Chapter 61 Arrhythmias and Electrolyte Disorders
Cardiac arrhythmias are an expression of the same fundamental electrophysiological principles that underlie the normal electrical behavior of the heart. The electrical activity of the heart depends on transmembrane ionic gradients and the time-dependent and voltage-dependent alterations of their conductance. The resting membrane potential (Vm) is calculated by the Goldman constant field equation1:
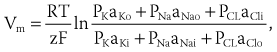
which incorporates the permeability (P) and activity (a) of all ionic species that contribute to it.
Potassium
Potassium is the most abundant intracellular cation and the most important determinant of the resting membrane potential (RMP). The electrophysiological effects of potassium depend not only on its extracellular concentration, but also on the direction (hypokalemia versus hyperkalemia) and rate of change. Hoffman and Suckling have noted that the effect of potassium on the RMP is modulated by the simultaneous calcium concentration.2 The interrelationship is such that an elevated calcium level decreases the depolarizing effect of an elevated potassium level, and low calcium levels diminish the depolarization produced by hypokalemia. When extracellular potassium levels are higher than normal, the cell membrane behaves as a potassium electrode, as described by the Nernst equation:
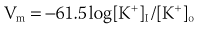
At levels of less than ∼3 mmol/L, the transmembrane potential (Vm) is less than that predicted by the Nernst equation, because hypokalemia reduces membrane permeability to potassium (Pk).3
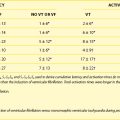
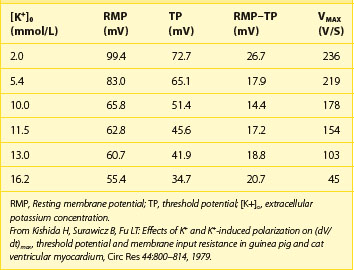
Indeed, potassium currents are modulated by the potassium gradient itself and other electrolytes as well (Table 61-1). The conductance of the inward rectifier current (iK1) is proportional to the square root of the extracellular potassium concentration [K+]o.4,5 The dependence of the activation of the delayed rectifier current (IKr) on the extracellular potassium concentration [K+]o helps explain why the action potential duration (APD) is shorter at higher [K+]o and longer at low [K+]o concentrations (see Table 61-1).6 As important as the time factor may be on the electrophysiological impact of different potassium levels, it is equally important to note that rapid fluctuations in extracellular potassium levels do occur, especially through transcellular shifts (Table 61-2). Insulin, β-adrenergic agonists, aldosterone, and changes in blood pH may independently affect serum potassium levels.6
Table 61-1 Modulation of Potassium Currents by Electrolyte Concentration
| IK1, Inward rectifier | Its conductance is proportional to the square root of [K+]o. The instantaneous inward rectification on depolarization is caused by the Mg2+ block at physiological [Mg2+]i.* |
| IKr, Delayed rectifier | Low [K+]o decreases the delayed rectifier current (IKr). |
| Ito, Transient outward | One type is voltage activated and modulated by neurotransmitters. The other type is activated by intracellular calcium. |
| IK(Ca) | Opens in the presence of high levels of intracellular calcium. |
| IK(Na) | Opens in the presence of high levels of intracellular sodium. |
* From Ishihara K, Mitsuiye T, Noma A, Takano M: The Mg2+ block and intrinsic gating underlying inward rectification of the K+ current in guinea-pig cardiac myocytes, J Physiol (Lond) 419:297–320, 1989.
