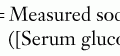21 Approach to the Child with Primary Immunodeficiency
Etiology and Pathogenesis
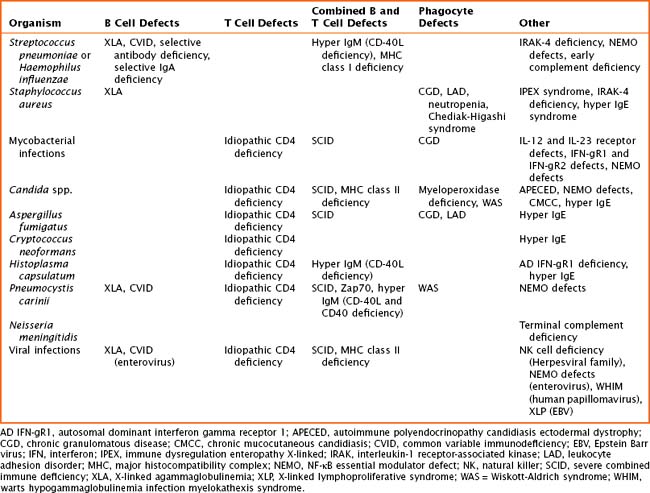
The pathogenesis of primary immune defects is related to the underlying cellular defect, which may be further divided into problems with the innate and adaptive immune system. Immune defects are distinguished by the cellular mechanism involved, including B cells and humoral or antibody defects, T cells and cell-mediated defects, combined B and T cell defects, phagocytic defects, complement defects, and newly described defects in pattern recognition molecules (Toll-like receptors [TLRs] and signaling molecules). The innate immune system is composed of phagocytes (dendritic cells, macrophages, and neutrophils), complement components, natural killer (NK) cells, TLRs, and signaling molecules. These cells are the first line of defense and respond to pathogens in a nonspecific manner. The adaptive immune system includes B cells, T cells, and combined defects. These cells recognize and respond to pathogens in a specific manner, leading to long-lasting immunity. There are also genetic disorders of immune regulation that cross both arms of the immune system. Specific disorders are often associated with particular infectious organisms (Table 21-1).
Disorders of the Innate Immune System
Complement
The complement system shares responsibility in host defense, induction of the adaptive immune system, inflammation, and clearance of apoptotic cells and immune complexes. The clinical manifestations of deficiencies early in the complement cascade are susceptibility to infections with Streptococcus pneumoniae and Haemophilus influenzae and rheumatologic abnormalities, such as systemic lupus erythematosus (see Chapter 29). Neisseria infections are more common with defects in later components of the complement cascade. Hereditary angioedema results from a defect in C1 esterase inhibitor enzyme function.
Clinical Presentation
History
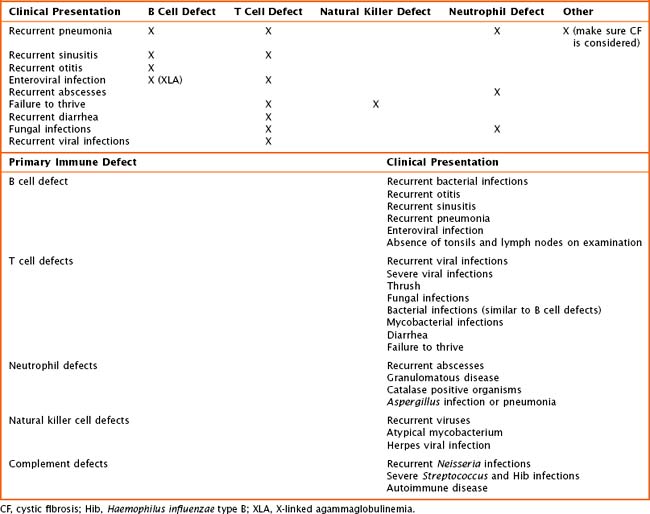
Children with primary immune deficiencies can present in a variety of ways (Table 21-2). Classically, the most severe defects occur early on with recurrent or persistent viral illnesses, failure to thrive, or chronic diarrhea. Older toddlers and school-aged children may present with recurrent ear infections, sinus infections, pneumonias, or poor growth. When obtaining the history, one must pay particular attention to patient’s age at time of infection, type(s) of infection, site of infection, and ability to treat infection with oral or intravenous (IV) antimicrobials, as well as the number of hospitalizations. The overall health, growth, and development of the patient are important. The birth history and maternal prenatal history must be obtained. Prior miscarriages may be an indication of a genetic problem. The presence of maternal infection during pregnancy may lead to an immunodeficiency in a newborn. The immunization history is crucial when evaluating for an antibody defect or a T cell abnormality. The child’s response to immunizations is a way to look at the function of the immune system. Also, it is important to recognize that children with suspected or known severe immune defects should not receive live viral vaccines. Included in the history should be a comprehensive family history. Many of the primary immunodeficiencies are X-linked or associated with known genetic mutations. At times, there is a family history of autoimmune disease. A list of medications should be obtained, including any that may have been given in the past such as chemotherapeutics or antiepileptics. Finally, a detailed review of systems may be helpful detailing concomitant medical or development concerns and phenotypes leading to the diagnosis of a genetic syndrome.
