Chapter 59 Arrhythmogenic Right Ventricular Cardiomyopathy
Historical Summary
In 1977, Fontaine et al reported six cases of sustained ventricular tachycardia (VT) in patients with enlarged right ventricles and hypothesized that this was a specific cardiomyopathy limited to the right ventricle.1 In 1982, Marcus et al coined the term arrhythmogenic right ventricular dysplasia (ARVD) in a report of 24 cases characterized by left bundle branch block (LBBB) pattern VT, wall motion abnormalities of the right ventricle (RV), and replacement of the RV myocardium by adipose and fibrous tissue.2 In 1994, the first diagnostic criteria were proposed. These were based on the identification of structural abnormalities, fibro-fatty replacement of the RV myocardium, particular electrocardiographic changes, arrhythmias of RV origin, and familial disease.3 This led to ARVC being recognized as a separate entity in the reclassification of cardiomyopathies by the World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies.4–6 Over the following decade, various groups mapped familial forms of ARVC to a number of genetic loci.7–14 Molecular studies in a recessive, syndromic variant of ARVC, known as Naxos disease, led to identification of the first disease-causing mutation in the gene encoding plakoglobin, a major constituent of cell-to-cell junctions.15 This paved the way for the discovery of disease-causing mutations in other genes encoding desmosomal proteins in the more common autosomal dominant forms of ARVC.
Epidemiology
The prevalence of ARVC in the general population is difficult to determine because of the challenging nature of the diagnosis.16 Various studies in Europe reported a prevalence of between 0.6 and 4.4 per 1000, but it is unclear whether these represent local interest and expertise in the disease. ARVC is reported as a cause of SCD in 11% to 27% of individuals 35 years old and younger.17–19 The prevalence in one Italian cohort was found to be higher in athletes (22.4%) compared with nonathletes (8.2%).17
Pathology
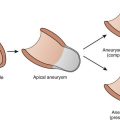
In the original descriptions of ARVC, the RV cavity was described as typically dilated, and the RV wall was characterized by dome-shaped aneurysms, occurring particularly on the anterior surface of the pulmonary infundibulum, at the RV apex, and on the inferior wall of the RV (subsequently called the triangle of dysplasia). These macroscopic changes were accompanied by abundant subepicardial fat and endocardial thickening (Figure 59-1). Microscopically, a marked decrease was seen in myocardial fibers, with replacement of the myocardium with fatty tissue and fibrosis (Figure 59-2).2 Later, pathologic descriptions divided ARVC into two morphologic patterns: the fatty and the fibro-fatty forms, but fatty infiltration alone can be a normal phenomenon, particularly in older women.20
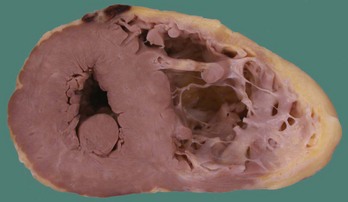
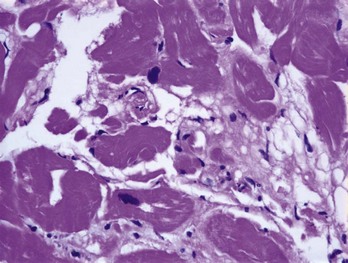
FIGURE 59-2 Histologic specimen of the right ventricle showing fatty infiltration and fibrosis within the myocardium.
(Courtesy Dr. Antonis Pantazis, Consultant Cardiologist, University College London Hospitals, UK.)
In a small series of studies on deaths due to ARVC, apoptosis was reported in 75% of patients.21 These apoptotic myocardial cells were frequently found in the myocardium not affected by fibro-fatty replacement, which suggests that this could account for the progressive loss of myocardial cells in the disease.22 Endomyocardial biopsy in 20 patients with ARVC revealed apoptosis in 35%. This was significantly associated with acute symptoms and signs and with a shorter (<6 months) clinical history, indicating that apoptosis is present in an early symptomatic stage of the disease.23
A number of postmortem reports described inflammatory infiltrates, and endomyocardial biopsies from some ARVC cases have been found to contain enteroviral ribonucleic acid (RNA) with homology to Coxsackie virus type B.21,24–26 In one study, the frequency of viral deoxyribonucleic acid (DNA) was similar to that observed in patients with myocarditis or dilated cardiomyopathy (DCM), but subsequent reports have failed to confirm these findings, perhaps reflecting patient selection and differences among inherited, sporadic, and nonfamilial forms of ARVC.27
Left Ventricular Involvement
By definition, the right ventricle must be affected to make a clinical diagnosis of ARVC, but it is well recognized that left ventricular involvement is common in patients fulfilling current diagnostic criteria. Pathologic examination of heart specimens at autopsy or cardiac transplantation revealed macroscopic and microscopic evidence of left ventricular involvement in up to 76% of cases.28 More recently, a study of 200 patients with ARVC undergoing cardiac magnetic resonance imaging (MRI) reported biventricular involvement in 56% of cases and predominant left-sided disease in 5%.29
Genetics
Systematic family studies have shown that ARVC is inherited in up to 50% of cases. Numerous genetic loci have been identified in linkage studies (Table 59-1). The mode of transmission is usually autosomal dominant, with a male predominance of 3 : 1 and variable penetrance, but autosomal recessive forms are well recognized and have provided the first insights into the genetic basis of ARVC.16
Table 59-1 Chromosomal Location, Involved Genes, and Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy
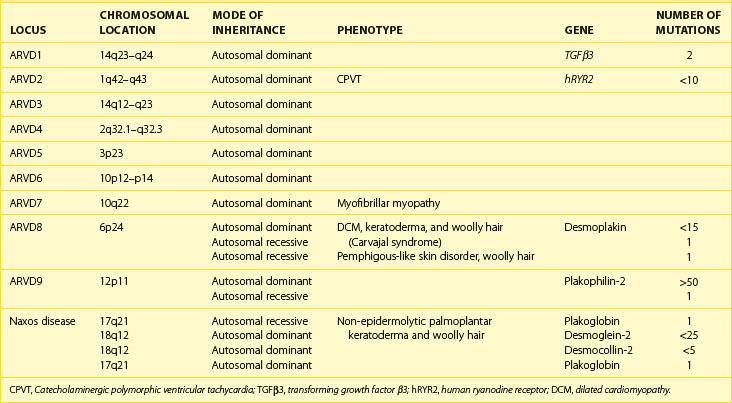
The first disease causing mutation was identified in an autosomal recessive syndromic form of ARVC, characterized by diffuse palmo-plantar keratoderma, woolly hair, and cardiomyopathy (Naxos disease).30 A homozygous mutation was identified in the gene encoding plakoglobin, a member of the armadillo protein family found in the adherens and desmosomal junctions between cardiac myocytes.15 Mutations in another desmosomal protein, desmoplakin, were subsequently identified in similar autosomal recessive syndromic forms of ARVC in South American and Arab families.31,32 Since then, heterozygous mutations in genes encoding these and other desmosomal proteins have been identified in patients with nonsyndromic autosomal dominant forms of ARVC.