Table 61-2 Factors That Affect the Transcellular Shift of Potassium
| FROM INSIDE TO OUTSIDE | FROM OUTSIDE TO INSIDE |
|---|---|
| Acidosis | Alkalosis |
| β-Adrenergic receptor stimulation | β2-Adrenergic receptor stimulation |
| Digitalis | Insulin |
| Solvent drag |
Hypokalemia
Hypokalemia is the most common electrolyte abnormality encountered in clinical practice. Potassium values of <3.6 mmol/L are seen in over 20% of hospitalized patients.8 As many as 10% to 40% of patients on thiazide diuretics and almost 50% of patients resuscitated from out-of-hospital ventricular fibrillation have low potassium levels. Hypokalemia results from decreased potassium intake, transcellular shift, and, most commonly, increased renal or extra-renal losses (Table 61-3).9,10
| Decreased intake | |
| Potassium shift into the cell (see Table 61-2) | |
| RENAL POTASSIUM LOSS | |
| Increased mineralocorticoid effects | Increased flow to distal nephron |
| Primary or secondary aldosteronism | Diuretics |
| Ectopic adrenocorticotropic hormone–producing tumor or Cushing syndrome | Salt-losing nephropathy |
| Hypomagnesemia | |
| Bartter syndrome | Nonresorbable anion |
| Licorice | Carbenicillin, penicillin |
| Renovascular or malignant hypertension | Renal tubular acidosis (type I or II) |
| Congenital abnormality of steroid metabolism | Congenital defect of distal nephron |
| Renin-producing tumor | Liddle syndrome |
| EXTRARENAL POTASSIUM LOSS | |
| Vomiting, diarrhea, laxative abuse, villous adenoma, Zollinger-Ellison syndrome | |
Electrophysiological Effects of Hypokalemia
Hypokalemia leads to a higher (more negative) RMP and, at least during electrical diastole, a decrease in membrane excitability as a result of widening of the RMP and the threshold potential (TP) difference. Low extracellular potassium decreases the delayed rectifier current (IKr), resulting in an increase in the APD and a delay in repolarization. It has been suggested that extracellular K+ ions are required to open the delayed rectifier channel.7
Most importantly, hypokalemia alters the configuration of the action potential (AP), with the duration of phase 2 first increasing and subsequently decreasing, whereas the slope of phase 3 decelerates. The latter effect leads to an AP with a long “tail,” resulting in an increase in the relative refractory period (RRP) and a decrease in the difference of the RMP from the TP during the terminal phase of the AP. Thus, cardiac tissue demonstrates increased excitability with associated ectopy for a considerable portion of the AP. Conduction slows because depolarization begins in incompletely repolarized fibers. Furthermore, hypokalemia prolongs the plateau in the Purkinje fibers but shortens it in the ventricular fibers.11 The AP tail of the conducting system prolongs more than that of the ventricles, increasing the dispersion of repolarization. Hypokalemia increases diastolic depolarization in Purkinje fibers, thereby increasing automaticity.
In summary, the electrophysiological effects of hypokalemia are (1) a decrease in conduction velocity; (2) shortening of the effective refractory period (ERP); (3) prolongation of the RRP; (4) increased automaticity; and (5) early after-depolarizations (EADs) (Box 61-1).
Electrocardiographic Manifestations of Hypokalemia
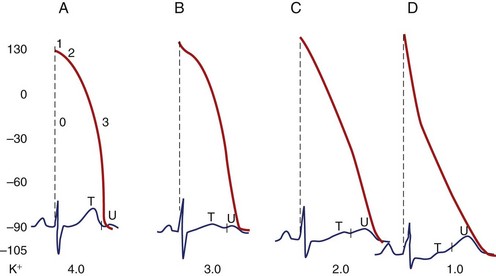
The electrocardiographic manifestations of hypokalemia can be conceptualized as those caused by its effects on repolarization and those emanating from its effects on conduction (Box 61-2).12 The electrocardiogram (ECG) changes resulting from its effects on repolarization include (1) decreased amplitude and broadening of the T waves; (2) prominent U waves; (3) ST-segment depression; and (4) T and U wave fusions, all of which is seen in severe hypokalemia (Figure 61-1). When the U wave exceeds the T wave in amplitude, the serum potassium is less than 3 mmol/L. Electrocardiographic changes caused by conduction abnormalities are seen in the more advanced stages of hypokalemia and include (1) increase in QRS duration without a concomitant change in the QRS configuration; (2) atrioventricular (AV) block; (3) cardiac arrest; (4) increase in P-wave amplitude and duration; and (5) slight prolongation of the P-R interval.
Arrhythmogenic Potential and Clinical Implications of Hypokalemia
Hypokalemia-induced hyperexcitability is clinically manifested by an increase in supraventricular and ventricular ectopy. In the Framingham Offspring Study, potassium and magnesium levels were inversely related to the occurrence of complex or frequent ventricular premature complexes (VPCs) after adjustment for covariates.13
Hypokalemia facilitates re-entry by slowing conduction during the prolonged RRP and by causing an increase in the dispersion of refractoriness. Its suppressant effect on the sodium-potassium (Na-K) pump leads to intracellular Ca2+ overload and this facilitates the development of delayed after-depolarizations (DADs) via a transient inward current (Iti). Hypokalemia enhances the propensity for ventricular fibrillation in the normal as well as the ischemic canine heart.14 An association between hypokalemia and ventricular fibrillation in patients with acute myocardial infarction has been well established.15–17
Dofetilide and quinidine were found to exert increased block of Ikr in the setting of low [K+]o providing a mechanism that explains the link between hypokalemia and torsades de pointes (Figures 61-2 and 61-3).
