Chapter 24 Arterial hypertension, angina pectoris, myocardial infarction and heart failure







Hypertension: how drugs act
Consider the following relationship:
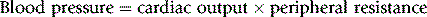
This being true, drugs can lower blood pressure by:
• Dilating arteriolar resistance vessels; achieved through direct relaxation of vascular smooth muscle cells, indirect relaxation by stimulating nitric oxide (NO) production, or by blocking the production or action of endogenous vasconstrictors, such as noradrenaline/norepinephrine and angiotensin.
• Dilating venous capacitance vessels; reduced venous return to the heart (preload) leads to reduced cardiac output, especially in the upright position.
• Reduction of cardiac contractility and heart rate.
• Depletion of body sodium. This reduces plasma volume (transiently), and reduces arteriolar response to noradrenaline/norepinephrine.
Angina pectoris: how drugs act
The supply of myocardial oxygen can be increased by:
• slowing the heart (coronary flow, uniquely, occurs in diastole, which lengthens as heart rate falls).
Drugs used in hypertension and angina
Vasodilators
Organic nitrates
Organic nitrates (and nitrite) were introduced into medicine in the 19th century.1 De-nitration in the smooth muscle cell releases nitric oxide (NO), which is the main physiological vasodilator, normally produced by endothelial cells. Nitrodilators (a generic term for drugs that release or mimic the action of NO) activate the soluble guanylate cyclase in vascular smooth muscle cells and cause an increase in intracellular cyclic guanosine monophosphate (GMP) concentrations. This is the second messenger which alters calcium fluxes in the cell, decreases stored calcium and induces relaxation. The result is a generalised dilatation of venules (capacitance vessels) and to a lesser extent of arterioles (resistance vessels), causing a fall of blood pressure that is postural at first; the larger coronary arteries especially dilate. Whereas some vasodilators can ‘steal’ blood away from atheromatous arteries, with their fixed stenoses, to other, healthier arteries, nitrates probably have the reverse effect as a result of their supplementing the endogenous NO. Atheroma is associated with impaired endothelial function, resulting in reduced release of NO and, possibly, its accelerated destruction by the oxidised low-density lipoprotein (LDL) in atheroma (see Ch. 26).
The nitrates are generally well absorbed across skin and the mucosal surface of the mouth or gut wall. Nitrates absorbed from the gut are subject to extensive first-pass metabolism in the liver, as shown by the substantially higher doses required by that route compared with sublingual application (and explains why swallowing a sublingual tablet of glyceryl trinitrate terminates its effect). They are first de-nitrated and then conjugated with glucuronic acid. The t½ periods vary (see below), but for glyceryl trinitrate (GTN) it is 1–4 min. The de-nitration of GTN is in fact genetically determined as the enzyme responsible, a mitochondrial alcohol dehydrogenase, ALDH2, is polymorphic and in subjects carrying a common coding variant (E504K) sublingual GTN has reduced efficacy.2
to the characteristic vasodilator headache comes and goes quickly (hours).3 Ensuring that a continuous steady-state plasma concentration is avoided prevents tolerance. This is easy with occasional use of GTN, but with nitrates having longer t½ (see below) and sustained-release formulations it is necessary to plan the dosing to allow a low plasma concentration for 4–8 h, e.g. overnight; alternatively, transdermal patches may be removed for a few hours if tolerance is suspected.
An important footnote to the use of nitrates (and NO dilators generally) has been the marked potentiation of their vasodilator effects observed in patients taking phosphodiesterase (PDE) inhibitors, such as sildenafil (Viagra) and tadalafil (Cialis). These agents target an isoform of PDE (PDE-5) expressed in the blood vessel wall. Other methylaxanthine PDE inhibitors, such as theophylline, do not cause a similar interaction because they are rather weak inhibitors of PDE-5, even at the doses effective in asthma. A number of pericoital deaths reported in patients taking sildenafil have been attributed to the substantial fall in blood pressure that occurs when used with a nitrate. This is an ironic twist for an agent in first-line use in erectile dysfunction that was originally developed as a drug to treat angina.4
Glyceryl trinitrate (see also above)
Calcium channel blockers
Calcium is involved in the initiation of smooth muscle and cardiac cell contraction, and in the propagation of the cardiac impulse. Actions on cardiac pacemaker cells and conducting tissue are described in Chapter 25.
Contraction of these cells requires an influx of calcium across the cell membrane. This occurs through voltage-operated ion channels (VOCs) and this influx is able to trigger further release of calcium from intracellular stores in the sarcoplasmic reticulum. The VOCs have relatively long opening times and carry large fluxes; hence they are usually referred to as L-type channels.6 The rise in intracellular free calcium results in activation of the contractile proteins, myosin and actin, with shortening of the myofibril and contraction of smooth muscle. During relaxation calcium is released from the myofibril and either pumped back into the sarcoplasm or lost through Na/Ca exchange at the cell surface.
There are three structurally distinct classes of calcium channel blocker:
Indications for use
• Hypertension: amlodipine, isradipine, nicardipine, nifedipine, verapamil.
• Angina: amlodipine, diltiazem, nicardipine, nifedipine, verapamil.
• Cardiac arrhythmia: verapamil.
• Raynaud’s disease: nifedipine.
• Prevention of ischaemic neurological damage following subarachnoid haemorrhage: nimodipine.
There has been some concern that the shorter-acting calcium channel blockers may adversely affect the risk of myocardial infarction and cardiac death. The evidence is based on case–control studies which cannot escape the possibility that sicker patients, i.e. with worse hypertension or angina, received calcium channel blockade. The safety and efficacy of the class has been strengthened by the recent findings of two prospective comparisons with other antihypertensives.7
Individual calcium blockers
(t½ 2 h) is the prototype dihydropyridine. It selectively dilates arteries with little effect on veins; its negative myocardial inotropic and chronotropic effects are much less than those of verapamil. There are sustained-release formulations of nifedipine that permit once-daily dosing, minimising peaks and troughs in plasma concentration so that adverse effects due to rapid fluctuation of concentrations are lessened. Various methods have been used to prolong, and smooth, drug delivery, and bio-equivalence between these formulations cannot be assumed; prescribers should specify the brand to be dispensed. The adverse effects of calcium blockers with a short duration of action may include the hazards of activating the sympathetic system each time a dose is taken. The dose range for nifedipine is 30–90 mg daily. In addition to the adverse effects listed above, gum hypertrophy may occur. Nifedipine can be taken ‘sublingually’, by biting a capsule and squeezing the contents under the tongue. In point of fact, absorption is still largely from the stomach after this manoeuvre, and it should not be used in a hypertensive emergency because the blood pressure reduction is unpredictable and sometimes large enough to cause cerebral ischaemia (see p. 417).
has a t½ (40 h) sufficient to permit the same benefits as the longest-acting formulations of nifedipine without requiring a special formulation. Its slow association with L-channels and long duration of action render it unsuitable for emergency reduction of blood pressure where frequent dose adjustment is needed. On the other hand, an occasional missed dose is of little consequence. Amlodipine differs from all other dihydropyridines listed in this chapter in being safe to use in patients with cardiac failure (the PRAISE study).8
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin (AT) II receptor blockers (ARBs) and renin inhibitors
Uses
(see p. 406). ACE inhibitors have a useful vasodilator and diuretic-sparing (but not diuretic-substitute) action that is critical to the treatment of all grades of heart failure. Mortality reduction here may result from their being the only vasodilator that does not reflexly activate the sympathetic system.
The ARBs are at least as effective as ACE inhibitors in patients with heart failure and they can be substituted if patients are intolerant of an ACE inhibitor. Based on the Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity (CHARM) trial, they may also benefit patients with heart failure and a low ejection fraction when added to treatment with a β-blocker and ACE inhibitor.9
In patients with type I (insulin-dependent) diabetes, hypertension often accompanies the diagnosis of frank nephropathy, and aggressive blood pressure control is essential to slow the otherwise inexorable decline in renal function that follows. ACE inhibitors appear to have a specific renoprotective effect, probably because of the role of angiotensin II in driving the underlying glomerular hyperfiltration.10 These drugs are now first-line treatment for hypertensive type I diabetics, although most patients will need a second or third agent to reach the rigorous blood pressure targets for this condition (see below). Their role in preventing the progression of the earliest manifestation of renal damage, microalbuminuria, is more complicated. Here the evidence suggests that ACE inhibitors do not slow the incidence of microalbuminuria in type I diabetics and an ARB may actually substantially increase it.10 In contrast, an ACE inhibitor halves the incidence of microalbuminuria in type 2 diabetics with hypertension and normal renal function on follow-up. A parallel group on verapamil did not show any protection confirming that inhibition of the renin–angiotensin–aldosterone (RAAS) axis is required for this effect, not simply lowering the blood pressure.11 For hypertensive type 2 diabetics with established nephropathy, both ARBs and ACE inhibitors protect against a decline in renal function and reduce macroproteinuria.10 The evidence suggests they are interchangeable in this respect. Whether combining the two classes of drugs (‘dual block’) confers further protection of renal function is not yet resolved, although ‘dual block’ does produce substantially better urine protein sparing than either agent alone.10
Following a myocardial infarction, the left ventricle may fail acutely from the loss of functional tissue or in the long term from a process of ‘remodelling’ due to thinning and enlargement of the scarred ventricular wall (see p. 425). Angiotensin II plays a key role in both of these processes and an ACE inhibitor given after MI markedly reduces the incidence of heart failure. The effect is seen even in patients without overt signs of cardiac failure, but who have low left ventricular ejection fractions (< 40%) during the convalescent phase (3–10 days) following the MI. Such patients receiving captopril in the SAVE trial,12 had a 37% reduction in progressive heart failure over the 60-month follow-up period compared with placebo. The benefits of ACE inhibition after MI are additional to those conferred by thrombolysis, aspirin and β-blockers. ARBs also prevent remodelling and heart failure in post-MI patients, but there is no additional benefit from ‘dual blockade’.13
Cautions
Certain constraints apply to the use of ACE inhibitors:
• Heart failure: severe hypotension may result in patients taking diuretics, or who are hypovolaemic, hyponatraemic, elderly, have renal impairment or with systolic blood pressure of less than 100 mmHg. A test dose of captopril 6.25 mg by mouth may be given because its effect lasts for only 4–6 h. If tolerated, the preferred long-acting ACE inhibitor may then be initiated in low dose.