The Intercellular Junction
Intercalated discs are specialized structures that provide mechanical and electrical coupling between adjacent cardiomyocytes. The intercalated disc is made up of three distinct structures: (1) gap junctions, (2) adherens junctions, and (3) desmosomes. The gap junction mediates the transfer of ions between cells, whereas the adherens junction (composed of cadherins, β-catenin, and γ-catenin [plakoglobin]) mediates the transmission of force between cells. The desmosome also mediates mechanical attachment between cells by linking the desmosomal cadherins, desmocollin, and desmoglein with the intermediate filament cytoskeleton.33,34 The intracellular components of the desmosomal cadherins interact with plakoglobin and plakophilin, which, in turn, bind to the N-terminal domain of desmoplakin, a plakin protein. The C-terminal of desmoplakin anchors desmin intermediate filaments to the cell surface.35 Another protein of the plakin family, plectin, is also present in desmosomes and contributes to the mechanical strength of cells.36,37 As well as providing cells with mechanical strength, the desmosome also plays an important role in tissue morphogenesis and differentiation.38 A simplified schematic representation of the organization of the cardiac desmosome is shown in Figure 59-3.
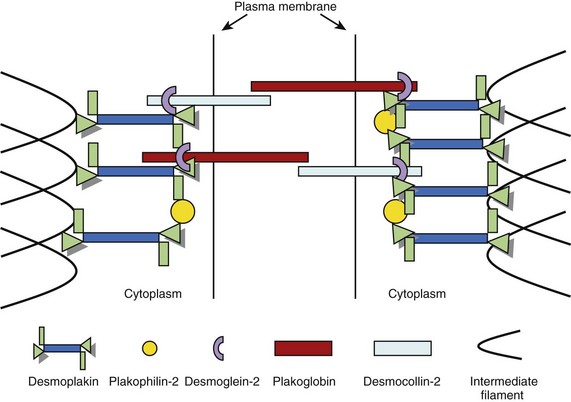
FIGURE 59-3 A schematic representation of the molecular desmosome.
(Data adapted from Green KJ, Gaudry CA: Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol 1:208–216, 2000.)
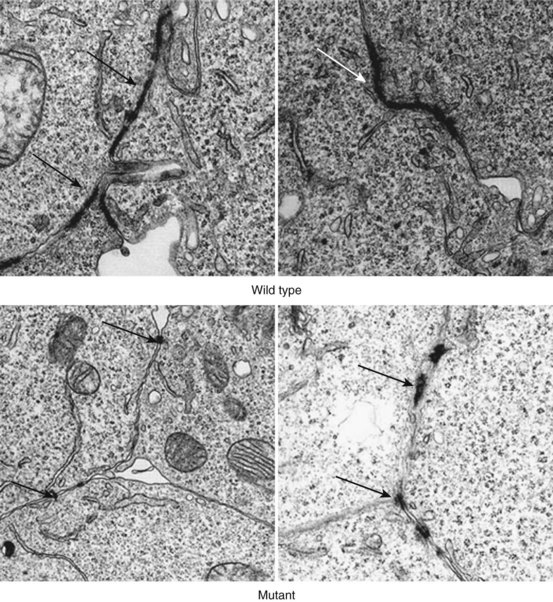
In 2002, the first mutation in the autosomal dominant form of ARVC was reported. This was found in the plakoglobin-binding domain of desmoplakin, and mutations in this gene have been reported in 6% to 16% of probands.14,39,40 Since then, heterozygous mutations have also been reported in plakoglobin, plakophilin-2 (PKP2), desmocollin-2 (DSC2), and desmoglein-2 (DSG2). PKP2 mutations are the most frequently found in patients with ARVC, with a reported prevalence between 27% and 43% of patents fulfilling diagnostic criteria.41 Over 50 individual mutations have been identified to date, but studies have not as yet revealed consistent genotype-phenotype correlations. The mechanism by which mutations result in disease is also unclear. It is suggested that mutations increase the susceptibility of the myocardium to the damaging effects of mechanical stress, thereby predisposing to cardiomyocyte detachment, death, and eventual replacement by fibro-fatty tissue.42 The predilection for the right ventricle has been explained by its thin wall and greater distensibility, but this is unlikely to be the sole explanation for disease in either ventricle.43 As desmosomal proteins interact with many other proteins, including key components of the cellular cytoskeleton and intermediate filaments, it is possible that ventricular dysfunction occurs as the result of reduced cytoskeletal integrity and impaired force transduction.37 Some desmosomal proteins (in particular plakoglobin) are also important signaling molecules that regulate the transcription of many other genes.44 Finally, a reduction in the number and size of gap junctions may result in an electrical coupling defect, thus increasing the propensity to arrhythmia, and could explain the occurrence of sudden death in patients without significant morphologic changes Figure 59-4.42
Nondesmosomal ARVC
Two nondesmosomal genes have been linked to ARVC: (1) the cardiac ryanodine receptor (hRYR2) and (2) transforming growth factor β3 (TGFβ3). Mutations in hRYR2 are more typically associated with effort-induced polymorphic VT and juvenile sudden cardiac death, without resting electrocardiogram (ECG) abnormalities or structural abnormalities, and mutations in this gene are no longer classified as a subtype of ARVC.45,46 Mutations in the intronic region of TGFβ3, which stimulates the production of extracellular matrix components, have been found to promoting myocardial fibrosis in vivo.47–49 Two ARVD1 kindred, however, lacked mutations in TGFβ3, which raises a question about its involvement in the pathogenesis of ARVC.50 Recently, a mutation in the transmembrane protein 43 (TNEM43) has been described (ARVD5). TNEM43 is predicted to be a cytoplasmic membrane protein without a cadherin domain, and the TNEM43 gene contains a response element for an adipogenic transcription factor.51
Phenocopies
When the left ventricle is involved and severe biventricular dysfunction is present, the differentiation from DCM can be difficult.52 Cardiac sarcoidosis can also mimic some of the clinical and structural abnormalities of ARVC but can be differentiated on endomyocardial biopsy.53 Right ventricular outflow tract VT can be difficult to distinguish from ARVC but generally has a good prognosis and can be successfully treated with catheter ablation therapy.54 Right ventricular myocarditis can mimic ARVC, and significant overlap between these two conditions exists, as inflammatory infiltrates are a feature of both.55,56 The histopathologic features of the heart in X-linked Emery-Dreifuss muscular dystrophy (EDMD) can mirror those of ARVC, but the two can be distinguished by family history and the presence of skeletal muscle involvement in EDMD.57
