[level-membership-for-cardiovascular-category]
CHAPTER 65 Arrhythmogenic Right Ventricular Dysplasia
DEFINITION
ARVD is a genetic cardiomyopathy principally involving the right ventricle that is characterized clinically by ventricular arrhythmias and progressive structural alterations in right ventricular size and function. Fibrofatty replacement of the right ventricular myocardium forms the histopathologic hallmark of this disease. There are three patterns of disease expression: (1) classic, characterized by disease limited to the right ventricle with either no or minimal involvement of the left ventricle, which appears late in the course of the disease; (2) left dominant, characterized by early or predominant left ventricular involvement; and (3) biventricular, characterized by parallel involvement of both ventricles from the beginning of disease process.1
PREVALENCE AND EPIDEMIOLOGY
Although the exact incidence of ARVD is unknown, the prevalence has been estimated to be 0.02% of the general population.2 ARVD reportedly is more prevalent (0.8%) in certain parts of northern Italy (Veneto) and Greece (Naxos Island).3,4 ARVD most commonly manifests after puberty and before age 50 years.5,6 ARVD is more common in highly athletic individuals. It is an important cause of sudden cardiac death in young individuals, particularly in athletes, accounting for 5% of sudden deaths in individuals younger than 35 years in the United States, and 25% of deaths in athletes in the Veneto region of Italy.7 ARVD is believed to be more common than reported because of its frequently asymptomatic clinical course, difficulty in diagnosis using conventional noninvasive methods, and frequent misdiagnosis.
ETIOLOGY AND PATHOPHYSIOLOGY
The exact pathogenesis of ARVD remains speculative, but it is most widely believed to be due to genetic abnormalities mainly affecting cardiac desmosomal structure and function. In the United States, the most common reported genetic abnormality in ARVD is a mutation in plakophillin-2 (PKP2), which is a desmosomal protein and is present in more than one third of patients. Desmosomal mutations are believed to disrupt cell-cell adhesion, which provokes myocyte detachment and death under conditions of high mechanical stress, as suggested by its frequent occurrence in athletics and the thinnest portions of the right ventricle, such as the inferior subtricuspid region, right ventricular apex, and right ventricular infundibulum—together termed the triangle of dysplasia (Fig. 65-1).8 This leads to a compensatory repair process characterized by inflammation and fibrofatty substitution. Familial ARVD accounts for 30% to 80% of cases, and most commonly shows an autosomal dominant inheritance, with age-dependent penetrance and variable clinical expression, which can be attributed to the complex interplay of age, gender effects, and environmental influences such as exposure to viruses and athletic activity with the underlying genetic background in the ultimate disease causation and progression. Mere inheritance of a genetic abnormality does not imply “disease” for the same reason. To date, at least 11 distinct subtypes of autosomal dominant ARVD are known, and at least seven genes have been implicated in ARVD (Table 65-1).9
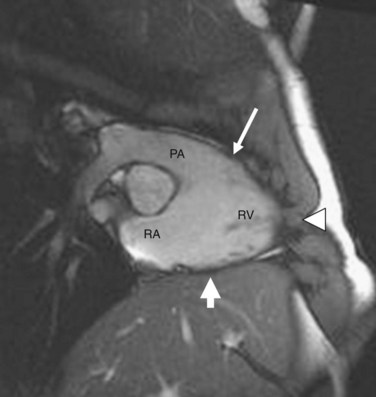
 FIGURE 65-1
FIGURE 65-1
Genetic mutations are also implicated in determining the ventricular predominance in ARVD. Chain-termination mutations causing truncation of the C-terminal domain of desmoplakin can disrupt cytoskeletal integrity, predisposing to predominant left ventricular involvement. Conversely, defects in the N-terminal domain of desmoplakin can result in a primary dysfunction of cell adhesion, leading to predominant right ventricular disease.8
In addition to genetic causes, other important proposed etiologic mechanisms of ARVD are (1) apoptosis (programmed cell death), (2) inherited metabolic or ultrastructural defects leading to myocardial dystrophy, and (3) secondary complication of a prior primary right ventricular myocarditis on the basis of frequently found myocardial inflammatory infiltrates and isolation of coxsackie B and enteroviral RNA viruses in some biopsy samples.10
Pathologically, ARVD is characterized by gradual myocyte loss and fibrofatty infiltration, which starts in the subepicardial region and gradually involves the endocardium, often sparing the trabecular myocardium. Plexiform arrangement of the residual myocardial fibers, separated by fat and fibrosis, leads to electrical instability and provides a conduit for a reentrant phenomenon. This situation explains the arrhythmogenic propensity characteristic of this disease, which can be worsened further by superimposed myocarditis that occurs frequently in ARVD because of the genetic susceptibility.11 A myocardial dysplastic process and the associated loss of its strength and integrity are responsible for the varying degrees of structural derangement and functional disturbances of the heart seen in ARVD.
MANIFESTATIONS OF DISEASE
Clinical Presentation
ARVD usually manifests between the second and fifth decades (mean age at diagnosis approximately 30 years). Patients most commonly present with symptomatic ventricular arrhythmias or sudden cardiac death, or may remain asymptomatic, with ARVD discovered only incidentally on routine examination. The most common symptoms are palpitations, syncope, atypical chest pain, and dyspnea. Ventricular arrhythmias are often precipitated by exercise or stress, and range from isolated ventricular premature beats to sustained monomorphic or polymorphic ventricular tachycardia with left bundle branch block morphology, or abrupt ventricular fibrillation, which is the most common underlying cause of sudden cardiac death in ARVD.12
In addition to ventricular arrhythmias, about 25% of patients may also show supraventricular arrhythmias, which include atrial fibrillation, atrial tachycardia, and atrial flutter, in that order of frequency.13 Left ventricular involvement is associated with an increased incidence of arrhythmic events, more severe right ventricular wall thinning, and heart failure. Frank left-sided heart failure is rare. Figures 65-2, 65-4, and 65-6 illustrate left ventricular involvement in ARVD.
