[level-membership-for-surgery-category]Chapter 9
Arterial Aneurysms
Nathan Airhart, John A. Curci
Based on a chapter in the seventh edition by Maureen M. Tedesco and Ronald L. Dalman
Definition
An aneurysm is defined as a focal dilatation of an artery that exceeds the normal diameter by at least 50%. Aneurysms can develop at multiple locations throughout the arterial tree as a result of diverse pathologies that are often specific to that region. A “true” aneurysm results from a progressive weakening of the structural elements of the arterial wall, and both radial and longitudinal lengthening involving all three mural layers (intima, media, and adventitia). In comparison, “false” aneurysms, or “pseudoaneurysms,” form as a result of injury to the aortic wall and the flow of extraluminal blood, which is contained by surrounding tissue. Aneurysms are also classified according to their shape. “Fusiform” aneurysms are characterized by a symmetric dilatation of the complete circumference of the aortic wall. Aneurysms are classified as “saccular” when they exhibit an outpouching of only a portion of the circumference of the aortic wall.
Large elastic arteries appear to be particularly prone to aneurysmal degeneration. By far, the abdominal aortic aneurysm (AAA) is the most common type of arterial aneurysm, with an estimated prevalence of 5% to 16% in men older than 65 years. The disease in women is less common, with an estimated prevalence of 0.7% to 2.2%.1–3 AAAs can be associated with aneurysmal dilatation of the common and internal iliac arteries as well as the popliteal arteries. The AAA is the best-studied aneurysm and often serves as a reference point for studies of other, less common aneurysms.
The dissection is another arterial pathology that is often confused with primary aneurysm disease. An aortic dissection occurs acutely as a result of a mechanical separation of the layers of the aortic wall. Injury to the intima allows blood to enter the media. The laminar structure of the media is excellent for containing the normal radial force of blood pressure but poses little resistance to the intralaminar force of blood flow, resulting in predominantly longitudinal propagation of the “tear” proximally or distally. The subsequent inflammatory response to the dissection injury often leads to further structural weakening of the remaining wall, and secondary aneurysmal dilatation can then occur.
The term “atherosclerotic aneurysm” has often been used, particularly to describe aneurysms of the abdominal aorta and its branches to the lower extremities. This term is misleading in that it suggests that atherosclerosis is the cause of aneurysm disease. It is true that patients with AAA often have atherosclerosis, and population-based studies have demonstrated a strong association of coronary heart disease and peripheral atherosclerosis with AAA.3 There is very little evidence suggesting that aneurysmal degeneration is a direct consequence of atherosclerosis, however, and most research has concluded that these are independent disease processes. Supporting this hypothesis is the fact that epidemiologic data consistently demonstrate that diabetes, which is very strongly associated with atherosclerosis, has a protective effect on AAA risk. Further, there are sites along the vascular tree that are prone to atherosclerotic lesions but in which aneurysms very rarely develop (i.e., external iliac artery and superficial femoral artery).
The Aortic Wall in Health and Disease
As noted previously, the reference pathology for arterial aneurysm disease is the AAA. The normal aorta has three layers: the tunica intima, tunica media, and tunica adventitia. The tunica intima, the innermost layer of the aortic wall, is composed of endothelial cells and a thin layer of smooth muscle cells (SMCs) and connective tissue. The intima is the site of atherosclerotic disease in the aorta, and the manifestations of atherosclerosis are generally limited to this layer.
Tunica Media
The tunica media contains extracellular connective elements (elastin, collagen types I and III, proteoglycans, and glycosaminoglycans) and vascular SMCs. The architecture of the tunica media provides resistance against structural failure of the aorta. Concentric sheets of elastic membranes alternating with layers of vascular SMCs, known as lamina, are critical to the distensibility and tensile strength of the vessel. In comparison with the thoracic segment of the aorta, the abdominal aorta contains a smaller number of medial elastic lamina and a higher collagen to elastin ratio.
Elastin and Collagen
Elastin and collagen are the primary load-bearing elements of the aorta, and deficiencies or derangements of these extracellular matrix components can lead to aneurysmal disease. Normal elastic fibers are crucial to the maintenance of arterial wall integrity. Elastin, which is very distensible, functions to evenly distribute stress throughout the aortic wall. The elastic fiber is the most stable extracellular matrix component in the arterial wall, with a very long biologic half-life, typically measured in decades. During aneurysm pathogenesis, elastin fibers are dramatically depleted. This remarkable feature of the AAA media is what distinguishes the disease from other arterial pathologies and is the focus of the attempts to stabilize or restore the tensile strength of the abdominal aortic wall. Although alterations in the normal patterns of elastic fibers are observed in thoracic aortic aneurysms (TAAs), the loss of elastic fibers is not as severe as that seen in AAAs. The implication of this distinction between the two types of aortic aneurysms is not fully understood (Fig. 9-1).
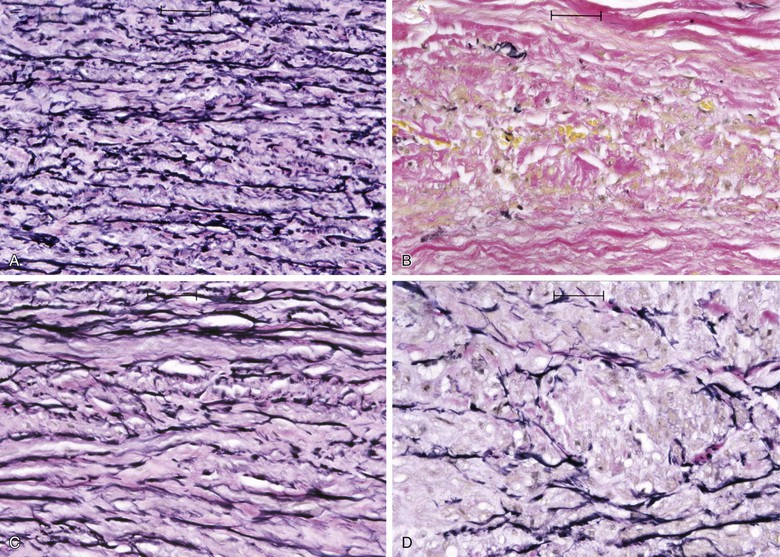
Figure 9-1 Photomicrographs (×200) of elastin-specific staining in the media of the aortic wall. They demonstrate the normal lamellar pattern of the elastin fibers in a nondilated age-matched aorta (A) and the absence of essentially any stainable elastin fibers in the abdominal aortic aneurysm (B). C and D, The media of the aneurysmal thoracic aorta, however, typically demonstrates fractures and thinning of the elastic fibers of the aortic wall.
Collagen, which has a much higher tensile strength but is less distensible than elastin, is recruited at elevated pressures, adding to resistance against further dilatation. Rupture occurs when residual and newly synthesized medial and adventitial collagen fibers fail to maintain structural integrity. The precise sequence of events leading to rupture remains unknown but almost certainly involves alterations in wall stress, medial inflammation, and proteolysis, resulting in critical reductions in mural tensile strength.
Vascular SMCs
Vascular SMCs play a very important role in normal aortic wall physiology. They are responsible for producing the components of the extracellular matrix, including elastin and collagen, and are crucial to the vascular remodeling process that occurs in response to changing stresses on the artery. The importance of normally functioning SMCs has been illustrated in studies using murine AAA models, in which repopulation of the aorta with normal vascular SMCs protected against progression of aneurysmal disease. Aneurysm pathology is associated with a depletion of medial SMCs and apparent dysfunction of those that remain. There is evidence that SMCs residing within the media of aortic aneurysms directly participate in aneurysm progression with production of matrix-degrading proteases such as matrix metalloproteinases MMP-9 and MMP-2.4,5
Adventitia
The adventitia, which is the outermost layer of the aorta, is composed of interstitial collagen fibers, fibroblasts, nerve fibers, and a network of small blood vessels that supply the adventitia and outer region of the tunica media known as the vasa vasorum. The density of the vasa vasorum decreases along the length of the aorta from aortic root to bifurcation. Speculation has existed regarding a potential relationship between reduced adventitial vasa density and the proclivity for increased aneurysm formation in the distal aorta. Evidence of aneurysm causality related to regional differences in aortic adventitial vascularity remains inconclusive, however. Increased inflammation-driven adventitial neocapillary formation, or “neovascularity,” has been recognized in surgical specimens obtained at the time of aneurysm repair (Fig. 9-2) and found to be most prominent at sites of aortic rupture.6 Though this neovascularity is clearly present, whether it actively promotes progression of aneurysm disease and rupture or simply represents evidence of progressive mural inflammation remains uncertain.
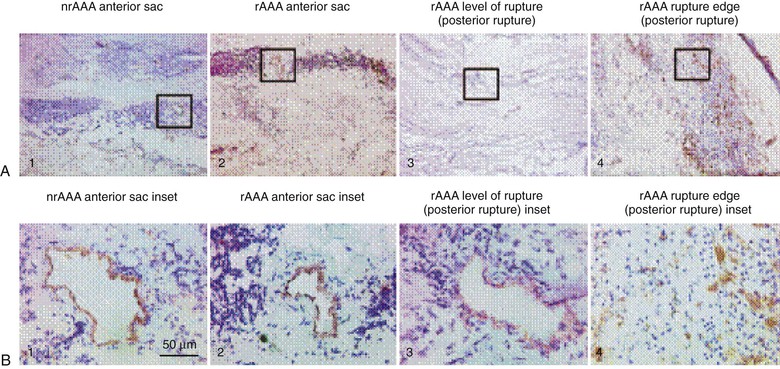
Figure 9-2 Low-power (×50) (A) and high-power (×400) (B) photomicrographs show higher-density and smaller-diameter CD31+ microvessels in the tunica media of a ruptured abdominal aortic aneurysm (rAAA) (rupture edge, posterior rupture; A4 and B4) than in a nonruptured AAA (nrAAA) anterior sac (A1 and B1), a ruptured AAA anterior sac (A2 and B2), or rAAA level of rupture (posterior rupture) (A3 and B3). (Reproduced with permission from Choke E, et al: Abdominal aortic aneurysm rupture is associated with increased medial neovascularization and overexpression of proangiogenic cytokines. Arterioscler Thromb Vasc Biol 26:2077, 2006.)
The Epidemiology of Aortic Aneurysms
Aneurysms may develop at a number of specific locations throughout the arterial tree, with corresponding risk factors, pathology, and natural history unique to each segment. Large-scale, multicenter observational series completed during the last 15 years have provided new insight into the complex and interrelated nature of aortic aneurysm pathobiology.1–3,7,8 Although epidemiologic and demographic risk factors for the development of aneurysms have been established in various populations, these relationships have proved less reliable in predicting significant clinical events such as expansion and rupture with any clinically meaningful precision. A significant reason for failing to identify predictors of disease progression is that most epidemiologic studies are based on comparisons across cohorts rather than longitudinal observations. Even longitudinal follow-up is limited by censoring of observational endpoints by surgical intervention, which obscures the natural history of advanced disease. In addition, aneurysms cannot be diagnosed until the disease process has become sufficiently advanced to result in a measurable dilatation of the artery. Pathologic studies in humans are limited to specimens obtained very late in the natural history of the disease, typically after an intervention has taken place to prevent rupture.
Risk Factors for the Development of Aortic Aneurysms
Epidemiologic risk factors for the development of abdominal aortic aneurysms include cigarette smoking, advanced age, family history, hypertension, central obesity, hypercholesterolemia, and the presence of atherosclerotic occlusive disease such as coronary artery disease.
Cigarette smoking is by far the strongest predictor of AAA in both men and women, with reported odds ratios ranging from 3.0 to 12.0.1–3 There is a strong dose-response relationship, with the risk of AAA increasing directly with the number of packs per day and number of years smoked.7 Smoking confers at least a 3.5-fold greater increase in relative risk than any other recognized AAA risk factor, and the excess prevalence associated with smoking accounts for 75% of all AAAs 4.0 cm or larger. AAA disease has the closest relationship of any disease to cigarette smoking except pulmonary malignancy and emphysema. Although cessation of smoking is associated with a decline in the risk for AAA, individuals with a remote history of smoking still have a higher risk for AAA than individuals who have never smoked. The effect of “ever smoking” is thought to be quite durable, lasting decades.9 The association of a history of “ever smoking” with the development of an aortic aneurysm in men is 2.5 times greater than the association of “ever smoking” with the development of coronary artery disease and is 3.5 times greater than the association of “ever-smoking” with cerebrovascular disease.9
Female gender, African-American race, diabetes mellitus, and regular exercise (in men) are protective against AAA disease. Although the prevalence of AAAs is much lower in women than in men, with a male-to-female ratio of approximately 4 : 1 to 6 : 1, several studies have reported that women with AAAs tend to have poorer disease outcomes than men. Abdominal aneurysms in women also exhibit an elevated risk of rupture, especially if surgical therapy is reserved for AAAs more than 5.5 cm in diameter.10 Recognized risk factors for AAA disease are summarized in Table 9-1.