Symptoms and Physical Signs
Patients can present at any age with syncope, presyncope, or palpitations, In one case series, approximately one third of live probands gave a history of syncope, with over half reporting palpitations and presyncope.58 Other symptoms include chest pain or dyspnea often precipitated by exercise.59 In approximately 50% of cases, the first manifestation of ARVC is SCD, but in living patients, aborted SCD is the first manifestation in less than 10% of cases.28,60 Heart failure as a first presentation is probably uncommon; it accounted for less than 10% of cases in one series.58 Increasingly, patients are identified through family screening and are often asymptomatic. Cardiac examination in symptomatic and asymptomatic individuals is usually unremarkable.
Diagnosis
Because of the nonspecific nature of the clinical findings and the absence of a single diagnostic test, the diagnosis of ARVC is challenging. Recognition of these difficulties resulted in the formation of an International Task Force, which proposed standardized criteria for the diagnosis of ARVC in 1994.3 These criteria are based on the identification of structural, histologic, ECG, arrhythmic, and familial features, which are subdivided into major and minor criteria according to specificity. The presence of two major criteria, one major criterion plus two minor criteria, or four minor criteria from different categories is considered diagnostic (Table 59-2).3
Table 59-2 Task Force Criteria for the Diagnosis of Right Ventricular Dysplasia/Cardiomyopathy
| MAJOR CRITERIA | MINOR | |
|---|---|---|
| Global and/or regional dysfunction and structural alterations | Severe dilation and reduction of RV ejection fraction Localized RV aneurysms (akinetic or dyskinetic areas with diastolic bulging) Severe segmental dilation of the RV |
Mild global RV dilation and/or ejection fraction reduction with normal LV Mild segmental dilation of the RV Regional RV hypokinesia |
| Tissue characterization of wall | Fibro-fatty replacement of myocardium on endomyocardial biopsy | |
| Repolarization abnormalities | Inverted T waves in right precordial leads (V2 and V3) in people aged >12 years, in absence of RBBB | |
| Depolarization/conduction abnormalities | Epsilon waves or localized prolongation (>110 ms) of QRS complex in right precordial leads (V1–V3) | Late potentials (SAECG) |
| Arrhythmias | LBBB-type ventricular tachycardia (sustained and non-sustained) by ECG, Holter, or exercise testing Frequent ventricular extrasystoles (>1000/24 hours on Holter) |
|
| Family history | Familial disease confirmed at necropsy or surgery | Family history of premature sudden death (<35 years) from suspected RV dysplasia Familial history (clinical diagnosis based on present criteria) |
RV, Right ventricle; LV, left ventricle; RBBB, right bundle branch block; SAECG, signal-averaged electrocardiogram.
From McKenna WJ, Thiene G, Nava A, et al: Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy, Br Heart J 71:215–218, 1994.
The main goal of these guidelines was to establish a definitive diagnosis in probands, but tertiary referral center experience suggests that while the criteria are highly specific, they lack sensitivity for early disease.59 This led to the proposal of modified familial criteria for the diagnosis of ARVC in relatives of index cases. According to these criteria, the presence of a single disease feature (right precordial T-wave inversion, late potentials on signal averaged ECG, VT with LBBB morphology, or minor functional or structural abnormalities of the RV on imaging) in a relative of an index case should be considered diagnostic of ARVC (Table 59-3).58
Table 59-3 Familial Criteria for the Diagnosis of Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy
| Meet the familial criteria for a diagnosis of ARVC if ARVC is present in a first-degree relative plus one of the following: | |
| ECG | T-wave inversion in right precordial leads (V2 and V3) |
| SAECG | Late potentials seen on signal-averaged ECG |
| Arrhythmia | LBBB-type VT on ECG, Holter or during exercise testing Extrasystoles >200 over 24-hour period |
| Structural or functional abnormalities of the RV | Mild global RV dilation or EF reduction with normal LV Mild segmental dilation of the RV Regional RV hypokinesia |
ARVC, Arrhythmogenic right ventricular cardiomyopathy; ECG, electrocardiogram; SAECG, signal-averaged electrocardiogram; LBBB, left bundle branch block; VT, ventricular tachycardia; RV, right ventricle; EF, ejection fraction.
Modified from Hamid MS, Norman M, Quaraishi A, et al: Prospective evaluation of relatives for familial arrhythmogenic right ventricular dysplasia/cardiomyopathy reveals a need to broaden diagnostic criteria, J Am Coll Cardiol 40(8):1445–1450, 2002.
