related to liver disease and cirrhosis.6 It is associated with profound changes in the splanchnic and systemic circulation and with renal abnormalities (Figure 97-2). However, the pathogenesis of renal sodium retention and ascites formation in cirrhosis remains a subject of much controversy.
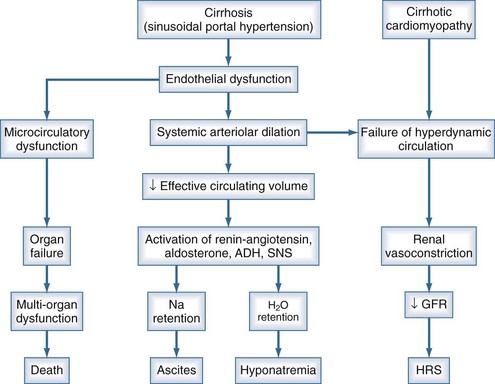
One accepted theory of ascites formation is the forward theory, which states that the development of ascites is related to the existence of severe sinusoidal portal hypertension that causes marked splanchnic arterial vasodilation and a forward increase in the splanchnic production of lymph.7 Splanchnic arterial vasodilation also produces arterial vascular underfilling, a significant reduction of the effective blood volume, and arterial hypotension. These pathophysiologic changes lead to compensatory activation of sodium- and water-retaining mechanisms (the renin-angiotensin-aldosterone system, sympathetic nervous system, and nonosmotic release of vasopressin) and promote ascites formation. Therefore, according to this theory, derangements in the splanchnic arterial circulation rather than the venous portal system are primary in the pathogenesis of ascites formation.8
Patients with advanced cirrhosis and portal hypertension often show an abnormal regulation of extracellular fluid volume, resulting in the accumulation of fluid as ascites, pleural effusion, or edema. The mechanisms responsible for ascites formation include alterations in the splanchnic circulation as well as renal functional abnormalities that favor sodium and water retention.9 The renal functional abnormalities occur in the setting of a hyperdynamic circulatory state that is characterized by increased cardiac output, decreased systemic vascular resistance, and activation of neurohormonal vasoactive systems. This circulatory dysfunction, due mainly to intense arterial vasodilation in the splanchnic circulation, is considered to be a primary feature in the pathogenesis of ascites.
The peripheral arterial vasodilation hypothesis incriminates relative underfilling of the arterial vascular compartment as the primary problem. Relative arterial underfilling leads to the same neurohumoral responses that occur in states characterized by low cardiac output (e.g., chronic congestive heart failure).10 Activation of the renin-angiotensin-aldosterone axis and the sympathetic system, as well as nonosmotic release of vasopressin, are well documented in cases of cirrhosis. This sequence of events results in renal water and sodium retention, failure to escape from the sodium-retaining effect of aldosterone, and renal resistance to atrial natriuretic peptide. Dilutional hyponatremia is the strongest predictor of the occurrence of hepatorenal syndrome.
The pathogenesis of peripheral arterial vasodilation in cirrhosis is not completely elucidated, but there is evidence for a major role of NO.11 Increased vascular NO production has been demonstrated in cirrhosis. In patients with ascites, the hepatic artery produces more NO than it does in patients without ascites. In a rat model of cirrhosis, normalization of vascular NO production with administration of a NO synthase inhibitor corrects the hyperdynamic circulation, improves sodium and water excretion, and decreases neurohumoral activation. This insight into the mechanisms of the peripheral arterial vasodilation in cirrhosis should provide new tools in the treatment of edema and ascites, a major cause of morbidity and mortality in patients with cirrhosis.
The generally accepted peripheral arterial vasodilation hypothesis seems to best explain the mechanism of sodium retention and other clinical findings such as hyperdynamic circulation in patients with cirrhosis. However, recent data in patients with pre-ascites or early ascites do not seem to conform to the peripheral arterial vasodilation hypothesis.12 Renal sodium handling abnormalities can be demonstrated in patients with cirrhosis prior to the development of ascites when these individuals are challenged with a sodium load. These changes are apparent even in the absence of systemic vasodilation or arterial underfilling. Therefore, an alternative hypothesis with a direct hepatorenal interaction, acting via sinusoidal portal hypertension and/or hepatic dysfunction as the effector mechanism, is proposed to be the initiating event promoting renal sodium retention in patients with cirrhosis. The second and later process is the development of systemic arterial vasodilation, possibly due to the presence of excess systemic vasodilators and/or decreased responsiveness of the vasculature to endogenous vasoconstrictors. These changes in turn lead to a relatively underfilled circulation with consequent activation of neurohumoral systems, promoting further renal sodium retention as described by the peripheral arterial vasodilation hypothesis. When compensatory natriuretic mechanisms fail, refractory ascites develops and hepatorenal syndrome sets in. Thus renal sodium retention in patients with cirrhosis is the result of an interplay of many factors; direct hepatorenal interaction predominates in the earlier stages of the cirrhotic process, whereas systemic vasodilation becomes a more important pathogenetic mechanism as the disease progresses.
 Etiology
Etiology
Most cases of ascites are due to liver disease. However, a number of disorders may be associated with ascites, and these include portal vein thrombosis, cardiac disorders (constrictive pericarditis, congestive heart failure), liver cancer, nephrotic syndrome, protein-losing enteropathy, and pancreatitis (see Table 97-1). Nonhepatic causes include cardiac failure, malignancy, renal failure, and intraabdominal inflammation. It is important to diagnose nonhepatic causes of ascites such as malignancy, tuberculosis, and pancreatic ascites, since these occur with increased frequency in patients with liver disease.
 Management
Management
Ascites is the most common presentation of decompensated cirrhosis. It occurs in more than half of all patients with cirrhosis, and its development heralds a poor prognosis (50% 2-year survival rate). Ascites is characterized by three grades of severity, and treatment is based on grade (Table 97-2). Effective first-line medical therapy for ascites includes dietary sodium restriction (2 g/d) and use of diuretics.13
| Grade | Definition | Treatment |
|---|---|---|
| Grade 1 | Mild ascites only detectable by ultrasonographic examination | No specific treatment Dietary sodium restriction Careful follow-up |
| Grade 2 | Moderate ascites manifest by moderate symmetrical distention of the abdomen | Dietary sodium restriction Diuretics (spironolactone with or without furosemide, amiloride for patients with nonactivated renin-angiotensin-aldosterone system) |
| Grade 3 | Large or gross ascites with marked abdominal distention | Paracentesis (total or large-volume, with colloid volume expansion) Dietary sodium restriction Diuretics |
Adapted from Moore KP, Wong F, Gines P, et al. The management of ascites in cirrhosis: report on the Consensus Conference of the International Ascites Club. Hepatology 2003;38:258-66.