• Renal artery stenosis (RAS, whether unilateral, bilateral renal or suspected from the presence of generalised atherosclerosis): an ACE inhibitor may cause renal failure and is contraindicated. ARBs are not necessarily any safer in this situation, because angiotensin II-mediated constriction of the efferent arteriole is thought to be crucial to the maintenance of glomerular perfusion in RAS.
• Aortic stenosis/left ventricular outflow tract obstruction: an ACE inhibitor may cause severe, sudden hypotension and, depending on severity, is relatively or absolutely contraindicated.
• Pregnancy represents an absolute contraindication (see below).
Adverse effects
• Persistent dry cough occurs in 10–15% of patients.
• Urticaria and angioedema (less than 1 in 100 patients) are much rarer, occurring usually in the first weeks of treatment. The angioedema varies from mild swelling of the tongue to life-threatening tracheal obstruction, when subcutaneous adrenaline/epinephrine should be given. The basis of the reaction is probably pharmacological rather than allergic, due to reduced breakdown of bradykinin.
• Impaired renal function may result from reduced glomerular filling pressure, systemic hypotension or glomerulonephritis, and plasma creatinine levels should be checked before and during treatment.
• Hyponatraemia may develop, especially where a diuretic is also given; clinically significant hyperkalaemia (see effect on aldosterone above) is confined to patients with impaired renal function.
• ACE inhibitors cause major malformations in the first trimester and are fetotoxic in the second trimester, causing reduced renal perfusion, hypotension, oligohydramnios and fetal death (see Pregnancy hypertension, p. 417).
• Neutropenia and other blood dyscrasias occur. Other reported reactions include rashes, taste disturbance (dysguesia), musculoskeletal pain, proteinuria, liver injury and pancreatitis.
Individual drugs
include cilazapril, fosinopril, imidapril, lisinopril, moexipril, perindopril, quinapril, ramipril and trandolapril. Of these, lisinopril has a marginally longer t½ than enalapril (it is the lysine analogue of enalaprilat), probably justifying its popularity as a once-daily ACE inhibitor. Some of the others are longer acting, with quinapril and ramipril also having a higher degree of binding to ACE in vascular tissue. The clinical significance of these differences is disputed. In the Heart Outcomes Prevention Evaluation (HOPE) study of 9297 patients, ramipril reduced, by 20–30%, the rates of death, myocardial infarction and stroke in a broad range of high-risk patients who were not known to have a low ejection fraction or heart failure.14 The authors considered (probably erroneously) that the results could not be explained entirely by blood pressure reduction.
in clinical use include candesartan, eprosartan, irbesartan, telmisartan, valsartan and olmesartan. Some of these may be marginally more effective than losartan at lowering blood pressure, but few if any comparisons have been performed at maximal dose of each drug. Losartan is generally used in combination with hydrochlorothiazide. In a landmark study this combination was 25% more effective than atenolol plus hydrochlorothiazide in preventing stroke.15
The cautions listed for the use of ACE inhibitors (above) apply also to AT1-receptor blockers.
Individual drugs
is the only orally active non-peptide renin inhibitor licensed (t½ 40 h). The agent is well tolerated apart from dose-dependent diarrhoea; it is not clear if this is a class side-effect. It produces additive effects on blood pressure with ACE inhibitors, ARBs, calcium channel blockers and thiazide diuretics. There are currently no outcome data in terms of preventing hypertension-related cardiovascular events, so it should be reserved for inhibiting the RAAS where an ACE inhibitor or ARB is not tolerated.16
Other vasodilators
Sodium nitroprusside is used in hypertensive emergencies, refractory heart failure and for controlled hypotension in surgery. An infusion17 may begin at 0.3–1.0 micrograms/kg/min, and control of blood pressure is likely to be established at 0.5–6.0 micrograms/kg/min; close monitoring of blood pressure is mandatory, usually by direct arterial monitoring; rate changes of infusion may be made every 5–10 min.
is effective through two actions: it acts as a nitrate by activating cyclic GMP (see above) but also opens the ATP-dependent potassium channel to allow potassium efflux and hyperpolarisation of the membrane, which reduces calcium ion entry and induces muscular relaxation. It is indicated for use in angina, where it has similar efficacy to β-blockade, nitrates or calcium channel blockade. It is administered orally and is an alternative to nitrates when tolerance is a problem, or to the other classes when these are contraindicated by asthma or cardiac failure. Adverse effects to nicorandil are similar to those of nitrates, with headache reported in 35% of patients. It is the only antianginal drug for which at least one trial has demonstrated a beneficial influence on outcome.18
is an alkaloid present in opium, but is structurally unrelated to morphine. It inhibits phosphodiesterase and its principal action is to relax smooth muscle throughout the body, especially in the vascular system. It is occasionally injected into an area where local vasodilatation is desired, especially into and around arteries and veins to relieve spasm during vascular surgery and when setting up intravenous infusions. It is also used to treat male erectile dysfunction (see p. 465).
Vasodilators in peripheral vascular disease
Night cramps occur in the disease and quinine has a somewhat controversial reputation in their prevention. Nevertheless, meta-analysis of six double-blind trials of nocturnal cramps (not necessarily associated with peripheral vascular disease) shows that the number, but not severity or duration of episodes, is reduced by a night-time dose.19 The benefit may not be seen for 4 weeks.
Adrenoceptor-blocking drugs
Adrenoceptor-blocking drugs occupy the adrenoceptor in competition with adrenaline/epinephrine and noradrenaline/norepinephrine (and other sympathomimetic amines) whether released from stores in nerve terminals or injected. There are two principal classes of adrenoceptor, α and β: for details of receptor effects see Table 23.1.
α-Adrenoceptor-blocking drugs
There are two main subtypes of α adrenoceptor:
• ‘Classic’ α1 adrenoceptors, on the effector organ (post-synaptic), mediate vasoconstriction.
• α2 Adrenoceptors are present both on some effector tissues (post-synaptic) and on the nerve ending (pre-synaptic). The pre-synaptic receptors (or autoreceptors) inhibit release of chemotransmitter (noradrenaline/norepinephrine), i.e. they provide negative feedback control of transmitter release. They are also present in the CNS.
For use in prostatic hypertrophy, see page 619.


The converse of the benefit in the treatment of prostatism is the adverse effect of urinary incontinence in women. Other adverse effects of α-adrenoceptor blockade are postural hypotension, nasal stuffiness, red sclerae and, in the male, failure of ejaculation. They may also exacerbate symptoms of angina.20 Effects peculiar to each drug are mentioned below.
Notes on individual drugs
is an irreversible non-selective α-adrenoceptor-blocking drug whose effects may last for 2 days or longer. The daily dose must therefore be increased slowly. It is impossible to reverse the circulatory effects by secreting noradrenaline/norepinephrine or other sympathomimetic drugs because its effects are insurmountable. This makes it the preferred α-blocker for treating phaeochromocytoma (see p. 419).
Indigestion and nausea can occur with oral therapy, which is best given with food.
β-Adrenoceptor-blocking drugs
Actions
β-Adrenoceptor selectivity
Some β-adrenoceptor blockers have higher affinity for cardiac β1 receptors than for cardiac and peripheral β2 receptors (Table 24.1). The ratio of the amount of drug required to block the two receptor subtypes is a measure of the selectivity of the drug. (See note to Table 23.1, p. 384, regarding the use of the terms ‘β1 selective’ and ‘cardioselective’.) The question is whether the differences between selective and non-selective β-blockers confer clinical advantages. In theory β1-blockers are less likely to cause bronchoconstriction, but in practice few available β1-blockers are sufficiently selective to be safely recommended in asthma. Bisoprolol and nebivolol may be exceptions that can be tried at low doses in patients with mild asthma and a strong indication for β-blockade. There are unlikely ever to be satisfactory safety data to support such use. The main practical use of β1-selective blockade is in diabetics, where β2 receptors mediate both the symptoms of hypoglycaemia and the counter-regulatory metabolic responses that reverse the hypoglycaemia.
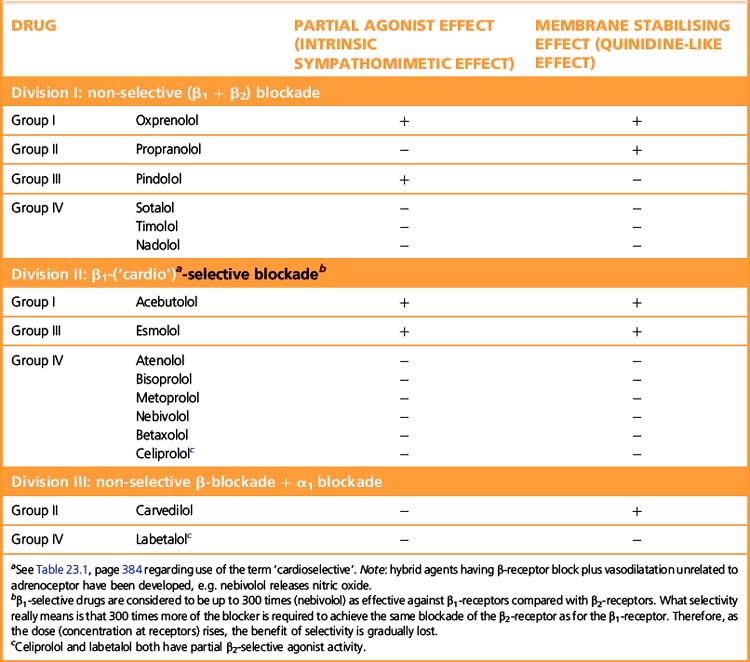
Some β-blockers (antagonists) also have agonist action or ISA, i.e. they are partial agonists. These agents cause less fall in resting heart rate than do the pure antagonists and may thus be less effective in severe angina pectoris where reduction of heart rate is particularly important. The fall in cardiac output may be less, and fewer patients may experience unpleasantly cold extremities. Intermittent claudication may be worsened by β-blockade whether or not there is partial agonist effect. Both classes of drug can precipitate heart failure, and indeed no important difference is to be expected because patients with heart failure already have high sympathetic drive (but note that β-blockade can be used to treat cardiac failure, p. 406).
