[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 23 Approach to the diagnosis and classification of blood diseases
Initial screening tests
Although the range of haematological tests available to support clinical and public health services is broad, it is often the simplest investigations that are most useful in indicating the diagnosis. Even poorly-resourced laboratories are usually able to provide an initial panel of tests such as haemoglobin concentration (Hb), white blood cell count (WBC) and platelet count (Chapter 26) and examination of a peripheral blood film for a differential leucocyte count (Chapter 3) and cellular morphology (Chapter 5). These screening tests will often enable the underlying pathological processes to be suspected promptly and point to a few key diagnostic tests. The investigation of specific haematological problems is covered in detail in Chapters 9 (iron deficiency anaemia), 10 (megaloblastic anaemia), 11, 12 and 13 (haemolytic anaemias), 14 (haemoglobinopathies) and 18, 19 and 20 (coagulation disorders).
Interpretation of Screening Tests
Results of laboratory screening tests should always be interpreted with an understanding of the limitations of the tests and the physiological variations that occur with sex, age and conditions such as pregnancy and exercise. Physiological variations in cell counts are detailed in Chapter 2. Abnormalities of red cells, white cells or platelets may be quantitative (increased or reduced numbers) or qualitative (abnormal appearance and/or function).
Quantitative Abnormalities of Blood Cells
Erythrocytosis
Secondary polycythaemia can generally be excluded by the clinical history and examination, assessment of serum erythropoietin concentration and arterial oxygen saturation, haemoglobin electrophoresis plus oxygen dissociation curve and abdominal ultrasound examination. The presence of splenomegaly is suggestive of PV and this diagnosis can be confirmed by demonstrating the JAK2 V617F mutation, which is present in 95% of patients.2 Only if this mutation (or one of the much less common JAK2 exon 12 mutations) is not detected is the measurement of total red cell and plasma volume necessary (Chapter 17).
Leucocytosis
Neutrophilia
Neutrophils are commonly increased during pregnancy and in acute infections, inflammation, alcohol intoxication, corticosteroid therapy and acute blood loss or red cell destruction. Neutrophilia with the neutrophils showing heavy cytoplasmic granulation (‘toxic’ granulation) is a common finding in severe bacterial infections. In the absence of any underlying cause, a high neutrophil count with immature myeloid cells suggests chronic myelogenous leukaemia (CML); cytogenetic and molecular studies to look for t(9;22) and the BCR–ABL1 fusion gene are indicated (Chapter 8).
Lymphocytosis
Lymphocytosis is a feature of certain infections, particularly infections in children. It may be especially marked in pertussis, infectious mononucleosis, cytomegalovirus infection, infectious hepatitis, tuberculosis and brucellosis. Lymphocytosis is also a common transient reaction to severe physical stress. Elderly patients with lymphoproliferative disorders, including chronic lymphocytic leukaemia and lymphomas, often present with lymphadenopathy and a lymphocytosis. Morphology and immunophenotyping of the cells combined with histological examination of a bone marrow trephine biopsy specimen (and if necessary other tissue biopsy) are used to classify these disorders and to give an indication of management and prognosis.3 It is occasionally difficult to differentiate between a reactive and a neoplastic lymphocytosis. In this situation, immunophenotyping, to provide evidence of light chain restriction and polymerase chain reaction for immunoglobulin or T-cell receptor gene rearrangements, may indicate the presence of a monoclonal population of lymphocytes, thereby supporting a diagnosis of neoplastic, rather than reactive, lymphoproliferation. If lymph nodes are enlarged, a lymph node biopsy for histology and immunohistochemistry may be helpful in diagnosis.
Monocytosis
A slight to moderate monocytosis may be associated with some protozoal, rickettsial and bacterial infections including malaria, typhus and tuberculosis. High levels of monocytes (monocyte count >1 × 109/l) in an elderly patient suggest chronic myelomonocytic leukaemia or, sometimes, atypical chronic myeloid leukaemia. Because these conditions fall into the myelodysplastic/myeloproliferative neoplasm group of disorders,4 the diagnosis would be supported by finding splenomegaly, quantitative and qualitative abnormalities in other cell lines or a clonal cytogenetic abnormality.
Eosinophilia
Eosinophilia is typically associated with allergic disorders including drug sensitivity, skin diseases and parasitic infections. In most cases, the cause is indicated by the clinical history, which should include details of all medications and foreign travel, and by examination of the stool and urine for parasites, cysts and ova. A diagnosis of chronic eosinophilic leukaemia is made if there is dominant eosinophilia with an increase in blast cells in the blood or marrow, or cytogenetic or molecular evidence of an abnormal myeloid clone.5 If no other cause for eosinophilia is found it is important, because of the therapeutic implications (i.e. responsiveness to imatinib), to confirm or exclude a diagnosis of eosinophilic leukaemia related to rearrangement of PDGFRA or PDGFRB (see p. 559).6 The idiopathic hypereosinophilic syndrome is an unusual cause of eosinophilia in which release of the contents of eosinophil granules results in damage to the heart, lungs and other tissues. It is defined by the presence of a peripheral blood eosinophil count of 1.5 × 109/l or greater for at least 6 months with resultant tissue damage. This is a diagnosis of exclusion, made only when detailed investigations exclude other possible causes of eosinophilia including systemic mastocytosis, eosinophilic leukaemia and eosinophilia associated with a phenotypically aberrant T-cell population or a neoplastic clone of T cells.
