Atypical presentation and pain patterns are common, especially in the very old or the very young. Diagnosing appendicitis in children can be especially challenging because they tend to respond so dramatically to stimulation and obtaining an accurate history may be difficult. In addition, it is important to remember that the smaller omentum found in children may be less likely to wall off an appendiceal perforation. Observing the child in a quiet surrounding may be helpful.
Signs and symptoms of appendicitis can be subtle in the elderly who may not react as vigorously to appendicitis as younger people. Pain, if noticed, may be minimal and have originated in the right lower quadrant or, otherwise, where the appendix is located. It may never have been noticed to be intermittent, or there may only be significant discomfort with deep palpation. Nausea, anorexia, and emesis may be the predominant complaints. The rare patient may even present with signs and symptoms of distal bowel obstruction secondary to appendiceal inflammation and phlegmon or abscess formation.
LABORATORY TESTING
Laboratory testing does not identify patients with appendicitis but can help the clinician work through the differential diagnosis. The white blood cell count is only mildly to moderately elevated in approximately 70% of patients with simple appendicitis (with a leukocytosis of 10,000–18,000 cells/μL). A “left shift” toward immature polymorphonuclear leukocytes is present in >95% of cases. A sickle cell preparation may be prudent to obtain in those of African, Spanish, Mediterranean, or Indian ancestry. Serum amylase and lipase levels should be measured.
Urinalysis is indicated to help exclude genitourinary conditions that may mimic acute appendicitis, but a few red or white blood cells may be present as a nonspecific finding. However, an inflamed appendix that abuts the ureter or bladder may cause sterile pyuria or hematuria. Every woman of childbearing age should have a pregnancy test. Cervical cultures are indicated if pelvic inflammatory disease is suspected. Anemia and guaiac-positive stools should raise concern about the presence of other diseases or complications such as cancer.
IMAGING
Plain films of the abdomen are rarely helpful and so are not routinely obtained unless the clinician is worried about other conditions such as intestinal obstruction, perforated viscus, or ureterolithiasis. Less than 5% of patients will present with an opaque fecalith in the right lower quadrant. The presence of a fecalith is not diagnostic of appendicitis, although its presence in an appropriate location where the patient complains of pain is suggestive.
The effectiveness of ultrasonography as a tool to diagnosis appendicitis is highly operator dependent. Even in very skilled hands, the appendix may not be visualized. Its overall sensitivity is 0.86, with a specificity of 0.81. Ultrasonography, especially intravaginal techniques, appears to be most useful for identifying pelvic pathology in women. Ultrasonographic findings suggesting the presence of appendicitis include wall thickening, an increased appendiceal diameter, and the presence of free fluid.
The sensitivity and specificity of computed tomography (CT) are 0.94 and 0.95, respectively. Thus, CT imaging, given its high negative predictive value, may be helpful if the diagnosis is in doubt, although studies performed early in the course of disease may not have any typical radiographic findings. Suggestive findings on CT examination include dilatation >6 mm with wall thickening, a lumen that does not fill with enteric contrast, and fatty tissue stranding or air surrounding the appendix, which suggests inflammation (Figs. 356-3 and 356-4). The presence of luminal air or contrast is not consistent with a diagnosis of appendicitis. Furthermore, nonvisualization of the appendix is a nonspecific finding that should not be used to rule out the presence of appendiceal or periappendiceal inflammation.
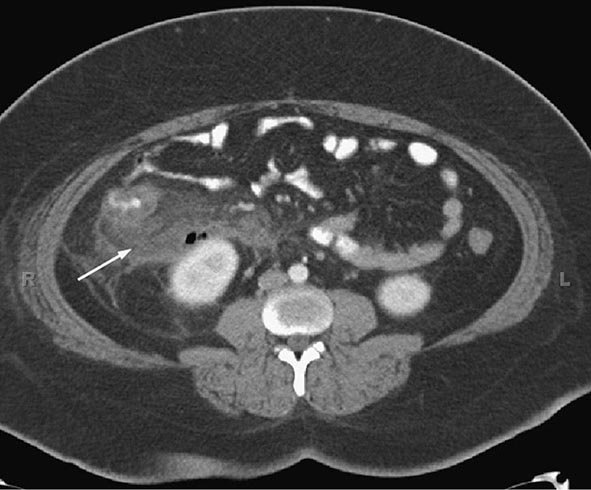
FIGURE 356-3 Computed tomography with oral and intravenous contrast of acute appendicitis. There is thickening of the wall of the appendix and periappendiceal stranding (arrow).
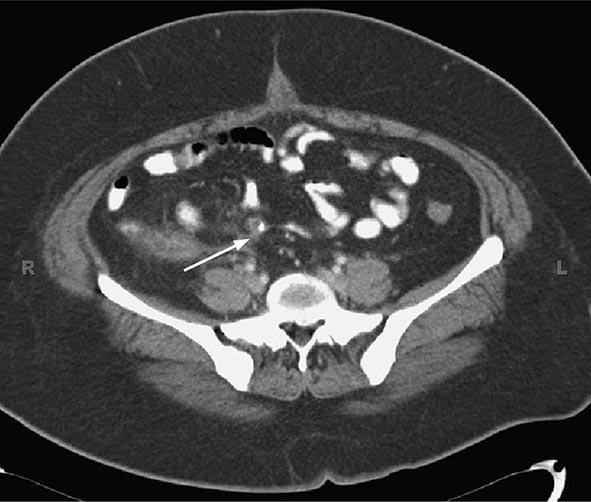
FIGURE 356-4 Appendiceal fecalith (arrow).
SPECIAL PATIENT POPULATIONS
Appendicitis in the most common extrauterine general surgical emergency observed during pregnancy. Early symptoms of appendicitis such as nausea and anorexia may be overlooked. Diagnosing appendicitis in pregnant patients may be especially difficult because as the uterus enlarges the appendix may be pushed higher along the right flank even to the right upper quadrant or because the gravid uterus may obscure typical physical findings. Ultrasonography may facilitate early diagnosis. A high index of suspicion is required because of the effects of unrecognized and untreated appendicitis on the fetus. For example, the fetal mortality rate is four times greater (from 5 to 20%) in patients with perforation.
Immunocompromised patients may present with only mild tenderness and may have many other disease processes in their differential diagnosis, including atypical infections from mycobacteria, Cytomegalovirus, or other fungi. Enterocolitis is a concern and may be present in patients who present with abdominal pain, fever, and neutropenia due to chemotherapy. CT imaging may be very helpful, although it is important not to be overly cautious and delay operative intervention for those patients who are believed to have appendicitis.
ACUTE PERITONITIS
Acute peritonitis, or inflammation of the visceral and parietal peritoneum, is most often but not always infectious in origin, resulting from perforation of a hollow viscus. This is called secondary peritonitis, as opposed to primary or spontaneous peritonitis, when a specific intraabdominal source cannot be identified. In either instance, the inflammation can be localized or diffuse.
ETIOLOGY
Infective organisms may contaminate the peritoneal cavity after spillage from a hollow viscus, because of a penetrating wound of the abdominal wall, or because of the introduction of a foreign object like a peritoneal dialysis catheter or port that becomes infected. Secondary peritonitis most commonly results from perforation of the appendix, colonic diverticuli, or the stomach and duodenum. It may also occur as a complication of bowel infarction or incarceration, cancer, inflammatory bowel disease, and intestinal obstruction or volvulus. Conditions that may cause secondary bacterial peritonitis and their mechanisms are listed in Table 356-5. Over 90% of the cases of primary or spontaneous bacterial peritonitis occur in patients with ascites or hypoproteinemia (<1 g/L).
|
CONDITIONS LEADING TO SECONDARY BACTERIAL PERITONITIS |
Aseptic peritonitis is most commonly caused by the abnormal presence of physiologic fluids like gastric juice, bile, pancreatic enzymes, blood, or urine. It can also be caused by the effects of normally sterile foreign bodies like surgical sponges or instruments. More rarely, it occurs as a complication of systemic diseases like lupus erythematosus, porphyria, and familial Mediterranean fever. The chemical irritation caused by stomach acid and activated pancreatic enzymes is extreme and secondary bacterial infection may occur.
CLINICAL FEATURES
The cardinal signs and symptoms of peritonitis are acute, typically severe, abdominal pain with tenderness and fever. How the patient’s complaints of pain are manifested depends on their overall physical health and whether the inflammation is diffuse or localized. Elderly and immunosuppressed patients may not respond as aggressively to the irritation. Diffuse, generalized peritonitis is most often recognized as diffuse abdominal tenderness with local guarding, rigidity, and other evidence of parietal peritoneal irritation. Physical findings may only be identified in a specific region of the abdomen if the intraperitoneal inflammatory process is limited or otherwise contained as may occur in patients with uncomplicated appendicitis or diverticulitis. Bowel sounds are usually absent to hypoactive.
Most patients present with tachycardia and signs of volume depletion with hypotension. Laboratory testing typically reveals a significant leukocytosis, and patients may be severely acidotic. Radiographic studies may show dilatation of the bowel and associated bowel wall edema. Free air, or other evidence of leakage, requires attention and could represent a surgical emergency. In stable patients in whom ascites is present, diagnostic paracentesis is indicated, where the fluid is tested for protein and lactate dehydrogenase and the cell count is measured.
THERAPY AND PROGNOSIS
Whereas mortality rates can be less than 10% for reasonably healthy patients with relatively uncomplicated, localized peritonitis, mortality rates >40% have been reported for the elderly or immunocompromised. Successful treatment depends on correcting any electrolyte abnormalities, restoration of fluid volume and stabilization of the cardiovascular system, appropriate antibiotic therapy, and surgical correction of any underlying abnormalities.
ACKNOWLEDGMENT
The wisdom and expertise of Dr. William Silen is gratefully acknowledged in this updated chapter on acute appendicitis and peritonitis.
SECTION 2 |
LIVER AND BILIARY TRACT DISEASE |
357 |
Approach to the Patient with Liver Disease |
A diagnosis of liver disease usually can be made accurately by careful elicitation of the patient’s history, physical examination, and application of a few laboratory tests. In some circumstances, radiologic examinations are helpful or, indeed, diagnostic. Liver biopsy is considered the criterion standard in evaluation of liver disease but is now needed less for diagnosis than for grading and staging of disease. This chapter provides an introduction to diagnosis and management of liver disease, briefly reviewing the structure and function of the liver; the major clinical manifestations of liver disease; and the use of clinical history, physical examination, laboratory tests, imaging studies, and liver biopsy.
LIVER STRUCTURE AND FUNCTION
The liver is the largest organ of the body, weighing 1–1.5 kg and representing 1.5–2.5% of the lean body mass. The size and shape of the liver vary and generally match the general body shape—long and lean or squat and square. This organ is located in the right upper quadrant of the abdomen under the right lower rib cage against the diaphragm and projects for a variable extent into the left upper quadrant. It is held in place by ligamentous attachments to the diaphragm, peritoneum, great vessels, and upper gastrointestinal organs. The liver receives a dual blood supply; ~20% of the blood flow is oxygen-rich blood from the hepatic artery, and 80% is nutrient-rich blood from the portal vein arising from the stomach, intestines, pancreas, and spleen.
The majority of cells in the liver are hepatocytes, which constitute two-thirds of the organ’s mass. The remaining cell types are Kupffer cells (members of the reticuloendothelial system), stellate (Ito or fat-storing) cells, endothelial and blood vessel cells, bile ductular cells, and cells of supporting structures. Viewed by light microscopy, the liver appears to be organized in lobules, with portal areas at the periphery and central veins in the center of each lobule. However, from a functional point of view, the liver is organized into acini, with both hepatic arterial and portal venous blood entering the acinus from the portal areas (zone 1) and then flowing through the sinusoids to the terminal hepatic veins (zone 3); the intervening hepatocytes constitute zone 2. The advantage of viewing the acinus as the physiologic unit of the liver is that this perspective helps to explain the morphologic patterns and zonality of many vascular and biliary diseases not explained by the lobular arrangement.
Portal areas of the liver consist of small veins, arteries, bile ducts, and lymphatics organized in a loose stroma of supporting matrix and small amounts of collagen. Blood flowing into the portal areas is distributed through the sinusoids, passing from zone 1 to zone 3 of the acinus and draining into the terminal hepatic veins (“central veins”). Secreted bile flows in the opposite direction—i.e., in a counter-current pattern from zone 3 to zone 1. The sinusoids are lined by unique endothelial cells that have prominent fenestrae of variable sizes, allowing the free flow of plasma but not of cellular elements. The plasma is thus in direct contact with hepatocytes in the subendothelial space of Disse.
Hepatocytes have distinct polarity. The basolateral side of the hepatocyte lines the space of Disse and is richly lined with microvilli; it exhibits endocytotic and pinocytotic activity, with passive and active uptake of nutrients, proteins, and other molecules. The apical pole of the hepatocyte forms the canalicular membranes through which bile components are secreted. The canaliculi of hepatocytes form a fine network, which fuses into the bile ductular elements near the portal areas. Kupffer cells usually lie within the sinusoidal vascular space and represent the largest group of fixed macrophages in the body. The stellate cells are located in the space of Disse but are not usually prominent unless activated, when they produce collagen and matrix. Red blood cells stay in the sinusoidal space as blood flows through the lobules, but white blood cells can migrate through or around endothelial cells into the space of Disse and from there to portal areas, where they can return to the circulation through lymphatics.
Hepatocytes perform numerous and vital roles in maintaining homeostasis and health. These functions include the synthesis of most essential serum proteins (albumin, carrier proteins, coagulation factors, many hormonal and growth factors), the production of bile and its carriers (bile acids, cholesterol, lecithin, phospholipids), the regulation of nutrients (glucose, glycogen, lipids, cholesterol, amino acids), and the metabolism and conjugation of lipophilic compounds (bilirubin, anions, cations, drugs) for excretion in the bile or urine. Measurement of these activities to assess liver function is complicated by the multiplicity and variability of these functions. The most commonly used liver “function” tests are measurements of serum bilirubin, serum albumin, and prothrombin time. The serum bilirubin level is a measure of hepatic conjugation and excretion; the serum albumin level and prothrombin time are measures of protein synthesis. Abnormalities of bilirubin, albumin, and prothrombin time are typical of hepatic dysfunction. Frank liver failure is incompatible with life, and the functions of the liver are too complex and diverse to be subserved by a mechanical pump; a dialysis membrane; or a concoction of infused hormones, proteins, and growth factors.
LIVER DISEASES
While there are many causes of liver disease (Table 357-1), these disorders generally present clinically in a few distinct patterns and are usually classified as hepatocellular, cholestatic (obstructive), or mixed. In hepatocellular diseases (such as viral hepatitis and alcoholic liver disease), features of liver injury, inflammation, and necrosis predominate. In cholestatic diseases (such as gallstone or malignant obstruction, primary biliary cirrhosis, and some drug-induced liver diseases), features of inhibition of bile flow predominate. In a mixed pattern, features of both hepatocellular and cholestatic injury are present (such as in cholestatic forms of viral hepatitis and many drug-induced liver diseases). The pattern of onset and prominence of symptoms can rapidly suggest a diagnosis, particularly if major risk factors are considered, such as the age and sex of the patient and a history of exposure or risk behaviors.
|
LIVER DISEASES |
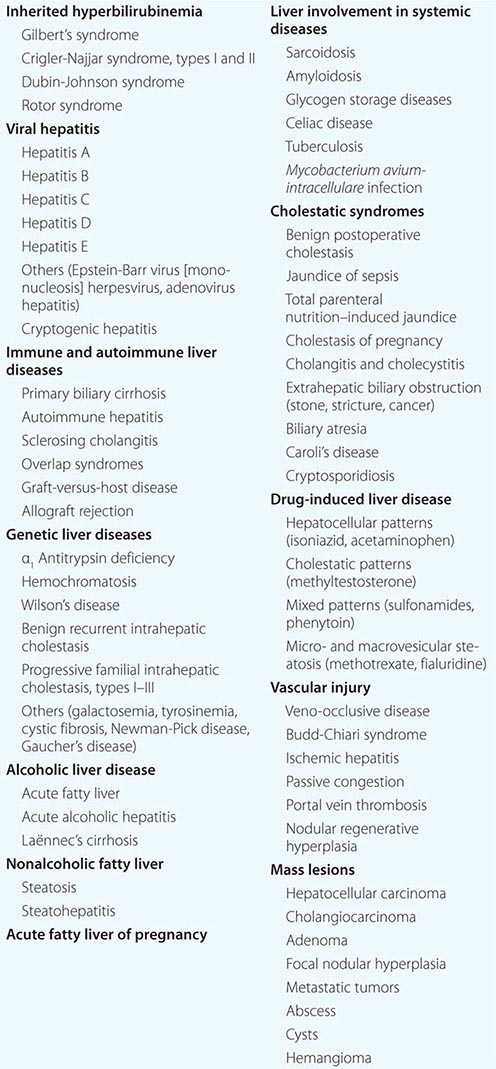
Typical presenting symptoms of liver disease include jaundice, fatigue, itching, right-upper-quadrant pain, nausea, poor appetite, abdominal distention, and intestinal bleeding. At present, however, many patients are diagnosed with liver disease who have no symptoms and who have been found to have abnormalities in biochemical liver tests as a part of a routine physical examination or screening for blood donation or for insurance or employment. The wide availability of batteries of liver tests makes it relatively simple to demonstrate the presence of liver injury as well as to rule it out in someone in whom liver disease is suspected.
Evaluation of patients with liver disease should be directed at (1) establishing the etiologic diagnosis, (2) estimating disease severity (grading), and (3) establishing the disease stage (staging). Diagnosis should focus on the category of disease (hepatocellular, cholestatic, or mixed injury) as well as on the specific etiologic diagnosis. Grading refers to assessment of the severity or activity of disease—active or inactive as well as mild, moderate, or severe. Staging refers to estimation of the point in the course of the natural history of the disease, whether early or late; or precirrhotic, cirrhotic, or end-stage. This chapter introduces general, salient concepts in the evaluation of patients with liver disease that help lead to the diagnoses discussed in subsequent chapters.
CLINICAL HISTORY
The clinical history should focus on the symptoms of liver disease—their nature, patterns of onset, and progression—and on potential risk factors for liver disease. The manifestations of liver disease include constitutional symptoms such as fatigue, weakness, nausea, poor appetite, and malaise and the more liver-specific symptoms of jaundice, dark urine, light stools, itching, abdominal pain, and bloating. Symptoms can also suggest the presence of cirrhosis, end-stage liver disease, or complications of cirrhosis such as portal hypertension. Generally, the constellation of symptoms and their patterns of onset rather than a specific symptom points to an etiology.
Fatigue is the most common and most characteristic symptom of liver disease. It is variously described as lethargy, weakness, listlessness, malaise, increased need for sleep, lack of stamina, and poor energy. The fatigue of liver disease typically arises after activity or exercise and is rarely present or severe after adequate rest; i.e., it is “afternoon” rather than “morning” fatigue. Fatigue in liver disease is often intermittent and variable in severity from hour to hour and day to day. In some patients, it may not be clear whether fatigue is due to the liver disease or to other problems such as stress, anxiety, sleep disturbance, or a concurrent illness.
Nausea occurs with more severe liver disease and may accompany fatigue or be provoked by smelling food odors or eating fatty foods. Vomiting can occur but is rarely persistent or prominent. Poor appetite with weight loss occurs frequently in acute liver disease but is rare in chronic disease except when cirrhosis is present and advanced. Diarrhea is uncommon in liver disease except with severe jaundice, in which a lack of bile acids reaching the intestine can lead to steatorrhea.
Right-upper-quadrant discomfort or ache (“liver pain”) occurs in many liver diseases and is usually marked by tenderness over the liver area. The pain arises from stretching or irritation of Glisson’s capsule, which surrounds the liver and is rich in nerve endings. Severe pain is most typical of gallbladder disease, liver abscess, and severe veno-occlusive disease but is also an occasional accompaniment of acute hepatitis.
Itching occurs with acute liver disease, appearing early in obstructive jaundice (from biliary obstruction or drug-induced cholestasis) and somewhat later in hepatocellular disease (acute hepatitis). Itching also occurs in chronic liver diseases—typically the cholestatic forms such as primary biliary cirrhosis and sclerosing cholangitis, in which it is often the presenting symptom, preceding the onset of jaundice. However, itching can occur in any liver disease, particularly once cirrhosis develops.
Jaundice is the hallmark symptom of liver disease and perhaps the most reliable marker of severity. Patients usually report darkening of the urine before they notice scleral icterus. Jaundice is rarely detectable with a bilirubin level <43 μmol/L (2.5 mg/dL). With severe cholestasis, there will also be lightening of the color of the stools and steatorrhea. Jaundice without dark urine usually indicates indirect (unconjugated) hyperbilirubinemia and is typical of hemolytic anemia and the genetic disorders of bilirubin conjugation, the common and benign form being Gilbert’s syndrome and the rare and severe form being Crigler-Najjar syndrome. Gilbert’s syndrome affects up to 5% of the general population; the jaundice in this condition is more noticeable after fasting and with stress.
Major risk factors for liver disease that should be sought in the clinical history include details of alcohol use, medication use (including herbal compounds, birth control pills, and over-the-counter medications), personal habits, sexual activity, travel, exposure to jaundiced or other high-risk persons, injection drug use, recent surgery, remote or recent transfusion of blood or blood products, occupation, accidental exposure to blood or needlestick, and familial history of liver disease.
For assessing the risk of viral hepatitis, a careful history of sexual activity is of particular importance and should include the number of lifetime sexual partners and, for men, a history of having sex with men. Sexual exposure is a common mode of spread of hepatitis B but is rare for hepatitis C. A family history of hepatitis, liver disease, and liver cancer is also important. Maternal-infant transmission occurs with both hepatitis B and C. Vertical spread of hepatitis B can now be prevented by passive and active immunization of the infant at birth. Vertical spread of hepatitis C is uncommon, but there are no reliable means of prevention. Transmission is more common among HIV-co-infected mothers and is also linked to prolonged and difficult labor and delivery, early rupture of membranes, and internal fetal monitoring. A history of injection drug use, even in the remote past, is of great importance in assessing the risk for hepatitis B and C. Injection drug use is now the single most common risk factor for hepatitis C. Transfusion with blood or blood products is no longer an important risk factor for acute viral hepatitis. However, blood transfusions received before the introduction of sensitive enzyme immunoassays for antibody to hepatitis C virus in 1992 is an important risk factor for chronic hepatitis C. Blood transfusion before 1986, when screening for antibody to hepatitis B core antigen was introduced, is also a risk factor for hepatitis B. Travel to a developing area of the world, exposure to persons with jaundice, and exposure to young children in day-care centers are risk factors for hepatitis A. Tattooing and body piercing (for hepatitis B and C) and eating shellfish (for hepatitis A) are frequently mentioned but are actually types of exposure that quite rarely lead to the acquisition of hepatitis.
![]() Hepatitis E is one of the more common causes of jaundice in Asia and Africa but is uncommon in developed nations. Recently, non-travel-related (autochthonous) cases of hepatitis E have been described in developed countries, including the United States. These cases appear to be due to strains of hepatitis E virus that are endemic in swine and some wild animals (genotypes 3 and 4). While occasional cases are associated with eating raw or undercooked pork or game (deer and wild boars), most cases of hepatitis E occur without known exposure, predominantly in elderly man without typical risk factors for viral hepatitis. Hepatitis E infection can become chronic in immunosuppressed individuals (such as transplant recipients, patients receiving chemotherapy, or patients with HIV infection), in whom it presents with abnormal serum enzymes in the absence of markers of hepatitis B or C.
Hepatitis E is one of the more common causes of jaundice in Asia and Africa but is uncommon in developed nations. Recently, non-travel-related (autochthonous) cases of hepatitis E have been described in developed countries, including the United States. These cases appear to be due to strains of hepatitis E virus that are endemic in swine and some wild animals (genotypes 3 and 4). While occasional cases are associated with eating raw or undercooked pork or game (deer and wild boars), most cases of hepatitis E occur without known exposure, predominantly in elderly man without typical risk factors for viral hepatitis. Hepatitis E infection can become chronic in immunosuppressed individuals (such as transplant recipients, patients receiving chemotherapy, or patients with HIV infection), in whom it presents with abnormal serum enzymes in the absence of markers of hepatitis B or C.
A history of alcohol intake is important in assessing the cause of liver disease and also in planning management and recommendations. In the United States, for example, at least 70% of adults drink alcohol to some degree, but significant alcohol intake is less common; in population-based surveys, only 5% of individuals have more than two drinks per day, the average drink representing 11–15 g of alcohol. Alcohol consumption associated with an increased rate of alcoholic liver disease is probably more than two drinks (22–30 g) per day in women and three drinks (33–45 g) in men. Most patients with alcoholic cirrhosis have a much higher daily intake and have drunk excessively for ≥10 years before onset of liver disease. In assessing alcohol intake, the history should also focus on whether alcohol abuse or dependence is present. Alcoholism is usually defined by the behavioral patterns and consequences of alcohol intake, not by the amount. Abuse is defined by a repetitive pattern of drinking alcohol that has adverse effects on social, family, occupational, or health status. Dependence is defined by alcohol-seeking behavior, despite its adverse effects. Many alcoholics demonstrate both dependence and abuse, and dependence is considered the more serious and advanced form of alcoholism. A clinically helpful approach to diagnosis of alcohol dependence and abuse is the use of the CAGE questionnaire (Table 357-2), which is recommended for all medical history-taking.
|
CAGE QUESTIONSa |

Family history can be helpful in assessing liver disease. Familial causes of liver disease include Wilson’s disease; hemochromatosis and α1 antitrypsin deficiency; and the more uncommon inherited pediatric liver diseases—i.e., familial intrahepatic cholestasis, benign recurrent intrahepatic cholestasis, and Alagille syndrome. Onset of severe liver disease in childhood or adolescence in conjunction with a family history of liver disease or neuropsychiatric disturbance should lead to investigation for Wilson’s disease. A family history of cirrhosis, diabetes, or endocrine failure and the appearance of liver disease in adulthood suggests hemochromatosis and should prompt investigation of iron status. Abnormal iron studies in adult patients warrant genotyping of the HFE gene for the C282Y and H63D mutations typical of genetic hemochromatosis. In children and adolescents with iron overload, other non-HFE causes of hemochromatosis should be sought. A family history of emphysema should provoke investigation of α1 antitrypsin levels and, if levels are low, for protease inhibitor (Pi) genotype.
PHYSICAL EXAMINATION
The physical examination rarely uncovers evidence of liver dysfunction in a patient without symptoms or laboratory findings, nor are most signs of liver disease specific to one diagnosis. Thus, the physical examination complements rather than replaces the need for other diagnostic approaches. In many patients, the physical examination is normal unless the disease is acute or severe and advanced. Nevertheless, the physical examination is important in that it can yield the first evidence of hepatic failure, portal hypertension, and liver decompensation. In addition, the physical examination can reveal signs—related either to risk factors or to associated diseases or findings—that point to a specific diagnosis.
Typical physical findings in liver disease are icterus, hepatomegaly, hepatic tenderness, splenomegaly, spider angiomata, palmar erythema, and excoriations. Signs of advanced disease include muscle wasting, ascites, edema, dilated abdominal veins, hepatic fetor, asterixis, mental confusion, stupor, and coma. In male patients with cirrhosis, particularly that related to alcohol use, signs of hyperestrogenemia such as gynecomastia, testicular atrophy, and loss of male-pattern hair distribution may be found.
