Chapter 57 Arrhythmias in Coronary Artery Disease
Other than hypertensive heart disease, coronary artery disease (CAD) is the most common cause of structural heart disease in the United States. Most patients who experience life-threatening arrhythmias have underlying structural heart disease, and the majority of patients presenting with sustained ventricular arrhythmias have underlying CAD.1 Patients with coronary disease may also have various less severe forms of arrhythmias, including bradyarrhythmias and supraventricular and ventricular arrhythmias.
Arrhythmias Associated with Acute Ischemia and Myocardial Infarction
Acute myocardial ischemia (AMI) usually results from partial or total coronary occlusion with a subsequent imbalance between myocardial oxygen supply and demand. Acute coronary syndromes (ACS) include myocardial infarction (MI) (ST-segment elevation and depression, Q wave and non–Q wave) as well as unstable angina.2,3 ACS may result in arrhythmias during acute coronary occlusion, reperfusion, myocardial infarct evolution, or the healing phase after infarction. Ischemia-induced changes in ions, metabolites, ion channels, gap junctions, and cellular and tissue architecture result in profound changes in the electrophysiological properties of the affected myocardium, which interact with modulating factors (autonomic nervous system, electrolytes, ischemic preconditioning, changes in heart rate) and the presence of concurrent structural heart disease (scar, hypertrophy, depressed ejection fraction), leading to generation of cardiac arrhythmia.
Mechanisms
Acute ischemia following coronary artery occlusion results in local tissue hypoxia and a loss of function of the adenosine triphosphate (ATP)-dependent sodium-potassium (Na+-K+) pump. Cellular membrane permeability is altered, pH falls, and a net K+ leakage from the myocyte and a rise in extracellular K+ occur. The normal cardiac resting membrane potential decreases from −80 mV to around −50 mV.4,5 The action potential (AP) amplitude falls, and maximal upstroke velocity (dV/dt max) decreases.6 Within the first 2 minutes, the fall in resting membrane potential results in an increase in conduction velocity.7 As the AP upstroke velocity falls over the next 10 minutes, conduction velocities decrease by up to 50%.8 Importantly, the effects of ischemia on the electrophysiological properties of myocardial cells are heterogeneous. AP duration and upstroke velocity are more reduced in subepicardial cells than in subendocardial cells.9 Within the central zone of ischemia, refractory periods are prolonged, and conduction velocity is decreased.10 In the surrounding nonischemic tissue, the refractory period may become shortened, and the conduction velocity may increase—possibly as a result of local catecholamines, circulating catecholamines, or both. Intermediate or mixed changes occur in the border zone between ischemic and nonischemic tissues, which results in a marked heterogeneity of electrophysiological properties. Changes in the degree of cell-to-cell coupling and tissue architecture also cause slowing and then failure of electrical propagation. The extracellular compartment shrinks, and extracellular resistance increases.11 Gap junction disruption results in cellular uncoupling. Heterogeneities in the changes in intracellular and extracellular resistance are particularly pronounced in the border zone.
Autonomic nervous system changes occur during MI. While infarcted areas show sympathetic denervation, surrounding and distant areas develop hyperinnervation.12 In addition, subepicardial sympathetic fibers traveling from base to apex may be damaged by transmural infarctions, which results in denervation of areas located more apically; these areas may show denervation hypersensitivity to catecholamines.13–18 Autonomic and neurohumoral influences can thus modify the electrophysiological properties of the substrate and can also result in arrhythmic triggers (premature ventricular contractions) via enhanced automaticity or after-depolarizations.
If the acute ischemia resolves, further myocardial injury occurs during reperfusion; this includes vascular damage, myocardial stunning, and further necrosis, mediated by intracellular calcium overload and oxygen free radicals. Calcium-dependent arrhythmias resulting from triggered activity, such as delayed after-depolarizations, may develop. Premature ventricular contractions (PVCs) and accelerated idioventricular rhythms are the most common rhythms associated with this phase, and they do not portend an adverse prognosis.19,20
Recurrent ischemia may result in alterations of the cellular metabolism and local biochemical environment and thus modulate arrhythmogenicity. Short, repetitive coronary occlusions have been shown experimentally to result in decreased incidence of ventricular fibrillation (VF) during reperfusion—a phenomenon termed ischemic preconditioning.21
Electrolyte abnormalities, particularly hypokalemia and hypomagnesemia, can alter myocardial electrophysiological properties and can also generate arrhythmia triggers. Circulating fatty acid levels have also been associated with an increased risk of sudden death as a manifestation of coronary disease.22,23
Genetic factors may play a significant role. In two retrospective studies, MI patients who experienced VF or sudden death were more likely to have a family history of sudden cardiac death (SCD).24,25 Candidate genes include genes that predispose to the development of the underlying substrate (coronary disease as well as acute plaque rupture, thrombosis, or both) and genes that directly influence the electrical properties of the myocardium and its vulnerability to ventricular fibrillation. A number of monogenic arrhythmic disorders have been well characterized (long QT syndromes, short QT syndromes, Brugada syndrome, catecholaminergic polymorphic VT syndromes).26 In addition, several genetic variants (polymorphisms) have been associated with sudden death or arrhythmia in general populations (SCN5A gene, β2-adrenergic receptor gene27,28). These genetic abnormalities and variants, and others still undiscovered, clinically apparent or subclinical, are likely to influence an individual’s susceptibility to develop arrhythmias during both acute and chronic coronary ischemia.
Stages of Ventricular Arrhythmogenesis Following Coronary Artery Occlusion
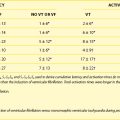
Two distinct phases of arrhythmogenesis (Table 57-1) occur during the initial 30 minutes (“acute phase”) of ischemia after experimental coronary artery ligation.8 Phase 1a arrhythmias occur 2 to 10 minutes following coronary artery occlusion (peak 5 to 6 minutes) and are caused by re-entry within the ischemic myocardium resulting from the inhomogeneity of refractory periods in normal and ischemic tissue. Mapping studies have revealed the presence of low-amplitude fractionated electrograms.7,8
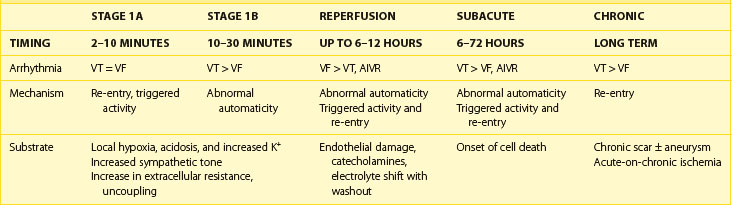
Phase 1b arrhythmias occur 10 to 30 minutes following coronary artery occlusion (peak 15 to 20 minutes). The precise mechanism of type 1b ventricular arrhythmias is unclear. By this stage, the inhomogeneities in subepicardial refractoriness and conduction have improved to near-normal values.8 Because of the important role of catecholamines in arrhythmogenesis, it has been postulated that abnormal automaticity is the underlying mechanism.29 Myocardial stretch mechanisms have also been implicated in the generation of abnormal automaticity.30 Studies in canine hearts during the first 30 minutes following coronary artery occlusion have suggested that up to 60% of ventricular tachycardias (VT) are focal in origin, arising from Purkinje fibers.31 Generally, during these two phases, spanning the first 30 minutes following occlusion, no permanent structural damage occurs. On reperfusion, ischemic cells survive and generally recover function. However, toward the end of phase 1b, changes in the internal axial resistance of cardiac tissue are first noted, indicating the onset of irreversible cellular and gap junction damage.32
The subacute or delayed phase occurs 6 to 72 hours following coronary artery occlusion (peak 12 to 24 hours).33 It coincides with the onset of cell death; reperfusion at this stage does not reduce the amount of cell damage. Although substantial myocardial cell death occurs in the infarcted region, subendocardial Purkinje fibers survive with altered electrophysiological properties predisposing to arrhythmia generation.34 A reduced resting membrane potential and spontaneous membrane depolarizations lead to abnormal automaticity. Delayed after-depolarizations resulting in triggered activity have also been demonstrated.35 In addition, heterogeneity of conduction and refractoriness at the border zone, which is the interface between the dead myocardium and the still-viable myocardium, may lead to re-entrant arrhythmias.
Reperfusion Arrhythmias
Reperfusion arrhythmias are more common after short ischemic episodes than after long ischemic periods.36 In the canine model, reperfusion arrhythmias have been shown to occur in two stages. Immediately following restoration of perfusion after coronary artery occlusion, VF may occur due to multiple wavelet re-entry. This occurs as a result of a rapid but inhomogeneous return of APs to previously unexcitable cells within the ischemic zone and a shortening of refractory periods in the border zone brought about by the washout of K+ and metabolites from the extracellular space.37 In addition, premature depolarizations may be induced by triggered activity. Although overall electrical function can return to normal at this stage, gap junction injury may persist with a corresponding inhomogeneous delay in conduction properties.
Accelerated idioventricular rhythms are commonly seen following reperfusion in the canine model. This arrhythmia may be due to the increased adrenergic stimulation of Purkinje fibers near the ischemic region causing enhanced automaticity or triggered activity.38 As accumulation of catecholamines is required, these arrhythmias typically occur after 20 to 30 minutes of occlusion. Compared with the canine model, the incidence of early reperfusion arrhythmias in the human population is significantly lower. This probably reflects the longer occlusion times and less rapid or incomplete reperfusion typically seen in patients presenting with AMI.