Thrombocytosis
Thrombocytosis is often associated with infectious and inflammatory conditions such as osteomyelitis and rheumatoid arthritis. Haematological causes of thrombocytosis include chronic blood loss, red cell destruction, splenectomy and rebound following recovery from marrow suppression. Under these circumstances, a moderately increased platelet count (e.g. 400–800 × 109/l) does not usually have any pathological implications. Primary thrombocythaemia is usually the result of a myeloproliferative neoplasm; rarely, it is an inherited condition. When there is isolated persistent thrombocytosis in a myeloproliferative neoplasm the diagnosis is essential thrombocythaemia (as long as a BCR–ABL1 fusion gene has been excluded). Thrombotic or haemorrhagic complications can occur but often the diagnosis is an incidental one.7 A significant proportion of individuals with essential thrombocythaemia have the JAK2 V617F mutation, which is associated with an increased risk of thrombosis.8 The criteria for this diagnosis are discussed on p. 559.
Anaemia
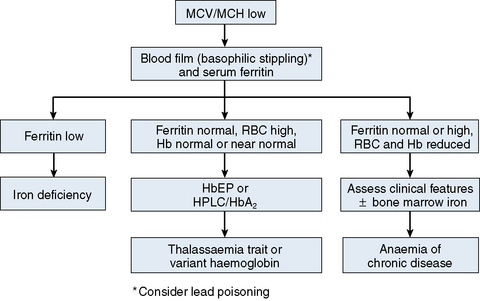
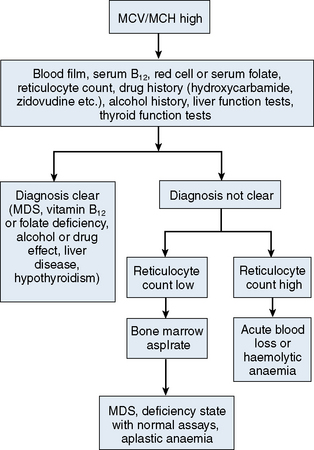
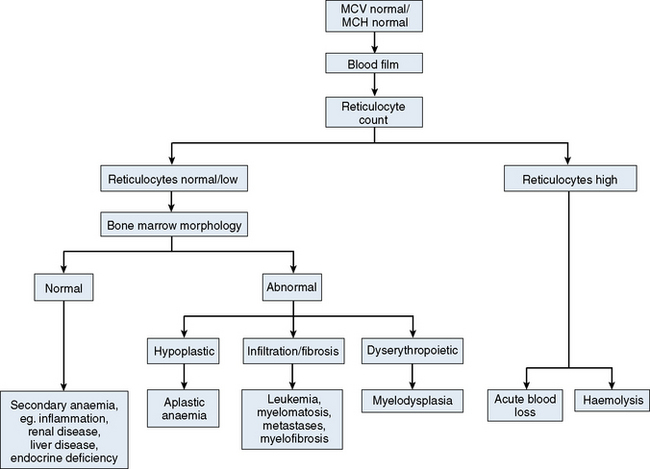
The mechanisms which result in anaemia are decreased production, reduced red cell lifespan, blood loss and splenic pooling. Anaemia is broadly divided into three types: microcytic (low MCV), macrocytic (high MCV) and normocytic (normal MCV). The choice of investigations is guided by the MCV and red cell morphology in addition to clinical features. Figures 23.1–23.3 are flow charts that provide an orderly sequence of investigations for the different types of anaemia on the basis of these indices. Examination of a blood film will usually suggest the quickest route to the diagnosis; confirmation may require the more specific tests, which are given in the text. The presence of basophilic stippling in a patient with microcytic red cells suggests thalassaemia trait or, much less often, lead poisoning. A dimorphic blood film is typical of congenital sideroblastic anaemia but is more often the result of iron deficiency responding to treatment. Pappenheimer bodies suggest that a microcytic anaemia is the result of sideroblastic erythropoiesis.
Microcytic Anaemia
The most common cause of anaemia worldwide is iron deficiency, which can be suspected from a low MCV (Fig. 23.1) and the presence of hypochromic, microcytic red cells. Laboratory confirmation of iron deficiency may be based on measurements of serum ferritin, serum iron plus either total iron-binding capacity or transferrin assay, red cell protoporphyrin and staining of bone marrow aspirates for iron (see Chapter 4). Assay of soluble transferrin receptors has good sensitivity and specificity for iron deficiency and may be useful in the presence of inflammation when interpretation of ferritin levels is difficult.9 A diagnosis of iron deficiency must be followed by a search for the cause. This should include specific questions relating to blood loss and dietary insufficiency and may require stool examination for parasites and occult blood, endoscopic examination of the gastrointestinal tract to exclude occult malignancy and tests for coeliac disease. The differential diagnosis of iron deficiency anaemia includes anaemia of chronic disease. Clinical and laboratory features of inflammation or chronic infection may suggest this diagnosis, which is confirmed by demonstration of normal or high serum ferritin and reduced serum iron, transferrin and iron-binding capacity.
The thalassaemias also cause microcytosis, but both α and β thalassaemia trait are usually associated with an increased red blood cell count (RBC) and a normal or near-normal Hb despite a considerable reduction of the MCV and MCH. In contrast, in iron deficiency the MCV and MCH do not fall until the Hb is significantly reduced. Further investigations, such as high-performance liquid chromatography (HPLC) or haemoglobin electrophoresis supplemented by measurement of Hb A2 and Hb F usually confirm the diagnosis of β thalassaemia trait. The diagnosis of α thalassaemia trait is more difficult; detection of infrequent Hb H inclusions is usually possible in α0 thalassaemia trait, but definitive diagnosis requires DNA analysis.10 A diagnosis of α0 thalassaemia heterozygosity can be clinically important for prediction of haemoglobin Bart’s hydrops fetalis since, if both parents have α0 thalassaemia, this very serious condition can occur in a fetus. DNA analysis is therefore indicated when a pregnant woman of appropriate ethnic origin has an MCH of <25 pg. Hb H inclusions may not be detected in α+ thalassaemia trait but this diagnosis is of less clinical importance and confirmation is thus not usually required.