Medical Management
Management of uncomplicated ascites includes salt restriction, diuretics, and large-volume paracentesis (LVP) (Table 97-3). Diuretics are the mainstay of medical therapy in the treatment of ascites. Initially, an aldosterone antagonist (spironolactone) is used. Spironolactone competes with aldosterone for receptor sites in the distal renal tubules, increasing salt and water excretion and promoting retention of potassium and hydrogen ions. Spironolactone is usually initiated at a dose of 100 mg per day. The addition of a loop diuretic (e.g., furosemide) may be necessary in some cases to increase the natriuretic effect. The dosage of both the aldosterone antagonist and the loop diuretic should be increased sequentially until an adequate diuretic response is observed. Sodium restriction and diuretic therapy are initially effective in approximately 95% of patients. Water restriction is used only if persistent hyponatremia is present.
| General Management | Treat ascites once complications have been treated. | |
| Avoid NSAIDs. | ||
| Norfloxacin prophylaxis (400 mg PO once daily) in patients with an ascites protein level of <1.5 g/dL, impaired renal function (serum creatinine level ≥ 1.2 mg/dL, BUN ≥ 25 mg/dL, serum sodium level ≤ 130 mEq/L, or severe liver failure (CTP score ≥ 9 points with serum bilirubin level ≥ 3 mg/dL) | ||
| Specific Management | Salt restriction | 1-2 g/day Liberalize if restriction results in poor food intake. |
| Diuretics | Spironolactone based: spironolactone alone (start at 50-100 mg once daily, single morning dose) or: Spironolactone (50-100 mg once daily) + furosemide (start 20-40 mg once daily, single morning dose) |
|
| LVP | Use as initial therapy only in patients with tense ascites; administer intravenous albumin (6-8 g/L of ascites removed). | |
| Follow-up and Goals | Adjustment of diuretic dosage should be performed every 4-7 days. | |
| Patient should be weighed at least weekly, and BUN, creatinine, and electrolytes measured every 1-2 weeks while adjusting dosage. | ||
| Double dosage of diuretics if: Weight loss < 4 lb (2 kg) a week and BUN, creatinine, and electrolytes stable |
||
| Halve the dosage of diuretics or discontinue if: Weight loss ≥ 1 lb (0.5 kg/day) or if there are abnormalities in BUN, creatinine, or electrolytes |
||
| Maximum diuretic dosage is spironolactone, 400 mg once daily, and furosemide, 160 mg once daily. | ||
BUN, blood urea nitrogen; CTP, Child-Turcotte-Pugh; LVP, large volume paracentesis; NSAIDs, nonsteroidal anti-inflammatory drugs; PO, orally.
Data from Garcia-Tsao G, Lim JK; Members of the Veterans Affairs Hepatitis C Resource Center Program. Management and treatment of patients with cirrhosis and portal hypertension: recommendations from the Department of Veterans Affairs Hepatitis C Resource Center Program and the National Hepatitis C Program. Am J Gastroenterol 2009;104:1802-29.
Paracentesis
In the treatment of ascites, paracentesis is reserved for patients refractory to medical management. As a therapeutic intervention, abdominal paracentesis is usually performed to drain a large volume of abdominal ascites.14 When tense or refractory ascites is present, LVP (removal of more than 5 L of ascitic fluid) is safe and effective and has the advantage of producing immediate relief from ascites and its associated symptoms.15 Total paracentesis—removal of all ascites (even >20 L)—usually can be performed safely. Recent studies demonstrate that intravenous (IV) infusion of 5 g of albumin for each liter of ascites removed (>5 L) decreases complications of paracentesis such as electrolyte imbalances and increased serum creatinine concentration secondary to large shifts of intravascular volume. LVP provides rapid resolution of symptoms with minimal complications and is well tolerated by most patients. Paracentesis-induced circulatory dysfunction (PICD) may occur after LVP and is characterized by hyponatremia, azotemia, and increased plasma renin activity. PICD is associated with increased mortality and may be prevented by administration of albumin IV (6–8 g/L of ascites removed).
The International Ascites Club, representing the spectrum of clinical practice from North America to Europe, has developed consensus guidelines for the management of cirrhotic ascites from the stage of early ascites to the stage of refractory ascites.16 Mild to moderate ascites should be managed by modest salt restriction and diuretic therapy with spironolactone or an equivalent. Diuretics should be added in a stepwise fashion while maintaining sodium restriction. Gross ascites should be treated with therapeutic paracentesis followed by colloid volume expansion and diuretic therapy. Refractory ascites is managed by repeated LVP or insertion of a TIPS shunt. Successful placement of TIPS results in improved renal function, sodium excretion, and general well-being but has not been shown to improve survival. Clinicians caring for these patients should be aware of the potential complications of each treatment modality and be prepared to discontinue diuretics, or not proceed with TIPS placement, if complications or contraindications develop. Liver transplantation should be considered for all patients with ascites and cirrhosis. Ideally, liver transplantation should be performed prior to the development of renal dysfunction to minimize the risk of mortality.
Refractory Ascites
Refractory or recurrent ascites is a clinical challenge frequently encountered in patients with cirrhosis.17,18 Ascites becomes refractory to medical treatment in 10% of cirrhotic patients. The diagnosis of refractory ascites recently has been revised (Table 97-4). Refractory ascites is a poor prognostic sign; as many as 50% of patients with this condition die within 6 months of its development.
TABLE 97-4 Revised Diagnostic Criteria of Refractory Ascites
| Treatment duration: Patients must be on intensive diuretic therapy (spironolactone 400 mg/d and furosemide 160 mg/d) for at least 1 week and on a salt-restricted diet of less than 90 mmol or 5.2 g of salt/day. |
| Lack of response: Mean weight loss of <0.8 kg over 4 days and urinary sodium output less than the sodium intake |
| Early ascites recurrence: Reappearance of grade 2 or 3 ascites within 4 weeks of initial mobilization |
| Diuretic-Induced Complications: |
| Diuretic-induced hepatic encephalopathy is the development of encephalopathy in the absence of any other precipitating factor. |
| Diuretic-induced renal impairment is an increase of serum creatinine by >100% to a value >2 mg/dL in patients with ascites responding to treatment. |
| Diuretic-induced hyponatremia is a decrease of serum sodium by >10 mmol/L to a serum sodium concentration of <125 mmol/L. |
| Diuretic-induced hypo- or hyperkalemia is a change in serum potassium to <3 mmol/L or >6 mmol/L despite appropriate measures. |
Adapted from Moore KP, Wong F, Gines P et al. The management of ascites in cirrhosis: report on the Consensus Conference of the International Ascites Club. Hepatology 2003;38:258-66.