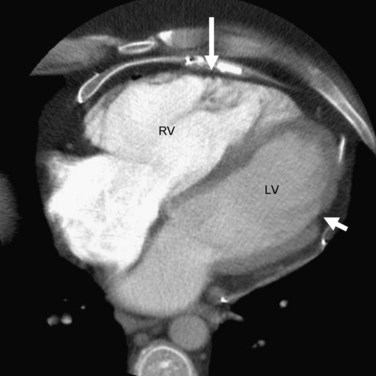
 FIGURE 65-2
FIGURE 65-2Diagnosis
Diagnosis of ARVD is challenging and is currently based on family history and morphofunctional, ECG, histopathologic, and clinical findings as proposed by the Task Force of Cardiomyopathies in 1994 (Table 65-2).14 The diagnosis is made in the presence of two major criteria, one major criterion and two minor criteria, or four minor criteria. Various modifications of the existing Task Force criteria are being increasingly proposed and emphasize the increment in the diagnostic sensitivity of these criteria for less severe forms of the disease and asymptomatic first-degree relatives of affected probands. These modifications include more comprehensive characterization of ECG and echocardiographic criteria, inclusion of mutation analysis, and descriptions of left ventricular involvement.15,16
TABLE 65-2 Task Force Criteria for the Diagnosis of Arrhythmogenic Right Ventricular Dysplasia
| Major Criteria | Minor Criteria | |
|---|---|---|
| Global or regional dysfunction and structural alterations* | Severe dilation and reduction of right ventricular ejection fraction with no (or only mild) left ventricular impairment | Mild global right ventricular dilation or ejection fraction reduction with normal left ventricle |
| Localized right ventricular aneurysms (akinetic or dyskinetic areas with diastolic bulging) | Mild segmental dilation of right ventricle | |
| Severe segmental dilation of right ventricle | Regional right ventricular hypokinesia | |
| Tissue characterization of wall | Fibrofatty replacement of myocardium on endomyocardial biopsy specimen | |
| Repolarization abnormalities | Inverted T waves in right precordial leads (V2 and V3) in individuals >12 yr old in absence of right bundle branch block | |
| Depolarization or conduction abnormalities | Epsilon waves or localized prolongation (>110 ms) of the QRS complex in right precordial leads (V1-V3) | Late potentials (signal-averaged ECG) |
| Arrhythmias | Left bundle branch block ventricular tachycardia (sustained and nonsustained) by ECG, Holter, or exercise testing | |
| Frequent ventricular extrasystoles (>1000/24 hr) (Holter) | ||
| Family history | Familial disease confirmed at necropsy or surgery | Family history of premature sudden death (<35 yr old) owing to suspected right ventricular dysplasia |
| Familial history (clinical diagnosis based on present criteria) |
* Detected by echocardiography, angiography, MRI, or radionuclide scintigraphy.
Electrocardiogram Evaluation
At least one ECG abnormality (Table 65-3) is detectable in more than 90% of patients with ARVD. A newly proposed marker of delayed right ventricular activation, prolonged S wave upstroke (≥55 ms) in V1 to V3, is the most prevalent ECG marker and correlates with disease severity and ventricular tachycardia induction on electrophysiology testing.17 Signal-averaged ECG is used to detect delayed ventricular activation owing to slowed propagation in the fibrofatty myocardium of ARVD. Late potentials on signal-averaged ECG (two or more of the following: filtered QRS duration ≥114 ms, duration of the low-amplitude signal <40 µV in the terminal portion of filtered QRS (LAS40) ≥38 ms, and root mean square voltage of the terminal 40 ms of filtered QRS (RMS40) <20 µV) form a minor diagnostic criterion for ARVD as per the Task Force criteria. Similar to extent of right precordial T wave inversion, late potentials on signal-averaged ECG are correlated with the extent of right ventricular involvement in ARVD. More recently, our group found that using filtered QRS 110 ms or greater identifies ARVD patients with inducible ventricular tachycardia more accurately than does conventional signal-averaged ECG criteria.18 T wave inversions and signal-averaged ECG abnormalities are more commonly seen in left-sided involvement in ARVD.
TABLE 65-3 Electrocardiogram Features of Arrhythmogenic Right Ventricular Dysplasia‡
* Minor Task Force criterion in the absence of right bundle branch block in individuals >12 yr old.
‡ These electrical abnormalities are believed to be caused by intraventricular myocardial defect (parietal block) rather than definite alteration of conduction in the bundle branch (septal block).
Imaging Techniques and Findings
Chest Radiograph
Chest radiographs generally show cardiac enlargement depending on the stage of the disease, with the cardiothoracic ratio generally less than 0.6. The heart can also show a convexity between the aortic knob and the left ventricle, and there is no pulmonary vascular redistribution.11 Radiographs are not a sensitive modality to diagnose ARVD.
Cine Angiography
Right ventricular cine angiography has been regarded as the traditional gold standard, with high specificity and positive and negative predictive values in ARVD diagnosis. It is performed using two orthogonal views in 30 degrees right anterior oblique and 60 degrees left anterior oblique during sinus rhythm with careful evaluation of structure and function of the right ventricle. Coexistence of subtricuspid and anterior infundibular wall bulging and hypertrophic trabeculae has been shown to have 96% sensitivity and 87.5% specificity in angiographic diagnosis of ARVD.10 Invasiveness of the procedure, frequent ventricular ectopy during catheter manipulation and contrast injection, and interobserver variability in the visual assessment of right ventricular wall motion abnormalities are important limitations. Because of these limitations, and the availability of other, more quantitative imaging tools such as MRI, right ventricular cine angiography plays only a small role today in the evaluation of patients with suspected ARVD. Table 65-4 lists major angiographic features of ARVD.
TABLE 65-4 Angiographic Features of Arrhythmogenic Right Ventricular Dysplasia
Nuclear Ventriculoscintigraphy
Nuclear ventriculoscintigraphy provides information about the size, ejection fraction, contraction pattern of the ventricles, and site of origin of ventricular tachycardia. In most cases of ARVD, the size of the right ventricle is increased, and ejection fraction is diminished. Contraction pattern of the right ventricular wall is asynchronous as a result of the ongoing dysplastic process. Use of specific radionuclide tracers has shown abnormal sympathetic myocardial innervation in ARVD patients, providing a new insight into the arrhythmogenicity of the disease. Myocardial imaging with iodine 123 MIBG and thallium 201 has detected left ventricular involvement early in the course of the disease, suggesting that it may be potentially more sensitive in the diagnosis of ARVD.19,20 Although nuclear imaging can be used in the evaluation of patients with suspected ARVD, it is rarely employed today.
