22 Arrhythmias
Normal cardiac electrophysiology
Cardiac action potential
The resting membrane potential of -60 to -90 mV occurs because the intracellular potassium (K+) concentration is much higher than the extracellular K+ concentration as a result of a transmembrane pump known as Na+–K+–ATPase, which pumps K+ ions into the cell in exchange for sodium (Na+) ions. K+ ions diffuse out of the cell through selective K+ channels (the inward rectifier current or IK1) unaccompanied by anions, resulting in a net loss of charge and thus a negative resting, diastolic or phase 4, transmembrane potential (Fig. 22.1A).
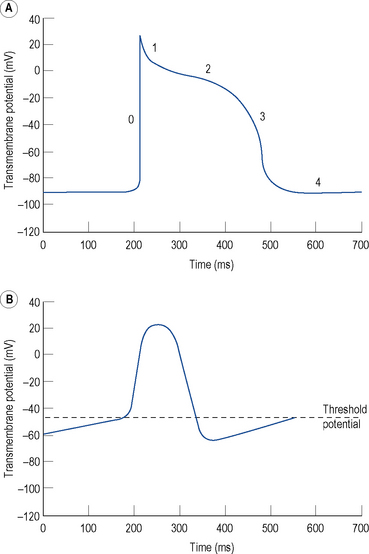
Certain specialised myocytes form the cardiac conduction system and these cells have pacemaker activity, that is, they are capable of generating their own action potentials due to gradual depolarisation of the transmembrane potential during diastole (phase 4), referred to as the pacemaker potential (Fig. 22.1B). The pacemaker potential occurs as a result of (i) a gradual reduction in an outward K+ current called the delayed rectifier (IK) current, (ii) increasing dominance of an inward current of Na+ and some Ca2+ ions known as If (f stands for ‘funny’) and (iii) an inward calcium current ICa through voltage-gated calcium channels. As a result of the pacemaker potential, the transmembrane potential gradually becomes less negative until a threshold potential is reached at which an action potential is triggered. The rate of depolarisation of the pacemaker potential, and hence the heart rate, is influenced by the autonomic nervous system. Sympathetic nervous system activation and circulating catecholamines increase the heart rate by binding to ß1-adrenoreceptors leading to an increase in intracellular cyclic AMP, which results in changes to the permeability of the various ion channels responsible for the pacemaker current. Parasympathetic nervous system activation, mediated by muscarinic cholinergic receptors, has the opposite effect.
The rapid depolarisation of the cardiac action potential (Fig. 22.1A, phase 0) occurs because of a rapid increase in the permeability of the cell membrane to Na+ ions, which enter rapidly through ‘fast’ Na+ channels in a current known as INa. The INa current is brief as the ‘fast’ Na+ channels inactivate rapidly. The early phase of repolarisation (phase 1) is due to closure of the fast Na+ channels, an outward K+ current known as Ito (to – transient outward) and a further K+ current known as the ultra-rapid component of the delayed rectifier current or IKur. The plateau phase (phase 2) of the cardiac action potential occurs because the inward movement of Ca2+ ions (ICa) is balanced by the outward movement of K+ ions. Repolarisation (phase 3) occurs as ICa diminishes and two further components of the delayed rectifier (IK) current known as the rapid (IKr) and slow (IKs) components predominate, with an important contribution from IK1.
There is considerable variation in the expression of trans-membrane ion channels in different parts of the heart, with corresponding variation in the morphology of the action potential. The most marked example is that myocytes in the sinus and AV nodes contain few Na+ channels. The upstroke of the action potential in these cells is due, predominantly, to the influx of Ca2+ ions and, therefore, is considerably slower than the upstroke in other myocytes (Fig. 22.1B). The variation in ion channel expression throughout the heart is essential for normal cardiac function, helps to explain the pathophysiology of many inherited and acquired diseases complicated by cardiac arrhythmia and accounts for the relative selectivity of antiarrhythmic and other drugs for certain parts of the heart.