Classification of β-adrenoceptor-blocking drugs
• Pharmacokinetic: lipid soluble, water soluble, see above.
• Pharmacodynamic (see Table 24.1). The associated properties (partial agonist action and membrane-stabilising action) have only minor clinical importance with current drugs at doses ordinarily used and may be insignificant in most cases. But it is desirable that they be known, for they can sometimes matter and they may foreshadow future developments.
Uses of β-adrenoceptor-blocking drugs
Angina pectoris: β-blockade reduces cardiac work and oxygen consumption.
Cardiac tachyarrhythmias: β-blockade reduces drive to cardiac pacemakers: subsidiary properties (see Table 25.1, p. 430) may also be relevant.
• Early use within 6 h (or at most 12 h) of onset (intravenously for 24 h then orally for 3–4 weeks). Benefit has been demonstrated only for atenolol. Cardiac work is reduced, resulting in a reduction in infarct size by up to 25% and protection against cardiac rupture. Surprisingly, tachyarrhythmias are not less frequent, perhaps because the cardiac β2 receptor is not blocked by atenolol. Maximum benefit is in the first 24–36 h, but mortality remains lower for up to 1 year. Contraindications to early use include bradycardia (< 55 beats/min), hypotension (systolic < 90 mmHg) and left ventricular failure. A patient already taking a β-blocker may be given additional doses.
• Late use for secondary prevention of another myocardial infarction. The drug is started between 4 days and 4 weeks after the onset of the infarct and is continued for at least 2 years.21
• Choice of drug. The agent should be a pure antagonist, i.e. without ISA.
Hepatic portal hypertension and oesophageal variceal bleeding: reduction of portal pressure (see p. 548).
Cardiac failure (see also Ch. 25): there is now clear evidence from prospective trials that β-blockade is beneficial in terms of mortality for patients with all grades of moderate heart failure. Data support the use of both non-selective (carvedilol, α-blocker as well) and β1-selective (metoprolol and bisoprolol) agents. Survival benefit exceeds that provided by ACE inhibitors over placebo. The negative inotropic effects can still be significant, so the starting dose is low (e.g. bisoprolol 1.25 mg orally daily in the morning or carvedilol 3.125 mg twice daily, with food) and may be tolerated only with additional antifailure therapy, e.g. diuretic.





Adverse reactions due to β-adrenoceptor blockade
Bronchoconstriction (β2 receptor) occurs as expected, especially in patients with asthma22 (in whom even eye drops are dangerous23). In elderly chronic bronchitics there may be gradually increasing bronchoconstriction over weeks (even with eye drops). Plainly, risk is greater with non-selective agents, but β1-receptor-selective members can still have significant β2-receptor occupancy and may precipitate asthma.
Hypotension when the drug is given after myocardial infarction.
Sexual function: interference is unusual and generally not supported in placebo-controlled trials.
Adverse reactions not certainly due to β-adrenoceptor blockade
Oculomucocutaneous syndrome occurred with chronic use of practolol (now obsolete) and even occasionally after cessation of use.24 Other members either do not cause it, or so rarely do so that they are under suspicion only and, properly prescribed, the benefits of their use far outweigh such a very low risk. The mechanism of the syndrome is uncertain but appears immunological.
Overdose
Overdose, including self-poisoning, causes bradycardia, heart block, hypotension and low-output cardiac failure that can proceed to cardiogenic shock; death is more likely with agents that have a membrane-stabilising action (see Table 24.1). Bronchoconstriction can be severe, even fatal, in patients subject to any bronchospastic disease; loss of consciousness may occur with lipid-soluble agents that penetrate the CNS. Receptor blockade will outlast the persistence of the drug in the plasma.
• Atropine (1–2 mg i.v. as one or two bolus doses) to eliminate the unopposed vagal activity that contributes to bradycardia. Most patients also require direct cardiac pacing.
• Glucagon, which has cardiac inotropic and chronotropic actions independent of the β-adrenoceptor (dose 50–150 micrograms/kg in glucose 5% i.v., repeated if necessary) to be used at the outset in severe cases (an unlicensed indication).
• If there is no response, intravenous injection or infusion of a β-adrenoceptor agonist is an alternative, e.g. isoprenaline (4 micrograms/min, increasing at 1–3-min intervals until the heart rate is 50–70 beats/min).
• Other sympathomimetics may be used as judgement counsels, according to the desired receptor agonist actions (β1, β2, α) required by the clinical condition, e.g. dobutamine, dopamine, dopexamine, noradrenaline/norepinephrine, adrenaline/epinephrine.
• For bronchoconstriction, salbutamol may be used; aminophylline has non-adrenergic cardiac inotropic and bronchodilator actions and should be given intravenously very slowly to avoid precipitating hypotension.
• A cardiac pacemaker may be used to increase the heart rate.
Treatment may be needed for days. With prompt treatment, death is unusual.
Interactions
Combined β1– and α-adrenoceptor-blocking drug
is a racemic mixture: one isomer is a β-adrenoceptor blocker (non-selective), another blocks α-adrenoceptors. Its dual effect on blood vessels minimises the vasoconstriction characteristic of non-selective β-blockade so that, for practical purposes, the outcome is similar to that of a β1-selective β-blocker (see Table 24.1). It is less effective than drugs such as atenolol or bisoprolol for the routine treatment of hypertension, but is useful for some specific indications.
Labetalol reduces the hypertensive response to orgasm in women.
Peripheral sympathetic nerve terminal
Adrenergic neurone-blocking drugs
Adrenergic neurone-blocking drugs are taken up into adrenergic nerve endings by the active noradrenaline/norepinephrine reuptake mechanism (uptake 1) (see Fig. 23.1). They are relatively ineffective in reducing blood pressure except in the erect position, and their use to control hypertension is now obsolete. Guanethidine is still licensed in the UK as an option for the rapid control of blood pressure, and may also be used for regional intravenous sympathetic blockade in patients with intractable Raynaud’s disease.
Meta-iodobenzylguanidine (MIBG) is used diagnostically as a radio-iodinated tracer, to locate or confirm chromaffin tumours (phaeochromocytoma and neuroblastoma), which accumulate with drugs in this class (see p. 419).
Depletion of stored transmitter (noradrenaline/norepinephrine)
Reserpine is an alkaloid from plants of the genus Rauwolfia, used in medicine since ancient times for insanity. Reserpine depletes adrenergic nerves of noradrenaline/norepinephrine, primarily by blocking the transport of noradrenaline/norepinephrine into storage vesicles (see Fig. 23.1). Its antihypertensive action is chiefly a peripheral action, but it enters the CNS and depletes central catecholamine stores; this explains the sedation, depression and parkinsonian side-effects that can accompany its use. Reserpine is rarely used now that its low cost is matched by many superior classes.
Central nervous system
α2-Adrenoceptor agonists
Its most serious handicap is that abrupt or even gradual withdrawal causes rebound hypertension. This is characterised by plasma catecholamine concentrations as high as those seen in hypertensive attacks of phaeochromocytoma. The onset may be rapid (a few hours) or delayed for as long as 2 days; it subsides over 2–3 days. The treatment is either to reinstitute clonidine, intramuscularly if necessary, or to treat as for a phaeochromocytoma (see below). Clonidine should never be used with a β-adrenoceptor blocker that exacerbates withdrawal hypertension (see phaeochromocytoma, p. 419). Other common adverse effects include sedation and dry mouth.
Drug treatment of angina, myocardial infarction and hypertension
Angina pectoris25
An attack of angina pectoris26 occurs when myocardial demand for oxygen exceeds supply from the coronary circulation. The principal forms relevant to choice of drug therapy are angina of exercise (commonest) and its worsening form, unstable (preinfarction or crescendo) angina (see below), which occurs at rest. Variant (Prinzmetal) angina (very uncommon) results from spasm of a large coronary artery.
• Organic nitrates reduce preload and afterload and dilate the main coronary arteries (rather than the arterioles).
• β-Adrenoceptor-blocking drugs reduce myocardial contractility and slow the heart rate. They may increase coronary artery spasm in variant angina.
• Calcium channel-blocking drugs reduce cardiac contractility, dilate the coronary arteries (where there is evidence of spasm) and reduce afterload (dilate peripheral arterioles).
These classes of drug complement one another and can be used together. The combined nitrate and potassium channel activator nicorandil is an alternative when any of the other drugs is contraindicated.
Summary of treatment
• Any contributory cause is treated when possible, e.g. anaemia, arrhythmia.
• Lifestyle is changed so as to reduce the number of attacks. Weight reduction can be very helpful; stop smoking.
• For immediate pre-exertional prophylaxis: glyceryl trinitrate sublingually or nifedipine (bite the capsule and hold the liquid in the mouth or swallow it).
• For an acute attack: glyceryl trinitrate (sublingual) or nifedipine (bite capsule, as above).
• A β1–adrenoceptor-blocking drug, e.g. bisoprolol, is given regularly (not merely when an attack is expected). Dosage is adjusted by response. Some put an arbitrary upper limit to dose, but others recommend that, if complete relief is not obtained, the dose should be raised to the maximum tolerated, provided the resting heart rate is not reduced below 55 beats/min; or raise the dose to a level at which an increase causes no further inhibition of exercise tachycardia. In severe angina a pure antagonist, i.e. an agent lacking partial agonist activity, is preferred, as the latter may not slow the heart sufficiently. Warn the patient of the risk of abrupt withdrawal.
• A calcium channel-blocking drug, e.g. nifedipine or diltiazem, is an alternative to a β-adrenoceptor blocker; use especially if coronary spasm is suspected or if the patient has myocardial insufficiency or any reversible airflow obstruction. It can also be used with a β-blocker, or
• A long-acting nitrate, isosorbide dinitrate or mononitrate: use so as to avoid tolerance (see p. 394).
• Nicorandil, a long-acting potassium channel activator, does not cause tolerance like the nitrates.
• Drug therapy may be adapted to the time of attacks, e.g. nocturnal (transdermal glyceryl trinitrate, or isosorbide mononitrate orally at night).