Echocardiography
Two-dimensional echocardiography is an important first-line imaging tool for diagnosis and follow-up in ARVD because of its noninvasiveness, low cost, wide availability, and ease of performance. Echocardiographic abnormalities associated with the Task Force criteria for ARVD are summarized in Table 65-5. A right ventricular outflow tract long-axis diameter greater than 30 mm on echocardiography has been found to have the highest sensitivity and specificity for the diagnosis of ARVD. Right ventricular dysfunction has been defined as a fractional area contraction less than 32%, or the presence of segmental right ventricular wall motion abnormalities (mostly observed in anterior and apical walls) for the purpose of echocardiographic diagnosis of ARVD.21
TABLE 65-5 Echocardiography Features Associated with Task Force Criteria for Arrhythmogenic Right Ventricular Dysplasia
We evaluated three-dimensional echocardiography, tissue Doppler echocardiography, and strain echocardiography, and found that these imaging modalities show high feasibility and reproducibility in ARVD. Three-dimensional echocardiography measurement of right ventricular volumes and ejection fraction showed close correlation with MRI, the current reference standard, and three-dimensional echocardiography may be potentially more useful in the assessment of right ventricular function and follow-up of patients with ARVD, in contrast to two-dimensional echocardiography. Tissue Doppler echocardiography and strain echocardiography showed higher sensitivity in detection of mild cases of ARVD with normal right ventricle on conventional echocardiography. In our opinion, these new echocardiographic modalities may have a potentially greater diagnostic utility in ARVD than conventional methods.22,23 Contrast echocardiography using saline injections may outline the right ventricle better, enabling better evaluation of right ventricular volumes and regional and global function.11
Computed Tomography
Electron-beam CT and multidetector CT can yield accurate and reliable qualitative and quantitative assessment of the right ventricle at an excellent spatial and temporal resolution. They can also provide tissue characterization of the myocardium and detect fatty changes, similar to MRI. Electron-beam CT can directly assess left ventricular adipose tissue changes and can even detect subclinical cases of ARVD without left ventricular wall motion abnormality.24 Major abnormalities in ARVD reported on CT include (1) dilated right ventricle, (2) abundant epicardial fatty tissue, (3) intramyocardial fat deposits, (4) scalloped appearance (bulging) of the right ventricular free wall, and (5) conspicuous trabeculae with low attenuation (Figs. 65-2 and 65-3).
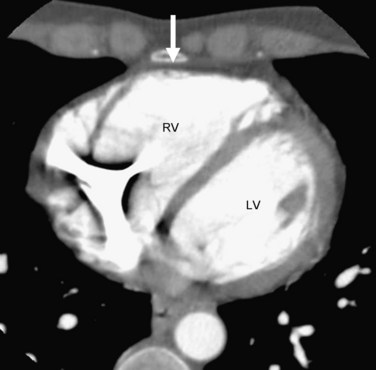
 FIGURE 65-3
FIGURE 65-3CT is less expensive, faster, technologically easier to perform, less operator-dependent, and more reliable in terms of image quality compared with MRI. It is less susceptible to artifacts resulting from respiratory motion and patient movement because of shorter image acquisition times on the order of milliseconds. CT is the diagnostic modality of choice in ARVD patients with claustrophobia and frequent arrhythmic events. Although the presence of an implantable cardioverter defibrillator (ICD) was previously considered to be an indication for a CT scan versus MRI, more recent studies have shown that MRI can be safely performed in patients with ICDs.25 Limitations of CT include its fixed axial plane for image acquisition, requirement for ionizing radiation exposure, use of nephrotoxic contrast agent, and relatively lower temporal resolution compared with MRI.7
Magnetic Resonance Imaging
MRI has been increasingly recognized as the imaging technique of choice for ARVD. Its three-dimensional, multiplanar visualization of the heart along with excellent spatial and temporal resolution enables highly reproducible assessment of structural morphology and function of the right ventricle without using any geometric assumptions. The improved blood-myocardial image contrast allows optimal visualization of the right ventricle. For myocardial tissue characterization, MRI can specifically locate and quantify intramyocardial adipose infiltration, which forms the pathologic hallmark of the disease (Fig. 65-4).26 MRI has the potential to visualize fibrous tissue replacement of right ventricular myocardium in ARVD by myocardial delayed enhancement (Fig. 65-5).27
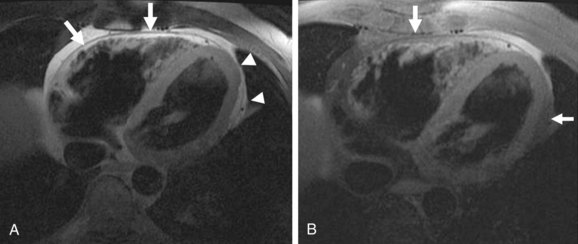
 FIGURE 65-4
FIGURE 65-4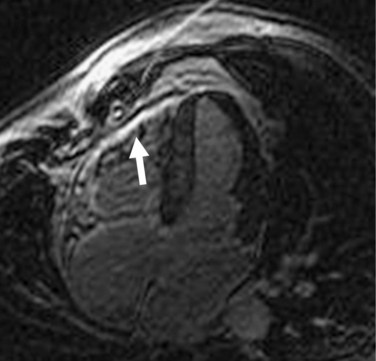
 FIGURE 65-5
FIGURE 65-5MRI can be uniquely applied for the identification of regional and diastolic dysfunction of the right ventricle and minor changes in right ventricular volume over time and other functional details, such as velocity mapping of the tricuspid flow and right ventricular strain measurements using myocardial tagging. MRI also has prognostic significance because of its ability to predict arrhythmia-free survival in ARVD.28 MRI is noninvasive and devoid of radiation hazards. These features make MRI highly suited and superior to other imaging investigations for the detection and serial follow-up of patients with clinically suspected ARVD.
MRI findings in ARVD can be broadly divided into two groups: (1) morphologic abnormalities, including intramyocardial fat infiltration, right ventricular wall thinning (Fig. 65-6), trabecular hypertrophy (Fig. 65-7), and right ventricular outflow tract enlargement (Fig. 65-8), and (2) functional abnormalities, including right ventricular wall motion abnormalities, aneurysms (Fig. 65-9), right ventricular dilation (Fig. 65-10), and right atrial enlargement (Fig. 65-11). These findings are listed in Table 65-6.