Normal cardiac conduction
During normal sinus rhythm (Fig. 22.2), an activation wavefront begins in the sinus node, a group of cells with pacemaker activity on the upper free wall of the right atrium. The rate of diastolic depolarisation and hence the rate of discharge of the sinus node is increased by sympathetic nerve stimulation, circulating catecholamines or sympathomimetic drugs mediated by ß1-adrenoreceptors on the cell membranes of the sinus node myocytes. Parasympathetic (vagus) nerve stimulation exerts the opposite effect, mediated by muscarinic cholinergic receptors.
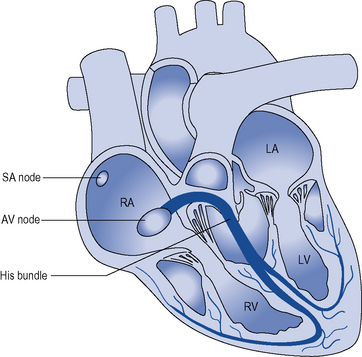
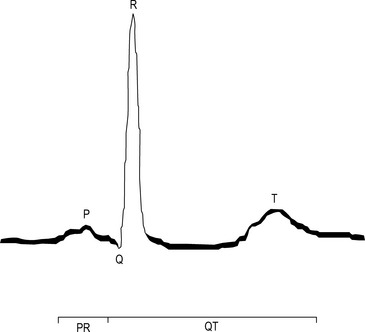
An activation wavefront spreads across the atrial myocardium, leading to atrial contraction and generating the P wave on the surface electrocardiogram (ECG; Fig. 22.3). The last part of the atria to be activated is the atrioventricular (AV) node, the electrical and structural properties of which result in a slow conduction velocity, allowing atrial emptying to be completed before ventricular contraction begins and represented by the PR interval on the ECG. Conduction velocity in the AV node is increased by sympathetic nerve stimulation, circulating catecholamines or sympathomimetic drugs, mediated by ß1-adrenoreceptors while parasympathetic (vagus) nerve stimulation exerts the opposite effect via muscarinic cholinergic receptors.
Arrhythmia mechanisms
Cardiac arrhythmias occur because of abnormalities of impulse formation or propagation.
Abnormal impulse formation
Abnormal automaticity
Automaticity is another term for pacemaker activity, a characteristic possessed by all cells of the specialised cardiac conduction system during health and, potentially, by other cardiac myocytes during certain disease states. The rate of firing of a pacemaker cell is largely determined by the duration of the phase 4 diastolic interval (Fig. 22.4). This in turn is determined by (i) the maximum diastolic potential following repolarisation of the preceding action potential, (ii) the slope of diastolic depolarisation due to pacemaker currents and (iii) the threshold potential for generation of a new action potential. In the healthy state, there is a hierarchy of firing rates within the specialised conduction system with the highest rate in the sinus node followed by the AV node and then the His–Purkinje system. The sinus node is, therefore, the dominant pacemaker and determines the heart rate, while the pacemaker activity in the distal conduction system is ‘overdriven’ by the sinus node. Abnormal automaticity describes either accelerated pacemaker activity in cells of the distal cardiac conduction system such that they escape from overdrive suppression by the sinus node, or the development of pacemaker activity in cells that do not form part of the cardiac conduction system.
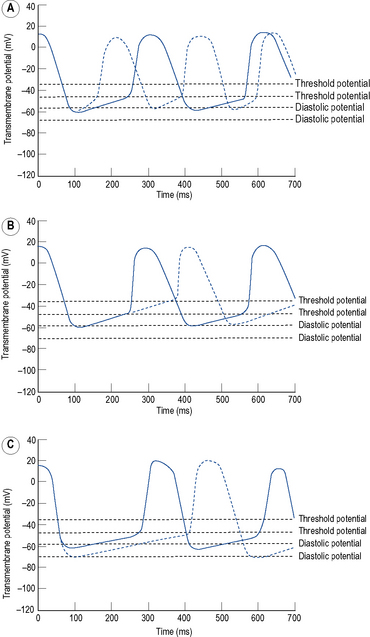
Triggered activity
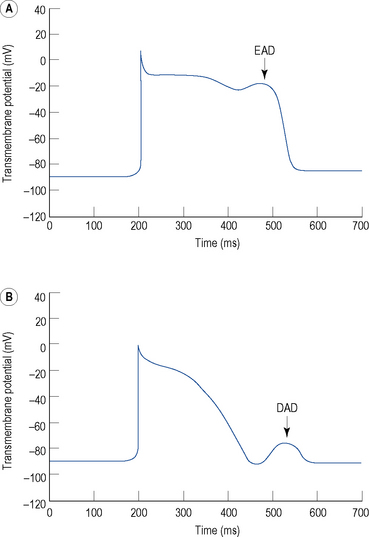
Triggered activity describes impulse formation dependent upon afterdepolarisations. Early afterdepolarisations (EADs) occur during phase 2 or 3 of the cardiac action potential whereas delayed afterdepolarisations (DADs) occur during phase 4 (Fig. 22.5). In both cases, afterdepolarisation may reach the threshold potential required for generation of a new action potential.