Electrocardiographic Features of Arrhythmogenic Right Ventricular Cardiomyopathy
A wide range of ECG changes are seen in ARVC.59 ECG changes also change with disease progression and may be dynamic.61 The most common abnormality is T-wave inversion in the right precordial leads (V1-V3) in the absence of right bundle branch block (RBBB), which has been reported in 54% to 85% of probands.62,63 This constitutes a minor diagnostic criterion for ARVC but is more common in cases with severe RV dysfunction and overt right ventricular dilation.64 Complete and incomplete RBBB are commonly observed but are also common among normal subjects and are therefore not included in the diagnostic criteria.59
The presence of a post-excitation epsilon wave, reflecting delayed right ventricular activation, is a major diagnostic criterion.3,65 This has been reported in around 30% of cases, but its sensitivity can be increased by using a highly amplified, modified recording technique (Figure 59-5).63,64 Another major ECG criterion is localized prolongation (>110 ms) of the QRS complex in the right precordial leads (V1-V3).3 In some series, this has been reported in 70% to 75% of patients.62,64 A more recently proposed criterion of prolonged (≥55 ms) S-wave upstroke in V1-V3 has been reported in 84% to 95% of ARVC patients without RBBB.63,64 This parameter has been correlated with disease severity, and in one study, it was an independent marker for VT induction at electrophysiology study.
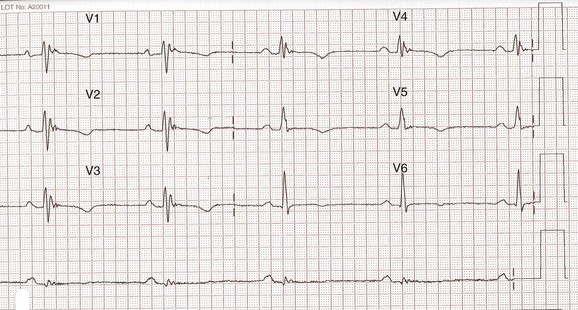
FIGURE 59-5 Highly amplified electrocardiogram showing epsilon wave in V1-V4.
(Courtesy Dr. Antonis Pantazis, Consultant Cardiologist, University College London Hospitals, UK.)
The signal-averaged ECG (SAECG) detects late potentials, a sign of delayed after-depolarizations. It is considered abnormal when two or more of the following parameters are met66:
An abnormal SAECG has been reported in 58% of probands and 45% of affected relatives.58,63
Arrhythmia
Ventricular arrhythmias with LBBB morphology are reported in 42% to 64% of patients on ECG, ambulatory ECG monitoring, or exercise testing.29,58,60 The presence of more than 1000 extrasystoles in a 24-hour period is a minor diagnostic criterion and has been reported in 22% to 42%.58,63 However, the cutoff 1000 extrasystoles is arbitrary; Hamid et al proposed a lower burden of ventricular ectopy (>200 per 24 hours) as a manifestation of the disease in relatives of affected individuals.58
Electroanatomic Mapping
Electroanatomic mapping combines electrophysiological and spatial information to allow real-time three-dimensional anatomic reconstruction of the cardiac chamber in sinus rhythm, with electrophysiological information color-coded and superimposed on the reconstruction (electroanatomic map). In a small study, Baulos et al demonstrated that patients with ARVC could be differentiated from controls by the presence of discrete areas of low-amplitude electrograms.67 In a larger study of 31 patients who fulfilled diagnostic criteria, three-dimensional electroanatomic voltage mapping of the RV increased accuracy in the diagnosis of ARVC.54 Mapping may help differentiate RVOT VT from early ARVC by detecting electroanatomic scars, which correlate with fibro-fatty replacement on endomyocardial biopsy.68
Echocardiography
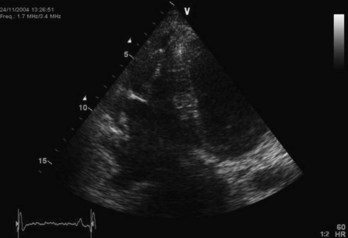
The greatest challenge when using echocardiography to assess the RV is the chamber’s complex three-dimensional structure. Considering the segmental nature of the disease, this also means that structure and function must be assessed using multiple (standardized) views. Initial reports described echocardiographic abnormalities in 100% of affected patients (64% mild, 30% moderate, and 6% severe), but contemporary studies in individuals with disease-causing genetic mutations have shown that echocardiography results are often normal.60 Major echocardiographic criteria for ARVC include severe dilation and reduced RV function, RV aneurysms, and severe segmental dilation of the RV (Figure 59-6). Minor criteria include mild RV dilation and regional RV hypokinesis. Other features of uncertain significance include increased echogenicity of the moderator band and RV apical hypertrabeculation.69
Magnetic Resonance Imaging
Three-dimensional imaging and the ability to characterize tissue give cardiac magnetic resonance (CMR) an important advantage over echocardiography in the assessment of ARVC.69,70 However, both these techniques have in common a high level of interobserver variability and subjectivity with regard to interpretation of wall thinning, localized functional abnormalities, and fatty replacement.71 Early studies had suggested that high intramyocardial T1 signals indicative of fat replacement were present in 75% of ARVC patients meeting Task Force criteria, but these studies failed to account for normal epicardial fat that is seen in older adults, in obese patients, and in conditions other than ARVC.72–75 Intramyocardial fibrosis can be assessed by late gadolinium enhancement (LGE) techniques but can be difficult to detect in the thin RV wall. The presence of LGE in the LV is more readily apparent and is common in patients fulfilling diagnostic criteria. It is not, however, included in the current diagnostic criteria (Figure 59-7).76
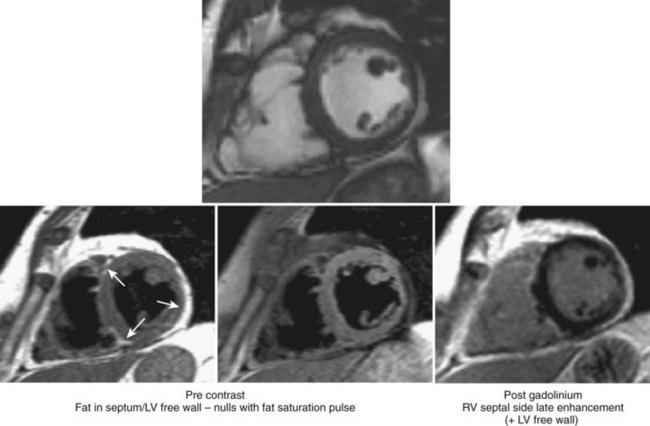
Endomyocardial Biopsy
A definitive or gold standard for the diagnosis of ARVC is histologic demonstration of transmural fibro-fatty replacement of the RV myocardium at autopsy or surgery.3 These alterations can also be demonstrated on RV endomyocardial biopsy (EMB), which, however, lacks sensitivity because of the segmental nature of the disease and because tissue is usually obtained from the septum, which is rarely involved in the disease process. Some studies show that biopsy guided by electroanatomic voltage mapping to areas of low voltage increases the diagnostic accuracy of biopsy.77