Icterus is best appreciated when the sclera is inspected under natural light. In fair-skinned individuals, a yellow tinge to the skin may be obvious. In dark-skinned individuals, examination of the mucous membranes below the tongue can demonstrate jaundice. Jaundice is rarely detectable if the serum bilirubin level is <43 μmol/L (2.5 mg/dL) but may remain detectable below this level during recovery from jaundice (because of protein and tissue binding of conjugated bilirubin).
Spider angiomata and palmar erythema occur in both acute and chronic liver disease; these manifestations may be especially prominent in persons with cirrhosis but can develop in normal individuals and are frequently found during pregnancy. Spider angiomata are superficial, tortuous arterioles and—unlike simple telangiectases—typically fill from the center outward. Spider angiomata occur only on the arms, face, and upper torso; they can be pulsatile and may be difficult to detect in dark-skinned individuals.
Hepatomegaly is not a highly reliable sign of liver disease because of variability in the liver’s size and shape and the physical impediments to assessment of liver size by percussion and palpation. Marked hepatomegaly is typical of cirrhosis, veno-occlusive disease, infiltrative disorders such as amyloidosis, metastatic or primary cancers of the liver, and alcoholic hepatitis. Careful assessment of the liver edge may also reveal unusual firmness, irregularity of the surface, or frank nodules. Perhaps the most reliable physical finding in the liver examination is hepatic tenderness. Discomfort when the liver is touched or pressed upon should be carefully sought with percussive comparison of the right and left upper quadrants.
Splenomegaly, which occurs in many medical conditions, can be a subtle but significant physical finding in liver disease. The availability of ultrasound methods for assessment of the spleen allows confirmation of the physical finding.
Signs of advanced liver disease include muscle wasting and weight loss as well as hepatomegaly, bruising, ascites, and edema. Ascites is best appreciated by attempts to detect shifting dullness by careful percussion. Ultrasound examination will confirm the finding of ascites in equivocal cases. Peripheral edema can occur with or without ascites. In patients with advanced liver disease, other factors frequently contribute to edema formation, including hypoalbuminemia, venous insufficiency, heart failure, and medications.
Hepatic failure is defined as the occurrence of signs or symptoms of hepatic encephalopathy in a person with severe acute or chronic liver disease. The first signs of hepatic encephalopathy can be subtle and nonspecific—change in sleep patterns, change in personality, irritability, and mental dullness. Thereafter, confusion, disorientation, stupor, and eventually coma supervene. In acute liver failure, excitability and mania may be present. Physical findings include asterixis and flapping tremors of the body and tongue. Fetor hepaticus refers to the slightly sweet, ammoniacal odor that can develop in patients with liver failure, particularly if there is portal-venous shunting of blood around the liver. Other causes of coma and disorientation should be excluded, mainly electrolyte imbalances, sedative use, and renal or respiratory failure. The appearance of hepatic encephalopathy during acute hepatitis is the major criterion for diagnosis of fulminant hepatitis and indicates a poor prognosis. In chronic liver disease, encephalopathy is usually triggered by a medical complication such as gastrointestinal bleeding, over-diuresis, uremia, dehydration, electrolyte imbalance, infection, constipation, or use of narcotic analgesics.
A helpful measure of hepatic encephalopathy is a careful mental-status examination and use of the trail-making test, which consists of a series of 25 numbered circles that the patient is asked to connect as rapidly as possible using a pencil. The normal range for the connect-the-dot test is 15–30 sec; it is considerably longer in patients with early hepatic encephalopathy. Other tests include drawing of abstract objects or comparison of a signature to previous examples. More sophisticated testing—e.g., with electroencephalography and visual evoked potentials—can detect mild forms of encephalopathy but are rarely clinically useful.
Other signs of advanced liver disease include umbilical hernia from ascites, hydrothorax, prominent veins over the abdomen, and caput medusa, a condition that consists of collateral veins radiating from the umbilicus and results from recanulation of the umbilical vein. Widened pulse pressure and signs of a hyperdynamic circulation can occur in patients with cirrhosis as a result of fluid and sodium retention, increased cardiac output, and reduced peripheral resistance. Patients with long-standing cirrhosis and portal hypertension are prone to develop the hepatopulmonary syndrome, which is defined by the triad of liver disease, hypoxemia, and pulmonary arteriovenous shunting. The hepatopulmonary syndrome is characterized by platypnea and orthodeoxia: shortness of breath and oxygen desaturation that occur paradoxically upon the assumption of an upright position. Measurement of oxygen saturation by pulse oximetry is a reliable screening test for hepatopulmonary syndrome.
Several skin disorders and changes are common in liver disease. Hyperpigmentation is typical of advanced chronic cholestatic diseases such as primary biliary cirrhosis and sclerosing cholangitis. In these same conditions, xanthelasma and tendon xanthomata occur as a result of retention and high serum levels of lipids and cholesterol. Slate-gray pigmentation of the skin is also seen with hemochromatosis if iron levels are high for a prolonged period. Mucocutaneous vasculitis with palpable purpura, especially on the lower extremities, is typical of cryoglobulinemia of chronic hepatitis C but can also occur in chronic hepatitis B.
Some physical signs point to specific liver diseases. Kayser-Fleischer rings occur in Wilson’s disease and consist of a golden-brown copper pigment deposited in Descemet’s membrane at the periphery of the cornea; they are best seen by slit-lamp examination. Dupuytren contracture and parotid enlargement are suggestive of chronic alcoholism and alcoholic liver disease. In metastatic liver disease or primary hepatocellular carcinoma, signs of cachexia and wasting as well as firm hepatomegaly and a hepatic bruit may be prominent.
DIAGNOSIS OF LIVER DISEASE
The major causes of liver disease and key diagnostic features are outlined in Table 357-3, and an algorithm for evaluation of the patient with suspected liver disease is shown in Fig. 357-1. Specifics of diagnosis are discussed in later chapters. The most common causes of acute liver disease are viral hepatitis (particularly hepatitis A, B, and C), drug-induced liver injury, cholangitis, and alcoholic liver disease. Liver biopsy usually is not needed in the diagnosis and management of acute liver disease, exceptions being situations where the diagnosis remains unclear despite thorough clinical and laboratory investigation. Liver biopsy can be helpful in diagnosing drug-induced liver disease and acute alcoholic hepatitis.
|
IMPORTANT DIAGNOSTIC TESTS IN COMMON LIVER DISEASES |
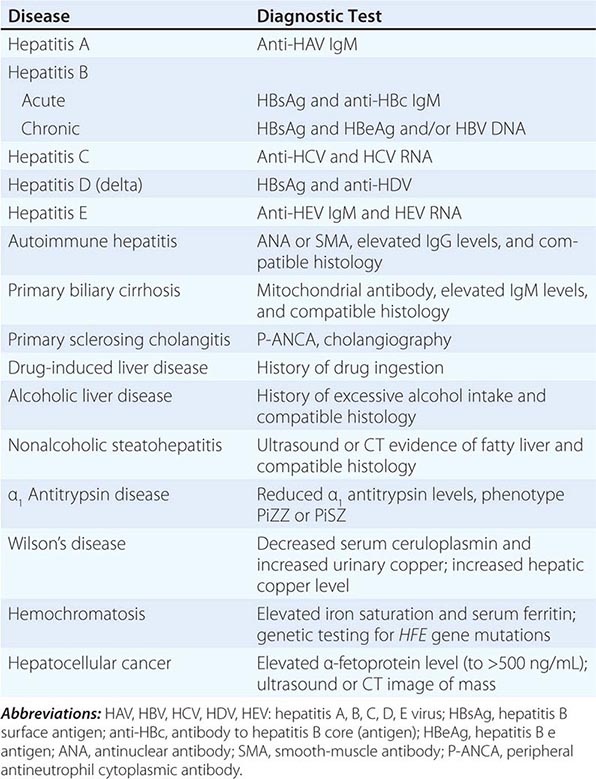
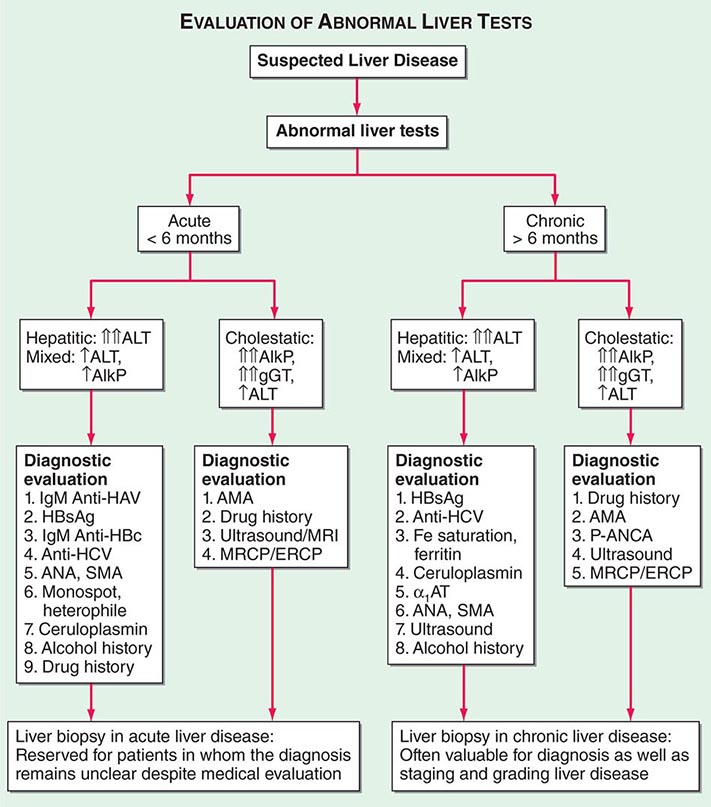
FIGURE 357-1 Algorithm for evaluation of abnormal liver tests. For patients with suspected liver disease, an appropriate approach to evaluation is initial routine liver testing—e.g., measurement of serum bilirubin, albumin, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (AlkP). These results (sometimes complemented by testing of γ-glutamyl transpeptidase; gGT) will establish whether the pattern of abnormalities is hepatic, cholestatic, or mixed. In addition, the duration of symptoms or abnormalities will indicate whether the disease is acute or chronic. If the disease is acute and if history, laboratory tests, and imaging studies do not reveal a diagnosis, liver biopsy is appropriate to help establish the diagnosis. If the disease is chronic, liver biopsy can be helpful not only for diagnosis but also for grading of the activity and staging the progression of disease. This approach is generally applicable to patients without immune deficiency. In patients with HIV infection or recipients of bone marrow or solid organ transplants, the diagnostic evaluation should also include evaluation for opportunistic infections (e.g., with adenovirus, cytomegalovirus, Coccidioides, hepatitis E virus) as well as for vascular and immunologic conditions (veno-occlusive disease, graft-versus-host disease). HAV, hepatitis A virus; HCV, hepatitis C virus; HBsAg, hepatitis B surface antigen; anti-HBc, antibody to hepatitis B core (antigen); ANA, antinuclear antibody; SMA, smooth-muscle antibody; MRCP, magnetic resonance cholangiopancreatography; ERCP, endoscopic retrograde cholangiopancreatography; α1 AT, α1 antitrypsin; AMA; antimitochondrial antibody; P-ANCA, peripheral antineutrophil cytoplasmic antibody.
The most common causes of chronic liver disease, in general order of frequency, are chronic hepatitis C, alcoholic liver disease, nonalcoholic steatohepatitis, chronic hepatitis B, autoimmune hepatitis, sclerosing cholangitis, primary biliary cirrhosis, hemochromatosis, and Wilson’s disease. Hepatitis E virus is a rare cause of chronic hepatitis, with cases occurring mostly in persons who are immunosuppressed or immunodeficient. Strict diagnostic criteria have not been developed for most liver diseases, but liver biopsy plays an important role in the diagnosis of autoimmune hepatitis, primary biliary cirrhosis, nonalcoholic and alcoholic steatohepatitis, and Wilson’s disease (with a quantitative hepatic copper level in the last instance).
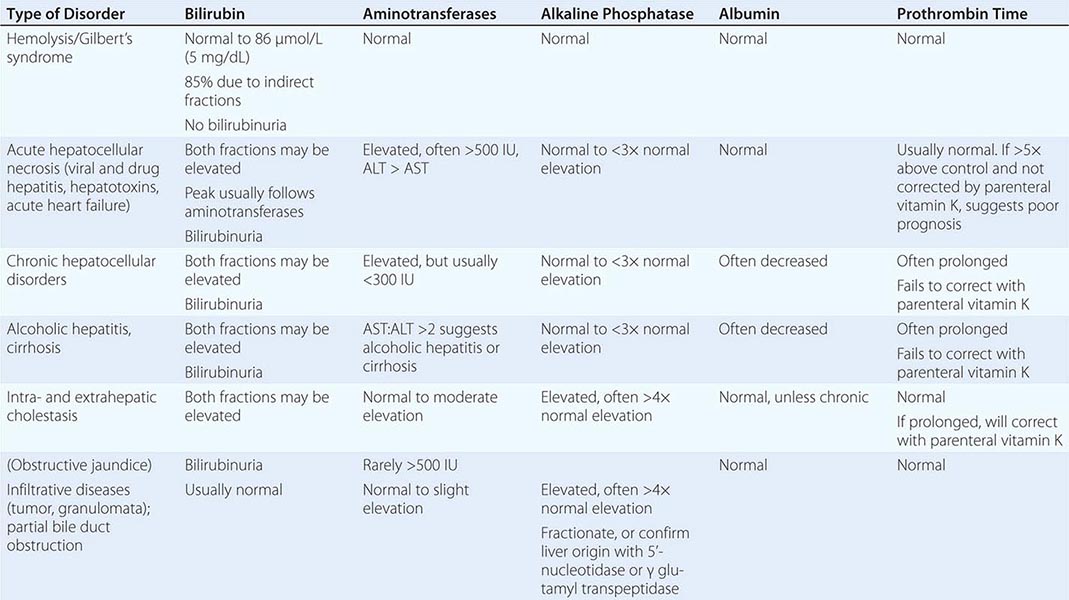
Laboratory Testing Diagnosis of liver disease is greatly aided by the availability of reliable and sensitive tests of liver injury and function. A typical battery of blood tests used for initial assessment of liver disease includes measurement of levels of serum alanine and aspartate aminotransferases, alkaline phosphatase, direct and total serum bilirubin and albumin, and prothrombin time. The pattern of abnormalities generally points to hepatocellular versus cholestatic liver disease and helps determine whether the disease is acute or chronic and whether cirrhosis and hepatic failure are present. On the basis of these results, further testing over time may be necessary. Other laboratory tests may be helpful, such as γ-glutamyl transpeptidase to define whether alkaline phosphatase elevations are due to liver disease; hepatitis serology to define the type of viral hepatitis; and autoimmune markers to diagnose primary biliary cirrhosis (antimitochondrial antibody), sclerosing cholangitis (peripheral antineutrophil cytoplasmic antibody), and autoimmune hepatitis (antinuclear, smooth-muscle, and liver-kidney microsomal antibody). A simple delineation of laboratory abnormalities and common liver diseases is given in Table 357-3.
The use and interpretation of liver function tests are summarized in Chap. 358.
Diagnostic Imaging Great advances have been made in hepatobiliary imaging, although no method is adequately accurate in demonstrating underlying cirrhosis. Of the many modalities available for imaging the liver, ultrasound, CT, and MRI are the most commonly employed and are complementary to one another. In general, ultrasound and CT are highly sensitive for detecting biliary duct dilation and are the first-line options for investigating cases of suspected obstructive jaundice. All three modalities can detect a fatty liver, which appears bright on imaging studies. Modifications of CT and MRI can be used to quantify liver fat, and this information may ultimately be valuable in monitoring therapy in patients with fatty liver disease. Magnetic resonance cholangiopancreatography (MRCP) and endoscopic retrograde cholangiopancreatography (ERCP) are the procedures of choice for visualization of the biliary tree. MRCP offers several advantages over ERCP: there is no need for contrast media or ionizing radiation, images can be acquired faster, the procedure is less operator dependent, and it carries no risk of pancreatitis. MRCP is superior to ultrasound and CT for detecting choledocholithiasis but is less specific. MRCP is useful in the diagnosis of bile duct obstruction and congenital biliary abnormalities, but ERCP is more valuable in evaluating ampullary lesions and primary sclerosing cholangitis. ERCP permits biopsy, direct visualization of the ampulla and common bile duct, and intraductal ultrasonography. It also provides several therapeutic options in patients with obstructive jaundice, such as sphincterotomy, stone extraction, and placement of nasobiliary catheters and biliary stents. Doppler ultrasound and MRI are used to assess hepatic vasculature and hemodynamics and to monitor surgically or radiologically placed vascular shunts, including transjugular intrahepatic portosystemic shunts. Multidetector or spiral CT and MRI with contrast-enhancement are the procedures of choice for the identification and evaluation of hepatic masses, the staging of liver tumors, and preoperative assessment. With regard to mass lesions, the sensitivity of hepatic imaging continues to increase; unfortunately, specificity remains a problem, and often two and sometimes three studies are needed before a diagnosis can be reached. Recently, ultrasound transient elastography has been approved for the measurement of hepatic stiffness—providing an indirect assessment of cirrhosis; this technique can eliminate the need for liver biopsy if the only indication is the assessment of disease stage. Magnetic resonance elastography is now undergoing evaluation for its ability to detect different degrees of hepatic fibrosis. Studies are ongoing to determine whether hepatic elastography is an appropriate means of monitoring fibrosis and disease progression. Finally, interventional radiologic techniques allow the biopsy of solitary lesions, the radiofrequency ablation and chemoembolization of cancerous lesions, the insertion of drains into hepatic abscesses, the measurement of portal pressure, and the creation of vascular shunts in patients with portal hypertension. Which modality to use depends on factors such as availability, cost, and experience of the radiologist with each technique.
Liver Biopsy Liver biopsy remains the criterion standard in the evaluation of patients with liver disease, particularly chronic liver disease. Liver biopsy is necessary for diagnosis in selected instances but is more often useful for assessment of the severity (grade) and stage of liver damage, prediction of prognosis, and monitoring of the response to treatment. The size of the liver biopsy sample is an important determinant of reliability; a length of 1.5–2 cm is necessary for accurate assessment of fibrosis. In the future, noninvasive means of assessing disease activity (batteries of blood tests) and fibrosis (elastography and fibrosis markers) may replace liver biopsy for the staging and grading of disease.
GRADING AND STAGING OF LIVER DISEASE
Grading refers to an assessment of the severity or activity of liver disease, whether acute or chronic; active or inactive; and mild, moderate, or severe. Liver biopsy is the most accurate means of assessing severity, particularly in chronic liver disease. Serum aminotransferase levels serve as convenient and noninvasive markers for disease activity but do not always reliably reflect disease severity. Thus, normal serum aminotransferase levels in patients with hepatitis B surface antigen in serum may indicate the inactive carrier state or may reflect mild chronic hepatitis B or hepatitis B with fluctuating disease activity. Serum testing for hepatitis B e antigen and hepatitis B virus DNA can help sort out these different patterns, but these markers can also fluctuate and change over time. Similarly, in chronic hepatitis C, serum aminotransferase levels can be normal despite moderate disease activity. Finally, in both alcoholic and nonalcoholic steatohepatitis, aminotransferase levels are quite unreliable in reflecting severity. In these conditions, liver biopsy is helpful in guiding management and identifying appropriate therapy, particularly if treatment is difficult, prolonged, and expensive, as is often the case in chronic viral hepatitis. Of the several well-verified numerical scales for grading activity in chronic liver disease, the most commonly used are the histology activity index and the Ishak histology scale.
Liver biopsy is also the most accurate means of assessing stage of disease as early or advanced, precirrhotic, and cirrhotic. Staging of disease pertains largely to chronic liver diseases in which progression to cirrhosis and end-stage disease can occur but may require years or decades. Clinical features, biochemical tests, and hepatic imaging studies are helpful in assessing stage but generally become abnormal only in the middle to late stages of cirrhosis. Noninvasive tests that suggest advanced fibrosis include mild elevations of bilirubin, prolongation of prothrombin time, slight decreases in serum albumin, and mild thrombocytopenia (which is often the first indication of worsening fibrosis). Combinations of blood test results have been used to create models for predicting advanced liver disease, but these models are not reliable enough to use on a regular basis and only separate advanced from early disease. Recently, elastography and noninvasive breath tests using 13C-labeled compounds have been proposed as a means of detecting early stages of fibrosis and liver dysfunction, but their reliability and reproducibility remain to be proven. Thus, at present, mild to moderate stages of hepatic fibrosis are detectable only by liver biopsy. In the assessment of stage, the degree of fibrosis is usually used as the quantitative measure. The amount of fibrosis is generally staged on a scale of 0 to 4+ (Metavir scale) or 0 to 6+ (Ishak scale). The importance of staging relates primarily to prognosis and to optimal management of complications. Patients with cirrhosis are candidates for screening and surveillance for esophageal varices and hepatocellular carcinoma. Patients without advanced fibrosis need not undergo screening.
Cirrhosis can also be staged clinically. A reliable staging system is the modified Child-Pugh classification, with a scoring system of 5–15: scores of 5 and 6 represent Child-Pugh class A (consistent with “compensated cirrhosis”), scores of 7–9 represent class B, and scores of 10–15 represent class C (Table 357-4). This scoring system was initially devised to stratify patients into risk groups before portal decompressive surgery. The Child-Pugh score is a reasonably reliable predictor of survival in many liver diseases and predicts the likelihood of major complications of cirrhosis, such as bleeding from varices and spontaneous bacterial peritonitis. This classification scheme was used to assess prognosis in cirrhosis and to provide standard criteria for listing a patient as a candidate for liver transplantation (Child-Pugh class B). Recently, the Child-Pugh system has been replaced by the Model for End-Stage Liver Disease (MELD) system for the latter purpose. The MELD score is a prospectively derived system designed to predict the prognosis of patients with liver disease and portal hypertension. This score is calculated from three noninvasive variables: the prothrombin time expressed as the international normalized ratio (INR), the serum bilirubin level, and the serum creatinine concentration. (http://optn.transplant.hrsa.gov/resources/MeldPeldCalculator.asp?index=98).
|
CHILD-PUGH CLASSIFICATION OF CIRRHOSIS |
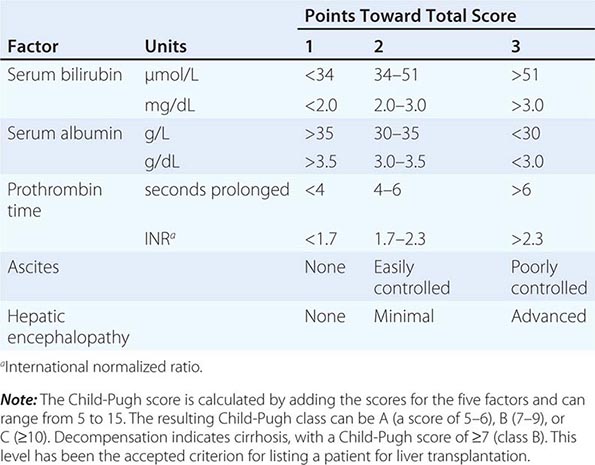
The MELD system provides a more objective means of assessing disease severity and has less center-to-center variation than the Child-Pugh score as well as a wider range of values. MELD is currently used to establish priority listing for liver transplantation in the United States. A similar system, PELD (pediatric end-stage liver disease), is based on bilirubin, INR, serum albumin, age, and nutritional status and is used for children <12 years of age.
Thus, liver biopsy is helpful not only in diagnosis but also in management of chronic liver disease and assessment of prognosis. Because liver biopsy is an invasive procedure and not without complications, it should be used only when it will contribute materially to decisions about management and therapy.
NONSPECIFIC ISSUES IN THE MANAGEMENT OF PATIENTS WITH LIVER DISEASE
Specifics on the management of different forms of acute or chronic liver disease are supplied in subsequent chapters, but certain issues are applicable to any patient with liver disease. These issues include advice regarding alcohol use, medication use, vaccination, and surveillance for complications of liver disease. Alcohol should be used sparingly, if at all, by patients with liver disease. Abstinence from alcohol should be encouraged for all patients with alcohol-related liver disease, patients with cirrhosis, and patients receiving interferon-based therapy for hepatitis B or C. With regard to vaccinations, all patients with liver disease should receive hepatitis A vaccine, and those with risk factors should receive hepatitis B vaccine as well. Influenza and pneumococcal vaccination should also be encouraged, with adherence to the recommendations of the Centers for Disease Control and Prevention. Patients with liver disease should exercise caution in using any medications other than those that are most necessary. Drug-induced hepatotoxicity can mimic many forms of liver disease and can cause exacerbations of chronic hepatitis and cirrhosis; drugs should be suspected in any situation in which the cause of exacerbation is unknown. Finally, consideration should be given to surveillance for complications of chronic liver disease such as variceal hemorrhage and hepatocellular carcinoma. Cirrhosis warrants upper endoscopy to assess the presence of varices, and the patient should receive chronic therapy with beta blockers or should be offered endoscopic obliteration if large varices are found. Moreover, cirrhosis warrants screening and long-term surveillance for development of hepatocellular carcinoma. While the optimal regimen for such surveillance has not been established, an appropriate approach is ultrasound of the liver at 6- to 12-month intervals.
358 |
Evaluation of Liver Function |
Several biochemical tests are useful in the evaluation and management of patients with hepatic dysfunction. These tests can be used to (1) detect the presence of liver disease, (2) distinguish among different types of liver disorders, (3) gauge the extent of known liver damage, and (4) follow the response to treatment.
Liver tests have shortcomings. They can be normal in patients with serious liver disease and abnormal in patients with diseases that do not affect the liver. Liver tests rarely suggest a specific diagnosis; rather, they suggest a general category of liver disease, such as hepatocellular or cholestatic, which then further directs the evaluation.
The liver carries out thousands of biochemical functions, most of which cannot be easily measured by blood tests. Laboratory tests measure only a limited number of these functions. In fact, many tests, such as the aminotransferases or alkaline phosphatase, do not measure liver function at all. Rather, they detect liver cell damage or interference with bile flow. Thus, no one test enables the clinician to accurately assess the liver’s total functional capacity.
To increase both the sensitivity and the specificity of laboratory tests in the detection of liver disease, it is best to use them as a battery. Tests usually employed in clinical practice include the bilirubin, aminotransferases, alkaline phosphatase, albumin, and prothrombin time tests. When more than one of these tests provide abnormal findings or the findings are persistently abnormal on serial determinations, the probability of liver disease is high. When all test results are normal, the probability of missing occult liver disease is low.
When evaluating patients with liver disorders, it is helpful to group these tests into general categories as outlined below.
TESTS BASED ON DETOXIFICATION AND EXCRETORY FUNCTIONS
Serum Bilirubin (See also Chap. 58) Bilirubin, a breakdown product of the porphyrin ring of heme-containing proteins, is found in the blood in two fractions—conjugated and unconjugated. The unconjugated fraction, also termed the indirect fraction, is insoluble in water and is bound to albumin in the blood. The conjugated (direct) bilirubin fraction is water soluble and can therefore be excreted by the kidney. When measured by modifications of the original van den Bergh method, normal values of total serum bilirubin are reported between 1 and 1.5 mg/dL with 95% of a normal population falling between 0.2 and 0.9 mg/dL. If the direct-acting fraction is less than 15% of the total, the bilirubin can be considered to all be indirect. The most frequently reported upper limit of normal for conjugated bilirubin is 0.3 mg/dL.