The only definitive therapy for refractory ascites with cirrhosis is orthotopic liver transplantation. The other options that are available include LVP, peritoneovenous shunts, and TIPS (Table 97-5). TIPS is contraindicated in patients who have advanced liver failure, because it can hasten death in such individuals. Peritoneovenous shunts are associated with a high incidence of complications and frequent occlusion. They are therefore rarely used for management of refractory ascites. The initial treatment option for refractory ascites is repetitive LVP or total paracentesis.19,20
TABLE 97-5 Management of Refractory Ascites
| Definitions | Ascites that is not eliminated even with maximum diuretic therapy |
| Ascites that is not eliminated because maximum dosages of diuretics cannot be attained, given the development of diuretic-induced complications | |
| Recommended Therapy | Total paracentesis + IV albumin (6-8 g/L of ascites removed) |
| If <5 L of ascites is removed, a synthetic plasma volume expander may be used instead of albumin. | |
| Continue with salt restriction and diuretic therapy as tolerated. | |
| Alternative Therapy | TIPS for patients who require frequent paracenteses (every 1-2 weeks) and whose CTP score is ≤11 |
| Peritoneovenous shunt for patients who are not TIPS or transplant candidates |
CTP, Child-Turcotte-Pugh; IV, intravenous; TIPS, transjugular intrahepatic portosystemic shunt.
Data from Garcia-Tsao G, Lim JK; Members of the Veterans Affairs Hepatitis C Resource Center Program. Management and treatment of patients with cirrhosis and portal hypertension: recommendations from the Department of Veterans Affairs Hepatitis C Resource Center Program and the National Hepatitis C Program. Am J Gastroenterol 2009;104:1802–29.
An early report describing experience with TIPS management of refractory ascites documented that ascites was markedly reduced after this procedure.21 In responders, plasma aldosterone and renin activity decreased, serum creatinine concentration decreased, and urinary sodium excretion increased. However, new-onset hepatic encephalopathy was seen in 14 of 30 patients studied. Severe disabling chronic encephalopathy occurred in 5 patients but was successfully reversed by balloon occlusion of the shunt in 3 patients. Cumulative survival in this study was 41% and 34% at 1 and 2 years, respectively.
Clinicians would find it useful to have a way to predict a favorable clinical response to TIPS for refractory ascites. Accordingly, a prospective cohort study of 53 cirrhotic patients without organic renal disease and with refractory ascites was conducted.22 Some of the patients were “responders” to TIPS. Responders included patients who survived for more than 6 months without severe chronic hepatic encephalopathy and with good control of ascites. The following parameters were examined for prognostic value: age, creatinine clearance, plasma renin activity, plasma aldosterone concentration, and Pugh score. Good control of ascites was obtained in 90% of patients, and 47% were responders to TIPS. The cumulative survival rate was 54% at 6 months, 48% at 1 year, and 39% at 2 years. The majority of patients died of complications of hepatic insufficiency. Severe chronic hepatic encephalopathy developed in 26% of patients. Creatinine clearance was the only factor that was a significant independent predictor of good clinical response to TIPS for refractory ascites. In patients with poor renal function, therefore, TIPS should not be considered.
A randomized prospective trial compared LVP and TIPS in 60 patients with cirrhosis and refractory ascites in Germany.23 Multivariate analysis confirmed that TIPS was independently associated with survival without the need for transplantation (P = .02), with a mean follow-up of 45 months. At 3 months, 61% of the TIPS patients had no ascites, compared to 18% of the paracentesis group (P = .006).
A similar study performed in Spain randomized 70 patients with cirrhosis and refractory ascites to TIPS or repeated paracentesis plus IV albumin.24 Recurrence of ascites and development of hepatorenal syndrome were lower in the TIPS group compared with the paracentesis group, whereas the frequency of severe hepatic encephalopathy was greater in the TIPS group. TIPS did not improve survival and was associated with higher costs.
The North American Study for the Treatment of Refractory Ascites multicenter clinical trial enrolled a larger sample size (n = 109) than prior studies.25 Patients with refractory ascites were randomized to medical therapy (sodium restriction, diuretics, and total paracentesis, n = 57) or medical therapy plus TIPS (n = 52). The principal endpoints were recurrence of tense symptomatic ascites and mortality. A technically adequate shunt was created in 49 of 52 subjects. TIPS plus medical therapy was significantly superior to medical therapy alone in preventing recurrence of ascites (P < .001), but no difference in mortality was identified (21 deaths occurred in each group). There was a higher incidence of moderate to severe encephalopathy in the TIPS group (20 of 52 versus 12 of 57, P = .058), but no difference in the incidence of liver failure, variceal hemorrhage, or acute renal failure. No differences in frequency of emergency department visits, medically indicated hospitalizations, or quality of life were identified. Although TIPS plus medical therapy was superior to medical therapy alone for the control of ascites, TIPS did not improve survival, affect hospitalization rates, or improve quality of life.
The Cochrane Database Systematic Review of TIPS versus paracentesis for cirrhotic patients with refractory ascites included 5 randomized trials, which collectively enrolled 330 patients.26 Mortality at 30 days (OR 1.00, 95% CI 0.10–10.06, P = 1.0) and 24 months (OR 1.29, 95% CI 0.65–2.56, P = 0.5) did not differ significantly between TIPS and paracentesis. Patients randomized to TIPS has significantly decreased reaccumulation of ascites at 3 months (OR 0.07, 95% CI 0.03–0.18, P < .01) and 12 months (OR 0.14, 95% CI 0.06–0.28, P < .01). Hepatic encephalopathy occurred significantly more often in the TIPS group (OR 2.24, 95% CI 1.39–3.6, P < .01), but gastrointestinal bleeding, infection, and acute renal failure did not differ significantly between the two groups. This meta-analysis supports that TIPS was more effective at removing ascites than paracentesis, without a significant difference in mortality, gastrointestinal bleeding, infection, and acute renal failure. However, TIPS patients developed hepatic encephalopathy significantly more often.
The unavailability of individual data, however, precluded the possibility of analyzing survival as a time-dependent variable and separating the confounding effect liver transplantation had on survival of patients with advanced cirrhosis. The proportion of patients who underwent liver transplantation in the five randomized trials ranged from 5% to 20%. A new meta-analysis used individual patient data of four randomized trials27 but excluded one trial28 because refractory ascites was not defined according to the International Ascites Club criteria, and mortality was not the primary endpoint of the study. This meta-analysis (305 patients: 149 TIPS, 156 paracentesis) documented that TIPS significantly reduced the recurrence of tense ascites (42% versus 89%, P < .0001) and significantly improved transplant-free survival of cirrhotic patients with refractory ascites (Figure 97-3). The average number of hepatic encephalopathy episodes was significantly higher in the TIPS group, although the cumulative probability of developing the first episodes of hepatic encephalopathy was similar between the groups.