Table 9-1
Epidemiologic Factors Associated with Abdominal Aortic Aneurysms
| Factor | Odds Ratio |
| Hyperhomocysteinemia | 7.8 |
| Current smoker | 7.4 |
| Ever smoked | 5.1 |
| Hernia | 3.9 |
| Low vitamin B6 levels | 3.75 |
| Age 75-84 years | 3.3 |
| Family history of abdominal aortic aneurysms | 1.9 |
| Age (per 7-year increase) | 1.7 |
| Symptomatic atherosclerosis | 1.7 |
| Hypercholesterolemia | 1.4 |
| Serum resistin concentration | 1.53 |
| Waist-to-hip ratio | 1.22 |
| Waist circumference | 1.14 |
| Black race | 0.5 |
| Diabetes mellitus | 0.5 |
| Female sex | 0.2 |
The negative association observed between diabetes mellitus and AAA has been a focus of much attention. A number of studies have demonstrated this apparent protective effect, with a decreased incidence of AAA disease in diabetics as well as decreased aneurysm growth and rupture rates. These observations reinforce the idea that the pathogenesis of AAA is independent from the development of atherosclerosis and aorto-occlusive disease.11
Risk for Expansion and Rupture of Aortic Aneurysms
Once aneurysm pathology is initiated, genetic and environmental factors influence further dilatation of the aorta and progression toward rupture. Although rates of enlargement vary with time and aortic diameter, the average abdominal aortic aneurysm grows at the rate of 2 to 3 mm/yr. Smoking is a major risk factor for progression and rupture of AAA. A meta-analysis using data from patients with small AAAs demonstrated that the rate of expansion increases with current smoking status by approximately 20%. This meta-analysis reported that current smoking status doubled the risk of rupture in individuals with AAAs (hazard ratio 2.02; 95% confidence interval [CI] 1.33-3.06).12
The baseline diameter of an aneurysm strongly influences its growth rate (larger AAAs grow more rapidly). Data from the UK small aneurysm trial demonstrated that AAAs 5 cm in diameter grow approximately 70% faster than those 4 cm in diameter.13 As the size of the aneurysm increases, so does the risk of rupture. Several large population studies have demonstrated that the risk of aneurysm rupture is very low (0%-2.5% after 5 years) for aneurysms less than 5.0 cm in diameter. For aneurysms measuring more than 5.0 cm, the 5-year risk of rupture rises well above 20%.14,15 Data from the UK Small Aneurysm Trial and the Aneurysm Detection and Management (ADAM) study did not show an advantage of surgical repair for AAA measuring less than 5.5 cm.2,13 For this reason, elective repair of AAAs is most often indicated for aneurysms larger than 5.5 cm.
After initial diagnosis, risk for expansion and rupture of AAAs differs between men and women. Multiple studies have demonstrated that women have a higher risk of AAA rupture than men.1,16 Mortality following repair of AAA is also higher in women than in men, a difference that is most pronounced with endovascular repair (EVAR).17 There are several proposed causes of this discrepancy. The first is that women often have smaller normal aortic diameters than men, so the absolute indication for surgical repair of an AAA (5.5 cm) may reflect a later stage of disease development in women. Another factor relates to the reduced cardiovascular disease awareness in women compared with men, which may lead to delayed diagnosis and intervention. Finally, inferior outcomes following endovascular repair of AAAs in women may relate to the actual endovascular devices used to stabilize the aorta, because these devices are designed for and tested based mainly in male anatomy.17
Recognized AAA risk factors, including peripheral vascular disease (as indicated by reduced ankle-brachial indices), hypertension, and hyperlipidemia, have not been consistently associated with rates of expansion. As mentioned, the presence of diabetes mellitus has a protective effect on aneurysm growth (reduction by 30%).2,3,11–13
Pathophysiology of the Aneurysm Wall
Because abdominal aortic aneurysms are the most common type of aneurysm disease, research efforts to determine the pathobiology of arterial aneurysms have traditionally focused on AAAs. The pathobiology of AAAs also serves as a useful reference point for studies on other types of aneurysms. Since the first investigations on AAAs as a disease process potentially distinct from atherosclerosis more than 20 years ago, there has been an exponential growth in our understanding of the pathobiology underlying AAAs. This growth has been due to increasingly detailed study of human AAA tissues and has been facilitated by development of successful animal models.
Animal Models of Aneurysm Pathogenesis
The aneurysm is, by nature, a degenerative disease that is evident only when the structural failure leads to dilatation. Therefore, the advanced state of aneurysmal degeneration present in most surgical specimens provides little opportunity for analytic insight into the mechanisms responsible for initiation or early progression of disease. Toward the goal of understanding the mechanisms that result in the late-stage disease seen in human pathology, a variety of experimental aneurysms have been created in rodent models that share important pathologic, cellular, and humoral features with human AAA disease.19,20
Temporary introduction of porcine pancreatic elastase into the infrarenal aorta reliably promotes aneurysmal degeneration in mice, rats, rabbits, and pigs. After a very brief dwell time within an isolated segment of murine aorta, porcine pancreatic elastase alone results in limited damage to the aorta. However, progressive medial elastolysis, transmural mononuclear infiltration, and SMC apoptosis ensues over days to weeks (Fig. 9-3). The most important feature of this model is the transient nature of the elastase effect; within 24 hours no trace of exogenous elastase is present, whereas peak inflammation occurs days later. Relative strengths of this model include the development of circumferential aneurysmal degeneration in vessels of varying size and precise localization in the infrarenal segment of the aorta. Although dilatation is very consistently achievable, the aneurysms in these models nearly always stabilize structurally and do not proceed to rupture. Other limitations include the need for direct manipulation and injury to the aorta.
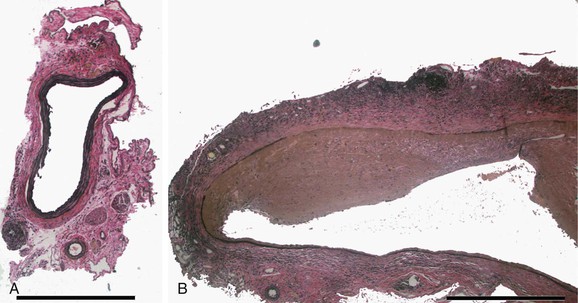
Figure 9-3 Histologic changes after elastase perfusion. Photomicrographs at the same magnification of a normal preperfusion mouse aorta (A) and a mouse aorta perfused with active porcine pancreatic elastase 14 days after perfusion (B). After elastase perfusion, there is a fusiform aneurysm of the aorta with extensive inflammation and medial elastin loss.
Long-term subcutaneous infusion of angiotensin II (Ang II) in congenitally hyperlipidemic mice deficient in either apolipoprotein E or low-density lipoprotein receptors promotes AAA development in the perivisceral segment of the mouse aorta. The dilatation occurs over the course of several days with only mild increases in blood pressure; aneurysm formation in these mice is frequently preceded by focal aortic dissections stimulating inflammation, degradation of elastin, and new vessel formation (Fig. 9-4).20 Relative strengths of this model include the co-occurrence of fatty streaks and early atherosclerosis in the proximal aorta and distal arterial beds (as is commonly present in the human condition), as well as the development in the absence of direct aortic manipulation. Relative limitations include the uncertain relevance of hyperlipidemia to human AAA disease, the typically suprarenal location of the resulting aneurysms (with uncertainty regarding the “appropriate” location for AAAs in quadruped mammalian models), and the failure of the end-stage diseased pathology to closely resemble the appearance of human pathology.
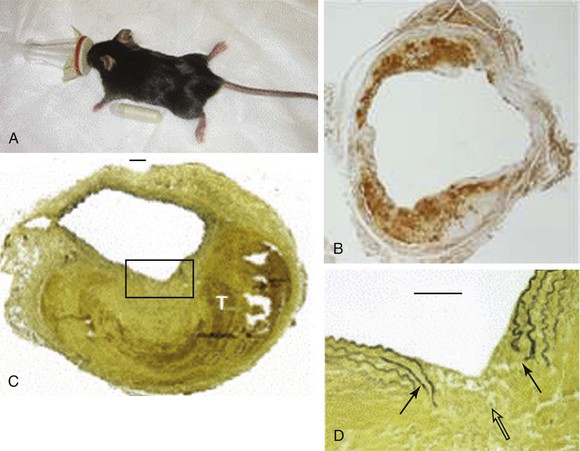
Figure 9-4 Angiotensin II model of abdominal aortic aneurysm disease. A, An osmotic pump filled with angiotensin II is inserted into the subcutaneous tissue of the mouse for continuous delivery. B, Mac-2 stain of an angiotensin II–induced aneurysm illustrating transmural inflammation. C, Verhoeff-van Giessen–stained section demonstrating elastin rupture; D, enlarged view of square in C. The medial elastin bands (black arrows in D) display an area of complete rupture (open arrow in D) leading to thrombus formation (T in C). (Reprinted with permission from Gavrila D, et al: Vitamin E inhibits abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 25:1671, 2005.)
The calcium chloride model induces AAA formation through periaortic (in rats and mice) or intraaortic (in rabbits) application of 250 mmol/L of CaCl2. In this model, focal aortic enlargement is induced through a chemical injury initiated from outside the adventitia. Though this method is reliable and focally reproducible, the AAAs it creates tend to stabilize after several days and rarely proceed to rupture.
Each of the three models incorporates features that are remarkably similar to individual facets of clinical disease, but no one model faithfully recreates the human condition in its entirety. Other, less frequently used models, such as the xenograft transplant model, help to further elucidate the mechanisms of aneurysmal degeneration. Understanding of the specific features and limitations inherent in each model is essential for interpreting the significance and potential generalizability of results obtained from any one model alone. Familiarity with the limitations inherent in rodent modeling is also critical for understanding why so many promising therapeutic strategies found to limit progression of experimental aneurysms have failed to make the transition to effective clinical treatments. Readers interested in a more in-depth comparison between available AAA model systems are referred to a comprehensive review by Daugherty and Cassis.19
Proteolytic Activity within the Aneurysm Wall
The medial destruction characteristic of the abdominal aortic aneurysm is remarkable for the near-complete elimination of the normal structural elements, particularly the typical elastic fiber sheets. Because the elastin is normally incredibly durable, the investigations into the pathophysiology of the AAA have focused on the limited number of enzymatic processes capable of elastolysis. Elastolytic enzymes noted to be substantially elevated within the aneurysm wall include neutrophil elastase as well as several members of the MMP class. Subsequently, a number of other elastolytic enzymes have also been identified in aneurysm tissue and implicated in the destructive process. However, the biology of MMPs in the AAA have been very well characterized and likely play a supporting, if not central, role in the disease.
Matrix Metalloproteinases and AAA
MMPs are a family of extracellular matrix–degrading enzymes essential for a range of homeostatic physiologic processes, including wound healing, angiogenesis, tissue remodeling, and bone resorption. The active sites of MMPs have substantially similar amino acid sequences, contain a zinc ion vital for their enzymatic activity, and are inhibited by chelating agents. In vivo, they are inhibited by the expression and local release of biologic inhibitors of MMP activity such as tissue inhibitors of metalloproteinases (TIMPs). Pro-MMPs are secreted by neutrophils, macrophages, fibroblasts, and vascular SMCs. Enzymatic cleavage and activation are catalyzed by extracellular proteases such as plasmin, plasminogen activators, and other MMPs. MMPs are controlled at several levels, including the induction and suppression of MMP gene transcription, extracellular activation, and interaction with natural inhibitors. They were originally categorized according to substrate (collagenase, elastase, and other constituents), but later classifications are numbered in recognition of the considerable substrate overlap that exists between functionally distinct MMPs.
Several MMPs have been implicated in AAA disease, including types 1, 2, 3, 8, 9, 12, and 13 and membrane type 1 MMP (MT1-MMP), with molecular weights ranging from 52 to 92 kD (Table 9-2).21–35 Targeted deletions of genes encoding for TIMP expression lead to the development of larger aneurysms, thus suggesting the possibility that aneurysmal disease may also result from a local or systemic imbalance between TIMP and MMP production. The relationship between MMP and TIMP production is almost certainly more nuanced in the human disease condition; tissue analysis has consistently demonstrated increased TIMP production in AAA surgical specimens.
Table 9-2
Summary of Known Matrix Metalloproteinases (MMPs) Implicated in the Pathogenesis of Abdominal Aortic Aneurysms (AAAs)
Data from references 20-34.
MMP-9, also known as gelatinase B or 92-kD gelatinase, cleaves elastin, collagen types I and IV, and fibrinogen. Several in vitro studies have demonstrated that MMP-9 is the predominant gelatinase produced in AAA tissue. It is more highly expressed in aneurysmal compared to normal aortic tissue, with infiltrating macrophages being its dominant source. However, it has also been shown that vascular SMCs within the tunica media, as well as adventitial fibroblasts, are capable of expressing MMP-9, especially under inflammatory conditions. Plasma MMP-9 levels are also increased in AAA disease. Patients with intermediate-sized AAAs (between 5 and 6.9 cm) have higher circulating levels than patients with smaller (<4 cm) or larger (>7 cm) AAAs.29 MMP-9 knockout mice are protected against aneurysm formation and exhibit morphologic preservation of elastic lamella.30
Like MMP-9, MMP-2 is capable of degrading elastin and type IV collagen. It is constitutively expressed by SMCs and fibroblasts. Aortic samples obtained from smaller aneurysms demonstrate relatively more MMP-2 than MMP-9 activity, suggesting that increased MMP-2 activity may represent an early event in the time course of AAA pathogenesis. Like mice deficient in MMP-9 production, those deficient in MMP-2 production are protected from aneurysm formation. Experimental data also suggest that both MMP-2 and MMP-9 need to be present and active for maximal aneurysm progression to be achieved, thus implying a synergistic and codependent relationship.31
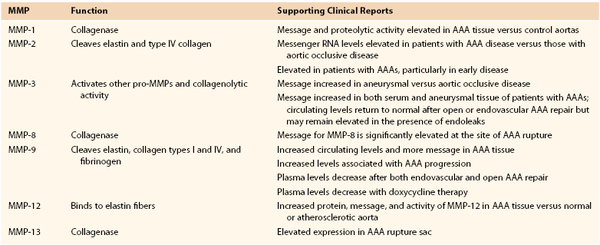
One of the most potent elastolytic enzymes in this class is MMP-12, or human macrophage elastase (HME). The murine analog was shown to be critical to smoke-induced emphysema in mice. In an evaluation of human aortic aneurysm specimens, the enzyme and its activity were found to be substantially increased. Moreover, the enzyme was specifically localized by immunohistology to the residual elastic fibers within the wall of the aneurysm (Fig. 9-5).32
Figure 9-5 Histologic and zymographic evidence of representative active elastolytic enzymes of the matrix metalloprotease (MMP) family in human aneurysm wall. A, The enzyme MMP-12 is localized by immunohistology to the residual elastic fibers in the wall of the abdominal aortic aneurysm by in-situ hybridization to the inflammatory cells of the aortic wall, and by casein zymography. B, The presence of MMP-9 is also seen in the wall of the aorta by immunohistology, in-situ hybridization, and gelatin zymography.