Elevation of the unconjugated fraction of bilirubin is rarely due to liver disease. An isolated elevation of unconjugated bilirubin is seen primarily in hemolytic disorders and in a number of genetic conditions such as Crigler-Najjar and Gilbert’s syndromes (Chap. 58). Isolated unconjugated hyperbilirubinemia (bilirubin elevated but <15% direct) should prompt a workup for hemolysis (Fig. 358-1). In the absence of hemolysis, an isolated, unconjugated hyperbilirubinemia in an otherwise healthy patient can be attributed to Gilbert’s syndrome, and no further evaluation is required.
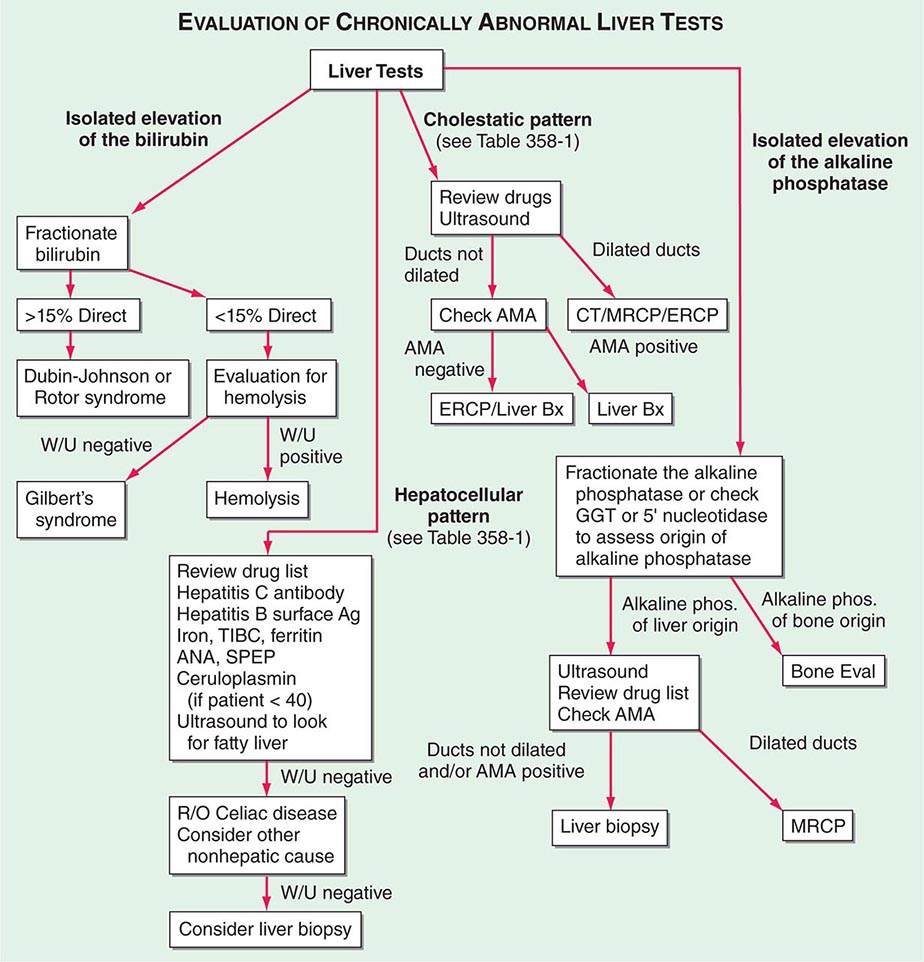
FIGURE 358-1 Algorithm for the evaluation of chronically abnormal liver tests. AMA, antimitochondrial antibody; ANA, antinuclear antibody; Bx, biopsy; CT, computed tomography; ERCP, endoscopic retrograde cholangiopancreatography; GGT, γ glutamyl transpeptidase; MRCP, magnetic resonance cholangiopancreatography; R/O, rule out; SPEP, serum protein electrophoresis; TIBC, total iron-binding capacity; W/U, workup.
In contrast, conjugated hyperbilirubinemia almost always implies liver or biliary tract disease. The rate-limiting step in bilirubin metabolism is not conjugation of bilirubin, but rather the transport of conjugated bilirubin into the bile canaliculi. Thus, elevation of the conjugated fraction may be seen in any type of liver disease. In most liver diseases, both conjugated and unconjugated fractions of the bilirubin tend to be elevated. Except in the presence of a purely unconjugated hyperbilirubinemia, fractionation of the bilirubin is rarely helpful in determining the cause of jaundice.
Although the degree of elevation of the serum bilirubin has not been critically assessed as a prognostic marker, it is important in a number of conditions. In viral hepatitis, the higher the serum bilirubin, the greater is the hepatocellular damage. Total serum bilirubin correlates with poor outcomes in alcoholic hepatitis. It is also a critical component of the Model for End-Stage Liver Disease (MELD) score, a tool used to estimate survival of patients with end-stage liver disease and assess operative risk of patients with cirrhosis. An elevated total serum bilirubin in patients with drug-induced liver disease indicates more severe injury.
Urine Bilirubin Unconjugated bilirubin always binds to albumin in the serum and is not filtered by the kidney. Therefore, any bilirubin found in the urine is conjugated bilirubin; the presence of bilirubinuria implies the presence of liver disease. A urine dipstick test can theoretically give the same information as fractionation of the serum bilirubin. This test is almost 100% accurate. Phenothiazines may give a false-positive reading with the Ictotest tablet. In patients recovering from jaundice, the urine bilirubin clears prior to the serum bilirubin.
Blood Ammonia Ammonia is produced in the body during normal protein metabolism and by intestinal bacteria, primarily those in the colon. The liver plays a role in the detoxification of ammonia by converting it to urea, which is excreted by the kidneys. Striated muscle also plays a role in detoxification of ammonia, where it is combined with glutamic acid to form glutamine. Patients with advanced liver disease typically have significant muscle wasting, which likely contributes to hyperammonemia in these patients. Some physicians use the blood ammonia for detecting encephalopathy or for monitoring hepatic synthetic function, although its use for either of these indications has problems. There is very poor correlation between either the presence or the severity of acute encephalopathy and elevation of blood ammonia; it can be occasionally useful for identifying occult liver disease in patients with mental status changes. There is also a poor correlation of the blood serum ammonia and hepatic function. The ammonia can be elevated in patients with severe portal hypertension and portal blood shunting around the liver even in the presence of normal or near-normal hepatic function. Elevated arterial ammonia levels have been shown to correlate with outcome in fulminant hepatic failure.
Serum Enzymes The liver contains thousands of enzymes, some of which are also present in the serum in very low concentrations. These enzymes have no known function in the serum and behave like other serum proteins. They are distributed in the plasma and in interstitial fluid and have characteristic half-lives, which are usually measured in days. Very little is known about the catabolism of serum enzymes, although they are probably cleared by cells in the reticuloendothelial system. The elevation of a given enzyme activity in the serum is thought to primarily reflect its increased rate of entrance into serum from damaged liver cells.
Serum enzyme tests can be grouped into three categories: (1) enzymes whose elevation in serum reflects damage to hepatocytes, (2) enzymes whose elevation in serum reflects cholestasis, and (3) enzyme tests that do not fit precisely into either pattern.
ENZYMES THAT REFLECT DAMAGE TO HEPATOCYTES The aminotransferases (transaminases) are sensitive indicators of liver cell injury and are most helpful in recognizing acute hepatocellular diseases such as hepatitis. They include aspartate aminotransferase (AST) and alanine aminotransferase (ALT). AST is found in the liver, cardiac muscle, skeletal muscle, kidneys, brain, pancreas, lungs, leukocytes, and erythrocytes in decreasing order of concentration. ALT is found primarily in the liver and is therefore a more specific indicator of liver injury. The aminotransferases are normally present in the serum in low concentrations. These enzymes are released into the blood in greater amounts when there is damage to the liver cell membrane resulting in increased permeability. Liver cell necrosis is not required for the release of the aminotransferases, and there is a poor correlation between the degree of liver cell damage and the level of the aminotransferases. Thus, the absolute elevation of the aminotransferases is of no prognostic significance in acute hepatocellular disorders.
The normal range for aminotransferases varies widely among laboratories, but generally ranges from 10–40 IU/L. The interlaboratory variation in normal range is due to technical reasons; no reference standards exist to establish upper limits of normal for ALT and AST. Some have recommended revisions of normal limits of the aminotransferases to adjust for sex and body mass index, but others have noted the potential costs and unclear benefits of implementing this change.
Any type of liver cell injury can cause modest elevations in the serum aminotransferases. Levels of up to 300 IU/L are nonspecific and may be found in any type of liver disorder. Minimal ALT elevations in asymptomatic blood donors rarely indicate severe liver disease; studies have shown that fatty liver disease is the most likely explanation. Striking elevations—i.e., aminotransferases >1000 IU/L—occur almost exclusively in disorders associated with extensive hepatocellular injury such as (1) viral hepatitis, (2) ischemic liver injury (prolonged hypotension or acute heart failure), or (3) toxin- or drug-induced liver injury.
The pattern of the aminotransferase elevation can be helpful diagnostically. In most acute hepatocellular disorders, the ALT is higher than or equal to the AST. Whereas the AST:ALT ratio is typically <1 in patients with chronic viral hepatitis and nonalcoholic fatty liver disease, a number of groups have noted that as cirrhosis develops, this ratio rises to >1. An AST:ALT ratio >2:1 is suggestive, whereas a ratio >3:1 is highly suggestive, of alcoholic liver disease. The AST in alcoholic liver disease is rarely >300 IU/L, and the ALT is often normal. A low level of ALT in the serum is due to an alcohol-induced deficiency of pyridoxal phosphate.
The aminotransferases are usually not greatly elevated in obstructive jaundice. One notable exception occurs during the acute phase of biliary obstruction caused by the passage of a gallstone into the common bile duct. In this setting, the aminotransferases can briefly be in the 1000–2000 IU/L range. However, aminotransferase levels decrease quickly, and the liver function tests rapidly evolve into those typical of cholestasis.
ENZYMES THAT REFLECT CHOLESTASIS The activities of three enzymes—alkaline phosphatase, 5′-nucleotidase, and γ-glutamyl transpeptidase (GGT)—are usually elevated in cholestasis. Alkaline phosphatase and 5′-nucleotidase are found in or near the bile canalicular membrane of hepatocytes, whereas GGT is located in the endoplasmic reticulum and in bile duct epithelial cells. Reflecting its more diffuse localization in the liver, GGT elevation in serum is less specific for cholestasis than are elevations of alkaline phosphatase or 5′-nucleotidase. Some have advocated the use of GGT to identify patients with occult alcohol use. Its lack of specificity makes its use in this setting questionable.
The normal serum alkaline phosphatase consists of many distinct isoenzymes found in the liver; bone; placenta; and, less commonly, small intestine. Patients over age 60 can have a mildly elevated alkaline phosphatase (1–1.5 times normal), whereas individuals with blood types O and B can have an elevation of the serum alkaline phosphatase after eating a fatty meal due to the influx of intestinal alkaline phosphatase into the blood. It is also nonpathologically elevated in children and adolescents undergoing rapid bone growth because of bone alkaline phosphatase, and late in normal pregnancies due to the influx of placental alkaline phosphatase.
Elevation of liver-derived alkaline phosphatase is not totally specific for cholestasis, and a less than threefold elevation can be seen in almost any type of liver disease. Alkaline phosphatase elevations greater than four times normal occur primarily in patients with cholestatic liver disorders, infiltrative liver diseases such as cancer and amyloidosis, and bone conditions characterized by rapid bone turnover (e.g., Paget’s disease). In bone diseases, the elevation is due to increased amounts of the bone isoenzymes. In liver diseases, the elevation is almost always due to increased amounts of the liver isoenzyme.
If an elevated serum alkaline phosphatase is the only abnormal finding in an apparently healthy person, or if the degree of elevation is higher than expected in the clinical setting, identification of the source of elevated isoenzymes is helpful (Fig. 358-1). This problem can be approached in two ways. First, and most precise, is the fractionation of the alkaline phosphatase by electrophoresis. The second, best substantiated, and most available approach involves the measurement of serum 5′-nucleotidase or GGT. These enzymes are rarely elevated in conditions other than liver disease.
In the absence of jaundice or elevated aminotransferases, an elevated alkaline phosphatase of liver origin often, but not always, suggests early cholestasis and, less often, hepatic infiltration by tumor or granulomata. Other conditions that cause isolated elevations of the alkaline phosphatase include Hodgkin’s disease, diabetes, hyperthyroidism, congestive heart failure, amyloidosis, and inflammatory bowel disease.
The level of serum alkaline phosphatase elevation is not helpful in distinguishing between intrahepatic and extrahepatic cholestasis. There is essentially no difference among the values found in obstructive jaundice due to cancer, common duct stone, sclerosing cholangitis, or bile duct stricture. Values are similarly increased in patients with intrahepatic cholestasis due to drug-induced hepatitis; primary biliary cirrhosis; rejection of transplanted livers; and, rarely, alcohol-induced steatohepatitis. Values are also greatly elevated in hepatobiliary disorders seen in patients with AIDS (e.g., AIDS cholangiopathy due to cytomegalovirus or cryptosporidial infection and tuberculosis with hepatic involvement).
TESTS THAT MEASURE BIOSYNTHETIC FUNCTION OF THE LIVER
Serum Albumin Serum albumin is synthesized exclusively by hepatocytes. Serum albumin has a long half-life: 18–20 days, with ~4% degraded per day. Because of this slow turnover, the serum albumin is not a good indicator of acute or mild hepatic dysfunction; only minimal changes in the serum albumin are seen in acute liver conditions such as viral hepatitis, drug-related hepatotoxicity, and obstructive jaundice. In hepatitis, albumin levels <3 g/dL should raise the possibility of chronic liver disease. Hypoalbuminemia is more common in chronic liver disorders such as cirrhosis and usually reflects severe liver damage and decreased albumin synthesis. One exception is the patient with ascites in whom synthesis may be normal or even increased, but levels are low because of the increased volume of distribution. However, hypoalbuminemia is not specific for liver disease and may occur in protein malnutrition of any cause, as well as protein-losing enteropathies, nephrotic syndrome, and chronic infections that are associated with prolonged increases in levels of serum interleukin 1 and/or tumor necrosis factor, cytokines that inhibit albumin synthesis. Serum albumin should not be measured for screening in patients in whom there is no suspicion of liver disease. A general medical clinic study of consecutive patients in whom no indications were present for albumin measurement showed that although 12% of patients had abnormal test results, the finding was of clinical importance in only 0.4%.
Serum Globulins Serum globulins are a group of proteins made up of γ globulins (immunoglobulins) produced by B lymphocytes and α and β globulins produced primarily in hepatocytes. γ globulins are increased in chronic liver disease, such as chronic hepatitis and cirrhosis. In cirrhosis, the increased serum γ globulin concentration is due to the increased synthesis of antibodies, some of which are directed against intestinal bacteria. This occurs because the cirrhotic liver fails to clear bacterial antigens that normally reach the liver through the hepatic circulation.
Increases in the concentration of specific isotypes of γ globulins are often helpful in the recognition of certain chronic liver diseases. Diffuse polyclonal increases in IgG levels are common in autoimmune hepatitis; increases >100% should alert the clinician to this possibility. Increases in the IgM levels are common in primary biliary cirrhosis, whereas increases in the IgA levels occur in alcoholic liver disease.
COAGULATION FACTORS
With the exception of factor VIII, which is produced by vascular endothelial cells, the blood clotting factors are made exclusively in hepatocytes. Their serum half-lives are much shorter than albumin, ranging from 6 h for factor VII to 5 days for fibrinogen. Because of their rapid turnover, measurement of the clotting factors is the single best acute measure of hepatic synthetic function and helpful in both diagnosis and assessing the prognosis of acute parenchymal liver disease. Useful for this purpose is the serum prothrombin time, which collectively measures factors II, V, VII, and X. Biosynthesis of factors II, VII, IX, and × depends on vitamin K. The international normalized ratio (INR) is used to express the degree of anticoagulation on warfarin therapy. The INR standardizes prothrombin time measurement according to the characteristics of the thromboplastin reagent used in a particular lab, which is expressed as an International Sensitivity Index (ISI); the ISI is then used in calculating the INR.
The prothrombin time may be elevated in hepatitis and cirrhosis as well as in disorders that lead to vitamin K deficiency such as obstructive jaundice or fat malabsorption of any kind. Marked prolongation of the prothrombin time, >5 s above control and not corrected by parenteral vitamin K administration, is a poor prognostic sign in acute viral hepatitis and other acute and chronic liver diseases. The INR, along with the total serum bilirubin and creatinine, are components of the MELD score, which is used as a measure of hepatic decompensation and to allocate organs for liver transplantation.
OTHER DIAGNOSTIC TESTS
Although tests may direct the physician to a category of liver disease, additional radiologic testing and procedures are often necessary to make the proper diagnosis, as shown in Fig. 358-1. The most commonly used ancillary tests are reviewed here, as are the noninvasive tests available for assessing hepatic fibrosis.
Percutaneous Liver Biopsy Percutaneous biopsy of the liver is a safe procedure that can be easily performed at the bedside with local anesthesia and ultrasound guidance. Liver biopsy is of proven value in the following situations: (1) hepatocellular disease of uncertain cause, (2) prolonged hepatitis with the possibility of autoimmune hepatitis, (3) unexplained hepatomegaly, (4) unexplained splenomegaly, (5) hepatic filling defects by radiologic imaging, (6) fever of unknown origin, (7) and staging of malignant lymphoma. Liver biopsy is most accurate in disorders causing diffuse changes throughout the liver and is subject to sampling error in focal infiltrative disorders such as hepatic metastases. Liver biopsy should not be the initial procedure in the diagnosis of cholestasis. The biliary tree should first be assessed for signs of obstruction. Contraindications to performing a percutaneous liver biopsy include significant ascites and prolonged INR. Under these circumstances, the biopsy can be performed via the transjugular approach.
Noninvasive Tests to Detect Hepatic Fibrosis Although liver biopsy is the standard for the assessment of hepatic fibrosis, noninvasive measures of hepatic fibrosis have been developed and show promise. These measures include multiparameter tests aimed at detecting and staging the degree of hepatic fibrosis and imaging techniques. FibroTest (marketed as FibroSure in the United States) is the best evaluated of the multiparameter blood tests. The test incorporates haptoglobin, bilirubin, GGT, apolipoprotein A-I, and α2-macroglobulin and has been found to have high positive and negative predictive values for diagnosing advanced fibrosis in patients with chronic hepatitis C, chronic hepatitis B, and alcoholic liver disease and patients taking methotrexate for psoriasis. Transient elastography (TE), marketed as FibroScan, and magnetic resonance elastography (MRE) both have gained U.S. Food and Drug Administration approval for use in the management of patients with liver disease. TE uses ultrasound waves to measure hepatic stiffness noninvasively. TE has been shown to be accurate for identifying advanced fibrosis in patients with chronic hepatitis C, primary biliary cirrhosis, hemochromatosis, nonalcoholic fatty liver disease, and recurrent chronic hepatitis after liver transplantation. MRE has been found to be superior to TE for staging liver fibrosis in patients with a variety of chronic liver diseases, but requires access to a magnetic resonance imaging scanner.
Ultrasonography Ultrasonography is the first diagnostic test to use in patients whose liver tests suggest cholestasis, to look for the presence of a dilated intrahepatic or extrahepatic biliary tree or to identify gallstones. In addition, it shows space-occupying lesions within the liver, enables the clinician to distinguish between cystic and solid masses, and helps direct percutaneous biopsies. Ultrasound with Doppler imaging can detect the patency of the portal vein, hepatic artery, and hepatic veins and determine the direction of blood flow. This is the first test ordered in patients suspected of having Budd-Chiari syndrome.
USE OF LIVER TESTS
As previously noted, the best way to increase the sensitivity and specificity of laboratory tests in the detection of liver disease is to employ a battery of tests that includes the aminotransferases, alkaline phosphatase, bilirubin, albumin, and prothrombin time along with the judicious use of the other tests described in this chapter. Table 358-1 shows how patterns of liver tests can lead the clinician to a category of disease that will direct further evaluation. However, it is important to remember that no single set of liver tests will necessarily provide a diagnosis. It is often necessary to repeat these tests on several occasions over days to weeks for a diagnostic pattern to emerge. Figure 358-1 is an algorithm for the evaluation of chronically abnormal liver tests.
|
LIVER TEST PATTERNS IN HEPATOBILIARY DISORDERS |
GLOBAL CONSIDERATIONS
![]() The tests and principles presented in this chapter are applicable worldwide. The causes of liver test abnormalities vary according to region. In developing nations, infectious diseases are more commonly the etiology of abnormal serum liver tests than in developed nations.
The tests and principles presented in this chapter are applicable worldwide. The causes of liver test abnormalities vary according to region. In developing nations, infectious diseases are more commonly the etiology of abnormal serum liver tests than in developed nations.
ACKNOWLEDGMENT
This chapter represents a revised version of a chapter in previous editions of Harrison’s in which Marshall M. Kaplan was a co-author.
359 |
The Hyperbilirubinemias |
BILIRUBIN METABOLISM
The details of bilirubin metabolism are presented in Chap. 58. However, the hyperbilirubinemias are best understood in terms of perturbations of specific aspects of bilirubin metabolism and transport, and these will be briefly reviewed here as depicted in Fig. 359-1.
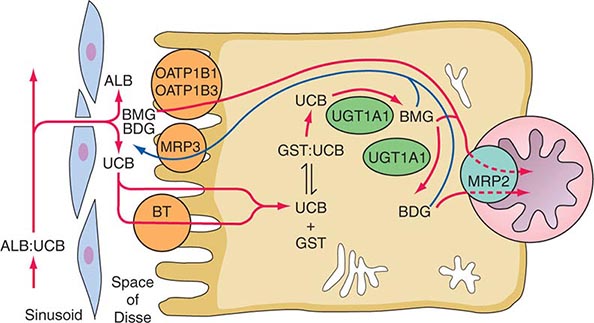
FIGURE 359-1 Hepatocellular bilirubin transport. Albumin-bound bilirubin in sinusoidal blood passes through endothelial cell fenestrae to reach the hepatocyte surface, entering the cell by both facilitated and simple diffusional processes. Within the cell, it is bound to glutathione-S-transferases and conjugated by bilirubin-UDP-glucuronosyltransferase (UGT1A1) to mono- and diglucuronides, which are actively transported across the canalicular membrane into the bile. In addition to this direct excretion of bilirubin glucuronides, a portion are transported into the portal circulation by MRP3 and subjected to reuptake into the hepatocyte by OATP1B1 and OATP1B3. ALB, albumin; BDG, bilirubin diglucuronide; BMG, bilirubin monoglucuronide; BT, proposed bilirubin transporter; GST, glutathione-S-transferase; MRP2 and MRP3, multidrug resistance–associated proteins 2 and 3; OATP1B1 and OATP1B3, organic anion transport proteins 1B1 and 1B3; UCB, unconjugated bilirubin; UGT1A1, bilirubin-UDP-glucuronosyltransferase.
Bilirubin is the end product of heme degradation. Some 70–90% of bilirubin is derived from degradation of the hemoglobin of senescent red blood cells. Bilirubin produced in the periphery is transported to the liver within the plasma, where, due to its insolubility in aqueous solutions, it is tightly bound to albumin. Under normal circumstances, bilirubin is removed from the circulation rapidly and efficiently by hepatocytes. Transfer of bilirubin from blood to bile involves four distinct but interrelated steps (Fig. 359-1).
1. Hepatocellular uptake: Uptake of bilirubin by the hepatocyte has carrier-mediated kinetics. Although a number of candidate bilirubin transporters have been proposed, the actual transporter remains elusive.
2. Intracellular binding: Within the hepatocyte, bilirubin is kept in solution by binding as a nonsubstrate ligand to several of the glutathione-S-transferases, formerly called ligandins.
3. Conjugation: Bilirubin is conjugated with one or two glucuronic acid moieties by a specific UDP-glucuronosyltransferase to form bilirubin mono- and diglucuronide, respectively. Conjugation disrupts the internal hydrogen bonding that limits aqueous solubility of bilirubin, and the resulting glucuronide conjugates are highly soluble in water. Conjugation is obligatory for excretion of bilirubin across the bile canalicular membrane into bile. The UDP-glucuronosyltransferases have been classified into gene families based on the degree of homology among the mRNAs for the various isoforms. Those that conjugate bilirubin and certain other substrates have been designated the UGT1 family. These are expressed from a single gene complex by alternative promoter usage. This gene complex contains multiple substrate-specific first exons, designated A1, A2, etc. (Fig. 359-2), each with its own promoter and each encoding the amino-terminal half of a specific isoform. In addition, there are four common exons (exons 2–5) that encode the shared carboxyl-terminal half of all of the UGT1 isoforms. The various first exons encode the specific aglycone substrate binding sites for each isoform, while the shared exons encode the binding site for the sugar donor, UDP-glucuronic acid, and the transmembrane domain. Exon A1 and the four common exons, collectively designated the UGT1A1 gene (Fig. 359-2), encode the physiologically critical enzyme bilirubin-UDP-glucuronosyltransferase (UGT1A1). A functional corollary of the organization of the UGT1 gene is that a mutation in one of the first exons will affect only a single enzyme isoform. By contrast, a mutation in exons 2–5 will alter all isoforms encoded by the UGT1 gene complex.
FIGURE 359-2 Structural organization of the human UGT1 gene complex. This large complex on chromosome 2 contains at least 13 substrate-specific first exons (A1, A2, etc.). Since four of these are pseudogenes, nine UGT1 isoforms with differing substrate specificities are expressed. Each exon 1 has its own promoter and encodes the amino-terminal substrate-specific ~286 amino acids of the various UGT1-encoded isoforms, and common exons 2–5 that encode the 245 carboxyl-terminal amino acids common to all of the isoforms. mRNAs for specific isoforms are assembled by splicing a particular first exon such as the bilirubin-specific exon A1 to exons 2 to 5. The resulting message encodes a complete enzyme, in this particular case bilirubin-UDP-glucuronosyltransferase (UGT1A1). Mutations in a first exon affect only a single isoform. Those in exons 2–5 affect all enzymes encoded by the UGT1 complex.
4. Biliary excretion: It has been thought until recently that bilirubin mono- and diglucuronides are excreted directly across the canalicular plasma membrane into the bile canaliculus by an ATP-dependent transport process mediated by a canalicular membrane protein called multidrug resistance–associated protein 2 (MRP2). Mutations of MRP2 result in the Dubin-Johnson syndrome (see below). However, studies in patients with Rotor syndrome (see below) indicate that after formation, a portion of the glucuronides are transported into the portal circulation by a sinusoidal membrane protein called multidrug resistance–associated protein 3 (MRP3) and subjected to reuptake into the hepatocyte by the sinusoidal membrane uptake transporters organic anion transport protein 1B1 (OATP1B1) and OATP1B3.
