375e |
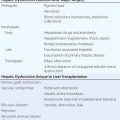
Primary Immunodeficiencies Associated with (or Secondary to) Other Diseases |
There are an increasing number of conditions in which a primary immunodeficiency (PID) has been described as one facet of a more complex disease setting. It is essential to consider associated diseases when a PID is identified as the primary manifestation and, conversely, not neglect the potentially harmful consequences of a PID that could be masked by other manifestations of a particular syndrome.
Below is a short description of these syndromes in which the PID is classified according to the arm of the immune system that is affected.
1. Primary Immunodeficiencies of the Innate Immune System
a) Several severe congenital neutropenia (SCN) syndromes can be associated with malformations. The recently described SCN disease caused by glucose-6-phosphatase deficiency (G6PC3) can be associated with heart and urogenital malformations. The related glycogenesis Ib disease combines SCN with hypoglycemia and hepatosplenomegaly. Some HAX-1 gene mutations lead to neurocognitive impairments as well as SCN. Barth syndrome combines SCN with cardiomyopathy. Lastly, Shwachman syndrome is a known autosomal recessive entity (caused by mutation of the SBDS gene) in which the defect in granulopoiesis can extend to the other hematopoietic lineages; short stature, bone metaphyseal dysplasia, and exocrine pancreatic insufficiency are known hallmarks of this condition.
b) Syndromic asplenia combines the risk of infection with heart defects and situs inversus.
c) Leukocyte adhesion deficiency (LAD) type II includes growth retardation and impairment of cognitive development.
d) A few patients with X-linked chronic granulomatous disease present with a contiguous gene deletion syndrome that can include the McLeod phenotype, which is characterized by anemia, acanthocytosis, and a severe risk of immune reaction against donor red cells because the patient’s red cells do not express the Kell antigen. The McLeod phenotype also can result in a neurologic disease.
e) X-linked nuclear factor-κB (NF-κB) essential modulator (NEMO) deficiency provokes not only a variable set of deficiencies of both innate and adaptive immunity but also mild osteopetrosis, lymphedema, and, more frequently, anhydrotic ectodermal dysplasia, dysmorphic facies, and abnormal conical teeth. The last finding is often helpful in the diagnosis of that condition.
2. Primary Immunodeficiencies of the Adaptive Immune System
a) T cell primary immunodeficiencies. Reticular dysgenesis, a rare severe combined immunodeficiency (SCID) characterized by T lymphopenia and agranulocytosis, can cause sensorineural deafness. Coronin A deficiency is another SCID variant that can be associated with behavioral disorders because the Coronin A gene is located in a genome area known to have been deleted in some patients with this disorder. The lack of enzymes of purine metabolism (adenosine deaminase and purine nucleoside phosphorylase) provokes not only profound T cell lymphocytopenia but also neurologic impairment, including dysautonomia and abnormalities of cognitive development of variable intensity, in many patients. The neurologic impairment can persist after hematopoietic stem cell transplantation (HSCT). Mild chondrodysplasia is a common finding in adenosine deaminase (ADA) deficiency and, indeed, can help the physician arrive at a final diagnosis.
b) Primary thymic defects. DiGeorge syndrome is a complex embryopathy that is caused by hemizygous interstitial deletion of chromosome 22, leading to multiple developmental defects, including conotruncal defects, hypoparathyroidism, and dysmorphic syndrome. Although a profound T cell immunodeficiency is rare in DiGeorge syndrome (∼1% of cases), failure to recognize this feature is likely to have a fatal outcome. Similarly, some forms of the related CHARGE syndrome (mutation of the CHD7 gene) also cause a profound T cell immunodeficiency.
c) T cell primary immunodeficiencies related to calcium influx defects. Recently, rare T cell PIDs were found to be caused by defective store-operated entry of calcium ions into T and B lymphocytes after antigen stimulation. These defects (caused by ORA-1 and STIM-1 deficiencies) also lead to anhydrotic ectodermal dysplasia, abnormal teeth, and, above all, a nonprogressive muscle disease characterized by excessive fatigue.
d) DNA repair defects. Several genetic defects impair DNA repair pathways. Many lead to combined T and B lymphocyte PIDs in a syndromal setting of varying complexity. The most common is ataxia-telangiectasia (AT), an autosomal recessive disorder with an incidence of 1 in 40,000 live births; AT causes a B cell immunodeficiency (low IgA, IgG2 deficiency, and low antibody production) that often requires immunoglobulin replacement therapy.
AT is associated with a progressive T cell immunodeficiency. As the condition’s name suggests, the hallmark features are telangiectasia and cerebellar ataxia.
These manifestations may not be detectable before age 3–4 years, and so AT should be considered in young children with IgA deficiency and problematic infections. Diagnosis is based on a cytogenetic analysis showing excessive chromosomal rearrangements (mostly affecting chromosomes 7 and 14) in lymphocytes. AT is caused by mutation of the gene encoding the ATM protein, a kinase that plays a major role in the detection of DNA lesions and the organization of DNA repair (or cell death if the lesions are too numerous) by triggering several different pathways. Overall, AT is a progressive disease that carries a very high risk of lymphoma, leukemia, and (during adulthood) carcinomas. A variant of AT (AT-like disease) is caused by mutation of the MRE11 gene.
Nijmegen breakage syndrome (NBS) is a less common condition that also results from chromosome instability (and the same cytogenetic abnormalities as in AT). It is characterized by a severe T and B cell combined immunodeficiency with autosomal recessive inheritance. Subjects with NBS exhibit microcephaly and a birdlike face but neither ataxia nor telangiectasia. The risk of malignancies is also very high. Nijmegen breakage syndrome results from a deficiency in Nibrin (NBS1, a protein associated with MRE11 and Rad50 that is involved in checking DNA lesions) caused by hypomorphic mutations.
Severe forms of dyskeratosis congenita (also known as Hoyeraal-Hreidersson syndrome) combine a progressive immunodeficiency that can include an absence of B and natural killer cell (NK) lymphocytes, progressive bone marrow failure, microcephaly, in utero growth retardation, and gut disease. The disease can be X-linked or, more rarely, autosomal recessive. It is caused by the mutation of genes encoding telomere maintenance proteins, including dyskerin (DKC1).
Bloom syndrome (helicase deficiency) combines a typical dysmorphic syndrome with growth retardation, skin lesions, and a mild immunodeficiency that also can be found in some patients with Fanconi’s anemia.
Rare forms of combined T and B cell immunodeficiencies with autosomal recessive inheritance are associated in more complex syndromes with microcephaly, failure to grow, and a variable dysmorphic syndrome. These disorders are caused by mutation of the genes that encode DNA ligase 4 and Cernunnos (XLF), both of which are members of the nonhomologous end-joining DNA repair pathway.
The Vici syndrome combines callosal agenesis, cataracts, cardiomyopathy, hyperpigmentation, and a combined immunodeficiency. It is caused by biallelic EPG5 gene mutation that results in defective autophagy.
Lastly, immunodeficiency, centromere instability, and facial anomalies (ICF) syndrome is a complex autosomal recessive syndrome that variably combines a mild T cell immunodeficiency and a more severe B cell immunodeficiency with a coarse face, intestinal disease, and mild mental retardation. A cytogenetic diagnostic feature is the presence of multiradial chromosomes (most frequently chromosomes 1, 9, and 16) caused by DNA defective methylation. The syndrome is a result of either DNA methyltransferase DNMT3B deficiency or ZBTB24 deficiency.
e) Growth hormone insensitivity syndrome (Laron dwarfism) with combined primary immunodeficiency. Mutations in the STAT5b gene, which encodes a transcription factor involved in signaling downstream of the growth hormone receptor and the interleukin 2 (IL-2) receptor, lead to susceptibility to infection because of a partial, functional T cell immunodeficiency associated with autoimmune manifestations. The autoimmune manifestations probably result from defective generation/activation of regulatory T cells.
f) Hyper-IgE syndrome (autosomal dominant form). Hyper-IgE syndrome is a complex disorder that combines skin infections, inflammation, and susceptibility to bacterial and fungal infections of skin and lungs, often with pneumatoceles, with characteristic syndromic signs such as facial dysmorphy, defective loss of primary teeth, hyperextensibility, scoliosis, and osteoporosis. Elevated serum IgE levels are typical of hyper-IgE syndrome. The recently reported defects in TH17 effector responses account, at least in part, for the vulnerability to specific infections. This condition is caused by heterozygous (dominant) mutation of the gene encoding the transcription factor STAT3, which is required in a number of signaling pathways downstream of cytokine/cytokine receptor interactions (notably for IL-6 and IL21).
g) Primary immunodeficiencies with bone disease. The autosomal recessive cartilage hair hypoplasia (CHH) disease is characterized by short-limb dwarfism, metaphyseal dysostosis, and sparse hair, together with a combined T and B cell PID of variable intensity, ranging from quasi-SCID to an absence of clinically significant immunodeficiency. The condition can predispose to erythroblastopenia, autoimmunity, and tumors. It is caused by mutations in the RMRP gene for a noncoding ribosome-associated RNA.
Schimke immunoosseous dysplasia is a rare autosomal recessive condition characterized by severe T and B cell immunodeficiency with spondyloepiphyseal dysplasia, growth retardation, and kidney and vascular diseases. It is the consequence of mutations in the SMARCAL1 gene. The function of the gene product may be related to DNA repair.
h) Venoocclusive disease with immunodeficiency (VODI syndrome) is a rare autosomal recessive condition predominantly found in populations originating from Lebanon. It combines severe hepatic venoocclusive disease with usually mild T cell immunodeficiency and panhypogammaglobulinemia. It is caused by a deficiency in a nuclear protein, Sp110.
3. B Cell Primary Immunodeficiencies Hypogammaglobulinemia can be associated with chromosomal defects such as trisomy 18 and Jacobsen syndrome (hemizygous deletion of part of the long arm of chromosome 11). A rare biallelic deficiency of the mismatch repair protein PMS2 leads to a partial deficiency in Ig class switch recombination in patients at a very high risk of cancer in general and colon carcinomas and lymphomas in particular. Transcobalamin deficiency disturbs vitamin B12 transport and therefore impairs hematopoiesis. Hypogammaglobulinemia is easily corrected by vitamin B12 administration and can be a characteristic of this very rare disorder.
4. Primary Immunodeficiencies Affecting Regulatory Pathways Several inherited disorders that lead to hemophagocytic lymphohistiocytosis (HLH) also have features that are important in terms of both diagnosis and prognosis. Three of these disorders—Griscelli syndrome, Chédiak-Higashi syndrome, and the Hermansky-Pudlak type II syndrome—are characterized by partial albinism and silvery hair appearance that can facilitate diagnosis. Hermansky-Pudlak type II also can be a bleeding disorder if platelet aggregation is defective. Chédiak-Higashi syndrome also is characterized by an early-onset progressive neurologic disorder with impaired cognitive development and motor and sensory deficiencies, culminating in a generalized encephalopathy. The encephalopathy is not prevented or arrested by allogeneic HSCT even when the HLH risk is controlled.