Other Proteinases in Abdominal Aortic Aneurysms
Several proteases from different classes have been demonstrated to contribute to AAA disease either through direct destruction of extracellular matrix or by activating latent elastolytic MMPs. Cathepsins are cysteine proteases that catalyze elastin degradation and depletion in experimental AAA disease. Increased amounts of cathepsins K, L, and S are present in the human aortic wall. As is the case for several key MMPs, human aortic aneurysm specimens show a greater activity of cathepsins B, H, L, and S than do surgical specimens obtained from patients with occlusive aortic disease. Expression of cystatin C, a cysteine protease inhibitor, is reduced in human and experimental aneurysm tissue. Targeted deletion of cystatin C in experimental aneurysm disease increases aneurysm size and the rate of expansion.
The cysteine protease dipeptidyl peptidase I (DPPI) appears to facilitate model aneurysm development through the recruitment of neutrophils, which appear to be critical for the development of aneurysms in the elastase model.36 Expression of tenascin-X, an extracellular matrix protein absent in Ehlers-Danlos syndrome, is also markedly diminished in AAA tissue despite elevated serum levels. Serine proteases, particularly those from the plasminogen activator family, urokinase plasminogen activator (u-PA) and tissue plasminogen activator (t-PA), have also been demonstrated to be involved in AAA pathogenesis. These enzymes cleave plasminogen, forming plasmin, which is capable of directly degrading extracellular matrix components and also of activating metalloproteinases.37
Parenchymal Cell Changes Contributing to the Aortic Abdominal Aneurysm
Diminished density of the vascular SMCs in the media of the aortic wall is a pronounced pathologic feature of advanced AAA disease. Evidence of SMC apoptosis, including enhanced p53 production, is more pronounced in aneurysmal than in occlusive aortic disease specimens at the time of surgical repair. The remaining SMCs exhibit diminished proliferative ability. Smooth muscle depletion is also a feature of some models of experimental aneurysm. The xenograft models of AAA can be altered by population of the aortic wall with recipient cells, which stabilize aortic integrity and limit aneurysm growth. Pharmaceutical inhibition of proapoptotic pathways with fasudil, a Rho-kinase inhibitor, has also been shown to attenuate AAA formation in the apolipoprotein E–deficient angiotensin II infusion model.
Failure of the reparative capacity may not be the only means by which vascular SMCs participate in the matrix changes of aneurysms. Aortic medial SMCs may produce and secrete MMPs in response to inflammatory stimuli and may directly contribute to the progression of AAA disease. Direct participation of the vascular SMCs in the process of AAA development may be related to acquired or intrinsic enhancement of the matrix-modifying capabilities of these cells. Developmentally, the distal aorta and adjacent iliac and hypogastric vessels are generally derived from the same set of precursors during development of the paired dorsal aortae. The independent development of the external iliac arteries later in gestation from a different set of precursor cells may help account for the unique resistance of these vessels to aneurysmal degeneration.
Inflammation, Immunity, and Cell Signaling in Aneurysm Disease
Besides the conspicuous loss of medial elastin, an exuberant medial inflammatory infiltrate is the most prominent feature of the aneurysm wall, and considerable study has gone into understanding this component of the disease. The initiating events of inflammation in human AAA disease remain uncertain. Persistent recruitment and proliferation of these predominantly chronic inflammatory cells is likely due to increased production of chemotactic cytokines, including interleukin-8 (IL-8), macrophage inflammatory protein-1α, and monocyte chemotactic protein-1. There is experimental evidence suggesting that elastin degradation products, or hydrophilic peptides released from early elastolytic events, stimulate and perpetuate mononuclear cell localization and activation within the aortic wall.38 Elastin degradation products bind to cell surface elastin-binding proteins and stimulate mononuclear chemotaxis, activation, and phagocytosis. Serum concentrations of elastin degradation products are elevated in patients with AAA, and levels correlate with risk for disease progression (Fig. 9-6).
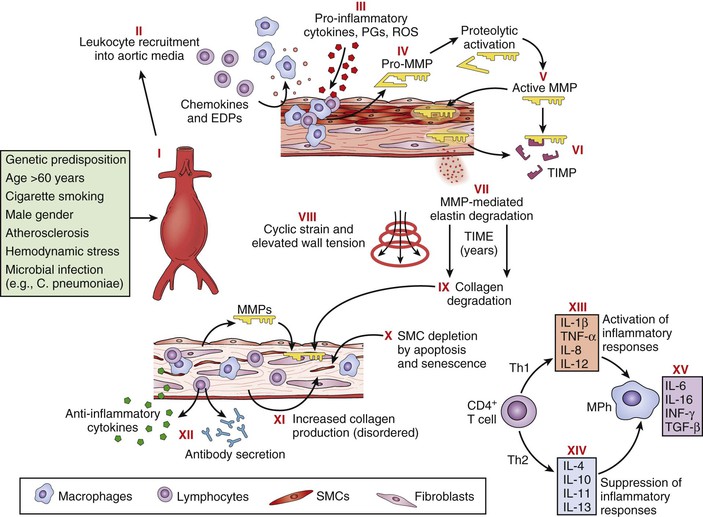
Figure 9-6 Pathophysiology of abdominal aortic aneurysms. Schematic diagram illustrating events thought to contribute to the development and progression of abdominal aortic aneurysms. Injury to the aortic wall, either as a consequence of or in association with known risk factors (I), leads to recruitment of leukocytes into the aortic media (II), macrophage (MPh) activation, and production of proinflammatory molecules (III). Macrophages also produce proenzyme forms of matrix metalloproteinases (MMPs) (pro-MMPs) (IV), which are activated in the extracellular space (V). Tissue inhibitors of matrix metalloproteinases (TIMPs) may neutralize MMP activity (VI), but this neutralization appears insufficient to prevent degradation of structural matrix proteins (elastin and interstitial collagens) (VII). Over a period of years, elastin degradation, cyclic strain, and elevated wall tension bring about progressive aortic dilatation (VIII). Collagen degradation further weakens the aortic wall (IX); although medial smooth muscle cells (SMCs) and fibroblasts might promote structural repair, apoptosis, and cellular senescence cause SMC depletion (X), and interstitial collagen appears disorganized (XI). Aneurysm tissues exhibit infiltration by T cells, B lymphocytes, plasma cells, and dendritic cells and local deposition of immunoglobulins, reflecting a cellular and humoral immune response (XII). Understanding the adaptive cellular immune response in abdominal aortic aneurysms may reveal how different T-cell subsets (i.e., helper T cell type 1 [Th1] versus Th2) interact with macrophages to promote or suppress aneurysmal degeneration, on the basis of the local balance of proinflammatory (XIII) and anti-inflammatory (XIV) molecules. Some cytokines produced within aneurysm tissue, such as interleukin-6 (IL-6) and interferon-γ (IFN-γ), may have dual and opposing functions depending on the specific context (XV). EDPs, Elastin degradation peptides; PGs, prostaglandins; ROS, reactive oxygen species; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α. (Reproduced with permission from Curci JA, et al: Adaptive cellular immunity in aortic aneurysms: cause, consequence or context? J Clin Invest 114:168-171, 2004.)
Characterization of Immune Responses in Abdominal Aortic Aneurysm
Inflammatory cells present in surgical AAA specimens include T cells, B cells, macrophages, mast cells, neutrophils, dendritic cells, and plasma cells. The role of these various cells and immune pathways in the disease is clearly complex. For example, the chronic inflammatory pathways involving helper T cell type 1 (Th1), Th2, and Th17 all appear to play a role in the animal models of the disease, but the predominant inflammatory process in the human disease remains unknown. Later studies have also shown a more active role for the innate immune responses in the development of the disease as well. It is likely that a more detailed understanding of the role of these cells in not only the destruction but also in the stabilization of the damaged aortic wall will be important to our ability to favorably modify the disease process.
Oxidation and Oxidative Stress
Oxidative stress may also contribute to proinflammatory signaling events that lead to tissue damage and apoptosis. Increased production of reactive oxygen species, including superoxide (O2−) and hydrogen peroxide (H2O2), by infiltrating leukocytes, fibroblasts, and native SMCs has multiple effects on AAA pathology, including further recruitment of inflammatory cells into the aortic wall and the inhibition of plasminogen activator inhibitor-1 (PAI-1), an enzyme that limits MMP activation. Oxidative stress levels are markedly higher in AAA surgical specimens than in uninvolved aortic tissue.39 Mouse models deficient in enzymatic sources of reactive oxygen species, such as inducible nitric oxide synthase and NADPH oxidase 1 (NOX1), have attenuated development of AAAs with preservation of aortic wall structure. In addition, interventions that reduce levels of reactive oxygen species, including upregulation of redox-sensitive enzymes such as heme oxygenase-1 and pharmacologic scavenging with vitamin E, inhibit AAA progression in animal studies. Despite promising experimental evidence, clinical trials with antioxidants have failed to limit progression of early and presurgical aneurysm disease, findings consistent with those seen in trials testing other cardiovascular disease endpoints.
Cell Signaling Pathways
Inflammation and inflammatory cell signaling and chemotaxis are mediated by cytokines and chemokines, small (molecular weight of 8-30 kDa) extracellular proteins, and glycoproteins produced by lymphocytes and macrophages. Damaged matrix itself also results in proinflammatory cellular signals. Cytokines include interleukins, interferons, and inflammatory mediators such as tumor necrosis factor and granulocyte-macrophage colony-stimulating factor. Representative chemokines include RANTES (regulated upon activation, normally T cell expressed and secreted), a chemokine that recruits leukocytes to the aortic wall, and macrophage inflammatory proteins 1α and 1β. Cytokines and chemokines are differentially expressed in aneurysmal and healthy aortic tissue samples (Table 9-3).40
Table 9-3
Mediators Upregulated in Abdominal Aortic Aneurysm Tissue versus Control Tissue
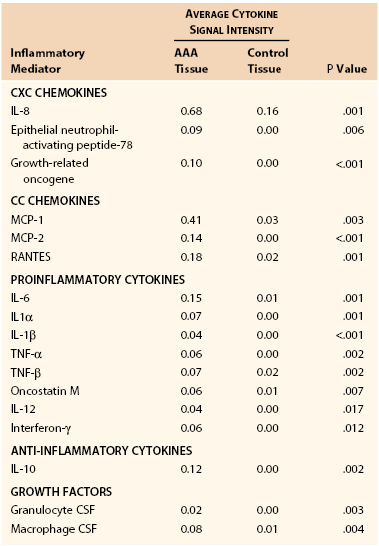
CSF, Colony-stimulating factor; IL, interleukin; MCP, monocyte chemotactic factor; RANTES, regulated upon activation, normally T-cell expressed, and presumably secreted; TNF, tumor necrosis factor.
Prostaglandins may also play an important role in AAA pathophysiology. Prostaglandins stimulate production of the proinflammatory cytokine IL-6 by macrophages through activation of the cell surface EP4 receptor. Pharmacologic inhibition of prostaglandin production via indomethacin has been shown to reduce production of cyclooxygenase-2, prostaglandin E2, and MMP-9 as well as limit expansion of experimental AAAs, although later evidence has questioned the significance of this pathway.41
Many inflammatory responses in vascular SMCs and macrophages include activation of the Jun N-terminal kinase (JNK) pathway. Also known as stress-activated protein kinase, JNK is a proximal signaling molecule that regulates the proteolytic and synthetic functions of vascular SMCs as well as proinflammatory cytokine production and the proteolytic activity of macrophages. Production and activation of more than 20 proteins considered relevant to AAA pathogenesis, including MMP-9 and IL-1α, are regulated to some extent by JNK activity. JNK expression is increased in experimental as well as human AAA tissue samples. Pharmacologic inhibition of JNK in the CaCl2 model prevents aneurysm formation, and multimodality JNK inhibition promotes regression of existing AAAs in both the CaCl2−/− and apolipoprotein E−/− angiotensin II murine models.42
Proteins classically associated with thrombosis and regulation of the clotting cascade may also participate in the pathogenesis of AAAs. Activated protein C (APC) and protein C inhibitor (PCI) concentrations correspond to increased generation of thrombin. In one study, patients with aortic aneurysms had a threefold higher median concentration of activated protein C and protein C inhibitors than did controls. Levels of both of these substances also correlate with AAA diameter.43 Osteopontin, a pluripotential mediator of bone metabolism, inflammation, and vascular calcification, may also play a pivotal role in AAA disease. Serum and tissue concentrations of osteopontin are increased in human disease, and mice deficient in osteopontin demonstrate attenuated AAA formation in experimental models.44,45
In addition to providing clues related to pathogenic mechanisms in AAA disease, biomarkers such as osteopontin and C-reactive protein may ultimately prove useful in monitoring disease status, disease progression, or response to medical therapies during suppressive treatment of early-stage disease. Serum levels of high-sensitivity C-reactive protein, a nonspecific marker of systemic inflammation, are increased in patients with AAAs in a stepwise fashion related to aneurysm size.46 The utility of high-sensitivity C-reactive protein as a single marker for monitoring disease, however, is limited by the finding that although levels correlate with size, they do not predict rates of subsequent AAA progression.