EXTRAHEPATIC ASPECTS OF BILIRUBIN DISPOSITION
Bilirubin in the Gut Following secretion into bile, conjugated bilirubin reaches the duodenum and passes down the gastrointestinal tract without reabsorption by the intestinal mucosa. An appreciable fraction is converted by bacterial metabolism in the gut to the water-soluble colorless compound urobilinogen. Urobilinogen undergoes enterohepatic cycling. Urobilinogen not taken up by the liver reaches the systemic circulation, from which some is cleared by the kidneys. Unconjugated bilirubin ordinarily does not reach the gut except in neonates or, by ill-defined alternative pathways, in the presence of severe unconjugated hyperbilirubinemia (e.g., Crigler-Najjar syndrome, type I [CN-I]). Unconjugated bilirubin that reaches the gut is partly reabsorbed, amplifying any underlying hyperbilirubinemia. Recent reports suggest that oral administration of calcium phosphate with or without the lipase inhibitor orlistat may be an efficient means to interrupt bilirubin enterohepatic cycling to reduce serum bilirubin levels in this situation. Although orlistat administration for 4–6 weeks to 16 patients with Crigler-Najjar syndrome was associated with a 10–20% decrease in serum bilirubin in 7 patients, the cost and side effects (i.e., diarrhea) may obviate the small benefit achievable with this treatment.
Renal Excretion of Bilirubin Conjugates Unconjugated bilirubin is not excreted in urine, as it is too tightly bound to albumin for effective glomerular filtration and there is no tubular mechanism for its renal secretion. In contrast, the bilirubin conjugates are readily filtered at the glomerulus and can appear in urine in disorders characterized by increased bilirubin conjugates in the circulation.
DISORDERS OF BILIRUBIN METABOLISM LEADING TO UNCONJUGATED HYPERBILIRUBINEMIA
INCREASED BILIRUBIN PRODUCTION
Hemolysis Increased destruction of erythrocytes leads to increased bilirubin turnover and unconjugated hyperbilirubinemia; the hyperbilirubinemia is usually modest in the presence of normal liver function. In particular, the bone marrow is only capable of a sustained eightfold increase in erythrocyte production in response to a hemolytic stress. Therefore, hemolysis alone cannot result in a sustained hyperbilirubinemia of more than ~68 μmol/L (4 mg/dL). Higher values imply concomitant hepatic dysfunction. When hemolysis is the only abnormality in an otherwise healthy individual, the result is a purely unconjugated hyperbilirubinemia, with the direct-reacting fraction as measured in a typical clinical laboratory being ≤15% of the total serum bilirubin. In the presence of systemic disease, which may include a degree of hepatic dysfunction, hemolysis may produce a component of conjugated hyperbilirubinemia in addition to an elevated unconjugated bilirubin concentration. Prolonged hemolysis may lead to the precipitation of bilirubin salts within the gallbladder or biliary tree, resulting in the formation of gallstones in which bilirubin, rather than cholesterol, is the major component. Such pigment stones may lead to acute or chronic cholecystitis, biliary obstruction, or any other biliary tract consequence of calculous disease.
Ineffective Erythropoiesis During erythroid maturation, small amounts of hemoglobin may be lost at the time of nuclear extrusion, and a fraction of developing erythroid cells is destroyed within the marrow. These processes normally account for a small proportion of bilirubin that is produced. In various disorders, including thalassemia major, megaloblastic anemias due to folate or vitamin B12 deficiency, congenital erythropoietic porphyria, lead poisoning, and various congenital and acquired dyserythropoietic anemias, the fraction of total bilirubin production derived from ineffective erythropoiesis is increased, reaching as much as 70% of the total. This may be sufficient to produce modest degrees of unconjugated hyperbilirubinemia.
Miscellaneous Degradation of the hemoglobin of extravascular collections of erythrocytes, such as those seen in massive tissue infarctions or large hematomas, may lead transiently to unconjugated hyperbilirubinemia.
DECREASED HEPATIC BILIRUBIN CLEARANCE
Decreased Hepatic Uptake Decreased hepatic bilirubin uptake is believed to contribute to the unconjugated hyperbilirubinemia of Gilbert syndrome (GS), although the molecular basis for this finding remains unclear (see below). Several drugs, including flavaspidic acid, novobiocin, and rifampin, as well as various cholecystographic contrast agents, have been reported to inhibit bilirubin uptake. The resulting unconjugated hyperbilirubinemia resolves with cessation of the medication.
Impaired Conjugation • PHYSIOLOGIC NEONATAL JAUNDICE Bilirubin produced by the fetus is cleared by the placenta and eliminated by the maternal liver. Immediately after birth, the neonatal liver must assume responsibility for bilirubin clearance and excretion. However, many hepatic physiologic processes are incompletely developed at birth. Levels of UGT1A1 are low, and alternative excretory pathways allow passage of unconjugated bilirubin into the gut. Since the intestinal flora that convert bilirubin to urobilinogen are also undeveloped, an enterohepatic circulation of unconjugated bilirubin ensues. As a consequence, most neonates develop mild unconjugated hyperbilirubinemia between days 2 and 5 after birth. Peak levels are typically <85–170 μmol/L (5–10 mg/dL) and decline to normal adult concentrations within 2 weeks, as mechanisms required for bilirubin disposition mature. Prematurity, often associated with more profound immaturity of hepatic function and hemolysis, can result in higher levels of unconjugated hyperbilirubinemia. A rapidly rising unconjugated bilirubin concentration, or absolute levels >340 μmol/L (20 mg/dL), puts the infant at risk for bilirubin encephalopathy, or kernicterus. Under these circumstances, bilirubin crosses an immature blood-brain barrier and precipitates in the basal ganglia and other areas of the brain. The consequences range from appreciable neurologic deficits to death. Treatment options include phototherapy, which converts bilirubin into water-soluble photoisomers that are excreted directly into bile, and exchange transfusion. The canalicular mechanisms responsible for bilirubin excretion are also immature at birth, and their maturation may lag behind that of UGT1A1; this can lead to transient conjugated neonatal hyperbilirubinemia, especially in infants with hemolysis.
ACQUIRED CONJUGATION DEFECTS A modest reduction in bilirubin conjugating capacity may be observed in advanced hepatitis or cirrhosis. However, in this setting, conjugation is better preserved than other aspects of bilirubin disposition, such as canalicular excretion. Various drugs, including pregnanediol, novobiocin, chloramphenicol, and gentamicin, may produce unconjugated hyperbilirubinemia by inhibiting UGT1A1 activity. Bilirubin conjugation may be inhibited by certain fatty acids that are present in breast milk but not serum of mothers whose infants have excessive neonatal hyperbilirubinemia (breast milk jaundice). Alternatively, there may be increased enterohepatic circulation of bilirubin in these infants. A recent study has correlated epidermal growth factor (EGF) content of breast milk with elevated bilirubin levels in these infants; however, a cause-and-effect relationship remains to be established. The pathogenesis of breast milk jaundice appears to differ from that of transient familial neonatal hyperbilirubinemia (Lucey-Driscoll syndrome), in which there is a UGT1A1 inhibitor in maternal serum.
HEREDITARY DEFECTS IN BILIRUBIN CONJUGATION
Three familial disorders characterized by differing degrees of unconjugated hyperbilirubinemia have long been recognized. The defining clinical features of each are described below (Table 359-1). While these disorders have been recognized for decades to reflect differing degrees of deficiency in the ability to conjugate bilirubin, recent advances in the molecular biology of the UGT1 gene complex have elucidated their interrelationships and clarified previously puzzling features.
|
PRINCIPAL DIFFERENTIAL CHARACTERISTICS OF GILBERT AND CRIGLER-NAJJAR SYNDROMES |
Crigler-Najjar Syndrome, Type I CN-I is characterized by striking unconjugated hyperbilirubinemia of about 340–765 μmol/L (20–45 mg/dL) that appears in the neonatal period and persists for life. Other conventional hepatic biochemical tests such as serum aminotransferases and alkaline phosphatase are normal, and there is no evidence of hemolysis. Hepatic histology is also essentially normal except for the occasional presence of bile plugs within canaliculi. Bilirubin glucuronides are virtually absent from the bile, and there is no detectable constitutive expression of UGT1A1 activity in hepatic tissue. Neither UGT1A1 activity nor the serum bilirubin concentration responds to administration of phenobarbital or other enzyme inducers. In the absence of conjugation, unconjugated bilirubin accumulates in plasma, from which it is eliminated very slowly by alternative pathways that include direct passage into the bile and small intestine. These account for the small amounts of urobilinogen found in feces. No bilirubin is found in the urine. First described in 1952, the disorder is rare (estimated prevalence, 0.6–1.0 per million). Many patients are from geographically or socially isolated communities in which consanguinity is common, and pedigree analyses show an autosomal recessive pattern of inheritance. The majority of patients (type IA) exhibit defects in the glucuronide conjugation of a spectrum of substrates in addition to bilirubin, including various drugs and other xenobiotics. These individuals have mutations in one of the common exons (2–5) of the UGT1 gene (Fig. 359-2). In a smaller subset (type IB), the defect is limited largely to bilirubin conjugation, and the causative mutation is in the bilirubin-specific exon A1. Estrogen glucuronidation is mediated by UGT1A1 and is defective in all CN-I patients. More than 30 different genetic lesions of UGT1A1 responsible for CN-I have been identified, including deletions, insertions, alterations in intron splice donor and acceptor sites, exon skipping, and point mutations that introduce premature stop codons or alter critical amino acids. Their common feature is that they all encode proteins with absent or, at most, traces of bilirubin-UDP-glucuronosyltransferase enzymatic activity.
Prior to the availability of phototherapy, most patients with CN-I died of bilirubin encephalopathy (kernicterus) in infancy or early childhood. A few lived as long as early adult life without overt neurologic damage, although more subtle testing usually indicated mild but progressive brain damage. In the absence of liver transplantation, death eventually supervened from late-onset bilirubin encephalopathy, which often followed a nonspecific febrile illness. Although isolated hepatocyte transplantation has been used in a small number of cases of CN-I, early liver transplantation (Chap. 368) remains the best hope to prevent brain injury and death.
Crigler-Najjar Syndrome, Type II (CN-II) This condition was recognized as a distinct entity in 1962 and is characterized by marked unconjugated hyperbilirubinemia in the absence of abnormalities of other conventional hepatic biochemical tests, hepatic histology, or hemolysis. It differs from CN-I in several specific ways (Table 359-1): (1) Although there is considerable overlap, average bilirubin concentrations are lower in CN-II; (2) accordingly, CN-II is only infrequently associated with kernicterus; (3) bile is deeply colored, and bilirubin glucuronides are present, with a striking, characteristic increase in the proportion of monoglucuronides; (4) UGT1A1 in liver is usually present at reduced levels (typically ≤10% of normal) but may be undetectable by older, less sensitive assays; and (5) while typically detected in infancy, hyperbilirubinemia was not recognized in some cases until later in life and, in one instance, at age 34. As with CN-I, most CN-II cases exhibit abnormalities in the conjugation of other compounds, such as salicylamide and menthol, but in some instances, the defect appears limited to bilirubin. Reduction of serum bilirubin concentrations by >25% in response to enzyme inducers such as phenobarbital distinguishes CN-II from CN-I, although this response may not be elicited in early infancy and often is not accompanied by measurable UGT1A1 induction. Bilirubin concentrations during phenobarbital administration do not return to normal but are typically in the range of 51–86 μmol/L (3–5 mg/dL). Although the incidence of kernicterus in CN-II is low, instances have occurred, not only in infants but also in adolescents and adults, often in the setting of an intercurrent illness, fasting, or another factor that temporarily raises the serum bilirubin concentration above baseline and reduces serum albumin levels. For this reason, phenobarbital therapy is widely recommended, a single bedtime dose often sufficing to maintain clinically safe serum bilirubin concentrations.
Over 77 different mutations in the UGT1 gene have been identified as causing CN-I or CN-II. It was found that missense mutations are more common in CN-II patients, as would be expected in this less severe phenotype. Their common feature is that they encode for a bilirubin-UDP-glucuronosyltransferase with markedly reduced, but detectable, enzymatic activity. The spectrum of residual enzyme activity explains the spectrum of phenotypic severity of the resulting hyperbilirubinemia. Molecular analysis has established that a large majority of CN-II patients are either homozygotes or compound heterozygotes for CN-II mutations and that individuals carrying one mutated and one entirely normal allele have normal bilirubin concentrations.
Gilbert Syndrome (GS) This syndrome is characterized by mild unconjugated hyperbilirubinemia, normal values for standard hepatic biochemical tests, and normal hepatic histology other than a modest increase of lipofuscin pigment in some patients. Serum bilirubin concentrations are most often <51 μmol/L (<3 mg/dL), although both higher and lower values are frequent. The clinical spectrum of hyperbilirubinemia fades into that of CN-II at serum bilirubin concentrations of 86–136 μmol/L (5–8 mg/dL). At the other end of the scale, the distinction between mild cases of GS and a normal state is often blurred. Bilirubin concentrations may fluctuate substantially in any given individual, and at least 25% of patients will exhibit temporarily normal values during prolonged follow-up. More elevated values are associated with stress, fatigue, alcohol use, reduced caloric intake, and intercurrent illness, while increased caloric intake or administration of enzyme-inducing agents produces lower bilirubin levels. GS is most often diagnosed at or shortly after puberty or in adult life during routine examinations that include multichannel biochemical analyses. UGT1A1 activity is typically reduced to 10–35% of normal, and bile pigments exhibit a characteristic increase in bilirubin monoglucuronides. Studies of radiobilirubin kinetics indicate that hepatic bilirubin clearance is reduced to an average of one-third of normal. Administration of phenobarbital normalizes both the serum bilirubin concentration and hepatic bilirubin clearance; however, failure of UGT1A1 activity to improve in many such instances suggests the possible coexistence of an additional defect. Compartmental analysis of bilirubin kinetic data suggests that GS patients have a defect in bilirubin uptake as well as in conjugation. Defect(s) in the hepatic uptake of other organic anions that at least partially share an uptake mechanism with bilirubin, such as sulfobromophthalein and indocyanine green (ICG), are observed in a minority of patients. The metabolism and transport of bile acids that do not utilize the bilirubin uptake mechanism are normal. The magnitude of changes in the serum bilirubin concentration induced by provocation tests such as 48 hours of fasting or the IV administration of nicotinic acid have been reported to be of help in separating GS patients from normal individuals. Other studies dispute this assertion. Moreover, on theoretical grounds, the results of such studies should provide no more information than simple measurements of the baseline serum bilirubin concentration. Family studies indicate that GS and hereditary hemolytic anemias such as hereditary spherocytosis, glucose-6-phosphate dehydrogenase deficiency, and β-thalassemia trait sort independently. Reports of hemolysis in up to 50% of GS patients are believed to reflect better case finding, since patients with both GS and hemolysis have higher bilirubin concentrations, and are more likely to be jaundiced, than patients with either defect alone.
GS is common, with many series placing its prevalence at ≥8%. Males predominate over females by reported ratios ranging from 1.5:1 to >7:1. However, these ratios may have a large artifactual component since normal males have higher mean bilirubin levels than normal females, but the diagnosis of GS is often based on comparison to normal ranges established in men. The high prevalence of GS in the general population may explain the reported frequency of mild unconjugated hyperbilirubinemia in liver transplant recipients. The disposition of most xenobiotics metabolized by glucuronidation appears to be normal in GS, as is oxidative drug metabolism in the majority of reported studies. The principal exception is the metabolism of the antitumor agent irinotecan (CPT-11), whose active metabolite (SN-38) is glucuronidated specifically by bilirubin-UDP-glucuronosyltransferase. Administration of CPT-11 to patients with GS has resulted in several toxicities, including intractable diarrhea and myelosuppression. Some reports also suggest abnormal disposition of menthol, estradiol benzoate, acetaminophen, tolbutamide, and rifamycin SV. Although some of these studies have been disputed, and there have been no reports of clinical complications from use of these agents in GS, prudence should be exercised in prescribing them, or any agents metabolized primarily by glucuronidation, in this condition. It should also be noted that the HIV protease inhibitors indinavir and atazanavir (Chap. 226) can inhibit UGT1A1, resulting in hyperbilirubinemia that is most pronounced in patients with preexisting GS.
Most older pedigree studies of GS were consistent with autosomal dominant inheritance with variable expressivity. However, studies of the UGT1 gene in GS have indicated a variety of molecular genetic bases for the phenotypic picture and several different patterns of inheritance. Studies in Europe and the United States found that nearly all patients had normal coding regions for UGT1A1 but were homozygous for the insertion of an extra TA (i.e., A[TA]7TAA rather than A[TA]6TAA) in the promoter region of the first exon. This appeared to be necessary, but not sufficient, for clinically expressed GS, since 15% of normal controls were also homozygous for this variant. While normal by standard criteria, these individuals had somewhat higher bilirubin concentrations than the rest of the controls studied. Heterozygotes for this abnormality had bilirubin concentrations identical to those homozygous for the normal A[TA]6TAA allele. The prevalence of the A[TA]7TAA allele in a general Western population is 30%, in which case 9% would be homozygotes. This is slightly higher than the prevalence of GS based on purely phenotypic parameters. It was suggested that additional variables, such as mild hemolysis or a defect in bilirubin uptake, might be among the factors enhancing phenotypic expression of the defect.
Phenotypic expression of GS due solely to the A[TA]7TAA promoter abnormality is inherited as an autosomal recessive trait. A number of CN-II kindreds have been identified in whom there is also an allele containing a normal coding region but the A[TA]7TAA promoter abnormality. CN-II heterozygotes who have the A[TA]6TAA promoter are phenotypically normal, whereas those with the A[TA]7TAA promoter express the phenotypic picture of GS. GS in such kindreds may also result from homozygosity for the A[TA]7TAA promoter abnormality. Seven different missense mutations in the UGT1 gene that reportedly cause GS with dominant inheritance have been found in Japanese individuals. Another Japanese patient with mild unconjugated hyperbilirubinemia was homozygous for a missense mutation in exon 5. GS in her family appeared to be recessive. Missense mutations causing GS have not been reported outside of certain Asian populations.
DISORDERS OF BILIRUBIN METABOLISM LEADING TO MIXED OR PREDOMINANTLY CONJUGATED HYPERBILIRUBINEMIA
In hyperbilirubinemia due to acquired liver disease (e.g., acute hepatitis, common bile duct stone), there are usually elevations in the serum concentrations of both conjugated and unconjugated bilirubin. Although biliary tract obstruction or hepatocellular cholestatic injury may present on occasion with a predominantly conjugated hyperbilirubinemia, it is generally not possible to differentiate intrahepatic from extrahepatic causes of jaundice based on the serum levels or relative proportions of unconjugated and conjugated bilirubin. The major reason for determining the amounts of conjugated and unconjugated bilirubin in the serum is for the initial differentiation of hepatic parenchymal and obstructive disorders (mixed conjugated and unconjugated hyperbilirubinemia) from the inheritable and hemolytic disorders discussed above that are associated with unconjugated hyperbilirubinemia.
FAMILIAL DEFECTS IN HEPATIC EXCRETORY FUNCTION
Dubin-Johnson Syndrome (DJS) This benign, relatively rare disorder is characterized by low-grade, predominantly conjugated hyperbilirubinemia (Table 359-2). Total bilirubin concentrations are typically between 34 and 85 μmol/L (2 and 5 mg/dL) but on occasion can be in the normal range or as high as 340–430 μmol/L (20–25 mg/dL) and can fluctuate widely in any given patient. The degree of hyperbilirubinemia may be increased by intercurrent illness, oral contraceptive use, and pregnancy. Because the hyperbilirubinemia is due to a predominant rise in conjugated bilirubin, bilirubinuria is characteristically present. Aside from elevated serum bilirubin levels, other routine laboratory tests are normal. Physical examination is usually normal except for jaundice, although an occasional patient may have hepatosplenomegaly.
|
PRINCIPAL DIFFERENTIAL CHARACTERISTICS OF INHERITABLE DISORDERS OF BILE CANALICULAR FUNCTION |
Patients with DJS are usually asymptomatic, although some may have vague constitutional symptoms. These latter patients have usually undergone extensive and often unnecessary diagnostic examinations for unexplained jaundice and have high levels of anxiety. In women, the condition may be subclinical until the patient becomes pregnant or receives oral contraceptives, at which time chemical hyperbilirubinemia becomes frank jaundice. Even in these situations, other routine liver function tests, including serum alkaline phosphatase and transaminase activities, are normal.
A cardinal feature of DJS is the accumulation in the lysosomes of centrilobular hepatocytes of dark, coarsely granular pigment. As a result, the liver may be grossly black in appearance. This pigment is thought to be derived from epinephrine metabolites that are not excreted normally. The pigment may disappear during bouts of viral hepatitis, only to reaccumulate slowly after recovery.
Biliary excretion of a number of anionic compounds is compromised in DJS. These include various cholecystographic agents, as well as sulfobromophthalein (Bromsulphalein, BSP), a synthetic dye formerly used in a test of liver function. In this test, the rate of disappearance of BSP from plasma was determined following bolus IV administration. BSP is conjugated with glutathione in the hepatocyte; the resulting conjugate is normally excreted rapidly into the bile canaliculus. Patients with DJS exhibit characteristic rises in plasma concentrations at 90 minutes after injection, due to reflux of conjugated BSP into the circulation from the hepatocyte. Dyes such as ICG that are taken up by hepatocytes but are not further metabolized prior to biliary excretion do not show this reflux phenomenon. Continuous BSP infusion studies suggest a reduction in the time to maximum plasma concentration (tmax) for biliary excretion. Bile acid disposition, including hepatocellular uptake and biliary excretion, is normal in DJS. These patients have normal serum and biliary bile acid concentrations and do not have pruritus.
By analogy with findings in several mutant rat strains, the selective defect in biliary excretion of bilirubin conjugates and certain other classes of organic compounds, but not of bile acids, that characterizes DJS in humans was found to reflect defective expression of MRP2, an ATP-dependent canalicular membrane transporter. Several different mutations in the MRP2 gene produce the Dubin-Johnson phenotype, which has an autosomal recessive pattern of inheritance. Although MRP2 is undoubtedly important in the biliary excretion of conjugated bilirubin, the fact that this pigment is still excreted in the absence of MRP2 suggests that other, as yet uncharacterized, transport proteins may serve in a secondary role in this process.
Patients with DJS also have a diagnostic abnormality in urinary coproporphyrin excretion. There are two naturally occurring coproporphyrin isomers, I and III. Normally, ~75% of the coproporphyrin in urine is isomer III. In urine from DJS patients, total coproporphyrin content is normal, but >80% is isomer I. Heterozygotes for the syndrome show an intermediate pattern. The molecular basis for this phenomenon remains unclear.
Rotor Syndrome This benign, autosomal recessive disorder is clinically similar to DJS (Table 359-2), although it is seen even less frequently. A major phenotypic difference is that the liver in patients with Rotor syndrome has no increased pigmentation and appears totally normal. The only abnormality in routine laboratory tests is an elevation of total serum bilirubin, due to a predominant rise in conjugated bilirubin. This is accompanied by bilirubinuria. Several additional features differentiate Rotor syndrome from DJS. In Rotor syndrome, the gallbladder is usually visualized on oral cholecystography, in contrast to the nonvisualization that is typical of DJS. The pattern of urinary coproporphyrin excretion also differs. The pattern in Rotor syndrome resembles that of many acquired disorders of hepatobiliary function, in which coproporphyrin I, the major coproporphyrin isomer in bile, refluxes from the hepatocyte back into the circulation and is excreted in urine. Thus, total urinary coproporphyrin excretion is substantially increased in Rotor syndrome, in contrast to the normal levels seen in DJS. Although the fraction of coproporphyrin I in urine is elevated, it is usually <70% of the total, compared with ≥80% in DJS. The disorders also can be distinguished by their patterns of BSP excretion. Although clearance of BSP from plasma is delayed in Rotor syndrome, there is no reflux of conjugated BSP back into the circulation as seen in DJS. Kinetic analysis of plasma BSP infusion studies suggests the presence of a defect in intrahepatocellular storage of this compound. This has never been demonstrated directly. Recent studies indicate that the molecular basis of Rotor syndrome results from simultaneous deficiency of the plasma membrane transporters OATP1B1 and OATP1B3. This results in reduced reuptake of conjugated bilirubin that has been pumped out of the cell into the portal circulation by MRP3 (Fig. 359-1).
Benign Recurrent Intrahepatic Cholestasis (BRIC) This rare disorder is characterized by recurrent attacks of pruritus and jaundice. The typical episode begins with mild malaise and elevations in serum aminotransferase levels, followed rapidly by rises in alkaline phosphatase and conjugated bilirubin and onset of jaundice and itching. The first one or two episodes may be misdiagnosed as acute viral hepatitis. The cholestatic episodes, which may begin in childhood or adulthood, can vary in duration from several weeks to months, followed by a complete clinical and biochemical resolution. Intervals between attacks may vary from several months to years. Between episodes, physical examination is normal, as are serum levels of bile acids, bilirubin, transaminases, and alkaline phosphatase. The disorder is familial and has an autosomal recessive pattern of inheritance. BRIC is considered a benign disorder in that it does not lead to cirrhosis or end-stage liver disease. However, the episodes of jaundice and pruritus can be prolonged and debilitating, and some patients have undergone liver transplantation to relieve the intractable and disabling symptoms. Treatment during the cholestatic episodes is symptomatic; there is no specific treatment to prevent or shorten the occurrence of episodes.
A gene termed FIC1 was recently identified and found to be mutated in patients with BRIC. Curiously, this gene is expressed strongly in the small intestine but only weakly in the liver. The protein encoded by FIC1 shows little similarity to those that have been shown to play a role in bile canalicular excretion of various compounds. Rather, it appears to be a member of a P-type ATPase family that transports aminophospholipids from the outer to the inner leaflet of a variety of cell membranes. Its relationship to the pathobiology of this disorder remains unclear. A second phenotypically identical form of BRIC, termed BRIC type 2, has been described resulting from mutations in the bile salt excretory protein (BSEP), the protein that is defective in progressive familial intrahepatic cholestasis type 2 (Table 359-2). How some mutations in this protein result in the episodic BRIC phenotype is unknown.
Progressive Familial Intrahepatic Cholestasis (FIC) This name is applied to three phenotypically related syndromes (Table 359-2). Progressive FIC type 1 (Byler disease) presents in early infancy as cholestasis that may be initially episodic. However, in contrast to BRIC, Byler disease progresses to malnutrition, growth retardation, and end-stage liver disease during childhood. This disorder is also a consequence of a FIC1 mutation. The functional relationship of the FIC1 protein to the pathogenesis of cholestasis in these disorders is unknown. Two other types of progressive FIC (types 2 and 3) have been described. Progressive FIC type 2 is associated with a mutation in the protein originally named sister of p-glycoprotein, now known as bile salt excretory protein, which is the major bile canalicular exporter of bile acids. As noted above, some mutations of this protein are associated with BRIC type 2, rather than the progressive FIC type 2 phenotype. Progressive FIC type 3 has been associated with a mutation of MDR3, a protein that is essential for normal hepatocellular excretion of phospholipids across the bile canaliculus. Although all three types of progressive FIC have similar clinical phenotypes, only type 3 is associated with high serum levels of γ-glutamyltransferase activity. In contrast, activity of this enzyme is normal or only mildly elevated in symptomatic BRIC and progressive FIC types 1 and 2.