Clinical Characteristics of Ventricular Arrhythmias in Acute Coronary Syndromes
Ventricular arrhythmias are present in 64.1% of patients following acute ST-segment elevation myocardial infarction (STEMI).20 More than 10 PVCs per hour may be seen in 19.7% and nonsustained VT (NSVT) in 6.8% of patients. Sustained VT or VF occurs in 10.2% of admissions, with an incidence of 1.9% within the first 24 hours and 3.7% to 4.4% in the first 48 hours.39–42 Older age, systemic hypertension, previous MI, Killip class, anterior infarct, and depressed ejection fraction are associated with a higher risk of sustained VT and VF.39 Ventricular arrhythmias are more common in patients with signs of extensive left ventricular damage. However, early mortality is increased in patients who develop VT and fibrillation, even in the absence of congestive heart failure and hypotension. The incidence of VF in AMI seems to have declined over the last 20 years, whereas the incidence of VT has not changed much.43
Ventricular arrhythmias also occur in the setting of unstable angina (UA) or non–ST-elevation MI (NSTEMI), both during episodes of pain and when patients are pain free.44 In a pooled analysis of over 25,000 patients with UA or NSTEMI from four trials, the incidence of sustained VT or VF was 2.1%.45
Premature Ventricular Contractions
PVCs are seen in the majority of cases of acute MI. Early PVCs (within the first 48 hours) do not appear to affect the prognosis, but frequent or complex PVCs occurring beyond 48 hours after AMI may be associated with increased arrhythmic risk. In the human heart, R-on-T PVCs are rarely observed, accounting for only 1.8% of PVCs during the first 24 hours of admission, and most PVCs do not trigger severe ventricular tachyarrhythmias.46–48 However, in a canine model, 24% of PVCs occurring between 12 and 30 minutes (phase 1b) resulted in R-on-T and were responsible for the initiation of 34% of spontaneous episodes of VT and fibrillation.
Several studies from the prethrombolytic era have suggested that frequent PVCs (>10 PVCs per hour), complex PVCs (ventricular bigeminy, couplets, or multiform ventricular premature beats), or both are a risk factor independent of the degree of myocardial damage and left ventricular systolic dysfunction,20,49 but in another trial, PVC frequency had no independent predictive value in multivariate analysis.50 Antiarrhythmic suppressive therapy (lidocaine) has not been shown to improve outcomes, and class Ic antiarrhythmics may increase mortality. Electrophysiology study for risk stratification is currently not recommended for either early or late post-MI PVCs.
Accelerated Idioventricular Rhythm
Accelerated idioventricular rhythm (AIVR) is commonly witnessed in the first 12 hours after admission for AMI. Although more common in patients with successful reperfusion therapy, it is not a specific marker, with 63% of patients with occluded arteries still demonstrating the arrhythmia.51 The presence of AIVR does not affect the prognosis.
Nonsustained Ventricular Tachycardia
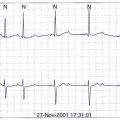
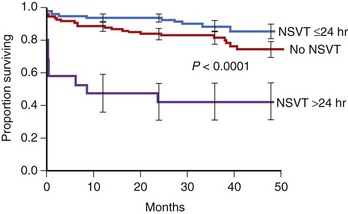
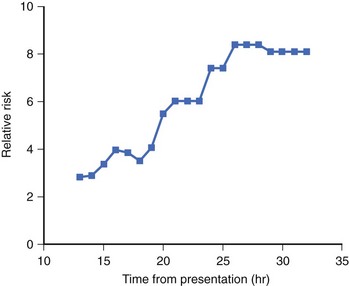
The presence of NSVT identifies patients at risk of in-hospital cardiac arrest. NSVT that occurs within the first 2 to 3 hours does not carry an adverse prognosis, whereas NSVT that occurs beyond several hours after admission does, particularly in patients with prior MI. NSVT in the setting of AMI occurs in 1% to 7% and possibly in as many as 75% of patients (Figure 57-1).52 NSVT occurring 24 hours after AMI carries a worse prognosis than NSVT occurring within the first 24 hours following AMI (Figure 57-2). This is contrary to the commonly held belief that arrhythmias occurring within the first 48 hours following MI do not carry an adverse long-term prognosis. NSVT in the setting of healing MI (7 to 10 days following MI) is also associated with a poorer prognosis. Aside from β-blockers, antiarrhythmic therapy is not currently recommended for either early or late post-MI asymptomatic NSVT. Electrophysiology testing is not currently recommended for risk stratification of NSVT in the first several weeks after AMI but is considered “reasonable” for risk stratification in patients with remote MI, NSVT, and left ventricular ejection fraction (LVEF) 40% or less.53
Ventricular Tachycardia, Polymorphic Ventricular Tachycardia, and Ventricular Fibrillation
The incidence of documented “early” sustained monomorphic VT (SMVT) within the first 48 hours of AMI is in the range of 2% to 3% in STEMI and less than 0.9% in NSTEMI.39,45,54 It may indicate extensive myocardial damage and serve as an independent predictor of mortality.54,55 As discussed above, arrhythmia mechanisms undergo dynamic changes in the early minutes and hours after onset of ischemia and may involve both re-entry (within ischemic areas of slowed conduction and increased refractoriness) or non–re-entrant mechanisms (triggered activity or increased automaticity). SMVT, however, implies stability of the ventricular depolarization pattern, which is most readily provided by a stable re-entry circuit. Thus, it is likely that SMVT, even in the early hours following MI, occurs in the presence of an already established permanent substrate (developing necrosis or pre-existing scar). Electrolyte abnormalities or ischemia that can cause the events that initiate re-entry (PVCs, NSVT) should be corrected; however, SMVT should be addressed as it would be even in the absence of these factors. From the currently available data, it is unclear that SMVT can be lumped together with other early post-MI arrhythmias in terms of its effect on the long-term prognosis—as most studies have not analyzed it separately from VF, polymorphic VT, or NSVT. In the GUSTO-I (Global Utilization of Streptokinase and t-PA for Occluded Coronary Arteries) study, patients with early (<48 hours) sustained VT had a 7.1% 1-year mortality among 30-day survivors, compared with 6.1% in patients with “late” (>48 hours) VF, and 2.6% in patients without any early or late VT or VF.39 Therefore, SMVT, occurring even “early” after MI, is generally considered by many experts to be an indicator of high risk for future arrhythmia and SCD, warranting further investigation and intervention.53,56
Polymorphic VT, which occurs in 0.3% to 2% of patients, may be a marker of ongoing ischemia; therefore, it can often be effectively managed by anti-ischemic interventions. It is more often seen in patients who also develop VF.57 In general, efforts are made to correct potential triggering factors such as hypokalemia, hypomagnesemia, abnormal serum calcium, or bradycardia (in those with bradycardia or pause-dependent onset).53 In a case series of 11 patients with polymorphic VT, none had sinus bradycardia, but 3 of 11 had a sinus pause preceding the onset.58 None had prolonged Q-T interval, hypokalemia, or abnormal serum magnesium or calcium. Nine of eleven had signs of recurrent ischemia immediately before arrhythmia onset. VF occurs in 3.7% of all acute STEMIs in the first 48 hours, and this is likely an underestimation, as prehospital events are not included.39,41 Of these, most VF episodes occur within the first 4 hours (3.1%).41 When all VF events, before and after 48 hours, were included, VF was found to occur in 6.7% of STEMI patients and in 1.3% of NSTEMI patients.39,45 In the first 4 hours of admission, VF was more likely to occur in the setting of hypokalemia, low blood pressure, larger infarct size, current smoking, and a younger age. VF was more common in inferoposterior infarcts, possibly because of greater autonomic upset. The association of initial bradycardia with early fibrillatory risk fits with the observation that vagal overactivity may precede VF. VF at all stages of infarct evolution is more common in patients with larger infarcts as determined by serial cardiac enzyme measurements.59
Traditionally, primary VF has referred to VF that occurs during the first 48 hours of an uncomplicated MI (without recurrent ischemia or heart failure), and in the GISSI (Gruppo Italiano per lo Studio della Streptochinasi nell’Infarto Miocardico)-2 trial, it was associated with increased in-hospital mortality; however, no statistically significant association with post-discharge mortality (1-year mortality of those who survived to hospital discharge) was observed. These effects of early ventricular arrhythmias on early mortality were confirmed in the percutaneous coronary intervention era.60,61 In contrast, nonprimary VF (VF occurring in the setting of recurrent ischemia or heart failure or beyond the first 48 hours following MI) was associated with marked increases in both 30-day mortality and 6-month mortality.41 In the GUSTO trial, all “late” (>48 hours following MI) sustained VT, VF episodes, or both were associated with markedly increased long-term mortality at 1 year among 30-day survivors.39 More recently published data from the GUSTO-V trial found that “early” VT or VF (<48 hours) was associated with increased in-hospital mortality but not with 1-year mortality among 30-day survivors42; however, all arrhythmias (VF, all VT) were pooled in this analysis. The temporal cutoff between “early” and “late” arrhythmias at 48 hours following MI is arbitrary to some extent; data to suggest that this should be at 24 hours or even earlier exist52; clearly, decisions should be individualized and based on expert evaluation and judgment. Moreover, additional tools for risk stratification are needed, and this is an area of active investigation; in the future, these may include a combination of electrophysiological testing, genetic evaluation, scar imaging, autonomic evaluation, and so on.