Quantitative diagnostic histomorphometric parameters have been proposed to increase the specificity of the histopathologic diagnosis of ARVC at biopsy. A fat percentage greater than 3%, fibrous tissue greater than 40%, and residual myocytes less than 45% has a sensitivity and specificity of 67% and 92%, respectively.78 Nither septal nor LV EMB has diagnostic value, as the dystrophic process in the LV is usually segmental and restricted to the subepicardial or midmural layers of the thick free wall, which are not accessible with the EMB approach.79 Endomyocardial biopsy with immunohistochemical staining has been proposed as a diagnostic test for ARVC to identify patients before the development of extensive fibro-fatty infiltration or structural abnormalities of the RV. Diffuse reduction in the plakoglobin signal has been observed not only in areas of the RV showing typical pathologic features of ARVC but also in the LV and interventricular septum, which otherwise appear structurally normal; this suggests that a conventional EMB of the right side of the septum may reliably show this diagnostic change. This has been shown to be a consistent feature of ARVC but not other forms of severe heart disease and suggests that a shift in plakoglobin from junctional pools to intracellular or intranuclear pools may play a role in disease pathogenesis through signaling changes that could contribute to myocyte injury. This diagnostic approach requires further validation in larger cohorts.80
Disease Management
The main treatment goal in patients with ARVC and their relatives is the prevention of sudden arrhythmic death and heart failure. The recommended workup for patients with suspected ARVC and for their family members includes annual review of personal and family histories, 12-lead ECG, SAECG, exercise testing, ambulatory ECG monitoring, and echocardiography. CMR can be performed to aid in the assessment of cardiac morphology; invasive, intracardiac electrophysiological studies should be reserved for patients with symptomatic VT, ventricular fibrillation (VF), or syncope with negative noninvasive investigations.59,70
Prevention of Sudden Cardiac Death
The overall mortality of ARVC has been estimated in one study at 18.5% over 8.1 ± 7.8 years, with an annual mortality rate of 2.3%.81 In another study, the incidence of sudden death declined after the fourth decade of life. Evidence from prospective controlled trials assessing clinical markers that can predict SCD is lacking. Retrospective analyses of clinical and pathologic series have identified a number of possible predictors of adverse outcome in probands. These include an onset of symptoms at an early age, involvement in competitive sports, a significant family history of SCD, severe RV dilation, LV involvement, syncope, episodes of complex ventricular arrhythmias or VT, and increased QRS dispersion on 12-lead ECG.30,59,82,83
The AHA/ACC/ESC 2006 guidelines for the management of patients with ventricular arrhythmias and the prevention of SCD state that implantable cardioverter-defibrillator (ICD) implantation is recommended for the prevention of SCD in patients with ARVC with documented sustained VT or VF, who are receiving optimal medical therapy and have reasonable expectation of survival with a good functional status for more than 1 year (class I recommendation). It can also be effective in the prevention of SCD in patients with extensive disease, including those with LV involvement, one or more affected family members with SCD, or undiagnosed syncope when VT/VF has not been excluded as the cause of syncope (class IIa recommendation).84
Management of Arrhythmia
Evidence for the efficacy of antiarrhythmic drugs in ARVC is largely anecdotal, and the impact of medical therapy on mortality is unknown.84–86 Pharmacologic treatment is the first-choice therapy for patients with well tolerated, non–life-threatening ventricular arrhythmias such as frequent ventricular extrasystoles. As arrhythmias are often precipitated by an increase in heart rate, a reasonable first-line agent is a β-blocker; a number of studies have suggested that sotalol may be the most effective.85,87
Catheter ablation is not routinely carried out in ARVC patients unless drug-refractory, incessant ventricular arrhythmia is present or VT recurs frequently after ICD implantation. This is caused by the low efficacy of the therapy and the recurrence of further arrhythmia with disease progression.88 In one study of 32 patients, acute success was achieved in 75%, with the remainder having only partial acute success. After a successful primary procedure, 21% experienced recurrent VT within the first 3 months, and 63% of those with only partial success from the primary procedure had recurrent arrhythmia. None of the patients, however, experienced syncope or sudden death during a mean follow-up of 28 months.89
Management of Heart Failure
Studies have reported an incidence of heart failure in up to 20% of patients.74,86 In one study, death from heart failure occurred twice as frequently as from SCD, whereas in a large American cohort, only 6% of patients developed right-sided failure, and of these, only one progressed to biventricular failure and death while awaiting cardiac transplantation.81,90 Standard therapy for heart failure is indicated in patients in whom ARVC has progressed to severe heart failure or biventricular systolic dysfunction, including diuretics, angiotensin-converting enzyme inhibitors, and β-blockers. Anticoagulation should be considered in the presence of atrial fibrillation, marked ventricular dilation, or ventricular aneurysms. In patients in whom congestive heart failure is refractory, cardiac transplantation should be considered.59,70
Genetic Testing
The discovery of gene mutations involved in the pathogenesis of ARVC has introduced the possibility of molecular genetic diagnosis in clinical care. The aim of molecular genetic screening is to identify family members at risk. As the first presentation of ARVC can be with SCD, genetic screening has the potential to identify at-risk individuals at an early stage and thus save lives.16 However, the value of genetic testing in predicting outcomes is limited by the fact that many sequence variants are private (i.e., confined to single families), which makes it difficult to determine the pathogenicity of these variants. Even when mutations have been described previously, either an absence of data on genotype-phenotype relations or variations in disease expression is an issue.16 Finally, more than 10% of patients are homozygous, double-heterozygotes, or compound-heterozygotes, which underscores the need to screen every coding region of all known disease-causing genes, even after the identification of a first mutation.50
Advice on Lifestyle Changes
Patients in whom a diagnosis of ARVC is confirmed or strongly suspected should be discouraged from participating in competitive sports and endurance training. This advice is based on animal models and on reports suggesting that individuals who perform intensive, regular sports activities have more symptoms at an earlier age and a greater risk of SCD.91,92
Key References
Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360(11):1075-1084.