Abnormal impulse propagation
Re-entry
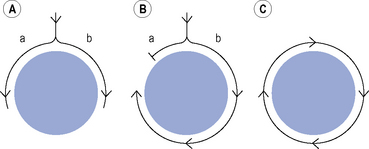
Many clinically important arrhythmias are due to re-entry, in which an activation wavefront rotates continuously around a circuit. Re-entry depends upon a trigger in the form of a premature beat, and a substrate, that is, the re-entry circuit itself. A precise set of electrophysiological conditions must be met in order for re-entry to occur (Fig. 22.6): (i) there must be a central non-conducting obstacle around which the re-entry circuit develops, (ii) a premature beat must encounter unidirectional conduction block in one limb (a) of the re-entry circuit, (iii) conduction must proceed slowly enough down the other limb (b) of the re-entry circuit that electrical excitability has returned in the original limb (a), allowing the activation wavefront to propagate in a retrograde direction along that limb, and (iv) the circulating activation wavefront must continue to encounter electrically excitable tissue. This is a function of the length of the re-entry circuit, the conduction velocity of the activation wavefront and the effective refractory period of the myocardium throughout the circuit. Class I antiarrhythmic drugs block sodium channels and, therefore, reduce the amplitude and rate of rise of the cardiac action potential and in so doing, reduce the conduction velocity of an activation wavefront. Class I antiarrhythmic drugs may exert their major antiarrhythmic effect by abolishing conduction altogether in areas of diseased myocardium forming part of a re-entry circuit in which conduction is already critically depressed. Class III antiarrhythmic drugs prolong cardiac APD and hence the refractory period. If previously activated cells in a re-entry circuit (the ‘tail’) remain refractory when the re-entrant wavefront (the ‘head’) returns to that area, conduction will fail and re-entry will be abolished. Drug-induced prolongation of the refractory period may, therefore, terminate and/or prevent re-entrant arrhythmias.
Clinical problems
Patients with a cardiac arrhythmia may present with a number of symptoms:
Diagnosis
Management
Supraventricular tachycardias
These are tachycardias arising from or involving the atria.
Atrial flutter
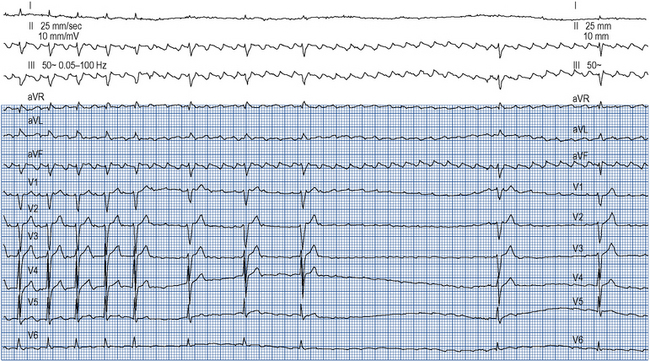
Atrial flutter confers a risk of thromboembolism similar to that of AF and this risk should be managed in the same way. Emergency management of atrial flutter is dictated by the clinical presentation but may include d.c. cardioversion or ventricular rate control with drugs which increase the refractory period of the AV node such as ß-blockers, verapamil, diltiazem or digoxin. ß-Blockers, verapamil and digoxin may be given intravenously. As the re-entry circuit is confined to the right atrium and does not involve the AV node, adenosine will not terminate atrial flutter but will produce transient AV block, allowing the characteristic flutter waves to be seen on the ECG (Fig. 22.7).