Macrocytic Anaemia
A high MCV (Fig. 23.2) with oval macrocytes and hypersegmented neutrophils suggests folate or vitamin B12 deficiency and is an indication for assays of these vitamins (see Chapter 10); subsequent investigations could include malabsorption studies, tests for coeliac disease and tests for intrinsic factor and gastric parietal cell antibodies. In patients with these blood film findings and normal vitamin assays, haematinic deficiency is not completely excluded and further investigation is indicated. A Schilling test permits a definitive diagnosis of pernicious anaemia but currently this test is not available due to a lack of reagents. In the absence of intrinsic factor antibodies, the diagnosis of pernicious anaemia may be presumptive. Pernicious anaemia is commonly associated with autoimmune thyroid disease and other autoimmune disorders, such as diabetes mellitus. A high MCV may also be associated with alcohol excess and liver disease or use of drugs such as hydroxycarbamide or zidovudine. Macrocytosis resulting from chronic haemolysis is associated with increased numbers of immature red cells, which appear slightly larger and bluer than normal red cells (polychromatic macrocytes) on a Romanowsky-stained peripheral blood film. Supravital staining of blood films (see p. 33) or an automated reticulocyte count can be used to confirm reticulocytosis. Untreated anaemia associated with polychromasia is likely to indicate blood loss or haemolysis. The combination of red cell fragments, thrombocytopenia and polychromasia indicates a microangiopathic haemolytic anaemia and should trigger further tests such as a platelet count, coagulation studies, assessment of renal function and a search for infection or neoplastic disease. This further assessment is urgent because these may be features of thrombotic thrombocytopenic purpura, which requires immediate treatment by plasma exchange.
Normocytic Anaemia
Normochromic, normocytic anaemia (Fig. 23.3) is frequently the result of an underlying chronic, non-haematological disease. Investigations should include screening for renal insufficiency, subclinical infections, autoimmune diseases and neoplasia. In the presence of anaemia, a lack of polychromasia, confirmed by reticulocytopenia, points toward a primary failure of erythropoiesis or lack of compensatory increased red cell production in blood loss or haemolysis. Examination of the bone marrow may be helpful in demonstrating haematological causes for a normochromic, normocytic anaemia such as aplastic anaemia or MDS.11 Staining for iron may also show that there is a block in iron metabolism suggestive of anaemia associated with chronic inflammatory disease.
Leucopenia
Neutropenia
Once physiological variation, ethnicity and familial or cyclic neutropenia have been excluded (see p. 19), the non-haematological causes of isolated neutropenia to be considered include overwhelming infection, autoimmune disorders such as systemic lupus erythematosus, irradiation, drugs (particularly anticancer agents) and large granular lymphocyte leukaemia. Bone marrow examination may assist in determining whether the problem is the result of peripheral destruction (increased marrow myeloid precursors) or stem cell failure (lack of marrow myeloid precursors). Typical marrow appearances occur in drug-induced neutropenia, in which there is a relative paucity of mature neutrophils and in Kostmann’s syndrome (infant genetic agranulocytosis) where there is maturation arrest at the promyelocytic stage.
Qualitative Abnormalities of Blood Cells
Abnormalities of Individual Cell Lines
Red cells
Congenital abnormalities of the red cell affecting the structure (e.g. spherocytosis, elliptocytosis) and content (e.g. haemoglobinopathies, enzymopathies) often produce typical morphological changes (see Chapter 5). The type of changes will guide further investigations toward analysis of structural proteins, haemoglobin electrophoresis or HPLC, or enzyme assays. Acquired red cell abnormalities may also help to indicate underlying pathology. For example, target cells may prompt investigation of liver function, whereas increased rouleaux formation may indicate the need for investigations for multiple myeloma or inflammatory conditions.
White cells
Congenital abnormalities of neutrophils are unusual, but similar morphological abnormalities (e.g. Pelger–Huët cells) may be seen in acquired conditions such as MDS. Reactive changes in lymphocytes, including basophilic, faceted cytoplasm, are typically seen in infectious mononucleosis, which can be diagnosed using an appropriate serological screening test (see p. 105) or, if this is negative, by demonstration of immunoglobulin M (IgM) antibodies to the Epstein–Barr virus. These atypical lymphocytes can sometimes be difficult to differentiate from circulating lymphoma cells. Bone marrow histology, combined with immunophenotyping studies and determination of lymphocyte clonality by demonstration of light chain restriction or by gene rearrangement studies, may be needed to reach a firm conclusion.
Specific tests for common haematological disorders
Red Cell Disorders
Microcytic Hypochromic Anaemias
For more information, see Chapters 9 and 14.
Macrocytic Anaemias
If macrocytic, megaloblastic erythroid maturation is demonstrated, further investigations should be undertaken as described in Chapter 10. If the blood film is typical of megaloblastic anaemia, relevant assays and further investigations can often indicate the diagnosis without the need for a bone marrow aspirate. Macrocytosis may also be secondary to conditions such as alcohol excess, liver disease, MDS, hydroxycarbamide administration and hypothyroidism. Reticulocytosis from any cause can also increase the MCV.