360 |
Acute Viral Hepatitis |
Acute viral hepatitis is a systemic infection affecting the liver predominantly. Almost all cases of acute viral hepatitis are caused by one of five viral agents: hepatitis A virus (HAV), hepatitis B virus (HBV), hepatitis C virus (HCV), the HBV-associated delta agent or hepatitis D virus (HDV), and hepatitis E virus (HEV). All these human hepatitis viruses are RNA viruses, except for hepatitis B, which is a DNA virus but replicates like a retrovirus. Although these agents can be distinguished by their molecular and antigenic properties, all types of viral hepatitis produce clinically similar illnesses. These range from asymptomatic and inapparent to fulminant and fatal acute infections common to all types, on the one hand, and from subclinical persistent infections to rapidly progressive chronic liver disease with cirrhosis and even hepatocellular carcinoma, common to the bloodborne types (HBV, HCV, and HDV), on the other.
VIROLOGY AND ETIOLOGY
Hepatitis A HAV is a nonenveloped 27-nm, heat-, acid-, and ether-resistant RNA virus in the Hepatovirus genus of the picornavirus family (Fig. 360-1). Its virion contains four capsid polypeptides, designated VP1 to VP4, which are cleaved posttranslationally from the polyprotein product of a 7500-nucleotide genome. Inactivation of viral activity can be achieved by boiling for 1 min, by contact with formaldehyde and chlorine, or by ultraviolet irradiation. Despite nucleotide sequence variation of up to 20% among isolates of HAV, and despite the recognition of four genotypes affecting humans, all strains of this virus are immunologically indistinguishable and belong to one serotype. Hepatitis A has an incubation period of ~4 weeks. Its replication is limited to the liver, but the virus is present in the liver, bile, stools, and blood during the late incubation period and acute preicteric/presymptomatic phase of illness. Despite slightly longer persistence of virus in the liver, fecal shedding, viremia, and infectivity diminish rapidly once jaundice becomes apparent. HAV can be cultivated reproducibly in vitro.
FIGURE 360-1 Electron micrographs of hepatitis A virus particles and serum from a patient with hepatitis B. Left: 27-nm hepatitis A virus particles purified from stool of a patient with acute hepatitis A and aggregated by antibody to hepatitis A virus. Right: Concentrated serum from a patient with hepatitis B, demonstrating the 42-nm virions, tubular forms, and spherical 22-nm particles of hepatitis B surface antigen. 132,000×. (Hepatitis D resembles 42-nm virions of hepatitis B but is smaller, 35–37 nm; hepatitis E resembles hepatitis A virus but is slightly larger, 32–34 nm; hepatitis C has been visualized as a 55-nm particle.)
Antibodies to HAV (anti-HAV) can be detected during acute illness when serum aminotransferase activity is elevated and fecal HAV shedding is still occurring. This early antibody response is predominantly of the IgM class and persists for several (~3) months, rarely for 6–12 months. During convalescence, however, anti-HAV of the IgG class becomes the predominant antibody (Fig. 360-2). Therefore, the diagnosis of hepatitis A is made during acute illness by demonstrating anti-HAV of the IgM class. After acute illness, anti-HAV of the IgG class remains detectable indefinitely, and patients with serum anti-HAV are immune to reinfection. Neutralizing antibody activity parallels the appearance of anti-HAV, and the IgG anti-HAV present in immune globulin accounts for the protection it affords against HAV infection.
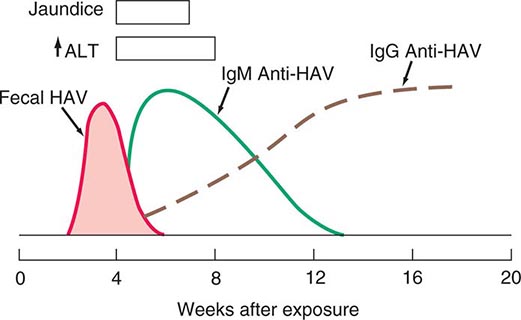
FIGURE 360-2 Scheme of typical clinical and laboratory features of hepatitis A virus (HAV). ALT, alanine aminotransferase.
Hepatitis B HBV is a DNA virus with a remarkably compact genomic structure; despite its small, circular, 3200-bp size, HBV DNA codes for four sets of viral products with a complex, multiparticle structure. HBV achieves its genomic economy by relying on an efficient strategy of encoding proteins from four overlapping genes: S, C, P, and × (Fig. 360-3), as detailed below. Once thought to be unique among viruses, HBV is now recognized as one of a family of animal viruses, hepadnaviruses (hepatotropic DNA viruses), and is classified as hepadnavirus type 1. Similar viruses infect certain species of woodchucks, ground and tree squirrels, and Pekin ducks, to mention the most carefully characterized. Like HBV, all have the same distinctive three morphologic forms, have counterparts to the envelope and nucleocapsid virus antigens of HBV, replicate in the liver but exist in extrahepatic sites, contain their own endogenous DNA polymerase, have partially double-strand and partially single-strand genomes, are associated with acute and chronic hepatitis and hepatocellular carcinoma, and rely on a replicative strategy unique among DNA viruses but typical of retroviruses. Instead of DNA replication directly from a DNA template, hepadnaviruses rely on reverse transcription (effected by the DNA polymerase) of minus-strand DNA from a “pregenomic” RNA intermediate. Then plus-strand DNA is transcribed from the minus-strand DNA template by the DNA-dependent DNA polymerase and converted in the hepatocyte nucleus to a covalently closed circular DNA, which serves as a template for messenger RNA and pregenomic RNA. Viral proteins are translated by the messenger RNA, and the proteins and genome are packaged into virions and secreted from the hepatocyte. Although HBV is difficult to cultivate in vitro in the conventional sense from clinical material, several cell lines have been transfected with HBV DNA. Such transfected cells support in vitro replication of the intact virus and its component proteins.
FIGURE 360-3 Compact genomic structure of hepatitis B virus (HBV). This structure, with overlapping genes, permits HBV to code for multiple proteins. The S gene codes for the “major” envelope protein, HBsAg. Pre-S1 and pre-S2, upstream of S, combine with S to code for two larger proteins, “middle” protein, the product of pre-S2 + S, and “large” protein, the product of pre-S1 + pre-S2 + S. The largest gene, P, codes for DNA polymerase. The C gene codes for two nucleocapsid proteins, HBeAg, a soluble, secreted protein (initiation from the pre-C region of the gene), and HBcAg, the intracellular core protein (initiation after pre-C). The × gene codes for HBxAg, which can transactivate the transcription of cellular and viral genes; its clinical relevance is not known, but it may contribute to carcinogenesis by binding to p53.
VIRAL PROTEINS AND PARTICLES Of the three particulate forms of HBV (Table 360-1), the most numerous are the 22-nm particles, which appear as spherical or long filamentous forms; these are antigenically indistinguishable from the outer surface or envelope protein of HBV and are thought to represent excess viral envelope protein. Outnumbered in serum by a factor of 100 or 1000 to 1 compared with the spheres and tubules are large, 42-nm, double-shelled spherical particles, which represent the intact hepatitis B virion (Fig. 360-1). The envelope protein expressed on the outer surface of the virion and on the smaller spherical and tubular structures is referred to as hepatitis B surface antigen (HBsAg). The concentration of HBsAg and virus particles in the blood may reach 500 μg/mL and 10 trillion particles per milliliter, respectively. The envelope protein, HBsAg, is the product of the S gene of HBV.
|
NOMENCLATURE AND FEATURES OF HEPATITIS VIRUSES |
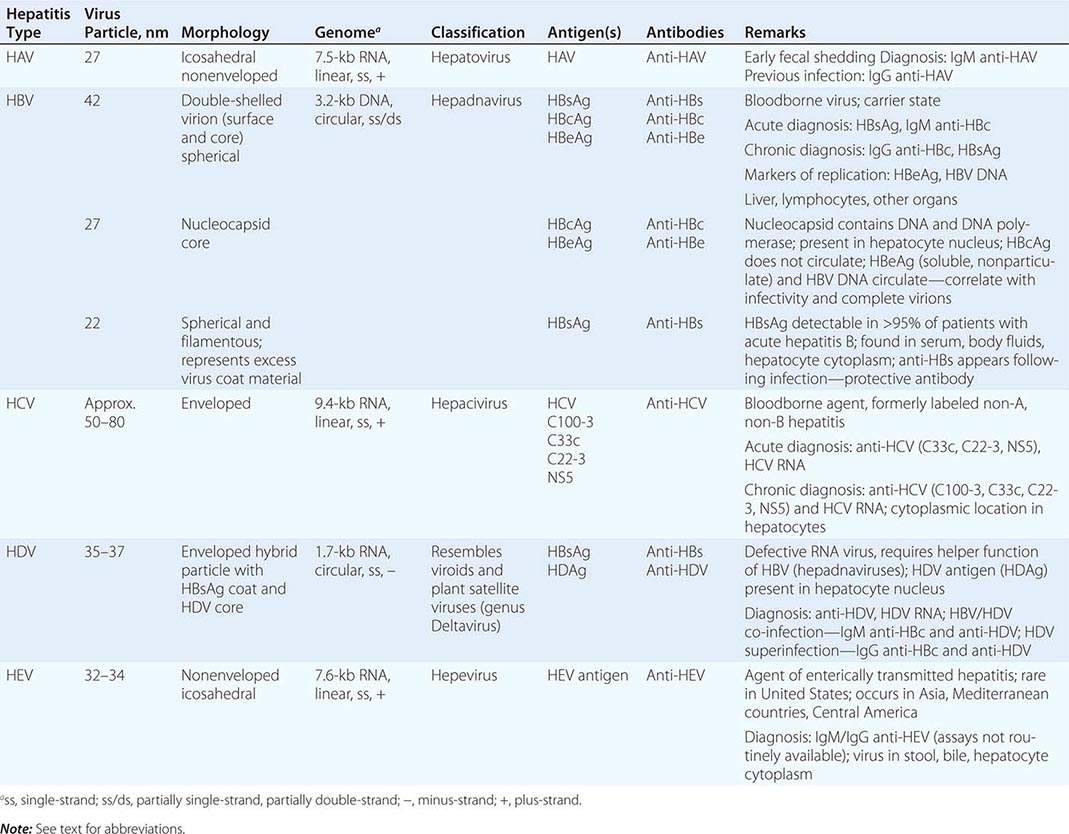
Envelope HBsAg subdeterminants include a common group-reactive antigen, a, shared by all HBsAg isolates and one of several subtype-specific antigens—d or y, w or r—as well as other specificities. Hepatitis B isolates fall into one of at least eight subtypes and ten genotypes (A–J). Geographic distribution of genotypes and subtypes varies; genotypes A (corresponding to subtype adw) and D (ayw) predominate in the United States and Europe, whereas genotypes B (adw) and C (adr) predominate in Asia. Clinical course and outcome are independent of subtype, but genotype B appears to be associated with less rapidly progressive liver disease and cirrhosis and a lower likelihood, or delayed appearance, of hepatocellular carcinoma than genotype C or D. Patients with genotype A are more likely to clear circulating viremia and to achieve HBeAg and HBsAg seroconversion, both spontaneously and in response to antiviral therapy. In addition, “precore” mutations are favored by certain genotypes (see below).
Upstream of the S gene are the pre-S genes (Fig. 360-3), which code for pre-S gene products, including receptors on the HBV surface for polymerized human serum albumin and for hepatocyte membrane proteins. The pre-S region actually consists of both pre-S1 and pre-S2. Depending on where translation is initiated, three potential HBsAg gene products are synthesized. The protein product of the S gene is HBsAg (major protein), the product of the S region plus the adjacent pre-S2 region is the middle protein, and the product of the pre-S1 plus pre-S2 plus S regions is the large protein. Compared with the smaller spherical and tubular particles of HBV, complete 42-nm virions are enriched in the large protein. Both pre-S proteins and their respective antibodies can be detected during HBV infection, and the period of pre-S antigenemia appears to coincide with other markers of virus replication, as detailed below; however, pre-S proteins have little clinical relevance and are not included in routine serologic testing repertoires.
The intact 42-nm virion contains a 27-nm nucleocapsid core particle. Nucleocapsid proteins are coded for by the C gene. The antigen expressed on the surface of the nucleocapsid core is hepatitis B core antigen (HBcAg), and its corresponding antibody is anti-HBc. A third HBV antigen is hepatitis B e antigen (HBeAg), a soluble, nonparticulate, nucleocapsid protein that is immunologically distinct from intact HBcAg but is a product of the same C gene. The C gene has two initiation codons, a precore and a core region (Fig. 360-3). If translation is initiated at the precore region, the protein product is HBeAg, which has a signal peptide that binds it to the smooth endoplasmic reticulum, the secretory apparatus of the cell, leading to its secretion into the circulation. If translation begins at the core region, HBcAg is the protein product; it has no signal peptide, it is not secreted, but it assembles into nucleocapsid particles, which bind to and incorporate RNA, and which, ultimately, contain HBV DNA. Also packaged within the nucleocapsid core is a DNA polymerase, which directs replication and repair of HBV DNA. When packaging within viral proteins is complete, synthesis of the incomplete plus strand stops; this accounts for the single-strand gap and for differences in the size of the gap. HBcAg particles remain in the hepatocyte, where they are readily detectable by immunohistochemical staining and are exported after encapsidation by an envelope of HBsAg. Therefore, naked core particles do not circulate in the serum. The secreted nucleocapsid protein, HBeAg, provides a convenient, readily detectable, qualitative marker of HBV replication and relative infectivity.
HBsAg-positive serum containing HBeAg is more likely to be highly infectious and to be associated with the presence of hepatitis B virions (and detectable HBV DNA, see below) than HBeAg-negative or anti-HBe-positive serum. For example, HBsAg-positive mothers who are HBeAg-positive almost invariably (>90%) transmit hepatitis B infection to their offspring, whereas HBsAg-positive mothers with anti-HBe rarely (10–15%) infect their offspring.
Early during the course of acute hepatitis B, HBeAg appears transiently; its disappearance may be a harbinger of clinical improvement and resolution of infection. Persistence of HBeAg in serum beyond the first 3 months of acute infection may be predictive of the development of chronic infection, and the presence of HBeAg during chronic hepatitis B tends to be associated with ongoing viral replication, infectivity, and inflammatory liver injury (except during the early decades after perinatally acquired HBV infection; see below).
The third and largest of the HBV genes, the P gene (Fig. 360-3), codes for HBV DNA polymerase; as noted above, this enzyme has both DNA-dependent DNA polymerase and RNA-dependent reverse transcriptase activities. The fourth gene, X, codes for a small, nonparticulate protein, hepatitis B × antigen (HBxAg), that is capable of transactivating the transcription of both viral and cellular genes (Fig. 360-3). In the cytoplasm, HBxAg effects calcium release (possibly from mitochondria), which activates signal-transduction pathways that lead to stimulation of HBV reverse transcription and HBV DNA replication. Such transactivation may enhance the replication of HBV, leading to the clinical association observed between the expression of HBxAg and antibodies to it in patients with severe chronic hepatitis and hepatocellular carcinoma. The transactivating activity can enhance the transcription and replication of other viruses besides HBV, such as HIV. Cellular processes transactivated by × include the human interferon γ gene and class I major histocompatibility genes; potentially, these effects could contribute to enhanced susceptibility of HBV-infected hepatocytes to cytolytic T cells. The expression of × can also induce programmed cell death (apoptosis). The clinical relevance of HBxAg is limited, however, and testing for it is not part of routine clinical practice.
SEROLOGIC AND VIROLOGIC MARKERS After a person is infected with HBV, the first virologic marker detectable in serum within 1–12 weeks, usually between 8 and 12 weeks, is HBsAg (Fig. 360-4). Circulating HBsAg precedes elevations of serum aminotransferase activity and clinical symptoms by 2–6 weeks and remains detectable during the entire icteric or symptomatic phase of acute hepatitis B and beyond. In typical cases, HBsAg becomes undetectable 1–2 months after the onset of jaundice and rarely persists beyond 6 months. After HBsAg disappears, antibody to HBsAg (anti-HBs) becomes detectable in serum and remains detectable indefinitely thereafter. Because HBcAg is intracellular and, when in the serum, sequestered within an HBsAg coat, naked core particles do not circulate in serum, and therefore, HBcAg is not detectable routinely in the serum of patients with HBV infection. By contrast, anti-HBc is readily demonstrable in serum, beginning within the first 1–2 weeks after the appearance of HBsAg and preceding detectable levels of anti-HBs by weeks to months. Because variability exists in the time of appearance of anti-HBs after HBV infection, occasionally a gap of several weeks or longer may separate the disappearance of HBsAg and the appearance of anti-HBs. During this “gap” or “window” period, anti-HBc may represent the only serologic evidence of current or recent HBV infection, and blood containing anti-HBc in the absence of HBsAg and anti-HBs has been implicated in transfusion-associated hepatitis B. In part because the sensitivity of immunoassays for HBsAg and anti-HBs has increased, however, this window period is rarely encountered. In some persons, years after HBV infection, anti-HBc may persist in the circulation longer than anti-HBs. Therefore, isolated anti-HBc does not necessarily indicate active virus replication; most instances of isolated anti-HBc represent hepatitis B infection in the remote past. Rarely, however, isolated anti-HBc represents low-level hepatitis B viremia, with HBsAg below the detection threshold, and, occasionally, isolated anti-HBc represents a cross-reacting or false-positive immunologic specificity. Recent and remote HBV infections can be distinguished by determination of the immunoglobulin class of anti-HBc. Anti-HBc of the IgM class (IgM anti-HBc) predominates during the first 6 months after acute infection, whereas IgG anti-HBc is the predominant class of anti-HBc beyond 6 months. Therefore, patients with current or recent acute hepatitis B, including those in the anti-HBc window, have IgM anti-HBc in their serum. In patients who have recovered from hepatitis B in the remote past as well as those with chronic HBV infection, anti-HBc is predominantly of the IgG class. Infrequently, in ≤1–5% of patients with acute HBV infection, levels of HBsAg are too low to be detected; in such cases, the presence of IgM anti-HBc establishes the diagnosis of acute hepatitis B. When isolated anti-HBc occurs in the rare patient with chronic hepatitis B whose HBsAg level is below the sensitivity threshold of contemporary immunoassays (a low-level carrier), anti-HBc is of the IgG class. Generally, in persons who have recovered from hepatitis B, anti-HBs and anti-HBc persist indefinitely.
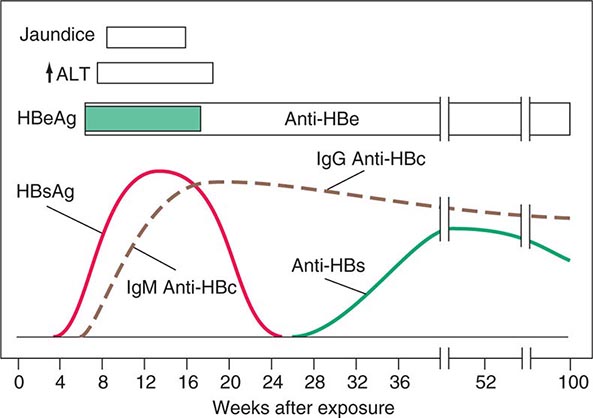
FIGURE 360-4 Scheme of typical clinical and laboratory features of acute hepatitis B. ALT, alanine aminotransferase.
The temporal association between the appearance of anti-HBs and resolution of HBV infection as well as the observation that persons with anti-HBs in serum are protected against reinfection with HBV suggests that anti-HBs is the protective antibody. Therefore, strategies for prevention of HBV infection are based on providing susceptible persons with circulating anti-HBs (see below). Occasionally, in ~10% of patients with chronic hepatitis B, low-level, low-affinity anti-HBs can be detected. This antibody is directed against a subtype determinant different from that represented by the patient’s HBsAg; its presence is thought to reflect the stimulation of a related clone of antibody-forming cells, but it has no clinical relevance and does not signal imminent clearance of hepatitis B. These patients with HBsAg and such nonneutralizing anti-HBs should be categorized as having chronic HBV infection.
The other readily detectable serologic marker of HBV infection, HBeAg, appears concurrently with or shortly after HBsAg. Its appearance coincides temporally with high levels of virus replication and reflects the presence of circulating intact virions and detectable HBV DNA (with the notable exception of patients with precore mutations who cannot synthesize HBeAg—see “Molecular Variants”). Pre-S1 and pre-S2 proteins are also expressed during periods of peak replication, but assays for these gene products are not routinely available. In self-limited HBV infections, HBeAg becomes undetectable shortly after peak elevations in aminotransferase activity, before the disappearance of HBsAg, and anti-HBe then becomes detectable, coinciding with a period of relatively lower infectivity (Fig. 360-4). Because markers of HBV replication appear transiently during acute infection, testing for such markers is of little clinical utility in typical cases of acute HBV infection. In contrast, markers of HBV replication provide valuable information in patients with protracted infections.
Departing from the pattern typical of acute HBV infections, in chronic HBV infection, HBsAg remains detectable beyond 6 months, anti-HBc is primarily of the IgG class, and anti-HBs is either undetectable or detectable at low levels (see “Laboratory Features”) (Fig. 360-5). During early chronic HBV infection, HBV DNA can be detected both in serum and in hepatocyte nuclei, where it is present in free or episomal form. This relatively highly replicative stage of HBV infection is the time of maximal infectivity and liver injury; HBeAg is a qualitative marker and HBV DNA a quantitative marker of this replicative phase, during which all three forms of HBV circulate, including intact virions. Over time, the relatively replicative phase of chronic HBV infection gives way to a relatively nonreplicative phase. This occurs at a rate of ~10% per year and is accompanied by seroconversion from HBeAg to anti-HBe. In many cases, this seroconversion coincides with a transient, usually mild, acute hepatitis-like elevation in aminotransferase activity, believed to reflect cell-mediated immune clearance of virus-infected hepatocytes. In the nonreplicative phase of chronic infection, when HBV DNA is demonstrable in hepatocyte nuclei, it tends to be integrated into the host genome. In this phase, only spherical and tubular forms of HBV, not intact virions, circulate, and liver injury tends to subside. Most such patients would be characterized as inactive HBV carriers. In reality, the designations replicative and nonreplicative are only relative; even in the so-called nonreplicative phase, HBV replication can be detected at levels of approximately ≤103 virions with highly sensitive amplification probes such as the polymerase chain reaction (PCR); below this replication threshold, liver injury and infectivity of HBV are limited to negligible. Still, the distinctions are pathophysiologically and clinically meaningful. Occasionally, nonreplicative HBV infection converts back to replicative infection. Such spontaneous reactivations are accompanied by reexpression of HBeAg and HBV DNA, and sometimes of IgM anti-HBc, as well as by exacerbations of liver injury. Because high-titer IgM anti-HBc can reappear during acute exacerbations of chronic hepatitis B, relying on IgM anti-HBc versus IgG anti-HBc to distinguish between acute and chronic hepatitis B infection, respectively, may not always be reliable; in such cases, patient history is invaluable in helping to distinguish de novo acute hepatitis B infection from acute exacerbation of chronic hepatitis B infection.
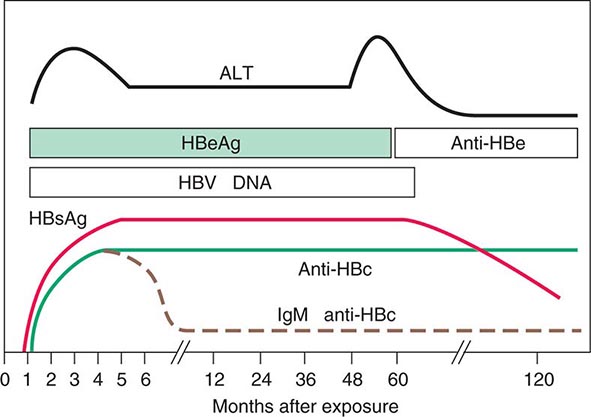
FIGURE 360-5 Scheme of typical laboratory features of wild-type chronic hepatitis B. HBeAg and hepatitis B virus (HBV) DNA can be detected in serum during the relatively replicative phase of chronic infection, which is associated with infectivity and liver injury. Seroconversion from the replicative phase to the relatively nonreplicative phase occurs at a rate of ~10% per year and is heralded by an acute hepatitis–like elevation of alanine aminotransferase (ALT) activity; during the nonreplicative phase, infectivity and liver injury are limited. In HBeAg-negative chronic hepatitis B associated with mutations in the precore region of the HBV genome, replicative chronic hepatitis B occurs in the absence of HBeAg.
MOLECULAR VARIANTS Variation occurs throughout the HBV genome, and clinical isolates of HBV that do not express typical viral proteins have been attributed to mutations in individual or even multiple gene locations. For example, variants have been described that lack nucleocapsid proteins (commonly), envelope proteins (very rarely), or both. Two categories of naturally occurring HBV variants have attracted the most attention. One of these was identified initially in Mediterranean countries among patients with severe chronic HBV infection and detectable HBV DNA but with anti-HBe instead of HBeAg. These patients were found to be infected with an HBV mutant that contained an alteration in the precore region rendering the virus incapable of encoding HBeAg. Although several potential mutation sites exist in the pre-C region, the region of the C gene necessary for the expression of HBeAg (see “Virology and Etiology”), the most commonly encountered in such patients is a single base substitution, from G to A in the second to last codon of the pre-C gene at nucleotide 1896. This substitution results in the replacement of the TGG tryptophan codon by a stop codon (TAG), which prevents the translation of HBeAg. Another mutation, in the core-promoter region, prevents transcription of the coding region for HBeAg and yields an HBeAg-negative phenotype. Patients with such mutations in the precore region and who are unable to secrete HBeAg may have severe liver disease that progresses more rapidly to cirrhosis, or alternatively, they are identified clinically later in the course of the natural history of chronic hepatitis B, when the disease is more advanced. Both “wild-type” HBV and precore-mutant HBV can coexist in the same patient, or mutant HBV may arise late during wild-type HBV infection. In addition, clusters of fulminant hepatitis B in Israel and Japan were attributed to common-source infection with a precore mutant. Fulminant hepatitis B in North America and western Europe, however, occurs in patients infected with wild-type HBV, in the absence of precore mutants, and both precore mutants and other mutations throughout the HBV genome occur commonly, even in patients with typical, self-limited, milder forms of HBV infection. HBeAg-negative chronic hepatitis with mutations in the precore region is now the most frequently encountered form of hepatitis B in Mediterranean countries and in Europe. In the United States, where HBV genotype A (less prone to G1896A mutation) is prevalent, precore-mutant HBV is much less common; however, as a result of immigration from Asia and Europe, the proportion of HBeAg-negative hepatitis B–infected individuals has increased in the United States, and they now represent approximately 30–40% of patients with chronic hepatitis B. Characteristic of such HBeAg-negative chronic hepatitis B are lower levels of HBV DNA (usually ≤105 IU/mL) and one of several patterns of aminotransferase activity—persistent elevations, periodic fluctuations above the normal range, and periodic fluctuations between the normal and elevated range.