Junctional re-entry tachycardia
Two mechanisms account for most junctional re-entry tachycardias: both involve a macroreentry circuit (Fig. 22.8). AV nodal re-entry tachycardia (AVNRT) rotates around a circuit including the AV node itself and the so-called AV nodal fast and slow pathways, which feed into the AV node. Atrioventricular re-entry tachycardia (AVRT) comprises a re-entry circuit involving the atrial myocardium, the AV node, the ventricular myocardium and an accessory pathway, a congenital abnormality providing a second electrical connection between the atria and ventricles in addition to the His bundle, thus forming a potential re-entry circuit.
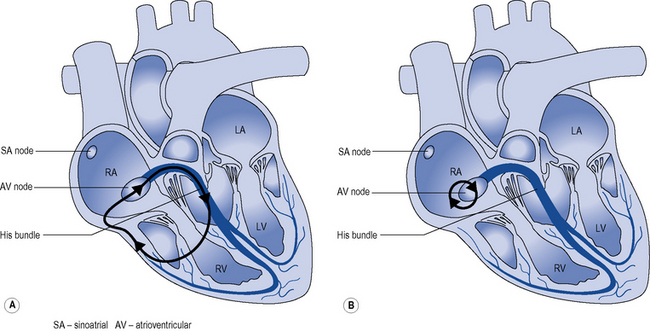
Many accessory pathways conduct only retrogradedly from the ventricles to the atrium. In these cases, the ECG during sinus rhythm appears normal and the accessory pathway is described as ‘concealed’. Other accessory pathways conduct anterogradely and retrogradely. In these cases, the ECG during sinus rhythm is abnormal and is described as having a Wolff–Parkinson–White pattern (Fig. 22.9). This abnormality is characterised by a short PR interval as the conduction velocity of an accessory pathway is usually faster than that of the AV node, and a delta wave, a slurred onset to the QRS complex which occurs because an accessory pathway inserts into ventricular myocardium which conducts more slowly than the His–Purkinje system.
Atrial fibrillation
The clinical importance of AF results from:
Paroxysmal: self-limiting episodes of AF lasting no more than 7 days
Persistent: AF lasting more than 7 days or requiring cardioversion
Longstanding persistent: continuous AF for more than 1 year
Permanent: where a decision has been made not to attempt cure of persistent AF.
Stroke risk
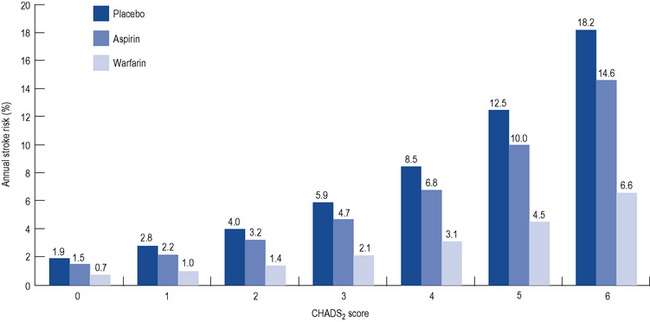
All patients presenting with AF (or atrial flutter) should undergo assessment of their risk of stroke. Although various risk stratification schemes exist, they are exemplified by the CHADS2 score, which assigns one point each for Congestive cardiac failure, Hypertension, Age >75 years, and Diabetes mellitus and two points for if there is a history of previous Stroke. Surprisingly, the frequency of AF episodes does not seem to influence stroke risk. The risk of stroke is directly proportional to the CHADS2 score. Meta-analysis of numerous trials of stroke prevention in AF has suggested a relative risk reduction for stroke of 64% with warfarin and 22% with aspirin (Fig. 22.10).