Aplastic Anaemia11
White Cell Disorders
Acute Leukaemia
Neutropenia
Chronic Myelogenous Leukaemia
Chronic Lymphoproliferative Disorders/Lymphadenopathy
Diagnosis may be made from various specimens including lymph nodes, bone marrow aspirates, trephine biopsy cores and peripheral blood and other fluids such as cerebrospinal fluid, ascitic fluid and pleural aspirates.12
Myelomatosis (Plasma Cell Myeloma)
Classification of haematological neoplasms
Previous classifications for haematological neoplasms have now been largely supplanted by the World Health Organization’s Classification of Tumours of Haematopoietic and Lymphoid Tissues,3 which outlines the international standards for assessment and diagnosis of haematological neoplasms. Application of the WHO criteria requires the results of history and physical examination, morphology (cytology or histology), immunophenotyping, cytogenetic analysis and, in some circumstances, molecular genetic analysis. The French–American–British (FAB) group classifications (see previous editions) continue to have a place (a) when these techniques are not all available and (b) in making a provisional morphological diagnosis, e.g. in acute leukaemia, while awaiting the results of further tests. Whichever classification is used the criteria should be strictly observed so that there is consistency between different centres and countries. To avoid confusion, FAB terminology (e.g. M1, M2) should not be applied if the WHO classification is being used. Details of the FAB classifications can be found in specialized textbooks.14
The WHO classification of haematological neoplasms has several major categories (Table 23.1)
Table 23.1 WHO classification of myeloid neoplasms and acute leukaemias15
| Myeloproliferative neoplasms (MPN) |
| Myeloid and lymphoid neoplasms associated with eosinophilia and abnormalities of PDGFRA, PDGFRB or FGFR1 |
| Myelodysplastic/myeloproliferative neoplasms (MDS/MPN) |
| Myelodysplastic syndrome (MDS) |
| Acute myeloid leukaemia and related neoplasms |
| Acute leukaemias of ambiguous lineage |
| B lymphoblastic leukaemia/lymphoma |
| T lymphoblastic leukaemia/lymphoma |
Classification of Acute Myeloid Leukaemia
The WHO classification categorizes cases as AML (Table 23.2) if the following criteria are met:
Table 23.2 WHO Classification of acute myeloid leukaemia and related neoplasms15
| Therapy-related myeloid neoplasms |
| Acute myeloid leukaemia with recurrent genetic abnormalities |
| AML with t(8;21)(q22;q22); RUNX1-RUNX1T1 |
| AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11 |
| APL with t(15;17)(q22;q12); PML-RARA |
| AML with t(9;11)(p22;q23); MLLT3-MLL |
| AML with t(6;9)(p23;q34); DEK-NUP214 |
| AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1 |
| AML (megakaryoblastic) with t(1;22)(p13;q13); RBM15-MKL1 |
| Provisional entity: AML with mutated NPM1 |
| Provisional entity: AML with mutated CEBPA |
| Acute myeloid leukaemia with myelodysplasia-related changes |
| Acute myeloid leukaemia, not otherwise specified |
| AML with minimal differentiation |
| AML without maturation |
| AML with maturation |
| Acute myelomonocytic leukaemia |
| Acute monoblastic/monocytic leukaemia |
| Acute erythroid leukaemia |
| Pure erythroid leukaemia |
| Erythroleukaemia, erythroid/myeloid |
| Acute megakaryoblastic leukaemia |
| Acute basophilic leukaemia |
| Acute panmyelosis with myelofibrosis |
| Myeloid sarcoma |
| Myeloid proliferations related to Down syndrome |
| Transient abnormal myelopoiesis |
| Myeloid leukaemia associated with Down syndrome |
| Blastic plasmacytoid dendritic cell neoplasm |
It should be noted that the WHO classification is hierarchical. If appropriate, cases are first assigned to the category of therapy-related leukaemia. Next, cases are assigned, if appropriate, to the category of AML with recurrent genetic abnormalities. Cases continue to be assigned to successive categories in the order shown in Table 23.2, with remaining cases finally being categorized as ‘AML not otherwise specified.’ This final group is further subdivided into categories resembling those of the FAB classification (but defined in quite a different manner). Blastic plasmacytoid dendritic cell neoplasm and myeloid neoplasms associated with Down syndrome are recognized as specific entities.
The WHO classification of acute leukaemia15 lists cytogenetic abnormalities that, in combination with ≥20 blasts, indicate a diagnosis of AML with myelodysplasia-related changes; assignment to this category can also be based on a previous history of MDS or on morphological evidence of dysplasia.
Classification of the Myelodysplastic Syndromes
The WHO classification of MDS (Table 23.3) requires evidence for a myeloid neoplasm with ineffective and, generally, dysplastic haemopoiesis; blasts must be <20% in both blood and bone marrow (Table 23.4). It will be noted that cytogenetic analysis is essential for the application of the WHO classification because cases of the 5q− syndrome cannot otherwise be recognized. Like the classification of AML, this is a hierarchical classification. Therapy-related MDS is categorized with therapy-related AML. Remaining cases are then assessed as to whether they meet the criteria for the 5q− syndrome. If they do not, they are assigned to the remaining categories, depending on the number of lineages showing dysplasia, the percentage of ring sideroblasts, the presence or absence of Auer rods and the percentage of blast cells in the blood and marrow. The WHO classification differs from the FAB classification in not regarding chronic myelomonocytic leukaemia as MDS; instead it assigns it to the MDS/MPN group.