5. Primary Immunodeficiencies Associated with Other Conditions Predisposition to infection, notably severe disseminated opportunistic infections including nontuberculous mycobacterial infections, can be associated with autoantibodies against interferon γ as observed in Asia.
A number of conditions can cause PIDs indirectly. For example, hypercatabolism in patients with Steinert’s disease may cause hypogammaglobulinemia. Intestinal lymphangiectasia that includes both immunoglobulin and naive T cell loss and can expose the patient to a significant infectious risk. Urinary IgG loss may result from severe nephritic syndromes.
A number of drugs, including antimalarials, captopril, penicillamine, phenytoin, and sulfasalazine, can induce predominantly IgA hypogammaglobulinemia in (probably predisposed) adults.
One also should also consider (1) diseases that are not thought to be PIDs but include the occurrence of recurrent infections and (2) genetic defects of the immune system that lead to other clinical manifestations. A very good example of the first group is cystic fibrosis (CF). Despite having a functionally normal immune system, patients with CF develop protracted bacterial respiratory tract infections, notably Pseudomonas aeruginosa colonization. This bacterium can incapacitate innate immune responses and cause unremitting inflammation that further facilitates infection. An example of the second group is primary alveolar proteinosis, which is caused by a defect in surfactant clearance by alveolar macrophages. The condition results from mutation of the gene encoding the granulocyte-macrophage colony-stimulating factor receptor α.
SECTION 2 |
DISORDERS OF IMMUNE-MEDIATED INJURY |
376 |
Allergies, Anaphylaxis, and Systemic Mastocytosis |
The term atopy implies a tendency to manifest asthma, rhinitis, urticaria, and atopic dermatitis alone or in combination, in association with the presence of allergen-specific IgE. However, individuals without an atopic background may also develop hypersensitivity reactions, particularly urticaria and anaphylaxis, associated with the presence of IgE. Inasmuch as the mast cell is a key effector cell in allergic rhinitis and asthma, and the dominant effector in urticaria, anaphylaxis, and systemic mastocytosis, its developmental biology, activation pathway, product profile, and target tissues will be considered in the introduction to these clinical disorders.
The binding of IgE to human mast cells and basophils, a process termed sensitization, prepares these cells for subsequent antigen-specific activation. The high-affinity Fc receptor for IgE, designated FcεRI, is composed of one α, one β, and two disulfide-linked γ chains, which together cross the plasma membrane seven times. The α chain is responsible for IgE binding, and the β and γ chains provide for signal transduction that follows the aggregation of the sensitized tetrameric receptors by polymeric antigen. The binding of IgE stabilizes the α chain at the plasma membrane, thus increasing the density of FcεRI receptors at the cell surface while sensitizing the cell for effector responses. This accounts for the correlation between serum IgE levels and the numbers of FcεRI receptors detected on circulating basophils.
Signal transduction is initiated through the action of a Src family–related tyrosine kinase termed Lyn that is constitutively associated with the β chain. Lyn transphosphorylates the canonical immunoreceptor tyrosine-based activation motifs (ITAMs) of the β and γ chains of the receptor, resulting in recruitment of more active Lyn to the β chain and of Syk tyrosine kinase. The phosphorylated tyrosines in the ITAMs function as binding sites for the tandem src homology two (SH2) domains within Syk. Syk activates not only phospholipase Cγ, which associates with the linker of activated T cells at the plasma membrane, but also phosphatidylinositol 3-kinase to provide phosphatidylinositol-3,4,5-trisphosphate, which allows membrane targeting of the Tec family kinase Btk and its activation by Lyn. In addition, the Src family tyrosine kinase Fyn becomes activated after aggregation of IgE receptors and phosphorylates the adapter protein Gab2 that enhances activation of phosphatidylinositol 3-kinase. Indeed, this additional input is essential for mast cell activation, but it can be partially inhibited by Lyn, indicating that the extent of mast cell activation is in part regulated by the interplay between these Src family kinases. Activated phospholipase Cγ cleaves phospholipid membrane substrates to provide inositol-1,4,5-trisphosphate (IP3) and 1,2-diacylglycerols (1,2-DAGs) so as to mobilize intracellular calcium and activate protein kinase C, respectively. The subsequent opening of calcium-regulated activated channels provides the sustained elevations of intracellular calcium required to recruit the mitogen-activated protein kinases, ERK, JNK, and p38 (serine/threonine kinases), which provide cascades to augment arachidonic acid release and to mediate nuclear translocation of transcription factors for various cytokines. The calcium ion–dependent activation of phospholipases cleaves membrane phospholipids to generate lysophospholipids, which, like 1,2-DAG, may facilitate the fusion of the secretory granule perigranular membrane with the cell membrane, a step that releases the membrane-free granules containing the preformed mediators of mast cell effects.
The secretory granule of the human mast cell has a crystalline structure, unlike mast cells of lower species. IgE-dependent cell activation results in solubilization and swelling of the granule contents within the first minute of receptor perturbation; this reaction is followed by the ordering of intermediate filaments about the swollen granule, movement of the granule toward the cell surface, and fusion of the perigranular membrane with that of other granules and with the plasmalemma to form extracellular channels for mediator release while maintaining cell viability.
In addition to exocytosis, aggregation of FcεRI initiates two other pathways for generation of bioactive products, namely, lipid mediators and cytokines. The biochemical steps involved in expression of such cytokines as tumor necrosis factor α (TNF-α), interleukin (IL) 1, IL-6, IL-4, IL-5, IL-13, granulocyte-macrophage colony-stimulating factor (GM-CSF), and others, including an array of chemokines, have not been specifically defined for mast cells. Inhibition studies of cytokine production (IL-1β, TNF-α, and IL-6) in mouse mast cells with cyclosporine or FK506 reveal binding to the ligand-specific immunophilin and attenuation of the calcium ion- and calmodulin-dependent serine/threonine phosphatase, calcineurin.
Lipid mediator generation (Fig. 376-1) involves translocation of calcium ion–dependent cytosolic phospholipase A2 to the outer nuclear membrane, with subsequent release of arachidonic acid for metabolic processing by the distinct prostanoid and leukotriene pathways. The constitutive prostaglandin endoperoxide synthase-1 (PGHS-1/cyclooxygenase-1) and the de novo inducible PGHS-2 (cyclooxygenase-2) convert released arachidonic acid to the sequential intermediates, prostaglandins G2 and H2. The glutathione-dependent hematopoietic prostaglandin D2 (PGD2) synthase then converts PGH2 to PGD2, the predominant mast cell prostanoid. The PGD2 receptor DP1 is expressed by platelets and epithelial cells, whereas DP2 is expressed by TH2 lymphocytes, eosinophils, and basophils. Mast cells also generate thromboxane A2 (TXA2), a short lived but powerful mediator that induces bronchoconstriction and platelet activation through the T prostanoid (TP) receptor.
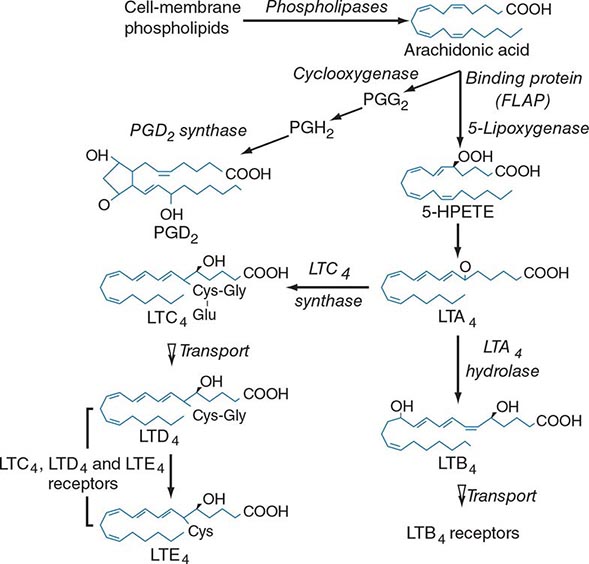
FIGURE 376-1 Pathways for biosynthesis and release of membrane-derived lipid mediators from mast cells. In the 5-lipoxygenase pathway, leukotriene A4 (LTA4) is the intermediate from which the terminal-pathway enzymes generate the distinct final products, leukotriene C4 (LTC4) and leukotriene B4 (LTB4), which leave the cell by separate saturable transport systems. Gamma glutamyl transpeptidase and a dipeptidase then cleave glutamic acid and glycine from LTC4 to form LTD4 and LTE4, respectively. The major mast cell product of the cyclooxygenase system is PGD2.
For leukotriene biosynthesis, the released arachidonic acid is metabolized by 5-lipoxygenase (5-LO) in the presence of an integral nuclear membrane protein, 5-LO activating protein (FLAP). The calcium ion–dependent translocation of 5-LO to the nuclear membrane converts the arachidonic acid to the sequential intermediates, 5-hydroperoxyeicosatetraenoic acid (5-HPETE) and leukotriene (LT) A4. LTA4 is conjugated with reduced glutathione by LTC4 synthase, an integral nuclear membrane protein homologous to FLAP. Intracellular LTC4 is released by a carrier-specific export step for extracellular metabolism to the additional cysteinyl leukotrienes, LTD4 and LTE4, by the sequential removal of glutamic acid and glycine. Alternatively, cytosolic LTA4 hydrolase converts some LTA4 to the dihydroxy leukotriene LTB4, which also undergoes specific export. Two receptors for LTB4, BLT1 and BLT2, mediate chemotaxis of human neutrophils. Two receptors for the cysteinyl leukotrienes, CysLT1 and CysLT2, are present on smooth muscle of the airways and the microvasculature and on hematopoietic cells such as macrophages, eosinophils, and mast cells. Whereas the CysLT1 receptor has a preference for LTD4 and is blocked by the receptor antagonists in clinical use, the CysLT2 receptor is equally responsive to LTD4 and LTC4, is unaffected by these antagonists, and is a negative regulator of the function of the CysLT1 receptor. LTD4, acting at CysLT1 receptors, is the most potent known bronchoconstrictor, whereas LTE4 induces a vascular leak and mediates the recruitment of eosinophils to the bronchial mucosa. Studies in gene-deleted mice indicate the existence of additional receptors for LTE4. The lysophospholipid formed during the release of arachidonic acid from 1-O-alkyl-2-acyl-sn-glyceryl-3-phosphorylcholine can be acetylated in the second position to form platelet-activating factor (PAF). Serum levels of PAF correlated positively with the severity of anaphylaxis to peanut in a recent study, whereas the levels of PAF acetyl hydrolase (a PAF-degrading enzyme) were inversely related to the same outcome.