Evaluation and Management
Laboratory Studies
If one is concerned about an underlying immunodeficiency, screening blood tests are necessary. For most patients, this includes obtaining a complete blood count (CBC) with differential, quantitative immunoglobulins (IgG, IgA, IgM, and IgE), and antibody titers to immunizations (diphtheria, tetanus, pneumococcal serotypes, and H. influenzae B) or naturally occurring antibodies (isohemagglutinin titers). When obtaining a CBC with differential, calculating the absolute lymphocyte count and the absolute neutrophil count is important. “Normal” lymphocyte values change with the age of the child, and thus appropriate references should be consulted for normal values when obtaining these tests. If there are further concerns or abnormalities in this screening laboratory evaluation, additional testing can be performed, and referral to a specialist is warranted (Table 21-3). For T cell defects, lymphocyte subsets along with functional assays, including mitogens or antigens, are important. In children where a neutrophil defect is suspected, absolute neutrophil number, morphology, and functional assays should be performed. A dihydrorodamine reduction (DHR) assay or nitrotetrazolium blue (NBT) assay is available to assess oxidative burst, the abnormality in chronic granulomatous disease (CGD). Complement deficiencies can be screened with a CH50 and C4 levels. Some assays look at absolute numbers and function of NK cells. DNA mutation analysis is available for some of the immune deficiencies with known genetic mutations. If a child has an abnormal immune evaluation or if there is a suspected defect or concern, immediate referral to an immunologist may be necessary.
| Laboratory Test | Abnormality Seen | Possible Immune Defect |
|---|---|---|
| CBC | ||
| Absolute lymphocyte count | Changes as patient ages; <2800 cells/microliter in an infant is concerning | SCID |
| Absolute neutrophil count | Decreased | Congenital neutropenia |
| Platelet count | Decreased | WAS (small platelets, eczema, and T cell immunodeficiency) |
| DHR assay | Assesses neutrophil oxidative burst; absent burst is diagnostic | Chronic granulomatous disease |
| CH50 assay | If negative | Complement deficiency |
| T cell markers (CD3, CD4, CD8) | Low | Multiple immunodeficiencies, including SCID, CVID, selective deficiency |
| B cell markers (CD19, CD20) | Absent mature B cells only | XLA |
| Absent mature B cells with decreased or absent T cells or NK cells | SCID | |
| IgG | Low or absent |
CBC, complete blood count; Con A, concanavalin A; CVID, common variable immune deficiency; DHR, dihydrorodamine reduction; Ig, immunoglobulin; NK, natural killer; PHA, phytohaemagglutinin; PWM, Pokweed mitogen; SCID, severe combined immunodeficiency; WAS, Wiskott-Aldrich syndrome; XLA, X-linked agammaglobulinemia.
Ballow M. Approach to the patient with recurrent infections. Clin Rev Allergy Immunol. 2008;34:129-140.
Bonilla FA, Bernstein IL, Khan DA, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94(suppl):S1-S63.
Carneiro-Sampaio M, Coutnho A. Immunity to microbes: lessons from primary immunodeficiencies. Infect Immun. 2007;75(4):1545-1555.
Slatter MA, Gennery AR. Recurrent infections in childhood. Clin Exp Immunol. 2008;152:389-396.
Stiehm ER, Ochs HD, Winkelstein JA. Immunodeficiency Disorders in Infants and Children, ed 5. Philadelphia: Elsevier Saunders; 2004.