• Antiplatelet therapy (aspirin or clopidogrel) reduces the incidence of fatal and non-fatal myocardial infarction in patients with unstable angina, used alone or with low-dose heparin.
• Revascularisation in selected cases (largely by percutaneous coronary intervention (PCI) and stenting).
Myocardial infarction (MI)
An overview
• Morphine or diamorphine (2.5 or 5 mg i.v. because of the certainty of haematoma formation when intramuscular injections are followed by thrombolytic therapy).
The immediate objectives are relief of pain and initiation of treatment demonstrated to reduce mortality. Subsequent management of proven MI is concerned with treatment of complications, arrhythmias, heart failure and thromboemboli, and then prevention of further infarctions.
The choice of thrombolytic is in most places dictated first by a wealth of comparative outcome data from well-designed trials, and second by relative costs. So, for a first MI, patients should receive streptokinase 1 500 000 units infused over 1 h, unless they are in cardiogenic shock. For subsequent infarcts, the presence of antistreptokinase antibodies dictates the use of the recombinant tissue plasminogen activator (rtPA), alteplase (or reteplase). Both alteplase and streptokinase bind plasminogen and convert it to plasmin, which lyses fibrin. Alteplase has a much higher affinity for plasminogen bound to fibrin than in the circulation. This selectivity does not confer any therapeutic advantage as was originally anticipated, as severe haemorrhage following thrombolysis is almost always due to lysis of an appropriate clot at previous sites of bleeding or trauma. Indeed, the tendency for some lysis of circulating fibrinogen as well as fibrin gives streptokinase anticoagulant activity, which is lacking with alteplase, use of which needs to be accompanied and followed by heparin (for further details of thrombolytics, see p. 490).
For a discussion about the role of aspirin, see p. 246.
A third treatment reduces mortality in MI, namely β–blockade. In the ISIS-1 study,27 atenolol 5 mg was given intravenously, followed by 50 mg orally. The reduction in mortality is due mainly to prevention of cardiac rupture, which appears interestingly to remain the only complication of MI that is not reduced by thrombolysis. The usual contraindications to β-blockade apply, but most patients with a first MI should be able to receive this treatment.
The final common pathway to platelet aggregation and thrombus formation involves the expression of the glycoprotein IIb/IIIa receptor at the cell surface. This receptor binds fibrinogen with high affinity and can be blocked using either a specific monoclonal antibody (abciximab) or one of a rapidly expanding class of specific antagonists, e.g. eptifibatide and tirofiban. Another agent, clopidogrel, acts by inhibiting ADP-dependent platelet aggregation. It is more effective than aspirin for the prevention of ischaemic stroke, cardiovascular death in patients at high risk (see p. 492) or following a STEMI or non-STEMI event.
Principal contraindications to thrombolysis
• Recent symptoms of peptic ulcer, or gastrointestinal bleeding.
• Recent stroke (previous 3 months).
• Recent surgery (previous 10–14 days), especially neurosurgery.
• Prolonged cardiopulmonary resuscitation (during current presentation).
• Proliferative diabetic retinopathy.
• Severe, uncontrolled hypertension (diastolic blood pressure > 120 mmHg).
Drugs for secondary prevention
All patients should receive aspirin (see Ch. 4, Fig. 4.3), an ACE inhibitor and a β-blocker for at least 2 years, unless contraindicated. The commonest contraindications to β-blockade after MI are transient heart failure, which should now be uncommon after a first MI, and various degrees of heart block or bradyarrythmias. These are, however, usually transient, so the β-blocker can be introduced during convalescence.
Any of these agents, aspirin, a β-blocker or an ACE inhibitor,28 will reduce the incidence of reinfarction by 20–25%, although their benefit has not been shown to be additive.
In addition to these drugs, most patients should receive a statin, regardless of their plasma cholesterol concentration. Long-term benefit from LDL reduction after MI has been shown for simvastatin (20–40 mg/day) and pravastatin (40 mg/day).29
Arterial hypertension
Clinical evaluation of antihypertensive drugs seeks to answer two types of question:
1. Whether long-term reduction of blood pressure benefits the patient by preventing complications and prolonging life; these studies take years, require enormous numbers of patients and are extremely costly.
2. Whether a drug is capable of effective, safe and comfortable control of blood pressure for about 1 year. There is now sufficient evidence of the benefit of reducing raised blood pressure that regulatory authorities do not demand trials of the first kind for all new drugs. Shorter studies are therefore deemed sufficient to allow the introduction of a new drug.
Threshold and targets for treatment
The joint NICE/British Hypertension Society guidelines30 require that antihypertensive drug therapy be initiated:
• when sustained blood pressure exceeds 160/100 mmHg, or
• when blood pressure is in the range 140–159/90–99 mmHg and there is evidence of target organ damage, cardiovascular disease or a 10-year cardiovascular risk greater than 20%
The optimal target is to lower blood pressure to 140/85 mmHg or less in all patients except those with renal impairment, in diabetics or in established cardiovascular disease where there is a lower target of less than 130/80 mmHg.
Relative risk refers to the increased likelihood of a patient having a complication, compared with a normotensive patient of the same age and sex. Absolute risk refers to the number of patients out of 100, with the same age, sex and blood pressure, predicted to have a complication over the next 10 years (see p. 50). So, the relative risk of MI due to hypertension is fixed, but substantial reduction in the absolute risk of MI is possible by reducing the level of cholesterol and blood pressure, i.e. both factors contribute independently to the risk of MI whereas hypertension is a more important risk factor for stroke than hypercholesterolaemia.
Principles of antihypertensive therapy
General measures may be sufficient to control mild cases as follows:
• Alcohol: stay within recommended limits, e.g. 14 units/week for women, 21 units/week for men.
• Diet: of proven value for the short-term reduction in blood pressure is reduction in fat content, and increase in fruit, vegetables and fibre. There is additional benefit from reducing intake of salt (< 6 g/day): avoidance of highly salted foods, and omission of added salt from freshly prepared food.31
• Relaxation therapy: worth considering for highly motivated borderline patients.
Drug therapy
Blood pressure may be reduced by any one or more of the actions listed at the beginning of this chapter (see p. 393). The large number of different drug classes for hypertension reduces, paradoxically, the likelihood of a randomly selected drug being the best for an individual patient. Patients and drugs can be divided broadly into two groups depending on their renin status and drug effect on this (Fig. 24.1).
• Type 1, or high-renin patients, are the younger, non-black patients (aged < 55 years); they respond better to an ACE inhibitor, ARB, or β-blocker.
• Type 2, or low-renin patients, in whom diuretics or calcium blockers are more likely to be effective as single agents.
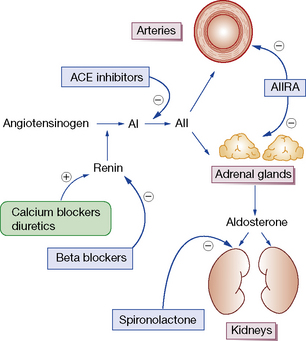
1. Increase in blood volume: this occurs with any drug that reduces peripheral resistance (increases intravascular volume) or cardiac output (reduces glomerular flow) due to activation of the renin–angiotensin system. The result is that cardiac output and blood pressure rise. Adding a diuretic in combination with the other drug can prevent this compensatory effect.
2. Baroreceptor reflexes: a fall in blood pressure evokes reflex activity of the sympathetic system, causing increased peripheral resistance and cardiac activity (rate and contractility).
Therefore, whenever high blood pressure is proving difficult to control and whenever a number of antihypertensives are used in combination, the drugs chosen should between them act on all three main determinants of blood pressure, namely:
• Maximise antihypertensive efficacy by exerting actions at three different points in the cardiovascular system.
• Minimise the opposing homeostatic effects by blocking the compensatory changes in blood volume, vascular tone and cardiac function.
• Minimise adverse effects by permitting smaller doses of each drug each acting at a different site and having different unwanted effects.
Treating hypertension
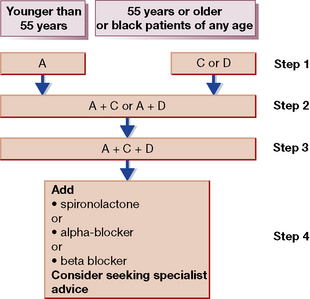
A simple stepped regimen in keeping with the 2006 revision of the National Institute for Health and Clinical Excellence (NICE)/British Hypertension Society guidelines32 is the ‘A/CD’ schema illustrated in Figure 24.2.33
1. If the patient is young (age < 55 years) or non-black, use either an ACE inhibitor (or ARB) (A). For older patients start with either a Calcium channel blocker or thiazide Diuretic as first-line therapy (C or D). If a drug is effective but not tolerated, switch to the other member of the pair.
2. If the blood pressure is not controlled at 4 weeks, a second agent should be added, using the opposite pair to the first drug, e.g. if the patient is on an ACE inhibitor add a calcium channel blocker or thiazide diuretic (A + C or A + D), as both vasodilatation or diuresis will stimulate the renin–angiotensin system and turn non-renin-dependent hypertension into renin-dependent hypertension.
3. If blood pressure control is still inadequate on dual therapy, A + C + D is the ideal triple regimen.
4. Patients whose blood pressure remains substantially above target on triple therapy should have either escalated diuretic therapy, e.g. by addition of spironolactone; or addition of α- or β-blockade. There is some evidence that a trial-and-error approach at this point can be avoided by measurement of plasma renin, with low plasma renin indicating the need for further diuretic.
Treatment and severity
A single drug may adequately treat mild hypertension, and a few patients with hypertension due to pure vasoconstriction or pure sodium excess. In most patients, the target systolic blood pressure of < 140 mmHg recommended by most guidelines requires two or more drugs. While convention until recently has been to ‘step-care’ from single to combination therapy, the risks of adverse effects from treatment, and of early complications from uncontrolled hypertension, may be diminished by starting with combination therapy.34
Monitoring
The potassium-losing (kaliuretic) diuretics used in hypertension can deplete body potassium by up to 10–15%. However, at low doses, e.g. bendroflumethiazide 2.5 mg/day, significant hypokalaemia is unusual, and should raise suspicion of Conn’s syndrome (see p. 420). Vulnerable patients, e.g. the elderly, should be monitored for potassium loss at 3 months and thereafter every 6–12 months. If required, for correction of hypokalaemia, a potassium-sparing diuretic (amiloride) in a fixed-dose combination with a thiazide (co-amilozide) is preferred over the use of fixed-dose diuretic/potassium chloride formulations (most supplements, typically 8 mmol KCl, are in any case inadequate).