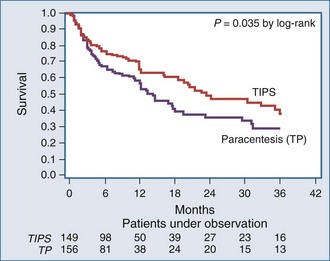
Figure 97-3 Cumulative probability of transplant-free survival according to treatment with TIPS or total paracentesis.
(From Salerno F, Camma C, Enea M, Rossle M, Wong F. Transjugular intrahepatic portosystemic shunt for refractory ascites: a meta-analysis of individual patient data. Gastroenterology 2007;133:825–34.)
Malignant Ascites
Malignant ascites is associated with intraabdominal and pelvic malignancies, and the pathophysiology includes lymphatic obstruction by tumor cells, excess vascular permeability, hormonal effects, and excess metalloproteinase production. Palliative therapies included fluid restriction, diuretics, paracentesis, implantation of drainage catheters (including the PleurX catheter approved by the U.S. Food and Drug Administration [FDA] in 2005), and surgical shunting techniques (peritoneovenous shunts.)29 New approaches to the management of malignant ascites include administration of octreotide as an antisecretory agent, administration of metalloproteinase inhibitors such as batimastat, intraperitoneal immunotherapy (interferon, tumor necrosis factor alpha), and intraperitoneal administration of trifunctional antibodies (e.g., catumaxomab)30,31 that attach to specific overexpressed surface markers on tumor cells and trigger an immune response leading to cytoreductive effects.
 Complications
Complications
Paracentesis-Induced Circulatory Dysfunction
A study randomized 72 patients to receive albumin or saline after total paracentesis.32 The incidence of PICD was significantly higher in the saline-treated group as compared with the albumin-treated group (33.3% versus 11.4%, P = .03). However, no significant differences were found when 6 L of ascitic fluid was evacuated (6.7% versus 5.6%, P = .9). Significant increases in plasma renin activity were found 24 hours and 6 days after paracentesis when saline was used, whereas no changes were observed when albumin was infused. Albumin was more effective than saline for the prevention of PICD but is not required when less than 6 L of ascitic fluid is evacuated. Therefore, administration of IV albumin (6 to 8 g/L of ascites removed) is recommended with LVP.
Nine randomized controlled trials (n = 806 procedures) have examined the use of plasma expanders for therapeutic paracentesis.33–38 This systematic review identified no significant differences between therapeutic paracentesis with and without volume expansion with albumin, nor with nonalbumin plasma expanders compared with albumin for hyponatremia, renal impairment, encephalopathy, or death. However, these studies did not specifically examine prevention of PICD (defined by an increase in plasma renin activity or aldosterone concentration), and some studies determined that albumin prevented PICD more effectively than synthetic plasma expanders.25
Spontaneous Bacterial Peritonitis
SBP is a common complication of cirrhotic ascites.39 It can precipitate hepatorenal syndrome. The overall mortality rate from an episode of SBP is approximately 20%, and following an episode, the 1-year mortality rate approaches 70%.
To diagnose SBP, ascitic fluid should be examined by microscopy and inoculated directly into blood culture bottles. An ascitic fluid neutrophil count ≥ 250 polymorphonuclear cells/µL is diagnostic of SBP, but a Gram stain of the ascitic fluid is usually uninformative (Table 97-6).40
TABLE 97-6 Diagnosis and Management of Spontaneous Bacterial Peritonitis (SBP)
| Diagnosis | Consider SBP and perform diagnostic paracentesis if: Symptoms/signs (abdominal pain, fever, chills) Patient is in emergency room or admitted Worsening renal function or encephalopathy SBP present if ascites PMN count > 250 cells/µL (if fluid bloody, subtract 1 PMN per 250 RBC/µL) |
| General Management | Avoid therapeutic paracenteses during active infection. IV albumin (1 g/kg of body weight) if BUN > 30 mg/dL, bilirubin > 4 mg/dL; repeat at day 3 if renal dysfunction persists. Avoid aminoglycosides. |
| Specific Management | Cefotaxime (2 g IV every 12 h) or Ceftriaxone (2 g every 24 h) or Ampicillin/sulbactam (2 g/L g IV every 6 h) |
| Follow-up | Continue therapy for 7 days. Repeat diagnostic paracentesis at day 2. If ascites PMN count decreases by at least 25% at day 2, IV therapy can be switched to oral therapy (quinolone such as ciprofloxacin or levofloxacin, 250 mg PO BID) to complete 7 days of therapy. |
BID, twice a day; BUN, blood urea nitrogen; IV, intravenous; PMN, polymorphonuclear (neutrophil) cell count; PO, orally; RBC, red blood cell count.
Data from: Garcia-Tsao G, Lim JK; Members of the Veterans Affairs Hepatitis C Resource Center Program. Management and treatment of patients with cirrhosis and portal hypertension: recommendations from the Department of Veterans Affairs Hepatitis C Resource Center Program and the National Hepatitis C Program. Am J Gastroenterol 2009;104:1802-29.
Gram-negative aerobic bacteria are the most common organisms isolated from ascites.41 The three most common isolates are Escherichia coli, Klebsiella pneumoniae, and Streptococcus pneumoniae. Although the number of bacteria present in an episode of SBP is very low, they excite an intensive inflammatory response. Hospitalized patients should be treated with appropriate IV antibiotics.
Patients who have survived an episode of SBP have a 40% to 70% 1-year probability of a further episode. A randomized placebo-controlled trial examined the efficacy of antibiotic treatment purely for secondary prophylaxis of SBP.42 Long-term treatment with norfloxacin reduced the recurrence of SBP at 1 year from 68% to 20%. The treatment effect was mostly due to a reduction of SBP secondary to gram-negative pathogens.
A meta-analysis of 8 prospective studies with a total of 647 patients randomized to oral antibiotic prophylaxis for SBP compared with placebo or no intervention documented an overall mortality benefit (RR = 0.65; 95% CI 0.48-0.88) for antibiotic treatment groups. The overall mortality rate was 16% for treated patients and 25% for the control cohort. Groups treated with prophylactic antibiotics also demonstrated a lower incidence of all infections (including SBP) of 6.2% compared with the control group rate of 22.2% (RR = 0.32; 95% CI 0.20-0.51).43 A Cochrane meta-analysis of 9 trials concluded that antibiotic prophylaxis might be prudent among cirrhotic patients with ascites, but poor trial methodology, concern for systematic bias in publication and design, and concern regarding potential development of resistant pathogens for both the patient and society were articulated clearly.44 On the basis of these results, long-term oral antibiotics are advised for patients recovering from an episode of SBP until resolution of ascites, transplantation, or death (International Ascites Club recommendations). Furthermore, specific patients at high risk of a first episode of SBP (patients with a protein level < 1 g/dL in ascitic fluid and those hospitalized with gastrointestinal hemorrhage) should also receive prophylaxis with orally administered antibiotics, usually a fluoroquinolone, as a management strategy for prevention of SBP (Table 97-7).36,37
TABLE 97-7 Management Strategy for the Prevention of Recurrent SBP
| Recommended Therapy | Oral norfloxacin, 400 mg PO once daily (preferred) or |
| Oral ciprofloxacin, 250-500 mg once daily* or | |
| Oral levofloxacin 250 mg once daily* | |
| Alternative Therapy | TMP-SMX 1 double-strength tablet PO once daily (Patients who develop quinolone-resistant organisms may also have resistance to TMP-SMX.) |
| Duration | Prophylaxis should be continued until the disappearance of ascites or until liver transplantation. |
PO, orally; SBP, spontaneous bacterial peritonitis; TMP-SMX, trimethoprim-sulfamethoxazole.