Preliminary data from a post hoc analysis suggest that ranolazine, an antianginal that inhibits the late inward Na+ current, may decrease the incidence of VT or VF (as well as SVT or AF), but this requires further study.62 A large pooled analysis has suggested that early administration of intravenous β-blockers in acute myocardial infarction may decrease the mortality and incidence rates of VF, but the Clopidogrel and Metoprolol in Myocardial Infarction Trial (COMMIT) performed on 46,000 patients with AMI did not show the mortality benefit of this intervention.63,64
In summary, the available data on the outcomes of ventricular arrhythmias in AMI have limitations. These data come mostly from thrombolytic trials, retrospective analyses, limited numbers of events, and analyses of multiple types of arrhythmia, rather than specific arrhythmias, pooled together. Mainly on the basis of large thrombolytic trials, “early” sustained VF or polymorphic VT occurring within the first 24 to 48 hours of an uncomplicated AMI is associated with increased in-hospital and 30-day mortality but appears to have little effect on long-term mortality in patients surviving hospital discharge; however, this may be because high-risk patients die during their initial hospital stay. Conversely, all sustained ventricular arrhythmias that are “late” (>24 to 48 hours following MI) or in the context of complicated MI and any sustained monomorphic VT are considered indicators of high risk of arrhythmia and SCD, and patients are considered survivors of cardiac arrest. The temporal cutoff between “early” and “late” arrhythmias is unclear and may be close to 24 hours or even earlier. Occasionally, complete revascularization may be achievable with sufficient treatment—in the absence of prior MI, residual scar, SMVT, or systolic dysfunction—but most of these patients should be considered for defibrillator implantation, and expert, individualized decisions should be made. Detailed acute and chronic management guidelines for ventricular arrhythmias associated with AMI have been published.2,3,53,56
Supraventricular Arrhythmias in the Setting of Acute Ischemia
Incidence
Holter monitoring has revealed that 15% of patients recovering from MI have supraventricular tachycardia, ranging from single atrial premature beats (APBs) to sustained AF during their hospital stay.65 The prevalence of SVT increases during the first month after MI. Overall, the incidence of AF in AMI in the modern era is 7.8% to 28%.66–71
Mechanism of Atrial Fibrillation During Acute Myocardial Ischemia
The pathophysiology of AF that occurs in the course of AMI has many components. Inflammation (pericarditis), changes in hemodynamics (atrial stretch and dilation), and atrial ischemia may all play a role.72–75 Following significant ventricular damage, end-diastolic volume and pressure rise, causing an increase in atrial pressure and wall tension. This predisposes to AF and also explains the close relationship between heart failure and AF in the setting of MI. In an angiographic study, AF that occurred during inferior MI was shown to more likely occur in the setting of an occluded proximal left circumflex artery, with or without right CAD, if it was combined with impaired perfusion of the AV nodal artery.76 AF did not occur in patients with right coronary artery occlusions if the circumflex artery was unobstructed. In a series of 266 patients, all 12 who developed atrial arrhythmias had inferior infarction. In the vast majority of these patients, the sinus node artery was distal to the site of right coronary occlusion, which suggests that sinus node ischemia may also play a role.77 Evidence of atrial infarction in the 12-lead electrocardiogram (ECG; manifesting as PR-segment displacement) may also predict the onset of AF during AMI.78 Other risk factors include advanced age, the presence of congestive heart failure, three-vessel coronary disease, right coronary artery (RCA) occlusion, female gender, anterior Q-wave MI, previous MI, and previous coronary artery bypass graft (CABG).66,79
Consequences of Atrial Fibrillation During Acute Myocardial Infarction
The development of AF results in the loss of atrial contraction and rapid, irregular heart rates, which, in turn, will cause impaired diastolic filling and increased myocardial oxygen demand. Atrial contraction is an important component of ventricular filling, particularly in failing hearts. In the ischemic canine heart, induced AF was shown to cause a reduction in cardiac output, a fall in mean aortic pressure, and a fall in mean myocardial blood flow.80 This may precipitate a vicious downward spiral, with AF exacerbating heart failure, which, in turn, promotes AF. Both will increase the ischemic burden and the likelihood of ventricular arrhythmias.
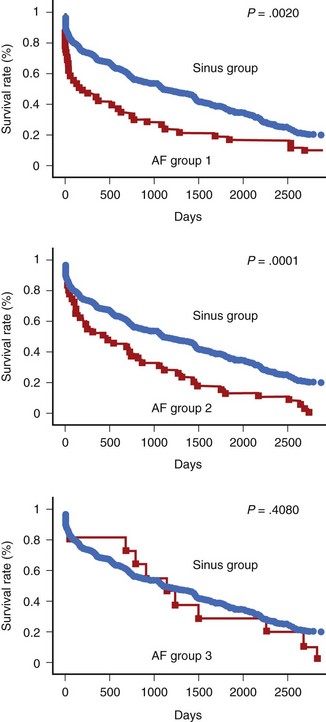
In patients who sustain an AMI, hospital mortality is significantly higher in those with AF than in those without it (Figure 57-3).66,68,69,71,72 It has been suggested that AF may be a risk factor for VF.81 AF occurs in patients with signs of heart failure and larger infarctions. In large-scale trials, the negative impact of AF has been shown to be independent of other variables.70,79 However, it is possible that the increase in in-hospital mortality is restricted to those patients with new-onset AF (after admission) rather than pre-existing AF (see Figure 57-3).70,82
Bradyarrhythmias in the Setting of Acute Ischemia
High-degree atrioventricular (AV) block is seen in a significant proportion of patients presenting with acute inferior MI. The incidence of advanced (second-degree and third-degree) AV block in the thrombolytic era ranges from 5.6% to 3.7% of all AMI patients.83 In inferior wall MI, the reported incidence ranges from 7.3% to 9.8% of patients developing advanced AV block to 13% of patients having complete heart block, compared with 3.2% advanced AV block in patients with acute anterior wall infarction.84–86
All studies examining patients with heart block after infarction have found an association with a greater degree of myocardial damage, whether measured by cardiac enzymes, echocardiography, or nuclear scintigraphy.85,87–90 As it has long been recognized that heart block is most prevalent in patients with inferior wall MI (two- to threefold increase compared with anterior AMI patients), the majority of studies were done in this population of patients.84,86 Within this group, right ventricular involvement also appears to be associated with the development of advanced AV block.88
Patients with inferior MI and coexisting left anterior descending coronary artery obstruction have a sixfold greater chance of developing heart block in the acute phase of infarction than do patients with inferior infarction without such obstruction.91 The site of left anterior descending artery occlusion is usually proximal to the origin of the first septal perforator. These findings suggest that the proximal AV conduction system has a dual arterial blood supply from both the right and left anterior descending coronary arteries and may explain the transient behavior of heart block and lack of necrosis of the AV node seen in many patients with inferior MI. A histopathologic study of hearts with posteroinferior MI has shown a strong correlation with atrial infarction in the region of the inputs to the AV node but a lack of correlation with infarction of the specialized conducting system.92
In the setting of inferior infarction, patients with CHB have higher mortality; more episodes of VF or tachycardia; and sustained hypotension, pulmonary edema, pericarditis, and atrial fibrillation than do patients without heart block.71,84,85,89,90 In contrast, in those with inferior MI who survive to hospital discharge, the presence of heart block has no effect on long-term mortality.84,85,89
Differences between patients who develop heart block early and those who develop it late in the course of their AMI do exist. Different studies, however, reveal conflicting data. Sclarovsky et al reported that patients who develop early advanced block—defined as that occurring with continuing hyperacute changes of AMI on the ECG—had CHB that was of short duration, was unresponsive to atropine, and often required pacemaker therapy.93 Symptoms of syncope, heart failure, and cardiogenic shock were frequently present. Patients with late block typically had second-degree heart block of longer duration, had a positive response to atropine, and rarely required pacemaker therapy. The mortality rate was high in the early group (23%) compared with that of the late group (7%). In another study, using a 6-hour cutoff time limit from admission, patients with inferior MI were divided into those who developed second- or third-degree block early and late.94 In the early group, all patients had transient AV block that appeared suddenly, disappeared by 24 hours, and displayed a positive response to atropine. In the late group, heart block was often preceded by first-degree block, lasted longer, had a relatively fast ventricular escape rhythm, and had little response to atropine. A third study, dividing patients on the basis of AV block appearing before or after 24 hours from admission, found no significant difference in hospital mortality.95
The mechanisms responsible for AV block during acute inferior MI would, therefore, appear to be multiple and related to the time course. Along with acute necrosis of the perinodal atrial myocardium or specialized conduction tissue, increased parasympathetic tone is a factor that is usually postulated; however, persistence of AV block after atropine administration is frequently observed. It has been demonstrated that endogenously released adenosine in the oxygen-deprived myocardium can cause AV block.96 Thus, not surprisingly, it has been reported that aminophylline may be successful in restoring sinus rhythm in atropine-resistant patients with inferior infarction.97–99
Arrhythmias in Chronic Coronary Artery Disease
Patients with chronic CAD may develop ventricular arrhythmias during episodes of AMI. In addition, prior MI may provide a nidus for the development of tachyarrhythmias in the setting of chronic CAD. It is often challenging for the clinician to determine the extent to which chronic infarction or acute ischemia contributes to a particular arrhythmic event. However, a determination as to whether ischemia, chronic scarring, or a combination is responsible for arrhythmia in patients with CAD can help direct therapy.100
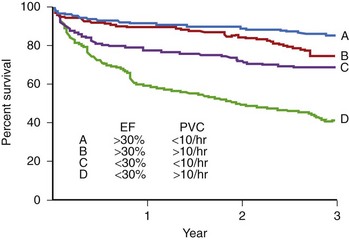
Patients with chronic CAD may present with PVCs and NSVT that are either asymptomatic or associated with mild palpitations.101 However, more serious forms of arrhythmia include nonsustained polymorphic VT, nonsustained or sustained monomorphic VT, and VF (Figure 57-4).
Mechanisms
The mechanisms of ventricular arrhythmias in patients with chronic coronary disease are diverse. The contribution of ischemia to arrhythmogenesis were discussed earlier. The development of myocardial fibrosis can lead to VT through a number of mechanisms. In both experimental models and human tissue, activation in healed infarction takes a slow, “zigzag” course, in which fibrous septa separate bundles of muscle and the gap junction number is decreased to produce slow conduction, creating the substrate for re-entry.102–106 This phenomenon is associated with the recording of fractionated electrograms. The poor coupling among surviving myocardial cells may also allow the development of unidirectional block.