Treatment of hypertensive emergencies
It is important to distinguish three circumstances that may exist separately or together; see the Venn diagram (Fig. 24.3)35 which emphasises the following:
• Severe hypertension is not on its own an indication for urgent (or large) reductions in blood pressure.
• Blood pressure can occasionally require urgent (emergency) reduction even when the hypertension is not severe, especially where the blood pressure has risen rapidly.
• Accelerated phase (malignant) hypertension rarely requires urgent reduction, and should instead be regarded as an indication for slow reduction in blood pressure during the first few days.
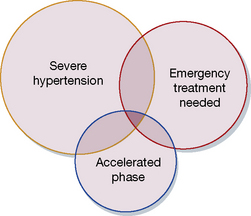
Fig. 24.3 Venn diagram illustrating intersections of three overlapping clinical states defined in the text.
The indications for emergency reduction of blood pressure are rare. They are:
• Hypertensive encephalopathy (including eclampsia).
A theoretically preferable, but often impractical, alternative is intravenous infusion of the vasodilator, nitroprusside (see p. 400). In dissecting aneurysm, vasodilators should not be used unless patients are first β-blocked, because any increase in the rate of rise of the pulse stroke is undesirable. Labetalol provides a convenient method of treating all patients within the three circles (except asthmatics), using either oral or parenteral therapy as appropriate. That said, it is not the most effective therapy and should be combined with a long-acting formulation of nifedipine, orally, where further blood pressure reduction is required.
Pregnancy hypertension
ACE inhibitors and angiotensin II receptor blockers (ARBs) are absolutely contraindicated during pregnancy. They cause major malformations after first-trimester exposure36 and fetal death, typically mid-trimester. For this reason they are probably best avoided in women of child-bearing age, especially where there is no effective contraception, since it is not uncommon for women to discover their pregnancy late into its first trimester. If they are used, women should be counselled to stop an ACE or ARB as soon as they suspect they are pregnant.
Raised blood pressure and proteinuria (pre-eclampsia) complicates 2–8% of pregnancies and may proceed to fitting (eclampsia), a major cause of mortality in mother and child. Magnesium sulphate halves the risk of progress to eclampsia (typically 4 g i.v. over 5–10 min followed by 1 g/h by i.v. infusion for 24 h after the last seizure).37 Additionally, if a woman has one fit (treat with diazepam), then the magnesium regimen is superior to diazepam or phenytoin in preventing further fits.38
Sexual function and cardiovascular drugs
Sexual intercourse and the cardiovascular system
There are few, if any, records of sudden cardiovascular death among women under these circumstances.
Patients subject to angina pectoris should also use glyceryl trinitrate or isosorbide dinitrate as usual for pre-exertional prophylaxis 10 min before intercourse. But they should be aware of the potentially fatal interaction of sildenafil (Viagra) and other PDE5 inhibitors with nitrates (see p. 394).
• The treatment of both hypertension and angina requires drugs that reduce the work of the heart either directly or by lowering peripheral vascular resistance.
• β-Blockade, which acts mainly through reduced cardiac output, and calcium channel blockade, acting by selective arterial dilatation, may be used in either condition.
• Other vasodilators are suited preferentially to hypertension (ACE inhibitors, angiotensin (AT1) receptor blockers (ARBs) and α-adrenoceptor blockers) or to angina (nitrates).
• The treatment of myocardial infarction requires thrombolysis, aspirin and β-adrenoceptor blockade acutely, with the latter two continued for at least 2 years as secondary prevention of a further infarction.
• Other important steps in secondary prevention include ACE inhibitors for cardiac failure and statins for hypercholesterolaemia in selected patients.
Pulmonary hypertension
Idiopathic (primary) pulmonary arterial hypertension (PAH)
Verapamil may give symptomatic benefit. Prostanoid formulations used to treat PAH include intravenous epoprostenol (prostacyclin) and inhaled iloprost.39 The prostanoid formulations have the limitations of a short half-life and a heterogeneous response to therapy. Evidence suggests that endothelin, a powerful endogenous vasoconstrictor, may play a pathogenic role; antagonists, bosentan, ambrisentan and sitoxsentan, improve symptoms and haemodyamic measurements, but dose is limited by hepatic toxicity. The PDE5 inhibitors, sildenafil, tadalafil, and vardenafil, are an alternative. Only tadalafil has been shown to improve survival, although in a comparison of the three drugs only sildenafil increased exercise tolerance. Heart and lung transplantation is recommended for younger patients.
Phaeochromocytoma
In cases of borderline biochemistry, pharmacological suppression tests are useful. Either the ganglion-blocking drug pentolinium or centrally acting α2-agonist clonidine suppresses physiological elevations of metaphrines, but not autonomous secretion from a tumour.40,41 Provocation tests should not be deliberately employed; but the initial search for phaeochromocytoma may be prompted by a history of hypertensive crisis induced by dopamine antagonists (e.g. metoclopramide) or any drug that releases histamine (opioids, curare, trimetaphan).
Conn’s syndrome
This refers to benign adenomas of the adrenal cortex, which secrete the sodium-retaining hormone aldosterone, and are present in 2–3% of patients with hypertension. Synonyms include primary hyperaldosteronism, although this term can extend to a larger number of patients with elevated plasma aldosterone to renin ratios but no lateralisation of aldosterone secretion. Conn’s adenomas are diagnosed by finding a suppressed plasma renin, without suppression of aldosterone, and an adrenal adenoma on CT or MRI. Since 5% of adults have incidental non-functional adrenal adenomas, the key step in diagnosis is lateralisation: the demonstration that the adenoma is responsible for excess aldosterone secretion. Conventionally this is done by adrenal venous sampling. An alternative, in specialist centres, is a PET-CT using a tracer dose of the anaesthetic drug metomidate labelled with F18, which has high affinity binding to the steroid synthases (Fig. 24.4).
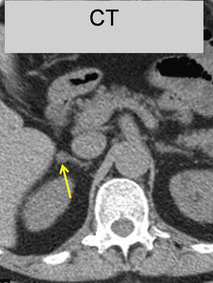
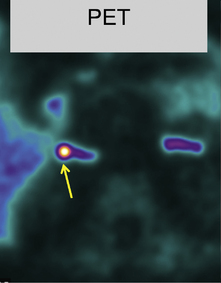
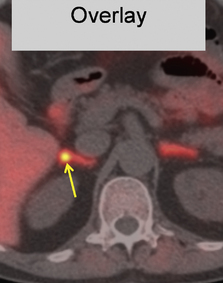
Fig. 24.4 A PET-CT of a 0.5 cm right adrenal adenoma. The radio-tracer is 11 C-metomidate
(From Burton TJ, MacKenzie IS, Balan K et al 2012. Evaluation of the Sensitivity and Specificity of 11C-Metomidate Positron Emission Tomography (PET)-CT for Lateralizing Aldosterone Secretion by Conn’s Adenomas. J Clin Endocrinol Metab 97:100–109, with permission).
Heart failure and its treatment
Some physiology and pathophysiology
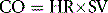
The three factors that regulate the stroke volume are preload, afterload and contractility:
• Preload is the load on the heart created by the volume of blood received into the left ventricle from the left atrium (at the end of ventricular diastole) and that it must eject with each contraction. It can also be viewed as the amount of stretch to which the left ventricle is subject. As the preload rises so also do the degree of stretch and the length of cardiac muscle fibres. Preload is thus a volume load and can be excessive, e.g. when there is valvular incompetence.
• Afterload refers to the load on the contracting ventricle created by the resistance to the blood projected by the ventricle into the arterial system, i.e. the total peripheral resistance. Afterload is thus a pressure load and is excessive, e.g. in arterial hypertension.
• Contractility refers to the capacity of the myocardium to generate the force necessary to respond to preload and to overcome afterload.
Definition of chronic heart failure
The therapeutic importance of recognising this pathophysiology is that many of the neuroendocrine abnormalities of heart failure – particularly the increased renin output and sympathetic activity – can be a consequence of drug treatment, as well as the disease. Renal perfusion is normal in early heart failure, whereas diuretics and vasodilators stimulate renin and noradrenaline/norepinephrine production through actions at the juxtaglomerular apparatus in the kidney and on the arterial baroreflex, respectively. The earliest endocrine abnormality in almost all types of cardiac disease is increased release of the heart’s own hormones, the natriuretic peptides ANP and BNP (A for atrial, B for brain, where it was first discovered). The concentration in plasma of BNP provides a strong prognostic indicator for patients with all stages of heart failure. These peptides normally suppress renin and aldosterone production, but heart failure overrides this control, and measurement of BNP now aids the diagnosis of heart failure, with a raised plasma concentration being a sensitive indicator of the disease.42
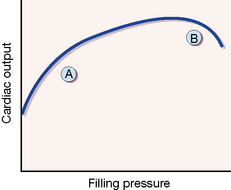
The Starling curve and heart failure
The Starling43 curve originally described increased contractility of cardiac muscle fibres in response to increased stretch but, applied to the whole ventricle, it can explain the normal relationship between filling pressure and cardiac output (Fig. 24.5). Most patients with heart failure present in phase ‘A’ of the relationship, and before the ‘decompensated’ phase (B), in which there is gross dilatation of the ventricle. Diuretic therapy improves the congestive symptoms of heart failure, which are due to the increased filling pressure (preload), but actually reduces cardiac output in most patients. Depending on whether their predominant symptom is dyspnoea (due to pulmonary venous congestion) or fatigue (due to reduced cardiac output), patients feel better or worse. It is likely that a principal benefit of using angiotensin-converting enzyme (ACE) inhibitors in heart failure is their diuretic sparing effect.