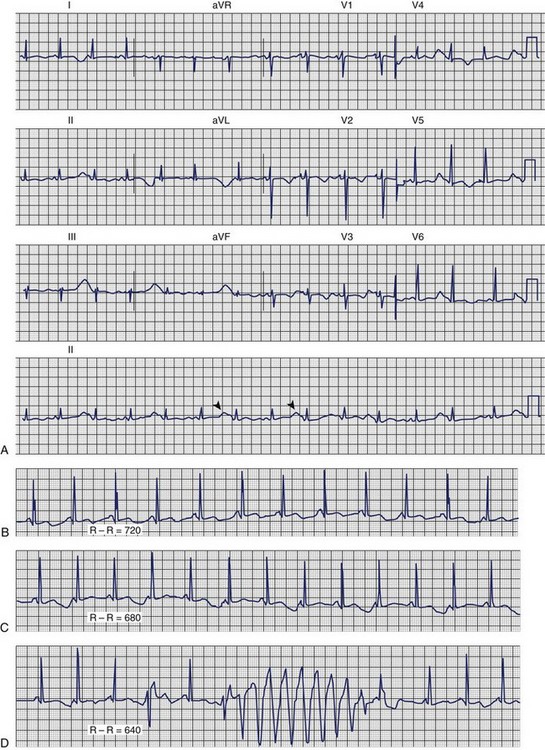
Hyperkalemia
Although less common than hypokalemia, hyperkalemia may affect approximately 8% of hospitalized patients in the United States. Hyperkalemia is seen mainly in the setting of compromised renal function, particularly in association with the administration of a variety of nephrotoxic medications. Hyperkalemia may result from either decreased excretion or a shift of potassium from within the cell (Box 61-3).
Electrophysiological Effects of Hyperkalemia
The disproportional effects of varying levels of hyperkalemia on the RMP and the TP explain the initial increase in excitability and conduction velocity and then their decrease as the potassium level increases further (Table 61-4). Mild-to-moderate levels of hyperkalemia decrease the RMP (less negative) more than the TP, thereby diminishing the difference between the two and increasing excitability. The decrease in the slope of the upstroke of the AP (dV/dT), one of the major determinants of conduction velocity, is counterbalanced by a decrease in the difference between the RMP and the TP, resulting in an ultimate increase in conduction velocity. Severe hyperkalemia is associated with an increase in the difference between the RMP and the TP, leading to a decrease in excitability. Further decrement in the AP upstroke overwhelms the positive effect of the TP decrease on conduction velocity, resulting in a definitive decrease of the latter.
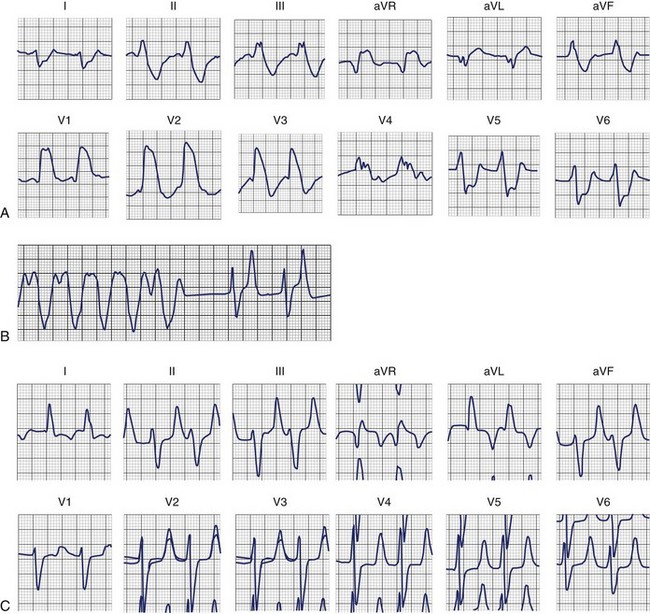
Hyperkalemia is associated with increased membrane permeability to potassium, a consequence of an increase of the inward-going rectifier current [ik1] and the delayed rectifier current (IKr).4–6 This accelerates the rate of repolarization and shortens the AP duration. Hyperkalemia preferentially shortens the plateau of the Purkinje fibers, thereby decreasing the dispersion of repolarization in the ventricle.11 Furthermore, it slows the diastolic depolarization of the Purkinje fibers.
The effects of hyperkalemia depend on the tissue involved, with the atrial myocardium being the most sensitive, the ventricular myocardium less sensitive, and the specialized tissue (sinoatrial node and His bundle) the least sensitive. In other words, the depression of excitability and conduction in the atrium occur at lower extracellular potassium levels than in other types of myocardial tissue, as exemplified by the occasional pacemaker case of atrial noncapture and ventricular capture.18 The sympathetic nervous system seems to contribute to the sinus node resistance to hyperkalemia.19
Many investigators have observed regional differences in repolarization time in the nonischemic myocardium in response to hyperkalemia. Sutton and colleagues have recorded monophasic action potentials (MAPs) from the endocardium and the epicardium in open-chest canine studies during graded intravenous infusion of potassium to a plasma level of 9 mmol/L. Their results suggested that the regional differences in repolarization times are mainly a result of local changes in activation times rather than a direct effect on the APD.20
An interesting phenomenon, the Zwaardemaker-Libbrecht effect, is the result of a change from a low extracellular potassium level to a high level and manifests itself by a transient arrest of pacemaker cells, abbreviation of APD, and hyperpolarization.21–23 This phenomenon underscores the fact that the rate of intravenous administration of potassium is more important—from a proarrhythmic standpoint—than the absolute amount of potassium administered and the final level of extracellular potassium.