The mechanism of beats initiating ventricular tachyarrhythmias in patients with CAD remains unclear. In experimental models, triggered activity, abnormal automaticity, and re-entry due to functional block in sinus rhythm all have occurred in healed MI.7 One clinical study of monomorphic VT has demonstrated that the QRS configuration of the beat that initiates sustained VT is similar to that occurring in sustained tachycardia. This suggests the possibility that the initiating beat is, indeed, re-entrant. One careful intraoperative three-dimensional mapping study of NSVT has been performed. Ten patients with NSVT in the setting of CAD were studied, and in one half of these patients, a re-entrant circuit was identified, and in the other half, a focal origin to the tachycardia appeared to be present.107,108 However, even with advanced mapping techniques, the mechanisms of isolated PVCs, short episodes of NSVT, and the beats that initiate VT or VF have not been established with certainty in humans because of the difficulty in mapping isolated premature beats and because of the inferential nature of the evidence required to determine the mechanism of isolated premature beats.
More is known about the mechanism of sustained VT. Mapping studies in the electrophysiology laboratory and in the operating room and inferential studies using pacing techniques suggest that most cases of myocardial sustained VT in patients or experimental animals with healed MI are caused by re-entry.5,102,109,110 Both fixed and functional blocks may contribute to sustained VT in patients with coronary disease.33,106,111 A number of experimental studies have suggested that VF, at least shortly after its origin, is caused by re-entry.112–115 The mechanism of initiating beats is unclear and likely not uniform, but VF in most experimental models appears to be maintained by re-entry. Further study of human VF is required to better clarify these mechanisms.
Ventricular Arrhythmias in Chronic Coronary Artery Disease
Premature Ventricular Contractions and Nonsustained Ventricular Tachycardia
PVCs are common in patients with healed MI.116 The prevalence of PVCs varies in different studies and is weakly related to the extent of left ventricular dysfunction. In the GISSI-2 study, over 64% of patients had some PVCs, as seen on Holter monitors, and obtained a mean of 17 days following MI.20 Twenty percent had more than 10 PVCs per hour, and 6.8% had NSVT. In this and other studies, frequent PVCs (>10 premature beats per hour) and complex ventricular arrhythmias were shown to increase the risk of sudden cardiac death.20,117–119 By multivariate analysis, the presence of more than 10 PVCs per hour in a week was associated with an odds ratio of 1.62 for total mortality and an odds ratio of 2.22 for sudden mortality.20 In this study, NSVT (defined as three beats to 30 seconds of VT) was associated with an increased mortality during follow-up by univariate analysis but not multivariate analysis. In contrast, only a small fraction of post-infarction patients (<10%) with tachyarrhythmias during Holter monitoring die suddenly, which gives a low positive predictive value. However, the European Infarct Study Group showed that fewer than 1% of patients in whom Holter monitor readings were normal died suddenly during the first year after MI.120 Thus, the absence of arrhythmia in the healing phase of infarction predicts a good prognosis.
The anatomic and electrophysiological characteristics of MI evolve in the first several months after infarction.121 Thus, findings on the prognostic significance of spontaneous arrhythmia shortly after infarction may not apply to healed infarction. Data on patients with PVCs or NSVT after the subacute phase of MI are based on studies of smaller numbers of patients and are inconsistent.101 Nonetheless, several studies have used NSVT in association with left ventricular dysfunction and inducible sustained VT at electrophysiological testing to risk stratify patients with chronic CAD.122,123
Monomorphic Ventricular Tachycardia
The clinical presentation of monomorphic VT is also variable. Some patients, especially those with large MIs, may have stable monomorphic VT at slow rates that is hemodynamically reasonably well tolerated. This is particularly true if patients are treated with antiarrhythmic drugs. In other patients, sustained VT is associated with presyncope, syncope, or cardiac arrest.124 A variety of factors may affect the hemodynamic tolerance of sustained VT. These include the rate of the tachycardia, atrial synchrony, and left ventricular function.125
Data on the prognostic significance of patients with sustained, hemodynamically well-tolerated VT are conflicting.126 It is most likely that while presentation with sustained, hemodynamically well-tolerated VT does indicate a substantial increase in the risk of sudden cardiac death, this risk may not be as high as in patients who present with a cardiac arrest. However, in an AVID (Antiarrhythmics Versus Implantable Defibrillators) substudy, patients who presented with sustained, well-tolerated VT had at least as poor a prognosis as patients presenting with cardiac arrest.127 This subanalysis of a large, prospective study strongly suggests that patients who present with stable VT are also at risk for life-threatening tachyarrhythmias.
Cardiac Arrest and Ventricular Fibrillation
Despite a decline in the incidence of cardiovascular disease, 300,000 to 350,000 sudden deaths still occur every year in the United States.53,128 The precise arrhythmia initiating sudden death in patients with chronic CAD (and in patients with other types of structural heart disease) is not completely known. Data from the Seattle Heart Watch Project and from several Holter monitoring studies from the late 1970s and 1980s have demonstrated that VT, VF, or both may be responsible for 40% to 50% of cardiac arrests and that Q-wave infarction is only present in 20% of these.129,130 It has been postulated that in many of the 40% of patients with cardiac arrests who present with asystole, this actually represents a terminal rhythm following prior VT or VF.131,132 Holter monitoring data support this contention.133 One study by Luu and colleagues in patients with advanced heart failure demonstrated that bradycardia, electromechanical dissociation, or both may be responsible for 50% or more of cardiac arrests, but it is likely that these data do not apply to the majority of patients having a cardiac arrest.134 Thus, most patients with chronic CAD who suffer cardiac arrest have VT or VF as the mechanism of death. Even in the absence of infarction, ischemia may be a frequent contributing factor.135
Polymorphic Ventricular Tachycardia
Polymorphic VT is defined as VT in which QRS configuration varies from beat to beat but a clearly defined QRS complex (as opposed to ventricular flutter or fibrillation) can be detected. Polymorphic VT is often associated with a congenital or acquired long QT syndrome, which is rarely caused by CAD.136 However, isolated case reports have described long QT syndrome and associated polymorphic VT in patients with coronary disease. AMI can also classically cause polymorphic VT. However, some patients with healed MI may also present with nonsustained polymorphic VT or episodes of polymorphic VT degenerating to VF.133,137
Electrical Storm
Implantable cardioverter-defibrillators (ICDs) have led to the recognition of a subset of patients who develop multiple recurrent episodes of VT or VF leading to cardiac arrest, multiple ICD shocks, or both in a short period. Electrical storm has been defined as two or more VT or VF episodes occurring in less than 24 hours, but some patients develop numerous arrhythmic episodes. The etiology of such temporal clustering of VT or VF episodes is not completely clear. Arrhythmic substrate (scar) is almost always present in patients with CAD, but other types of arrhythmia susceptibility may play a role, as VF storms also develop in patients without apparent structural heart disease (“idiopathic”) or with channelopathies. An arrhythmic storm in a patient with CAD should prompt a search for and treatment of AMI, particularly with polymorphic VT storm. Other possible precipitating factors include electrolyte abnormalities, drug toxicity, biventricular pacing, bradycardia or pauses (pause-dependent VT initiation), and decompensated heart failure (though it is frequently difficult to discern cause from effect in these situations).138,139 The autonomic nervous system appears to play a prominent role, and this is illustrated by the observed effectiveness of autonomic modulation (β-blockers, sedation, sympathetic nervous block, or stellate ganglion resection) and by the transient nature of the storm.53,140 It is likely that arrhythmia, hemodynamic instability, ICD shocks, or all result in sympatho-adrenergic activation, which, in turn, sets up a vicious spiral. On the basis of anecdotal reports and case series, the treatment of VT or VF storm, includes correcting the precipitating factors, intravenous β-blockers and other antiarrhythmic drugs, overdrive pacing (for pause-dependent VT), autonomic modulation, and ablation.53 When monomorphic PVC triggers of VF are observed, these are frequently mapped to infarct border zones and are preceded by Purkinje-like potentials, which suggests that they are caused by locally enhanced automaticity of the Purkinje fibers.141 Ablation of these PVCs by targeting the Purkinje-like potentials has been reported to be effective.141 While the short-term prognosis can be improved with treatment, the longer-term prognosis is unclear and is generally guarded.142–144
Exercise-Induced Arrhythmias
Exercise-induced arrhythmias represent a potentially life-threatening problem in patients with CAD. While physical training in general decreases total mortality from heart disease, the relative risk of sudden death during exercise is increased.145 Data on the prognostic significance of PVCs or NSVT during exercise are controversial.146 PVCs and NSVT that occur during exercise are likely multifactorial, including myocardial ischemia, the presence of scar substrate, and catecholamine and other autonomic effects. The extent to which exertional ventricular arrhythmias indicate myocardial ischemia in patients with CAD and their prognostic significance are still not completely clear.