Natural history of chronic heart failure
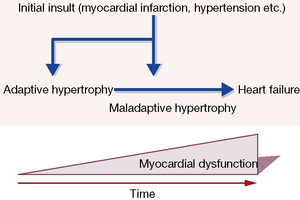
Injury to the heart, e.g. myocardial infarction, hypertension, leads to adaptive (‘compensatory’) molecular, cellular and interstitial changes that alter its size, shape and function. Myocardial hypertophy and ‘remodelling’ takes place over weeks or months in response to haemodynamic load, neurohormonal activation and other factors, and the resulting pattern differs according to whether the stimulus is a pressure or volume overload. With the passage of time, and with maladaption, the heart ‘decompensates’ and heart failure worsens. The process is outlined in Figure 24.6.
The degree of activity that the patient can undertake without becoming dyspnoeic provides one useful classification of the severity of heart failure. The New York Heart Association (NYHA) classification44 offers also an approximate prognosis, with that of the worst grade (Class IV) being as bad as most cancers. Many patients with heart failure die from an arrhythmia, rather than from terminal decompensation, and drugs that avoid increasing the heart’s exposure to increased catecholamine concentrations, as do some vasodilators (but see below), appear best for improving prognosis.
Classification of drugs
Reduction of preload
increase salt and water loss, reduce blood volume and lower excessive venous filling pressure (see Ch. 27). They are almost invariably required to relieve the congestive features of oedema, in the lungs and the periphery; when the heart is grossly enlarged, cardiac output will also increase (see discussion of Starling curve, above). They are used flexibly, starting with a low dose; the usual sequence would be to begin with a thiazide, then move to furosemide, and in the most extreme cases then judiciously add metolazone.
Reduction of preload and afterload
ACE inhibitors and angiotensin receptor II blockers (ARBs)
• reduction of afterload, by preventing the conversion of angiotensin I to the active form, angiotensin II, or by blocking the effects of angiotensin II, which is a powerful arterioconstrictor and is present in the plasma in high concentration in heart failure
• reduction of preload, because the formation of aldosterone, and thus retention of salt and water (increased blood volume), is prevented by reducing the effects of angiotensin II.
ACE inhibitors are the only drugs that reduce peripheral resistance (afterload) without causing a reflex activation of the sympathetic system. The landmark CONSENSUS study compared enalapril with placebo in patients with NYHA class IV heart failure; after 6 months 26% of the enalapril group had died, compared with 44% in the control group. The reduction in mortality occurred among patients with progressive heart failure.45 There is now strong evidence from long-term studies that ACE inhibitors46 and ARBs47 improve survival in and reduce hospital admissions for heart failure.
A common practice has been to give a test dose of a short-acting ACE inhibitor (e.g. ramipril 1.25 mg by mouth) to patients who are in heart failure or on diuretic therapy for another reason, e.g. hypertension. Maintenance of blood pressure in such individuals may depend greatly on an activated renin–angiotensin–aldosterone system, and a standard dose of an ACE inhibitor or ARB can cause a sudden fall in blood pressure. That said, some of the many ACE inhibitors now available (see p. 399) have a sufficiently prolonged action that the initial doses have a cumulative effect on blood pressure over several days. Long-acting ACE inhibitors such as lisinopril (t½ 12 h) and perindopril (t½ 31 h) avoid the risk of sudden falls in blood pressure or renal function (glomerular filtration) after the first dose. Such drugs can be initiated outside hospital in patients who are unlikely to have a high plasma renin (absence of gross oedema or widespread atherosclerotic disease), although it is prudent to arrange for the first dose to be taken just before going to bed. Therapy begins with an ACE inhibitor, and an ARB is substituted if there is intolerance, or added if symptoms continue.
Stimulation of the myocardium
improves myocardial contractility (positive inotropic effect) most effectively in the dilated, failing heart and, in the longer term, after an episode of heart failure has been brought under control. This effect occurs in patients in sinus rhythm and is distinct from its (negative chronotropic) action of reducing ventricular rate and thus improving ventricular filling in atrial fibrillation. Over 200 years after the first use of digitalis for dropsy, the DIG trial provided relief for doctors seeking evidence of long-term benefit.48 Unlike all other positive inotropes, digoxin does not increase overall mortality or arrhythmias.
Drug management of heart failure
Chronic heart failure
A scheme for the stepwise drug management of chronic heart failure appears in Figure 24.7. Points to emphasise in this scheme are that all patients, even those with mild heart failure, should receive an ACE inhibitor as first-line therapy. Several long-term studies have demonstrated improved survival even when cardiac failure is mild.49
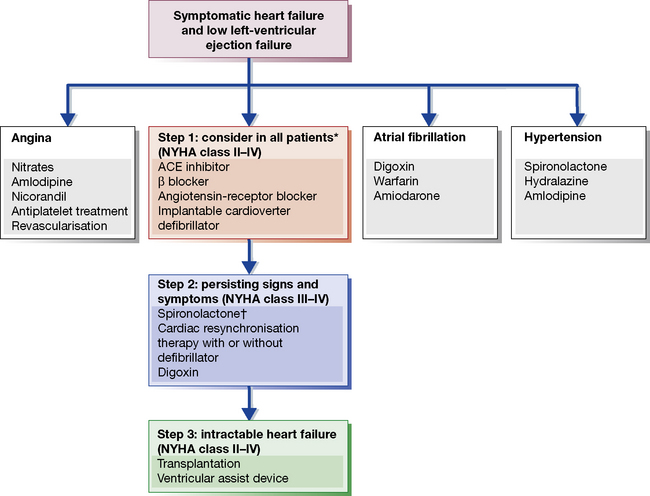
Black patients have a less activated renin system than other ethnic groups. In a landmark study, following subgroup analyses of earlier trials, 1050 black patients who had New York Heart Association class III or IV heart failure with dilated ventricles were randomly assigned to receive a combination of isosorbide dinitrate 120 mg daily plus hydralazine 225 mg daily or placebo in addition to standard therapy for heart failure. The study was terminated early owing to a significantly higher mortality rate in the placebo group, 10.2%, than in the group receiving the active combination, 6.2%, P = 0.02.50
There is now overwhelming evidence for the benefit of β-blockers in chronic heart failure, despite the long-held belief that their negative inotropic effect was a contraindication. Early trials were underpowered but a meta-analysis did suggest a 31% reduction in the mortality rate. Subsequently, the CIBIS-2 and MERIT-HF trials, have independently confirmed that chronic β-blockade has a survival effect of this size in moderate to severe (NHYA III/IV) heart failure.51 Both studies confirmed the one-third reduction in mortality. In MERIT-HF a life was saved for just 27 patient-years of treatment, i.e. it was unusually cost effective – more so than ACE inhibitor therapy. The action is probably a class effect of β-blockade, given the divergent pharmacology of the drugs used to date.
The use of spironolactone has received considerable support from the RALES trial,52 which implies that ACE inhibition even at high dose does not effectively suppress hyperaldosteronism in heart failure. The benefit occurs at a surprisingly low dose of spironolactone (25 mg/day); it probably reflects both improved potassium and magnesium conservation (both are antiarrhythmic) and reversal of fibrosis in the myocardium by aldosterone.
Acute left ventricular failure
Although there may be a case for short-term use of inotropic drugs (see Ch. 25) for heart failure where low output is a dominating feature, most such drugs substantially increase the risk of arrhythmias when the heart is hypoxic. The pharmacokinetics of digoxin does not favour emergency use. The possibility of assisted ventilation should be considered; where pulmonary oedema is the main problem, ventilation is likely to be both safer and more effective than inotropic drugs.
Surgery for heart failure
Although these options lie outside the scope of clinical pharmacology, an important element in meeting the objectives of treatment (see p. 422) is to recognise when further drug treatment is unlikely to improve symptoms or prognosis. Then, the physician must consider the possibility of a surgical intervention. Increasingly this may involve procedures short of transplantation itself, e.g. bypass grafting or stenting where stenosed vessels contribute to the heart failure or even a left ventricular assist device (LVAD) or totally artificial heart. On occasion, it can help to make the patient aware that failure of both the heart and the drugs is not necessarily the end of the road.
• Heart failure is present when the heart cannot provide all organs with the blood supply appropriate to demand.
• Stroke volume is regulated by preload, afterload and contractility.
• In chronic heart failure, diuretics and nitrates reduce preload and provide symptomatic relief without affecting outcome.
• ACE inhibitors reduce both preload and afterload, and reduce morbidity and mortality by about one-third in all patients.
• β-Adrenoceptor blockers, gradually introduced, have an effect equivalent to that of ACE inhibitors in patients with moderate or severe heart failure (NYHA III or IV).
• Spironolactone, in low dose, adds further benefit.
• Digoxin improves myocardial contractility most effectively in the dilated, failing heart but also in the longer term, including in patients in sinus rhythm.
• The principal agents for treating acute left ventricular failure are furosemide, diamorphine and oxygen.
Armstrong P.W. Aldosterone antagonists – last man standing? N. Engl. J. Med.. 2011;364:79–80.
Ashrafian H., Williams L., Frenneaux M.P. The pathophysiology of heart failure: a tale of two paradigms revisited. Clin. Med. (Northfield Il). 2008;8(2):192–197.
Braunwald E. Biomarkers in heart failure. N. Engl. J. Med.. 2009;358(20):2148–2159.
Brown M.J. Hypertension and ethnic group. Br. Med. J.. 2006;332:833–836.
Brown M.J. Renin: friend or foe? Heart. 2007;93:1026–1033.
Brown M.J. Heterogeneity of blood pressure response to therapy. Am. J. Hypertens.. 2010;23:926–928.
Brown M.J., Secondary hypertension: Warrell. D., Cox T., Firth. J. Oxford Textbook of Medicine, fifth ed, Oxford: Oxford University Press, 2010. (Chapter 16.17.3)
Brown M.J. Aliskiren. Circulation. 2008;118:773–784.
Brown M.J., Cruickshank J.K., Macdonald T.M. Navigating the shoals in hypertension: discovery and guidance. Br. Med. J.. 2012;344:23–26.
Camm A.J., Kirchhof P., Lip G.Y., et al. Guidelines for the management of atrial fibrillation: the Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Eur. Heart J.. 2010;31:2369–2429.