Chlamydia pneumoniae
The role of Chlamydia pneumoniae in aneurysm pathology is controversial. Patients with AAAs frequently express anti–C. pneumoniae antibodies, and immunoglobulin A (IgA) antibody titers correlate positively with aneurysm expansion rates. Polymerase chain reaction and enzyme-linked immunosorbent assays of aortic tissue have documented the presence of C. pneumoniae DNA in AAAs. Although C. pneumoniae may be present in AAA tissue, insufficient evidence exists to date to confirm its direct pathogenicity.47
Pathobiologic Mechanisms of Risk Factors Associated with Abdominal Aortic Aneurysms
Smoking
Despite the incredibly strong association of smoking with AAA development, the mechanism of the causal effect of smoking on AAA formation and progression is only beginning to be understood. The effects of smoking have been presumed to be mediated through alterations in the inflammatory response, vascular SMC function, and enhanced damage to the extracellular matrix of the vessel wall. The link between exposure to tobacco smoke and extracellular matrix destruction has been well studied in the lung. Much like what is observed in the aneurysmal aorta, emphysema results from destruction of elastic alveolar septa due to elevated production of elastolytic proteases, chronic inflammation, and the loss of parenchymal cells capable of matrix deposition and repair.
Studying the effects of tobacco smoke on the aortic wall has proved to be difficult. Insights into this process have been largely derived from in vitro studies examining the effects of nicotine and other isolated tobacco smoke components on cultured vascular cells. Cultured vascular SMCs exposed to nicotine exhibit enhanced production of fibroblast growth factor, MMP-2, MMP-7, and tissue factors. These cells are also more likely to adapt from the contractile to the synthetic phenotype.49–51 Exposure to water-soluble cigarette smoke extract suppresses collagen production by vascular SMCs, potentially inhibiting the ability of these cells to repair structural damage to the aorta in vivo.52
Later studies have utilized murine models of AAA to examine the in vivo effect of nicotine exposure on AAA development. One important study, utilizing apolipoprotein E knockout mice, demonstrated that nicotine exposure significantly increases aneurysm formation, even without angiotensin II infusion. Increased aneurysm formation was attributed to nicotine-induced activation of adenosine monophosphate–activated protein kinase α2 (AMPK-α2), which resulted in increased MMP-2 expression and matrix destruction.53 A similar enhancement of AAA development with nicotine exposure has been observed in the elastase-perfusion model.54
Models examining the in vivo effects of cigarette smoke exposure, rather than mere components of smoke, are emerging and may allow new insights in to the development of the aneurysm. Aneurysm formation is enhanced with cigarette smoke exposure in both the elastase-perfusion and angiotensin II–treated apolipoprotein E knockout models. It has also been reported that in the elastase-perfusion model, a 6-week exposure to smoke results in increased AAA development, and this effect lasted for at least 8 weeks after cessation of smoke exposure.55 Surprisingly, the activity of elastolytic proteases, such as MMP-9 and MMP-12, does not appear to be necessary for the enhancement of AAA development by smoke exposure. This observation has important implications for the potential development of AAA therapies that rely on inhibition of elastolytic proteases (Fig. 9-7).
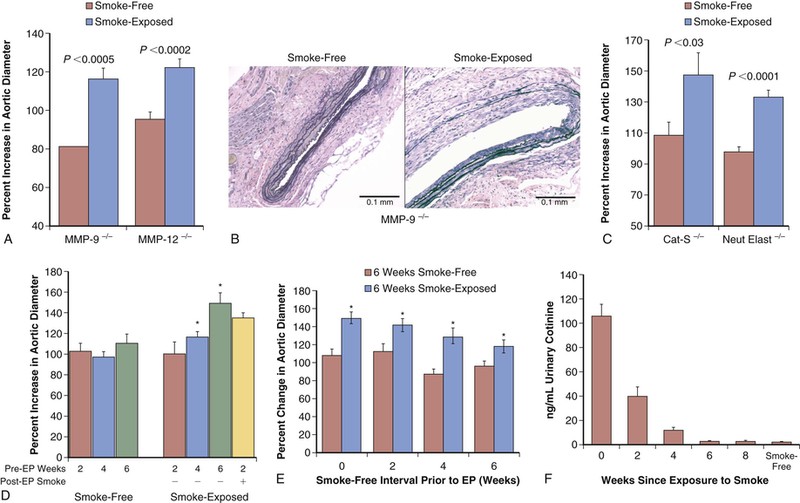
Figure 9-7 Effect of tobacco smoke exposure on the development of model aneurysms. A, After elastase perfusion (EP), smoke exposure resulted in significantly increased abdominal aortic aneurysm diameter in both matrix metalloproteinase-negative (MMP-9−/− and MMP-12−/−) mice. B, Representative sections of mouse aorta were stained with Verhoff–von Giesen stain; in smoke-free MMP-9−/− animals, aortic elastin fibers were relatively intact with minimal inflammation, whereas in smoke-exposed animals there was considerable elastin degradation. C, As with MMP elastases, the absence of either cathepsin-S (Cat-S) or neutrophil elastase (Neut Elast) did not inhibit the increased percent change in aortic diameter induced by smoke exposure. D, Aortic dilatation was evaluated in mice exposed to 2, 4, or 6 weeks of tobacco smoke and smoke-free controls that underwent EP and were maintained in smoke-free conditions until harvest. Four weeks of tobacco smoke was sufficient to enhance abdominal aortic aneurysms after EP. E, Animals exposed to 6 weeks of tobacco smoke (or 6 weeks of smoke-free conditions) were then maintained in smoke-free conditions for 0, 2, 4, or 6 weeks before EP and were harvested at day 14. Remote smoke exposure resulted in larger aneurysms in all groups. F, The cotinine level in the urine of the animals declined rapidly after smoke cessation and was indistinguishable from that in smoke-free mice by 6 weeks after perfusion. (Reproduced with permission from Jin J, et al: Novel mechanism of aortic aneurysm development in mice associated with smoking and leukocytes. Arterioscler Thromb Vasc Biol 32:2901-2909, 2012.)
The mechanisms of smoke-enhanced aneurysm development in this model appear to be unique. It has been shown that the transfer of a broad population of leukocytes from a smoke-exposed animal can induce an enhanced aneurysm phenotype in a recipient animal that has never been exposed to tobacco smoke. On the basis of this and other data, the effects of smoke on AAAs seem, therefore, to be mediated, at least in part, by altered leukocyte (possibly T-cell) function.
Gender
As discussed earlier, epidemiologic studies have consistently demonstrated female gender to be protective against AAA formation. Other risk factors for this disease, including smoking, age, hypertension, and family history, are fairly equally distributed between males and females. The protective effect of female gender has been demonstrated in animal models of AAA as well. Angiotensin II–infused, male apolipoprotein E knockout mice are more susceptible to aneurysm development than females. Male elastase-perfused mice and rats are also more prone to aneurysm formation than identically treated females. In both models, females consistently demonstrate decreased expression of various cytokines and chemokines, MMP-2, and MMP-9 and an overall decreased inflammatory response in the aortic wall.56
The effect of estrogen in females with regard to inflammation in cardiovascular disease is thought to play a role in protection from aneurysm formation. Estrogen treatment significantly reduces the severity of elastase-induced AAAs in mice.56 Estrogen is known to have broad anti-inflammatory actions, including suppression of monocyte adhesion, inhibition of vascular cellular adhesion molecule-1 (VCAM-1), and expression of monocyte chemotactic protein-1 (MCP-1). There is also evidence that estrogen interferes with signaling by nuclear factor κB (NF-κB), affecting the production of MMPs by macrophages and SMCs.57
Diabetes
Several mechanisms have been proposed for the protective effect of diabetes, including hyperinsulinemia and hyperglycemia, which can alter the metabolism of the arterial matrix, and the effects of pharmacologic therapy used in diabetes management, which can stabilize mural thrombi and decrease inflammation.58
The consistently observed protective effect of diabetes in AAA has led to a number of studies that aim to elucidate the mechanism of protection. Golledge et al59 demonstrated that cross-linking of collagen lattices in the aortic media due to advanced glycation leads to a marked reduction in secretion of MMP-9, MMP-2, and IL-6 by activated monocytes. Hyperglycemia has been demonstrated to upregulate plasminogen activator inhibitor-1, which is important in the regulation of matrix metalloproteinase activity.60
Biomechanical Effects
Hemodynamic Forces on the Aorta
Aortic hemodynamic forces are essential contributors to progressive aneurysmal degeneration and ultimate rupture. In the consideration of biomechanical forces in the context of a pressurized tube, the term stress refers to the amount of force exerted on the arterial wall per unit area, whereas strain constitutes stress-induced mural deformation per unit area. Wall shear stress (WSS) refers to the drag exerted on the arterial wall by moving blood as a function of local flow conditions (laminar versus turbulent), as distinguished from outward, perpendicular stress applied to the aortic wall as a result of pressure loading during the cardiac cycle. Although both shear and perpendicular stress contribute to progression of disease, outward forces are far greater (10,000-fold higher) than WSS and are of paramount importance in precipitating rupture.
Hemodynamic stresses and their resulting strain have been studied both in physical models and by computational modeling via finite element methods.61 With use of these methods, focal peak wall strain has been found to be higher in symptomatic AAAs and in ruptured AAAs than in aneurysms in patients undergoing elective AAA repair, even after matching for diameter, thus suggesting that luminal conditions may play an equal or greater role in influencing rupture than the tensile integrity of the wall itself.
Laminar intraluminal thrombus also influences aortic responses to hemodynamic conditions. Substantial layers of laminar thrombus may accumulate during aneurysmal degeneration, with the volume and density varying considerably among similarly sized AAAs in individual patients. These differences probably reflect local variations in flow conditions, WSS, and progression of mural inflammation and degeneration. In addition to being an indirect marker of disease progression, intraluminal thrombus may also directly mediate progression of aneurysms through activation of proteolytic MMPs by plasmin. Furthermore, accumulation of thrombus may impair diffusion of oxygen across the aortic wall, resulting in relative hypoxia and potential SMC apoptosis/necrosis. Laminar thrombus may also alter peak wall strain in AAAs. The balance of thrombus-related influences on AAA disease remains uncertain, with most investigators concluding that laminar thrombus generally augments progression of disease and risk for rupture of AAAs of similar diameter.
Altered Hemodynamics
One well-supported theory to explain the fivefold greater incidence of AAAs than of TAAs is the presence of differential hemodynamic conditions along the length of the aorta. When compared with the suprarenal segments, the infrarenal aorta experiences increased peripheral resistance, increased oscillatory WSS, and reduced flow during resting conditions. These conditions are recognized to predispose arteries to degenerative disease.62 The potential for hemodynamic forces to influence AAA risk has been demonstrated in both experimental and clinical investigations. Expression of MMP-9 in the intact murine infrarenal aorta is significantly greater than in more proximal aortic segments during resting conditions. Transposition of the abdominal aorta to the thoracic position, however, reduces mural MMP-9 expression in the transposed segment, whereas reciprocal transposition of the thoracic aorta to the abdominal position increases levels to those seen in the infrarenal aorta in situ.63
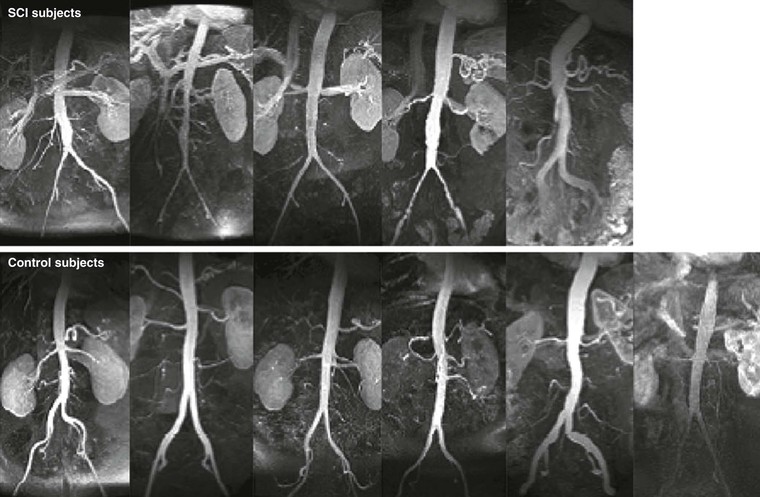
AAA diameter varies inversely with aortic flow in experimental rodent preparations: Increasing flow either before or after creation of an aneurysm by surgical distal arteriovenous fistula formation or by increased daily exercise wheel access reduces aneurysm size, whereas reductions in aortic flow augment aneurysm size, all without measurable influences on aortic pressure.64,65 Clinical relevance is underscored by the recognition that major lower limb amputation and, in later studies, chronic spinal cord injury predispose patients to an increased risk for late AAA formation independent of other recognized systemic factors, such as cigarette smoking or obesity (Fig. 9-8).66,67 Mechanisms proposed for the regulatory effect of WSS on AAA disease include flow-related influences on aortic SMC cellularity, production of reactive oxygen species, circulating mononuclear inflammatory cell localization and activation, and circulating vascular progenitor cell localization and differentiation.