Data from Garcia-Tsao G, Lim JK; Members of the Veterans Affairs Hepatitis C Resource Center Program. Management and treatment of patients with cirrhosis and portal hypertension: recommendations from the Department of Veterans Affairs Hepatitis C Resource Center Program and the National Hepatitis C Program. Am J Gastroenterol 2009;10:1802-29.
Hepatorenal Syndrome
Hepatorenal syndrome (HRS) is a serious complication of end-stage liver disease, occurring mainly in patients with advanced cirrhosis and ascites who have marked circulatory dysfunction. In spite of its functional nature, HRS is associated with a poor prognosis45 HRS can be precipitated by management of ascites with LVP, and therefore knowledge regarding pathophysiology and treatment of HRS is of paramount importance in treating the patient with ascites related to liver disease. For an in-depth discussion of hepatorenal syndrome, see Chapter 99.
 Prognosis and Outcomes
Prognosis and Outcomes
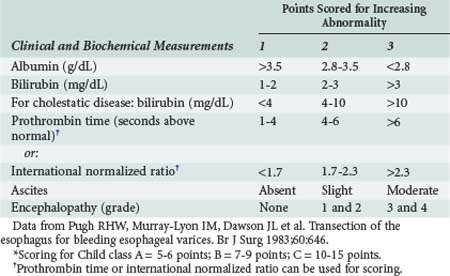
The short-term prognosis of acutely ill patients with cirrhosis is influenced by the degree of hepatic insufficiency and by dysfunction of extrahepatic organ systems. The Child-Turcot-Pugh classification system (Table 97-8) was initially described for estimating outcome in cirrhotic patients undergoing surgery. One important component of this classification is the degree of ascites present, graded as absent, slight, or moderate.
A study46 compared the Child-Pugh classification, the Acute Physiology and Chronic Health Evaluation (APACHE) II system, and the Sequential Organ Failure Assessment (SOFA) for predicting hospital mortality in patients (n = 143) with cirrhosis when used 24 hours after admission to a medical intensive care unit (ICU). Cumulative mortality rates were 36% in the ICU, 46% in the hospital, and 56% at 6-month follow-up. By using the area under receiver operating characteristic (ROC) curves, the SOFA score showed an excellent discriminative power (0.94) which was clearly superior to the APACHE II (0.79) and the Child-Pugh system (0.74). Hospital mortality rates below and above a cutoff of 8 SOFA points were 4% and 88%, respectively (P < .0005). The SOFA score also reflected resource use during the ICU treatment, as measured by daily workload and length of stay. The SOFA score is an easily applied tool with excellent prognostic abilities and can be used to enhance clinical judgment of prognosis as well as to provide patients and families with objective information. A similar study in 111 critically ill cirrhotic patients compared organ system failure scores obtained on the first day of ICU admission to the Child-Pugh classification in predicting hospital mortality.47 The overall hospital mortality rate was 64.9%. Similarly, the organ system failure score (ROC 0.901) was superior to the Child-Pugh score (ROC 0.748) in prediction of hospital mortality in these ICU patients with cirrhosis.
In contrast, the prognostic accuracy of the Child-Pugh score was superior to either the APACHE II or III scores in prediction of short-term hospital mortality of patients with liver cirrhosis (n = 147) admitted to a medical ward and not the ICU.48 Overall mortality in this study was 11.5%. Discrimination was excellent for Child-Pugh (ROC 0.859) and APACHE III (ROC 0.816) scores and acceptable for APACHE II score (ROC 0.759). Although the Hosmer-Lemeshow statistic revealed adequate goodness-of-fit for Child-Pugh score (P = .192), such was not the case for APACHE II and III scores (P = .004 and .003, respectively). This study documented that of the three models, the Child-Pugh score had the least statistically significant discrepancy between predicted and observed mortality.
Key Points
Garcia-Tsao G, Lim JK, Members of Veterans Affairs Hepatitis C Resource Center Program. Management and treatment of patients with cirrhosis and portal hypertension: recommendations from the Department of Veterans Affairs Hepatitis C Resource Center Program and the National Hepatitis C Program. Am J Gastroenterol. 2009;104:1802-1829.
Salerno F, Camma C, Enea M, Rossle M, Wong F. Transjugular intrahepatic portosystemic shunt for refractory ascites: a meta-analysis of individual patient data. Gastroenterology. 2007;133:825-834.
Wong CL, Holroyd-Leduc J, Thorpe KE, Straus SE. Does this patient have bacterial peritonitis or portal hypertension? How do I perform a paracentesis and analyze the results? JAMA. 2008;299:1166-1178.
Saab S, Hernandez JC, Chi AC, Tong MJ. Oral antibiotic prophylaxis reduces spontaneous bacterial peritonitis occurrence and improves short-term survival in cirrhosis: a meta-analysis. Am J Gastroenterol. 2009;104:993-1001.
1 Hou W, Sanyal AJ. Ascites: diagnosis and management. Med Clin North Am. 2009 Jul;93(4):801-817.
2 Thomsen TW, Shaffer RW, White B, Setnik GS. N Engl J Med. 2006 Nov 9;355(19):e21. Videos in clinical medicine. Paracentesis. Erratum in: N Engl J Med. 2007 Feb 15;356(7):760.
3 Moller S, Henriksen JH, Bendtsen F. Ascites: pathogenesis and therapeutic principles. Scand J Gastroenterol. 2009;44(8):902-911.
4 Dittrich S, Yordi LM, de Mattos AA. The value of serum-ascites albumin gradient for the determination of portal hypertension in the diagnosis of ascites. Hepatogastroenterology. 2001;48:166-168.
5 Runyon BA, Montano AA, Akriviadis EA, et al. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med. 1992;117:215-220.
6 Moller S, Henriksen JH, Bendtsen F. Pathogenetic background for treatment of ascites and hepatorenal syndrome. Hepatol Int. 2008 Dec;2(4):416-428.
7 De Franchis R, Salerno F. Pathogenesis of ascites and predictors of resistance to therapy. J Gastroenterol Hepatol. 2002;17(Suppl 3):S242-S247.