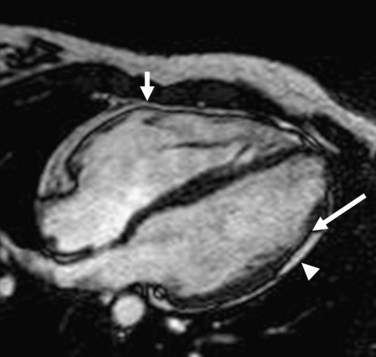
 FIGURE 65-6
FIGURE 65-6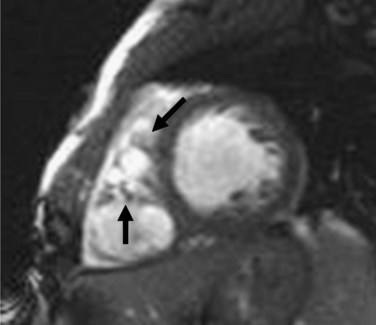
 FIGURE 65-7
FIGURE 65-7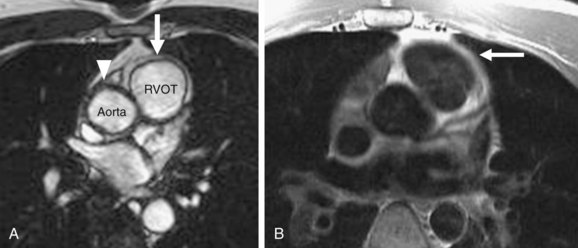
 FIGURE 65-8
FIGURE 65-8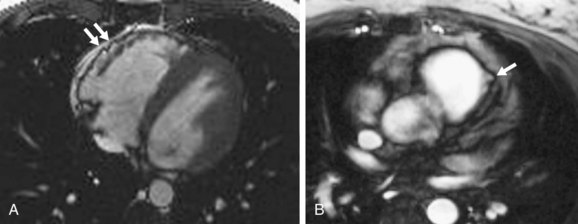
 FIGURE 65-9
FIGURE 65-9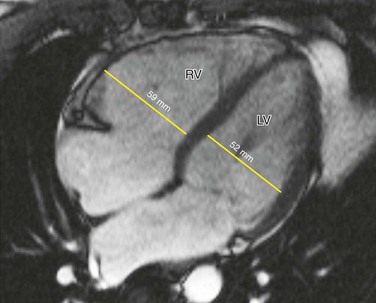
 FIGURE 65-10
FIGURE 65-10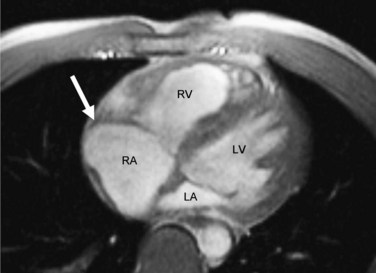
 FIGURE 65-11
FIGURE 65-11TABLE 65-6 Magnetic Resonance Imaging Features of Arrhythmogenic Right Ventricular Dysplasia
Black blood breath-hold fast spin-echo MRI (see Figs. 65-4A and 65-8B) with dual magnetization preparation pulse (double inversion recovery) is the preferred modality to detail cardiac morphology. Inversion recovery pulses are used to null (darken) the signal from a tissue of interest to highlight (enhance) the surrounding pathology. Breath-holding reduces motion artifacts and improves overall image quality of myocardial detail. Black blood single-shot spin-echo techniques (e.g., HASTE, turboFLASH, SSFSE) are ultrafast imaging modalities that substantially reduce image acquisition times and motion artifacts, but at the expense of blurring of detail. They are not recommended, unless a severe arrhythmia is present that results in failure of other black blood sequences. Incorporation of fat-suppressed sequences (see Fig. 65-4B) to conventional T1-weighted spin-echo imaging has been found to increase the interobserver agreement and confidence in the diagnosis of intramyocardial fat infiltration.29,30 Table 65-7 presents a detailed MRI protocol recommended for ARVD.
TABLE 65-7 Magnetic Resonance Imaging Protocol for Arrhythmogenic Right Ventricular Dysplasia
| Patient preparation: If the patient is known to have frequent ventricular ectopy, use oral metoprolol, 50 mg, 1 hr before the procedure, provided that the patient has no contraindications. If ventricular arrhythmias are frequent and would have a substantial impact on image quality, the examination should be terminated at this point. |
Right ventricular intramyocardial fat on MRI has a low sensitivity for detection of ARVD compared with other abnormalities, such as regional right ventricular dysfunction. Normal presence of epicardial fat, especially in areas such as the atrioventricular sulcus and anterolateral and apical portions of the right ventricle, sometimes makes distinction from pathologic fat difficult, particularly given the thinness of normal right ventricular wall and limited spatial and contrast resolution achievable by even state-of-the-art MRI protocols.31 Fat was also found to be the least specific and least reproducible of any of the other parameters in the MRI evaluation of ARVD.32,33 High intramyocardial T1 signals are not specific for ARVD because isolated fatty infiltration of the right ventricle has also been reported in elderly individuals, obese individuals, patients on long-term steroid therapy, patients with alcohol abuse, patients with idiopathic right ventricular outflow tract tachycardia, and patients with inherited myopathies, and more recently as a separate disorder in itself, clinicopathologically distinct from ARVD.34 False-positive high signal intensities potentially misconstrued as fat may be produced by various artifacts related to motion, respiration, blood flow, arrhythmia, or truncation band.
The ability of MRI stands out in the diagnosis of early and subtle cases of ARVD because of its potential to detect regional and diastolic ventricular dysfunction, which are thought to precede global and systolic dysfunction during the progressive course of the disease.35 This capability of MRI can add to the overall sensitivity of diagnosis of ARVD because these patients may otherwise be clinically inapparent and less sensitive to diagnosis by Task Force criteria. In one of our studies, we found that despite preserved systolic function, 37% of patients with early or limited forms of ARVD show focal discoordinate contraction of right ventricular free basal wall on MRI, which was unique in its appearance as focal crinkling of the right ventricular wall in peak systole (Fig. 65-12).