Genetic Determinants
In association with environmental and acquired risk factors such as smoking, genetic predisposition influences risk for and progression of aneurysm disease. The association between variations in genomic sequence (base-pair polymorphisms and mutations) and risk for thoracic aneurysm disease is much more established than that related to abdominal aneurysm disease. This situation is primarily due to major differences in underlying disease pathogenesis. Aortic root and ascending aortic aneurysms commonly develop as a consequence of cystic medial degeneration, often in younger patients with Marfan syndrome, Ehlers-Danlos syndrome, Turner’s syndrome, bicuspid aortic valves, or familial thoracic aortic aneurysm syndromes.
Inherited Connective Tissue Disorders
Inherited connective tissue disorders are a common cause of aortic aneurysms in younger patients. Approximately 20% of TAAs are attributable to syndromes associated with single-gene mutations. In total, seven monogenic mutations have been described to cause TAA disease, including fibrillin 1 (FBN1), transforming growth factor-β (TGF-β) receptors 1 and 2 (TGFBR1, TGFBR2), myosin light chain kinase (MYLK), smooth muscle myosin heavy chain 11 (MYH11), smooth muscle alpha actin 2 (ACTA2), and SMAD family member 3 (SMAD3). Mutations in any one of these genes is usually highly penetrant and is inherited in an autosomal dominant pattern. Currently, no known monogenic disorders have been described as causing aneurysms specific to the abdominal aorta.
In Marfan syndrome, abnormalities in the fibrillin-1 gene product impair collagen cross-linking, predisposing patients to premature, severe cystic medial degeneration. More than 600 distinct mutations in the fibrillin-1 gene on chromosome 15 have been identified in patients with Marfan syndrome.68 Fibrillin deficiency leads to dysregulation of TGF-β signaling, resulting in abnormal activation of vascular SMCs, excess deposition of extracellular matrix components, excess metalloproteinase release, and infiltration of macrophages.69,70 Thoracic aortic aneurysms, dissection, aortic valvular incompetence, and mitral valve prolapse can develop in patients with Marfan syndrome. Dilatation of the aortic root, which may be followed by dissection and rupture, is seen in approximately 75% of such patients. Nonvascular sequelae of Marfan syndrome include musculoskeletal, ocular, central nervous system, and pulmonary abnormalities.
Patients with type IV Ehlers-Danlos syndrome have a defect in type III collagen production (COL3A1) that leads to impaired arterial tensile strength, dissections, aneurysm formation, and arterial rupture most frequently involving medium-sized arteries. The life expectancy of patients with type IV Ehlers-Danlos syndrome is dramatically shortened, with a median life span of 48 years, largely owing to arterial rupture.71
Loeys-Dietz syndrome is an autosomal dominant disorder characterized by mutations of TGF-β receptor 1 or 2 (TGFBR1 or TGFBR2) leading to dysregulation of TGF-β signaling. The syndrome is characterized by widespread vascular dilatation and tortuosity, musculoskeletal pathology, facial dysmorphology, and skin abnormalities. Dilatation of the aortic root is present in almost all of patients with this syndrome, many of whom go on to have dissection.72
Turner syndrome, which results from a partial or complete loss of an X chromosome, is associated with an increased risk of aortic dilatation and dissection. Histologic examination of aortic tissue from patients with the syndrome reveals increased cystic medial necrosis. The presence of other risk factors in individuals with Turner’s syndrome, including bicuspid aortic valve and coarctation of the aorta, likely contributes to this process. Studies have demonstrated altered TGF-β signaling in Turner’s syndrome. Dissection is six times more common and occurs at a significantly younger age and smaller aortic diameters in the Turner’s syndrome population than in age-matched controls.73
Other inherited disorders, including gonadal dysgenesis, neurofibromatosis type 1, Menkes’ kinky hair syndrome, polycystic kidney disease, and tuberous sclerosis, all include characteristic extracellular matrix deficiencies that may predispose affected patients to the formation of arterial aneurysms.
Other Genes Related to the AAA
If patients with generalized matrix deficiency diseases such as Marfan syndrome and Ehlers-Danlos syndrome are excluded, relevant family histories of AAA disease can be obtained from 15% to 20% of patients with AAAs.74,75 The risk of AAAs in males with a first-degree relative affected by the disease is approximately fourfold higher than the risk in the general population, and twin-based studies estimate a heritability of approximately 60%.76
A number of candidate gene studies have been carried out to examine the genetic contribution to AAA disease. A meta-analysis of candidate genes demonstrated that single nucleotide polymorphisms (SNPs) in the genes encoding angiotensin-converting enzyme (ACE), angiotensin type 1 receptor (AT1R), MMP-9, and methlyenetetrahydrofolate reductase (MTHFR) have been consistently associated with AAAs.77 Weaker, less significant associations have been reported with certain HLA and apolipoprotein E genotypes and genes regulating expression of angiotensin II, various MMPs, plasminogen activator inhibitor-1, chemokine receptors, nitric oxide synthase, and heme oxygenase-1.78 Several potential limitations exist regarding the sensitivity and specificity of candidate gene studies. They are frequently underpowered and susceptible to type II errors, in that small effects require large sample sizes for differences to be recognized and confirmed. This study design is also particularly poorly suited to the study of AAA disease because of confounding influences and risk factors that overlap with other, more common diseases such as atherosclerosis.
Genome-wide association studies provide a more robust method for identifying genes associated with disease processes such as AAA. These studies scan the entire genome, in an unbiased fashion, SNPs associated with a particular disease. With the relative ease of whole genome sequencing, relatively large sample sizes can be used. These studies have identified SNPs associated with AAA disease. Currently, the strongest genomic association with AAA that has been described relates to an SNP on 9p21 (rs10757278-G), previously associated with coronary artery disease. This SNP has been consistently been shown to associate with AAA (odds ratio 1.31; P = 1.2 × 10−12) and intracranial aneurysms (odds ratio 1.29; P = 2.5 × 10−6) across several diverse populations.79 Another locus for AAA has been identified on 9q33.2 (rs7025486), within the DAB2IP gene (odds ratio 1.21; P = 4.6 × 10−10). This SNP also demonstrated an association with coronary artery disease, peripheral artery disease, and pulmonary embolism but not with intracranial aneurysms.80 Finally, in another large genome-wide association study, a specific association between a variant in low-density-lipoprotein receptor–related protein 1 (LRP1) and AAA was reported (odds ratio 1.15; P = 4.52 × 10−10).81
Despite several polymorphisms recognized to influence disease risk, the risk attributable to these SNPs is relatively small. It is unlikely that any single gene is essential for initiation of AAA. A combination of predisposing polymorphisms is probably responsible for the majority of heritable risk for the development of AAA in any individual patient.
Potential for Nonsurgical Treatment of Abdominal Aortic Aneurysms
Ultimately, it is anticipated that a better mechanistic understanding of the development and progression of AAA will lead to the availability of disease-modifying therapies that may reduce or eliminate the need for physical aneurysm resection or repair. Potential translational hurdles limiting clinical utility include questions regarding the validity and fidelity of the mammalian models used to investigate AAA disease; lack of effective biomarkers or other biologically relevant endpoints (other than diameter alone) to monitor as surrogate endpoints; uncertainties related to the optimal dosage, treatment intervals, and delivery methods most appropriate for treatment of human disease; and lack of consensus surrounding definitions of treatment success (e.g., reduction in the rate of enlargement versus the rate of surgical conversion versus rupture). Current indications for medical therapy and the strength of evidence supporting its use in early AAA disease have been reviewed in detail.82
Inhibiting Matrix Proteolysis
Proteolytic inhibition via systemic or local administration of a variety of protease inhibitors, particularly those targeting MMP-class enzymes, has been consistently successful in the animal models. One of the most interesting of these MMP inhibitors is a drug long used as an antibiotic, doxycycline. Although many novel broad-spectrum MMP inhibitors have proved to have intolerable side effects in clinical trials, doxycycline has been well tolerated in the chronic treatment of other MMP-dependent diseases, such as gingivitis. Several small human trials have been conducted to test the efficacy of doxycycline in limiting small AAA progression as well as aneurysm sac and neck remodeling and persistence of type II endoleaks after endovascular exclusion. Although these studies have generally shown favorable disease modification, large multicenter trials are now being performed to determine whether this agent may be clinically useful.83,84
Altering Aortic Inflammation in the Abdominal Aortic Aneurysm
Anti-inflammatory/anti-immune strategies using angiotensin-converting enzyme (ACE) inhibitors, decoy oligodeoxynucleotide nuclear factor-κB suppression, steroids, rapamycin, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins), α-tocopherol, osteopontin knockout/inhibition, and increased TGF-β1 expression, as well as hemodynamic conditioning, neutrophil depletion, and mast cell or angiogenesis inhibition, have all proved effective in limiting or, in the case of JNK inhibition, causing regression of experimental AAAs. To a certain extent it has been recognized that generalized inhibition of inflammation, such as that used after solid organ transplantation, is not capable of inhibiting either aneurysm development or aneurysm progression. What this recognition ultimately suggests is that a targeted approach to inhibition of inflammation will be necessary to alter the natural history of the human AAA.
Other Aneurysms
Thoracic Aortic Aneurysms
The development of an aneurysm of the aorta proximal to the renal arteries occurs much less frequently than AAA. Data available from population-based studies estimate the incidence of TAAs to be around 10.4 cases per 100,000 person-years.85 A TAA may involve one or more aortic segments, including the aortic root, ascending aorta, arch, and descending aorta, and is classified according to its location. A majority of TAAs involve the aortic root and ascending aorta. Approximately 10% include the aortic arch or the thoracoabdominal aorta.
As noted previously, the degeneration of the thoracic aortic wall appears to be more commonly associated with inherited connective tissue disorders and is also found in association with a bicuspid aortic valve with or without aortic stenosis. Acquired risk factors for thoracic aneurysms include hypertension, smoking, and chronic obstructive pulmonary disease. The degenerative matrix change of the aneurysmal thoracic aortic wall includes proteolytic elastic fiber loss, inflammation, and focal SMC depletion. However, the elastic fiber depletion is not as complete as that seen in AAAs, and there is evidence that overall, SMC hyperplasia may be a distinguishing characteristic of the ascending aortic aneurysm.86
Risk factors for increased growth and rupture of TAAs include increasing age, female sex, chronic obstructive pulmonary disease, hypertension, cigarette smoking, the presence of dissection, and a positive family history of TAAs. In addition, growth rates of TAAs are associated with the size of the aneurysm, larger aneurysms exhibiting a more rapid rate of growth. The most important risk factor for aneurysm rupture is aortic diameter with the risk of rupture increasing sharply at 6.0 cm in the ascending aorta and 7.0 cm in the descending aorta.87
Currently, no medical therapy substantially alters the natural history of the TAA, although the potential for TGF-β–related therapies is being investigated.88 The treatment of TAAs has historically involved open surgical replacement of the large aneurysmal segments, although as for AAAs, endoluminal options are proliferating as technologically better graft components and techniques become available (see Chapter 136).
Nonaortic Axial Vessel Aneurysms
Degenerative arterial aneurysms also develop in large conduit arteries beyond the aorta. In descending order of frequency, aneurysms develop in the common iliac, popliteal, femoral, and carotid arteries. The external iliac arteries are remarkably resistant to aneurysmal degeneration, for reasons that are not known. Common iliac and lower extremity arterial aneurysms frequently develop either synchronously or metachronously with aneurysms of the infrarenal aorta. Because of the close clinical association of these peripheral aneurysms with AAAs, it has been presumed that the etiologies are similar. Histologically, the peripheral aneurysms are characterized by elastin fragmentation, inflammatory cell infiltrate, and loss of SMCs; however, they have not been studied to the same level of detail as AAAs.
Splanchnic Aneurysms
The apparent incidence of splanchnic artery aneurysms has increased significantly in the last 2 decades, largely because of the growing number of cross-sectional imaging examinations performed in the course of contemporary medical practice. Rupture of a splanchnic aneurysm is associated with comparable or greater mortality than that associated with AAA rupture. The true prevalence of splanchnic artery aneurysms remains unknown. They develop most commonly in the splenic (60%), proper hepatic (20%), and superior mesenteric (5.5%) arteries. Common underlying causes include infection and dissection, although the mechanisms of arterial degeneration in most cases are not known. Multiple synchronous splanchnic aneurysms are frequently found in association with systemic autoimmune diseases such as polyarteritis nodosa. Splenic aneurysms are commonly identified in men and women of all ages but are clinically most significant when present or recognized in women of childbearing age; along with renal artery aneurysms, they are predisposed to rupture during pregnancy, thereby potentially threatening the life of the mother as well as the fetus.