Corrado D, Basso C, Leoni L, et al. Three dimensional electroanatomical mapping and histological evaluation of myocardial substrate in right ventricular outflow tract tachycardia. J Am Coll Cardiol. 2005;51:731-739.
Corrado D, Basso C, Thiene G. Arrhythmogenic right ventricular cardiomyopathy: Diagnosis, prognosis and treatment. Heart. 2000;83:588-595.
Green KJ, Gaudry CA. Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol. 2000;1:208-216.
Hamid MS, Norman M, Quaraishi A, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular dysplasia/cardiomyopathy reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002;40(8):1445-1450.
Kaplan SR, Gard JJ, Protonotarios N, et al. Remodelling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm. 2004;1:3-11.
Marcus TF, Fontaine G, Guiraudon G, et al. Right ventricular dysplasia: A report of 24 adult cases. Circulation. 1982;65:384-398.
McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br Heart J. 1994;71:215-218.
Nasir K, Bomma C, Tandri H, et al. Electrocardiographic features of arrhythmogenic right ventricular dysplasia/cardiomyopathy according to disease severity. A need to broaden diagnostic criteria. Circulation. 2004;110(12):1527-1534.
Rampazzo A Nava A, Danieli GA, et al. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum Mol Genet. 1994;3(6):959-962.
Sen-Chowdhry S, Lowe MD, Sporton SC, McKenna WJ. Arrhythmogenic right ventricular cardiomyopathy: Clinical presentation, diagnosis and management. Am J Med. 2004;117:685-695.
Sen-Chowdhry S, Prasad SK, Syrris P, et al. Cardiac magnetic resonance in arrhythmogenic right ventricular cardiomyopathy revisited. J Am Coll Cardiol. 2006;48:2132-2140.
Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50(19):1813-1821.
Sen-Chowdhry S, Syris P, Ward D, et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into pattern if disease expression. Circulation. 2007;115:1710-1720.
Yoerger DM, Marcus F, Sherrill D, et al. Echocardiographic findings in patients meeting Task Force criteria of arrhythmogenic right ventricular dysplasia. J Am Coll Cardiol. 2005;45:860-865.
1 Fontaine G, Guiraudon G, Frank R, et al. Stimulation studies and epicardial mapping in ventricular tachycardia: A study of mechanism and selection for surgery. In: Kulbertus H, editor. Reentrant arrhythmias, mechanism and treatment. Lancaster: MTP Publishing, 1977.
2 Marcus TF, Fontaine G, Guiraudon G, et al. Right ventricular dysplasia: A report of 24 adult cases. Circulation. 1982;65:384-398.
3 McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br Heart J. 1994;71:215-218.
4 Richardson P, McKenna WJ, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. 1996;93:841-842.
5 Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies. Circulation. 2006;113:1807-1816.
6 Elliott P, Anderson B, Arbustini E, et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working group on myocardial and pericardial diseases. Eur Heart J. 2008;29:270-276.
7 Rampazzo A Nava A, Danieli GA, et al. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum Mol Genet. 1994;3(6):959-962.
8 Rampazzo A, Nava A, Erne P, et al. A new locus for arrhythmogenic right ventricular cardiomyopathy (ARVD2) maps to chromosome 1q42-q43. Hum Mol Genet. 1995;4:2151-2154.
9 Severini GM, Krajinovic M, Pinamonti B, et al. A new locus for arrhythmogenic right ventricular dysplasia on the long arm of chromosome 14. Genomics. 1996;31:193-200.
10 Rampazzo A, Nava A, Miorin M, et al. ARVD4, a new locus for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 2 long arm. Genomics. 1997;45:259-263.
11 Ahmad F, Li D, Karibe A, et al. Localization of a gene responsible for arrhythmogenic right ventricular dysplasia to chromosome 3p33. Circulation. 1998;98:2791-2795.
12 Li D, Ahmad F, Gardner MJ, et al. The locus of a novel gene responsible for arrhythmogenic right ventricular dysplasia characterized by early onset and high penetrance maps to chromosome 10p12-p14. Am J Hum Genet. 2000;66:148-156.
13 Melberg A, Oldfors A, Blomstrom-Lundqvist C, et al. Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy linked to chromosome 10q. Ann Neurol. 1999;46(5):684-692.
14 Rampazzo A, Nava A, Malacrida S, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71:1200-1206.
15 McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 2000;355:2119-2124.
16 Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Clinical impact of molecular genetic studies. Circulation. 2006;113:1634-1637.
17 Corrado D, Thiene G, Nava A, Rossi L, Pennelli N. Sudden death in young competitive athletes: Clinicopathologic correlations in 22 cases. Am J Med. 1990;89:588-596.
18 Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med. 1998;339:364-369.
19 Thiene G, Nava A, Corrado D, et al. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129-133.
20 Basso C, Thiene G, Corrado D, et al. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy or myocarditis? Circulation. 1996;94:983-991.
21 Bowles NE, Ni J, Marcus F, Towbin JA. The detection of cardiotropic viruses in the myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2002;39:892-895.
22 Mallat Z, Tedgui A, Fontaliran F, et al. Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med. 1996;335:1190-1196.
23 Valente M, Calabrese F, Thiene G, et al. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am J Pathol. 1998;152(2):479-484.