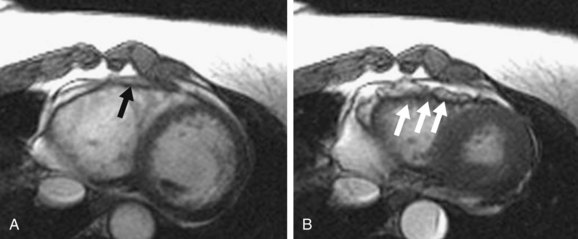
 FIGURE 65-12
FIGURE 65-12Despite the diagnostic utility of MRI in ARVD, its specificity and reliability remain in question because of the subjectivity in the interpretation of its findings, dependence on image quality, over-reliance on intramyocardial fat and wall thinning, and lack of standardized protocol and technical and clinical expertise in cardiovascular MRI. Observers without extensive experience in MRI for ARVD can overdiagnose this condition, particularly when the only abnormalities observed are with MRI. MRI can be a low-yield test for the diagnosis of ARVD in children compared with adults. MRI should be considered as only a part of the multidisciplinary diagnostic work-up of the patient, and the final diagnosis of ARVD should be based on an integrated assessment of all noninvasive and invasive testing in reference to Task Force criteria.35 Follow-up studies, in probands and suspected relatives, can be advantageous—keeping in mind the progressive and evolving nature of the disease.
DIFFERENTIAL DIAGNOSIS
Numerous clinically diverse conditions can mimic ARVD because of related histologic picture, ventricular tachycardia of right ventricular origin, or right ventricular enlargement and distorted right ventricular anatomy, and must be appropriately excluded (Table 65-8).11,36 Right ventricular outflow tract tachycardia is one of the most important differential diagnoses from the clinical standpoint (Table 65-9).37 A few other important conditions are discussed next.
TABLE 65-8 Differential Diagnosis of Arrhythmogenic Right Ventricular Dysplasia
* Whether right ventricular outflow tract tachycardia represents a form of arrhythmogenic right ventricular cardiomyopathies is still debated.
From Corrado D, Basso C, Thiene G. Arrhythmogenic right ventricular cardiomyopathy: diagnosis, prognosis, and treatment. Heart 2000; 83:588.
TABLE 65-9 Differences Between Right Ventricular Outflow Tract (RVOT) Tachycardia and Arrhythmogenic Right Ventricular Dysplasia
| Features | RVOT Tachycardia | ARVD |
|---|---|---|
| Clinical Presentation | ||
| Family history | No | Frequently yes |
| Arrhythmias | Premature ventricular contractions, nonsustained ventricular tachycardia, or sustained ventricular tachycardia at rest or with exercise | Same |
| Sudden cardiac death | Rare | 1%/yr |
| ECG Features | ||
| T wave morphology | T wave upright V2-V5 | T wave inverted beyond V1 |
| Parietal block | QRS duration <110 ms in V1-V3 | QRS duration >110 ms |
| Epsilon wave V1-V3 | Absent | Present in one third of cases |
| Prolonged S wave upstroke | Rarely seen | Usually present |
| Signal-averaged ECG | Normal | Usually abnormal |
| Electrophysiology Study | ||
| Presence of more than one ECG morphology during tachycardia | Absent | Seen frequently |
| Fractionated diastolic potentials during ventricular tachycardia | Absent | Seen frequently |
| Programmed premature stimulation–induced ventricular tachycardia | Absent | Seen frequently |
| Imaging Features | ||
| Echocardiogram | Normal | Usually abnormal |
| Right ventricular ventriculogram | Usually normal | Usually abnormal |
| MRI | Usually normal | Usually abnormal |
From Prakasa KR, Calkins H. Arrhythmogenic right ventricular dysplasia/cardiomyopathy. Curr Treat Options Cardiovasc Med 2005; 7:467.
Sarcoidosis
Involvement of the ventricle in cardiac sarcoidosis often can mimic the ECG and structural changes in ARVD.38 Presence of hilar lymphadenopathy should alert the physician to consider this diagnosis. MRI may reveal patchy left ventricular delayed hyperenhancement in a noncoronary distribution. Finally, an endomyocardial biopsy specimen of an affected region often reveals noncaseating granulomas, confirming the diagnosis of sarcoidosis.
TREATMENT OPTIONS
Medical
The pharmacologic agents commonly used to treat arrhythmias in ARVD include β blockers, calcium channel blockers, and class I and III antiarrhythmic agents. Sotalol and amiodarone (both class III agents) have been found to be the most effective antiarrhythmic drugs in this regard with low proarrhythmogenic risk. Their efficacy in preventing sudden cardiac death is unknown, however. Antiarrhythmic drugs represent a first-line treatment in patients with ARVD. Antiarrhythmic drugs do not prevent sudden cardiac death and are used merely to reduce the frequency of sustained ventricular arrhythmias, and in doing so, they improve quality of life. ARVD patients who have progressed to right ventricular/biventricular failure are treated with the usual regimen, which includes ACE inhibitors, diuretics, β blockers, and digitalis.12
Surgical/Interventional
Although it may result in acute procedural success, catheter radiofrequency ablation is seldom effective and is often accompanied by recurrence of ventricular tachycardia in the long-term because of the progressive nature of the disease. Its clinical role is limited as a short-term palliative procedure in cases of (1) ventricular arrhythmia when pharmacologic therapy is ineffectual, and (2) frequent ICD discharges owing to recurrent ventricular tachycardia refractory to antiarrhythmic therapy.12
INFORMATION FOR THE REFERRING PHYSICIAN



Ahmad F. The molecular genetics of arrhythmogenic right ventricular dysplasia-cardiomyopathy. Clin Invest Med. 2003;26:167.
Corrado D, Basso C, Thiene G. Arrhythmogenic right ventricular cardiomyopathy: diagnosis, prognosis, and treatment. Heart. 2000;83:588.
Fontaine G, Fontaliran F, Hebert JL, et al. Arrhythmogenic right ventricular dysplasia. Annu Rev Med. 1999;50:17.
Gemayel C, Pelliccia A, Thompson PD. Arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2001;38:1773.
Kayser HW, van der Wall EE, Sivananthan MU, et al. Diagnosis of arrhythmogenic right ventricular dysplasia: a review. RadioGraphics. 2002;22:639.
Moric-Janiszewska E, Markiewicz-Loskot G. Review on the genetics of arrhythmogenic right ventricular dysplasia. Europace. 2007;9:259.
Paul M, Schulze-Bahr E, Breithardt G, et al. Genetics of arrhythmogenic right ventricular cardiomyopathy—status quo and future perspectives. Z Kardiol. 2003;92:128.
Prakasa KR, Calkins H. Arrhythmogenic right ventricular dysplasia/cardiomyopathy. Curr Treat Options Cardiovasc Med. 2005;7:467.
Tandri H, Bomma C, Calkins H, et al. Magnetic resonance and computed tomography imaging of arrhythmogenic right ventricular dysplasia. J Magn Reson Imaging. 2004;19:848.