Electrocardiographic Manifestations of Hyperkalemia
The ECG is not a sensitive indicator of hyperkalemia; 50% of patients with potassium levels >6.5 mmol/L will not manifest any electrocardiographic changes (Box 61-4).24 The ECG changes caused by mild potassium elevations (K = 5.5 to 7.0 mmol/L) include tall, peaked, narrow-based T waves and fascicular blocks (left anterior fascicular block and left posterior fascicular block). Moderate hyperkalemia (K = 7.5 to 10.0 mmol/L) is associated with first-degree AV block and diminished P-wave amplitude. As the potassium level increases further, the P-wave disappears, and sinus arrest as well as ST-segment depression may develop. Atypical bundle branch blocks (left bundle branch block [LBBB] and right bundle branch block [RBBB]), intraventricular conduction delays (IVCDs), ventricular tachycardia (see Figure 61-3), ventricular fibrillation, and idioventricular rhythm are more commonly seen in cases of severe hyperkalemia (K+ >10.0 mmol/L). The bundle branch blocks associated with hyperkalemia are atypical, that is, they involve the initial and terminal forces of the QRS complex. Shifts in the QRS axis indicate disproportionate conduction delays in the left bundle fascicles. These manifestations of intraventricular conduction delay correlate with a prolonged intracardiac H-V interval.25 As hyperkalemia progresses, depolarization merges with repolarization, expressed on the ECG with QT shortening and apparent ST segment elevation simulating acute injury. The latter disappears with hemodialysis—dialyzable current of injury.26 The effect of hyperkalemia on QRS morphology of ventricular paced beats have also been studied.27 Hyperkalemia is the most common electrolyte abnormality to cause loss of capture. In patients with pacemakers, hyperkalemia causes two important clinical abnormalities: (1) widening of the paced QRS complex (and paced P wave) on the basis of delayed myocardial conduction. When the K level exceeds 7 mEq/L, the intraventricular conduction velocity is usually decreased and the paced QRS complex widens (Figure 61-4); (2) increased atrial and ventricular pacing thresholds with or without increased latency.27
Box 61-4 Electrocardiogram Manifestations of Hyperkalemia
LAFB, Left anterior fascicular block; LPFB, left posterior fascicular block; LBBB, left bundle branch block; RBBB, right bundle branch block; IVCD, intraventricular conduction delay.
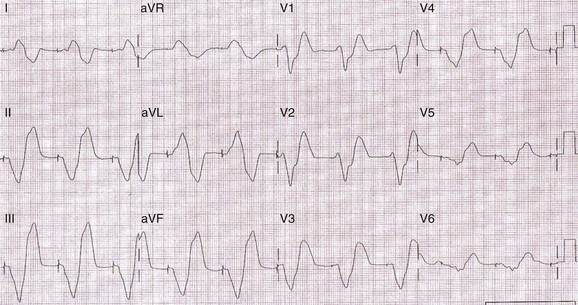
The parallel changes in the AP and ECG can be appreciated better by reviewing Figure 61-5.24
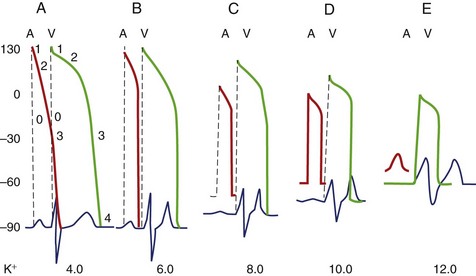
Arrhythmogenic Potential and Clinical Implications of Hyperkalemia
Potassium and Myocardial Ischemia
In the early phases of an ischemic insult, the cardiac membrane becomes increasingly permeable to potassium. After coronary ligation in dogs and pigs, the rise in extracellular potassium concentration was seen to result in currents of injury, refractory period shortening, conduction slowing, and ventricular fibrillation.28,29 During ischemia, APD shortening is more pronounced and the conduction velocity is slower in failing than in the control myocardium. Extracellular potassium [K+]o reaches higher values during acute ischemia in failing versus normal myocardium. Increased spatial dispersion in electrophysiological parameters and [K+]o over the ischemic border in failing hearts may explain the higher propensity for re-entrant arrhythmias during acute regional ischemia.30
Calcium
Isolated abnormalities of calcium concentration produce clinically significant electrophysiological effects only when they are extreme in either direction. As expected by reviewing the Goldman equation, extracellular calcium concentrations in the physiological range have no appreciable effect on the RMP. Hypocalcemia and hypercalcemia have opposing effects on the APD and ERP by affecting intracellular calcium concentration and modulating potassium currents.2
Hypocalcemia
Hypocalcemia is most frequently seen in the setting of chronic renal insufficiency and is usually associated with other electrolyte abnormalities. Generally, hypocalcemia may result from decreased intake or absorption or an increase in calcium loss (Box 61-5).