Unlike most other cells of bone marrow origin, mast cells circulate as committed progenitors lacking their characteristic secretory granules. These committed progenitors express c-kit, the receptor for stem cell factor (SCF). Unlike most other lineages, they retain and increase c-kit expression with maturation. The SCF interaction with c-kit is an absolute requirement for the development of constitutive tissue mast cells residing in skin and connective tissue sites and for the accumulation of mast cells at mucosal surfaces during TH2-type immune responses. Several T cell-derived cytokines (IL-3, IL-4, IL-5, and IL-9) can potentiate SCF-dependent mast cell proliferation and/or survival in vitro in mice and humans. Indeed, mast cells are absent from the intestinal mucosa in clinical T cell deficiencies, but are present in the submucosa. Based on the immunodetection of secretory granule neutral proteases, mast cells in the lung parenchyma and intestinal mucosa selectively express tryptase, and those in the intestinal and airway submucosa, perivascular spaces, skin, lymph nodes, and breast parenchyma express tryptase, chymase, and carboxypeptidase A (CPA). In the mucosal epithelium of severe asthmatics, mast cells can express tryptase and CPA without chymase. The secretory granules of mast cells selectively positive for tryptase exhibit closed scrolls with a periodicity suggestive of a crystalline structure by electron microscopy, whereas the secretory granules of mast cells with multiple proteases are scroll-poor, with an amorphous or lattice-like appearance.
Mast cells are distributed at cutaneous and mucosal surfaces and in submucosal tissues about venules and could influence the entry of foreign substances by their rapid response capability (Fig. 376-2). Upon stimulus-specific activation and secretory granule exocytosis, histamine and acid hydrolases are solubilized, whereas the neutral proteases, which are cationic, remain largely bound to the anionic proteoglycans, heparin and chondroitin sulfate E, with which they function as a complex. Histamine and the various lipid mediators (PGD2, LTC4/D4/E4, PAF) alter venular permeability, thereby allowing influx of plasma proteins such as complement and immunoglobulins, whereas LTB4 mediates leukocyte–endothelial cell adhesion and subsequent directed migration (chemotaxis). The accumulation of leukocytes and plasma opsonins facilitates defense of the microenvironment. The inflammatory response can also be detrimental, as in asthma, where the smooth-muscle constrictor activity of the cysteinyl leukotrienes is evident and much more potent than that of histamine.
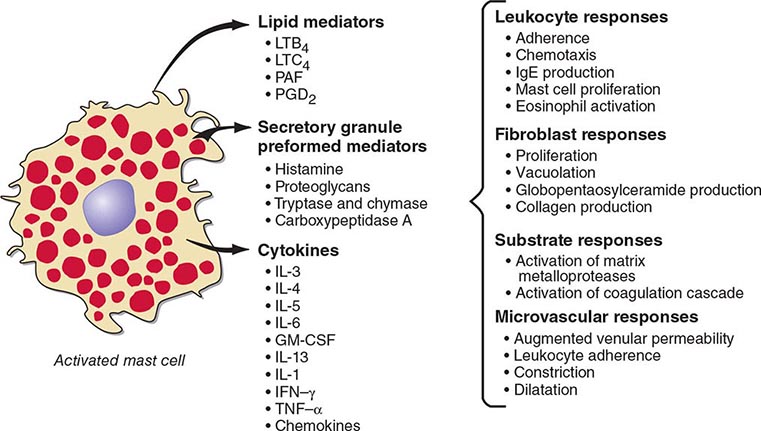
FIGURE 376-2 Bioactive mediators of three categories generated by IgE-dependent activation of murine mast cells can elicit common but sequential target cell effects leading to acute and sustained inflammatory responses. GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; IFN, interferon; LT, leukotriene; PAF, platelet-activating factor; PGD2, prostaglandin D2; TNF, tumor necrosis factor.
The cellular component of the mast cell–mediated inflammatory response is augmented and sustained by cytokines and chemokines. IgE-dependent activation of human skin mast cells in situ elicits TNF-α production and release, which in turn induces endothelial cell responses favoring leukocyte adhesion. Similarly, activation of purified human lung mast cells or cord blood–derived cultured mast cells in vitro results in substantial production of proinflammatory (TNF-α) and immunomodulatory cytokines (IL-4, IL-5, IL-13) and chemokines. Bronchial biopsy specimens from patients with asthma reveal that mast cells are immunohistochemically positive for IL-4 and IL-5, but that the predominant localization of IL-4, IL-5, and GM-CSF is to T cells, defined as TH2 by this profile. IL-4 modulates the T cell phenotype to the TH2 subtype, determines the isotype switch to IgE (as does IL-13), and upregulates FcεRI-mediated expression of cytokines by mast cells based on in vitro studies.
An immediate and late cellular phase of allergic inflammation can be induced in the skin, nose, or lung of some allergic humans with local allergen challenge. The immediate phase in the nose involves pruritus and watery discharge; in the lung, it involves bronchospasm and mucus secretion; and in the skin, it involves a wheal-and-flare response with pruritus. The reduced nasal patency, reduced pulmonary function, or erythema with swelling at the skin site in a late-phase response at 6–8 h is associated with biopsy findings of infiltrating and activated TH2 cells, eosinophils, basophils, and some neutrophils. The progression from early mast cell activation to late cellular infiltration has been used as an experimental surrogate of rhinitis or asthma. However, in asthma, there is an intrinsic hyperreactivity of the airways independent of the associated inflammation. Moreover, early- and late-phase responses (at least in the lung) are far more sensitive to blockade of IgE-dependent mast cell activation (or actions of histamine and cysteinyl leukotrienes) than are spontaneous or virally induced asthma exacerbations.
Consideration of the mechanism of immediate-type hypersensitivity diseases in the human has focused largely on the IgE-dependent recognition of otherwise innocuous substances. A region of chromosome 5 (5q23-31) contains genes implicated in the control of IgE levels including IL-4 and IL-13, as well as IL-3 and IL-9, which are involved in mucosal mast cell hyperplasia, and IL-5 and GM-CSF, which are central to eosinophil development and their enhanced tissue viability. Genes with linkage to the specific IgE response to particular allergens include those encoding the major histocompatibility complex (MHC) and certain chains of the T cell receptor (TCR-αδ). The complexity of atopy and the associated diseases includes susceptibility, severity, and therapeutic responses, each of which is among the separate variables modulated by both innate and adaptive immune stimuli.
The induction of allergic disease requires sensitization of a predisposed individual to a specific allergen. The greatest propensity for the development of atopic allergy occurs in childhood and early adolescence. The allergen is processed by antigen-presenting cells of the monocytic lineage (particularly dendritic cells) located throughout the body at surfaces that contact the outside environment, such as the nose, lungs, eyes, skin, and intestine. These antigen-presenting cells present the epitope-bearing peptides via their MHC to T helper cells and their subsets. The T cell response depends both on cognate recognition and on the cytokine microenvironment provided by the antigen-presenting dendritic cells, with IL-4 directing a TH2 subset, interferon (IFN) γ a TH1 profile, and IL-6 with transforming growth factor β (TGF-β) a TH17 subset. Allergens not only present antigenic epitopes via dendritic cells but also contain pattern recognition ligands that facilitate the immune response by direct initiation of cytokine generation from innate cell types such as basophils, mast cells, eosinophils, and others. The TH2 response is associated with activation of specific B cells that can also present allergens or that transform into plasma cells for antibody production. Synthesis and release into the plasma of allergen-specific IgE results in sensitization of FcεR1-bearing cells such as mast cells and basophils, which become activated on exposure to the specific allergen. In certain diseases, including those associated with atopy, the monocyte and eosinophil populations can express a trimeric FcεR1, which lacks the β chain, and yet respond to its aggregation. An additional recently recognized class of c-kit-expressing innate cells (termed nuocytes, natural helper cells, or group 2 innate lymphoid cells) can generate large quantities of IL-5 and IL-13 during antihelminth responses, are prominent in nasal polyps from humans, and could well contribute to inflammation in allergic diseases.
ANAPHYLAXIS
DEFINITION
Life-threatening anaphylactic responses of sensitized humans occur within minutes after systemic exposure to specific antigen. They are manifested by respiratory distress due to laryngeal edema and/or intense bronchospasm, often followed by vascular collapse, or by shock without antecedent respiratory difficulty. Cutaneous manifestations exemplified by pruritus and urticaria with or without angioedema are characteristic of such systemic anaphylactic reactions. Gastrointestinal manifestations include nausea, vomiting, crampy abdominal pain, and diarrhea.
PREDISPOSING FACTORS AND ETIOLOGY
There is no convincing evidence that age, sex, race, or geographic location predisposes a human to anaphylaxis except through exposure to specific immunogens. According to most studies, atopy does not predispose individuals to anaphylaxis from penicillin therapy or venom of a stinging insect but is a risk factor for allergens in food or latex. Risk factors for a poor outcome, however, include older age, use of beta blockers, and the presence of preexisting asthma. Severe hymenoptera anaphylaxis (generally with prominent hypotension) can be a presenting feature of underlying systemic mastocytosis. Additionally, some individuals suffering from recurrent episodes of idiopathic anaphylaxis possess morphologically aberrant mast cells in their bone marrow that express a mutant, constitutively active form of c-kit, even without evidence of frank mastocytosis.
The materials capable of eliciting the systemic anaphylactic reaction in humans include the following: heterologous proteins in the form of hormones (insulin, vasopressin, parathormone); enzymes (trypsin, chymotrypsin, penicillinase, streptokinase); pollen extracts (ragweed, grass, trees); nonpollen allergen extracts (dust mites, dander of cats, dogs, horses, and laboratory animals); food (peanuts, milk, eggs, seafood, nuts, grains, beans, gelatin in capsules); monoclonal antibodies; occupation-related products (latex rubber products); Hymenoptera venom (yellow jacket, yellow and white-faced hornets, paper wasp, honey bee, imported fire ants); polysaccharides such as dextran and thiomersal as a vaccine preservative; drugs such as protamine; antibiotics (penicillins, cephalosporins, amphotericin B, nitrofurantoin, quinolones); chemotherapy agents (carboplatin, paclitaxel, doxorubicin); local anesthetics (procaine, lidocaine); muscle relaxants (suxamethonium, gallamine, pancuronium); vitamins (thiamine, folic acid); diagnostic agents (sodium dehydrocholate, sulfobromophthalein); biologics (omalizumab, rituximab, etanercept); and occupation-related chemicals (ethylene oxide). Drugs function as haptens that form immunogenic conjugates with host proteins. The conjugating hapten may be the parent compound, a nonenzymatically derived storage product, or a metabolite formed in the host. Recombinant biologics can also induce the formation of IgE against the proteins or against glycosylated structures that serve as immunogens. Most recently, outbreaks of anaphylaxis to the anti-epidermal growth factor antibody cetuximab were reported in association with elevated titers of serum IgE to alpha-1,3-galactose, an oligosaccharide found on certain nonprimate proteins. Alpha-galactose antibodies also account for some episodes of delayed anaphylaxis to beef, lamb, and pork.