Mycotic Aneurysms
Approximately 1% of all aneurysms are associated with an arterial infection. These aneurysms, termed mycotic aneurysms (or microbial arteritis with aneurysm), in the past most commonly resulted from bacterial endocarditis and syphilis. Aneurysmal degeneration in the setting of infection results from local destruction of the arterial wall, either directly by the action of bacterial enzymes or indirectly by serine protease activity as a consequence of neutrophil infiltration. Mycotic aneurysms may develop after local deposition of septic emboli from intracardiac sources (usually gram-negative and anaerobic organisms, yeast, or fungi in immunocompromised patients), as a result of transient bacteremia (including most commonly Salmonella species), or by direct extension of adjacent extravascular infection. Iatrogenic or incidental penetrating arterial trauma remains a leading cause of gram-positive mycotic arterial aneurysms. Increases in medical procedures and recreational drug use underscore this prevalence. In most cases, streptococcal or staphylococcal species are cultured from affected arteries. Hematogenous spread of Salmonella species, most frequently associated with the ingestion of raw or undercooked poultry, eggs, and other farm products, is the cause of up to 10% of mycotic arterial aneurysms diagnosed in patients older than 50 years. Unlike the scourge of syphilitic aortitis in the pre-antibiotic era, Salmonella is the organism most commonly associated with mycotic aortic aneurysms today.89
Autoimmune Aneurysms
Aneurysms can also develop in the course of chronic autoimmune diseases. Giant cell (or temporal) arteritis is a necrotizing inflammatory condition that promotes aneurysmal degeneration or luminal obliteration of large muscular and elastic arteries. Takayasu’s disease, associated with aneurysmal disease in up to 31% of patients, precipitates chronic transmural inflammatory infiltrates and depletion of elastin and SMC cellularity, similar to that seen in degenerative aneurysm disease. Arterial aneurysms may also occur in patients with Wegener’s granulomatosis, Behçet’s syndrome, polyarteritis nodosa, and Kawasaki’s disease.
Selected Key References
Hinterseher I, Tromp G, Kuivaniemi H. Genes and abdominal aortic aneurysm. Annals of Vascular Surgery. 2011;25(3):388–412.
A comprehensive review of genetic association studies using single nucleotide polymorphisms recognized to influence the risk for AAA disease.
Kuivaniemi H, Elmore JR. Opportunities in abdominal aortic aneurysm research: epidemiology, genetics, and pathophysiology. Annals of Vascular Surgery. 2012;26(6):862–870.
A concise but thorough review of the newest concepts in aortic aneurysm disease.
Lederle FA, Johnson GR, Wilson SE, Gordon IL, Chute EP, Littooy FN, Krupski WC, Bandyk D, Barone GW, Graham LM, Hye RJ, Reinke DB. Relationship of age, gender, race, and body size to infrarenal aortic diameter. The Aneurysm Detection and Management (ADAM) Veterans Affairs Cooperative Study Investigators. J Vasc Surg. 1997;26:595–601.
First study to recognize a negative association between diabetes mellitus and AAA disease.
Urbonavicius S, Urbonaviciene G, Honoré B, Henneberg EW, Vorum H, Lindholt JS. Potential circulating biomarkers for abdominal aortic aneurysm expansion and rupture—a systematic review. European Journal of Vascular and Endovascular Surgery. 2008;36(3):273–280.
A comprehensive look at circulating biomarkers for aneurysm disease.
Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, Furutani A, Hoshii Y, Tanaka N, Ricci R, Ishihara T, Esato K, Hamano K, Matsuzaki M. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330–1338.
First study to report both inhibition and regression of experimental aneurysms.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Derubertis BG, et al. Abdominal aortic aneurysm in women: prevalence, risk factors, and implications for screening. J Vasc Surg. 2007;46(4):630–635.
2. Lederle FA, et al. The aneurysm detection and management study screening program: validation cohort and final results. Aneurysm Detection and Management Veterans Affairs Cooperative Study Investigators. Arch Intern Med. 2000;160(10):1425–1430.
3. Cornuz J, et al. Risk factors for asymptomatic abdominal aortic aneurysm: systematic review and meta-analysis of population-based screening studies. Eur J Public Health. 2004;14(4):343–349.
4. Goodall S, et al. Enhanced invasive properties exhibited by smooth muscle cells are associated with elevated production of MMP-2 in patients with aortic aneurysms. Eur J Vasc Endovasc Surg. 2002;24(1):72–80.
5. Crowther M, et al. Increased matrix metalloproteinase 2 expression in vascular smooth muscle cells cultured from abdominal aortic aneurysms. J Vasc Surg. 2000;32(3):575–583.
6. Choke E, et al. Abdominal aortic aneurysm rupture is associated with increased medial neovascularization and overexpression of proangiogenic cytokines. Arterioscler Thromb Vasc Biol. 2006;26:2077.
7. Kent KC, et al. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J Vasc Surg. 2010;52:539–548.
8. Forsdahl SH, et al. Risk factors for abdominal aortic aneurysms. A 7-year prospective study: the Tromsø Study, 1994-2001. Circulation. 2009;119:2201–2208.
9. Lederle FA, et al. Smokers’ relative risk for aortic aneurysm compared with other smoking-related diseases: a systematic review. J Vasc Surg. 2003;38:329.
10. Brown PM, et al. The risk of rupture in untreated aneurysms: the impact of size, gender, and expansion rate. J Vasc Surg. 2003;37(2):280–284.
11. Lederle FA. The strange relationship between diabetes and abdominal aortic aneurysm. Eur J Vasc Endovasc Surg. 2012;43(3):254–256.
12. Sweeting MJ, et al. meta-analysis of individual patient data to examine factors affecting growth and rupture of small abdominal aortic aneurysms. [RESCAN collaborators] Br J Surg. 2012;99:655–665.
13. Brady AR, et al. Abdominal aortic aneurysm expansion: risk factors and time intervals for surveillance. Circulation. 2004;110(1):16–21.
14. Glimåker H, et al. Natural history of patients with abdominal aortic aneurysm. Eur J Vasc Surg. 1991;5(2):125–130.
15. Nevitt MP, et al. Prognosis of abdominal aortic aneurysms. A population-based study. N Engl J Med. 1989;321(15):1009–1014.
16. Norman PE, et al. Abdominal aortic aneurysm: the prognosis in women is worse than in men. Circulation. 2007;115(22):2865–2869.
17. Grootenboer N, et al. Systematic review and meta-analysis of sex differences in outcome after intervention for abdominal aortic aneurysm. Br J Surg. 2010;97(8):1169–1179.
18. Deleted in page proofs.
19. Daugherty A, et al. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2004;24:429.
20. Saraff K, et al. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II–infused, apolipoprotein E–deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:1621.
21. McMillan WD, et al. Size matters: the relationship between MMP-9 expression and aortic diameter. Circulation. 1997;96:2228.
22. Lorelli DR, et al. Response of plasma matrix metalloproteinase-9 to conventional abdominal aortic aneurysm repair or endovascular exclusion: implications for endoleak. J Vasc Surg. 2002;35:916.
23. Irizarry E, et al. Demonstration of interstitial collagenase in abdominal aortic aneurysm disease. J Surg Res. 1993;54:571–574.
24. Annabi B, et al. Differential regulation of matrix metalloproteinase activities in abdominal aortic aneurysms. J Vasc Surg. 2002;35:539–546.
25. Davis V, et al. Matrix metalloproteinase-2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 1998;18:1625–1633.
26. Carrell TWG, et al. Stromelysin-1 (matrix metalloproteinase-3) and tissue inhibitor of metalloproteinase-3 are overexpressed in the wall of abdominal aortic aneurysms. Circulation. 2002;105:477–482.
27. Sangiorgi G, et al. Plasma levels of metalloproteinases-3 and -9 as markers of successful abdominal aortic aneurysm exclusion after endovascular graft treatment. Circulation. 2001;104(12 Suppl 1):I288–I295.
28. Wilson WRW, et al. Matrix metalloproteinase-8 and -9 are increased at the site of abdominal aortic aneurysm rupture. Circulation. 2006;113:438–445.
29. McMillan WD, et al. Increased plasma levels of metalloproteinase-9 are associated with abdominal aortic aneurysms. J Vasc Surg. 1999;29:122–127.
30. Pyo R, et al. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105(11):1641–1649.
31. Longo GM, et al. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625.
32. Curci JA, et al. Expression and localization of macrophage elastase (matrix metalloproteinase-12) in abdominal aortic aneurysms. J Clin Invest. 1998;102:1900–1910.
33. McMillan WD, et al. In situ localization and quantification of mRNA for 92-kD type IV collagenase and its inhibitor in aneurysmal, occlusive, and normal aorta. Arterioscler Thromb Vasc Biol. 1995;15:1139–1144.
34. Watanabe T, et al. The elevated level of circulating matrix metalloproteinase-9 in patients with abdominal aortic aneurysms decreased to levels equal to those of healthy controls after an aortic repair. Ann Vasc Surg. 2006;20:317–321.
35. Tromp G, et al. Elevated expression of matrix metalloproteinase-13 in abdominal aortic aneurysms. Ann Vasc Surg. 2004;18(4):414–420.
36. Pagano MB, et al. Complement-dependent neutrophil recruitment is critical for the development of elastase-induced abdominal aortic aneurysm. Circulation. 2009;119(13):1805–1813.
37. Wanhainen A, et al. Elevated tissue plasminogen activator in patients with screening-detected abdominal aortic aneurysm. J Vasc Surg. 2007;45(6):1109–1113.
38. Lindholt JS, et al. Chronic inflammation, immune response, and infection in abdominal aortic aneurysms. Eur J Vasc Endovasc Surg. 2006;31:453.
39. Miller FJ Jr, et al. Oxidative stress in human abdominal aortic aneurysms: a potential mediator of aneurysmal remodeling. Arterioscler Thromb Vasc Biol. 2002;22:560.
40. Middleton RK, et al. The pro-inflammatory and chemotactic cytokine microenvironment of the abdominal aortic aneurysm wall: a protein array study. J Vasc Surg. 2007;45:574.
41. Armstrong PJ, et al. Suppression of experimental aortic aneurysms: comparison of inducible nitric oxide synthase and cyclooxygenase inhibitors. Ann Vasc Surg. 2005;19:248.
42. Yoshimura K, et al. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330.
43. Kölbel T, et al. Activated protein C–protein C inhibitor complex in patients with abdominal aortic aneurysms: is it associated with diameter and growth rate? Vasc Endovasc Surg. 2008;42:135.
44. Golledge J, et al. Association between osteopontin and human abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2007;27:655.
45. Bruemmer D, et al. Angiotensin II–accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. J Clin Invest. 2003;112:1318.
46. Norman P, et al. C-reactive protein levels and the expansion of screen-detected abdominal aortic aneurysms in men. Circulation. 2004;110:862.
47. Sodeck G, et al. The role of Chlamydia pneumoniae in human aortic disease—a hypothesis revisited. Eur J Vasc Endovasc Surg. 2004;28:547.
48. Deleted in page proofs.
49. Lemaître V, et al. Cigarette smoke components induce matrix metalloproteinase-1 in aortic endothelial cells through inhibition of mTOR signaling. Toxicological Sciences. 2011;123:542–549.
51. Thyberg J. Effects of nicotine on phenotypic modulation and initiation of DNA synthesis in cultured arterial smooth muscle cells. Virchows Arch B Cell Pathol. 1986;52(1):25–32.
52. Raveendran M, et al. Cigarette suppresses the expression of P4Halpha and vascular collagen production. Biochem Biophys Res Commun. 2004;323(2):592–598.
53. Wang S, et al. Activation of AMP-activated protein kinase [alpha]2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med. 2012;18:902–910.
54. Maegdefessel L, et al. MicroRNA-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Science Translational Medicine. 2012;4:122ra22.
55. Jin J, et al. Novel mechanism of aortic aneurysm development in mice associated with smoking and leukocytes. Arterioscler Thromb Vasc Biol. 2012;32(12):2901–2909.
56. Ailawadi G, et al. Gender differences in experimental aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2004;24(11):2116–2122.
57. Evans MJ, et al. Reciprocal antagonism between estrogen receptor and NF-kappaB activity in vivo. Circ Res. 2001;89(9):823–830.
58. Astrand H, et al. Reduced aortic wall stress in diabetes mellitus. Eur J Vasc Endovasc Surg. 2007;33(5):592–598.
59. Golledge J, et al. Reduced expansion rate of abdominal aortic aneurysms in patients with diabetes may be related to aberrant monocyte-matrix interactions. Eur Heart J. 2008;29(5):665–672.
60. Dua MM, et al. Hyperglycemia modulates plasminogen activator inhibitor-1 expression and aortic diameter in experimental aortic aneurysm disease. Surgery. 2010;148(2):429–435.
61. Müller J, et al. Anisotropic adaptive finite element method for modeling blood flow. Comput Methods Biomech Biomed Engin. 2005;8:295.
62. Gimbrone MA Jr, et al. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci. 2000;902:230.
63. Ailawadi G, et al. A nonintrinsic regional basis for increased infrarenal aortic MMP-9 expression and activity. J Vasc Surg. 2003;37:1059.
64. Hoshina K, et al. Wall shear stress and strain modulate experimental aneurysm cellularity. J Vasc Surg. 2003;37:1067.
65. Greve JM, et al. Allometric scaling of wall shear stress from mice to humans: quantification using cine phase-contrast MRI and computational fluid dynamics. Am J Physiol Heart Circ Physiol. 2006;291:H1700.