Treatment
Detailed guidelines and reviews of the available literature on management of ventricular arrhythmias in CAD have been published, and they provide an excellent source.53,56
Although the treatment of serious ventricular arrhythmias is also discussed elsewhere in this text, a few principles regarding the approach to less serious arrhythmias in patients with chronic CAD may be useful. The two potential indications for treatment of patients with premature beats and NSVT are (1) improvement of symptoms and (2) prolongation of life. Most patients with isolated premature beats or NSVT are asymptomatic or have mild symptoms that are not clinically or hemodynamically significant. Holter monitoring studies have shown that up to 10% of older patients, even in the absence of structural heart disease, may have ventricular premature beats and that ventricular premature beats are extremely common after MI. This confirms that most patients with ventricular ectopy do not need treatment. However, a subset of patients with PVCs or NSVT have highly symptomatic palpitations or impairment in left ventricular function because of extremely frequent ventricular ectopy.147,148 In these patients, suppression of ventricular premature beats to control symptoms may be appropriate. In addition, ablation is an excellent option for patients who have significant symptoms, frequent PVCs resulting in cardiomyopathy, or PVCs that act as triggers for VF, as it offers the potential for long-term cure of arrhythmia.141
The therapy for arrhythmias is discussed in detail in a separate chapter. β-Blockers should be the therapy of first choice for the suppression of symptoms related to PVCs or NSVT in patients with CAD. Studies cited above have suggested that spontaneous ventricular arrhythmias may represent an independent risk factor for the prediction of sudden death in patients who have coronary disease and healed MI. Suppression of PVCs with antiarrhythmic drugs has failed to result in a decrease in mortality rates; encainide and flecainide have been, in fact, shown to increase mortality in the Cardiac Arrhythmia Suppression Trial (CAST).149 Amiodarone probably has a neutral effect on mortality in patients with CAD, although a meta-analysis suggested that amiodarone may decrease mortality by approximately 10% when administered prophylactically.150–152
Dofetilide, a K+ channel–blocking drug, has also been studied extensively in patients with prior MI and heart failure. Although the prognosis is not improved, dofetilide does not cause increased mortality in patients with CAD.153 However, dofetilide may have adverse effects in patients with baseline prolonged Q-T intervals.154 The available data do not support the routine use of antiarrhythmic drugs for the prevention of sudden death in patients with coronary disease and spontaneous arrhythmias. However, if drug therapy is required to suppress symptoms in patients with CAD, β-blockers, dofetilide, or amiodarone are all drugs that have been shown to have a beneficial or neutral effect on survival.
Atrial Fibrillation and Chronic Coronary Artery Disease
CAD is commonly cited as one of the principal causes of atrial fibrillation (AF). Although the role of AMI in the development of AF is undisputed (see earlier), the role of chronic CAD is much more controversial. It is likely that CHF, which is one of the potential consequences of CAD, predisposes to AF, but CAD itself does not.155–157
Advanced Atrioventricular Block and Chronic Coronary Artery Disease
Chronic CAD can be a cause of AV block, although it is a much less common cause than idiopathic fibrosis of the conduction system. AV block in coronary disease is usually related to extensive infarction and necrosis of the distal conduction system rather than ischemia. In 30 patients aged 45 to 65 years with CHB who were referred for pacing and had no symptoms of coronary disease, coronary angiography disclosed the presence of severe CAD in 13 patients (43%). Myocardial revascularization was undertaken in 6 patients but did not result in any sustained improvement in AV conduction.158 In contrast, multiple case reports in the literature have suggested that ischemia of the AV node may have a role in patients with paroxysmal or exercise-induced heart block having complete resolution of their symptoms after angioplasty of a lesion in the RCA.159–162 However, most patients with AV block and coronary disease require pacemaker therapy.
Key References
Albert CM, Mittleman MA, Chae CU, et al. Triggering of sudden death from cardiac causes by vigorous exertion. N Engl J Med. 2000;343(19):1355-1361.
Al-Khatib SM, Granger CB, Huang Y, et al. Sustained ventricular arrhythmias among patients with acute coronary syndromes with no ST-segment elevation: Incidence, predictors, and outcomes. Circulation. 2002;106(3):309-312.
Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non ST-elevation myocardial infarction. Circulation. 2007;116(7):e148-e304.
Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction. Circulation. 2004;110(9):e82-e292.
Cheema AN, Sheu K, Parker M, et al. Nonsustained ventricular tachycardia in the setting of acute myocardial infarction: Tachycardia characteristics and their prognostic implications. Circulation. 1998;98(19):2030-2036.
Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities. J Am Coll Cardiol. 2008;51(21):e1-e62.
Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. 1989;69(4):1049-1169.
Kaplinsky E, Ogawa S, Balke CW, Dreifus LS. Two periods of early ventricular arrhythmia in the canine acute myocardial infarction model. Circulation. 1979;60(2):397-403.
Maggioni AP, Zuanetti G, Franzosi MG, et al. Prevalence and prognostic significance of ventricular arrhythmias after acute myocardial infarction in the fibrinolytic era. GISSI-2 results. Circulation. 1993;87(2):312-322.
Marrouche NF, Verma A, Wazni O, et al. Mode of initiation and ablation of ventricular fibrillation storms in patients with ischemic cardiomyopathy. J Am Coll Cardiol. 2004;43(9):1715-1720.
Mont L, Cinca J, Blanch P, et al. Predisposing factors and prognostic value of sustained monomorphic ventricular tachycardia in the early phase of acute myocardial infarction. J Am Coll Cardiol. 1996;28(7):1670-1676.
Newby KH, Thompson T, Stebbins A, et al. Sustained ventricular arrhythmias in patients receiving thrombolytic therapy: Incidence and outcomes. The GUSTO Investigators. Circulation. 1998;98(23):2567-2573.
Passman R, Kadish A. Polymorphic ventricular tachycardia, long Q-T syndrome, and torsades de pointes. Med Clin North Am. 2001;85(2):321-341.
Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. J Am Coll Cardiol. 2006;48(5):e247-e346.
1 Chugh SS, Kelly KL, Titus JL. Sudden cardiac death with apparently normal heart. Circulation. 2000;102(6):649-654.
2 Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non– ST-elevation myocardial infarction. Circulation. 2007;116(7):e148-e304.
3 Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction. Circulation. 2004;110(9):e82-e292.
4 Kleber AG. Resting membrane potential, extracellular potassium activity, and intracellular sodium activity during acute global ischemia in isolated perfused guinea pig hearts. Circ Res. 1983;52(4):442-450.
5 Downar E, Janse MJ, Durrer D. The effect of acute coronary artery occlusion on subepicardial transmembrane potentials in the intact porcine heart. Circulation. 1977;56(2):217-224.
6 Kleber J. van Capelle: Mechanism and time course of ST and TQ segment changes during acute regional myocardial ischemia in the pig heart determined by extracellular and intracellular recordings. Circ Res. 1978;42:603-613.
7 Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. 1989;69(4):1049-1169.
8 Kaplinsky E, Ogawa S, Balke CW, Dreifus LS. Two periods of early ventricular arrhythmia in the canine acute myocardial infarction model. Circulation. 1979;60(2):397-403.
9 Gilmour RFJr, Zipes DP. Different electrophysiological responses of canine endocardium and epicardium to combined hyperkalemia, hypoxia, and acidosis. Circ Res. 1980;46(6):814-825.
10 Lazzara R, El-Sherif N, Hope RR, Scherlag BJ. Ventricular arrhythmias and electrophysiological consequences of myocardial ischemia and infarction. Circ Res. 1978;42(6):740-749.
11 Kleber AG, Riegger CB, Janse MJ. Electrical uncoupling and increase of extracellular resistance after induction of ischemia in isolated, arterially perfused rabbit papillary muscle. Circ Res. 1987;61(2):271-279.
12 Cao JM, Fishbein MC, Han JB, et al. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation. 2000;101(16):1960-1969.
13 Iwasaki T, Suzuki T, Tateno M, et al. Dual-tracer autoradiography with thallium-201 and iodine-125-metaiodobenzylguanidine in experimental myocardial infarction of rat. J Nucl Med. 1996;37(4):680-684.
14 Nishimura T, Oka H, Sago M, et al. Serial assessment of denervated but viable myocardium following acute myocardial infarction in dogs using iodine-123 metaiodobenzylguanidine and thallium-201 chloride myocardial single photon emission tomography. Eur J Nucl Med. 1992;19(1):25-29.
15 Stanton MS, Tuli MM, Radtke NL, et al. Regional sympathetic denervation after myocardial infarction in humans detected noninvasively using I-123-metaiodobenzylguanidine. J Am Coll Cardiol. 1989;14(6):1519-1526.
16 Inoue H, Zipes DP. Results of sympathetic denervation in the canine heart: Supersensitivity that may be arrhythmogenic. Circulation. 1987;75(4):877-887.
17 Kammerling JJ, Green FJ, Watanabe AM, et al. Denervation supersensitivity of refractoriness in noninfarcted areas apical to transmural myocardial infarction. Circulation. 1987;76(2):383-393.
18 Herre JM, Wetstein L, Lin YL, et al. Effect of transmural versus nontransmural myocardial infarction on inducibility of ventricular arrhythmias during sympathetic stimulation in dogs. J Am Coll Cardiol. 1988;11(2):414-421.
19 GISSI-2. Effectiveness of intravenous thrombolytic treatment in acute myocardial infarction. Gruppo Italiano per lo Studio della Streptochinasi nell’Infarto Miocardico (GISSI). Lancet. 1986;1(8478):397-402.
20 Maggioni AP, Zuanetti G, Franzosi MG, et al. Prevalence and prognostic significance of ventricular arrhythmias after acute myocardial infarction in the fibrinolytic era. GISSI-2 results. Circulation. 1993;87(2):312-322.
21 Vegh A, Komori S, Szekeres L, Parratt JR. Antiarrhythmic effects of preconditioning in anaesthetised dogs and rats. Cardiovasc Res. 1992;26(5):487-495.
22 Albert CM, Campos H, Stampfer MJ, et al. Blood levels of long-chain n-3 fatty acids and the risk of sudden death. N Engl J Med. 2002;346(15):1113-1118.
23 Jouven X, Charles MA, Desnos M, Ducimetiere P. Circulating nonesterified fatty acid level as a predictive risk factor for sudden death in the population. Circulation. 2001;104(7):756-761.
24 Dekker LR, Bezzina CR, Henriques JP, et al. Familial sudden death is an important risk factor for primary ventricular fibrillation: A case-control study in acute myocardial infarction patients. Circulation. 2006;114(11):1140-1145.
25 Kaikkonen KS, Kortelainen ML, Linna E, Huikuri HV. Family history and the risk of sudden cardiac death as a manifestation of an acute coronary event. Circulation. 2006;114(14):1462-1467.