PATHOPHYSIOLOGY AND MANIFESTATIONS
Individuals differ in the time of appearance of symptoms and signs, but the hallmark of the anaphylactic reaction is the onset of some manifestation within seconds to minutes after introduction of the antigen (with the exception of alpha-galactose allergy), generally by injection or less commonly by ingestion. There may be upper or lower airway obstruction or both. Laryngeal edema may be experienced as a “lump” in the throat, hoarseness, or stridor, whereas bronchial obstruction is associated with a feeling of tightness in the chest and/or audible wheezing. Patients with asthma are predisposed to severe involvement of the lower airways and increased mortality. Flushing with diffuse erythema and a feeling of warmth may occur. A characteristic feature is the eruption of well-circumscribed, discrete cutaneous wheals with erythematous, raised, serpiginous borders and blanched centers. These urticarial eruptions are intensely pruritic and may be localized or disseminated. They may coalesce to form giant hives, and they seldom persist beyond 48 h. A localized, nonpitting, deeper edematous cutaneous process, angioedema, may also be present. It may be asymptomatic or cause a burning or stinging sensation. Angioedema of the bowel wall may cause sufficient intravascular volume depletion to precipitate cardiovascular collapse.
In fatal cases with clinical bronchial obstruction, the lungs show marked hyperinflation on gross and microscopic examination. The microscopic findings in the bronchi, however, are limited to luminal secretions, peribronchial congestion, submucosal edema, and eosinophilic infiltration, and the acute emphysema is attributed to intractable bronchospasm that subsides with death. The angioedema resulting in death by mechanical obstruction occurs in the epiglottis and larynx, but the process also is evident in the hypopharynx and to some extent in the trachea. On microscopic examination, there is wide separation of the collagen fibers and the glandular elements; vascular congestion and eosinophilic infiltration also are present. Patients dying of vascular collapse without antecedent hypoxia from respiratory insufficiency have visceral congestion with a presumptive loss of intravascular fluid volume. The associated electrocardiographic abnormalities, with or without infarction, in some patients may reflect a primary cardiac event mediated by mast cells (which are prominent near the coronary vessels) or may be secondary to a critical reduction in blood volume.
The angioedematous and urticarial manifestations of anaphylaxis have been attributed to the release of endogenous histamine. A role for the cysteinyl leukotrienes in causing marked bronchiolar constriction seems likely. Vascular collapse without respiratory distress in response to experimental challenge with the sting of a hymenopteran was associated with marked and prolonged elevations in blood histamine and intravascular coagulation and kinin generation. The finding that patients with systemic mastocytosis and episodic vascular collapse excrete large amounts of PGD2 metabolites in addition to histamine suggests that PGD2 is also of importance in the hypotensive anaphylactic reactions. As noted, serum PAF levels correlate with severity of anaphylaxis and are inversely proportional to the constitutive level of the acetylhydrolase involved in PAF inactivation. The actions of the array of mast cell–derived mediators are likely additive or synergistic at the target tissues.
DIAGNOSIS
The diagnosis of an anaphylactic reaction depends on a history revealing the onset of symptoms and signs within minutes after the responsible material is encountered. It is appropriate to rule out a complement-mediated immune complex reaction, an idiosyncratic response to a nonsteroidal anti-inflammatory drug (NSAID), or the direct effect of certain drugs or diagnostic agents on mast cells. Intravenous administration of a chemical mast cell–degranulating agent, including opiate derivatives and radiographic contrast media, may elicit generalized urticaria, angioedema, and a sensation of retrosternal oppression with or without clinically detectable bronchoconstriction or hypotension. In the transfusion anaphylactic reaction that occurs in patients with IgA deficiency, the responsible specificity resides in IgG or IgE anti-IgA; the mechanism of the reaction mediated by IgG anti-IgA is presumed to be complement activation with secondary mast cell participation.
The presence of specific IgE in the blood of patients with systemic anaphylaxis was demonstrated historically by passive transfer of the serum intradermally into a normal recipient, followed 24 h later by antigen challenge into the same site, with subsequent development of a wheal and flare (the Prausnitz-Küstner reaction). In current clinical practice, immunoassays using purified or recombinant antigens can demonstrate the presence of specific IgE in the serum of patients with anaphylactic reactions, and skin testing may be performed after the patient has recovered to elicit a local wheal and flare in response to the putative antigen. Elevations of tryptase levels in serum implicate mast cell activation in a systemic reaction and are particularly informative for anaphylaxis with episodes of hypotension during general anesthesia or when there has been a fatal outcome. However, because of the short half-life of tryptase, elevated levels are best detected within 4 h of a systemic reaction. Moreover, anaphylactic reactions to foods characteristically are not associated with elevations in serum tryptase.
PREVENTION
Prevention of anaphylaxis must take into account the sensitivity of the individual, the dose and character of the diagnostic or therapeutic agent, and the effect of the route of administration on the rate of absorption. Beta blockers are relatively contraindicated in persons at risk for anaphylactic reactions, especially those sensitive to Hymenoptera venom or those undergoing immunotherapy for respiratory system allergy. If there is a definite history of a past anaphylactic reaction to a medication, it is advisable to select a structurally unrelated agent. A knowledge of cross-reactivity among agents is critical since, for example, cephalosporins have a cross-reactive ring structure with the penicillins. When skin testing, a prick or scratch skin test should precede an intradermal test, since the latter has a higher risk of causing anaphylaxis. These tests should be performed before the administration of certain materials that are likely to elicit anaphylactic reactions, such as allergenic extracts. Skin testing for antibiotics or chemotherapeutic agents should be performed only on patients with a positive clinical history consistent with an IgE-mediated reaction and in imminent need of the antibiotic in question; skin testing is of no value for non-IgE-mediated eruptions. With regard to penicillin, two-thirds of patients with a positive reaction history and positive skin tests to benzylpenicilloyl-polylysine (BPL) and/or the minor determinant mixture (MDM) of benzylpenicillin products experience allergic reactions with treatment, and these reactions are almost uniformly of the anaphylactic type in those patients with minor determinant reactivity. Even patients without a history of previous clinical reactions have a 2–6% incidence of positive skin tests to the two test materials, and about 3 per 1000 with a negative history experience anaphylaxis with therapy, with a mortality of about 1 per 100,000.
If an agent carrying a risk of eliciting an anaphylactic response is required because a non-cross-reactive alternative is not available, desensitization can be performed with most antibiotics and other classes of therapeutic agents by the IV, SC, or PO route. Typically, graded quantities of the drug are given by the selected route starting below the threshold dose for an adverse reaction and then doubling each dose until a therapeutic dosage is achieved. Due to the risk of systemic anaphylaxis during the course of desensitization, such a procedure should be performed under the supervision of a specialist and in a setting in which resuscitation equipment is at hand and an IV line is in place. Once a desensitized state is achieved, it is critical to continue administration of the therapeutic agent at regular intervals throughout the treatment period to prevent the reestablishment of a significant pool of sensitized cells.
A different form of protection involves the development of blocking antibody of the IgG class, which protects against Hymenoptera venom–induced anaphylaxis by interacting with antigen so that less reaches the sensitized tissue mast cells. The maximal risk for systemic anaphylactic reactions in persons with Hymenoptera sensitivity occurs in association with a currently positive skin test. Although there is little cross-reactivity between honey bee and yellow jacket venoms, there is a high degree of cross-reactivity between yellow jacket venom and the rest of the vespid venoms (yellow or white-faced hornets and wasps). Prevention involves modification of outdoor activities to exclude bare feet, wearing perfumed toiletries, eating in areas attractive to insects, clipping hedges or grass, and hauling away trash or fallen fruit. As with each anaphylactic sensitivity, the individual should wear an informational bracelet and have immediate access to an unexpired autoinjectable epinephrine kit. Venom immunotherapy for 5 years can induce a state of resistance to sting reactions that is independent of serum levels of specific IgG or IgE. For children under the age of 10 with a systemic reaction limited to skin, the likelihood of progression to more serious respiratory or vascular manifestations is low, and thus immunotherapy is not recommended.
URTICARIA AND ANGIOEDEMA
DEFINITION
Urticaria and angioedema may appear separately or together as cutaneous manifestations of localized nonpitting edema; a similar process may occur at mucosal surfaces of the upper respiratory or gastrointestinal tract. Urticaria involves only the superficial portion of the dermis, presenting as well-circumscribed wheals with erythematous raised serpiginous borders and blanched centers that may coalesce to become giant wheals. Angioedema is a well-demarcated localized edema involving the deeper layers of the skin, including the subcutaneous tissue, and can also involve the bowel wall. Recurrent episodes of urticaria and/or angioedema of less than 6 weeks’ duration are considered acute, whereas attacks persisting beyond this period are designated chronic.
PREDISPOSING FACTORS AND ETIOLOGY
Urticaria and angioedema probably occur more frequently than reported because of the evanescent, self-limited nature of such eruptions, which seldom require medical attention when limited to the skin. Although persons in any age group may experience acute or chronic urticaria and/or angioedema, these lesions increase in frequency after adolescence, with the highest incidence occurring in persons in the third decade of life; indeed, one survey of college students indicated that 15–20% had experienced a pruritic wheal reaction.
The classification of urticaria-angioedema presented in Table 376-1 focuses on the different mechanisms for eliciting clinical disease and can be useful for differential diagnosis; nonetheless, most cases of chronic urticaria are idiopathic. Urticaria and/or angioedema occurring during the appropriate season in patients with seasonal respiratory allergy or as a result of exposure to animals or molds is attributed to inhalation or physical contact with pollens, animal dander, and mold spores, respectively. However, urticaria and angioedema secondary to inhalation are relatively uncommon compared to urticaria and angioedema elicited by ingestion of fresh fruits, shellfish, fish, milk products, chocolate, legumes including peanuts, and various drugs that may elicit not only the anaphylactic syndrome with prominent gastrointestinal complaints but also urticaria alone.
|
CLASSIFICATION OF URTICARIA AND/OR ANGIOEDEMA |
Additional etiologies include physical stimuli such as cold, heat, solar rays, exercise, and mechanical irritation. The physical urticarias can be distinguished by the precipitating event and other aspects of the clinical presentation. Dermographism, which occurs in 1–4% of the population, is defined by the appearance of a linear wheal at the site of a brisk stroke with a firm object or by any configuration appropriate to the eliciting event (Fig. 376-3). Dermographism has a prevalence that peaks in the second to third decades. It is not influenced by atopy and has a duration generally of <5 years. Pressure urticaria, which often accompanies chronic idiopathic urticaria, presents in response to a sustained stimulus such as a shoulder strap or belt, running (feet), or manual labor (hands). Cholinergic urticaria is distinctive in that the pruritic wheals are of small size (1–2 mm) and are surrounded by a large area of erythema; attacks are precipitated by fever, a hot bath or shower, or exercise and are presumptively attributed to a rise in core body temperature. Exercise-induced anaphylaxis can be precipitated by exertion alone or can be dependent on prior food ingestion. There is an association with the presence of IgE specific for α-5 gliadin, a component of wheat. The clinical presentation can be limited to flushing, erythema, and pruritic urticaria but may progress to angioedema of the face, oropharynx, larynx, or intestine or to vascular collapse; it is distinguished from cholinergic urticaria by presenting with wheals of conventional size and by not occurring with fever or a hot bath. Cold urticaria is local at body areas exposed to low ambient temperature or cold objects but can progress to vascular collapse with immersion in cold water (swimming). Solar urticaria is subdivided into six groups by the response to specific portions of the light spectrum. Vibratory angioedema may occur after years of occupational exposure or can be idiopathic; it may be accompanied by cholinergic urticaria. Other rare forms of physical allergy, always defined by stimulus-specific elicitation, include local heat urticaria, aquagenic urticaria from contact with water of any temperature (sometimes associated with polycythemia vera), and contact urticaria from direct interaction with some chemical substance.