1 Sen-Chowdhry S, Syrris P, Ward D, et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710.
2 Norman MW, McKenna WJ. Arrhythmogenic right ventricular cardiomyopathy: perspectives on disease. Z Kardiol. 1999;88:550-554.
3 Thiene G, Basso C. Arrhythmogenic right ventricular cardiomyopathy: an update. Cardiovasc Pathol. 2001;10:109-117.
4 Kies P, Bootsma M, Bax J, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: screening, diagnosis, and treatment. Heart Rhythm. 2006;3:225-234.
5 Dalal D, Nasir K, Bomma C, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005;112:3823-3832.
6 Nava A, Bauce B, Basso C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36:2226-2233.
7 Tandri H, Bomma C, Calkins H, et al. Magnetic resonance and computed tomography imaging of arrhythmogenic right ventricular dysplasia. J Magn Reson Imaging. 2004;19:848.
8 Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2005;16:927.
9 Moric-Janiszewska E, Markiewicz-Loskot G. Review on the genetics of arrhythmogenic right ventricular dysplasia. Europace. 2007;9:259.
10 Gemayel C, Pelliccia A, Thompson PD. Arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2001;38:1773.
11 Fontaine G, Fontaliran F, Hebert JL, et al. Arrhythmogenic right ventricular dysplasia. Annu Rev Med. 1999;50:17.
12 Corrado D, Basso C, Thiene G. Arrhythmogenic right ventricular cardiomyopathy: diagnosis, prognosis, and treatment. Heart. 2000;83:588.
13 Tonet JL, Castro-Miranda R, Iwa T, et al. Frequency of supraventricular tachyarrhythmias in arrhythmogenic right ventricular dysplasia. Am J Cardiol. 1991;67:1153.
14 McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71:215.
15 Sen-Chowdhry S, Syrris P, McKenna WJ. Desmoplakin disease in arrhythmogenic right ventricular cardiomyopathy: early genotype-phenotype studies. Eur Heart J. 2005;26:1582.
16 Hamid MS, Norman M, Quraishi A, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002;40:1445.
17 Nasir K, Bomma C, Tandri H, et al. Electrocardiographic features of arrhythmogenic right ventricular dysplasia/cardiomyopathy according to disease severity: a need to broaden diagnostic criteria. Circulation. 2004;110:1527.
18 Nasir K, Tandri H, Rutberg J, et al. Filtered QRS duration on signal-averaged electrocardiography predicts inducibility of ventricular tachycardia in arrhythmogenic right ventricle dysplasia. Pacing Clin Electrophysiol. 2003;26:1955.
19 Takahashi N, Ishida Y, Maeno M, et al. Noninvasive identification of left ventricular involvements in arrhythmogenic right ventricular dysplasia: comparison of 123I-MIBG, 201TlCl, magnetic resonance imaging and ultrafast computed tomography. Ann Nucl Med. 1997;11:233.
20 Lindstrom L, Nylander E, Larsson H, et al. Left ventricular involvement in arrhythmogenic right ventricular cardiomyopathy—a scintigraphic and echocardiographic study. Clin Physiol Funct Imaging. 2005;25:171.
21 Yoerger DM, Marcus F, Sherrill D, et al. Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia: new insights from the multidisciplinary study of right ventricular dysplasia. J Am Coll Cardiol. 2005;45:860.
22 Prakasa KR, Wang J, Tandri H, et al. Utility of tissue Doppler and strain echocardiography in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol. 2007;100:507.
23 Prakasa KR, Dalal D, Wang J, et al. Feasibility and variability of three dimensional echocardiography in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol. 2006;97:703.
24 Tada H, Shimizu W, Ohe T, et al. Usefulness of electron-beam computed tomography in arrhythmogenic right ventricular dysplasia: relationship to electrophysiological abnormalities and left ventricular involvement. Circulation. 1996;94:437.
25 Nazarian S, Roguin A, Zviman MM, et al. Clinical utility and safety of a protocol for noncardiac and cardiac magnetic resonance imaging of patients with permanent pacemakers and implantable-cardioverter defibrillators at 1.5 tesla. Circulation. 2006;114:1277.
26 Fattori R, Tricoci P, Russo V, et al. Quantification of fatty tissue mass by magnetic resonance imaging in arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol. 2005;16:256.
27 Tandri H, Saranathan M, Rodriguez ER, et al. Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed-enhancement magnetic resonance imaging. J Am Coll Cardiol. 2005;45:98.
28 Keller DI, Osswald S, Bremerich J, et al. Arrhythmogenic right ventricular cardiomyopathy: diagnostic and prognostic value of the cardiac MRI in relation to arrhythmia-free survival. Int J Cardiovasc Imaging. 2003;19:537.
29 Tandri H, Friedrich MG, Calkins H, et al. MRI of arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Cardiovasc Magn Reson. 2004;6:557.
30 Abbara S, Migrino RQ, Sosnovik DE, et al. Value of fat suppression in the MRI evaluation of suspected arrhythmogenic right ventricular dysplasia. AJR Am J Roentgenol. 2004;182:587.
31 Castillo E, Tandri H, Rodriguez ER, et al. Arrhythmogenic right ventricular dysplasia: ex vivo and in vivo fat detection with black-blood MR imaging. Radiology. 2004;232:38.
32 Tandri H, Castillo E, Ferrari VA, et al. Magnetic resonance imaging of arrhythmogenic right ventricular dysplasia: sensitivity, specificity, and observer variability of fat detection versus functional analysis of the right ventricle. J Am Coll Cardiol. 2006;48:2277.
33 Bluemke DA, Krupinski EA, Ovitt T, et al. MR imaging of arrhythmogenic right ventricular cardiomyopathy: morphologic findings and interobserver reliability. Cardiology. 2003;99:153.
34 Macedo R, Prakasa K, Tichnell C, et al. Marked lipomatous infiltration of the right ventricle: MRI findings in relation to arrhythmogenic right ventricular dysplasia. AJR Am J Roentgenol. 2007;188:W423.
35 Bomma C, Dalal D, Tandri H, et al. Regional differences in systolic and diastolic function in arrhythmogenic right ventricular dysplasia/cardiomyopathy using magnetic resonance imaging. Am J Cardiol. 2005;95:1507.