Hypercalcemia
Hypercalcemia occurs most commonly in the setting of hyperparathyroidism or as a consequence of several malignancies. A plethora of other causes account for the remaining clinically encountered cases (Box 61-6).
Electrophysiological Effects and Electrocardiographic Manifestations of Hypercalcemia
High extracellular calcium levels shorten the AP plateau, the total APD, and, consequently, the duration of the ERP. A study of the effect of hypercalcemia on the guinea pig ventricular AP suggested that a decrease in the inward Na+/Ca2+ exchange current might be largely responsible for the shortening of the AP.31 Elevated extracellular Ca2+ concentration has a stabilizing effect on the membrane, increasing the extent of depolarization needed to initiate an AP. In addition, hypercalcemia has a positive inotropic effect, decreases excitability, and slightly increases the rate of diastolic depolarization in the Purkinje fibers.32 The electrocardiographic changes as a result of hypercalcemia are limited to shortening or elimination of the ST segment and decreased QTc interval.
Magnesium
Magnesium is the second most abundant intracellular cation after potassium. The significance of magnesium disorders has been debated because of difficulties in accurate measurement and their frequent association with other electrolyte abnormalities.33,34 It is an important cofactor in several enzymatic reactions that contribute to normal cardiovascular physiology. Magnesium deficiency is common, but its electrophysiological sequelae have escaped even the closest scrutiny. Magnesium therapy in pharmacologic doses has been beneficial in treating torsades de pointes. Magnesium toxicity rarely occurs except in patients with renal dysfunction.
Electrophysiological Effects and Electrocardiographic Manifestations of Hypomagnesemia and Hypermagnesemia
In the presence of extremely low extracellular calcium concentrations, magnesium exerts an effect on the current or currents that modulate the duration of the ventricular AP plateau. Hoffman and Suckling found that in the presence of normal calcium concentrations, magnesium deficiency had little effect on the AP of the canine papillary muscle.2 However, when the calcium concentration was lowered to one tenth of the normal, complete omission of magnesium in the superfusate prolonged the AP plateau, which was already increased in duration by low calcium, from a normal value of 100 to 150 ms to 1000 ms or more.
Magnesium blocks the calcium channel, shifts the steady-state inactivation curve of the fast sodium channel in the hyperpolarizing direction, modifies the effect of hyperkalemia, and exerts modulating effects on several potassium currents. Hypermagnesemia depresses AV and intraventricular conduction. DiCarlo and coworkers observed the following electrocardiographic effects of intravenous administration of magnesium in patients with normal baseline serum magnesium and other electrolyte levels35: (1) prolongation of sinus node recovery time (SNRT) and corrected sinus node recovery time (CSNRT); (2) prolongation of the AV nodal functional, relative, and effective refractory periods; (3) a small increase in QRS duration during ventricular pacing at cycle lengths of 250 and 500 ms; and (4) a significant increase in the atrium–His interval and the atrial paced cycle length causing AV node Wenckebach conduction.
Kulick and associates, in studying healthier hearts, noted the following ECG effects of intravenous magnesium administration36: (1) significant prolongation of the P-R interval from 145 to 155 ms after magnesium infusion; (2) prolongation of the atrium–His interval; (3) prolongation of the sinoatrial conduction time (SACT); (4) prolongation of the AV nodal effective refractory periods; and (5) no significant increase in SNRT in the atrial paced cycle length which causes AV node Wenckebach conduction or in QRS duration. Hypermagnesemia and hypomagnesemia do not produce specific ECG changes.