Table 23.3 WHO Classification of the myelodysplastic syndromes (MDS) (2008)
| Refractory cytopenia with unilineage dysplasia |
| Refractory anaemia |
| Refractory neutropenia |
| Refractory thrombocytopenia |
| Refractory anaemia with ring sideroblasts |
| Refractory cytopenia with multilineage dysplasia (with or without ring sideroblasts) |
| Refractory anaemia with excess blasts |
| Myelodysplastic syndrome with isolated del(5q) |
| Myelodysplastic syndrome, unclassifiable |
| Childhood myelodysplastic syndrome |
| Provisional entity: refractory cytopenia of childhood |
Table 23.4 WHO diagnostic criteria for the myelodysplastic syndromes (MDS)
| Disease | Blood findings | BM findings |
|---|---|---|
| Refractory cytopenia with unilineage dysplasia (RCUD): (refractory anaemia [RA]; refractory neutropenia [RN]; refractory thrombocytopenia [RT]) | Unicytopenia or bicytopeniaa No or rare blasts (<1%)b |
Unilineage dysplasia: ≥10% of the cells in one myeloid lineage <5% blasts <15% of erythroid precursors are ring sideroblasts |
| Refractory anaemia with ring sideroblasts (RARS) | Anaemia No blasts |
≥15% of erythroid precursors are ring sideroblasts Erythroid dysplasia only <5% blasts |
| Refractory cytopenia with multilineage dysplasia (RCMD) | Cytopenia(s) No or rare blasts (<1%)b No Auer rods <1 × 109/l monocytes |
Dysplasia in ≥10% of the cells in ≥2 myeloid lineages (neutrophil and/or erythroid precursors and/or megakaryocytes) <5% blasts in marrow No Auer rods ±15% ring sideroblasts |
| Refractory anaemia with excess blasts-1 (RAEB-1) | Cytopenia(s) <5% blastsb No Auer rods <1 × 109/l monocytes |
Unilineage or multilineage dysplasia 5–9% blastsb No Auer rods |
| Refractory anaemia with excess blasts-2 (RAEB-2) | Cytopenia(s) 5–19% blastsc Auer rods ±c <1 × 109/l monocytes |
Unilineage or multilineage dysplasia 10–19% blastsc Auer rods ±c |
| Myelodysplastic syndrome – unclassified (MDS-U) | Cytopenias ≤1% blastsb |
Unequivocal dysplasia in <10% of cells in one or more myeloid lineages when accompanied by a cytogenetic abnormality considered as presumptive evidence for a diagnosis of MDS (see Table 23.6) <5% blasts |
| MDS associated with isolated del(5q) | Anaemia Usually normal or increased platelet count No or rare blasts (<1%) |
Normal to increased megakaryocytes with hypolobated nuclei <5% blasts Isolated del(5q) cytogenetic abnormality No Auer rods |
a Bicytopenia may occasionally be observed. Cases with pancytopenia should be classified as MDS-U.
b If the marrow myeloblast percentage is <5% but there are 2–4% myeloblasts in the blood, the diagnostic classification is RAEB-1. Cases of RCUD and RCMD with 1% myeloblasts in the blood should be classified as MDS-U.
c Cases with Auer rods and <5% myeloblasts in the blood and <10% in the marrow should be classified as RAEB-2. Although the finding of 5–19% blasts in the blood is, in itself, diagnostic of RAEB-2, cases of RAEB-2 may have <5% blasts in the blood if they have Auer rods or 10–19% blasts in the marrow or both. Similarly, cases of RAEB-2 may have <10% blasts in the marrow but may be diagnosed by the other two findings, Auer rod + and/or 5–19% blasts in the blood.
From Vardiman et al (2009).15
Classification of Myeloproliferative Neoplasms and Related Conditions
The WHO classification of myeloproliferative neoplasms (previously called ‘disorders’) and related conditions (Table 23.5) is increasingly taking account of cytogenetic or molecular genetic analyses.
Table 23.5 Myeloproliferative neoplasms (MPN) and related conditions5,6,15,16
| Myeloproliferative neoplasms |
| Chronic myelogenous leukaemia |
| Polycythaemia vera |
| Essential thrombocythaemia |
| Primary myelofibrosis |
| Chronic neutrophilic leukaemia |
| Chronic eosinophilic leukaemia, not otherwise categorized |
| Mast cell disease |
| MPN, unclassifiable |
| Lymphoid and myeloid neoplasms associated with rearrangement of PDGFRA, PDGFRB or FGFR1 |
| Lymphoid and myeloid neoplasms associated with rearrangement of PDGFRA |
| Myeloid neoplasms associated with rearrangement of PDGFRB |
| Lymphoid and myeloid neoplasms associated with rearrangement of FGFR1 |
WHO criteria for a diagnosis of essential thrombocythaemia are: platelet count ≥450 × 109/l; megakaryocyte proliferation with large and mature megakaryocytes on examination of the bone marrow with little or no granulocyte or erythroid proliferation; not meeting WHO criteria for CML, PV, primary myelofibrosis, MDS or other myeloid neoplasm; demonstration of JAK2 V617F or other clonal marker or no evidence of reactive thrombocytosis.16
WHO criteria for a diagnosis of systemic mastocytosis are highly complex.3 A trephine biopsy with a mast cell tryptase stain is often crucial in the diagnosis. Molecular analysis for a KIT mutation can also be important.
The categorization of neoplasms with features of both myelodysplasia and myeloproliferation and their diagnostic criteria are listed in Table 23.6.