FIGURE 376-3 Dermographic urticarial lesion induced by stroking the forearm lightly with the edge of a tongue blade. The photograph, taken after 2 minutes, demonstrates a prominent wheal-and-flare reaction in the shape of an X. (From LA Goldsmith et al [eds]: Fitzpatrick’s Dermatology in General Medicine, 8th ed. New York, McGraw-Hill, 2012. Photograph provided by Allen P. Kaplan, MD, Medical University of South Carolina.)
Angioedema without urticaria due to the generation of bradykinin occurs with C1 inhibitor (C1INH) deficiency that may be inborn as an autosomal dominant characteristic or may be acquired through the appearance of an autoantibody. The angiotensin-converting enzyme (ACE) inhibitors can provoke a similar clinical presentation in 0.1–0.5% of hypertensive patients due to attenuated degradation of bradykinin. The urticaria and angioedema associated with classic serum sickness or with hypocomplementemic cutaneous necrotizing angiitis are believed to be immune-complex diseases. The drug reactions to mast cell granule–releasing agents and to NSAIDs may be systemic, resembling anaphylaxis, or limited to cutaneous sites.
PATHOPHYSIOLOGY AND MANIFESTATIONS
Urticarial eruptions are distinctly pruritic, may involve any area of the body from the scalp to the soles of the feet, and appear in crops of 12- to 36-h duration, with old lesions fading as new ones appear. Most of the physical urticarias (cold, cholinergic, dermatographism) are an exception, with individual lesions lasting less than 2 h. The most common sites for urticaria are the extremities and face, with angioedema often being periorbital and in the lips. Although self-limited in duration, angioedema of the upper respiratory tract may be life-threatening due to laryngeal obstruction, whereas gastrointestinal involvement may present with abdominal colic, with or without nausea and vomiting, and may result in unnecessary surgical intervention. No residual discoloration occurs with either urticaria or angioedema unless there is an underlying vasculitic process leading to superimposed extravasation of erythrocytes.
The pathology is characterized by edema of the superficial dermis in urticaria and of the subcutaneous tissue and deep dermis in angioedema. Collagen bundles in affected areas are widely separated, and the venules are sometimes dilated. Any perivenular infiltrate consists of lymphocytes, monocytes, eosinophils, and neutrophils that are present in varying combination and numbers.
Perhaps the best-studied example of IgE- and mast cell–mediated urticaria and angioedema is cold urticaria. Cryoglobulins or cold agglutinins are present in up to 5% of these patients. Immersion of an extremity in an ice bath precipitates angioedema of the distal portion with urticaria at the air interface within minutes of the challenge. Histologic studies reveal marked mast cell degranulation with associated edema of the dermis and subcutaneous tissues. The histamine level in the plasma of venous effluent of the cold-challenged and angioedematous extremity is markedly increased, but no such increase appears in the plasma of effluent of the contralateral normal extremity. Elevated levels of histamine have been found in the plasma of venous effluent and in the fluid of suction blisters at experimentally induced lesional sites in patients with dermographism, pressure urticaria, vibratory angioedema, light urticaria, and heat urticaria. By ultrastructural analysis, the pattern of mast cell degranulation in cold urticaria resembles an IgE-mediated response with solubilization of granule contents, fusion of the perigranular and cell membranes, and discharge of granule contents, whereas in a dermographic lesion, there is additional superimposed zonal (piecemeal) degranulation. There are several reports of resolution of cold urticaria by treatment with monoclonal anti-human IgE (omalizumab). Elevations of plasma histamine levels with biopsyproven mast cell degranulation have also been demonstrated with generalized attacks of cholinergic urticaria and exercise-related anaphylaxis precipitated experimentally in subjects exercising on a treadmill while wearing a wet suit; however, only subjects with cholinergic urticaria have a concomitant decrease in pulmonary function.
Up to 40% of patients with chronic urticaria have an autoimmune cause for their disease including autoantibodies to IgE (5–10%) or, more commonly, to the α chain of FcεRI (35–45%). In these patients, autologous serum injected into their own skin can induce a wheal-and-flare reaction involving mast cell activation. The presence of these antibodies can also be recognized by their capacity to release histamine or induce activation markers such as CD63 or CD203 on basophils. An association with antibodies to microsomal peroxidase and/or thyroglobulin has been observed often with clinically significant Hashimoto’s thyroiditis. In vitro studies reveal that these autoantibodies can mediate basophil degranulation with enhancement by serum as a source of the anaphylatoxic fragment, C5a.
Hereditary angioedema is an autosomal dominant disease due to a deficiency of C1INH (type 1) in about 85% of patients and to a dysfunctional protein (type 2) in the remainder. A third type of hereditary angioedema has been described in which C1INH function is normal, and the causal lesion is a mutant form of factor XII, which leads to generation of excessive bradykinin. In the acquired form of C1INH deficiency, there is excessive consumption due either to immune complexes formed between anti-idiotypic antibody and monoclonal IgG presented by B cell lymphomas or to an autoantibody directed to C1INH. C1INH blocks the catalytic function of activated factor XII (Hageman factor) and of kallikrein, as well as the C1r/C1s components of C1. During clinical attacks of angioedema, C1INH-deficient patients have elevated plasma levels of bradykinin, particularly in the venous effluent of an involved extremity, and reduced levels of prekallikrein and high-molecular-weight kininogen, from which bradykinin is cleaved. The parallel decline in the complement substrates C4 and C2 reflects the action of activated C1 during such attacks. Mice with targeted disruption of the gene for C1INH exhibit a chronic increase in vascular permeability. The pathobiology is aggravated by administration of an ACE inhibitor (captopril) and is attenuated by breeding the C1INH null strain to a bradykinin 2 receptor (Bk2R) null strain. As ACE is also described as kininase II, the use of blockers results in impaired bradykinin degradation and explains the angioedema that occurs idiosyncratically in hypertensive patients with a normal C1INH. Bradykinin-mediated angioedema, whether caused by ACE inhibitors or by C1INH deficiency, is noteworthy for the conspicuous absence of concomitant urticaria.
DIAGNOSIS
The rapid onset and self-limited nature of urticarial and angioedematous eruptions are distinguishing features. Additional characteristics are the occurrence of the urticarial crops in various stages of evolution and the asymmetric distribution of the angioedema. Urticaria and/or angioedema involving IgE-dependent mechanisms are often appreciated by historic considerations implicating specific allergens or physical stimuli, by seasonal incidence, and by exposure to certain environments. Direct reproduction of the lesion with physical stimuli is particularly valuable because it so often establishes the cause of the lesion. The diagnosis of an environmental allergen based on the clinical history can be confirmed by skin testing or assay for allergen-specific IgE in serum. IgE-mediated urticaria and/or angioedema may or may not be associated with an elevation of total IgE or with peripheral eosinophilia. Fever, leukocytosis, and an elevated sedimentation rate are absent.
The classification of urticarial and angioedematous states presented in Table 376-1 in terms of possible mechanisms necessarily includes some differential diagnostic points. Hypocomplementemia is not observed in IgE-mediated mast cell disease and may reflect either an acquired abnormality generally attributed to the formation of immune complexes or a genetic or acquired deficiency of C1INH. Chronic recurrent urticaria, generally in females, associated with arthralgias, an elevated sedimentation rate, and normo- or hypocomplementemia suggests an underlying cutaneous necrotizing angiitis. Vasculitic urticaria typically persists longer than 72 h, whereas conventional urticaria often has a duration of 12–36 h. Confirmation depends on a biopsy that reveals cellular infiltration, nuclear debris, and fibrinoid necrosis of the venules. The same pathobiologic process accounts for the urticaria in association with such diseases as systemic lupus erythematosus or viral hepatitis with or without associated arteritis. Serum sickness per se or a similar clinical entity due to drugs includes not only urticaria but also pyrexia, lymphadenopathy, myalgia, and arthralgia or arthritis. Urticarial reactions to blood products or intravenous administration of immunoglobulin are defined by the event and generally are not progressive unless the recipient is IgA-deficient in the former case or the reagent is aggregated in the latter.
The diagnosis of hereditary angioedema is suggested not only by family history but also by the lack of pruritus and of urticarial lesions, the prominence of recurrent gastrointestinal attacks of colic, and episodes of laryngeal edema. Laboratory diagnosis depends on demonstrating a deficiency of C1INH antigen (type 1) or a nonfunctional protein (type 2) by a catalytic inhibition assay. While levels of C1 are normal, its substrates, C4 and C2, are chronically depleted and fall further during attacks due to the activation of additional C1. Patients with the acquired forms of C1INH deficiency have the same clinical manifestations but differ in the lack of a familial element. Furthermore, their sera exhibit a reduction of C1 function and C1q protein as well as C1INH, C4, and C2. Inborn C1INH deficiency and ACE inhibitor–elicited angioedema are associated with elevated levels of bradykinin. Lastly, type 3 hereditary angioedema is associated with normal levels of complement proteins.
Urticaria and angioedema are distinct from contact sensitivity, a vesicular eruption that progresses to chronic thickening of the skin with continued allergenic exposure. They also differ from atopic dermatitis, a condition that may present as erythema, edema, papules, vesiculation, and oozing proceeding to a subacute and chronic stage in which vesiculation is less marked or absent and scaling, fissuring, and lichenification predominate in a distribution that characteristically involves the flexor surfaces. In cutaneous mastocytosis, the reddish brown macules and papules, characteristic of urticaria pigmentosa, urticate with pruritus upon trauma; and in systemic mastocytosis, without or with urticaria pigmentosa, there is episodic systemic flushing with or without urtication but no angioedema.
SYSTEMIC MASTOCYTOSIS
DEFINITION
Systemic mastocytosis is defined by a clonal expansion of mast cells that in most instances is indolent and nonmalignant. The mast cell expansion is generally recognized only in bone marrow and in the normal peripheral distribution sites of the cells, such as skin, gastrointestinal mucosa, liver, and spleen. Mastocytosis occurs at any age and has a slight preponderance in males. The prevalence of systemic mastocytosis is not known, a familial occurrence is rare, and atopy is not increased.