24 Fontaine G, Fontaliran F, Lascault G, et al. Congenital and acquired right ventricular dysplasia [translation]. Arch Mal Coeur Vaiss. 1990;83(7):915-922.
25 Pinamonti B, Miani D, Sinagra G, et al. Familial right ventricular dysplasia with biventricular involvement and inflammatory infiltration. Heart. 1996;76:66-69.
26 Chimenti C, Pieroni M, Maseri A, Frustaci A. Histologic findings in patients with clinical and instrumental diagnosis of sporadic arrhythmogenic right ventricular dysplasia. J Am Coll Cardiol. 2004;43:2305-2313.
27 Grumbach IM, Heim A, Vonhof S, et al. Coxsackievirus in myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Cardiology. 1998;89(4):241-245.
28 Corrado D, Basso C, Thiene G, et al. Spectrum if clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J Am Coll Cardiol. 1997;30(6):1512-1520.
29 Sen-Chowdhry S, Syris P, Ward D, et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into pattern of disease expression. Circulation. 2007;115:1710-1720.
30 Protonotarios N, Tsatsopoulou A, Anastasakis A, et al. Genotype-phenotype assessment in autosomal recessive arrhythmogenic right ventricular cardiomyopathy (Naxos disease) caused by a deletion in plakoglobin. J Am Coll Cardiol. 2001;38:1477-1484.
31 Norgett EE, Hatsell SJ, Carvajal-Huerta L, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761-2766.
32 Alcalai R, Metzger S, Rosenheck S, et al. A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder and woolly hair. J Am Coll Cardiol. 2003;42(2):319-327.
33 Huber O. Structure and function of desmosomal proteins and their role in development and disease. Cell Mol Life Sci. 2003;60:1872-1890.
34 Garrod DR, Merritt AJ, Nie Z. Desmosomal cadherins. Curr Opin Cell Biol. 2002;14:537-545.
35 Getsios S, Huen AC, Green KJ. Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol. 2004;5:271-281.
36 Andrä K, Lassmann H, Bittner R, et al. Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle and heart cytoarchitecture. Genes Dev. 1997;11:3143-3156.
37 Green KJ, Gaudry CA. Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol. 2000;1:208-216.
38 Charpentier E, Lavker R, Avquista E, Cowin P. Plakoglobin suppresses epithelial proliferation and hair growth in vivo. J Cell Biol. 2000;149:503-518.
39 Bauce B, Basso C, Rampazzo A, et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J. 2005;26:1666-1675.
40 Asimaki A, Syrris P, Wichter T, et al. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2007;81:964-973.
41 van Tintelen JP, Hofstra RMW, Wiesfeld ACP, et al. Molecular genetics of arrhythmogenic right ventricular cardiomyopathy: Emerging horizon? Curr Opin Cardiol. 2007;22:185-192.
42 Kaplan SR, Gard JJ, Protonotarios N, et al. Remodelling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm. 2004;1:3-11.
43 Basso C, Czarnowska E, Della Barbera M, et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: An electron microscopy investigation on endomyocardial biopsies. Eur Heart J. 2006;27:1847-1854.
44 Ko KS, Aroroa PD, McCulloch CA. Cadherins mediate intercellular mechanical signalling in fibroblasts by activation of stretch-sensitive calcium-permeable channels. J Biol Chem. 2001;276(38):35967-35977.
45 Nava A, Canciani B, Daliento L, et al. Juvenile sudden death and effort ventricular tachycardia in a family with right ventricular cardiomyopathy. Int J Cardiol. 1988;21:111-123.
46 Bauce B, Nava A, Rampazzo A, et al. Familial effort polymorphic ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy map to chromosome 1q42-43. Am J Cardiol. 2000;85:573-579.
47 Beffagna G, Occhi G, Nava A, et al. Regulatory mutations in transforming growth factor-β3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366-373.
48 Nattel S, Schott JJ. Arrhythmogenic right ventricular dysplasia type 1 and mutations in transforming growth factor β3 gene regulatory regions: A breakthrough? Cardiovasc Res. 2005;65:302-304.
49 Weber KT, Brilla CG, Campbell SE, Zhou G, Matsubara L, Guarda E. Pathologic hypertrophy with fibrosis: The structural basis for myocardial failure. Blood Press. 1992;1:75-85.
50 Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50(19):1813-1821.
51 Merner ND, Hodgkinson KA, Haywood AFM, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809-821.
52 Nemec J, Edwards BS, Osborn MJ, Edwards WD. Arrhythmogenic right ventricular dysplasia masquerading as dilated cardiomyopathy. Am J Cardiol. 1999;84:237-239.
53 Ott P, Marcus FI, Sobonya RE, et al. Cardiac sarcoidosis masquerading as right ventricular dysplasia. PACE. 2003;26:1498-1503.
54 Corrado D, Basso C, Leoni L, et al. Three dimensional electroanatomic voltage mapping increases the accuracy of diagnosing arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2005;111:3042-3050.
55 Pieroni M, Dello Russo A, Marzo F, et al. High prevalence of myocarditis mimicking arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2009;53:681-689.
56 Calabrese F, Basso C, Carturan E, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: Is there a role for viruses? Cardiovasc Path. 2006;15:11-17.
57 Fishbein MC, Siegel RJ, Thompson CE, Hopkins LC. Sudden death of a carrier of X-linked Emery-Dreifuss muscular dystrophy. Ann Intern Med. 1993;119:900-905.
58 Hamid MS, Norman M, Quaraishi A, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular dysplasia/cardiomyopathy reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002;40(8):1445-1450.
59 Sen-Chowdhry S, Lowe MD, Sporton SC, McKenna WJ. Arrhythmogenic right ventricular cardiomyopathy: Clinical presentation, diagnosis and management. Am J Med. 2004;117:685-695.
60 Nava A, Bauce B, Basso C, et al. Clinical profile and long-term follow up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36(7):2226-2233.