26 Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112(16):2517-2529.
27 Splawski I, Timothy KW, Tateyama M, et al. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297(5585):1333-1336.
28 Sotoodehnia N, Siscovick DS, Vatta M, et al. Beta2-adrenergic receptor genetic variants and risk of sudden cardiac death. Circulation. 2006;113(15):1842-1848.
29 Penny WJ. The deleterious effects of myocardial catecholamines on cellular electrophysiology and arrhythmias during ischaemia and reperfusion. Eur Heart J. 1984;5(12):960-973.
30 Coronel R, Wilms-Schopman FJ, deGroot JR. Origin of ischemia-induced phase 1b ventricular arrhythmias in pig hearts. J Am Coll Cardiol. 2002;39(1):166-176.
31 Arnar DO, Bullinga JR, Martins JB. Role of the Purkinje system in spontaneous ventricular tachycardia during acute ischemia in a canine model. Circulation. 1997;96(7):2421-2429.
32 Kieval RS, Spear JF, Moore EN. Gap junctional conductance in ventricular myocyte pairs isolated from postischemic rabbit myocardium. Circ Res. 1992;71(1):127-136.
33 Wit AL, Janse MJ. Experimental models of ventricular tachycardia and fibrillation caused by ischemia and infarction. Circulation. 1992;85(1 Suppl):I32-142.
34 Friedman PL, Stewart JR, Fenoglio JJJr. Wit AL: Survival of subendocardial Purkinje fibers after extensive myocardial infarction in dogs. Circ Res. 1973;33(5):597-611.
35 El-Sherif N, Gough WB, Zeiler RH, Mehra R. Triggered ventricular rhythms in 1-day-old myocardial infarction in the dog. Circ Res. 1983;52(5):566-579.
36 Birnbaum Y, Leor J, Kloner RA. Pathobiology and clinical impact of reperfusion injury. J Thromb Thrombolysis. 1997;4(2):185-195.
37 Ehlert FA, Goldberger JJ. Cellular and pathophysiological mechanisms of ventricular arrhythmias in acute ischemia and infarction. Pacing Clin Electrophysiol. 1997;20(4 Pt 1):966-975.
38 Kaplinsky E, Ogawa S, Michelson EL, Dreifus LS. Instantaneous and delayed ventricular arrhythmias after reperfusion of acutely ischemic myocardium: Evidence for multiple mechanisms. Circulation. 1981;63(2):333-340.
39 Newby KH, Thompson T, Stebbins A, et al. Sustained ventricular arrhythmias in patients receiving thrombolytic therapy: Incidence and outcomes. The GUSTO Investigators. Circulation. 1998;98(23):2567-2573.
40 Berger PB, Ruocco NA, Ryan TJ, et al. Incidence and significance of ventricular tachycardia and fibrillation in the absence of hypotension or heart failure in acute myocardial infarction treated with recombinant tissue-type plasminogen activator: Results from the Thrombolysis in Myocardial Infarction (TIMI) Phase II trial. J Am Coll Cardiol. 1993;22(7):1773-1779.
41 Volpi A, Cavalli A, Santoro L, Negri E. Incidence and prognosis of early primary ventricular fibrillation in acute myocardial infarction—results of the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI-2) database. Am J Cardiol. 1998;82(3):265-271.
42 Askari AT, Shishehbor MH, Kaminski MA, et al. The association between early ventricular arrhythmias, renin-angiotensin-aldosterone system antagonism, and mortality in patients with ST-segment-elevation myocardial infarction: Insights from Global Use of Strategies to Open coronary arteries (GUSTO) V. Am Heart J. 2009;158(2):238-243.
43 Henkel DM, Witt BJ, Gersh BJ, et al. Ventricular arrhythmias after acute myocardial infarction: A 20-year community study. Am Heart J. 2006;151(4):806-812.
44 Lopes MG, Harrison DC, Schroeder JS. Ventricular arrhythmias during unstable angina pectoris. Arch Intern Med. 1975;135(12):1548-1553.
45 Al-Khatib SM, Granger CB, Huang Y, et al. Sustained ventricular arrhythmias among patients with acute coronary syndromes with no ST-segment elevation: Incidence, predictors, and outcomes. Circulation. 2002;106(3):309-312.
46 Chiladakis JA, Karapanos G, Davlouros P, et al. Significance of R-on-T phenomenon in early ventricular tachyarrhythmia susceptibility after acute myocardial infarction in the thrombolytic era. Am J Cardiol. 2000;85(3):289-293.
47 Heidbuchel H, Tack J, Vanneste L, et al. Significance of arrhythmias during the first 24 hours of acute myocardial infarction treated with alteplase and effect of early administration of a beta-blocker or a bradycardiac agent on their incidence. Circulation. 1994;89(3):1051-1059.
48 Naito M, Michelson EL, Kaplinsky E, et al. Role of early cycle ventricular extrasystoles in initiation of ventricular tachycardia and fibrillation: Evaluation of the R on T phenomenon during acute ischemia in a canine model. Am J Cardiol. 1982;49(2):317-322.
49 Volpi A, De Vita C, Franzosi MG, et al. Determinants of 6-month mortality in survivors of myocardial infarction after thrombolysis. Results of the GISSI-2 data base. The Ad hoc Working Group of the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI)-2 Data Base. Circulation. 1993;88(2):416-429.
50 Rouleau JL, Talajic M, Sussex B, et al. Myocardial infarction patients in the 1990s—their risk factors, stratification and survival in Canada: The Canadian Assessment of Myocardial Infarction (CAMI) Study. J Am Coll Cardiol. 1996;27(5):1119-1127.
51 Miller FC, Krucoff MW, Satler LF, et al. Ventricular arrhythmias during reperfusion. Am Heart J. 1986;112(5):928-932.
52 Cheema AN, Sheu K, Parker M, et al. Nonsustained ventricular tachycardia in the setting of acute myocardial infarction: Tachycardia characteristics and their prognostic implications. Circulation. 1998;98(19):2030-2036.
53 Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. J Am Coll Cardiol. 2006;48(5):e247-e346.
54 Mont L, Cinca J, Blanch P, et al. Predisposing factors and prognostic value of sustained monomorphic ventricular tachycardia in the early phase of acute myocardial infarction. J Am Coll Cardiol. 1996;28(7):1670-1676.
55 Hatzinikolaou-Kotsakou E, Tziakas D, Hotidis A, et al. Could sustained monomorphic ventricular tachycardia in the early phase of a prime acute myocardial infarction affect patient outcome? J Electrocardiol. 2007;40(1):72-77.
56 Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities. J Am Coll Cardiol. 2008;51(21):e1-e62.
57 Bluzhas J, Lukshiene D, Shlapikiene B, Ragaishis J. Relation between ventricular arrhythmia and sudden cardiac death in patients with acute myocardial infarction: The predictors of ventricular fibrillation. J Am Coll Cardiol. 1986;8(1 Suppl A):69A-72A.
58 Wolfe CL, Nibley C, Bhandari A, et al. Polymorphous ventricular tachycardia associated with acute myocardial infarction. Circulation. 1991;84(4):1543-1551.
59 Herlitz J, Hjalmarson A, Swedberg K, et al. Relationship between infarct size and incidence of severe ventricular arrhythmias in a double-blind trial with metoprolol in acute myocardial infarction. Int J Cardiol. 1984;6(1):47-60.
60 Mehta RH, Starr AZ, Lopes RD, et al. Incidence of and outcomes associated with ventricular tachycardia or fibrillation in patients undergoing primary percutaneous coronary intervention. JAMA. 2009;301(17):1779-1789.
61 Piccini JP, Berger JS, Brown DL. Early sustained ventricular arrhythmias complicating acute myocardial infarction. Am J Med. 2008;121(9):797-804.
62 Scirica BM, Morrow DA, Hod H, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: Results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007;116(15):1647-1652.
63 Teo KK, Yusuf S, Furberg CD. Effects of prophylactic antiarrhythmic drug therapy in acute myocardial infarction. An overview of results from randomized controlled trials. JAMA. 1993;270(13):1589-1595.
64 Chen ZM, Pan HC, Chen YP, et al. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: Randomised placebo-controlled trial. Lancet. 2005;366(9497):1622-1632.
65 Jespersen CM, Vaage-Nilsen M, Hansen JF. The significance of myocardial ischaemia and verapamil treatment on the prevalence of supraventricular tachyarrhythmias in patients recovering from acute myocardial infarction. Danish Study Group on Verapamil in Myocardial Infarction. Eur Heart J. 1992;13(10):1427-1430.
66 Sakata K, Kurihara H, Iwamori K, et al. Clinical and prognostic significance of atrial fibrillation in acute myocardial infarction. Am J Cardiol. 1997;80(12):1522-1527.
67 Pizzetti F, Turazza FM, Franzosi MG, et al. Incidence and prognostic significance of atrial fibrillation in acute myocardial infarction: The GISSI-3 data. Heart. 2001;86(5):527-532.
68 Rathore SS, Berger AK, Weinfurt KP, et al. Acute myocardial infarction complicated by atrial fibrillation in the elderly: Prevalence and outcomes. Circulation. 2000;101(9):969-974.
69 Pedersen OD, Bagger H, Kober L, Torp-Pedersen C. Trandolapril reduces the incidence of atrial fibrillation after acute myocardial infarction in patients with left ventricular dysfunction. Circulation. 1999;100(4):376-380.
70 Crenshaw BS, Ward SR, Granger CB, et al. Atrial fibrillation in the setting of acute myocardial infarction: The GUSTO-I experience. Global Utilization of Streptokinase and TPA for Occluded Coronary Arteries. J Am Coll Cardiol. 1997;30(2):406-413.
71 Behar S, Zahavi Z, Goldbourt U, Reicher-Reiss H. Long-term prognosis of patients with paroxysmal atrial fibrillation complicating acute myocardial infarction. SPRINT Study Group. Eur Heart J. 1992;13(1):45-50.