CLASSIFICATION AND PATHOPHYSIOLOGY
A consensus classification for mastocytosis recognizes cutaneous mastocytosis with variants and four systemic forms (Table 376-2). Cutaneous mastocytosis is the most common diagnosis in children, whereas the form designated as indolent systemic mastocytosis (ISM) accounts for the majority of adult patients; it implies that there is no evidence of an associated hematologic disorder, liver disease, or lymphadenopathy and is not known to alter life expectancy. In systemic mastocytosis associated with clonal hematologic non–mast cell lineage disease (SM-AHNMD), the prognosis is determined by the nature of the associated disorder, which can range from dysmyelopoiesis to leukemia. In aggressive systemic mastocytosis (ASM), mast cell infiltration/proliferation in multiple organs such as liver, spleen, gut, and/or bone results in a poor prognosis; a subset of patients with this form has prominent eosinophilia with hepatosplenomegaly and lymphadenopathy. Mast cell leukemia (MCL) is the rarest form of the disease and is invariably fatal at present; the peripheral blood contains circulating, metachromatically staining, atypical mast cells. An aleukemic form of MCL is recognized without circulating mast cells when the percentage of high-grade immature mast cells in bone marrow smears exceeds 20% in a nonspicular area. Mast cell sarcoma and extracutaneous mastocytomas are rare solid mast cell tumors with malignant and benign features, respectively.
|
CLASSIFICATION OF MASTOCYTOSIS |
Source: Modified from SH Swerdlow et al (eds): World Health Organization Classification of Tumors: Pathology and Genetics in Tumors of Hematopoietic and Lymphoid Tissues. Lyon, IARC Press, 2008.
A point mutation of A to T at codon 816 of c-kit that causes an aspartic acid to valine substitution is found in multiple cell lineages in patients with mastocytosis, resulting in a somatic gain-in-function mutation. This substitution, as well as other rare mutations of c-kit, is characteristic of patients with all forms of systemic mastocytosis but is also present in some children with cutaneous mastocytosis, as might be anticipated because mast cells are of bone marrow lineage. The prognosis for patients with cutaneous mastocytosis and for almost all with ISM is a normal life expectancy, whereas that for patients with SM-AHNMD is determined by a non–mast cell component. ASM and MCL carry a poorer prognosis. In infants and children with cutaneous manifestations, namely, urticaria pigmentosa or bullous lesions, visceral involvement is usually lacking, and resolution is common.
CLINICAL MANIFESTATIONS
The clinical manifestations of systemic mastocytosis, distinct from a leukemic complication, are due to tissue occupancy by the mast cell mass, the tissue response to that mass, and the release of bioactive substances acting at both local and distal sites. The pharmacologically induced manifestations are pruritus, flushing, palpitations and vascular collapse, gastric distress, lower abdominal crampy pain, and recurrent headache. The increase in local cell burden is evidenced by the lesions of urticaria pigmentosa at skin sites and may be a direct local cause of bone pain and/or malabsorption. Mast cell–mediated fibrotic changes occur in liver, spleen, and bone marrow but not in gastrointestinal tissue or skin. Immunofluorescent analysis of bone marrow and skin lesions in ISM and of spleen, lymph node, and skin in ASM has revealed only one mast cell phenotype, namely, scroll-poor cells expressing tryptase, chymase, and CPA.
The cutaneous lesions of urticaria pigmentosa are reddish-brown macules or papules that respond to trauma with urtication and erythema (Darier’s sign). The apparent incidence of these lesions is ≥80% in patients with ISM and <50% in those with SM-AHNMD or ASM. Approximately 1% of patients with ISM have skin lesions that appear as tan-brown macules with striking patchy erythema and associated telangiectasia (telangiectasia macularis eruptiva perstans). In the upper gastrointestinal tract, gastritis and peptic ulcer are significant problems. In the lower intestinal tract, the occurrence of diarrhea and abdominal pain is attributed to increased motility due to mast cell mediators; this problem can be aggravated by malabsorption, which can also cause secondary nutritional insufficiency and osteomalacia. The periportal fibrosis associated with mast cell infiltration and a prominence of eosinophils may lead to portal hypertension and ascites. In some patients, flushing and recurrent vascular collapse are markedly aggravated by an idiosyncratic response to a minimal dosage of NSAIDs. The neuropsychiatric disturbances are clinically most evident as impaired recent memory, decreased attention span, and “migraine-like” headaches. Patients may experience exacerbation of a specific clinical sign or symptom with alcohol ingestion, temperature changes, stress, use of mast cell–interactive narcotics, or ingestion of NSAIDs.
DIAGNOSIS
Although the diagnosis of mastocytosis is generally suspected on the basis of the clinical history and physical findings, and can be supported by laboratory procedures, it can be established only by a tissue diagnosis. By convention, the diagnosis of systemic mastocytosis depends heavily on bone marrow biopsy to meet the criteria of one major plus one minor or three minor findings (Table 376-3). The bone marrow provides the major criterion by revealing aggregates of mast cells, often in paratrabecular and perivascular locations with lymphocytes and eosinophils, as well as the minor criteria of an abnormal mast cell morphology, an aberrant mast cell membrane immunophenotype, or a codon 816 mutation in any cell type. A serum total tryptase level and/or a 24-h urine collection for measurement of histamine, histamine metabolites, or metabolites of PGD2 are noninvasive approaches to consider before bone marrow biopsy. The pro-β and α forms of tryptase are elevated in more than one-half of patients with systemic mastocytosis and provide a minor criterion; the fully processed (“mature”) β form is increased in patients undergoing an anaphylactic reaction. Additional studies directed by the presentation include a bone densitometry, bone scan, or skeletal survey; contrast studies of the upper gastrointestinal tract with small-bowel follow-through, computed tomography scan, or endoscopy; and a neuropsychiatric evaluation. Osteoporosis is increased in mastocytosis and may lead to pathologic fractures.
|
DIAGNOSTIC CRITERIA FOR SYSTEMIC MASTOCYTOSISa |
aDiagnosis requires either the major criterion and one minor criterion or three minor criteria.
The differential diagnosis requires the exclusion of other flushing disorders. The 24-h urine assessment of 5-hydroxy-indoleacetic acid and metanephrines should exclude a carcinoid tumor or a pheochromocytoma. Some patients presenting with recurrent mast cell activation symptoms without an obvious increase in mast cell burden in skin or bone marrow have been shown to carry aberrant mast cells with clonality markers of D816C c-kit mutation or surface CD25 expression. Most patients with recurrent anaphylaxis, including the idiopathic group, present with angioedema and/or wheezing, which are not manifestations of systemic mastocytosis.
ALLERGIC RHINITIS
DEFINITION
Allergic rhinitis is characterized by sneezing; rhinorrhea; obstruction of the nasal passages; conjunctival, nasal, and pharyngeal itching; and lacrimation, all occurring in a temporal relationship to allergen exposure. Although commonly seasonal due to elicitation by airborne pollens, it can be perennial in an environment of chronic exposure to house dust mites, animal danders, or insect products. In North America, the incidence of allergic rhinitis is about 7%. The overall prevalence in North America is nearly 20%, with the peak prevalence of nearly 40% occurring in childhood and adolescence.
PREDISPOSING FACTORS AND ETIOLOGY
Allergic rhinitis generally occurs in atopic individuals, often in association with atopic dermatitis, food allergy, urticaria, and/or asthma (Chap. 309). Up to 40% of patients with rhinitis manifest asthma, whereas ∼70% of individuals with asthma experience rhinitis. Symptoms generally appear before the fourth decade of life and tend to diminish gradually with aging, although complete spontaneous remissions are uncommon. A relatively small number of weeds that depend on wind rather than insects for pollination, as well as grasses and some trees, produce sufficient quantities of pollen suitable for wide distribution by air currents to elicit seasonal allergic rhinitis. The dates of pollination of these species generally vary little from year to year in a particular locale but may be quite different in another climate. In the temperate areas of North America, trees typically pollinate from March through May, grasses in June and early July, and ragweed from mid-August to early October. Molds, which are widespread in nature because they occur in soil or decaying organic matter, propagate spores in a pattern that depends on climatic conditions. Perennial allergic rhinitis occurs in response to allergens that are present throughout the year, including animal dander, cockroach-derived proteins, mold spores, or dust mites such as Dermatophagoides farinae and Dermatophagoides pteronyssinus. Dust mites are scavengers of human skin and excrete cysteine protease allergens in their feces. In up to one-half of patients with perennial rhinitis, no clear-cut allergen can be demonstrated as causative. The ability of many allergens to cause rhinitis rather than lower respiratory tract symptoms (particularly pollens) may be attributed to their large size, 10–100 μm, and retention within the nose.
PATHOPHYSIOLOGY AND MANIFESTATIONS
Episodic rhinorrhea, sneezing, obstruction of the nasal passages with lacrimation, and pruritus of the conjunctiva, nasal mucosa, and oropharynx are the hallmarks of allergic rhinitis. The nasal mucosa is pale and boggy, the conjunctiva congested and edematous, and the pharynx generally unremarkable. Swelling of the turbinates and mucous membranes with obstruction of the sinus ostia and eustachian tubes precipitates secondary infections of the sinuses and middle ear, respectively. Nasal polyps, representing mucosal protrusions containing edema fluid with variable numbers of eosinophils and degranulated mast cells, can increase obstructive symptoms and can concurrently arise within the nasopharynx or sinuses. However, atopy is not a risk factor for nasal polyps, which instead may occur in the setting of the aspirin-intolerant triad of rhinosinusitis and asthma and in patients with chronic staphylococcal colonization, which produces superantigens leading to an intense TH2 inflammatory response.
The nose presents a large mucosal surface area through the folds of the turbinates and serves to adjust the temperature and moisture content of inhaled air and to filter out particulate materials >10 μm in size by impingement in a mucous blanket; ciliary action moves the entrapped particles toward the pharynx. Entrapment of pollen and digestion of the outer coat by mucosal enzymes such as lysozymes release protein allergens generally of 10,000–40,000 molecular weight. The initial interaction occurs between the allergen and intraepithelial mast cells and then proceeds to involve deeper perivenular mast cells, both of which are sensitized with specific IgE. During the symptomatic season when the mucosae are already swollen and hyperemic, there is enhanced adverse reactivity to the seasonal pollen. Biopsy specimens of nasal mucosa during seasonal rhinitis show submucosal edema with infiltration by eosinophils, along with some basophils and neutrophils.
The mucosal surface fluid contains IgA that is present because of its secretory piece and also IgE, which apparently arrives by diffusion from plasma cells in proximity to mucosal surfaces. IgE fixes to mucosal and submucosal mast cells, and the intensity of the clinical response to inhaled allergens is quantitatively related to the naturally occurring pollen dose. In sensitive individuals, the introduction of allergen into the nose is associated with sneezing, “stuffiness,” and discharge, and the fluid contains histamine, PGD2, and leukotrienes. Thus the mast cells of the nasal mucosa and submucosa generate and release mediators through IgE-dependent reactions that are capable of producing tissue edema and eosinophilic infiltration.