66. Vollmar JF, et al. Aortic aneurysms as late sequelae of above-knee amputation. Lancet. 1989;2:834.
67. Yeung JJ, et al. Aortoiliac hemodynamic and morphologic adaptation to chronic spinal cord injury. J Vasc Surg. 2006;44:1254.
68. Robinson PN, et al. The molecular genetics of Marfan syndrome and related disorders. J Med Genet. 2006;43:769.
69. Bunton TE, et al. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;88(1):37–43.
70. Neptune ER, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33(3):407–411.
71. Pepin M, et al. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342(10):673–680.
72. Loeys BL, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355(8):788–798.
73. Carlson M, et al. Moderate aortic enlargement and bicuspid aortic valve are associated with aortic dissection in Turner syndrome: report of the international Turner syndrome aortic dissection registry. Circulation. 2012;126(18):2220–2226.
74. Verloes A, et al. Aneruysms of the abdominal aorta: familial and genetic aspects in three hundred thirteen pedigrees. J Vasc Surg. 1995;21:646.
75. Kuivaniemi H, et al. Aortic aneurysms: an immune disease with a strong genetic component. Circulation. 2008;117:242.
76. Wahlgren CM, et al. Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J Vasc Surg. 2010;51(1):3–7.
77. Thompson AR, et al. Candidate gene association studies in abdominal aortic aneurysm disease: a review and meta-analysis. Eur J Vasc Endovasc Surg. 2008;35(1):19–30.
78. Hinterseher I, et al. Genes and abdominal aortic aneurysm. Ann Vasc Surg. 2011;25(3):388–412.
79. Helgadottir A, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217.
80. Gretarsdottir S, et al. Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat Genet. 2010;42(8):692–697.
81. Bown MJ, et al. Abdominal aortic aneurysm is associated with a variant in low-density lipoprotein receptor-related protein 1. Am J Hum Genet. 2011;89(5):619–627.
82. Baxter BT, et al. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117:1883–1889.
83. Høgh A, et al. Intermittent roxithromycin for preventing progression of small abdominal aortic aneurysms: long-term results of a small clinical trial. Vasc Endovascular Surg. 2009;43(5):452–456.
84. Mosorin M, et al. Use of doxycycline to decrease the growth rate of abdominal aortic aneurysms: a randomized, double-blind, placebo-controlled pilot study. J Vasc Surg. 2001;34(4):606–610.
85. Clouse WD, et al. Improved prognosis of thoracic aortic aneurysms: a population-based study. JAMA. 1998;280(22):1926–1929.
86. Tang PCY, et al. Hyperplastic cellular remodeling of the media in ascending thoracic aortic aneurysms. Circulation. 2005;112(8):1098–1105.
87. Kuzmik GA, et al. Natural history of thoracic aortic aneurysms. J Vasc Surg. 2012;56(2):565–571.
88. Matt P, et al. Novel pharmacological strategies to prevent aortic complications in Marfan syndrome. J Geriatr Cardiol. 2011;8(4):254–257.
89. Benenson S, et al. The risk of vascular infection in adult patients with nontyphi Salmonella bacteremia. Am J Med. 2001;110:60.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]Chapter 9
Arterial Aneurysms
Nathan Airhart, John A. Curci
Based on a chapter in the seventh edition by Maureen M. Tedesco and Ronald L. Dalman
Definition
An aneurysm is defined as a focal dilatation of an artery that exceeds the normal diameter by at least 50%. Aneurysms can develop at multiple locations throughout the arterial tree as a result of diverse pathologies that are often specific to that region. A “true” aneurysm results from a progressive weakening of the structural elements of the arterial wall, and both radial and longitudinal lengthening involving all three mural layers (intima, media, and adventitia). In comparison, “false” aneurysms, or “pseudoaneurysms,” form as a result of injury to the aortic wall and the flow of extraluminal blood, which is contained by surrounding tissue. Aneurysms are also classified according to their shape. “Fusiform” aneurysms are characterized by a symmetric dilatation of the complete circumference of the aortic wall. Aneurysms are classified as “saccular” when they exhibit an outpouching of only a portion of the circumference of the aortic wall.
Large elastic arteries appear to be particularly prone to aneurysmal degeneration. By far, the abdominal aortic aneurysm (AAA) is the most common type of arterial aneurysm, with an estimated prevalence of 5% to 16% in men older than 65 years. The disease in women is less common, with an estimated prevalence of 0.7% to 2.2%.1–3 AAAs can be associated with aneurysmal dilatation of the common and internal iliac arteries as well as the popliteal arteries. The AAA is the best-studied aneurysm and often serves as a reference point for studies of other, less common aneurysms.
The dissection is another arterial pathology that is often confused with primary aneurysm disease. An aortic dissection occurs acutely as a result of a mechanical separation of the layers of the aortic wall. Injury to the intima allows blood to enter the media. The laminar structure of the media is excellent for containing the normal radial force of blood pressure but poses little resistance to the intralaminar force of blood flow, resulting in predominantly longitudinal propagation of the “tear” proximally or distally. The subsequent inflammatory response to the dissection injury often leads to further structural weakening of the remaining wall, and secondary aneurysmal dilatation can then occur.
The term “atherosclerotic aneurysm” has often been used, particularly to describe aneurysms of the abdominal aorta and its branches to the lower extremities. This term is misleading in that it suggests that atherosclerosis is the cause of aneurysm disease. It is true that patients with AAA often have atherosclerosis, and population-based studies have demonstrated a strong association of coronary heart disease and peripheral atherosclerosis with AAA.3 There is very little evidence suggesting that aneurysmal degeneration is a direct consequence of atherosclerosis, however, and most research has concluded that these are independent disease processes. Supporting this hypothesis is the fact that epidemiologic data consistently demonstrate that diabetes, which is very strongly associated with atherosclerosis, has a protective effect on AAA risk. Further, there are sites along the vascular tree that are prone to atherosclerotic lesions but in which aneurysms very rarely develop (i.e., external iliac artery and superficial femoral artery).
The Aortic Wall in Health and Disease
As noted previously, the reference pathology for arterial aneurysm disease is the AAA. The normal aorta has three layers: the tunica intima, tunica media, and tunica adventitia. The tunica intima, the innermost layer of the aortic wall, is composed of endothelial cells and a thin layer of smooth muscle cells (SMCs) and connective tissue. The intima is the site of atherosclerotic disease in the aorta, and the manifestations of atherosclerosis are generally limited to this layer.
Tunica Media
The tunica media contains extracellular connective elements (elastin, collagen types I and III, proteoglycans, and glycosaminoglycans) and vascular SMCs. The architecture of the tunica media provides resistance against structural failure of the aorta. Concentric sheets of elastic membranes alternating with layers of vascular SMCs, known as lamina, are critical to the distensibility and tensile strength of the vessel. In comparison with the thoracic segment of the aorta, the abdominal aorta contains a smaller number of medial elastic lamina and a higher collagen to elastin ratio.
Elastin and Collagen
Elastin and collagen are the primary load-bearing elements of the aorta, and deficiencies or derangements of these extracellular matrix components can lead to aneurysmal disease. Normal elastic fibers are crucial to the maintenance of arterial wall integrity. Elastin, which is very distensible, functions to evenly distribute stress throughout the aortic wall. The elastic fiber is the most stable extracellular matrix component in the arterial wall, with a very long biologic half-life, typically measured in decades. During aneurysm pathogenesis, elastin fibers are dramatically depleted. This remarkable feature of the AAA media is what distinguishes the disease from other arterial pathologies and is the focus of the attempts to stabilize or restore the tensile strength of the abdominal aortic wall. Although alterations in the normal patterns of elastic fibers are observed in thoracic aortic aneurysms (TAAs), the loss of elastic fibers is not as severe as that seen in AAAs. The implication of this distinction between the two types of aortic aneurysms is not fully understood (Fig. 9-1).
Figure 9-1 Photomicrographs (×200) of elastin-specific staining in the media of the aortic wall. They demonstrate the normal lamellar pattern of the elastin fibers in a nondilated age-matched aorta (A) and the absence of essentially any stainable elastin fibers in the abdominal aortic aneurysm (B). C and D, The media of the aneurysmal thoracic aorta, however, typically demonstrates fractures and thinning of the elastic fibers of the aortic wall.
Collagen, which has a much higher tensile strength but is less distensible than elastin, is recruited at elevated pressures, adding to resistance against further dilatation. Rupture occurs when residual and newly synthesized medial and adventitial collagen fibers fail to maintain structural integrity. The precise sequence of events leading to rupture remains unknown but almost certainly involves alterations in wall stress, medial inflammation, and proteolysis, resulting in critical reductions in mural tensile strength.
Vascular SMCs
Vascular SMCs play a very important role in normal aortic wall physiology. They are responsible for producing the components of the extracellular matrix, including elastin and collagen, and are crucial to the vascular remodeling process that occurs in response to changing stresses on the artery. The importance of normally functioning SMCs has been illustrated in studies using murine AAA models, in which repopulation of the aorta with normal vascular SMCs protected against progression of aneurysmal disease. Aneurysm pathology is associated with a depletion of medial SMCs and apparent dysfunction of those that remain. There is evidence that SMCs residing within the media of aortic aneurysms directly participate in aneurysm progression with production of matrix-degrading proteases such as matrix metalloproteinases MMP-9 and MMP-2.4,5
Adventitia
The adventitia, which is the outermost layer of the aorta, is composed of interstitial collagen fibers, fibroblasts, nerve fibers, and a network of small blood vessels that supply the adventitia and outer region of the tunica media known as the vasa vasorum. The density of the vasa vasorum decreases along the length of the aorta from aortic root to bifurcation. Speculation has existed regarding a potential relationship between reduced adventitial vasa density and the proclivity for increased aneurysm formation in the distal aorta. Evidence of aneurysm causality related to regional differences in aortic adventitial vascularity remains inconclusive, however. Increased inflammation-driven adventitial neocapillary formation, or “neovascularity,” has been recognized in surgical specimens obtained at the time of aneurysm repair (Fig. 9-2) and found to be most prominent at sites of aortic rupture.6 Though this neovascularity is clearly present, whether it actively promotes progression of aneurysm disease and rupture or simply represents evidence of progressive mural inflammation remains uncertain.
Figure 9-2 Low-power (×50) (A) and high-power (×400) (B) photomicrographs show higher-density and smaller-diameter CD31+ microvessels in the tunica media of a ruptured abdominal aortic aneurysm (rAAA) (rupture edge, posterior rupture; A4 and B4) than in a nonruptured AAA (nrAAA) anterior sac (A1 and B1), a ruptured AAA anterior sac (A2 and B2), or rAAA level of rupture (posterior rupture) (A3 and B3). (Reproduced with permission from Choke E, et al: Abdominal aortic aneurysm rupture is associated with increased medial neovascularization and overexpression of proangiogenic cytokines. Arterioscler Thromb Vasc Biol 26:2077, 2006.)
The Epidemiology of Aortic Aneurysms
Aneurysms may develop at a number of specific locations throughout the arterial tree, with corresponding risk factors, pathology, and natural history unique to each segment. Large-scale, multicenter observational series completed during the last 15 years have provided new insight into the complex and interrelated nature of aortic aneurysm pathobiology.1–3,7,8 Although epidemiologic and demographic risk factors for the development of aneurysms have been established in various populations, these relationships have proved less reliable in predicting significant clinical events such as expansion and rupture with any clinically meaningful precision. A significant reason for failing to identify predictors of disease progression is that most epidemiologic studies are based on comparisons across cohorts rather than longitudinal observations. Even longitudinal follow-up is limited by censoring of observational endpoints by surgical intervention, which obscures the natural history of advanced disease. In addition, aneurysms cannot be diagnosed until the disease process has become sufficiently advanced to result in a measurable dilatation of the artery. Pathologic studies in humans are limited to specimens obtained very late in the natural history of the disease, typically after an intervention has taken place to prevent rupture.
Risk Factors for the Development of Aortic Aneurysms
Epidemiologic risk factors for the development of abdominal aortic aneurysms include cigarette smoking, advanced age, family history, hypertension, central obesity, hypercholesterolemia, and the presence of atherosclerotic occlusive disease such as coronary artery disease.
Cigarette smoking is by far the strongest predictor of AAA in both men and women, with reported odds ratios ranging from 3.0 to 12.0.1–3 There is a strong dose-response relationship, with the risk of AAA increasing directly with the number of packs per day and number of years smoked.7 Smoking confers at least a 3.5-fold greater increase in relative risk than any other recognized AAA risk factor, and the excess prevalence associated with smoking accounts for 75% of all AAAs 4.0 cm or larger. AAA disease has the closest relationship of any disease to cigarette smoking except pulmonary malignancy and emphysema. Although cessation of smoking is associated with a decline in the risk for AAA, individuals with a remote history of smoking still have a higher risk for AAA than individuals who have never smoked. The effect of “ever smoking” is thought to be quite durable, lasting decades.9 The association of a history of “ever smoking” with the development of an aortic aneurysm in men is 2.5 times greater than the association of “ever smoking” with the development of coronary artery disease and is 3.5 times greater than the association of “ever-smoking” with cerebrovascular disease.9
Female gender, African-American race, diabetes mellitus, and regular exercise (in men) are protective against AAA disease. Although the prevalence of AAAs is much lower in women than in men, with a male-to-female ratio of approximately 4 : 1 to 6 : 1, several studies have reported that women with AAAs tend to have poorer disease outcomes than men. Abdominal aneurysms in women also exhibit an elevated risk of rupture, especially if surgical therapy is reserved for AAAs more than 5.5 cm in diameter.10 Recognized risk factors for AAA disease are summarized in Table 9-1.