Table 23.6 Summary of the World Health Organization categories of myelodysplastic/myeloproliferative neoplasms
| Category | Criteria |
|---|---|
| Chronic myelomonocytic leukaemia (CMML) | A Ph-negative, BCR–ABL1-negative disorder with monocyte count >1 × 109/l |
| Fewer than 20% blasts plus promonocytes in peripheral blood (PB) or bone marrow (BM) | |
| Either dysplasia of one or more myeloid lineages or alternative criteria met (acquired clonal cytogenetic abnormality or monocytosis persisting for at least 3 months and alternative causes of monocytosis excluded) | |
| Atypical chronic myeloid leukaemia (aCML) | A Ph-negative, BCR–ABL1-negative disorder with leucocytosis resulting from an increase in neutrophils and their precursors, the precursors (promyelocyte to metamyelocytes) constituting a least 10% of PB white cells |
| Basophils <2% of white cells | |
| Monocytes <10% of white cells | |
| Hypercellular BM with granulocytic hyperplasia and dysplasia, with or without dysplasia of other lineages | |
| Fewer than 20% blasts plus promonocytes in peripheral blood or bone marrow | |
| Juvenile myelomonocytic leukaemia (JMML) | A Ph-negative, BCR–ABL1-negative disorder with monocyte count >1 × 109/l |
| Fewer than 20% blasts plus promonocytes in peripheral blood or bone marrow | |
| Plus two or more of the following | |
| Haemoglobin F increased for age | |
| Immature granulocytes in the PB | |
| WBC >10 × 109/l | |
| Clonal chromosomal abnormality (monosomy 7 not excluded) | |
| GM-CSF hypersensitivity of myeloid precursors in vitro | |
| Myelodysplastic/myeloproliferative neoplasm, unclassifiable | A myelodysplastic/myeloproliferative disorder in which the criteria of one of the myelodysplastic syndromes are met |
| There are prominent proliferative features (e.g. a platelet count of ≥450 × 109/l or a white cell count of ≥13 × 109/l) | |
| The condition has developed de novo | |
| The criteria for other MDS/MPD (CMML, aCML and JMML) are not met | |
| There is no Philadelphia chromosome, BCR–ABL1 fusion gene, 5q−, inv(3)(q21q26) or t(3;3)(q21;q26) |
1 McMullin M., Bareford D., Campbell P., et al. Guidelines for the diagnosis, investigation and management of polycythaemia/erythrocytosis. Br J Haematol. 2005;130:174-195.
2 McMullin M., Reilly J., Campbell P., et al. Amendment to the guideline for diagnosis and investigation of polycythaemia/erythrocytosis. Br J Haematol. 2007;138:812-823.
3 Swerdlow S., Campo E., Harris N., et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed., Lyon, France: IARC Press, 2008. WHO Classification of Tumours, Volume 2. IARC WHO Classification of Tumours, No 2
4 Orazi A., Germing U. The myelodysplastic/myeloproliferative neoplasms: myeloproliferative diseases with dysplastic features. Leukemia. 2008;22(7):1308-1319.
5 Bain B.J., Gilliland D.G., Vardiman J.W., et al. Chronic eosinophilic leukaemia, not otherwise specified. In: Swerdlow S.H., Campo E., Harris N.L., et al, editors. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissue. 4th ed. Lyon, France: IARC Press; 2008:51-53.
6 Bain B.J., Gilliland D.G., Horny H.-P., et al. Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB and FGFR1. In: Swerdlow S.H., Campo E., Harris N.L., et al, editors. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissue. 4th ed. Lyon, France: IARC Press; 2008:68-73.
7 Harrison C., Green A. Essential thrombocythaemia. Baillières Best Pract Clin Haematol. 2006;19(3):439-453.
8 Lussana F., Caberlon S., Pagani C., et al. Association of V617F Jak2 mutation with the risk of thrombosis among patients with essential thrombocythaemia or idiopathic myelofibrosis: a systematic review. Thromb Res. 2009;124(4):409-417.
9 Koulaouzidis A., Said E., Cottier R., et al. Soluble transferrin receptors and iron deficiency, a step beyond ferritin. A systematic review. J Gastrointestin Liver Dis. 2009;18(3):345-352.
10 Ryan K., Bain B., Worthington D., et al. Significant haemoglobinopathies: guidelines for screening and diagnosis. British Committee for Standards in Haematology, 2009. Available at: www.bcshguidelines.com/pdf/significant_final_sept09.pdf Accessed 3 3 2010
11 Marsh J., Ball S., Cavenagh J., et al. Guidelines for the diagnosis and management of aplastic anaemia. British Committee for Standards in Haematology, 2009. Available at: www.bcshguidelines.com/pdf/AplasticAnaemia_010409.pdf Accessed 3 3 2010
12 Parker A., Bain B., Devereux S., et al. Best Practice in Lymphoma Diagnosis and Reporting. British Committee for Standards in Haematology, 2008. Available at: www.bcshguidelines.com/publishedHO.asp?tf=Haemato-Oncology&status= Accessed 3 3 2010
13 Smith A., Wisloff F., Samson D. Guidelines on the diagnosis and management of multiple myeloma, 2005. Available at: www.bcshguidelines.com/pdf/multiplemyeloma0206.pdf Accessed 8 3 2010
14 Bain B.J. Leukaemia Diagnosis, 4th ed. Oxford: Blackwell Publishing; 2010.
15 Vardiman J., Thiele J., Arber D., et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937-951.
16 Tefferi A., Vardiman J. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22:14-22.
* The Ham test and sucrose lysis test are less sensitive and less quantitative than flow cytometry in the detection of PNH clones and when flow cytometry is available they have generally been abandoned as diagnostic tests for PNH.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 23 Approach to the diagnosis and classification of blood diseases
Initial screening tests
Although the range of haematological tests available to support clinical and public health services is broad, it is often the simplest investigations that are most useful in indicating the diagnosis. Even poorly-resourced laboratories are usually able to provide an initial panel of tests such as haemoglobin concentration (Hb), white blood cell count (WBC) and platelet count (Chapter 26) and examination of a peripheral blood film for a differential leucocyte count (Chapter 3) and cellular morphology (Chapter 5). These screening tests will often enable the underlying pathological processes to be suspected promptly and point to a few key diagnostic tests. The investigation of specific haematological problems is covered in detail in Chapters 9 (iron deficiency anaemia), 10 (megaloblastic anaemia), 11, 12 and 13 (haemolytic anaemias), 14 (haemoglobinopathies) and 18, 19 and 20 (coagulation disorders).