Long-term management
There has been considerable debate about the pros and cons of a rate-control strategy, focusing on achieving adequate control of the ventricular rate with drugs modifying the AV node versus a rhythm-control strategy, in which an attempt is made to restore and maintain sinus rhythm with antiarrhythmic drugs ± d.c. cardioversion. These strategies have been compared in several large-scale prospective randomised trials, which have failed to demonstrate an advantage to a rhythm-control strategy. Sinus rhythm has been associated with a 47% reduction in all-cause mortality, although this benefit was offset by a 49% increase in mortality associated with antiarrhythmic drugs (AFFIRM investigators, 2004). In that study, a wide variety of antiarrhythmic drugs were used at the discretion of the investigator. These included several class I antiarrhythmic drugs, sotalol and amiodarone. A rhythm-control strategy may be more appropriate in (i) younger patients, (ii) those with paroxysmal AF, (iii) AF associated with heart failure and (iv) patients who remain symptomatic despite adequate ventricular rate control.
Rhythm control
Although sinus rhythm can be restored in most patients through d.c. cardioversion or antiarrhythmic drugs, alone or in combination, most patients will revert to AF without further treatment. The SAFE-T trial examined 665 patients with AF of at least 72 h duration. Sinus rhythm was restored with antiarrhythmic drugs (sotalol or amiodarone) or placebo, supplemented where necessary by d.c. cardioversion (Singh et al., 2005). Patients were then maintained on placebo, sotalol or amiodarone. By 2 years, that probability of remaining in sinus rhythm was 10% (placebo), 30% (sotalol) and 50% (amiodarone). Another similar study (Roy et al., 2000) demonstrated equivalent efficacy of sotalol and the class Ic antiarrhythmic propafenone in the maintenance of sinus rhythm (40% at 2 years) and confirmed the superiority of amiodarone (60%). Most heart rhythm specialists consider that the toxicity of amiodarone precludes its long-term use for the management of AF. Dronedarone, it was hoped, would provide efficacy similar to amiodarone but it has been shown to be less effective, although it has far fewer side effects.
Ventricular tachyarrhythmias
Ventricular fibrillation
Channelopathies
Emergency management of ventricular arrhythmias
VF and pulseless VT should be managed according to Resuscitation Council (UK) guidelines for advanced life support, which are updated periodically and can be found at http://www.resus.org.uk.
Ongoing management of ventricular arrhythmias
Most patients with ischaemic heart disease should be treated with aspirin, statins, angiotensin converting enzyme (ACE) inhibitors or angiotensin II receptor antagonists and ß-blockers. ß-Blockers and ACE inhibitors reduce somewhat the risk of sudden cardiac death. Although these patients remain at high risk of sudden cardiac death due to recurrent ventricular tachyarrhythmias, there is no role for the routine use of antiarrhythmic drugs. In patients with complex ventricular ectopy and impaired left ventricular systolic function following acute myocardial infarction, class IC antiarrhythmic drugs including flecainide were shown in the CAST study (Echt et al., 1991) to increase mortality while in other high-risk groups amiodarone has been shown to have no effect on all-cause mortality (Cairns et al., 1997; Julian et al., 1997). All patients should be considered for an ICD. These devices have been shown to improve prognosis following:
Bradycardia
Third degree (‘complete heart block’). No P waves conduct to the ventricles.
The management of bradycardia is as follows:
In an emergency situation, drugs may be used in an attempt to support the heart rate until trans-venous pacing can be established. The most useful drugs in this situation are atropine in 500 μcg boluses up to a total of 3 mg, adrenaline infused at a rate of 2–10 μcg/min or isoprenaline 1–10 μcg/min, titrated against heart rate. External pacing is another useful measure until transvenous pacing can be established. More detailed guidance on the emergency management of bradycardia may be viewed at http://www.resus.org.uk.