36 Fontaine G, Fontaliran F, Frank R. Arrhythmogenic right ventricular cardiomyopathies: clinical forms and main differential diagnoses. Circulation. 1998;97:1532.
37 Prakasa KR, Calkins H. Arrhythmogenic right ventricular dysplasia/cardiomyopathy. Curr Treat Options Cardiovasc Med. 2005;7:467.
38 Ott P, Marcus FI, Sobonya RE, et al. Cardiac sarcoidosis masquerading as right ventricular dysplasia. Pacing Clin Electrophysiol. 2003;26:1498.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
CHAPTER 65 Arrhythmogenic Right Ventricular Dysplasia
DEFINITION
ARVD is a genetic cardiomyopathy principally involving the right ventricle that is characterized clinically by ventricular arrhythmias and progressive structural alterations in right ventricular size and function. Fibrofatty replacement of the right ventricular myocardium forms the histopathologic hallmark of this disease. There are three patterns of disease expression: (1) classic, characterized by disease limited to the right ventricle with either no or minimal involvement of the left ventricle, which appears late in the course of the disease; (2) left dominant, characterized by early or predominant left ventricular involvement; and (3) biventricular, characterized by parallel involvement of both ventricles from the beginning of disease process.1
PREVALENCE AND EPIDEMIOLOGY
Although the exact incidence of ARVD is unknown, the prevalence has been estimated to be 0.02% of the general population.2 ARVD reportedly is more prevalent (0.8%) in certain parts of northern Italy (Veneto) and Greece (Naxos Island).3,4 ARVD most commonly manifests after puberty and before age 50 years.5,6 ARVD is more common in highly athletic individuals. It is an important cause of sudden cardiac death in young individuals, particularly in athletes, accounting for 5% of sudden deaths in individuals younger than 35 years in the United States, and 25% of deaths in athletes in the Veneto region of Italy.7 ARVD is believed to be more common than reported because of its frequently asymptomatic clinical course, difficulty in diagnosis using conventional noninvasive methods, and frequent misdiagnosis.
ETIOLOGY AND PATHOPHYSIOLOGY
The exact pathogenesis of ARVD remains speculative, but it is most widely believed to be due to genetic abnormalities mainly affecting cardiac desmosomal structure and function. In the United States, the most common reported genetic abnormality in ARVD is a mutation in plakophillin-2 (PKP2), which is a desmosomal protein and is present in more than one third of patients. Desmosomal mutations are believed to disrupt cell-cell adhesion, which provokes myocyte detachment and death under conditions of high mechanical stress, as suggested by its frequent occurrence in athletics and the thinnest portions of the right ventricle, such as the inferior subtricuspid region, right ventricular apex, and right ventricular infundibulum—together termed the triangle of dysplasia (Fig. 65-1).8 This leads to a compensatory repair process characterized by inflammation and fibrofatty substitution. Familial ARVD accounts for 30% to 80% of cases, and most commonly shows an autosomal dominant inheritance, with age-dependent penetrance and variable clinical expression, which can be attributed to the complex interplay of age, gender effects, and environmental influences such as exposure to viruses and athletic activity with the underlying genetic background in the ultimate disease causation and progression. Mere inheritance of a genetic abnormality does not imply “disease” for the same reason. To date, at least 11 distinct subtypes of autosomal dominant ARVD are known, and at least seven genes have been implicated in ARVD (Table 65-1).9
Genetic mutations are also implicated in determining the ventricular predominance in ARVD. Chain-termination mutations causing truncation of the C-terminal domain of desmoplakin can disrupt cytoskeletal integrity, predisposing to predominant left ventricular involvement. Conversely, defects in the N-terminal domain of desmoplakin can result in a primary dysfunction of cell adhesion, leading to predominant right ventricular disease.8
In addition to genetic causes, other important proposed etiologic mechanisms of ARVD are (1) apoptosis (programmed cell death), (2) inherited metabolic or ultrastructural defects leading to myocardial dystrophy, and (3) secondary complication of a prior primary right ventricular myocarditis on the basis of frequently found myocardial inflammatory infiltrates and isolation of coxsackie B and enteroviral RNA viruses in some biopsy samples.10
Pathologically, ARVD is characterized by gradual myocyte loss and fibrofatty infiltration, which starts in the subepicardial region and gradually involves the endocardium, often sparing the trabecular myocardium. Plexiform arrangement of the residual myocardial fibers, separated by fat and fibrosis, leads to electrical instability and provides a conduit for a reentrant phenomenon. This situation explains the arrhythmogenic propensity characteristic of this disease, which can be worsened further by superimposed myocarditis that occurs frequently in ARVD because of the genetic susceptibility.11 A myocardial dysplastic process and the associated loss of its strength and integrity are responsible for the varying degrees of structural derangement and functional disturbances of the heart seen in ARVD.
MANIFESTATIONS OF DISEASE
Clinical Presentation
ARVD usually manifests between the second and fifth decades (mean age at diagnosis approximately 30 years). Patients most commonly present with symptomatic ventricular arrhythmias or sudden cardiac death, or may remain asymptomatic, with ARVD discovered only incidentally on routine examination. The most common symptoms are palpitations, syncope, atypical chest pain, and dyspnea. Ventricular arrhythmias are often precipitated by exercise or stress, and range from isolated ventricular premature beats to sustained monomorphic or polymorphic ventricular tachycardia with left bundle branch block morphology, or abrupt ventricular fibrillation, which is the most common underlying cause of sudden cardiac death in ARVD.12
In addition to ventricular arrhythmias, about 25% of patients may also show supraventricular arrhythmias, which include atrial fibrillation, atrial tachycardia, and atrial flutter, in that order of frequency.13 Left ventricular involvement is associated with an increased incidence of arrhythmic events, more severe right ventricular wall thinning, and heart failure. Frank left-sided heart failure is rare. Figures 65-2, 65-4, and 65-6 illustrate left ventricular involvement in ARVD.