Table 9-1
Epidemiologic Factors Associated with Abdominal Aortic Aneurysms
| Factor | Odds Ratio |
| Hyperhomocysteinemia | 7.8 |
| Current smoker | 7.4 |
| Ever smoked | 5.1 |
| Hernia | 3.9 |
| Low vitamin B6 levels | 3.75 |
| Age 75-84 years | 3.3 |
| Family history of abdominal aortic aneurysms | 1.9 |
| Age (per 7-year increase) | 1.7 |
| Symptomatic atherosclerosis | 1.7 |
| Hypercholesterolemia | 1.4 |
| Serum resistin concentration | 1.53 |
| Waist-to-hip ratio | 1.22 |
| Waist circumference | 1.14 |
| Black race | 0.5 |
| Diabetes mellitus | 0.5 |
| Female sex | 0.2 |
The negative association observed between diabetes mellitus and AAA has been a focus of much attention. A number of studies have demonstrated this apparent protective effect, with a decreased incidence of AAA disease in diabetics as well as decreased aneurysm growth and rupture rates. These observations reinforce the idea that the pathogenesis of AAA is independent from the development of atherosclerosis and aorto-occlusive disease.11
Risk for Expansion and Rupture of Aortic Aneurysms
Once aneurysm pathology is initiated, genetic and environmental factors influence further dilatation of the aorta and progression toward rupture. Although rates of enlargement vary with time and aortic diameter, the average abdominal aortic aneurysm grows at the rate of 2 to 3 mm/yr. Smoking is a major risk factor for progression and rupture of AAA. A meta-analysis using data from patients with small AAAs demonstrated that the rate of expansion increases with current smoking status by approximately 20%. This meta-analysis reported that current smoking status doubled the risk of rupture in individuals with AAAs (hazard ratio 2.02; 95% confidence interval [CI] 1.33-3.06).12
The baseline diameter of an aneurysm strongly influences its growth rate (larger AAAs grow more rapidly). Data from the UK small aneurysm trial demonstrated that AAAs 5 cm in diameter grow approximately 70% faster than those 4 cm in diameter.13 As the size of the aneurysm increases, so does the risk of rupture. Several large population studies have demonstrated that the risk of aneurysm rupture is very low (0%-2.5% after 5 years) for aneurysms less than 5.0 cm in diameter. For aneurysms measuring more than 5.0 cm, the 5-year risk of rupture rises well above 20%.14,15 Data from the UK Small Aneurysm Trial and the Aneurysm Detection and Management (ADAM) study did not show an advantage of surgical repair for AAA measuring less than 5.5 cm.2,13 For this reason, elective repair of AAAs is most often indicated for aneurysms larger than 5.5 cm.
After initial diagnosis, risk for expansion and rupture of AAAs differs between men and women. Multiple studies have demonstrated that women have a higher risk of AAA rupture than men.1,16 Mortality following repair of AAA is also higher in women than in men, a difference that is most pronounced with endovascular repair (EVAR).17 There are several proposed causes of this discrepancy. The first is that women often have smaller normal aortic diameters than men, so the absolute indication for surgical repair of an AAA (5.5 cm) may reflect a later stage of disease development in women. Another factor relates to the reduced cardiovascular disease awareness in women compared with men, which may lead to delayed diagnosis and intervention. Finally, inferior outcomes following endovascular repair of AAAs in women may relate to the actual endovascular devices used to stabilize the aorta, because these devices are designed for and tested based mainly in male anatomy.17
Recognized AAA risk factors, including peripheral vascular disease (as indicated by reduced ankle-brachial indices), hypertension, and hyperlipidemia, have not been consistently associated with rates of expansion. As mentioned, the presence of diabetes mellitus has a protective effect on aneurysm growth (reduction by 30%).2,3,11–13
Pathophysiology of the Aneurysm Wall
Because abdominal aortic aneurysms are the most common type of aneurysm disease, research efforts to determine the pathobiology of arterial aneurysms have traditionally focused on AAAs. The pathobiology of AAAs also serves as a useful reference point for studies on other types of aneurysms. Since the first investigations on AAAs as a disease process potentially distinct from atherosclerosis more than 20 years ago, there has been an exponential growth in our understanding of the pathobiology underlying AAAs. This growth has been due to increasingly detailed study of human AAA tissues and has been facilitated by development of successful animal models.
Animal Models of Aneurysm Pathogenesis
The aneurysm is, by nature, a degenerative disease that is evident only when the structural failure leads to dilatation. Therefore, the advanced state of aneurysmal degeneration present in most surgical specimens provides little opportunity for analytic insight into the mechanisms responsible for initiation or early progression of disease. Toward the goal of understanding the mechanisms that result in the late-stage disease seen in human pathology, a variety of experimental aneurysms have been created in rodent models that share important pathologic, cellular, and humoral features with human AAA disease.19,20
Temporary introduction of porcine pancreatic elastase into the infrarenal aorta reliably promotes aneurysmal degeneration in mice, rats, rabbits, and pigs. After a very brief dwell time within an isolated segment of murine aorta, porcine pancreatic elastase alone results in limited damage to the aorta. However, progressive medial elastolysis, transmural mononuclear infiltration, and SMC apoptosis ensues over days to weeks (Fig. 9-3). The most important feature of this model is the transient nature of the elastase effect; within 24 hours no trace of exogenous elastase is present, whereas peak inflammation occurs days later. Relative strengths of this model include the development of circumferential aneurysmal degeneration in vessels of varying size and precise localization in the infrarenal segment of the aorta. Although dilatation is very consistently achievable, the aneurysms in these models nearly always stabilize structurally and do not proceed to rupture. Other limitations include the need for direct manipulation and injury to the aorta.
Figure 9-3 Histologic changes after elastase perfusion. Photomicrographs at the same magnification of a normal preperfusion mouse aorta (A) and a mouse aorta perfused with active porcine pancreatic elastase 14 days after perfusion (B). After elastase perfusion, there is a fusiform aneurysm of the aorta with extensive inflammation and medial elastin loss.
Long-term subcutaneous infusion of angiotensin II (Ang II) in congenitally hyperlipidemic mice deficient in either apolipoprotein E or low-density lipoprotein receptors promotes AAA development in the perivisceral segment of the mouse aorta. The dilatation occurs over the course of several days with only mild increases in blood pressure; aneurysm formation in these mice is frequently preceded by focal aortic dissections stimulating inflammation, degradation of elastin, and new vessel formation (Fig. 9-4).20 Relative strengths of this model include the co-occurrence of fatty streaks and early atherosclerosis in the proximal aorta and distal arterial beds (as is commonly present in the human condition), as well as the development in the absence of direct aortic manipulation. Relative limitations include the uncertain relevance of hyperlipidemia to human AAA disease, the typically suprarenal location of the resulting aneurysms (with uncertainty regarding the “appropriate” location for AAAs in quadruped mammalian models), and the failure of the end-stage diseased pathology to closely resemble the appearance of human pathology.
Figure 9-4 Angiotensin II model of abdominal aortic aneurysm disease. A, An osmotic pump filled with angiotensin II is inserted into the subcutaneous tissue of the mouse for continuous delivery. B, Mac-2 stain of an angiotensin II–induced aneurysm illustrating transmural inflammation. C, Verhoeff-van Giessen–stained section demonstrating elastin rupture; D, enlarged view of square in C. The medial elastin bands (black arrows in D) display an area of complete rupture (open arrow in D) leading to thrombus formation (T in C). (Reprinted with permission from Gavrila D, et al: Vitamin E inhibits abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 25:1671, 2005.)
The calcium chloride model induces AAA formation through periaortic (in rats and mice) or intraaortic (in rabbits) application of 250 mmol/L of CaCl2. In this model, focal aortic enlargement is induced through a chemical injury initiated from outside the adventitia. Though this method is reliable and focally reproducible, the AAAs it creates tend to stabilize after several days and rarely proceed to rupture.
Each of the three models incorporates features that are remarkably similar to individual facets of clinical disease, but no one model faithfully recreates the human condition in its entirety. Other, less frequently used models, such as the xenograft transplant model, help to further elucidate the mechanisms of aneurysmal degeneration. Understanding of the specific features and limitations inherent in each model is essential for interpreting the significance and potential generalizability of results obtained from any one model alone. Familiarity with the limitations inherent in rodent modeling is also critical for understanding why so many promising therapeutic strategies found to limit progression of experimental aneurysms have failed to make the transition to effective clinical treatments. Readers interested in a more in-depth comparison between available AAA model systems are referred to a comprehensive review by Daugherty and Cassis.19
Proteolytic Activity within the Aneurysm Wall
The medial destruction characteristic of the abdominal aortic aneurysm is remarkable for the near-complete elimination of the normal structural elements, particularly the typical elastic fiber sheets. Because the elastin is normally incredibly durable, the investigations into the pathophysiology of the AAA have focused on the limited number of enzymatic processes capable of elastolysis. Elastolytic enzymes noted to be substantially elevated within the aneurysm wall include neutrophil elastase as well as several members of the MMP class. Subsequently, a number of other elastolytic enzymes have also been identified in aneurysm tissue and implicated in the destructive process. However, the biology of MMPs in the AAA have been very well characterized and likely play a supporting, if not central, role in the disease.
Matrix Metalloproteinases and AAA
MMPs are a family of extracellular matrix–degrading enzymes essential for a range of homeostatic physiologic processes, including wound healing, angiogenesis, tissue remodeling, and bone resorption. The active sites of MMPs have substantially similar amino acid sequences, contain a zinc ion vital for their enzymatic activity, and are inhibited by chelating agents. In vivo, they are inhibited by the expression and local release of biologic inhibitors of MMP activity such as tissue inhibitors of metalloproteinases (TIMPs). Pro-MMPs are secreted by neutrophils, macrophages, fibroblasts, and vascular SMCs. Enzymatic cleavage and activation are catalyzed by extracellular proteases such as plasmin, plasminogen activators, and other MMPs. MMPs are controlled at several levels, including the induction and suppression of MMP gene transcription, extracellular activation, and interaction with natural inhibitors. They were originally categorized according to substrate (collagenase, elastase, and other constituents), but later classifications are numbered in recognition of the considerable substrate overlap that exists between functionally distinct MMPs.
Several MMPs have been implicated in AAA disease, including types 1, 2, 3, 8, 9, 12, and 13 and membrane type 1 MMP (MT1-MMP), with molecular weights ranging from 52 to 92 kD (Table 9-2).21–35 Targeted deletions of genes encoding for TIMP expression lead to the development of larger aneurysms, thus suggesting the possibility that aneurysmal disease may also result from a local or systemic imbalance between TIMP and MMP production. The relationship between MMP and TIMP production is almost certainly more nuanced in the human disease condition; tissue analysis has consistently demonstrated increased TIMP production in AAA surgical specimens.
Table 9-2
Summary of Known Matrix Metalloproteinases (MMPs) Implicated in the Pathogenesis of Abdominal Aortic Aneurysms (AAAs)
Data from references 20-34.
MMP-9, also known as gelatinase B or 92-kD gelatinase, cleaves elastin, collagen types I and IV, and fibrinogen. Several in vitro studies have demonstrated that MMP-9 is the predominant gelatinase produced in AAA tissue. It is more highly expressed in aneurysmal compared to normal aortic tissue, with infiltrating macrophages being its dominant source. However, it has also been shown that vascular SMCs within the tunica media, as well as adventitial fibroblasts, are capable of expressing MMP-9, especially under inflammatory conditions. Plasma MMP-9 levels are also increased in AAA disease. Patients with intermediate-sized AAAs (between 5 and 6.9 cm) have higher circulating levels than patients with smaller (<4 cm) or larger (>7 cm) AAAs.29 MMP-9 knockout mice are protected against aneurysm formation and exhibit morphologic preservation of elastic lamella.30
Like MMP-9, MMP-2 is capable of degrading elastin and type IV collagen. It is constitutively expressed by SMCs and fibroblasts. Aortic samples obtained from smaller aneurysms demonstrate relatively more MMP-2 than MMP-9 activity, suggesting that increased MMP-2 activity may represent an early event in the time course of AAA pathogenesis. Like mice deficient in MMP-9 production, those deficient in MMP-2 production are protected from aneurysm formation. Experimental data also suggest that both MMP-2 and MMP-9 need to be present and active for maximal aneurysm progression to be achieved, thus implying a synergistic and codependent relationship.31
One of the most potent elastolytic enzymes in this class is MMP-12, or human macrophage elastase (HME). The murine analog was shown to be critical to smoke-induced emphysema in mice. In an evaluation of human aortic aneurysm specimens, the enzyme and its activity were found to be substantially increased. Moreover, the enzyme was specifically localized by immunohistology to the residual elastic fibers within the wall of the aneurysm (Fig. 9-5).32
Figure 9-5 Histologic and zymographic evidence of representative active elastolytic enzymes of the matrix metalloprotease (MMP) family in human aneurysm wall. A, The enzyme MMP-12 is localized by immunohistology to the residual elastic fibers in the wall of the abdominal aortic aneurysm by in-situ hybridization to the inflammatory cells of the aortic wall, and by casein zymography. B, The presence of MMP-9 is also seen in the wall of the aorta by immunohistology, in-situ hybridization, and gelatin zymography.
Other Proteinases in Abdominal Aortic Aneurysms
[/not-level-membership-for-surgery-category]