Magnesium and Torsades de Pointes
The administration of intravenous magnesium sulfate to patients with prolonged Q-T interval and torsades de pointes, whether the initial magnesium level is normal or low, may suppress ventricular tachycardia. Takanaka and colleagues studied the effects of magnesium and lidocaine on the APD and on barium-induced EADs in canine Purkinje fibers. Their data suggested that hypomagnesemia may be arrhythmogenic when combined with hypokalemia and bradycardia, and that magnesium administration may suppress triggered activity, mainly by directly preventing the development of triggered APs.37 In conclusion, magnesium sulfate is a very effective and safe treatment for torsades de pointes.38
Magnesium and Heart Failure
In a sample of ambulatory patients with heart failure, magnesium depletion in serum and tissue did not appear to occur more commonly in those with serious ventricular arrhythmias than in those without serious ventricular arrhythmias.39 In patients with moderate to severe heart failure, serum magnesium does not appear to be an independent risk factor for either sudden cardiac death or all-cause mortality.40 Hypomagnesemia was found to be associated with an increase in frequency of ventricular couplets, but it did not lead to a higher incidence of clinical events.40
Magnesium and Myocardial Infarction
Magnesium administration has been found to have a positive effect on the consequences of myocardial infarction in experimental models. Its effect in the clinical setting of myocardial infarction has been controversial. In the LIMIT-2 (Leicester Intravenous Magnesium Intervention Trial–2) study, magnesium administration was noted to have a positive effect on mortality rate, a finding that has not been reproduced in the ISIS-4 (International Study of Infarct Survival–4) trial.41–46
Relationship Among Potassium, Magnesium, and Cardiac Arrhythmias
No specific electrophysiological effects or arrhythmias have been linked to isolated magnesium deficiency. Nonetheless, magnesium may influence the incidence of cardiac arrhythmias through a direct effect by modulating the effects of potassium or through its action as a calcium channel blocker. Magnesium deficiency is thought to interfere with the normal functioning of membrane ATPase and, thus, the pumping of sodium out of the cell and potassium into the cell. This affects the transmembrane equilibrium of potassium, which may result in changes in the RMP, changes in potassium conductance across the cell membrane, and disturbances in the repolarization phase.47
Lithium
Although lithium is not a naturally occurring electrolyte, it is frequently encountered clinically in the treatment of manic-depressive disorders, and, as such, its potential adverse effects on the sinoatrial node should not be overlooked. Lithium is associated with sinoatrial node dysfunction (sinus bradycardia, sinoatrial arrest, or exit block, either type I or type II) and reversible T-wave changes.48–50
Key References
Barold S, Falkoff MD, Ong LS, Heinle RA. Hyperkalemia-induced failure of atrial capture during dual-chamber cardiac pacing. J Am Coll Cardiol. 1987;10:467-469.
Barold SS, Leonelli F, Herweg B. Hyperkalemia during cardiac pacing. PACE. 2007;30:1-3.
DiCarlo LAJr, Morady F, DeBuitleir M, et al. Effects of magnesium sulfate on cardiac conduction and refractoriness in humans. J Am Coll Cardiol. 1986;7:1356-1362.
Ettinger PO, Regan TJ, Oldewurtel HA. Hyperkalemia, cardiac conduction and the electrocardiogram: A review. Am Heart J. 1974;88:360-371.
Habbab MA, El-Sherif N. TU alternans, long QTU, and torsades de pointes. PACE. 1992;15:916-931.
Hoffman BF, Suckling EE. Effect of several cations on transmembrane potentials of cardiac muscle. Am J Physiol. 1956;186:317-324.
ISIS-4. A randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. ISIS-4 (Fourth International Study of Infarct Survival) Collaborative Group [see comments]. Lancet. 1995;345:669-685.
Keller PK, Aronson RS. The role of magnesium in cardiac arrhythmias. Prog Cardiovasc Dis. 1990;32:433-448.
Nordrehaug JE, Johannessen K, Von der Lippe G. Serum potassium concentration as a risk factor of ventricular arrhythmias early in acute myocardial infarction. Circulation. 1985;71:645-649.
Pelzer D, Trautwein W. Currents through ionic channels in multicellular cardiac tissue and single heart cells. Experientia. 1987;43:1153-1162.
Schulman M, Narins RG. Hypokalemia and cardiovascular disease. Am J Cardiol. 1990;65:4E-9E.
Sheu SS, Korth M, Lathrop DA, Fozzard HA. Intra- and extracellular K+ and Na+ activities and resting membrane potential in sheep cardiac Purkinje strands. Circ Res. 1980;47:692-700.
Surawicz B. Relation between electrocardiogram and electrolytes. Am Heart J. 1967;73:814-834.
Takanaka C, Ogunyankin KO, Sarma JS, Singh BN. Antiarrhythmic and arrhythmogenic actions of varying levels of extracellular magnesium: Possible cellular basis for the differences in the efficacy of magnesium and lidocaine in torsades de pointes. J Cardiovasc Pharmacol Ther. 1997;2:125-134.
Wellens HJ, Cats VM, Duren DR. Symptomatic sinus node abnormalities following lithium carbonate therapy. Am J Med. 1975;59:285-287.