DIAGNOSIS
The diagnosis of seasonal allergic rhinitis depends largely on an accurate history of occurrence coincident with the pollination of the offending weeds, grasses, or trees. The continuous character of perennial allergic rhinitis due to contamination of the home or place of work makes historic analysis difficult, but there may be variability in symptoms that can be related to exposure to animal dander, dust mite and/or cockroach allergens, fungal spores, or work-related allergens such as latex. Patients with perennial rhinitis commonly develop the problem in adult life, and manifest nasal congestion and a postnasal discharge, often associated with thickening of the sinus membranes demonstrated by radiography. Perennial nonallergic rhinitis with eosinophilia syndrome (NARES) occurs in the middle decades of life and is characterized by nasal obstruction, anosmia, chronic sinusitis, and frequent aspirin intolerance. The term vasomotor rhinitis or perennial nonallergic rhinitis designates a condition of enhanced reactivity of the nasopharynx in which a symptom complex resembling perennial allergic rhinitis occurs with nonspecific stimuli, including chemical odors, temperature and humidity variations, and position changes but occurs without tissue eosinophilia or an allergic etiology. Other entities to be excluded are structural abnormalities of the nasopharynx; exposure to irritants; gustatory rhinitis associated with cholinergic activation that occurs while eating or ingesting alcohol; hypothyroidism; upper respiratory tract infection; pregnancy with prominent nasal mucosal edema; prolonged topical use of α-adrenergic agents in the form of nose drops (rhinitis medicamentosa); and the use of certain therapeutic agents such as rauwolfia, β-adrenergic antagonists, estrogens, progesterone, ACE inhibitors, aspirin and other NSAIDS, and drugs for erectile dysfunction (phosphodiesterase-5 inhibitors).
The nasal secretions of allergic patients are rich in eosinophils, and a modest peripheral eosinophilia is a common feature. Local or systemic neutrophilia implies infection. Total serum IgE is frequently elevated, but the demonstration of immunologic specificity for IgE is critical to an etiologic diagnosis. A skin test by the intracutaneous route (puncture or prick) with the allergens of interest provides a rapid and reliable approach to identifying allergen-specific IgE that has sensitized cutaneous mast cells. A positive intracutaneous skin test with 1:10–1:20 weight/volume of extract has a high predictive value for the presence of allergy. An intradermal test with a 1:500–1:1000 dilution of 0.05 mL may follow if indicated by history when the intracutaneous test is negative, but while more sensitive, it is less reliable due to the reactivity of some asymptomatic individuals at the test dose. Skin testing by the intracutaneous route for food allergens can be supportive of the clinical history. A double-blind, placebo-controlled challenge may document a food allergy, but such a procedure does bear the risk of an anaphylactic reaction. An elimination diet is safer but is tedious and less definitive. Food allergy is uncommon as a cause of allergic rhinitis.
Newer methodology for detecting total IgE, including the development of enzyme-linked immunosorbent assays (ELISA) employing anti-IgE bound to either a solid-phase or a liquid-phase particle, provides rapid and cost-effective determinations. Measurements of specific anti-IgE in serum are obtained by its binding to an allergen and quantitation by subsequent uptake of labeled anti-IgE. As compared to the skin test, the assay of specific IgE in serum is less sensitive but has high specificity.
PREVENTION
Avoidance of exposure to the offending allergen is the most effective means of controlling allergic diseases; removal of pets from the home to avoid animal danders, utilization of air-filtration devices to minimize the concentrations of airborne pollens, elimination of cockroach-derived proteins by chemical destruction of the pest and careful food storage, travel to areas where the allergen is not being generated, and even a change of domicile to eliminate a mold spore problem may be necessary. Control of dust mites by allergen avoidance includes use of plastic-lined covers for mattresses, pillows, and comforters; using a filter-equipped vacuum cleaner; washing bedding and clothes at temperatures >54.5°C (above 130°F); and elimination of carpets and drapes.
|
TREATMENT |
ALLERGIC RHINITIS |
Although allergen avoidance is the most cost-effective means of managing allergic rhinitis, treatment with pharmacologic agents represents the standard approach to seasonal or perennial allergic rhinitis. Oral H1 antihistamines are effective for nasopharyngeal itching, sneezing, and watery rhinorrhea and for such ocular manifestations as itching, tearing, and erythema, but they are less efficacious for the nasal congestion. The older antihistamines are sedating, and they induce psychomotor impairment, including reduced eye-hand coordination and impaired automobile driving skills. Their anticholinergic (muscarinic) effects include visual disturbance, urinary retention, and constipation. Because the newer H1 antihistamines such as fexofenadine, loratadine, desloratadine, cetirizine, levocetirizine, olopatadine, bilastine, and azelastine are less lipophilic and more H1 selective, their ability to cross the blood-brain barrier is reduced, and thus their sedating and anticholinergic side effects are minimized. These newer antihistamines do not differ appreciably in efficacy for relief of rhinitis and/or sneezing. Azelastine nasal spray may benefit individuals with nonallergic vasomotor rhinitis, but it has an adverse effect of dysgeusia (taste perversion) in some patients. Because antihistamines have little effect on congestion, α-adrenergic agents such as phenylephrine or oxymetazoline are generally used topically to alleviate nasal congestion and obstruction. However, the duration of their efficacy is limited because of rebound rhinitis (i.e., 7- to 14-day use can lead to rhinitis medicamentosa) and such systemic responses as hypertension. Oral α-adrenergic agonist decongestants containing pseudoephedrine are standard for the management of nasal congestion, generally in combination with an antihistamine. While oral antihistamines typically reduce nasal and ocular symptoms by about one-third, pseudoephedrine must be added to achieve a similar reduction in nasal congestion. These pseudoephedrine combination products can cause insomnia and are precluded from use in patients with narrow angle glaucoma, urinary retention, severe hypertension, marked coronary artery disease, or a first-trimester pregnancy. The CysLT1 blocker montelukast is approved for treatment of both seasonal and perennial rhinitis, and it reduces both nasal and ocular symptoms by about 20%. Cromolyn sodium, a nasal spray, is essentially without side effects and is used prophylactically on a continuous basis during the season. Intranasal high-potency glucocorticoids are the most potent drugs available for the relief of established rhinitis, seasonal or perennial, and are effective in relieving nasal congestion. They provide efficacy with substantially reduced side effects as compared with this same class of agent administered orally. Their most frequent side effect is local irritation, with Candida overgrowth being a rare occurrence. The currently available intranasal glucocorticoids—beclomethasone, flunisolide, triamcinolone, budesonide, fluticasone propionate, fluticasone furoate, ciclesonide, and mometasone furoate—are equally effective for nasal symptom relief, including nasal congestion; these agents all achieve up to 70% overall symptom relief with some variation in the time period for onset of benefit. Topical ipratropium is an anticholinergic agent effective in reducing rhinorrhea, including that in patients with perennial symptoms, and it can be additionally efficacious when combined with intranasal glucocorticoids. Local treatment with cromolyn sodium is effective in treating mild allergic conjunctivitis. Topical antihistamines such as olopatadine, azelastine, ketotifen, or epinastine administered to the eye provide rapid relief of itching and redness and are more effective than oral antihistamines.
Immunotherapy, often termed hyposensitization, consists of repeated subcutaneous injections of gradually increasing concentrations of the allergen(s) considered to be specifically responsible for the symptom complex. Controlled studies of ragweed, grass, dust mite, and cat dander allergens administered for treatment of allergic rhinitis have demonstrated at least partial relief of symptoms and signs. The duration of such immunotherapy is 3–5 years, with discontinuation being based on minimal symptoms over two consecutive seasons of exposure to the allergen. Clinical benefit appears related to the administration of a high dose of relevant allergen, advancing from weekly to monthly intervals. Patients should remain at the treatment site for at least 20 minutes after allergen administration so that any anaphylactic consequence can be managed. Local reactions with erythema and induration are not uncommon and may persist for 1–3 days. Immunotherapy is contraindicated in patients with significant cardiovascular disease or unstable asthma and should be conducted with particular caution in any patient requiring β-adrenergic blocking therapy because of the difficulty in managing an anaphylactic complication. The response to immunotherapy is associated with a complex of cellular and humoral effects that includes a modulation in T cell cytokine production. Immunotherapy should be reserved for clearly documented seasonal or perennial rhinitis that is clinically related to defined allergen exposure with confirmation by the presence of allergen-specific IgE. Systemic treatment with a monoclonal antibody to IgE (omalizumab) that blocks mast cell and basophil sensitization has efficacy for allergic rhinitis and can be used with immunotherapy to enhance safety and efficacy. However, current approval is only for treatment of patients with persistent allergic asthma not controlled by inhaled glucocorticoid therapy. A sequence for the management of allergic or perennial rhinitis based on an allergen-specific diagnosis and stepwise management as required for symptom control would include the following: (1) identification of the offending allergen(s) by history with confirmation of the presence of allergen-specific IgE by skin test and/or serum assay; (2) avoidance of the offending allergen; and (3) medical management in a stepwise fashion (Fig. 376-4). Mild intermittent symptoms of allergic rhinitis are treated with oral antihistamines, oral CysLT1 receptor antagonists, intranasal antihistamines, or intranasal cromolyn prophylaxis. Moderate to more severe allergic rhinitis is managed with intranasal glucocorticoids plus oral antihistamines, oral CysLT1 receptor antagonists, or antihistamine-decongestant combinations. Persistent allergic rhinitis requiring the daily use of intranasal glucocorticoids with add-on interventions such as oral antihistamines, decongestant combinations, or topical ipratropium merits consideration of allergen-specific immunotherapy. Even a brief course of oral prednisone can be indicated for rapid relief of severe allergic rhinitis symptoms.
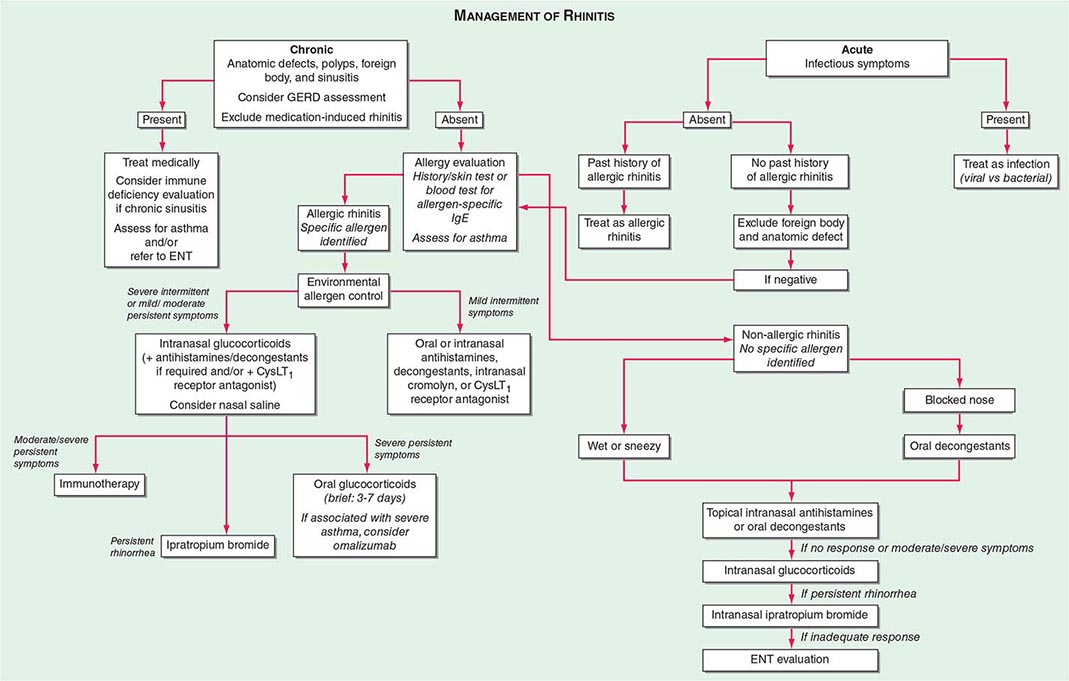
FIGURE 376-4 Algorithm for the diagnosis and management of rhinitis. ENT, ear, nose, and throat; GERD, gastroesophageal reflux disease.