The second important category of HBV mutants consists of escape mutants, in which a single amino acid substitution, from glycine to arginine, occurs at position 145 of the immunodominant a determinant common to all HBsAg subtypes. This HBsAg alteration leads to a critical conformational change that results in a loss of neutralizing activity by anti-HBs. This specific HBV/a mutant has been observed in two situations, active and passive immunization, in which humoral immunologic pressure may favor evolutionary change (“escape”) in the virus—in a small number of hepatitis B vaccine recipients who acquired HBV infection despite the prior appearance of neutralizing anti-HBs and in HBV-infected liver transplant recipients treated with a high-potency human monoclonal anti-HBs preparation. Although such mutants have not been recognized frequently, their existence raises a concern that may complicate vaccination strategies and serologic diagnosis.
Different types of mutations emerge during antiviral therapy of chronic hepatitis B with nucleoside analogues; such “YMDD” and similar mutations in the polymerase motif of HBV are described in Chap. 362.
EXTRAHEPATIC SITES Hepatitis B antigens and HBV DNA have been identified in extrahepatic sites, including lymph nodes, bone marrow, circulating lymphocytes, spleen, and pancreas. Although the virus does not appear to be associated with tissue injury in any of these extrahepatic sites, its presence in these “remote” reservoirs has been invoked (but is not necessary) to explain the recurrence of HBV infection after orthotopic liver transplantation. The clinical relevance of such extrahepatic HBV is limited.
Hepatitis D The delta hepatitis agent, or HDV, the only member of the genus Deltavirus, is a defective RNA virus that co-infects with and requires the helper function of HBV (or other hepadnaviruses) for its replication and expression. Slightly smaller than HBV, HDV is a formalin-sensitive, 35- to 37-nm virus with a hybrid structure. Its nucleocapsid expresses HDV antigen (HDAg), which bears no antigenic homology with any of the HBV antigens, and contains the virus genome. The HDV core is “encapsidated” by an outer envelope of HBsAg, indistinguishable from that of HBV except in its relative compositions of major, middle, and large HBsAg component proteins. The genome is a small, 1700-nucleotide, circular, single-strand RNA of negative polarity that is nonhomologous with HBV DNA (except for a small area of the polymerase gene) but that has features and the rolling circle model of replication common to genomes of plant satellite viruses or viroids. HDV RNA contains many areas of internal complementarity; therefore, it can fold on itself by internal base pairing to form an unusual, very stable, rodlike structure that contains a very stable, self-cleaving and self-ligating ribozyme. HDV RNA requires host RNA polymerase II for its replication in the hepatocyte nucleus via RNA-directed RNA synthesis by transcription of genomic RNA to a complementary antigenomic (plus strand) RNA; the antigenomic RNA, in turn, serves as a template for subsequent genomic RNA synthesis effected by host RNA polymerase I. HDV RNA has only one open reading frame, and HDAg, a product of the antigenomic strand, is the only known HDV protein; HDAg exists in two forms: a small, 195-amino-acid species, which plays a role in facilitating HDV RNA replication, and a large, 214-amino-acid species, which appears to suppress replication but is required for assembly of the antigen into virions. HDV antigens have been shown to bind directly to RNA polymerase II, resulting in stimulation of transcription. Although complete hepatitis D virions and liver injury require the cooperative helper function of HBV, intracellular replication of HDV RNA can occur without HBV. Genomic heterogeneity among HDV isolates has been described; however, pathophysiologic and clinical consequences of this genetic diversity have not been recognized. The clinical spectrum of hepatitis D is common to all eight genotypes identified, the predominant of which is genotype 1.
HDV can either infect a person simultaneously with HBV (coinfection) or superinfect a person already infected with HBV (superinfection); when HDV infection is transmitted from a donor with one HBsAg subtype to an HBsAg-positive recipient with a different subtype, HDV assumes the HBsAg subtype of the recipient, rather than the donor. Because HDV relies absolutely on HBV, the duration of HDV infection is determined by the duration of (and cannot outlast) HBV infection. HDV replication tends to suppress HBV replication; therefore, patients with hepatitis D tend to have lower levels of HBV replication. HDV antigen is expressed primarily in hepatocyte nuclei and is occasionally detectable in serum. During acute HDV infection, anti-HDV of the IgM class predominates, and 30–40 days may elapse after symptoms appear before anti-HDV can be detected. In self-limited infection, anti-HDV is low-titer and transient, rarely remaining detectable beyond the clearance of HBsAg and HDV antigen. In chronic HDV infection, anti-HDV circulates in high titer, and both IgM and IgG anti-HDV can be detected. HDV antigen in the liver and HDV RNA in serum and liver can be detected during HDV replication.
Hepatitis C Hepatitis C virus, which, before its identification was labeled “non-A, non-B hepatitis,” is a linear, single-strand, positive-sense, 9600-nucleotide RNA virus, the genome of which is similar in organization to that of flaviviruses and pestiviruses; HCV is the only member of the genus Hepacivirus in the family Flaviviridae. The HCV genome contains a single, large open reading frame (gene) that codes for a virus polyprotein of ~3000 amino acids, which is cleaved after translation to yield 10 viral proteins. The 5′ end of the genome consists of an untranslated region (containing an internal ribosomal entry site, IRES) adjacent to the genes for three structural proteins, the nucleocapsid core protein, C, and two structural envelope glycoproteins, E1 and E2. The 5′ untranslated region and core gene are highly conserved among genotypes, but the envelope proteins are coded for by the hypervariable region, which varies from isolate to isolate and may allow the virus to evade host immunologic containment directed at accessible virus-envelope proteins. The 3′ end of the genome also includes an untranslated region and contains the genes for seven nonstructural (NS) proteins, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B. p7 is a membrane ion channel protein necessary for efficient assembly and release of HCV. The NS2 cysteine protease cleaves NS3 from NS2, and the NS3-4A serine protease cleaves all the downstream proteins from the polyprotein. Important NS proteins involved in virus replication include the NS3 helicase; NS3-4A serine protease; the multifunctional membrane-associated phosphoprotein NS5A, an essential component of the viral replication membranous web (along with NS4B); and the NS5B RNA-dependent RNA polymerase (Fig. 360-6). Because HCV does not replicate via a DNA intermediate, it does not integrate into the host genome. Because HCV tends to circulate in relatively low titer, 103–107 virions/mL, visualization of the 50- to 80-nm virus particles remains difficult. Still, the replication rate of HCV is very high, 1012 virions per day; its half-life is 2.7 h. The chimpanzee is a helpful but cumbersome animal model. Although a robust, reproducible, small animal model is lacking, HCV replication has been documented in an immunodeficient mouse model containing explants of human liver and in transgenic mouse and rat models. Although in vitro replication is difficult, replicons in hepatocellular carcinoma–derived cell lines support replication of genetically manipulated, truncated, or full-length HCV RNA (but not intact virions); infectious pseudotyped retroviral HCV particles have been shown to yield functioning envelope proteins. In 2005, complete replication of HCV and intact 55-nm virions were described in cell culture systems. HCV entry into the hepatocyte occurs via the nonliver-specific CD81 receptor and the liver-specific tight junction protein claudin-1. A growing list of additional host receptors to which HCV binds on cell entry includes occludin, low-density lipoprotein receptors, glycosaminoglycans, scavenger receptor B1, and epidermal growth factor receptor, among others. Relying on the same assembly and secretion pathway as low-density and very-low-density lipoproteins, HCV is a lipoviroparticle and masquerades as a lipoprotein, which may limit its visibility to the adaptive immune system and which may explain its ability to evade immune containment and clearance. After viral entry and uncoating, translation is initiated by the IRES on the endoplasmic reticulum membrane, and the HCV polyprotein is cleaved during translation and posttranslationally by host cellular proteases as well as HCV NS2-3 and NS3-4A proteases. Host cofactors involved in HCV replication include cyclophilin A, which binds to NS5A and yields conformational changes required for viral replication, and liver-specific host microRNA miR-122.
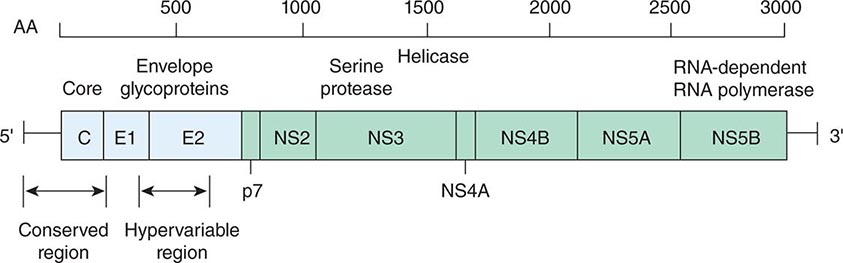
FIGURE 360-6 Organization of the hepatitis C virus genome and its associated, 3000-amino-acid (AA) proteins. The three structural genes at the 5’ end are the core region, C, which codes for the nucleocapsid, and the envelope regions, E1 and E2, which code for envelope glycoproteins. The 5’ untranslated region and the C region are highly conserved among isolates, whereas the envelope domain E2 contains the hypervariable region. At the 3’ end are seven nonstructural (NS) regions—p7, a membrane protein adjacent to the structural proteins that appears to function as an ion channel; NS2, which codes for a cysteine protease; NS3, which codes for a serine protease and an RNA helicase; NS4 and NS4B; NS5A, a multifunctional membrane-associated phosphoprotein, an essential component of the viral replication membranous web; and NS5B, which codes for an RNA-dependent RNA polymerase. After translation of the entire polyprotein, individual proteins are cleaved by both host and viral proteases.
At least six distinct major genotypes (and a minor genotype 7), as well as >50 subtypes within genotypes, of HCV have been identified by nucleotide sequencing. Genotypes differ from one another in sequence homology by ≥30%, and subtypes differ by approximately 20%. Because divergence of HCV isolates within a genotype or subtype and within the same host may vary insufficiently to define a distinct genotype, these intragenotypic differences are referred to as quasispecies and differ in sequence homology by only a few percent. The genotypic and quasispecies diversity of HCV, resulting from its high mutation rate, interferes with effective humoral immunity. Neutralizing antibodies to HCV have been demonstrated, but they tend to be short lived, and HCV infection does not induce lasting immunity against reinfection with different virus isolates or even the same virus isolate. Thus, neither heterologous nor homologous immunity appears to develop commonly after acute HCV infection. Some HCV genotypes are distributed worldwide, whereas others are more geographically confined (see “Epidemiology and Global Features”). In addition, differences exist among genotypes in responsiveness to antiviral therapy but not in pathogenicity or clinical progression (except for genotype 3, in which hepatic steatosis and clinical progression are more likely).
Currently available, third-generation immunoassays, which incorporate proteins from the core, NS3, and NS5 regions, detect anti-HCV antibodies during acute infection. The most sensitive indicator of HCV infection is the presence of HCV RNA, which requires molecular amplification by PCR or transcription-mediated amplification (TMA) (Fig. 360-7). To allow standardization of the quantification of HCV RNA among laboratories and commercial assays, HCV RNA is reported as international units (IUs) per milliliter; quantitative assays with a broad dynamic range are available that allow detection of HCV RNA with a sensitivity as low as 5 IU/mL. HCV RNA can be detected within a few days of exposure to HCV—well before the appearance of anti-HCV—and tends to persist for the duration of HCV infection. Application of sensitive molecular probes for HCV RNA has revealed the presence of replicative HCV in peripheral blood lymphocytes of infected persons; however, as is the case for HBV in lymphocytes, the clinical relevance of HCV lymphocyte infection is not known.
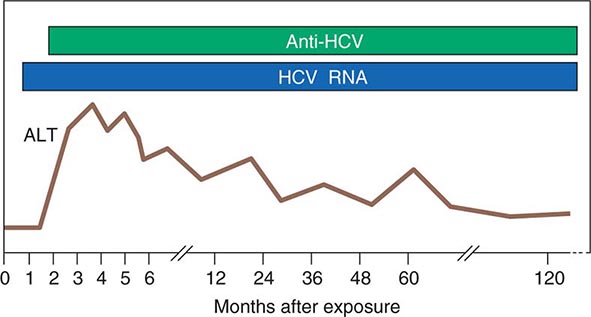
FIGURE 360-7 Scheme of typical laboratory features during acute hepatitis C progressing to chronicity. Hepatitis C virus (HCV) RNA is the first detectable event, preceding alanine aminotransferase (ALT) elevation and the appearance of anti-HCV.
Hepatitis E Previously labeled epidemic or enterically transmitted non-A, non-B hepatitis, HEV is an enterically transmitted virus that causes clinically apparent hepatitis primarily in India, Asia, Africa, and Central America; in those geographic areas, HEV is the most common cause of acute hepatitis; one-third of the global population appears to have been infected. This agent, with epidemiologic features resembling those of hepatitis A, is a 27- to 34-nm, nonenveloped, HAV-like virus with a 7200-nucleotide, single-strand, positive-sense RNA genome. HEV has three open reading frames (ORF) (genes), the largest of which, ORF1, encodes nonstructural proteins involved in virus replication. A middle-sized gene, ORF2, encodes the nucleocapsid protein, the major nonstructural protein, and the smallest, ORF3, encodes a structural protein whose function remains undetermined. All HEV isolates appear to belong to a single serotype, despite genomic heterogeneity of up to 25% and the existence of five genotypes, only four of which have been detected in humans; genotypes 1 and 2 appear to be more virulent, whereas genotypes 3 and 4 are more attenuated and account for subclinical infections. Contributing to the perpetuation of this virus are animal reservoirs, most notably in swine. No genomic or antigenic homology, however, exists between HEV and HAV or other picornaviruses; and HEV, although resembling caliciviruses, is sufficiently distinct from any known agent to merit its own classification as a unique genus, Hepevirus, within the family Hepeviridae. The virus has been detected in stool, bile, and liver and is excreted in the stool during the late incubation period. Both IgM anti-HEV during early acute infection and IgG anti-HEV predominating after the first 3 months can be detected. Currently, availability and reliability of serologic/virologic testing for HEV infection is limited but can be done in specialized laboratories (e.g., the Centers for Disease Control and Prevention).
PATHOGENESIS
Under ordinary circumstances, none of the hepatitis viruses is known to be directly cytopathic to hepatocytes. Evidence suggests that the clinical manifestations and outcomes after acute liver injury associated with viral hepatitis are determined by the immunologic responses of the host. Among the viral hepatitides, the immunopathogenesis of hepatitis B and C has been studied most extensively.
Hepatitis B For HBV, the existence of inactive hepatitis B carriers with normal liver histology and function suggests that the virus is not directly cytopathic. The fact that patients with defects in cellular immune competence are more likely to remain chronically infected rather than to clear HBV supports the role of cellular immune responses in the pathogenesis of hepatitis B–related liver injury. The model that has the most experimental support involves cytolytic T cells sensitized specifically to recognize host and hepatitis B viral antigens on the liver cell surface. Nucleocapsid proteins (HBcAg and possibly HBeAg), present on the cell membrane in minute quantities, are the viral target antigens that, with host antigens, invite cytolytic T cells to destroy HBV-infected hepatocytes. Differences in the robustness and broad polyclonality of CD8+ cytolytic T cell responsiveness; in the level of HBV-specific helper CD4+ T cells; in attenuation, depletion, and exhaustion of virus-specific T cells; in viral T cell epitope escape mutations that allow the virus to evade T cell containment; and in the elaboration of antiviral cytokines by T cells have been invoked to explain differences in outcomes between those who recover after acute hepatitis and those who progress to chronic hepatitis, or between those with mild and those with severe (fulminant) acute HBV infection.
Although a robust cytolytic T cell response occurs and eliminates virus-infected liver cells during acute hepatitis B, >90% of HBV DNA has been found in experimentally infected chimpanzees to disappear from the liver and blood before maximal T cell infiltration of the liver and before most of the biochemical and histologic evidence of liver injury. This observation suggests that components of the innate immune system and inflammatory cytokines, independent of cytopathic antiviral mechanisms, participate in the early immune response to HBV infection; this effect has been shown to represent elimination of HBV replicative intermediates from the cytoplasm and covalently closed circular viral DNA from the nucleus of infected hepatocytes. In turn, the innate immune response to HBV infection is mediated largely by natural killer (NK) cell cytotoxicity, activated by immunosuppressive cytokines (e.g., interleukin [IL] 10 and transforming growth factor [TGF] β), reduced signals from inhibitory receptor expression (e.g., major histocompatibility complex), or increased signals from activating receptor expression on infected hepatocytes. In addition, NK cells reduce helper CD4+ cells, which results in reduced CD8+ cells and exhaustion of the virus-specific T cell response to HBV infection. Ultimately, HBV-HLA-specific cytolytic T cell responses of the adaptive immune system are felt to be responsible for recovery from HBV infection.
Debate continues over the relative importance of viral and host factors in the pathogenesis of HBV-associated liver injury and its outcome. As noted above, precore genetic mutants of HBV have been associated with the more severe outcomes of HBV infection (severe chronic and fulminant hepatitis), suggesting that, under certain circumstances, relative pathogenicity is a property of the virus, not the host. The fact that concomitant HDV and HBV infections are associated with more severe liver injury than HBV infection alone and the fact that cells transfected in vitro with the gene for HDV antigen express HDV antigen and then become necrotic in the absence of any immunologic influences are also consistent with a viral effect on pathogenicity. Similarly, in patients who undergo liver transplantation for end-stage chronic hepatitis B, occasionally, rapidly progressive liver injury appears in the new liver. This clinical pattern is associated with an unusual histologic pattern in the new liver, fibrosing cholestatic hepatitis, which, ultrastructurally, appears to represent a choking of the cell with overwhelming quantities of HBsAg. This observation suggests that, under the influence of the potent immunosuppressive agents required to prevent allograft rejection, HBV may have a direct cytopathic effect on liver cells, independent of the immune system.
Although the precise mechanism of liver injury in HBV infection remains elusive, studies of nucleocapsid proteins have shed light on the profound immunologic tolerance to HBV of babies born to mothers with highly replicative (HBeAg-positive), chronic HBV infection. In HBeAg-expressing transgenic mice, in utero exposure to HBeAg, which is sufficiently small to traverse the placenta, induces T cell tolerance to both nucleocapsid proteins. This, in turn, may explain why, when infection occurs so early in life, immunologic clearance does not occur, and protracted, lifelong infection ensues.
An important distinction should be drawn between HBV infection acquired at birth, common in endemic areas, such as East Asia, and infection acquired in adulthood, common in the West. Infection in the neonatal period is associated with the acquisition of high-level immunologic tolerance to HBV and absence of an acute hepatitis illness, but the almost invariable establishment of chronic, often lifelong infection. Neonatally acquired HBV infection can culminate decades later in cirrhosis and hepatocellular carcinoma (see “Complications and Sequelae”). In contrast, when HBV infection is acquired during adolescence or early adulthood, the host immune response to HBV-infected hepatocytes tends to be robust, an acute hepatitis-like illness is the rule, and failure to recover is the exception. After adulthood-acquired infection, chronicity is uncommon, and the risk of hepatocellular carcinoma is very low. Based on these observations, some authorities categorize HBV infection into an “immunotolerant” phase, an “immunoreactive” phase, and an “inactive” phase. This somewhat simplistic formulation does not apply at all to the typical adult in the West with self-limited acute hepatitis B, in whom no period of immunologic tolerance occurs. Even among those with neonatally acquired HBV infection, in whom immunologic tolerance is established definitively, intermittent bursts of hepatic necroinflammatory activity punctuate the early decades of life during which liver injury appears to be quiescent (labeled by some as the “immunotolerant” phase). In addition, even when clinically apparent liver injury and progressive fibrosis emerge during later decades (the so-called immunoreactive, or immunointolerant, phase), the level of immunologic tolerance to HBV remains substantial. More accurately, in patients with neonatally acquired HBV infection, a dynamic equilibrium exists between tolerance and intolerance, the outcome of which determines the clinical expression of chronic infection. Persons infected as neonates tend to have a relatively higher level of immunologic tolerance during the early decades of life and a relatively lower level (but only rarely a loss) of tolerance in the later decades of life.
Hepatitis C Cell-mediated immune responses and elaboration by T cells of antiviral cytokines contribute to the multicellular innate and adaptive immune responses involved in the containment of infection and pathogenesis of liver injury associated with hepatitis C. The fact that HCV is so efficient in evading these immune mechanisms is a testament to its highly evolved ability to disrupt host immune responses at multiple levels. After exposure to HCV, the host cell identifies viral product motifs (pattern recognition receptors) that distinguish the virus from “self,” resulting in the elaboration of interferons and other cytokines that result in activation of innate and adaptive immune responses. Intrahepatic HLA class I restricted cytolytic T cells directed at nucleocapsid, envelope, and nonstructural viral protein antigens have been demonstrated in patients with chronic hepatitis C; however, such virus-specific cytolytic T cell responses do not correlate adequately with the degree of liver injury or with recovery. Yet, a consensus has emerged supporting a role in the pathogenesis of HCV-associated liver injury of virus-activated CD4+ helper T cells that stimulate, via the cytokines they elaborate, HCV-specific CD8+ cytotoxic T cells. These responses appear to be more robust (higher in number, more diverse in viral antigen specificity, more functionally effective, and more long lasting) in those who recover from HCV than in those who have chronic infection. Contributing to chronic infection are a CD4+ proliferative defect that results in rapid contraction of CD4+ responses, mutations in CD8+ T cell–targeted viral epitopes that allow HCV to escape immune-mediated clearance, and upregulation of inhibitory receptors on functionally impaired, exhausted T cells. Although attention has focused on adaptive immunity, HCV proteins have been shown to interfere with innate immunity by resulting in blocking of type 1 interferon responses and inhibition of interferon signaling and effector molecules in the interferon signaling cascade. Several HLA alleles have been linked with self-limited hepatitis C, the most convincing of which is the CC haplotype of the IL28B gene, which codes for interferon λ3, a component of innate immune antiviral defense. The IL28B association is even stronger when combined with HLA class II DQB1*03:01. The link between non-CC IL28B polymorphisms and failure to clear HCV infection has been explained by a chromosome 19q13.13 frameshift variant upstream of IL28B, the ΔG polymorphism of which creates an ORF in a novel interferon gene (IFN-λ4) associated with impaired HCV clearance. Also shown to contribute to limiting HCV infection are NK cells of the innate immune system that function when HLA class I molecules required for successful adaptive immunity are underexpressed. Both peripheral and intrahepatic NK cell cytotoxicity are dysfunctional in persistent HCV infection. Adding to the complexity of the immune response, HCV core, NS4B, and NS5B have been shown to suppress the immunoregulatory nuclear factor (NF)-κB pathway, resulting in reduced antiapoptotic proteins and a resultant increased vulnerability to tumor necrosis factor (TNF) α–mediated cell death. Patients with hepatitis C and unfavorable (non-CC, associated with reduced HCV clearance) IL28B alleles have been shown to have depressed NK cell/innate immune function. Of note, the emergence of substantial viral quasispecies diversity and HCV sequence variation allow the virus to evade attempts by the host to contain HCV infection by both humoral and cellular immunity.
Finally, cross-reactivity between viral antigens (HCV NS3 and NS5A) and host autoantigens (cytochrome P450 2D6) has been invoked to explain the association between hepatitis C and a subset of patients with autoimmune hepatitis and antibodies to liver-kidney microsomal (LKM) antigen (anti-LKM) (Chap. 362).
EXTRAHEPATIC MANIFESTATIONS
Immune complex–mediated tissue damage appears to play a pathogenetic role in the extrahepatic manifestations of acute hepatitis B. The occasional prodromal serum sickness–like syndrome observed in acute hepatitis B appears to be related to the deposition in tissue blood vessel walls of HBsAg-anti-HBs circulating immune complexes, leading to activation of the complement system and depressed serum complement levels.
In patients with chronic hepatitis B, other types of immune-complex disease may be seen. Glomerulonephritis with the nephrotic syndrome is observed occasionally; HBsAg, immunoglobulin, and C3 deposition has been found in the glomerular basement membrane. Whereas generalized vasculitis (polyarteritis nodosa) develops in considerably fewer than 1% of patients with chronic HBV infection, 20–30% of patients with polyarteritis nodosa have HBsAg in serum (Chap. 385). In these patients, the affected small- and medium-size arterioles contain HBsAg, immunoglobulins, and complement components. Another extrahepatic manifestation of viral hepatitis, essential mixed cryoglobulinemia (EMC), was reported initially to be associated with hepatitis B. The disorder is characterized clinically by arthritis, cutaneous vasculitis (palpable purpura), and occasionally, glomerulonephritis and serologically by the presence of circulating cryoprecipitable immune complexes of more than one immunoglobulin class (Chaps. 338 and 385). Many patients with this syndrome have chronic liver disease, but the association with HBV infection is limited; instead, a substantial proportion has chronic HCV infection, with circulating immune complexes containing HCV RNA. Immune-complex glomerulonephritis is another recognized extrahepatic manifestation of chronic hepatitis C.
PATHOLOGY
The typical morphologic lesions of all types of viral hepatitis are similar and consist of panlobular infiltration with mononuclear cells, hepatic cell necrosis, hyperplasia of Kupffer cells, and variable degrees of cholestasis. Hepatic cell regeneration is present, as evidenced by numerous mitotic figures, multinucleated cells, and “rosette” or “pseudoacinar” formation. The mononuclear infiltration consists primarily of small lymphocytes, although plasma cells and eosinophils occasionally are present. Liver cell damage consists of hepatic cell degeneration and necrosis, cell dropout, ballooning of cells, and acidophilic degeneration of hepatocytes (forming so-called Councilman or apoptotic bodies). Large hepatocytes with a ground-glass appearance of the cytoplasm may be seen in chronic but not in acute HBV infection; these cells contain HBsAg and can be identified histochemically with orcein or aldehyde fuchsin. In uncomplicated viral hepatitis, the reticulin framework is preserved.
In hepatitis C, the histologic lesion is often remarkable for a relative paucity of inflammation, a marked increase in activation of sinusoidal lining cells, lymphoid aggregates, the presence of fat (more frequent in genotype 3 and linked to increased fibrosis), and, occasionally, bile duct lesions in which biliary epithelial cells appear to be piled up without interruption of the basement membrane. Occasionally, microvesicular steatosis occurs in hepatitis D. In hepatitis E, a common histologic feature is marked cholestasis. A cholestatic variant of slowly resolving acute hepatitis A also has been described.
A more severe histologic lesion, bridging hepatic necrosis, also termed subacute or confluent necrosis or interface hepatitis, is observed occasionally in acute hepatitis. “Bridging” between lobules results from large areas of hepatic cell dropout, with collapse of the reticulin framework. Characteristically, the bridge consists of condensed reticulum, inflammatory debris, and degenerating liver cells that span adjacent portal areas, portal to central veins, or central vein to central vein. This lesion had been thought to have prognostic significance; in many of the originally described patients with this lesion, a subacute course terminated in death within several weeks to months, or severe chronic hepatitis and cirrhosis developed; however, the association between bridging necrosis and a poor prognosis in patients with acute hepatitis has not been upheld. Therefore, although demonstration of this lesion in patients with chronic hepatitis has prognostic significance (Chap. 362), its demonstration during acute hepatitis is less meaningful, and liver biopsies to identify this lesion are no longer undertaken routinely in patients with acute hepatitis. In massive hepatic necrosis (fulminant hepatitis, “acute yellow atrophy”), the striking feature at postmortem examination is the finding of a small, shrunken, soft liver. Histologic examination reveals massive necrosis and dropout of liver cells of most lobules with extensive collapse and condensation of the reticulin framework. When histologic documentation is required in the management of fulminant or very severe hepatitis, a biopsy can be done by the angiographically guided transjugular route, which permits the performance of this invasive procedure in the presence of severe coagulopathy.