1 Goldman DE. Potential, impedance and rectification in membranes. J Gen Physiol. 1943;27:37-60.
2 Hoffman BF, Suckling EE. Effect of several cations on transmembrane potentials of cardiac muscle. Am J Physiol. 1956;186:317-324.
3 Sheu SS, Korth M, Lathrop DA, Fozzard HA. Intra- and extracellular K+ and Na+ activities and resting membrane potential in sheep cardiac Purkinje strands. Circ Res. 1980;47:692-700.
4 Sakmann B, Trube G. Conductance properties of single inwardly rectifying potassium channels in ventricular cells from guinea pig heart. J Physiol. 1984;347:641-657.
5 Pelzer D, Trautwein W. Currents through ionic channels in multicellular cardiac tissue and single heart cells. Experientia. 1987;43:1153-1162.
6 Yang T, Roden DM. Extracellular potassium modulation of drug block of Ikr. Circulation. 1996;93:407-411.
7 Brown MJ, Brown DC, Murphy MB. Hypokalemia from beta2-stimulation by circulating epinephrine. N Engl J Med. 1983;309:1414-1419.
8 Paice BJ, Paterson KR, Onyanga-Omara F, et al. Record linkage study of hypokalemia in hospitalized patients. Postgrad Med J. 1986;62:187-191.
9 Schulman M, Narins RG. Hypokalemia and cardiovascular disease. Am J Cardiol. 1990;65:4E-9E.
10 Thompson RG, Gobb LA. Hypokalemia after resuscitation out- of-hospital ventricular fibrillation. JAMA. 1982;248:2860-2863.
11 Gettes L, Surawicz B. Effects of low and high concentrations of potassium on the simultaneously recorded Purkinje and ventricular action potentials of the perfused pig moderator band. Circ Res. 1968;23:717-729.
12 Surawicz B, Braun HA, Crum WB, et al. Quantitative analysis of the electrocardiographic pattern of hypopotassemia. Circulation. 1957;16:750-763.
13 Tsuji H, Venditti FJJr, Evans JC, et al. The associations of levels of serum potassium and magnesium with ventricular premature complexes (the Framingham Heart Study). Am J Cardiol (United States). 1994;74:232-235.
14 Hohnloser SH, Verrier RL, Lown B, Raeder EA. Effect of hypokalemia on susceptibility to ventricular fibrillation in the normal and ischemic canine heart. Am Heart J. 1986;112:32-35.
15 Nordrehaug JE, Johannessen K, Von der Lippe G. Serum potassium concentration as a risk factor of ventricular arrhythmias early in acute myocardial infarction. Circulation. 1985;71:645-649.
16 Cooper WD, Kuan P, Reuben SR, Vanderburg MJ. Cardiac arrhythmias following acute myocardial infraction: Association with serum potassium level and prior diuretic therapy. Eur Heart J. 1984;5:464-469.
17 Nordrehaug JE, Von der Lippe G. Hypokalemia and ventricular fibrillation in acute myocardial infarction. Br Heart J. 1983;50:525-529.
18 Barold S, Falkoff MD, Ong LS, Heinle RA. Hyperkalemia-induced failure of atrial capture during dual-chamber cardiac pacing. J Am Coll Cardiol. 1987;10:467-469.
19 Vassalle M, Greineder JK, Stuckey JH. Role of the sympathetic nervous system in the sinus node resistance to high potassium. Circ Res. 1973;32:348-355.
20 Sutton PM, Taggart P, Spear DW, et al. Monophasic action potential recordings in response to graded hyperkalemia in dogs. Am J Physiol. 1989;256(4 Pt2):H956-H961.
21 Zwaardemaker H. On physiological radio-activity. J Physiol. 1920;53:273-289.
22 Libbrecht W. Le paradoxe cardiaque. Arch Int Physiol. 1928;16:448-452.
23 Ito S, Surawicz B. Transient, “paradoxical” effects of increasing extracellular K+ concentration on transmembrane potential in canine cardiac Purkinje fibers. Circ Res. 1977;41:799-807.
24 Surawicz B. Relation between electrocardiogram and electrolytes. Am Heart J. 1967;73:814-834.
25 Ettinger PO, Regan TJ, Oldewurtel HA. Hyperkalemia, cardiac conduction and the electrocardiogram: A review. Am Heart J. 1974;88:360-371.
26 Levine HD, Wanzer SH, Merrill JP. Dialyzable currents of injury in potassium intoxication resembling acute myocardial infarction or pericarditis. Circulation. 1956;13:29-36.
27 Barold SS, Leonelli F, Herweg B. Hyperkalemia during cardiac pacing. PACE. 2007;30:1-3.
28 Hill JL, Gettes LS. Effect of acute coronary artery occlusion on local myocardial extracellular K+ activity in swine. Circulation. 1980;61:768-778.