72 Nagahama Y, Sugiura T, Takehana K, et al. The role of infarction-associated pericarditis on the occurrence of atrial fibrillation. Eur Heart J. 1998;19(2):287-292.
73 Widimsky P, Gregor P. Recent atrial fibrillation in acute myocardial infarction: A sign of pericarditis. Cor Vasa. 1993;35(6):230-232.
74 Sugiura T, Iwasaka T, Takahashi N, et al. Factors associated with atrial fibrillation in Q wave anterior myocardial infarction. Am Heart J. 1991;121(5):1409-1412.
75 Solti F, Kekesi V, Juhasz-Nagy A. The effect of atrial dilatation on reperfusion arrhythmias: Development of supraventricular tachycardias on reperfusion with atrial stretching. Acta Med Hung. 1992;49(3-4):159-170.
76 Hod H, Lew AS, Keltai M, et al. Early atrial fibrillation during evolving myocardial infarction: A consequence of impaired left atrial perfusion. Circulation. 1987;75(1):146-150.
77 Kyriakidis M, Barbetseas J, Antonopoulos A, et al. Early atrial arrhythmias in acute myocardial infarction. Role of the sinus node artery. Chest. 1992;101(4):944-947.
78 Nielsen FEAH, Gram-Hansen P, et al. The relationship between ECG signs of atrial infarction and the development of supraventricular arrhythmias in patients with acute myocardial infarction. Am Heart J. 1992;123:69-72.
79 Pedersen OD, Bagger H, Kober L, Torp-Pedersen C. The occurrence and prognostic significance of atrial fibrillation/-flutter following acute myocardial infarction. TRACE Study group. TRAndolapril Cardiac Evaluation. Eur Heart J. 1999;20(10):748-754.
80 Friedman HS, Kottmeier S, Melnicker L, et al. Effects of atrial fibrillation on myocardial blood flow in the ischemic heart of the dog. J Am Coll Cardiol. 1984;4(4):729-734.
81 Sankaranarayanan R, James MA, Nuta B, et al. Does atrial fibrillation beget ventricular fibrillation in patients with acute myocardial infarction? Pacing Clin Electrophysiol. 2008;31(12):1612-1619.
82 Schmitt J, Duray G, Gersh BJ, Hohnloser SH. Atrial fibrillation in acute myocardial infarction: A systematic review of the incidence, clinical features and prognostic implications. Eur Heart J. 2009;30(9):1038-1045.
83 Harpaz D, Behar S, Gottlieb S, et al. Complete atrioventricular block complicating acute myocardial infarction in the thrombolytic era. SPRINT Study Group and the Israeli Thrombolytic Survey Group. Secondary Prevention Reinfarction Israeli Nifedipine Trial. J Am Coll Cardiol. 1999;34(6):1721-1728.
84 Rathore SS, Gersh BJ, Berger PB, et al. Acute myocardial infarction complicated by heart block in the elderly: Prevalence and outcomes. Am Heart J. 2001;141(1):47-54.
85 Clemmensen P, Bates ER, Califf RM, et al. Complete atrioventricular block complicating inferior wall acute myocardial infarction treated with reperfusion therapy. TAMI Study Group. Am J Cardiol. 1991;67(4):225-230.
86 Meine TJ, Al-Khatib SM, Alexander JH, et al. Incidence, predictors, and outcomes of high-degree atrioventricular block complicating acute myocardial infarction treated with thrombolytic therapy. Am Heart J. 2005;149(4):670-674.
87 Tans AC, Lie KI, Durrer D. Clinical setting and prognostic significance of high degree atrioventricular block in acute inferior myocardial infarction: A study of 144 patients. Am Heart J. 1980;99(1):4-8.
88 Strasberg B, Pinchas A, Arditti A, et al. Left and right ventricular function in inferior acute myocardial infarction and significance of advanced atrioventricular block. Am J Cardiol. 1984;54(8):985-987.
89 Dubois C, Pierard LA, Smeets JP, et al. Long-term prognostic significance of atrioventricular block in inferior acute myocardial infarction. Eur Heart J. 1989;10(9):816-820.
90 Kaul U, Hari Haran V, Malhotra A, Bhatia ML. Significance of advanced atrioventricular block in acute inferior myocardial infarction—a study based on ventricular function and Holter monitoring. Int J Cardiol. 1986;11(2):187-193.
91 Bassan R, Maia IG, Bozza A, et al. Atrioventricular block in acute inferior wall myocardial infarction: Harbinger of associated obstruction of the left anterior descending coronary artery. J Am Coll Cardiol. 1986;8(4):773-778.
92 Bilbao FJ, Zabalza IE, Vilanova JR, Froufe J. Atrioventricular block in posterior acute myocardial infarction: A clinicopathologic correlation. Circulation. 1987;75(4):733-736.
93 Sclarovsky S, Strasberg B, Hirshberg A, et al. Advanced early and late atrioventricular block in acute inferior wall myocardial infarction. Am Heart J. 1984;108(1):19-24.
94 Feigl D, Ashkenazy J, Kishon Y. Early and late atrioventricular block in acute inferior myocardial infarction. J Am Coll Cardiol. 1984;4(1):35-38.
95 Altun A, Ozkan B, Gurcagan A, et al. Early and late advanced atrioventricular block in acute inferior myocardial infarction. Coron Artery Dis. 1998;9(1):1-4.
96 Belardinelli L, Mattos EC, Berne RM. Evidence for adenosine mediation of atrioventricular block in the ischemic canine myocardium. J Clin Invest. 1981;68(1):195-205.
97 Altun A, Kirdar C, Ozbay G. Effect of aminophylline in patients with atropine-resistant late advanced atrioventricular block during acute inferior myocardial infarction. Clin Cardiol. 1998;21(10):759-762.
98 Goodfellow J, Walker PR. Reversal of atropine-resistant atrioventricular block with intravenous aminophylline in the early phase of inferior wall acute myocardial infarction following treatment with streptokinase. Eur Heart J. 1995;16(6):862-865.
99 Bertolet BD, McMurtrie EB, Hill JA, Belardinelli L. Theophylline for the treatment of atrioventricular block after myocardial infarction. Ann Intern Med. 1995;123(7):509-511.
100 Mehta D, Curwin J, Gomes JA, Fuster V. Sudden death in coronary artery disease: Acute ischemia versus myocardial substrate. Circulation. 1997;96(9):3215-3223.
101 Denes P, Gillis AM, Pawitan Y, et al. Prevalence, characteristics and significance of ventricular premature complexes and ventricular tachycardia detected by 24-hour continuous electrocardiographic recording in the Cardiac Arrhythmia Suppression Trial. CAST Investigators. Am J Cardiol. 1991;68(9):887-896.
102 Gardner PI, Ursell PC, Fenoglio JJJr. Wit AL: Electrophysiologic and anatomic basis for fractionated electrograms recorded from healed myocardial infarcts. Circulation. 1985;72(3):596-611.
103 de Bakker JM, van Capelle FJ, Janse MJ, et al. Slow conduction in the infarcted human heart. ‘Zigzag’ course of activation. Circulation. 1993;88(3):915-926.
104 Kawara T, Derksen R, de Groot JR, et al. Activation delay after premature stimulation in chronically diseased human myocardium relates to the architecture of interstitial fibrosis. Circulation. 2001;104(25):3069-3075.
105 Kanno S, Saffitz JE. The role of myocardial gap junctions in electrical conduction and arrhythmogenesis. Cardiovasc Pathol. 2001;10(4):169-177.
106 Ursell PC, Gardner PI, Albala A, Fenoglio JJJr. Wit AL: Structural and electrophysiological changes in the epicardial border zone of canine myocardial infarcts during infarct healing. Circ Res. 1985;56(3):436-451.
107 Attin M, Ideker RE, Pogwizd SM. Mechanistic insights into ventricular arrhythmias from mapping studies in humans. Heart Rhythm. 2008;5(6 Suppl):S53-S58.
108 Pogwizd SM. Focal mechanisms underlying ventricular tachycardia during prolonged ischemic cardiomyopathy. Circulation. 1994;90(3):1441-1458.
109 Mickleborough LL, Harris L, Downar E, et al. A new intraoperative approach for endocardial mapping of ventricular tachycardia. J Thorac Cardiovasc Surg. 1988;95(2):271-280.
110 Josephson ME, Zimetbaum P, Huang D, et al. Pathophysiologic substrate for sustained ventricular tachycardia in coronary artery disease. Jpn Circ J. 1997;61(6):459-466.
111 Callans DJ, Zardini M, Gottlieb CD, Josephson ME. The variable contribution of functional and anatomic barriers in human ventricular tachycardia: An analysis with resetting from two sites. J Am Coll Cardiol. 1996;27(5):1106-1111.
112 Gray RA, Jalife J, Panfilov AV, et al. Mechanisms of cardiac fibrillation. Science. 1995;270(5239):1222-1223. author reply 1224–1225, 1995
113 Gray RA, Pertsov AM, Jalife J. Spatial and temporal organization during cardiac fibrillation. Nature. 1998;392(6671):75-78.
114 Damle RS, Kanaan NM, Robinson NS, et al. Spatial and temporal linking of epicardial activation directions during ventricular fibrillation in dogs. Evidence for underlying organization. Circulation. 1992;86(5):1547-1558.
115 Lee JJ, Kamjoo K, Hough D, et al. Reentrant wave fronts in Wiggers’ stage II ventricular fibrillation. Characteristics and mechanisms of termination and spontaneous regeneration. Circ Res. 1996;78(4):660-675.
116 Statters DJ, Malik M, Redwood S, et al. Use of ventricular premature complexes for risk stratification after acute myocardial infarction in the thrombolytic era. Am J Cardiol. 1996;77(2):133-138.