8 Cardenas A, Bataller R, Arroyo V. Mechanisms of ascites formation. Clin Liver Dis. 2000;4:447-465.
9 Cardenas A, Arroyo V. Mechanisms of water and sodium retention in cirrhosis and the pathogenesis of ascites. Best Pract Res Clin Endocrinol Metab. 2003;17:607-622.
10 Martin PY, Schrier RW. Pathogenesis of water and sodium retention in cirrhosis. Kidney Int Suppl. 1997;59:S43-S49.
11 Albornoz L, Motta A, Alvarez D, et al. Nitric oxide synthase activity in the splanchnic vasculature of patients with cirrhosis: Relationship with hemodynamic disturbances. J Hepatol. 2001;35:452-456.
12 Wong F, Girgrah N, Blendis L. Review: The controversy over the pathophysiology of ascites formation in cirrhosis. J Gastroenterol Hepatol. 1997;12:437-444.
13 Choudhury J, Sanyal AJ. Treatment of ascites. Curr Treat Options Gastroenterol. 2003;6:481-491.
14 Kuiper JJ, van Buuren HR, de Man RA. Ascites in cirrhosis: a review of management and complications. Neth J Med. 2007 Sep;65(8):283-288.
15 Kuiper JJ, de Man RA, van Buuren HR. Review article. Management of ascites and associated complications in patients with cirrhosis. Aliment Pharmacol. 2007 Dec;26(Suppl. 2):183-193.
16 Moore KP, Wong F, Gines P, et al. The management of ascites in cirrhosis: Report on the Consensus Conference of the International Ascites Club. Hepatology. 2003;38:258-266.
17 Arroyo V, Gines P, Gerbes AL, et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. Hepatology. 1996;23:164-176.
18 Velamati PG, Herlong HF. Treatment of refractory ascites. Curr Treat Options Gastroenterol. 2006;9(6):530-537.
19 Yu AS, Hu KQ. Management of ascites. Clin Liver Dis. 2001;5:541.
20 Zervos EE, Rosemurgy AS. Management of medically refractory ascites. Am J Surg. 2001;191:256.
21 Martinet JP, Fenyves D, Legault L, et al. Treatment of refractory ascites using transjugular intrahepatic portosystemic shunt (TIPS): A caution. Dig Dis Sci. 1997;42:161-166.
22 Deschenes M, Dufresne MP, Bui B, et al. Predictors of clinical response to transjugular intrahepatic portosystemic shunt (TIPS) in cirrhotic patients with refractory ascites. Am J Gastroenterol. 1999;94:1361-1365.
23 Rossle M, Ochs A, Gulberg V, et al. A comparison of paracentesis and transjugular intrahepatic portosystemic shunting in patients with ascites. N Engl J Med. 2000;342:1701.
24 Gines P, Uriz J, Calahorra B, et al. Transjugular intrahepatic portosystemic shunting versus paracentesis plus albumin for refractory ascites in cirrhosis. Gastroenterology. 2002;123:1839-1847.
25 Sanyal AJ, Genning C, Reddy KR, et al. The North American Study for the treatment of refractory ascites. Gastroenterology. 2003;124:634-641.
26 Saab S, Nieto JM, Lewis SK, Runyon BA. TIPS versus paracentesis for cirrhotic patients with refractory ascites. Cochrane Database Syst Rev. 2006 Oct 18;94:CD004889.
27 Sola-Vera J, Minana J, Ricart E, et al. Randomized trial comparing albumin and saline in the prevention of paracentesis-induced circulatory dysfunction in cirrhotic patients with ascites. Hepatology. 2003;37:1147-1153.
28 Salerno F, Camma C, Enea M, Rossle M, Wong F. Transjugular intrahepatic portosystemic shunt for refractory ascites: A meta-analysis of individual patient data. Gastroenterology. 2007;133:825-834.
29 Lebrec D, Giuily N, Hadengue A, et al. Transjugular intrahepatic portosystemic shunt: comparison with paracentesis in patients with cirrhosis and refractory ascites: a randomized trial. J Hepatol. 1996;25:135-144.
30 Becker G. Medical and palliative management of malignant ascites. Cancer Treat Res. 2007;134:459-467.
31 Ammouri L, Prommer EE. Palliative treatment of malignant ascites: profile of catumaxomab. Biologics. 2010 May 25;4:103-110.
32 Heiss MM, Murawa P, Koralewski P, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int J Cancer. 2010 Apr 27. [Epub ahead of print]
33 Gines A, Fernandez-Esparrach G, Monescillo A, et al. Randomized trial comparing albumin, dextran-70 and polygeline in cirrhotic patients with ascites treated by paracentesis. Gastroenterology. 1996;111:1002-1010.
34 Altman C, Bernard B, Roulot D, et al. Randomized comparative multi-center study of hydroxyethyl starch versus albumin as a plasma expander in cirrhotic patients with tense ascites treated with paracentesis. Eur J Gastroenterol Hepatol. 1998;10:5-10.
35 Planas R, Gines P, Arroyo V, et al. Dextran-70 versus albumin as plasma expanders in cirrhotic patients with ascites treated with total paracentesis: Results of a randomized study. Gastroenterology. 1990;99:1736-1744.
36 Salerno F, Badalamenti S, Lorenzano E, et al. Randomized comparative study of Haemaccel vs. albumin infusion after total paracentesis in cirrhotic patients with refractory ascites. Hepatology. 1991;13:707-713.
37 Fassio E, Terg R, Landeira G, et al. Paracentesis with dextran 70 vs paracentesis with albumin in cirrhosis with tense ascites: Results of a randomized study. J Hepatol. 1992;14:310-316.
38 Wong CL, Holroyd-Leduc J, Thorpe KE, Straus SE. Does this patient have bacterial peritonitis or portal hypertension? How do I perform a paracentesis and analyze the results? JAMA. 2008 Mar 12;299(10):1166-1178.
39 Garcia-Tsao G. Current management of the complications of cirrhosis and portal hypertension: Variceal hemorrhage, ascites, and spontaneous bacterial peritonitis. Gastroenterology. 2001;120:726-748.
40 Rimola A, Garcia-Tsao G, Navasa M, et al. Diagnosis, treatment and prophylaxis of spontaneous bacterial peritonitis: A consensus document. International Ascites Club. J Hepatol. 2000;32:142-153.
41 Wong CL, Holroyd-Leduc J, Thorpe KE, Straus SE. Does this patient have bacterial peritonitis or portal hypertension? How do I perform a paracentesis and analyze the results? JAMA. 2008 Mar 12;299(10):1166-1178.
42 Gines P, Rimola A, Planas R, et al. Norfloxacin prevents spontaneous bacterial peritonitis recurrence in cirrhosis: Results of a double-blind, placebo-controlled trial. Hepatology. 1990;12:716-724.