61 Quarta G, Ward D, Esteban MTT, et al. Dynamic electrocardiographic changes in patients with arrhythmogenic right ventricular cardiomyopathy. Heart. 2010;96(7):516-522.
62 Peters S, Trümmel M. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy: Value of standard electrocardiogram revisited. Ann Non-invas Electrocard. 2003;8(3):238-245.
63 Nasir K, Bomma C, Tandri H, et al. Electrocardiographic features of arrhythmogenic right ventricular dysplasia/cardiomyopathy according to disease severity. A need to broaden diagnostic criteria. Circulation. 2004;110:1527-1534.
64 Peters S, Trümmel M, Koehler B, Westermann KU. The value of different electrocardiographic depolarization criteria in the diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Electrocard. 2007;40:34-37.
65 Fontaine G, Umemura J, di Donna P, et al. Duration of QRS complexes in arrhythmogenic right ventricular dysplasia; a new non-invasive diagnostic marker. Ann Cardiol Angeiol (Paris). 1993;42:399-405.
66 Cain ME, Anderson JL, Arnsdorf MF. Signal-averaged electrocardiography. ACC Expert Consensus Document. J Am Coll Cardiol. 1996;27(1):238-249.
67 Boulos M, Lashevsky I, Reisnor S, Gepstein L. Electroanatomic mapping of arrhythmogenic right ventricular dysplasia. J Am Coll Cardiol. 2001;38(7):2020-2027.
68 Corrado D, Basso C, Leoni L, et al. Three dimensional electroanatomical mapping and histological evaluation of myocardial substrate in right ventricular outflow tract tachycardia. J Am Coll Cardiol. 2005;51:731-739.
69 Yoerger DM, Marcus F, Sherrill D, et al. Echocardiographic findings in patients meeting Task Force criteria of arrhythmogenic right ventricular dysplasia. J Am Coll Cardiol. 2005;45:860-865.
70 Maksimović R, Ekinci O, Reiner C, et al. The value of magnetic resonance imaging for the diagnosis of arrhythmogenic right ventricular cardiomyopathy. Eur Radiol. 2006;16:560-568.
71 Bluemke DA, Krupinski EA, Ovitt T, et al. MR imaging of arrhythmogenic right ventricular cardiomyopathy: Morphological findings and interobserver reliability. Cardiology. 2003;99:153-162.
72 Tandri H, Calkins H, Nasir K, et al. Magnetic resonance imaging findings in patients meeting Task Force criteria for arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol. 2003;14:476-482.
73 Kayser HWM, de Roos A, Schalij MJ, et al. Usefulness of magnetic resonance imaging in diagnosis of arrhythmogenic right ventricular dysplasia and agreement with electrocardiographic criteria. Am J Cardiol. 2003;91:365-367.
74 Blomström-Lunqvist C, Sabel KG, Olsson SB. A long term follow up of 15 patients with arrhythmogenic right ventricular dysplasia. Br Heart J. 1987;58:477-488.
75 Caruso G, Frassanito F, Serio G, Panella A. Is adipose tissue a normal component of the myocardium? Eur Heart J. 1989;10(Suppl D):89-92.
76 Sen-Chowdhry S, Prasad SK, Syrris P, et al. Cardiac magnetic resonance in arrhythmogenic right ventricular cardiomyopathy revisited. J Am Coll Cardiol. 2006;48:2132-2140.
77 Avella A, d’Amati G, Pappalardo A, et al. Diagnostic value of endomyocardial biopsy guided by electroanatomic voltage mapping in arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Cardiovasc Electrophysiol. 2008;19:1127-1134.
78 Angelini A, Basso C, Nava A, Thiene G. Endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy. Am Heart J. 1996;132:203-206.
79 Basso C, Ronco F, Marcus F, et al. Quantitative assessment of endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy/dysplasia: An in vitro validation of diagnostic criteria. Eur Heart J. 2008;29:2760-2771.
80 Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360(11):1075-1084.
81 Hulot JS, Jouven X, Empana JP, et al. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2004;110:1879-1884.
82 Turrini P, Corrado D, Basso C, et al. Dispersion of ventricular depolarization-repolarization: A non-invasive marker for risk stratification in arrhythmogenic right ventricular cardiomyopathy. Circulation. 2001;103:3075-3080.
83 Peters S, Peters H, Thierfelder L. Risk stratification of sudden cardiac death and malignant ventricular arrhythmias in right ventricular dysplasia-cardiomyopathy. Int J Cardiol. 1999;71:243-250.
84 ACC/AHA/ESC. 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Europace. 2006;8:746-837.
85 Wichter T, Borggrefe M, Haverkamp W, et al. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease: Results in patients with inducible and noninducible ventricular tachycardia. Circulation. 1992;86:29-37.
86 Marcus FI, Fontaine GH, Frank R, et al. Long-term follow-up in patients with arrhythmogenic right ventricular disease. Eur Heart J. 1989;10(Suppl D):68-73.
87 Leclercq JF, Potenza S, Maison-Blanche P, et al. Determinants of spontaneous occurrence of sustained monomorphic ventricular tachycardia in right ventricular dysplasia. J Am Coll Cardiol. 1996;28:720-724.
88 Corrado D, Basso C, Thiene G. Arrhythmogenic right ventricular cardiomyopathy: Diagnosis, prognosis and treatment. Heart. 2000;83:588-595.
89 Yao Y, Zhang S, Sheng D, et al. Radiofrequency ablation of the ventricular tachycardia with arrhythmogenic right ventricular cardiomyopathy using non-contact mapping. PACE. 2000;(30):526-533.
90 Dalal D, Nasir K, Bomma C, et al. Arrhythmogenic right ventricular dysplasia: A United States experience. Circulation. 2005;112:3823-3832.
91 Kirchhof P, Fabritz L, Zwiener M, et al. Age and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation. 2006;114:1799-1806.
92 Daubert C, Vauthier M, Carré F, et al. Influence of exercise and sport activity on functional symptoms and ventricular arrhythmias in arrhythmogenic right ventricular disease [abstract]. J Am Coll Cardiol. 1994;23(Suppl):34A.