Immunohistochemical and electron-microscopic studies have localized HBsAg to the cytoplasm and plasma membrane of infected liver cells. In contrast, HBcAg predominates in the nucleus, but, occasionally, scant amounts are also seen in the cytoplasm and on the cell membrane. HDV antigen is localized to the hepatocyte nucleus, whereas HAV, HCV, and HEV antigens are localized to the cytoplasm.
EPIDEMIOLOGY AND GLOBAL FEATURES
![]() Before the availability of serologic tests for hepatitis viruses, all viral hepatitis cases were labeled either as “infectious” or “serum” hepatitis. Modes of transmission overlap, however, and a clear distinction among the different types of viral hepatitis cannot be made solely on the basis of clinical or epidemiologic features (Table 360-2). The most accurate means to distinguish the various types of viral hepatitis involves specific serologic testing.
Before the availability of serologic tests for hepatitis viruses, all viral hepatitis cases were labeled either as “infectious” or “serum” hepatitis. Modes of transmission overlap, however, and a clear distinction among the different types of viral hepatitis cannot be made solely on the basis of clinical or epidemiologic features (Table 360-2). The most accurate means to distinguish the various types of viral hepatitis involves specific serologic testing.
|
CLINICAL AND EPIDEMIOLOGIC FEATURES OF VIRAL HEPATITIS |
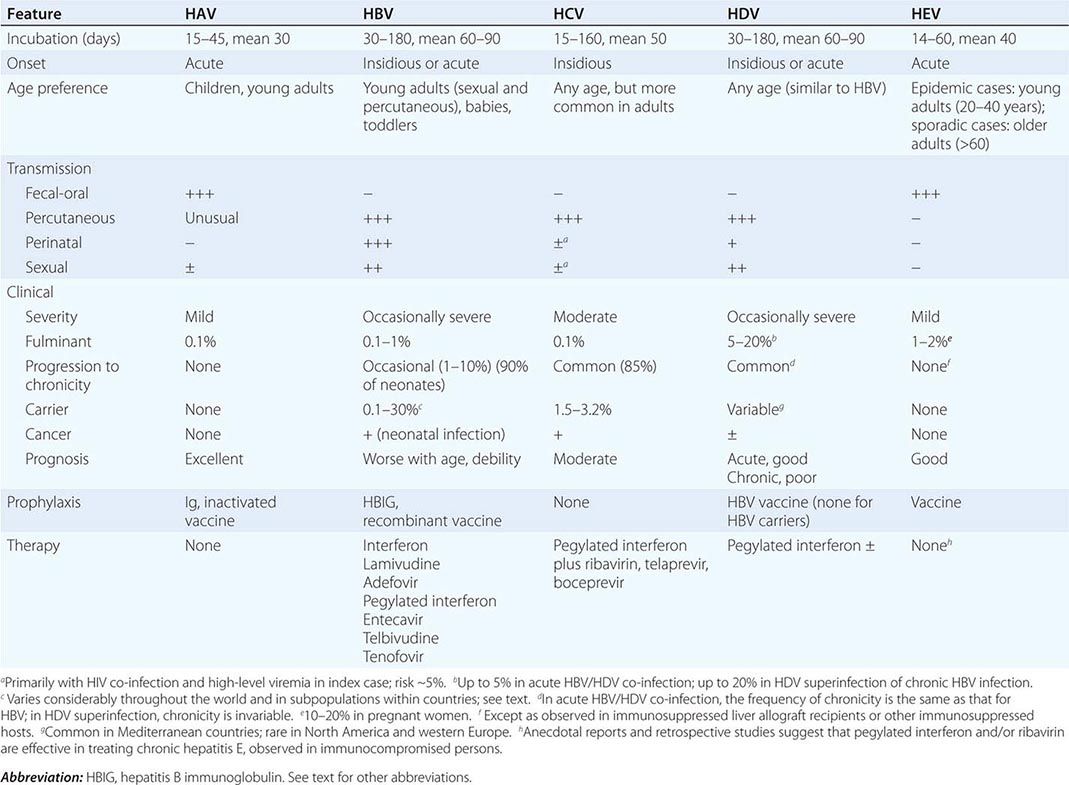
Hepatitis A This agent is transmitted almost exclusively by the fecal-oral route. Person-to-person spread of HAV is enhanced by poor personal hygiene and overcrowding; large outbreaks as well as sporadic cases have been traced to contaminated food, water, milk, frozen raspberries and strawberries, green onions imported from Mexico, and shellfish. Intrafamily and intrainstitutional spread are also common. Early epidemiologic observations supported a predilection for hepatitis A to occur in late fall and early winter. In temperate zones, epidemic waves have been recorded every 5–20 years as new segments of nonimmune population appeared; however, in developed countries, the incidence of hepatitis A has been declining, presumably as a function of improved sanitation, and these cyclic patterns are no longer observed. No HAV carrier state has been identified after acute hepatitis A; perpetuation of the virus in nature depends presumably on nonepidemic, inapparent subclinical infection, ingestion of contaminated food or water in, or imported from, endemic areas, and/or contamination linked to environmental reservoirs.
In the general population, anti-HAV, a marker for previous HAV infection, increases in prevalence as a function of increasing age and of decreasing socioeconomic status. In the 1970s, serologic evidence of prior hepatitis A infection occurred in ~40% of urban populations in the United States, most of whose members never recalled having had a symptomatic case of hepatitis. In subsequent decades, however, the prevalence of anti-HAV has been declining in the United States. In developing countries, exposure, infection, and subsequent immunity are almost universal in childhood. As the frequency of subclinical childhood infections declines in developed countries, a susceptible cohort of adults emerges. Hepatitis A tends to be more symptomatic in adults; therefore, paradoxically, as the frequency of HAV infection declines, the likelihood of clinically apparent, even severe, HAV illnesses increases in the susceptible adult population. Travel to endemic areas is a common source of infection for adults from nonendemic areas. More recently recognized epidemiologic foci of HAV infection include child care centers, neonatal intensive care units, promiscuous men who have sex with men, injection drug users, and unvaccinated close contacts of newly arrived international adopted children, most of whom emanate from countries with intermediate-to-high hepatitis A endemicity. Although hepatitis A is rarely bloodborne, several outbreaks have been recognized in recipients of clotting-factor concentrates. In the United States, the introduction of hepatitis A vaccination programs among children from high-incidence states has resulted in a >70% reduction in the annual incidence of new HAV infections and has shifted the burden of new infections from children to young adults. In the most recent, 1999–2006 U.S. Public Health Service National Health and Nutrition Examination Survey (NHANES), the prevalence of anti-HAV in the U.S. population was 35%, representing (compared to the 1988–1994 survey) a stable frequency of infection and natural immunity in adults >19 years old but an increase in vaccine-induced immunity for children age 6–19 years.
Hepatitis B Percutaneous inoculation has long been recognized as a major route of hepatitis B transmission, but the outmoded designation “serum hepatitis” is an inaccurate label for the epidemiologic spectrum of HBV infection. As detailed below, most of the hepatitis transmitted by blood transfusion is not caused by HBV; moreover, in approximately two-thirds of patients with acute type B hepatitis, no history of an identifiable percutaneous exposure can be elicited. We now recognize that many cases of hepatitis B result from less obvious modes of nonpercutaneous or covert percutaneous transmission. HBsAg has been identified in almost every body fluid from infected persons, and at least some of these body fluids—most notably semen and saliva—are infectious, albeit less so than serum, when administered percutaneously or nonpercutaneously to experimental animals. Among the nonpercutaneous modes of HBV transmission, oral ingestion has been documented as a potential but inefficient route of exposure. By contrast, the two nonpercutaneous routes considered to have the greatest impact are intimate (especially sexual) contact and perinatal transmission.
In sub-Saharan Africa, intimate contact among toddlers is considered instrumental in contributing to the maintenance of the high frequency of hepatitis B in the population. Perinatal transmission occurs primarily in infants born to mothers with chronic hepatitis B or (rarely) mothers with acute hepatitis B during the third trimester of pregnancy or during the early postpartum period. Perinatal transmission is uncommon in North America and western Europe but occurs with great frequency and is the most important mode of HBV perpetuation in East Asia and developing countries. Although the precise mode of perinatal transmission is unknown, and although ~10% of infections may be acquired in utero, epidemiologic evidence suggests that most infections occur approximately at the time of delivery and are not related to breast-feeding. The likelihood of perinatal transmission of HBV correlates with the presence of HBeAg and high-level viral replication; 90% of HBeAg-positive mothers but only 10–15% of anti-HBe-positive mothers transmit HBV infection to their offspring. In most cases, acute infection in the neonate is clinically asymptomatic, but the child is very likely to remain chronically infected.
The >350–400 million HBsAg carriers in the world constitute the main reservoir of hepatitis B in human beings. Whereas serum HBsAg is infrequent (0.1–0.5%) in normal populations in the United States and western Europe, a prevalence of up to 5–20% has been found in East Asia and in some tropical countries; in persons with Down’s syndrome, lepromatous leprosy, leukemia, Hodgkin’s disease, or polyarteritis nodosa; in patients with chronic renal disease on hemodialysis; and in injection drug users.
Other groups with high rates of HBV infection include spouses of acutely infected persons; sexually promiscuous persons (especially promiscuous men who have sex with men); health care workers exposed to blood; persons who require repeated transfusions especially with pooled blood-product concentrates (e.g., hemophiliacs); residents and staff of custodial institutions for the developmentally handicapped; prisoners; and, to a lesser extent, family members of chronically infected patients. In volunteer blood donors, the prevalence of anti-HBs, a reflection of previous HBV infection, ranges from 5–10%, but the prevalence is higher in lower socioeconomic strata, older age groups, and persons—including those mentioned above—exposed to blood products. Because of highly sensitive virologic screening of donor blood, the risk of acquiring HBV infection from a blood transfusion is 1 in 230,000.
Prevalence of infection, modes of transmission, and human behavior conspire to mold geographically different epidemiologic patterns of HBV infection. In East Asia and Africa, hepatitis B, a disease of the newborn and young children, is perpetuated by a cycle of maternal-neonatal spread. In North America and western Europe, hepatitis B is primarily a disease of adolescence and early adulthood, the time of life when intimate sexual contact and recreational and occupational percutaneous exposures tend to occur. To some degree, however, this dichotomy between high-prevalence and low-prevalence geographic regions has been minimized by immigration from high-prevalence to low-prevalence areas. The introduction of hepatitis B vaccine in the early 1980s and adoption of universal childhood vaccination policies in many countries resulted in a dramatic, ~90% decline in the incidence of new HBV infections in those countries as well as in the dire consequences of chronic infection, including hepatocellular carcinoma. Populations and groups for whom HBV infection screening is recommended are listed in Table 360-3.
|
HIGH-RISK POPULATIONS FOR WHOM HBV INFECTION SCREENING IS RECOMMENDED |
Hepatitis D Infection with HDV has a worldwide distribution, but two epidemiologic patterns exist. In Mediterranean countries (northern Africa, southern Europe, the Middle East), HDV infection is endemic among those with hepatitis B, and the disease is transmitted predominantly by nonpercutaneous means, especially close personal contact. In nonendemic areas, such as the United States and northern Europe, HDV infection is confined to persons exposed frequently to blood and blood products, primarily injection drug users and hemophiliacs. HDV infection can be introduced into a population through drug users or by migration of persons from endemic to nonendemic areas. Thus, patterns of population migration and human behavior facilitating percutaneous contact play important roles in the introduction and amplification of HDV infection. Occasionally, the migrating epidemiology of hepatitis D is expressed in explosive outbreaks of severe hepatitis, such as those that have occurred in remote South American villages as well as in urban centers in the United States. Ultimately, such outbreaks of hepatitis D—either of co-infections with acute hepatitis B or of superinfections in those already infected with HBV—may blur the distinctions between endemic and nonendemic areas. On a global scale, HDV infection declined at the end of the 1990s. Even in Italy, an HDV-endemic area, public health measures introduced to control HBV infection resulted during the 1990s in a 1.5%/year reduction in the prevalence of HDV infection. Still, the frequency of HDV infection during the first decade of the twenty-first century has not fallen below levels reached during the 1990s; the reservoir has been sustained by survivors infected during 1970–1980 and recent immigrants from still-endemic to less-endemic countries.
Hepatitis C Routine screening of blood donors for HBsAg and the elimination of commercial blood sources in the early 1970s reduced the frequency of, but did not eliminate, transfusion-associated hepatitis. During the 1970s, the likelihood of acquiring hepatitis after transfusion of voluntarily donated, HBsAg-screened blood was ~10% per patient (up to 0.9% per unit transfused); 90–95% of these cases were classified, based on serologic exclusion of hepatitis A and B, as “non-A, non-B” hepatitis. For patients requiring transfusion of pooled products, such as clotting factor concentrates, the risk was even higher, up to 20–30%.
During the 1980s, voluntary self-exclusion of blood donors with risk factors for AIDS and then the introduction of donor screening for anti-HIV reduced further the likelihood of transfusion-associated hepatitis to <5%. During the late 1980s and early 1990s, the introduction first of “surrogate” screening tests for non-A, non-B hepatitis (alanine aminotransferase [ALT] and anti-HBc, both shown to identify blood donors with a higher likelihood of transmitting non-A, non-B hepatitis to recipients) and, subsequently, after the discovery of HCV, first-generation immunoassays for anti-HCV reduced the frequency of transfusion-associated hepatitis even further. A prospective analysis of transfusion-associated hepatitis conducted between 1986 and 1990 showed that the frequency of transfusion-associated hepatitis at one urban university hospital fell from a baseline of 3.8% per patient (0.45% per unit transfused) to 1.5% per patient (0.19% per unit) after the introduction of surrogate testing and to 0.6% per patient (0.03% per unit) after the introduction of first-generation anti-HCV assays. The introduction of second-generation anti-HCV assays reduced the frequency of transfusion-associated hepatitis C to almost imperceptible levels—1 in 100,000—and these gains were reinforced by the application of third-generation anti-HCV assays and of automated PCR testing of donated blood for HCV RNA, which has resulted in a reduction in the risk of transfusion-associated HCV infection to 1 in 2.3 million transfusions.
In addition to being transmitted by transfusion, hepatitis C can be transmitted by other percutaneous routes, such as injection drug use. In addition, this virus can be transmitted by occupational exposure to blood, and the likelihood of infection is increased in hemodialysis units. Although the frequency of transfusion-associated hepatitis C fell as a result of blood-donor screening, the overall frequency of hepatitis C remained the same until the early 1990s, when the overall frequency fell by 80%, in parallel with a reduction in the number of new cases in injection drug users. After the exclusion of anti-HCV-positive plasma units from the donor pool, rare, sporadic instances have occurred of hepatitis C among recipients of immunoglobulin (Ig) preparations for intravenous (but not intramuscular) use.
Serologic evidence for HCV infection occurs in 90% of patients with a history of transfusion-associated hepatitis (almost all occurring before 1992, when second-generation HCV screening tests were introduced); hemophiliacs and others treated with clotting factors; injection drug users; 60–70% of patients with sporadic “non-A, non-B” hepatitis who lack identifiable risk factors; 0.5% of volunteer blood donors; and, in a survey conducted in the United States between 1999 and 2002, 1.6% of the general population in the United States, which translates into 4.1 million persons (3.2 million with viremia), the majority of whom are unaware of their infections. Moreover, such population surveys do not include higher-risk groups such as incarcerated prisoners and active injection drug users, indicating that the actual prevalence is even higher. Comparable frequencies of HCV infection occur in most countries around the world, with 170 million persons infected worldwide, but extraordinarily high prevalences of HCV infection occur in certain countries such as Egypt, where >20% of the population (as high as 50% in persons born prior to 1960) in some cities is infected. The high frequency in Egypt is attributable to contaminated equipment used for medical procedures and unsafe injection practices in the 1950s to 1980s (during a campaign to eradicate schistosomiasis with intravenous tartar emetic). In the United States, African Americans and Mexican Americans have higher frequencies of HCV infection than whites. Between 1988 and 1994, 30- to 40-year-old adult males had the highest prevalence of HCV infection; however, in a survey conducted between 1999 and 2002, the peak age decile had shifted to those age 40–49 years; an increase in hepatitis C–related mortality has paralleled this secular trend, increasing since 1995 predominantly in the 45- to 65-year age group. Thus, despite an 80% reduction in new HCV infections during the 1990s, the prevalence of HCV infection in the population was sustained by an aging cohort that had acquired their infections three to four decades earlier, during the 1960s and 1970s, as a result predominantly of self-inoculation with recreational drugs. As death resulting from HIV infection fell after 1999, age-adjusted mortality associated with HCV infection surpassed that of HIV infection in 2007; >70% of HCV-associated deaths occurred in the “baby boomer” cohort born between 1945 and 1965. Compared to the 1.6% prevalence of HCV infection in the population at large, the prevalence in the 1945–1965 birth cohort was 3.2%, representing three-quarters of all infected persons. Therefore, in 2012, the Centers for Disease Control and Prevention recommended that all persons born between 1945 and 1965 be screened for hepatitis C, without ascertainment of risk, a recommendation shown to be cost-effective and predicted to identify 800,000 infected persons. Because of the availability of highly effective antiviral therapy, such screening would have the potential to avert 200,000 cases of cirrhosis and 47,000 cases of hepatocellular carcinoma and to prevent 120,000 hepatitis-related deaths.
Hepatitis C accounts for 40% of chronic liver disease, is the most frequent indication for liver transplantation, and is estimated to account for 8000–10,000 deaths per year in the United States. The distribution of HCV genotypes varies in different parts of the world. Worldwide, genotype 1 is the most common. In the United States, genotype 1 accounts for 70% of HCV infections, whereas genotypes 2 and 3 account for the remaining 30%; among African Americans, the frequency of genotype 1 is even higher (i.e., 90%). Genotype 4 predominates in Egypt; genotype 5 is localized to South Africa, genotype 6 to Hong Kong, and genotype 7 to Central Africa. Most asymptomatic blood donors found to have anti-HCV and ~20–30% of persons with reported cases of acute hepatitis C do not fall into a recognized risk group; however, many such blood donors do recall risk-associated behaviors when questioned carefully.
As a bloodborne infection, HCV potentially can be transmitted sexually and perinatally; however, both of these modes of transmission are inefficient for hepatitis C. Although 10–15% of patients with acute hepatitis C report having potential sexual sources of infection, most studies have failed to identify sexual transmission of this agent. The chances of sexual and perinatal transmission have been estimated to be ~5% but shown in a prospective study to be only 1% between monogamous sexual partners, well below comparable rates for HIV and HBV infections. Moreover, sexual transmission appears to be confined to such subgroups as persons with multiple sexual partners and sexually transmitted diseases. Breast-feeding does not increase the risk of HCV infection between an infected mother and her infant. Infection of health workers is not dramatically higher than among the general population; however, health workers are more likely to acquire HCV infection through accidental needle punctures, the efficiency of which is ~3%. Infection of household contacts is rare as well.
Besides persons born between 1945 and 1965, other groups with an increased frequency of HCV infection are listed in Table 360-4. In immunosuppressed individuals, levels of anti-HCV may be undetectable, and a diagnosis may require testing for HCV RNA. Although new acute cases of hepatitis C are rare, newly diagnosed cases are common among otherwise healthy persons who experimented briefly with injection drugs, as noted above, three or four decades earlier. Such instances usually remain unrecognized for years, until unearthed by laboratory screening for routine medical examinations, insurance applications, and attempted blood donation. Although, overall, the annual incidence of new HCV infections has continued to fall, the rate of new infections has been increasing since 2002 in a new cohort of young injection drug users, age 15–24 years (accounting for more than two-thirds of all acute cases), who, unlike older cohorts, had not learned to take precautions to prevent bloodborne infections.
|
HIGH-RISK POPULATIONS FOR WHOM HCV-INFECTION SCREENING IS RECOMMENDED |
Hepatitis E This type of hepatitis, identified in India, Asia, Africa, the Middle East, and Central America, resembles hepatitis A in its primarily enteric mode of spread. The commonly recognized cases occur after contamination of water supplies such as after monsoon flooding, but sporadic, isolated cases occur. An epidemiologic feature that distinguishes HEV from other enteric agents is the rarity of secondary person-to-person spread from infected persons to their close contacts. Large waterborne outbreaks in endemic areas are linked to genotypes 1 and 2, arise in populations that are immune to HAV, favor young adults, and account for antibody prevalences of 30–80%. In nonendemic areas of the world, such as the United States, clinically apparent acute hepatitis E is extremely rare; however, during the 1988–1994 NHANES survey conducted by the U.S. Public Health Service, the prevalence of anti-HEV was 21%, reflecting subclinical infections, infection with genotypes 3 and 4, predominantly in older males (>60 years). In nonendemic areas, HEV accounts hardly at all for cases of sporadic hepatitis; however, cases imported from endemic areas have been found in the United States. Evidence supports a zoonotic reservoir for HEV primarily in swine, which may account for the mostly subclinical infections in nonendemic areas.
CLINICAL AND LABORATORY FEATURES
Symptoms and Signs Acute viral hepatitis occurs after an incubation period that varies according to the responsible agent. Generally, incubation periods for hepatitis A range from 15–45 days (mean, 4 weeks), for hepatitis B and D from 30–180 days (mean, 8–12 weeks), for hepatitis C from 15–160 days (mean, 7 weeks), and for hepatitis E from 14–60 days (mean, 5–6 weeks). The prodromal symptoms of acute viral hepatitis are systemic and quite variable. Constitutional symptoms of anorexia, nausea and vomiting, fatigue, malaise, arthralgias, myalgias, headache, photophobia, pharyngitis, cough, and coryza may precede the onset of jaundice by 1–2 weeks. The nausea, vomiting, and anorexia are frequently associated with alterations in olfaction and taste. A low-grade fever between 38° and 39°C (100°–102°F) is more often present in hepatitis A and E than in hepatitis B or C, except when hepatitis B is heralded by a serum sickness–like syndrome; rarely, a fever of 39.5°–40°C (103°–104°F) may accompany the constitutional symptoms. Dark urine and clay-colored stools may be noticed by the patient from 1–5 days before the onset of clinical jaundice.
With the onset of clinical jaundice, the constitutional prodromal symptoms usually diminish, but in some patients, mild weight loss (2.5–5 kg) is common and may continue during the entire icteric phase. The liver becomes enlarged and tender and may be associated with right upper quadrant pain and discomfort. Infrequently, patients present with a cholestatic picture, suggesting extrahepatic biliary obstruction. Splenomegaly and cervical adenopathy are present in 10–20% of patients with acute hepatitis. Rarely, a few spider angiomas appear during the icteric phase and disappear during convalescence. During the recovery phase, constitutional symptoms disappear, but usually some liver enlargement and abnormalities in liver biochemical tests are still evident. The duration of the posticteric phase is variable, ranging from 2–12 weeks, and is usually more prolonged in acute hepatitis B and C. Complete clinical and biochemical recovery is to be expected 1–2 months after all cases of hepatitis A and E and 3–4 months after the onset of jaundice in three-quarters of uncomplicated, self-limited cases of hepatitis B and C (among healthy adults, acute hepatitis B is self-limited in 95–99%, whereas hepatitis C is self-limited in only ~15%). In the remainder, biochemical recovery may be delayed. A substantial proportion of patients with viral hepatitis never become icteric.
Infection with HDV can occur in the presence of acute or chronic HBV infection; the duration of HBV infection determines the duration of HDV infection. When acute HDV and HBV infection occur simultaneously, clinical and biochemical features may be indistinguishable from those of HBV infection alone, although occasionally they are more severe. As opposed to patients with acute HBV infection, patients with chronic HBV infection can support HDV replication indefinitely, as when acute HDV infection occurs in the presence of a nonresolving acute HBV infection or, more commonly, when acute hepatitis D is superimposed on underlying chronic hepatitis B. In such cases, the HDV superinfection appears as a clinical exacerbation or an episode resembling acute viral hepatitis in someone already chronically infected with HBV. Superinfection with HDV in a patient with chronic hepatitis B often leads to clinical deterioration (see below).
In addition to superinfections with other hepatitis agents, acute hepatitis-like clinical events in persons with chronic hepatitis B may accompany spontaneous HBeAg to anti-HBe seroconversion or spontaneous reactivation (i.e., reversion from relatively nonreplicative to replicative infection). Such reactivations can occur as well in therapeutically immunosuppressed patients with chronic HBV infection when cytotoxic/immunosuppressive drugs are withdrawn; in these cases, restoration of immune competence is thought to allow resumption of previously checked cell-mediated immune cytolysis of HBV-infected hepatocytes. Occasionally, acute clinical exacerbations of chronic hepatitis B may represent the emergence of a precore mutant (see “Virology and Etiology”), and the subsequent course in such patients may be characterized by periodic exacerbations. Cytotoxic chemotherapy can lead to reactivation of chronic hepatitis C as well, and anti-TNF-α therapy can lead to reactivation of both hepatitis B and C.
Laboratory Features The serum aminotransferases aspartate aminotransferase (AST) and alanine aminotransferase (ALT) (previously designated SGOT and SGPT) increase to a variable degree during the prodromal phase of acute viral hepatitis and precede the rise in bilirubin level (Figs. 360-2 and 360-4). The level of these enzymes, however, does not correlate well with the degree of liver cell damage. Peak levels vary from 400–4000 IU or more; these levels are usually reached at the time the patient is clinically icteric and diminish progressively during the recovery phase of acute hepatitis. The diagnosis of anicteric hepatitis is based on clinical features and on aminotransferase elevations.
Jaundice is usually visible in the sclera or skin when the serum bilirubin value is >43 μmol/L (2.5 mg/dL). When jaundice appears, the serum bilirubin typically rises to levels ranging from 85–340 μmol/L (5–20 mg/dL). The serum bilirubin may continue to rise despite falling serum aminotransferase levels. In most instances, the total bilirubin is equally divided between the conjugated and unconjugated fractions. Bilirubin levels >340 μmol/L (20 mg/dL) extending and persisting late into the course of viral hepatitis are more likely to be associated with severe disease. In certain patients with underlying hemolytic anemia, however, such as glucose-6-phosphate dehydrogenase deficiency and sickle cell anemia, a high serum bilirubin level is common, resulting from superimposed hemolysis. In such patients, bilirubin levels >513 μmol/L (30 mg/dL) have been observed and are not necessarily associated with a poor prognosis.
Neutropenia and lymphopenia are transient and are followed by a relative lymphocytosis. Atypical lymphocytes (varying between 2 and 20%) are common during the acute phase. Measurement of the prothrombin time (PT) is important in patients with acute viral hepatitis, because a prolonged value may reflect a severe hepatic synthetic defect, signify extensive hepatocellular necrosis, and indicate a worse prognosis. Occasionally, a prolonged PT may occur with only mild increases in the serum bilirubin and aminotransferase levels. Prolonged nausea and vomiting, inadequate carbohydrate intake, and poor hepatic glycogen reserves may contribute to hypoglycemia noted occasionally in patients with severe viral hepatitis. Serum alkaline phosphatase may be normal or only mildly elevated, whereas a fall in serum albumin is uncommon in uncomplicated acute viral hepatitis. In some patients, mild and transient steatorrhea has been noted, as well as slight microscopic hematuria and minimal proteinuria.