43 Saab S, Hernandez JC, Chi AC, Mj Tong. Oral antibiotic prophylaxis reduces spontaneous bacterial peritonitis occurrence and improves short-term survival in cirrhosis: a meta-analysis. Am J Gastroenterol. 2009 Apr;104(4):993-1001.
44 Cohen MJ, Sahar T, Benenson S, Elinav E, Brezis M, Soares-Weiser K. Antibiotic prophylaxis for spontaneous bacterial peritonitis in cirrhotic patients with ascites, without gastro-intestinal bleeding. Cochrane Database Syst Rev.. 2009 Apr 15;2:CD004791.
45 Salerno F, Gerbes A, Gines P, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007 Sep;56(9):1310-1318.
46 Wehler M, Kokoska J, Reulbach U, et al. Short-term prognosis in critically ill patients with cirrhosis assessed by prognostic scoring systems. Hepatology. 2001;34:255-261.
47 Tsai MH, Chen YC, Ho YP, et al. Organ system failure scoring system can predict hospital mortality in critically ill cirrhotic patients. J Clin Gastroenterol. 2003;37:251-257.
48 Chatzicostas C, Roussomoustakaki M, Notas G, et al. A comparison of Child-Pugh, APACHE II and APACHE III scoring systems in predicting hospital mortality of patients with liver cirrhosis. BMC Gastroenterol. 2003;3:7.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
97 Ascites
Definition and Diagnosis
Ascites is the abnormal accumulation of fluid in the peritoneal cavity.1 Patients with ascites generally present on clinical examination with abdominal distention and a fluid wave or shifting dullness on abdominal percussion, but the abdominal examination findings also may be normal if the amount of ascites is not massive.
Diagnostic imaging can confirm the diagnosis of ascites. Ultrasonography is the easiest and most sensitive technique for the detection of ascitic fluid, being capable of visualizing very small volumes (5-10 mL). Computed tomography (CT) is also very sensitive for detecting ascites (Figure 97-1). Small amounts of ascitic fluid localize in the perihepatic area and in Morrison’s pouch (the hepatorenal space).
A diagnostic paracentesis (20 mL)2 is performed to determine the etiology of the ascites as well as to exclude or establish a diagnosis of spontaneous bacterial peritonitis (SBP). A diagnostic paracentesis should be performed in any person with new-onset ascites. Paracentesis to evaluate for SBP is also indicated for cirrhotic patients with known ascites who require hospitalization or sustain clinical deterioration, such as worsening encephalopathy or unexplained fever. A missed or delayed diagnosis of SBP can lead to sepsis and significant morbidity and mortality.
Peritoneal fluid from patients with new-onset ascites of unknown origin should be assayed for cell count, albumin level, culture, total protein concentration, Gram stain, and cytologic analysis.3 Serum albumin concentration should be measured as well.
The serum ascites albumin gradient (SAAG, serum albumin concentration—ascitic fluid albumin concentration) is the best diagnostic measure for classification of ascites (Table 97-1).4 The SAAG is very specific and sensitive for distinguishing ascites due to portal hypertension (SAAG > 1.1 g/dL) from that occurring as a result of other pathogenetic mechanisms such as inflammation or peritoneal malignancy (SAAG ≤ 1.1 g/dL). Ideally, specimens should be obtained simultaneously. In the past, ascites was classified as being an exudate (protein concentration ≥ 2.5 g/dL) or a transudate (protein concentration < 2.5 g/dL), but this classification scheme is no longer used because of its poor sensitivity and specificity.5 The total protein level may provide additional clues about diagnosis when used with the SAAG; that is, high SAAG and high protein concentration is seen in most cases of ascites due to hepatic congestion, whereas low serum ascites albumin gradient and high protein concentration characterizes malignant ascites. The terms high albumin gradient and low albumin gradient should replace the terms transudate and exudate in the description of ascites.
TABLE 97-1 Causes of Ascites Based on Normal or Diseased Peritoneum and Serum-to-Ascites Albumin Gradient (SAAG)
| Normal Peritoneum | |
| Portal Hypertension (SAAG > 1.1 g/dL) | Hypoalbuminemia (SAAG < 1.1 g/dL) |
| Hepatic congestion | Nephrotic syndrome |
| Congestive heart failure | Protein-losing enteropathy |
| Constrictive pericarditis | Severe malnutrition with anasarca |
| Tricuspid insufficiency | |
| Budd-Chiari syndrome | |
| Liver disease | Miscellaneous Conditions (SAAG < 1.1 g/dL) |
| Cirrhosis | Chylous ascites |
| Alcoholic hepatitis | Pancreatic ascites |
| Fulminant hepatic failure | Bile ascites |
| Massive hepatic metastases | Nephrogenic ascites |
| Urine ascites | |
| Ovarian disease | |
| Diseased Peritoneum (SAAG < 1.1 g/dL) | |
| Infections | Other Rare Conditions |
| Bacterial peritonitis | Familial Mediterranean fever |
| Tuberculous peritonitis | Vasculitis |
| Fungal peritonitis | Granulomatous peritonitis |
| HIV-associated peritonitis | Eosinophilic peritonitis |
| Malignant Conditions | |
| Peritoneal carcinomatosis | |
| Primary mesothelioma | |
| Pseudomyxoma peritonei | |
| Hepatocellular carcinoma | |
Pathophysiology
Ascites is the most common complication related to liver disease and cirrhosis.6 It is associated with profound changes in the splanchnic and systemic circulation and with renal abnormalities (Figure 97-2). However, the pathogenesis of renal sodium retention and ascites formation in cirrhosis remains a subject of much controversy.
One accepted theory of ascites formation is the forward theory, which states that the development of ascites is related to the existence of severe sinusoidal portal hypertension that causes marked splanchnic arterial vasodilation and a forward increase in the splanchnic production of lymph.7 Splanchnic arterial vasodilation also produces arterial vascular underfilling, a significant reduction of the effective blood volume, and arterial hypotension. These pathophysiologic changes lead to compensatory activation of sodium- and water-retaining mechanisms (the renin-angiotensin-aldosterone system, sympathetic nervous system, and nonosmotic release of vasopressin) and promote ascites formation. Therefore, according to this theory, derangements in the splanchnic arterial circulation rather than the venous portal system are primary in the pathogenesis of ascites formation.8
Patients with advanced cirrhosis and portal hypertension often show an abnormal regulation of extracellular fluid volume, resulting in the accumulation of fluid as ascites, pleural effusion, or edema. The mechanisms responsible for ascites formation include alterations in the splanchnic circulation as well as renal functional abnormalities that favor sodium and water retention.9 The renal functional abnormalities occur in the setting of a hyperdynamic circulatory state that is characterized by increased cardiac output, decreased systemic vascular resistance, and activation of neurohormonal vasoactive systems. This circulatory dysfunction, due mainly to intense arterial vasodilation in the splanchnic circulation, is considered to be a primary feature in the pathogenesis of ascites.