29 Coronel R, Fiolet JW, Wilms-Schopman FJ, et al. Distribution of extracellular potassium and its relation to electrophysiologic changes during acute myocardial ischemia in the isolated perfused porcine heart. Circulation. 1988;77:1125-1138.
30 Vermeulen JT, Tan HL, Rademaker H, et al. Electrophysiologic and extracellular ionic changes during acute ischemia in failing and normal rabbit myocardium. J Mol Cell Cardiol. 1996;28:123-131.
31 Leitch SP, Brown HF. Effect of raised extracellular calcium on characteristics of the guinea pig ventricular action potential. J Mol Cell Cardiol. 1996;28:541-551.
32 Weidmann S. Effects of calcium ions and local anesthetics on electrical properties of Purkinje fibers. J Physiol. 1955;129:568-582.
33 Keller PK, Aronson RS. The role of magnesium in cardiac arrhythmias. Prog Cardiovasc Dis. 1990;32:433-448.
34 Surawicz B, Lepeschkin E, Herrlich HC, Hoffman BF. Low and high magnesium concentrations at various calcium levels: Effect on the monophasic action potential, electrocardiogram and contractility of isolated rabbit hearts. Circ Res. 1959;9:811-818.
35 DiCarlo LAJr, Morady F, DeBuitleir M, et al. Effects of magnesium sulfate on cardiac conduction and refractoriness in humans. J Am Coll Cardiol. 1986;7:1356-1362.
36 Kulick DL, Hong R, Ryzen E. Electrophysiologic effects of intravenous magnesium in patients with normal conduction systems and no clinical evidence of significant cardiac disease. Am Heart J. 1988;115:367-373.
37 Takanaka C, Ogunyankin KO, Sarma JS, Singh BN. Antiarrhythmic and arrhythmogenic actions of varying levels of extracellular magnesium: Possible cellular basis for the differences in the efficacy of magnesium and lidocaine in torsades de pointes. J Cardiovasc Pharmacol Ther. 1997;2:125-134.
38 Tzivoni D, Banai S, Schuger C, et al. Treatment of torsades de pointes with magnesium sulfate. Circulation. 1988;77:392-397.
39 Ralston MA, Murname MR, Unverferth DV, Leier CV. Serum and tissue magnesium concentrations in patients with heart failure and serious ventricular arrhythmias. Ann Int Med. 1990;113:841-846.
40 Eichhorn EJ, Tandon PK, DiBianco R, et al. Clinical and prognostic significance of serum magnesium concentration in patients with severe chronic congestive heart failure: The PROMISE study. J Am Coll Cardiol. 1993;21:634-640.
41 Baxter GF, Sumeray MS, Walker JM. Infarct size and magnesium: Insights into LIMIT-2 and ISIS-4 from experimental studies [see comments]. Lancet. 1996;348:1424-1426.
42 Woods KL, Fletcher S. Long-term outcome after intravenous magnesium sulphate in suspected acute myocardial infarction: The second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2) [see comments]. Lancet. 1994;343:816-819.
43 Woods KL, Fletcher S, Roffe C, Haider Y. Intravenous magnesium sulphate in suspected acute myocardial infarction: Results of the second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2) [see comments]. Lancet. 1992;339:1553-1558.
44 ISIS-4. A randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. ISIS-4 (Fourth International Study of Infarct Survival) Collaborative Group [see comments]. Lancet. 1995;345:669-685.
45 Fourth International Study of Infarct Survival. Protocol for a large simple study of the effects of oral mononitrate, of oral captopril, and of intravenous magnesium. ISIS-4 collaborative group. Am J Cardiol. 1991;68:87D-100D.
46 Flather M, Pipilis A, Collins R, et al. Randomized controlled trial of oral captopril, of oral isosorbide mononitrate and of intravenous magnesium sulphate started early in acute myocardial infarction: Safety and haemodynamic effects. ISIS-4 (Fourth International Study of Infarct Survival) Pilot Study Investigators [see comments]. Eur Heart J. 1994;15:608-619.
47 Dyckner T, Wester PO. Relation between potassium, magnesium and cardiac arrhythmias. Acta Med Scand. 1981;647(Suppl):163-169.
48 Tilkian AG, Schroeder JS, Kao J, Hultgren H. Effect of lithium on cardiovascular performance: Report on extended ambulatory monitoring and exercise testing before and during lithium therapy. Am J Cardiol. 1976;38:701-708.
49 Wellens HJ, Cats VM, Duren DR. Symptomatic sinus node abnormalities following lithium carbonate therapy. Am J Med. 1975;59:285-287.
50 Carmeliet EE. Influence of lithium ions on transmembrane potential and cation content of cardiac cells. J Gen Physiol. 1964;47:501-530.