Drug therapy
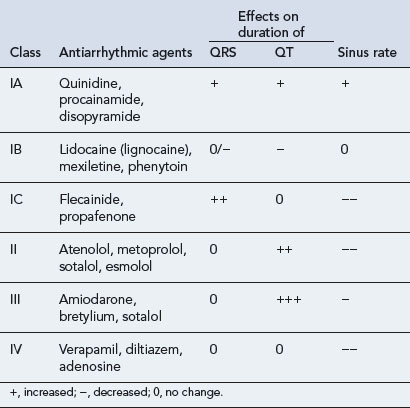
Vaughan–Williams Classification
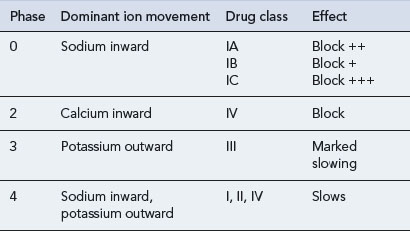
All antiarrhythmic drugs act by altering the movement of electrolytes across the myocardial cell membrane. The Vaughan–Williams classification groups drugs according to their ability to block the movement of one or more of these ions across the myocardial cell membrane. Most drugs have several modes of action, and their effectiveness as antiarrhythmic agents depends upon the summation of these effects. The effect of the different drug classes on the various phases of the action potential in His–Purkinje fibres are shown in Table 22.1. The choice of which drug to use is based upon the origin of the arrhythmia, regardless of its pattern. However, the preference of one class over another may vary, depending on a clinician’s experience with particular drugs, on the presentation of the arrhythmia and on patient characteristics. Such factors also govern the choice of drug within a class. The drug chosen should have the dosing schedule and adverse effect profile that best suit the patient or inconvenience them least (see Tables 22.2–22.4). Thus, for example, a patient with glaucoma or prostatism should not be given disopyramide which possesses marked anticholinergic properties, and a patient with obstructive airways disease should preferably not be prescribed a ß-blocker (class II), though if considered essential they could have a cardioselective agent.
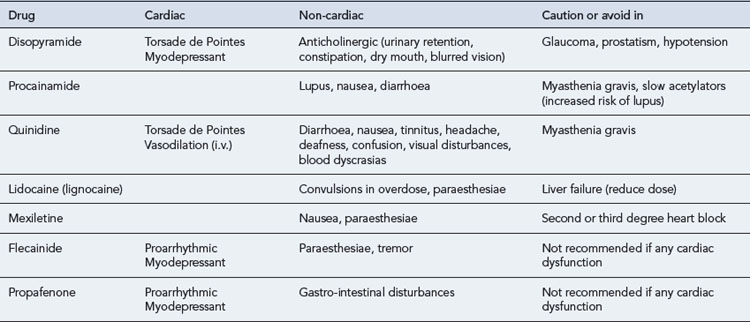
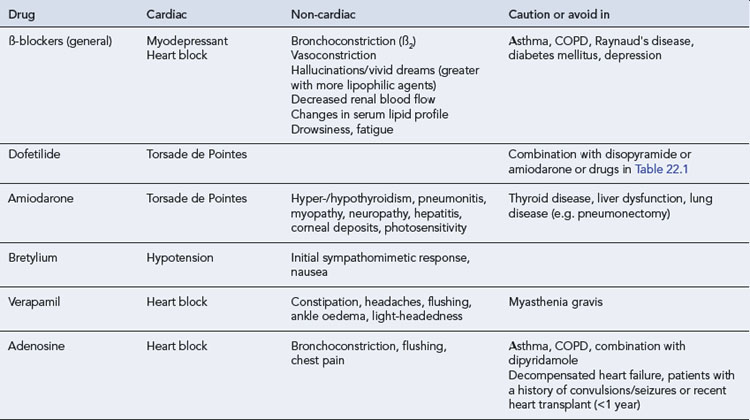
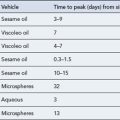
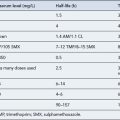
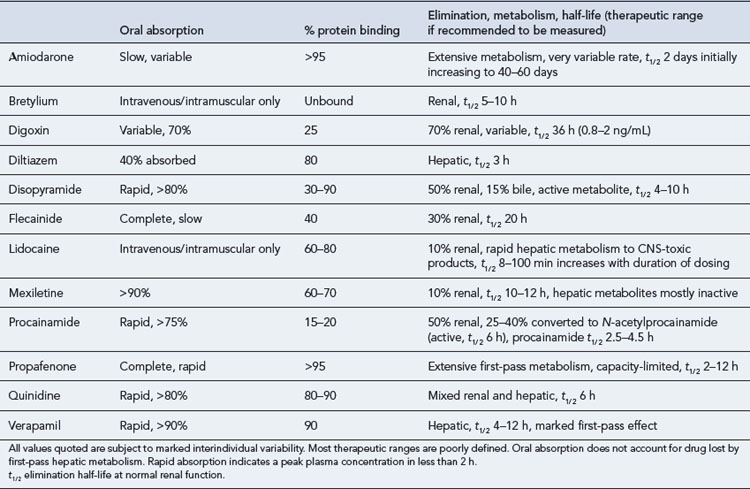
The pharmacokinetic profiles of selected antiarrhythmics are presented in Table 22.5.