Crystal E., Connolly S.J. Role of oral anticoagulation in management of atrial fibrillation. Heart. 2004;90:813–817.
Delacretaz E. Clinical practice. Supraventricular tachycardia. N. Engl. J. Med.. 2006;354:1039–1051.
Dobrev D., Nattel S. New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet. 2010;375:1212–1223.
Duley L., Meher S., Abalos A. Clinical review: management of pre-eclampsia. Br. Med. J.. 2006;332:463–468. Available online at: http://www.bmj.com/cgi/content/extract/332/7539/463 (accessed 2 August 2010)
Dworkin L.D., Cooper C.J. Clinical practice: renal artery stenosis. N. Engl. J. Med.. 2009;361:1972–1978. Available online at: http://www.nejm.org/doi/pdf/10.1056/NEJMcp0809200 (accessed 16 November 2011)
Gaziano T.A., Opie L.H., Weinstein M.S., et al. Cardiovascular disease prevention with a multidrug regimen in the developing world: a cost–effectiveness analysis. Lancet. 2006;368:679–686.
Hansson G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med.. 2005;352:1685–1695.
Hillis L.D., Lange R.A. Optimal management of acute coronary syndromes. N. Engl. J. Med.. 2009;360:2237–2239. Available online at: http://www.nejm.org/doi/pdf/10.1056/NEJMe0902632 (accessed 2 August 2010)
Huikuri H.V., Castellanos A., Myerburg R.J., et al. Sudden death due to cardiac arrhythmias. N. Engl. J. Med.. 2001;345(20):1473–1482.
Jarcho J.A. Resynchronizing ventricular contraction in heart failure. N. Engl. J. Med.. 2005;352:1594–1597.
JBS 2. Joint British Societies’ guidelines on prevention of cardiovascular disease in clinical practice. Heart. 91(Suppl. 5), 2005. Available online at: http://www.bcs.com/download/651/JBS2final.pdf (accessed 2 August 2010)
Kaplan N.M., Opie L.H. Controversies in hypertension. Lancet. 2006;367:168–176.
Krum H., Abraham W.T. Heart failure. Lancet. 2009;373:41–955.
Lip G.Y., Halperin J.L. Improving stroke risk stratification in atrial fibrillation. Am. J. Med.. 2010;123:484–488.
McMurray J.J. Systolic heart failure. N. Engl. J. Med.. 2010;362:228–238.
Messerli F.H. This day 50 years ago. N. Engl. J. Med.. 1995;332(15):1038–1039. [an account of the hypertension and stroke suffered by US President F D Roosevelt]
Morady F. Catheter ablation of supraventricular arrhythmias: state of the art. J. Cardiovasc. Electrophysiol.. 2004;15(1):124–139.
Neubauer S. The failing heart – an engine out of fuel. N. Engl. J. Med.. 2007;356:1140–1151.
Page R.L. Newly diagnosed atrial fibrillation. N. Engl. J. Med.. 2004;351(23):2408–2416.
Page R.L., Roden D.M. Drug therapy for atrial fibrillation: where do we go from here? Nat. Rev. Drug. Discov.. 2005;4(11):899–910.
Pickering T.G., Shimbo D., Haas D., et al. Ambulatory blood-pressure monitoring. N. Engl. J. Med.. 2006;354:2368–2374.
Schmieder R.E., Hilgers K.F., Schlaich M.P., Schmidt B.M.W. Renin-angiotensin system and cardiovascular risk. Lancet. 2007;369:1208–1219.
Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC7). Hypertension. 2003;42:1206. Available online at: http://www.nhlbi.nih.gov/guidelines/hypertension/ (accessed 2 August 2010)
Staessen J.A., Li Y., Richart T. Oral renin inhibitors. Lancet. 2006;368:1449–1456.
Torp-Pedersen C., Pedersen O.D., Kober L. Antiarrhythmic drugs: safety first. J. Am. Coll. Cardiol.. 2010;55:1577–1579.
Turnbull F. Effects of different blood-pressure-lowering regimens on major cardiovascular events: results of prospectively-designed overviews of randomised trials. Lancet. 2003;362:1527–1535.
Vaughan C.J., Delanty N. Hypertensive emergencies. Lancet. 2000;356:411–417.
Williams B., Poulter N.R., Brown M.J., et al. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. Br. Med. J.. 2004;328:634–640.
Zimetbaum P. Amiodarone for atrial fibrillation. N. Engl. J. Med.. 2007;356:935–941.
1 Murrell W 1879 Nitroglycerin as a remedy for angina pectoris. Lancet i:80–81. Nitroglycerin was actually first synthesised by Sobrero in 1847 who noted that, when he applied it to his tongue, it caused a severe headache.
2 Journal of Clinical Investigation 2006; 116:506–511. This same coding variant confers flushing to alcohol challenge and in parts of South-East Asia has a prevalence of almost 50%.
3 Explosives factory workers exposed to a nitrate-contaminated environment lost it over a weekend and some chose to maintain their intake by using nitrate-impregnated headbands (transdermal absorption) rather than have to accept the headaches and re-acquire tolerance so frequently. A recent study has also reported that patients with angina who develop a headache with GTN are less likely to have obstructive coronary artery disease (His D H, Roshandel A, Singh N, Szombathy T, Meszaros Z S 2005 Headache response to glyceryl trinitrate in patients with and without obstructive coronary artery disease. Heart 91:1164–1166).
4 It has been argued that deaths on sildenafil largely reflect the fact that it is used by patients at high cardiovascular risk. But post-marketing data show that death is 50 times more likely after sildenafil taken for erectile failure than alprostadil, the previous first-line agent (Mitka M 2000 Some men who take Viagra die – why? Journal of the American Medical Association 283:590–593).
5 Useful, but not always safe. Defibrillator paddles and nitrate patches make an explosive combination, and it is not always in the patient’s interest to have the patch as unobtrusive as possible (see Canadian Medical Association Journal 1993; 148:790).
6 Several calcium-selective channels have been described in different tissues, e.g. the N (present in neuronal tissue) and T (transient, found in brain, neuronal and cardiovascular pacemaker tissue); the drugs discussed here selectively target the L-channel for its cardiovascular importance.
7 Both the NORDIL and INSIGHT trials confirmed that a calcium channel blocker (diltiazem and nifedipine respectively) had the same efficacy as older therapies (diuretics and/or β-blockers) in hypertension, with no evidence of increased sudden death (Hansson L, Hedner T, Lund-Johansen P et al 2000 Randomised trial of effects of calcium antagonists compared with diuretics and beta-blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem [NORDIL] study. 356:359–365; Brown M J, Palmer C R, Castaigne A et al 2000 Morbidity and mortality in patients randomised to double-blind treatment with a long-acting calcium-channel blocker or diuretic in the International Nifedipine GITS study: Intervention as a Goal in Hypertension Treatment [INSIGHT]. Lancet 356:355–372).
8 PRAISE – Prospective Randomised Amlodipine Survival Evaluation (see Packer M, O’Connor C M, Ghali J K et al 1996 The effect of amlodipine on morbidity and mortality in severe chronic heart failure. New England Journal of Medicine 335:1107–1114).
9 Demers C, McMurray J J V, Swedberg K et al for the CHARM investigators 2005 Impact of candesartan on nonfatal myocardial infarction and cardiovascular death in patients with heart failure. Journal of the American Medical Association 294:1794–1798.
10 For a review see: Ruggenenti P, Cravedi P, Remuzzi G 2010 The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nature Reviews Nephrology 6:319–330. Available online at: http://www.nature.com/nrneph/journal/v6/n6/full/nrneph.2010.58.html (accessed 3 August 2010).
11 Ruggenenti P, Fassi A, Ilieva A P et al 2004 Preventing microalbuminuria in type 2 diabetes. New England Journal of Medicine 351:1941–1951. This was the BENEDICT trial comparing type 2 diabetics randomised to trandolopril, verapamil and the combination versus placebo with a 3.6 year follow-up.
12 Pfeffer M A, Braunwald E, Moye L A et al 1992 Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. New England Journal of Medicine 327:669–677.
13 Pfeffer M A, McMurray J V C, Velaquez E J et al for the Valsartan in Acute Myocardial Infarction Trial Investigators 2004 Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. New England Journal of Medicine 349:1893–1906.
14 Yusuf S, Sleight P, Pogue J et al 2000 Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. New England Journal of Medicine 342:145–153.
15 Dahlof B, Devereux R B, Kjeldsen S E et al 2002 Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 359:995–1010.
16 As we went to press, an outcome study (‘Altitude’) was stopped early because the hypothesised benefit of adding aliskiren to ACE inhibitors or ARBs in patients with diabetes showed no benefit. A borderline significant excess of non-fatal strokes led to a provisional warning against the combination in patients with diabetes.
17 Light causes sodium nitroprusside in solution to decompose; hence solutions should be made fresh and immediately protected by an opaque cover, e.g. aluminium foil. The fresh solution has a faint brown colour; if the colour intensifies it should be discarded.
18 The Impact Of Nicorandil in Angina (IONA) study was a double-blind, randomised, placebo-controlled trial conducted in the UK, in which high-risk patients with stable angina were assigned placebo or nicorandil 10–20 mg. Over a mean follow-up of 1.6 years significantly more placebo-treated patients suffered an acute coronary syndrome or coronary death (15.5% versus 13.1%, P = 0.01) (IONA Study Group 2002 Effect of nicorandil on coronary events in patients with stable angina: the Impact Of Nicorandil in Angina (IONA) randomised trial. Lancet 359:1269–1275).
19 Man-Son-Hing M, Wells G 1995 Meta-analysis of efficacy of quinine for treatment of nocturnal cramps in elderly people. British Medical Journal 310:13–17.
20 It can be the reflex sympathetic activation, as much as hypotension itself, that causes problems. Many cardiologists have had their efforts at controlling angina in elderly patients sabotaged when the patient visits a urologist for his prostatic symptoms, and is treated with a powerful α1-blocker.
21 In the first major study, sudden death occurred in 13.9% of placebo-treated and 7.7% of timolol-treated patients (Norwegian Multicentre Study Group 1981 Timolol-induced reduction in mortality and reinfarction in patients surviving myocardial infarction. New England Journal of Medicine 304:801–807).