117 Bigger JTJr, Fleiss JL, Kleiger R, et al. The relationships among ventricular arrhythmias, left ventricular dysfunction, and mortality in the 2 years after myocardial infarction. Circulation. 1984;69(2):250-258.
118 Hofmann ABF, Burckhardt D. High grade ventricular ectopic activity and 5-year survival in patients with chronic heart disease and in healthy subjects. Cardiology. 1983;70(Suppl. 1):82-87.
119 Hohnloser SH, Klingenheben T, Zabel M, et al. Prevalence, characteristics and prognostic value during long-term follow-up of nonsustained ventricular tachycardia after myocardial infarction in the thrombolytic era. J Am Coll Cardiol. 1999;33(7):1895-1902.
120 Andresen D, Bethge KP, Boissel JP, et al. Importance of quantitative analysis of ventricular arrhythmias for predicting the prognosis in low-risk postmyocardial infarction patients. European Infarction Study Group. Eur Heart J. 1990;11(6):529-536.
121 Kadish A. Polymorphic ventricular tachycardia. Card Electrophysiol Rev. 1999;3:140-142.
122 Buxton AE, Lee KL, DiCarlo L, et al. Electrophysiologic testing to identify patients with coronary artery disease who are at risk for sudden death. Multicenter Unsustained Tachycardia Trial Investigators. N Engl J Med. 2000;342(26):1937-1945.
123 Moss AJ, Hall WJ, Cannom DS, et al. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. Multicenter Automatic Defibrillator Implantation Trial Investigators. N Engl J Med. 1996;335(26):1933-1940.
124 Kolettis TM, Kyriakides ZS, Popov T, et al. Importance of the site of ventricular tachycardia origin on left ventricular hemodynamics in humans. Pacing Clin Electrophysiol. 1999;22(6 Pt 1):871-879.
125 Holmes J, Kubo SH, Cody RJ, Kligfield P. Arrhythmias in ischemic and nonischemic dilated cardiomyopathy: Prediction of mortality by ambulatory electrocardiography. Am J Cardiol. 1985;55(1):146-151.
126 Cherry JGM. Survival with sustained monomorphic ventricular tachycardia [abstract]. PACE. 1987;10:617.
127 Raitt MH, Renfroe EG, Epstein AE, et al. “Stable” ventricular tachycardia is not a benign rhythm: Insights from the antiarrhythmics versus implantable defibrillators (AVID) registry. Circulation. 2001;103(2):244-252.
128 Myerburg RJ, Interian AJr, Mitrani RM, et al. Frequency of sudden cardiac death and profiles of risk. Am J Cardiol. 1997;80(5B):10F-19F.
129 Eisenberg M, Bergner L, Hallstrom A. Paramedic programs and out-of-hospital cardiac arrest: I. Factors associated with successful resuscitation. Am J Public Health. 1979;69(1):30-38.
130 Pratt CM, Greenway PS, Schoenfeld MH, et al. Exploration of the precision of classifying sudden cardiac death. Implications for the interpretation of clinical trials. Circulation. 1996;93(3):519-524.
131 Peckova M, Fahrenbruch CE, Cobb LA, Hallstrom AP. Circadian variations in the occurrence of cardiac arrests: Initial and repeat episodes. Circulation. 1998;98(1):31-39.
132 De Maio VJ, Stiell IG, Wells GA, Spaite DW. Cardiac arrest witnessed by emergency medical services personnel: Descriptive epidemiology, prodromal symptoms, and predictors of survival. OPALS study group. Ann Emerg Med. 2000;35(2):138-146.
133 Kempf FCJr. Josephson ME: Cardiac arrest recorded on ambulatory electrocardiograms. Am J Cardiol. 1984;53(11):1577-1582.
134 Luu M, Stevenson WG, Stevenson LW, et al. Diverse mechanisms of unexpected cardiac arrest in advanced heart failure. Circulation. 1989;80(6):1675-1680.
135 Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345(20):1473-1482.
136 Kadish A, Quigg R, Schaechter A, et al. Defibrillators in nonischemic cardiomyopathy treatment evaluation. Pacing Clin Electrophysiol. 2000;23(3):338-343.
137 Passman R, Kadish A. Polymorphic ventricular tachycardia, long Q-T syndrome, and torsades de pointes. Med Clin North Am. 2001;85(2):321-341.
138 Shukla G, Chaudhry GM, Orlov M, et al. Potential proarrhythmic effect of biventricular pacing: Fact or myth? Heart Rhythm. 2005;2(9):951-956.
139 Nayak HM, Verdino RJ, Russo AM, et al. Ventricular tachycardia storm after initiation of biventricular pacing: incidence, clinical characteristics, management, and outcome. J Cardiovasc Electrophysiol. 2008;19(7):708-715.
140 Nademanee K, Taylor R, Bailey WE, et al. Treating electrical storm: sympathetic blockade versus advanced cardiac life support–guided therapy. Circulation. 2000;102(7):742-747.
141 Marrouche NF, Verma A, Wazni O, et al. Mode of initiation and ablation of ventricular fibrillation storms in patients with ischemic cardiomyopathy. J Am Coll Cardiol. 2004;43(9):1715-1720.
142 Carbucicchio C, Santamaria M, Trevisi N, et al. Catheter ablation for the treatment of electrical storm in patients with implantable cardioverter-defibrillators: Short- and long-term outcomes in a prospective single-center study. Circulation. 2008;117(4):462-469.
143 Verma A, Kilicaslan F, Marrouche NF, et al. Prevalence, predictors, and mortality significance of the causative arrhythmia in patients with electrical storm. J Cardiovasc Electrophysiol. 2004;15(11):1265-1270.
144 Hohnloser SH, Al-Khalidi HR, Pratt CM, et al. Electrical storm in patients with an implantable defibrillator: Incidence, features, and preventive therapy: Insights from a randomized trial. Eur Heart J. 2006;27(24):3027-3032.
145 Albert CM, Mittleman MA, Chae CU, et al. Triggering of sudden death from cardiac causes by vigorous exertion. N Engl J Med. 2000;343(19):1355-1361.
146 Elhendy A, Sozzi FB, van Domburg RT, et al. Relation among exercise-induced ventricular arrhythmias, myocardial ischemia, and viability late after acute myocardial infarction. Am J Cardiol. 2000;86(7):723-729.
147 A randomized trial of beta-blockade in heart failure. The Cardiac Insufficiency Bisoprolol Study (CIBIS). CIBIS Investigators and Committees. Circulation. 1994;90(4):1765-1773.
148 Viscoli CM, Horwitz RI, Singer BH. Beta-blockers after myocardial infarction: Influence of first-year clinical course on long-term effectiveness. Ann Intern Med. 1993;118(2):99-105.
149 Preliminary report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST) investigators. N Engl J Med. 1989;321(6):406-412.
150 Boutitie F, Boissel JP, Connolly SJ, et al. Amiodarone interaction with beta-blockers: Analysis of the merged EMIAT (European Myocardial Infarct Amiodarone Trial) and CAMIAT (Canadian Amiodarone Myocardial Infarction Trial) databases. The EMIAT and CAMIAT investigators. Circulation. 1999;99(17):2268-2275.
151 Cairns JA, Connolly SJ, Roberts R, Gent M. Randomised trial of outcome after myocardial infarction in patients with frequent or repetitive ventricular premature depolarisations: CAMIAT. Canadian Amiodarone Myocardial Infarction Arrhythmia Trial Investigators. Lancet. 1997;349(9053):675-682.
152 Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: Meta-analysis of individual data from 6500 patients in randomised trials. Lancet. 1997;350(9089):1417-1424.
153 Pedersen OD, Bagger H, Keller N, et al. Efficacy of dofetilide in the treatment of atrial fibrillation-flutter in patients with reduced left ventricular function: A Danish Investigations of Arrhythmia and Mortality on Dofetilide (DIAMOND) substudy. Circulation. 2001;104(3):292-296.
154 Brendorp B, Elming H, Jun L, et al. QTC interval as a guide to select those patients with congestive heart failure and reduced left ventricular systolic function who will benefit from antiarrhythmic treatment with dofetilide. Circulation. 2001;103(10):1422-1427.
155 Cameron A, Schwartz MJ, Kronmal RA, Kosinski AS. Prevalence and significance of atrial fibrillation in coronary artery disease (CASS Registry). Am J Cardiol. 1988;61(10):714-717.
156 Kannel WB, Abbott RD, Savage DD, McNamara PM. Coronary heart disease and atrial fibrillation: The Framingham Study. Am Heart J. 1983;106(2):389-396.
157 Onundarson PT, Thorgeirsson G, Jonmundsson E, et al. Chronic atrial fibrillation—epidemiologic features and 14 year follow-up: A case control study. Eur Heart J. 1987;8(5):521-527.
158 Ginks W, Sutton R, Siddons H, Leatham A. Unsuspected coronary artery disease as cause of chronic atrioventricular block in middle age. Br Heart J. 1980;44(6):699-702.
159 Moreyra AE, Horvitz L, Presant SB, Kostis JB. Resolution of complete heart block after right coronary artery angioplasty. Am Heart J. 1988;115(1 Pt 1):179-181.
160 Coplan NL, Morales MC, Romanello P, et al. Exercise-related atrioventricular block. Influence of myocardial ischemia. Chest. 1991;100(6):1728-1730.
161 Deaner A, Fluck D, Timmis AD. Exertional atrioventricular block presenting with recurrent syncope: Successful treatment by coronary angioplasty. Heart. 1996;75(6):640-641.
162 Kovac JD, Murgatroyd FD, Skehan JD. Recurrent syncope due to complete atrioventricular block, a rare presenting symptom of otherwise silent coronary artery disease: Successful treatment by PTCA. Cathet Cardiovasc Diagn. 1997;42(2):216-218.