Class I
The properties of class I antiarrhythmic drugs may be summarised as follows:
| Sodium channel blockade |
| IC > IA > IB |
| Effect on APD and ERP |
| IA prolong |
| IB shorten |
| IC no effect |
Digoxin
The positive inotropic effect of digoxin has long been used in the treatment of patients with systolic heart failure and sinus rhythm. As evidence emerged that other positive inotropic agents such as milrinone increase mortality in heart failure the role of digoxin was reexamined. Perhaps the largest and best designed of these trials, the Digitalis Investigation Group (1997) trial, established that digoxin had no effect on all-cause mortality in patients with stable congestive cardiac failure, a left ventricular ejection fraction of under 45% and sinus rhythm but significantly reduced a combined endpoint of CHF mortality and hospitalisation due to heart failure. With a large body of evidence attesting to the morbidity and mortality benefits of ACE inhibitors, ß-blockers and spironolactone the role of digoxin has diminished. Digoxin may still be useful in patients who remain symptomatic despite comprehensive therapy with these drugs.
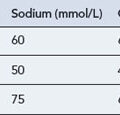
Both the therapeutic and toxic effects of digoxin are potentiated by hypokalaemia and hypercalcaemia. There are also numerous drug interactions (Table 22.6), some of which are pharmacokinetic and some of which are pharmacological.
| Effect | Offending agent or condition |
|---|---|
| Serum level increased by | Amiodarone, verapamil, diltiazem, quinidine, propafenone, clarithromycin, broad-spectrum antibiotics (erythromycin, tetracyclines), decreased renal blood flow (ß-blockers, NSAIDs), renal failure, heart failure |
| Serum level decreased by | Colestyramine, sulfasalazine, neomycin, rifampicin, antacids, improved renal blood flow (vasodilators), levothyroxine (thyroxine) |
| Therapeutic effect increased by | Hypokalaemia, hypercalcaemia, hypomagnesaemia, antiarrhythmic classes IA, II, IV, diuretics that cause hypokalaemia, corticosteroids, myxoedema, hypoxia (acute or chronic), acute myocardial ischaemia or myocarditis |
| Therapeutic effect decreased by | Hyperkalaemia, hypocalcaemia, thyrotoxicosis |
Patient care
Examples of some common therapeutic problems that may occur during the management of arrhythmias are set out in Table 22.7.