Diagnosis
Diagnosis of ARVD is challenging and is currently based on family history and morphofunctional, ECG, histopathologic, and clinical findings as proposed by the Task Force of Cardiomyopathies in 1994 (Table 65-2).14 The diagnosis is made in the presence of two major criteria, one major criterion and two minor criteria, or four minor criteria. Various modifications of the existing Task Force criteria are being increasingly proposed and emphasize the increment in the diagnostic sensitivity of these criteria for less severe forms of the disease and asymptomatic first-degree relatives of affected probands. These modifications include more comprehensive characterization of ECG and echocardiographic criteria, inclusion of mutation analysis, and descriptions of left ventricular involvement.15,16
TABLE 65-2 Task Force Criteria for the Diagnosis of Arrhythmogenic Right Ventricular Dysplasia
| Major Criteria | Minor Criteria | |
|---|---|---|
| Global or regional dysfunction and structural alterations* | Severe dilation and reduction of right ventricular ejection fraction with no (or only mild) left ventricular impairment | Mild global right ventricular dilation or ejection fraction reduction with normal left ventricle |
| Localized right ventricular aneurysms (akinetic or dyskinetic areas with diastolic bulging) | Mild segmental dilation of right ventricle | |
| Severe segmental dilation of right ventricle | Regional right ventricular hypokinesia | |
| Tissue characterization of wall | Fibrofatty replacement of myocardium on endomyocardial biopsy specimen | |
| Repolarization abnormalities | Inverted T waves in right precordial leads (V2 and V3) in individuals >12 yr old in absence of right bundle branch block | |
| Depolarization or conduction abnormalities | Epsilon waves or localized prolongation (>110 ms) of the QRS complex in right precordial leads (V1-V3) | Late potentials (signal-averaged ECG) |
| Arrhythmias | Left bundle branch block ventricular tachycardia (sustained and nonsustained) by ECG, Holter, or exercise testing | |
| Frequent ventricular extrasystoles (>1000/24 hr) (Holter) | ||
| Family history | Familial disease confirmed at necropsy or surgery | Family history of premature sudden death (<35 yr old) owing to suspected right ventricular dysplasia |
| Familial history (clinical diagnosis based on present criteria) |
* Detected by echocardiography, angiography, MRI, or radionuclide scintigraphy.
Electrocardiogram Evaluation
At least one ECG abnormality (Table 65-3) is detectable in more than 90% of patients with ARVD. A newly proposed marker of delayed right ventricular activation, prolonged S wave upstroke (≥55 ms) in V1 to V3, is the most prevalent ECG marker and correlates with disease severity and ventricular tachycardia induction on electrophysiology testing.17 Signal-averaged ECG is used to detect delayed ventricular activation owing to slowed propagation in the fibrofatty myocardium of ARVD. Late potentials on signal-averaged ECG (two or more of the following: filtered QRS duration ≥114 ms, duration of the low-amplitude signal <40 µV in the terminal portion of filtered QRS (LAS40) ≥38 ms, and root mean square voltage of the terminal 40 ms of filtered QRS (RMS40) <20 µV) form a minor diagnostic criterion for ARVD as per the Task Force criteria. Similar to extent of right precordial T wave inversion, late potentials on signal-averaged ECG are correlated with the extent of right ventricular involvement in ARVD. More recently, our group found that using filtered QRS 110 ms or greater identifies ARVD patients with inducible ventricular tachycardia more accurately than does conventional signal-averaged ECG criteria.18 T wave inversions and signal-averaged ECG abnormalities are more commonly seen in left-sided involvement in ARVD.
TABLE 65-3 Electrocardiogram Features of Arrhythmogenic Right Ventricular Dysplasia‡
* Minor Task Force criterion in the absence of right bundle branch block in individuals >12 yr old.
‡ These electrical abnormalities are believed to be caused by intraventricular myocardial defect (parietal block) rather than definite alteration of conduction in the bundle branch (septal block).
Imaging Techniques and Findings
Chest Radiograph
Chest radiographs generally show cardiac enlargement depending on the stage of the disease, with the cardiothoracic ratio generally less than 0.6. The heart can also show a convexity between the aortic knob and the left ventricle, and there is no pulmonary vascular redistribution.11 Radiographs are not a sensitive modality to diagnose ARVD.
Cine Angiography
Right ventricular cine angiography has been regarded as the traditional gold standard, with high specificity and positive and negative predictive values in ARVD diagnosis. It is performed using two orthogonal views in 30 degrees right anterior oblique and 60 degrees left anterior oblique during sinus rhythm with careful evaluation of structure and function of the right ventricle. Coexistence of subtricuspid and anterior infundibular wall bulging and hypertrophic trabeculae has been shown to have 96% sensitivity and 87.5% specificity in angiographic diagnosis of ARVD.10 Invasiveness of the procedure, frequent ventricular ectopy during catheter manipulation and contrast injection, and interobserver variability in the visual assessment of right ventricular wall motion abnormalities are important limitations. Because of these limitations, and the availability of other, more quantitative imaging tools such as MRI, right ventricular cine angiography plays only a small role today in the evaluation of patients with suspected ARVD. Table 65-4 lists major angiographic features of ARVD.
TABLE 65-4 Angiographic Features of Arrhythmogenic Right Ventricular Dysplasia
Nuclear Ventriculoscintigraphy
Nuclear ventriculoscintigraphy provides information about the size, ejection fraction, contraction pattern of the ventricles, and site of origin of ventricular tachycardia. In most cases of ARVD, the size of the right ventricle is increased, and ejection fraction is diminished. Contraction pattern of the right ventricular wall is asynchronous as a result of the ongoing dysplastic process. Use of specific radionuclide tracers has shown abnormal sympathetic myocardial innervation in ARVD patients, providing a new insight into the arrhythmogenicity of the disease. Myocardial imaging with iodine 123 MIBG and thallium 201 has detected left ventricular involvement early in the course of the disease, suggesting that it may be potentially more sensitive in the diagnosis of ARVD.19,20 Although nuclear imaging can be used in the evaluation of patients with suspected ARVD, it is rarely employed today.
Echocardiography
Two-dimensional echocardiography is an important first-line imaging tool for diagnosis and follow-up in ARVD because of its noninvasiveness, low cost, wide availability, and ease of performance. Echocardiographic abnormalities associated with the Task Force criteria for ARVD are summarized in Table 65-5. A right ventricular outflow tract long-axis diameter greater than 30 mm on echocardiography has been found to have the highest sensitivity and specificity for the diagnosis of ARVD. Right ventricular dysfunction has been defined as a fractional area contraction less than 32%, or the presence of segmental right ventricular wall motion abnormalities (mostly observed in anterior and apical walls) for the purpose of echocardiographic diagnosis of ARVD.21
TABLE 65-5 Echocardiography Features Associated with Task Force Criteria for Arrhythmogenic Right Ventricular Dysplasia
[/not-level-membership-for-cardiovascular-category]