Interpretation of Screening Tests
Results of laboratory screening tests should always be interpreted with an understanding of the limitations of the tests and the physiological variations that occur with sex, age and conditions such as pregnancy and exercise. Physiological variations in cell counts are detailed in Chapter 2. Abnormalities of red cells, white cells or platelets may be quantitative (increased or reduced numbers) or qualitative (abnormal appearance and/or function).
Quantitative Abnormalities of Blood Cells
Erythrocytosis
Secondary polycythaemia can generally be excluded by the clinical history and examination, assessment of serum erythropoietin concentration and arterial oxygen saturation, haemoglobin electrophoresis plus oxygen dissociation curve and abdominal ultrasound examination. The presence of splenomegaly is suggestive of PV and this diagnosis can be confirmed by demonstrating the JAK2 V617F mutation, which is present in 95% of patients.2 Only if this mutation (or one of the much less common JAK2 exon 12 mutations) is not detected is the measurement of total red cell and plasma volume necessary (Chapter 17).
Leucocytosis
Neutrophilia
Neutrophils are commonly increased during pregnancy and in acute infections, inflammation, alcohol intoxication, corticosteroid therapy and acute blood loss or red cell destruction. Neutrophilia with the neutrophils showing heavy cytoplasmic granulation (‘toxic’ granulation) is a common finding in severe bacterial infections. In the absence of any underlying cause, a high neutrophil count with immature myeloid cells suggests chronic myelogenous leukaemia (CML); cytogenetic and molecular studies to look for t(9;22) and the BCR–ABL1 fusion gene are indicated (Chapter 8).
Lymphocytosis
Lymphocytosis is a feature of certain infections, particularly infections in children. It may be especially marked in pertussis, infectious mononucleosis, cytomegalovirus infection, infectious hepatitis, tuberculosis and brucellosis. Lymphocytosis is also a common transient reaction to severe physical stress. Elderly patients with lymphoproliferative disorders, including chronic lymphocytic leukaemia and lymphomas, often present with lymphadenopathy and a lymphocytosis. Morphology and immunophenotyping of the cells combined with histological examination of a bone marrow trephine biopsy specimen (and if necessary other tissue biopsy) are used to classify these disorders and to give an indication of management and prognosis.3 It is occasionally difficult to differentiate between a reactive and a neoplastic lymphocytosis. In this situation, immunophenotyping, to provide evidence of light chain restriction and polymerase chain reaction for immunoglobulin or T-cell receptor gene rearrangements, may indicate the presence of a monoclonal population of lymphocytes, thereby supporting a diagnosis of neoplastic, rather than reactive, lymphoproliferation. If lymph nodes are enlarged, a lymph node biopsy for histology and immunohistochemistry may be helpful in diagnosis.
Monocytosis
A slight to moderate monocytosis may be associated with some protozoal, rickettsial and bacterial infections including malaria, typhus and tuberculosis. High levels of monocytes (monocyte count >1 × 109/l) in an elderly patient suggest chronic myelomonocytic leukaemia or, sometimes, atypical chronic myeloid leukaemia. Because these conditions fall into the myelodysplastic/myeloproliferative neoplasm group of disorders,4 the diagnosis would be supported by finding splenomegaly, quantitative and qualitative abnormalities in other cell lines or a clonal cytogenetic abnormality.
Eosinophilia
Eosinophilia is typically associated with allergic disorders including drug sensitivity, skin diseases and parasitic infections. In most cases, the cause is indicated by the clinical history, which should include details of all medications and foreign travel, and by examination of the stool and urine for parasites, cysts and ova. A diagnosis of chronic eosinophilic leukaemia is made if there is dominant eosinophilia with an increase in blast cells in the blood or marrow, or cytogenetic or molecular evidence of an abnormal myeloid clone.5 If no other cause for eosinophilia is found it is important, because of the therapeutic implications (i.e. responsiveness to imatinib), to confirm or exclude a diagnosis of eosinophilic leukaemia related to rearrangement of PDGFRA or PDGFRB (see p. 559).6 The idiopathic hypereosinophilic syndrome is an unusual cause of eosinophilia in which release of the contents of eosinophil granules results in damage to the heart, lungs and other tissues. It is defined by the presence of a peripheral blood eosinophil count of 1.5 × 109/l or greater for at least 6 months with resultant tissue damage. This is a diagnosis of exclusion, made only when detailed investigations exclude other possible causes of eosinophilia including systemic mastocytosis, eosinophilic leukaemia and eosinophilia associated with a phenotypically aberrant T-cell population or a neoplastic clone of T cells.
Thrombocytosis
Thrombocytosis is often associated with infectious and inflammatory conditions such as osteomyelitis and rheumatoid arthritis. Haematological causes of thrombocytosis include chronic blood loss, red cell destruction, splenectomy and rebound following recovery from marrow suppression. Under these circumstances, a moderately increased platelet count (e.g. 400–800 × 109/l) does not usually have any pathological implications. Primary thrombocythaemia is usually the result of a myeloproliferative neoplasm; rarely, it is an inherited condition. When there is isolated persistent thrombocytosis in a myeloproliferative neoplasm the diagnosis is essential thrombocythaemia (as long as a BCR–ABL1 fusion gene has been excluded). Thrombotic or haemorrhagic complications can occur but often the diagnosis is an incidental one.7 A significant proportion of individuals with essential thrombocythaemia have the JAK2 V617F mutation, which is associated with an increased risk of thrombosis.8 The criteria for this diagnosis are discussed on p. 559.