The peripheral arterial vasodilation hypothesis incriminates relative underfilling of the arterial vascular compartment as the primary problem. Relative arterial underfilling leads to the same neurohumoral responses that occur in states characterized by low cardiac output (e.g., chronic congestive heart failure).10 Activation of the renin-angiotensin-aldosterone axis and the sympathetic system, as well as nonosmotic release of vasopressin, are well documented in cases of cirrhosis. This sequence of events results in renal water and sodium retention, failure to escape from the sodium-retaining effect of aldosterone, and renal resistance to atrial natriuretic peptide. Dilutional hyponatremia is the strongest predictor of the occurrence of hepatorenal syndrome.
The pathogenesis of peripheral arterial vasodilation in cirrhosis is not completely elucidated, but there is evidence for a major role of NO.11 Increased vascular NO production has been demonstrated in cirrhosis. In patients with ascites, the hepatic artery produces more NO than it does in patients without ascites. In a rat model of cirrhosis, normalization of vascular NO production with administration of a NO synthase inhibitor corrects the hyperdynamic circulation, improves sodium and water excretion, and decreases neurohumoral activation. This insight into the mechanisms of the peripheral arterial vasodilation in cirrhosis should provide new tools in the treatment of edema and ascites, a major cause of morbidity and mortality in patients with cirrhosis.
The generally accepted peripheral arterial vasodilation hypothesis seems to best explain the mechanism of sodium retention and other clinical findings such as hyperdynamic circulation in patients with cirrhosis. However, recent data in patients with pre-ascites or early ascites do not seem to conform to the peripheral arterial vasodilation hypothesis.12 Renal sodium handling abnormalities can be demonstrated in patients with cirrhosis prior to the development of ascites when these individuals are challenged with a sodium load. These changes are apparent even in the absence of systemic vasodilation or arterial underfilling. Therefore, an alternative hypothesis with a direct hepatorenal interaction, acting via sinusoidal portal hypertension and/or hepatic dysfunction as the effector mechanism, is proposed to be the initiating event promoting renal sodium retention in patients with cirrhosis. The second and later process is the development of systemic arterial vasodilation, possibly due to the presence of excess systemic vasodilators and/or decreased responsiveness of the vasculature to endogenous vasoconstrictors. These changes in turn lead to a relatively underfilled circulation with consequent activation of neurohumoral systems, promoting further renal sodium retention as described by the peripheral arterial vasodilation hypothesis. When compensatory natriuretic mechanisms fail, refractory ascites develops and hepatorenal syndrome sets in. Thus renal sodium retention in patients with cirrhosis is the result of an interplay of many factors; direct hepatorenal interaction predominates in the earlier stages of the cirrhotic process, whereas systemic vasodilation becomes a more important pathogenetic mechanism as the disease progresses.
Etiology
Most cases of ascites are due to liver disease. However, a number of disorders may be associated with ascites, and these include portal vein thrombosis, cardiac disorders (constrictive pericarditis, congestive heart failure), liver cancer, nephrotic syndrome, protein-losing enteropathy, and pancreatitis (see Table 97-1). Nonhepatic causes include cardiac failure, malignancy, renal failure, and intraabdominal inflammation. It is important to diagnose nonhepatic causes of ascites such as malignancy, tuberculosis, and pancreatic ascites, since these occur with increased frequency in patients with liver disease.
Management
Ascites is the most common presentation of decompensated cirrhosis. It occurs in more than half of all patients with cirrhosis, and its development heralds a poor prognosis (50% 2-year survival rate). Ascites is characterized by three grades of severity, and treatment is based on grade (Table 97-2). Effective first-line medical therapy for ascites includes dietary sodium restriction (2 g/d) and use of diuretics.13
| Grade | Definition | Treatment |
|---|---|---|
| Grade 1 | Mild ascites only detectable by ultrasonographic examination | No specific treatment Dietary sodium restriction Careful follow-up |
| Grade 2 | Moderate ascites manifest by moderate symmetrical distention of the abdomen | Dietary sodium restriction Diuretics (spironolactone with or without furosemide, amiloride for patients with nonactivated renin-angiotensin-aldosterone system) |
| Grade 3 | Large or gross ascites with marked abdominal distention | Paracentesis (total or large-volume, with colloid volume expansion) Dietary sodium restriction Diuretics |
Adapted from Moore KP, Wong F, Gines P, et al. The management of ascites in cirrhosis: report on the Consensus Conference of the International Ascites Club. Hepatology 2003;38:258-66.
Medical Management
Management of uncomplicated ascites includes salt restriction, diuretics, and large-volume paracentesis (LVP) (Table 97-3). Diuretics are the mainstay of medical therapy in the treatment of ascites. Initially, an aldosterone antagonist (spironolactone) is used. Spironolactone competes with aldosterone for receptor sites in the distal renal tubules, increasing salt and water excretion and promoting retention of potassium and hydrogen ions. Spironolactone is usually initiated at a dose of 100 mg per day. The addition of a loop diuretic (e.g., furosemide) may be necessary in some cases to increase the natriuretic effect. The dosage of both the aldosterone antagonist and the loop diuretic should be increased sequentially until an adequate diuretic response is observed. Sodium restriction and diuretic therapy are initially effective in approximately 95% of patients. Water restriction is used only if persistent hyponatremia is present.
| General Management | Treat ascites once complications have been treated. | |
| Avoid NSAIDs. | ||
| Norfloxacin prophylaxis (400 mg PO once daily) in patients with an ascites protein level of <1.5 g/dL, impaired renal function (serum creatinine level ≥ 1.2 mg/dL, BUN ≥ 25 mg/dL, serum sodium level ≤ 130 mEq/L, or severe liver failure (CTP score ≥ 9 points with serum bilirubin level ≥ 3 mg/dL) | ||
| Specific Management | Salt restriction | 1-2 g/day Liberalize if restriction results in poor food intake. |
| Diuretics | Spironolactone based: spironolactone alone (start at 50-100 mg once daily, single morning dose) or: Spironolactone (50-100 mg once daily) + furosemide (start 20-40 mg once daily, single morning dose) |
|
| LVP | Use as initial therapy only in patients with tense ascites; administer intravenous albumin (6-8 g/L of ascites removed). | |
| Follow-up and Goals | Adjustment of diuretic dosage should be performed every 4-7 days. | |
| Patient should be weighed at least weekly, and BUN, creatinine, and electrolytes measured every 1-2 weeks while adjusting dosage. | ||
| Double dosage of diuretics if: Weight loss < 4 lb (2 kg) a week and BUN, creatinine, and electrolytes stable |
||