22 A 36-year-old patient with asthma collected, from a pharmacy, chlorphenamine for herself and oxprenolol for a friend. She took a tablet of oxprenolol by mistake. Wheezing began in 1 h and worsened rapidly; she experienced a convulsion, respiratory arrest and ventricular fibrillation. She was treated with positive-pressure ventilation (for 11 h) and intravenous salbutamol, aminophylline and hydrocortisone, and survived (Williams I P, Millard F J 1980 Severe asthma after inadvertent ingestion of oxprenolol. Thorax 35:160). There is a logical – or rather pharmacological – link between the use of timolol as eye drops and the risk of asthma. For local administration, a drug needs high potency, so that a high degree of receptor blockade is achieved using a physically small (and therefore locally administrable) dose of drug. Nevertheless, timolol is used topically as a 0.25–0.5% solution, which means the initial concentration of timolol in the tear film is up to 5 mg/mL (or > 10 mmol/L). As the majority of this will be swallowed and a few milligrams orally will block systemic β2 receptors, it is apparent why one drop of timolol down the lachrymal duct (of the wrong patient) is hazardous.
23 Müller M E, van der Velde N, Krulder J W M, van der Cammen T J M 2006 Syncope and falls due to timolol eye drops. British Medical Journal 332:960–961.
24 Practolol was developed to the highest current scientific standards; it was marketed in 1970 as the first cardioselective β-blocker, and only after independent review by the UK drug regulatory body. All seemed to go well for about 4 years, by which time there had accumulated about 200 000 patient-years of experience with the drug. It then became apparent that a small proportion of patients taking practolol could develop a bizarre syndrome that included conjunctival scarring, nasal and mucosal ulceration, fibrous peritonitis, pleurisy and cochlear damage (oculomucocutaneous syndrome). The condition was first recognised by an alert ophthalmologist who ran a special clinic for external eye diseases. (See Wright P 1975 Untoward effects associated with practolol administration: oculomucocutaneous syndrome. British Medical Journal i:595–589.)
25 Angina pectoris: angina, a strangling; pectoris, of the chest.
26 For a personal account by a physician of his experiences of angina pectoris, coronary bypass surgery, ventricular fibrillation and recovery, see Swyer G I M 1986 Personal view. British Medical Journal 292:337. Compelling and essential reading.
27 First International Study of Infarct Survival Collaborative Group 1986 Randomised trial of intravenous atenolol among 16027 cases of suspected acute myocardial infarction: ISIS-1. Lancet ii:57–66.
28 In the SAVE (Survival and Ventricular Enlargement) study, captopril 50 mg three times daily or placebo was started 3–16 days after MI in 2231 patients without overt cardiac failure but with a left ventricular ejection fraction of less than 40%. The captopril group had a lower incidence of recurrent myocardial infarction (133) and death (228) than the placebo group (170 and 275 respectively) (Rutherford J D, Moye L A, Pfeffer M A et al for the SAVE investigators 1994 Effects of captopril on ischemic events after myocardial infarction. Results of the Survival and Ventricular Enlargement trial. SAVE Investigators. Circulation 90:1731–1738). Several other trials of ACE inhibitors have provided similar results.
29 In the Heart Protection Study of 20 536 high-risk patients (one-third had previous MI), those randomly assigned to simvastatin 40 mg daily (compared with placebo) had a 12% reduction in all-cause mortality, and 24% reduction in strokes and coronary heart disease. The authors estimated that 5 years of statin treatment will prevent 100 major vascular events in every 1000 patients with previous MI, or 70 to 80 events in patients with other forms of coronary heart disease or diabetes. There was no upper age limit to this benefit, and no lower limit to the level of LDL at which benefit was seen (Heart Protection Study Collaborative Group 2002 MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20536 high-risk individuals. Lancet 360:7–22).
30 The British Hypertension Society Guidelines (BHS-IV) are summarised in British Medical Journal 2004; 328:634–640 or online at http://www.bhsoc.org. Joint guidance with the National Institute of Clinical Excellence (NICE) was issued in 2006. The update of 2011 is flawed and the Editors advise following the 2006 recommendations (see Brown MJ et al 2012, in Guide to Further Reading).
31 DASH-Sodium Collaborative Research Group 2001 Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. New England Journal of Medicine 344:3–10.
32 Available online at: http://www.nice.org.uk/CG034
33 The original schema included β-blockers, i.e. four drug groups AB/CD (Dickerson J E C, Hingorani A D, Ashby M J, Palmer C R, Brown M J 1999 Optimisation of anti-hypertensive treatment by crossover rotation of four major classes. Lancet 353:2008–2013). This is revised in the light of clinical trial evidence that β-blockers are usually less effective than other antihypertensives at reducing major cardiovascular events, particularly stroke, and are associated with an unacceptably high risk of diabetes especially in combination with diuretics. Hence they are no longer recommended either as monotherapy (B in the original schema) or in combination with diuretics (B + D) unless there is a second indication for prescribing the β-blocker, e.g. angina, or in women planning to have children.
34 Brown M J, McInnes G T, Papst C C et al 2011 Aliskiren and the calcium channel blocker amlodipine combination as an initial treatment strategy for hypertension control (ACCELERATE): a randomised, parallel-group trial. Lancet. 377:312–320.
35 J Venn (1834–1923), an English logician who ‘adopted the diagrammatic method of illustrating propositions by inclusive and exclusive circles’ (Dictionary of National Biography). A medical pilgrimage to Cambridge, where Venn worked, should take in Gonville and Caius College (named after its founder, Dr Caius, physician to the Tudor court and early president of the London College of Physicians in the 16th century); as well as stained glass windows celebrating Venn’s circles, the visitor can see a portrait of the most famous medical Caian, William Harvey.
36 Cooper W O, Hernandez-Diaz S, Arbogast P G et al 2006 Major congenital malformations after first-trimester exposure to ACE inhibitors. New England Journal of Medicine 354:2443–2451.
37 Magpie Trial Collaborative Group 2002 Do women with pre-eclampsia, and their babies, benefit from magnesium sulphate? The Magpie Trial: a randomised placebo-controlled trial. Lancet 359:1877–1890.
38 Eclampsia Trial Collaborative Group 1995 Which anticonvulsant for women with eclampsia? Evidence from the Collaborative Eclampsia Trial. Lancet 345:1455–1463.
39 Barst R J, Rubin L J, Long W A et al 1996 A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. New England Journal of Medicine 334:296–302. In a trial of 81 patients with severe PAH, a 12-week infusion of epoprostenol improved quality of life, mean pulmonary arterial pressure (− 8 versus + 3 %), pulmonary vascular resistance (− 21 versus + 9 %), and exercise capacity, as measured by a 6-min walk test (+ 47 versus − 66 meters). Eight patients died during the trial, all of whom were in the standard therapy group.
40 Brown M J, Allison D J, Jenner D A et al 1981 Increased sensitivity and accuracy of phaeochromocytoma diagnosis achieved by plasma adrenaline estimations and a pentolinium suppression test. Lancet ii:174–177.
41 Bravo E L, Tarazi R C, Fouad F M et al 1981 Clonidine-suppression test: a useful aid in the diagnosis of pheochromocytoma. New England Journal of Medicine 305:623–626.
42 Braunwald E 2008 Biomarkers in heart failure. New England Journal of Medicine 358:2148–2159.
43 Ernest Henry Starling, 1866–1927, Professor of Physiology, University College, London. He also coined the word ‘hormone’.
44 NYHA Class I, minimal dyspnoea (except after moderate exercise); Class II, dyspnoea while walking on the flat; Class III, dyspnoea on getting in/out of bed; Class IV, dyspnoea while lying in bed.
45 Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS) Trial Study Group 1987 Effects of enalapril on mortality in severe congestive heart failure. New England Journal of Medicine 316:1430–1435.
46 Flather M D, Yusuf S, Kober L et al for the ACE-Inhibitor Myocardial Infarction Collaborative Group 2000 Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: a systematic overview of data from individual patients. Lancet 355:1575–1587.
47 Demers C, McMurray J J V, Swedberg K et al for the CHARM investigators 2005 Impact of candesartan on nonfatal myocardial infarction and cardiovascular death in patients with heart failure. Journal of the American Medical Association 294:1794–1798.
48 This prospective randomised trial compared digoxin with placebo in 7788 patients in NYHA Class II–III heart failure and sinus rhythm, most of whom also received an ACE inhibitor and a diuretic. Overall mortality did not differ between the groups but patients who took digoxin had fewer episodes of hospitalisation for worsening heart failure (Digitalis Investigation Group 1997 The effect of digoxin on mortality and morbidity in patients with heart failure. New England Journal of Medicine 336:525–532).
49 In the Studies of Left Ventricular Dysfunction (SOLVD, enalapril was compared with placebo in patients with either clinical features of heart failure or reduced left ventricular function in the absence of symptoms. Treatment reduced serious events (myocardial infarction and unstable angina) by approximately 20% and hospital admissions with progressive heart failure by up to 40%. (SOLVD Investigators 1991 Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. New England Journal of Medicine 325:293–302).
50 Taylor A L, Ziesche S, Yancy C et al 2004 Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. New England Journal of Medicine 351:2049–2057.
51 Until 1997, 24 trials of β-blockade in heart failure provided just 3141 patients. MERIT-HF (1999 Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 353:2001–2007) alone contained 3991 patients and CIBIS-2 (1999 The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 353:9–13) provided a further 2467.
52 The RALES trial randomised 1663 patients with stable heart failure to either placebo or spironolactone. All patients maintained their ‘optimised’ therapy, which included ACE inhibitors. After 2 years of follow-up, the trial terminated prematurely following the demonstration of a 30% reduction in the mortality rate of spironolactone-treated patients, from sudden death as well as progressive pump failure. Gynaecomastia or breast discomfort occurred in 10% of patients receiving spironolactone (1% in controls), but significant hyperkalaemia occurred in surprisingly few patients. RALES was not adequately powered to decide whether the action of spironolactone was additive to that of a β-blocker (Pitt B, Zannad F, Remme W J et al 1999 The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. New England Journal of Medicine 341(10):709–717).