Table 22.7 Common therapeutic problems in the management of arrhythmias
| Problem | Comment |
|---|---|
| All antiarrhythmics are proarrhythmic | Prevention is better than cure. Minimise the requirement for drugs by careful attention to precipitating factors. Consider use of pacemakers or non-pharmacological therapies if appropriate |
| Nausea and vomiting with blurred vision and visual discolouration on digoxin | Symptoms and signs of digoxin toxicity noting digoxin has a narrow therapeutic range. Poor renal function may have also contributed |
| ß-Blockers are generally contraindicated in bronchial and peripheral vascular disease | Consider verapamil or diltiazem |
| Calcium channel blockers-induced constipation | If it occurs, give regular osmotic laxatives |
| Torsade de Pointes may be precipitated by taking other medication with amiodarone or disopyramide | Patients should remind members of health care team that they are taking antiarrhythmic drugs, as well as consider electrolyte disturbance such as hypokalaemia |
| Patients experiencing myopathy | Healthcare professionals involved in screening prescriptions for antiarrhythmics should be aware of the clinically relevant interactions and how to manage these. Patients who are taking statins will need regular monitoring for signs of myopathy, particularly those on high-intensity statin therapy |
| Severe asthmatic patient admitted with SVT | Adenosine is contraindicated due to the risk of bronchospasm. Verapamil is a suitable alternative |
| Amiodarone is commonly associated with an increased tendency to sunburn | Warn all patients to stay covered up when outdoors, use sun block or stay indoors |
| Acutely treated patient with AF cardioverted initially with i.v. amiodarone, but on converting to oral therapy, converted back to AF | While amiodarone has a long terminal half-life once saturated, amiodarone has a very large volume of distribution and since tissue stores are penetrated to a minimal extent during the early days of therapy, the effective elimination half-life (t1/ 2) is initially dependent upon a more rapidly exchanging compartment, with a t1/ 2 of 10–17 h, substantially shorter than the t1/ 2 seen during chronic administration. The shorter t1/ 2 becomes important during the acute phase and any intravenous to oral changeover period because the absorption of oral amiodarone is very slow, taking up to 15 h. The combination of a relatively fast elimination and a poor rate of absorption could lead to a significant fall in serum amiodarone levels if intravenous therapy is stopped abruptly when oral therapy is initiated, with the period of maximum risk being the first 24 h of oral therapy. It is, therefore, advisable to phase out intravenous therapy gradually and allow an intravenous/oral overlap period of at least 24 h |
SVT, supraventricular tachycardia; AF, atrial fibrillation.
Answers
| Chest X-ray: pulmonary | Baseline and if symptoms present |
| Thyroid panel | Baseline and every 3–6 months |
| Liver panel | Baseline and every 3–6 months |
| Eye examination | Baseline and every 12 months |
| ECG | Baseline and as required |
| Clinical evaluation | Baseline and every 3 months |
Answers
AFFIRM investigators. Relationship between sinus rhythm, treatment, and survival in the atrial fibrillation follow-up investigation of rhythm management (AFFIRM) study. Circulation. 2004;109:1509-1513.
Cairns J.A., Connolly S.J., Roberts R., et al. Randomised trial of outcome after myocardial infarction in patients with frequent or repetitive ventricular premature depolarisations: CAMIAT Canadian Amiodarone Myocardial Infarction Arrhythmia Trial Investigators. Lancet. 1997;349:675-682.
Crouch M.A., Limon L., Cassano A.T. Drug-related QT interval prolongation: drugs associated with QT prolongation. Pharmacotherapy. 2003;23:802-805.
Digitalis Investigation Group. The effect of digoxin on mortality and morbidity in patients with heart failure. N. Engl. J. Med.. 1997;336:5325-5333.
Echt D.S., Liebson P.R., Mitchell L.B., et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo: the Cardiac Arrhythmia Suppression Trial. N. Engl. J. Med.. 1991;324:781-788.
Julian D.G., Camm A.J., Frangin G., et al. Randomised trial of effect of amiodarone on mortality in patient with left ventricular dysfunction after recent myocardial infarction: EMIAT. European Myocardial Infarct Amiodarone Trial Investigators. Lancet. 1997;349:667-674.
Roy D., Talajic M., Doran P. Amiodarone to prevent recurrence of atrial fibrillation. N. Engl. J. Med.. 2000;342:913-920.
Singh B.N., Singh S.N., Reda D.J. Amiodarone versus sotalol for atrial fibrillation. N. Engl. J. Med.. 2005;352:1861-1872.
Billman G.E., editor. Novel Therapeutic Targets for Antiarrhythmic Drugs. Hoboken: Wiley, 2010.
Fogoros R.N. Antiarryhthmic Drugs: A Practical Guide, second ed. Oxford: Wiley-Blackwell; 2007.
Lip G., Nieuwlaat R., Pisters R., et al. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor based approach: the Euro Heart Survey on Atrial Fibrillation. Chest. 2010;137:263-272.