Anaemia
The mechanisms which result in anaemia are decreased production, reduced red cell lifespan, blood loss and splenic pooling. Anaemia is broadly divided into three types: microcytic (low MCV), macrocytic (high MCV) and normocytic (normal MCV). The choice of investigations is guided by the MCV and red cell morphology in addition to clinical features. Figures 23.1–23.3 are flow charts that provide an orderly sequence of investigations for the different types of anaemia on the basis of these indices. Examination of a blood film will usually suggest the quickest route to the diagnosis; confirmation may require the more specific tests, which are given in the text. The presence of basophilic stippling in a patient with microcytic red cells suggests thalassaemia trait or, much less often, lead poisoning. A dimorphic blood film is typical of congenital sideroblastic anaemia but is more often the result of iron deficiency responding to treatment. Pappenheimer bodies suggest that a microcytic anaemia is the result of sideroblastic erythropoiesis.
Microcytic Anaemia
The most common cause of anaemia worldwide is iron deficiency, which can be suspected from a low MCV (Fig. 23.1) and the presence of hypochromic, microcytic red cells. Laboratory confirmation of iron deficiency may be based on measurements of serum ferritin, serum iron plus either total iron-binding capacity or transferrin assay, red cell protoporphyrin and staining of bone marrow aspirates for iron (see Chapter 4). Assay of soluble transferrin receptors has good sensitivity and specificity for iron deficiency and may be useful in the presence of inflammation when interpretation of ferritin levels is difficult.9 A diagnosis of iron deficiency must be followed by a search for the cause. This should include specific questions relating to blood loss and dietary insufficiency and may require stool examination for parasites and occult blood, endoscopic examination of the gastrointestinal tract to exclude occult malignancy and tests for coeliac disease. The differential diagnosis of iron deficiency anaemia includes anaemia of chronic disease. Clinical and laboratory features of inflammation or chronic infection may suggest this diagnosis, which is confirmed by demonstration of normal or high serum ferritin and reduced serum iron, transferrin and iron-binding capacity.
The thalassaemias also cause microcytosis, but both α and β thalassaemia trait are usually associated with an increased red blood cell count (RBC) and a normal or near-normal Hb despite a considerable reduction of the MCV and MCH. In contrast, in iron deficiency the MCV and MCH do not fall until the Hb is significantly reduced. Further investigations, such as high-performance liquid chromatography (HPLC) or haemoglobin electrophoresis supplemented by measurement of Hb A2 and Hb F usually confirm the diagnosis of β thalassaemia trait. The diagnosis of α thalassaemia trait is more difficult; detection of infrequent Hb H inclusions is usually possible in α0 thalassaemia trait, but definitive diagnosis requires DNA analysis.10 A diagnosis of α0 thalassaemia heterozygosity can be clinically important for prediction of haemoglobin Bart’s hydrops fetalis since, if both parents have α0 thalassaemia, this very serious condition can occur in a fetus. DNA analysis is therefore indicated when a pregnant woman of appropriate ethnic origin has an MCH of <25 pg. Hb H inclusions may not be detected in α+ thalassaemia trait but this diagnosis is of less clinical importance and confirmation is thus not usually required.
Macrocytic Anaemia
A high MCV (Fig. 23.2) with oval macrocytes and hypersegmented neutrophils suggests folate or vitamin B12 deficiency and is an indication for assays of these vitamins (see Chapter 10); subsequent investigations could include malabsorption studies, tests for coeliac disease and tests for intrinsic factor and gastric parietal cell antibodies. In patients with these blood film findings and normal vitamin assays, haematinic deficiency is not completely excluded and further investigation is indicated. A Schilling test permits a definitive diagnosis of pernicious anaemia but currently this test is not available due to a lack of reagents. In the absence of intrinsic factor antibodies, the diagnosis of pernicious anaemia may be presumptive. Pernicious anaemia is commonly associated with autoimmune thyroid disease and other autoimmune disorders, such as diabetes mellitus. A high MCV may also be associated with alcohol excess and liver disease or use of drugs such as hydroxycarbamide or zidovudine. Macrocytosis resulting from chronic haemolysis is associated with increased numbers of immature red cells, which appear slightly larger and bluer than normal red cells (polychromatic macrocytes) on a Romanowsky-stained peripheral blood film. Supravital staining of blood films (see p. 33) or an automated reticulocyte count can be used to confirm reticulocytosis. Untreated anaemia associated with polychromasia is likely to indicate blood loss or haemolysis. The combination of red cell fragments, thrombocytopenia and polychromasia indicates a microangiopathic haemolytic anaemia and should trigger further tests such as a platelet count, coagulation studies, assessment of renal function and a search for infection or neoplastic disease. This further assessment is urgent because these may be features of thrombotic thrombocytopenic purpura, which requires immediate treatment by plasma exchange.
Normocytic Anaemia
Normochromic, normocytic anaemia (Fig. 23.3) is frequently the result of an underlying chronic, non-haematological disease. Investigations should include screening for renal insufficiency, subclinical infections, autoimmune diseases and neoplasia. In the presence of anaemia, a lack of polychromasia, confirmed by reticulocytopenia, points toward a primary failure of erythropoiesis or lack of compensatory increased red cell production in blood loss or haemolysis. Examination of the bone marrow may be helpful in demonstrating haematological causes for a normochromic, normocytic anaemia such as aplastic anaemia or MDS.11 Staining for iron may also show that there is a block in iron metabolism suggestive of anaemia associated with chronic inflammatory disease.
Leucopenia
Neutropenia
Once physiological variation, ethnicity and familial or cyclic neutropenia have been excluded (see p. 19
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