A diffuse but mild elevation of the γ globulin fraction is common during acute viral hepatitis. Serum IgG and IgM levels are elevated in about one-third of patients during the acute phase of viral hepatitis, but the serum IgM level is elevated more characteristically during acute hepatitis A. During the acute phase of viral hepatitis, antibodies to smooth muscle and other cell constituents may be present, and low titers of rheumatoid factor, nuclear antibody, and heterophile antibody can also be found occasionally. In hepatitis C and D, antibodies to LKM may occur; however, the species of LKM antibodies in the two types of hepatitis are different from each other as well as from the LKM antibody species characteristic of autoimmune hepatitis type 2 (Chap. 362). The autoantibodies in viral hepatitis are nonspecific and can also be associated with other viral and systemic diseases. In contrast, virus-specific antibodies, which appear during and after hepatitis virus infection, are serologic markers of diagnostic importance.
As described above, serologic tests are available routinely with which to establish a diagnosis of hepatitis A, B, D, and C. Tests for fecal or serum HAV are not routinely available. Therefore, a diagnosis of hepatitis A is based on detection of IgM anti-HAV during acute illness (Fig. 360-2). Rheumatoid factor can give rise to false-positive results in this test.
A diagnosis of HBV infection can usually be made by detection of HBsAg in serum. Infrequently, levels of HBsAg are too low to be detected during acute HBV infection, even with contemporary, highly sensitive immunoassays. In such cases, the diagnosis can be established by the presence of IgM anti-HBc.
The titer of HBsAg bears little relation to the severity of clinical disease. Indeed, an inverse correlation exists between the serum concentration of HBsAg and the degree of liver cell damage. For example, titers are highest in immunosuppressed patients, lower in patients with chronic liver disease (but higher in mild chronic than in severe chronic hepatitis), and very low in patients with acute fulminant hepatitis. These observations suggest that, in hepatitis B, the degree of liver cell damage and the clinical course are related to variations in the patient’s immune response to HBV rather than to the amount of circulating HBsAg. In immunocompetent persons, however, a correlation exists between markers of HBV replication and liver injury (see below).
Another important serologic marker in patients with hepatitis B is HBeAg. Its principal clinical usefulness is as an indicator of relative infectivity. Because HBeAg is invariably present during early acute hepatitis B, HBeAg testing is indicated primarily in chronic infection.
In patients with hepatitis B surface antigenemia of unknown duration (e.g., blood donors found to be HBsAg-positive) testing for IgM anti-HBc may be useful to distinguish between acute or recent infection (IgM anti-HBc-positive) and chronic HBV infection (IgM anti-HBc-negative, IgG anti-HBc-positive). A false-positive test for IgM anti-HBc may be encountered in patients with high-titer rheumatoid factor. Also, IgM anti-HBc may be reexpressed during acute reactivation of chronic hepatitis B.
Anti-HBs is rarely detectable in the presence of HBsAg in patients with acute hepatitis B, but 10–20% of persons with chronic HBV infection may harbor low-level anti-HBs. This antibody is directed not against the common group determinant, a, but against the heterotypic subtype determinant (e.g., HBsAg of subtype ad with anti-HBs of subtype y). In most cases, this serologic pattern cannot be attributed to infection with two different HBV subtypes, and the presence of this antibody is not a harbinger of imminent HBsAg clearance. When such antibody is detected, its presence is of no recognized clinical significance (see “Virology and Etiology”).
After immunization with hepatitis B vaccine, which consists of HBsAg alone, anti-HBs is the only serologic marker to appear. The commonly encountered serologic patterns of hepatitis B and their interpretations are summarized in Table 360-5. Tests for the detection of HBV DNA in liver and serum are now available. Like HBeAg, serum HBV DNA is an indicator of HBV replication, but tests for HBV DNA are more sensitive and quantitative. First-generation hybridization assays for HBV DNA had a sensitivity of 105–106 virions/mL, a relative threshold below which infectivity and liver injury are limited and HBeAg is usually undetectable. Currently, testing for HBV DNA has shifted from insensitive hybridization assays to amplification assays (e.g., the PCR-based assay, which can detect as few as 10 or 100 virions/mL); among the commercially available PCR assays, the most useful are those with the highest sensitivity (5–10 IU/mL) and the largest dynamic range (100–109 IU/mL). With increased sensitivity, amplification assays remain reactive well below the current 103–104 IU/mL threshold for infectivity and liver injury. These markers are useful in following the course of HBV replication in patients with chronic hepatitis B receiving antiviral chemotherapy (Chap. 362). Except for the early decades of life after perinatally acquired HBV infection (see above), in immunocompetent adults with chronic hepatitis B, a general correlation exists between the level of HBV replication, as reflected by the level of serum HBV DNA, and the degree of liver injury. High-serum HBV DNA levels, increased expression of viral antigens, and necroinflammatory activity in the liver go hand in hand unless immunosuppression interferes with cytolytic T cell responses to virus-infected cells; reduction of HBV replication with antiviral drugs tends to be accompanied by an improvement in liver histology. Among patients with chronic hepatitis B, high levels of HBV DNA increase the risk of cirrhosis, hepatic decompensation, and hepatocellular carcinoma (see “Complications and Sequelae”).
|
COMMONLY ENCOUNTERED SEROLOGIC PATTERNS OF HEPATITIS B INFECTION |
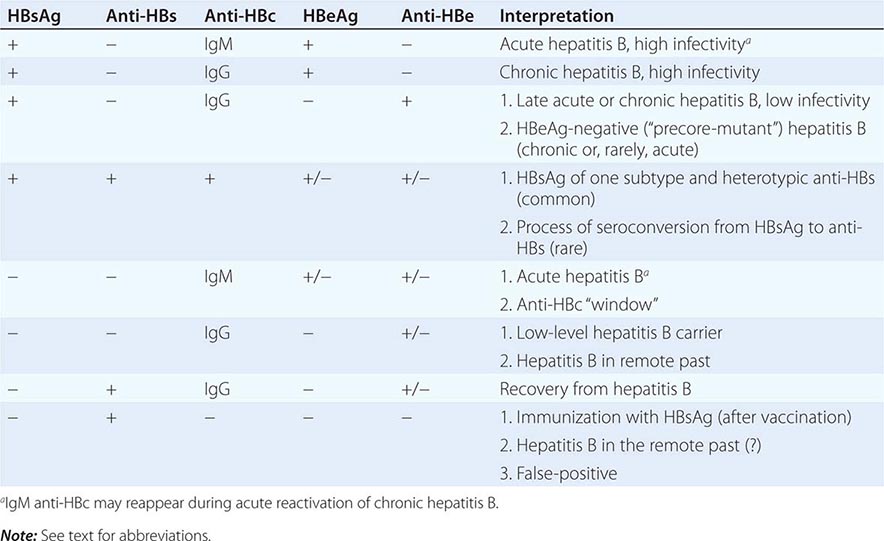
In patients with hepatitis C, an episodic pattern of aminotransferase elevation is common. A specific serologic diagnosis of hepatitis C can be made by demonstrating the presence in serum of anti-HCV. When contemporary immunoassays are used, anti-HCV can be detected in acute hepatitis C during the initial phase of elevated aminotransferase activity and remains detectable after recovery (rare) and during chronic infection (common). Nonspecificity can confound immunoassays for anti-HCV, especially in persons with a low prior probability of infection, such as volunteer blood donors, or in persons with circulating rheumatoid factor, which can bind nonspecifically to assay reagents; testing for HCV RNA can be used in such settings to distinguish between true-positive and false-positive anti-HCV determinations. Assays for HCV RNA are the most sensitive tests for HCV infection and represent the “gold standard” in establishing a diagnosis of hepatitis C. HCV RNA can be detected even before acute elevation of aminotransferase activity and before the appearance of anti-HCV in patients with acute hepatitis C. In addition, HCV RNA remains detectable indefinitely, continuously in most but intermittently in some, in patients with chronic hepatitis C (detectable as well in some persons with normal liver tests, i.e., inactive carriers). In the very small minority of patients with hepatitis C who lack anti-HCV, a diagnosis can be supported by detection of HCV RNA. If all these tests are negative and the patient has a well-characterized case of hepatitis after percutaneous exposure to blood or blood products, a diagnosis of hepatitis caused by an unidentified agent can be entertained.
Amplification techniques are required to detect HCV RNA, and two types are available. One is a branched-chain complementary DNA (bDNA) assay, in which the detection signal (a colorimetrically detectable enzyme bound to a complementary DNA probe) is amplified. The other involves target amplification (i.e., synthesis of multiple copies of the viral genome) by PCR or TMA, in which the viral RNA is reverse transcribed to complementary DNA and then amplified by repeated cycles of DNA synthesis. Both can be used as quantitative assays and a measurement of relative “viral load”; PCR and TMA, with a sensitivity of 10–102 IU/mL, are more sensitive than bDNA, with a sensitivity of 103 IU/mL; assays are available with a wide dynamic range (10–107 IU/mL). Determination of HCV RNA level is not a reliable marker of disease severity or prognosis but is helpful in predicting relative responsiveness to antiviral therapy. The same is true for determinations of HCV genotype (Chap. 362).
A proportion of patients with hepatitis C have isolated anti-HBc in their blood, a reflection of a common risk in certain populations of exposure to multiple bloodborne hepatitis agents. The anti-HBc in such cases is almost invariably of the IgG class and usually represents HBV infection in the remote past (HBV DNA undetectable); it rarely represents current HBV infection with low-level virus carriage.
The presence of HDV infection can be identified by demonstrating intrahepatic HDV antigen or, more practically, an anti-HDV seroconversion (a rise in titer of anti-HDV or de novo appearance of anti-HDV). Circulating HDV antigen, also diagnostic of acute infection, is detectable only briefly, if at all. Because anti-HDV is often undetectable once HBsAg disappears, retrospective serodiagnosis of acute self-limited, simultaneous HBV and HDV infection is difficult. Early diagnosis of acute infection may be hampered by a delay of up to 30–40 days in the appearance of anti-HDV.
When a patient presents with acute hepatitis and has HBsAg and anti-HDV in serum, determination of the class of anti-HBc is helpful in establishing the relationship between infection with HBV and HDV. Although IgM anti-HBc does not distinguish absolutely between acute and chronic HBV infection, its presence is a reliable indicator of recent infection and its absence a reliable indicator of infection in the remote past. In simultaneous acute HBV and HDV infections, IgM anti-HBc will be detectable, whereas in acute HDV infection superimposed on chronic HBV infection, anti-HBc will be of the IgG class. Tests for the presence of HDV RNA are useful for determining the presence of ongoing HDV replication and relative infectivity.
The serologic/virologic course of events during acute hepatitis E is entirely analogous to that of acute hepatitis A, with brief fecal shedding of virus and viremia and an early IgM anti-HEV response that predominates during approximately the first 3 months but is eclipsed thereafter by long-lasting IgG anti-HEV. Diagnostic tests of varying reliability for hepatitis E are commercially available but used routinely primarily outside the United States; in the United States, diagnostic serologic/virologic assays can be performed at the Centers for Disease Control and Prevention or other specialized reference laboratories.
Liver biopsy is rarely necessary or indicated in acute viral hepatitis, except when the diagnosis is questionable or when clinical evidence suggests a diagnosis of chronic hepatitis.
A diagnostic algorithm can be applied in the evaluation of cases of acute viral hepatitis. A patient with acute hepatitis should undergo four serologic tests, HBsAg, IgM anti-HAV, IgM anti-HBc, and anti-HCV (Table 360-6). The presence of HBsAg, with or without IgM anti-HBc, represents HBV infection. If IgM anti-HBc is present, the HBV infection is considered acute; if IgM anti-HBc is absent, the HBV infection is considered chronic. A diagnosis of acute hepatitis B can be made in the absence of HBsAg when IgM anti-HBc is detectable. A diagnosis of acute hepatitis A is based on the presence of IgM anti-HAV. If IgM anti-HAV coexists with HBsAg, a diagnosis of simultaneous HAV and HBV infections can be made; if IgM anti-HBc (with or without HBsAg) is detectable, the patient has simultaneous acute hepatitis A and B, and if IgM anti-HBc is undetectable, the patient has acute hepatitis A superimposed on chronic HBV infection. The presence of anti-HCV supports a diagnosis of acute hepatitis C. Occasionally, testing for HCV RNA or repeat anti-HCV testing later during the illness is necessary to establish the diagnosis. Absence of all serologic markers is consistent with a diagnosis of “non-A, non-B, non-C” hepatitis, if the epidemiologic setting is appropriate.
|
SIMPLIFIED DIAGNOSTIC APPROACH IN PATIENTS PRESENTING WITH ACUTE HEPATITIS |
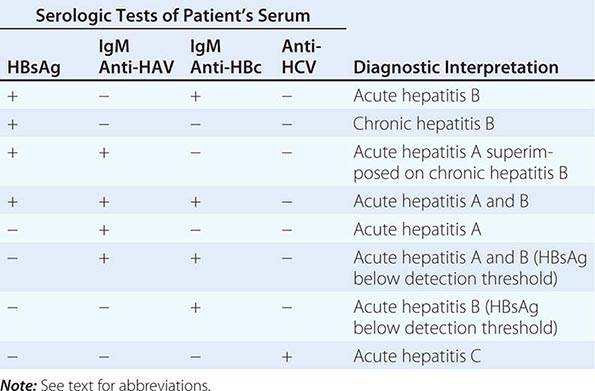
In patients with chronic hepatitis, initial testing should consist of HBsAg and anti-HCV. Anti-HCV supports and HCV RNA testing establishes the diagnosis of chronic hepatitis C. If a serologic diagnosis of chronic hepatitis B is made, testing for HBeAg and anti-HBe is indicated to evaluate relative infectivity. Testing for HBV DNA in such patients provides a more quantitative and sensitive measure of the level of virus replication and, therefore, is very helpful during antiviral therapy (Chap. 362). In patients with chronic hepatitis B and normal aminotransferase activity in the absence of HBeAg, serial testing over time is often required to distinguish between inactive carriage and HBeAg-negative chronic hepatitis B with fluctuating virologic and necroinflammatory activity. In persons with hepatitis B, testing for anti-HDV is useful in those with severe and fulminant disease, with severe chronic disease, with chronic hepatitis B and acute hepatitis-like exacerbations, with frequent percutaneous exposures, and from areas where HDV infection is endemic.
PROGNOSIS
Virtually all previously healthy patients with hepatitis A recover completely with no clinical sequelae. Similarly, in acute hepatitis B, 95–99% of previously healthy adults have a favorable course and recover completely. Certain clinical and laboratory features, however, suggest a more complicated and protracted course. Patients of advanced age and with serious underlying medical disorders may have a prolonged course and are more likely to experience severe hepatitis. Initial presenting features such as ascites, peripheral edema, and symptoms of hepatic encephalopathy suggest a poorer prognosis. In addition, a prolonged PT, low serum albumin level, hypoglycemia, and very high serum bilirubin values suggest severe hepatocellular disease. Patients with these clinical and laboratory features deserve prompt hospital admission. The case fatality rate in hepatitis A and B is very low (~0.1%) but is increased by advanced age and underlying debilitating disorders. Among patients ill enough to be hospitalized for acute hepatitis B, the fatality rate is 1%. Hepatitis C is less severe during the acute phase than hepatitis B and is more likely to be anicteric; fatalities are rare, but the precise case fatality rate is not known. In outbreaks of waterborne hepatitis E in India and Asia, the case fatality rate is 1–2% and up to 10–20% in pregnant women. Contributing to fulminant hepatitis E in endemic countries are instances of acute hepatitis E superimposed on underlying chronic liver disease (“acute-on-chronic” liver disease). Patients with simultaneous acute hepatitis B and hepatitis D do not necessarily experience a higher mortality rate than do patients with acute hepatitis B alone; however, in several outbreaks of acute simultaneous HBV and HDV infection among injection drug users, the case fatality rate was ~5%. When HDV superinfection occurs in a person with chronic hepatitis B, the likelihood of fulminant hepatitis and death is increased substantially. Although the case fatality rate for hepatitis D is not known definitively, in outbreaks of severe HDV superinfection in isolated populations with a high hepatitis B carrier rate, a mortality rate >20% has been recorded.
COMPLICATIONS AND SEQUELAE
A small proportion of patients with hepatitis A experience relapsing hepatitis weeks to months after apparent recovery from acute hepatitis. Relapses are characterized by recurrence of symptoms, aminotransferase elevations, occasionally jaundice, and fecal excretion of HAV. Another unusual variant of acute hepatitis A is cholestatic hepatitis, characterized by protracted cholestatic jaundice and pruritus. Rarely, liver test abnormalities persist for many months, even up to a year. Even when these complications occur, hepatitis A remains self-limited and does not progress to chronic liver disease. During the prodromal phase of acute hepatitis B, a serum sickness–like syndrome characterized by arthralgia or arthritis, rash, angioedema, and rarely, hematuria and proteinuria may develop in 5–10% of patients. This syndrome occurs before the onset of clinical jaundice, and these patients are often diagnosed erroneously as having rheumatologic diseases. The diagnosis can be established by measuring serum aminotransferase levels, which are almost invariably elevated, and serum HBsAg. As noted above, EMC is an immune-complex disease that can complicate chronic hepatitis C and is part of a spectrum of B cell lymphoproliferative disorders, which, in rare instances, can evolve to B cell lymphoma (Chap. 134). Attention has been drawn as well to associations between hepatitis C and such cutaneous disorders as porphyria cutanea tarda and lichen planus. A mechanism for these associations is unknown. Finally, related to the reliance of HCV on lipoprotein secretion and assembly pathways and on interactions of HCV with glucose metabolism, HCV infection may be complicated by hepatic steatosis, hypercholesterolemia, insulin resistance (and other manifestations of the metabolic syndrome), and type 2 diabetes mellitus; both hepatic steatosis and insulin resistance appear to accelerate hepatic fibrosis and blunt responsiveness to antiviral therapy (Chap. 362).
The most feared complication of viral hepatitis is fulminant hepatitis (massive hepatic necrosis); fortunately, this is a rare event. Fulminant hepatitis is seen primarily in hepatitis B, D, and E, but rare fulminant cases of hepatitis A occur primarily in older adults and in persons with underlying chronic liver disease, including, according to some reports, chronic hepatitis B and C. Hepatitis B accounts for >50% of fulminant cases of viral hepatitis, a sizable proportion of which are associated with HDV infection and another proportion with underlying chronic hepatitis C. Fulminant hepatitis is hardly ever seen in hepatitis C, but hepatitis E, as noted above, can be complicated by fatal fulminant hepatitis in 1–2% of all cases and in up to 20% of cases in pregnant women. Patients usually present with signs and symptoms of encephalopathy that may evolve to deep coma. The liver is usually small and the PT excessively prolonged. The combination of rapidly shrinking liver size, rapidly rising bilirubin level, and marked prolongation of the PT, even as aminotransferase levels fall, together with clinical signs of confusion, disorientation, somnolence, ascites, and edema, indicates that the patient has hepatic failure with encephalopathy. Cerebral edema is common; brainstem compression, gastrointestinal bleeding, sepsis, respiratory failure, cardiovascular collapse, and renal failure are terminal events. The mortality rate is exceedingly high (>80% in patients with deep coma), but patients who survive may have a complete biochemical and histologic recovery. If a donor liver can be located in time, liver transplantation may be life-saving in patients with fulminant hepatitis (Chap. 368).
Documenting the disappearance of HBsAg after apparent clinical recovery from acute hepatitis B is particularly important. Before laboratory methods were available to distinguish between acute hepatitis and acute hepatitis-like exacerbations (spontaneous reactivations) of chronic hepatitis B, observations suggested that ~10% of previously healthy patients remained HBsAg-positive for >6 months after the onset of clinically apparent acute hepatitis B. One-half of these persons cleared the antigen from their circulations during the next several years, but the other 5% remained chronically HBsAg-positive. More recent observations suggest that the true rate of chronic infection after clinically apparent acute hepatitis B is as low as 1% in normal, immunocompetent, young adults. Earlier, higher estimates may have been confounded by inadvertent inclusion of acute exacerbations in chronically infected patients; these patients, chronically HBsAg-positive before exacerbation, were unlikely to seroconvert to HBsAg-negative thereafter. Whether the rate of chronicity is 10% or 1%, such patients have IgG anti-HBc in serum; anti-HBs is either undetected or detected at low titer against the opposite subtype specificity of the antigen (see “Laboratory Features”). These patients may (1) be inactive carriers; (2) have low-grade, mild chronic hepatitis; or (3) have moderate to severe chronic hepatitis with or without cirrhosis. The likelihood of remaining chronically infected after acute HBV infection is especially high among neonates, persons with Down’s syndrome, chronically hemodialyzed patients, and immunosuppressed patients, including persons with HIV infection.
Chronic hepatitis is an important late complication of acute hepatitis B occurring in a small proportion of patients with acute disease but more common in those who present with chronic infection without having experienced an acute illness, as occurs typically after neonatal infection or after infection in an immunosuppressed host (Chap. 362). The following clinical and laboratory features suggest progression of acute hepatitis to chronic hepatitis: (1) lack of complete resolution of clinical symptoms of anorexia, weight loss, fatigue, and the persistence of hepatomegaly; (2) the presence of bridging/interface or multilobular hepatic necrosis on liver biopsy during protracted, severe acute viral hepatitis; (3) failure of the serum aminotransferase, bilirubin, and globulin levels to return to normal within 6–12 months after the acute illness; and (4) the persistence of HBeAg for >3 months or HBsAg for >6 months after acute hepatitis.
Although acute hepatitis D infection does not increase the likelihood of chronicity of simultaneous acute hepatitis B, hepatitis D has the potential for contributing to the severity of chronic hepatitis B. Hepatitis D superinfection can transform inactive or mild chronic hepatitis B into severe, progressive chronic hepatitis and cirrhosis; it also can accelerate the course of chronic hepatitis B. Some HDV superinfections in patients with chronic hepatitis B lead to fulminant hepatitis. As defined in longitudinal studies over three decades, the annual rates of cirrhosis and hepatocellular carcinoma in patients with chronic hepatitis D are 4% and 2.8%, respectively. Although HDV and HBV infections are associated with severe liver disease, mild hepatitis and even inactive carriage have been identified in some patients, and the disease may become indolent beyond the early years of infection.
After acute HCV infection, the likelihood of remaining chronically infected approaches 85–90%. Although many patients with chronic hepatitis C have no symptoms, cirrhosis may develop in as many as 20% within 10–20 years of acute illness; in some series of cases reported by referral centers, cirrhosis has been reported in as many as 50% of patients with chronic hepatitis C. Although chronic hepatitis C accounts for at least 40% of cases of chronic liver disease and of patients undergoing liver transplantation for end-stage liver disease in the United States and Europe, in the majority of patients with chronic hepatitis C, morbidity and mortality are limited during the initial 20 years after the onset of infection. Progression of chronic hepatitis C may be influenced by advanced age of acquisition, long duration of infection, immunosuppression, coexisting excessive alcohol use, concomitant hepatic steatosis, other hepatitis virus infection, or HIV coinfection. In fact, instances of severe and rapidly progressive chronic hepatitis B and C are being recognized with increasing frequency in patients with HIV infection (Chap. 226). In contrast, neither HAV nor HEV causes chronic liver disease in immunocompetent hosts; however, cases of chronic hepatitis E have been observed in immunosuppressed organ-transplant recipients, persons receiving cytotoxic chemotherapy, and persons with HIV infection.
Rare complications of viral hepatitis include pancreatitis, myocarditis, atypical pneumonia, aplastic anemia, transverse myelitis, and peripheral neuropathy. Persons with chronic hepatitis B, particularly those infected in infancy or early childhood and especially those with HBeAg and/or high-level HBV DNA, have an enhanced risk of hepatocellular carcinoma. The risk of hepatocellular carcinoma is increased as well in patients with chronic hepatitis C, almost exclusively in patients with cirrhosis, and almost always after at least several decades, usually after three decades of disease (Chap. 111). In children, hepatitis B may present rarely with anicteric hepatitis, a nonpruritic papular rash of the face, buttocks, and limbs, and lymphadenopathy (papular acrodermatitis of childhood or Gianotti-Crosti syndrome).
Rarely, autoimmune hepatitis (Chap. 362) can be triggered by a bout of otherwise self-limited acute hepatitis, as reported after acute hepatitis A, B, and C.
DIFFERENTIAL DIAGNOSIS
Viral diseases such as infectious mononucleosis; those due to cytomegalovirus, herpes simplex, and coxsackieviruses; and toxoplasmosis may share certain clinical features with viral hepatitis and cause elevations in serum aminotransferase and, less commonly, in serum bilirubin levels. Tests such as the differential heterophile and serologic tests for these agents may be helpful in the differential diagnosis if HBsAg, anti-HBc, IgM anti-HAV, and anti-HCV determinations are negative. Aminotransferase elevations can accompany almost any systemic viral infection; other rare causes of liver injury confused with viral hepatitis are infections with Leptospira, Candida, Brucella, Mycobacteria, and Pneumocystis. A complete drug history is particularly important because many drugs and certain anesthetic agents can produce a picture of either acute hepatitis or cholestasis (Chap. 361). Equally important is a past history of unexplained “repeated episodes” of acute hepatitis. This history should alert the physician to the possibility that the underlying disorder is chronic hepatitis. Alcoholic hepatitis must also be considered, but usually the serum aminotransferase levels are not as markedly elevated, and other stigmata of alcoholism may be present. The finding on liver biopsy of fatty infiltration, a neutrophilic inflammatory reaction, and “alcoholic hyaline” would be consistent with alcohol-induced rather than viral liver injury. Because acute hepatitis may present with right upper quadrant abdominal pain, nausea and vomiting, fever, and icterus, it is often confused with acute cholecystitis, common duct stone, or ascending cholangitis. Patients with acute viral hepatitis may tolerate surgery poorly; therefore, it is important to exclude this diagnosis, and in confusing cases, a percutaneous liver biopsy may be necessary before laparotomy. Viral hepatitis in the elderly is often misdiagnosed as obstructive jaundice resulting from a common duct stone or carcinoma of the pancreas. Because acute hepatitis in the elderly may be quite severe and the operative mortality high, a thorough evaluation including biochemical tests, radiographic studies of the biliary tree, and even liver biopsy may be necessary to exclude primary parenchymal liver disease. Another clinical constellation that may mimic acute hepatitis is right ventricular failure with passive hepatic congestion or hypoperfusion syndromes, such as those associated with shock, severe hypotension, and severe left ventricular failure. Also included in this general category is any disorder that interferes with venous return to the heart, such as right atrial myxoma, constrictive pericarditis, hepatic vein occlusion (Budd-Chiari syndrome), or venoocclusive disease. Clinical features are usually sufficient to distinguish among these vascular disorders and viral hepatitis. Acute fatty liver of pregnancy, cholestasis of pregnancy, eclampsia, and the HELLP (h emolysis, e levated l iver tests, and l ow p latelets) syndrome can be confused with viral hepatitis during pregnancy. Very rarely, malignancies metastatic to the liver can mimic acute or even fulminant viral hepatitis. Occasionally, genetic or metabolic liver disorders (e.g., Wilson’s disease, α1 antitrypsin deficiency) and nonalcoholic fatty liver disease are confused with acute viral hepatitis.