377e |
Autoimmunity and Autoimmune Diseases |
One of the central features of the immune system is the capacity to mount an inflammatory response to potentially harmful foreign materials while avoiding damage to self-tissues. Whereas recognition of self plays an important role in shaping the repertoires of immune receptors on both T and B cells and in clearing apoptotic and other tissue debris from sites throughout the body, the development of potentially harmful immune responses to self-antigens is, in general, prohibited. The essential feature of an autoimmune disease is that tissue injury is caused by the immunologic reaction of the organism against its own tissues. Autoimmunity, on the other hand, refers merely to the presence of antibodies or T lymphocytes that react with self-antigens and does not necessarily imply that the self-reactivity has pathogenic consequences. Autoimmunity is present in all individuals; however, autoimmune disease occurs only in those individuals in whom the breakdown of one or more of the basic mechanisms regulating immune tolerance results in self-reactivity that can cause tissue damage.
Autoimmunity is seen in normal individuals, with a higher frequency among normal older people. Polyreactive autoantibodies that recognize many host antigens are present throughout life. Expression of these autoantibodies may be increased after some inciting events. These antibodies are usually of the IgM heavy chain isotype and are encoded by nonmutated germline immunoglobulin variable region genes. When autoimmunity is induced by an inciting event, such as infection or tissue damage from trauma or ischemia, the autoreactivity is in general self-limited. When such autoimmunity does persist, however, pathology may or may not result. Even in the presence of organ pathology, it may be difficult to determine whether the damage is mediated by autoreactivity. After an inciting event, the development of self-reactivity may be the consequence of an ongoing pathologic process, may be nonpathogenic, or may contribute to tissue inflammation and damage. Individuals with autoimmune disease may have numerous autoantibodies, only some or even none of which may be pathogenic. Patients with systemic sclerosis may have a wide array of antinuclear antibodies that are important in disease classification but are not clearly pathogenic; patients with pemphigus may also exhibit a wide array of autoantibodies, only one of which (antibody to desmoglein) is known to be pathogenic.
MECHANISMS OF AUTOIMMUNITY
Since Ehrlich first postulated the existence of mechanisms to prevent the generation of self-reactivity in the early1900s, ideas concerning the nature of this prohibition have developed in parallel with a progressive increase in understanding of the immune system. Burnet’s clonal selection theory included the idea that interaction of lymphoid cells with their specific antigens during fetal or early postnatal life would lead to elimination of such “forbidden clones.” This idea was refuted, however, when it was shown that autoimmune diseases could be induced in experimental animals by simple immunization procedures, that autoantigen-binding cells could be demonstrated easily in the circulation of normal individuals, and that self-limited autoimmune phenomena frequently developed after tissue damage from infection or trauma. These observations indicated that clones of cells capable of responding to autoantigens were present in the repertoire of antigen-reactive cells in normal adults and suggested that mechanisms in addition to clonal deletion were responsible for preventing their activation.
Currently, three general processes are thought to be involved in the maintenance of selective unresponsiveness to autoantigens (Table 377e-1): (1) sequestration of self-antigens, rendering them inaccessible to the immune system; (2) specific unresponsiveness (tolerance or anergy) of relevant T or B cells; and (3) limitation of potential reactivity by regulatory mechanisms. Derangements of these normal processes may predispose to the development of autoimmunity (Table 377e-2). In general, these abnormal responses require both an exogenous trigger, such as bacterial or viral infection or cigarette smoking, and the presence of endogenous abnormalities in the cells of the immune system. Microbial superantigens, such as staphylococcal protein A and staphylococcal enterotoxins, are substances that can stimulate a broad range of T and B cells through specific interactions with selected families of immune receptors, irrespective of their antigen specificity. If autoantigen-reactive T and/or B cells express these receptors, autoimmunity may develop. Alternatively, molecular mimicry or cross-reactivity between a microbial product and a self-antigen may lead to activation of autoreactive lymphocytes. One of the best examples of autoreactivity and autoimmune disease resulting from molecular mimicry is rheumatic fever, in which antibodies to the M protein of streptococci cross-react with myosin, laminin, and other matrix proteins as well as with neuronal antigens. Deposition of these autoantibodies in the heart initiates an inflammatory response, whereas their penetration into the brain can result in Sydenham’s chorea. Molecular mimicry between microbial proteins and host tissues has been reported in type 1 diabetes mellitus, rheumatoid arthritis, celiac disease, and multiple sclerosis. It is presumed that infectious agents may be able to overcome self-tolerance because they possess pathogen-associated molecular patterns (PAMPs). These molecules (e.g., bacterial endotoxin, RNA, or DNA) exert adjuvant-like effects on the immune system by interacting with Toll-like receptors (TLRs) and other pattern recognition receptors (PRRs) that increase the immunogenicity and immunostimulatory capacity of the microbial material. The adjuvants activate dendritic cells through TLRs, which in turn stimulate the activation of previously quiescent lymphocytes that recognize both microbial antigens and self-antigens. Similarly, cellular and tissue damage due to the release of damage-associated molecular patterns (DAMPs), including DNA, RNA nucleosomes, and other tissue debris, may activate cells of the inflammatory and immune systems through engagement of the same array of PRRs.
|
MECHANISMS PREVENTING AUTOIMMUNITY |
Endogenous derangements of the immune system may also contribute to the loss of immunologic tolerance to self-antigens and the development of autoimmunity (Table 377e-2). Some autoantigens reside in immunologically privileged sites, such as the brain or the anterior chamber of the eye. These sites are characterized by the inability of engrafted tissue to elicit immune responses. Immunologic privilege results from a number of events, including the limited entry of proteins from those sites into lymphatics, the local production of immunosuppressive cytokines such as transforming growth factor β, and the local expression of molecules (including Fas ligand) that can induce apoptosis of activated T cells. Lymphoid cells remain in a state of immunologic ignorance (neither activated nor anergized) with regard to proteins expressed uniquely in immunologically privileged sites. If the privileged site is damaged by trauma or inflammation or if T cells are activated elsewhere, proteins expressed at this site can become immunogenic and also be the targets of immunologic assault. In multiple sclerosis and sympathetic ophthalmia, for example, antigens uniquely expressed in the brain and eye, respectively, become the target of activated T cells.
|
MECHANISMS OF AUTOIMMUNITY |
Alterations in antigen presentation may also contribute to autoimmunity. Peptide determinants (epitopes) of a self-antigen that are not routinely presented to lymphocytes may be recognized as a result of altered proteolytic processing of the molecule and the ensuing presentation of novel peptides (cryptic epitopes). When B cells rather than dendritic cells present self-antigen, they may also present cryptic epitopes that can activate autoreactive T cells. These cryptic epitopes will not previously have been available to effect the silencing of autoreactive lymphocytes. Furthermore, once there is immunologic recognition of one protein component of a multimolecular complex, reactivity may be induced to other components of the complex after internalization and presentation of all molecules within the complex (epitope spreading). Finally, inflammation, environmental agents, drug exposure, or normal senescence may cause a post-transitional alteration in proteins, resulting in the generation of immune responses that cross-react with normal self-proteins. For example, the induction and/or release of protein arginine deiminase enzymes results in the conversion of arginine residues to citrullines in a variety of proteins, thereby altering their capacity to induce immune responses. Production of antibodies to citrullinated proteins has been observed in rheumatoid arthritis and chronic lung disease as well as in normal smokers and may contribute to organ pathology. Alterations in the availability and presentation of autoantigens may be important components of immunoreactivity in certain models of organ-specific autoimmune diseases. In addition, these factors may be relevant to an understanding of the pathogenesis of various drug-induced autoimmune conditions. However, the diversity of autoreactivity manifesting in non-organ-specific systemic autoimmune diseases suggests that these conditions may result from a more general activation of the immune system rather than from an alteration in individual self-antigens.
Many autoimmune diseases are characterized by the presence of antibodies that react with apoptotic material. Defects in the clearance of apoptotic material have been shown to elicit auto-immunity and autoimmune disease in a number of animal models. Moreover, such defects have been found in patients with systemic lupus erythematosus (SLE). Apoptotic debris that is not cleared quickly by the immune system can function as endogenous ligands for a number of PRRs on dendritic cells and B cells. Under such circumstances, dendritic cells and/or B cells are activated, and an immune response to apoptotic debris can develop. In addition, the presence of extracellular apoptotic material within germinal centers of secondary lymphoid organs in patients with SLE may facilitate the direct activation of autoimmune B cell clones or may function to select such clones during immune responses.
Studies in a number of experimental models have suggested that intense stimulation of T lymphocytes can produce nonspecific signals that bypass the need for antigen-specific helper T cells and lead to polyclonal B cell activation with the formation of multiple autoantibodies. For example, antinuclear, antierythrocyte, and antilymphocyte antibodies are produced during the chronic graft-versus-host reaction. In addition, true autoimmune diseases, including autoimmune hemolytic anemia and immune complex–mediated glomerulonephritis, can be induced in this manner. While such diffuse activation of helper T cell activity clearly can cause autoimmunity, nonspecific stimulation of B lymphocytes can also lead to the production of autoantibodies. Thus, the administration of polyclonal B cell activators, such as bacterial endotoxin, to normal mice leads to the production of a number of autoantibodies, including those to DNA and IgG (rheumatoid factor). A variety of genetic modifications resulting in hyperresponsiveness of B cells also can lead to the production of autoantibodies and, in animals of appropriate genetic background, a lupus-like syndrome. Moreover, excess B cell activating factor (BAFF), a B cell survival factor, can cause T cell–independent B cell activation and the development of autoimmunity. SLE can also be induced in mice through exuberant dendritic cell activation, through a redundancy of TLR7 on the Y chromosome (as in BXSB-Yaa mice), or through exposure to CpG, a ligand for TLR9. The ensuing induction of inflammatory mediators can cause a switch from the production of nonpathogenic IgM autoantibodies to the production of pathogenic IgG autoantibodies in the absence of antigen-specific T cell help. Aberrant selection of the B or T cell repertoire at the time of antigen receptor expression can also predispose to autoimmunity. For example, B cell immunodeficiency caused by an absence of the B cell receptor–associated kinase (Bruton’s tyrosine kinase) leads to X-linked agammaglobulinemia. This syndrome is characterized by reduced B cell numbers. This leads to high levels of BAFF which alter B cell selection and lead to greater survival of autoreactive B cells. Likewise, negative selection of autoreactive T cells in the thymus requires expression of the autoimmune regulator (AIRE) gene that enables the expression of tissue-specific proteins in thymic medullary epithelial cells. Peptides from these proteins are expressed in the context of major histocompatibility complex (MHC) molecules and mediate the central deletion of autoreactive T cells. The absence of AIRE gene expression leads to a failure of negative selection of autoreactive cells, autoantibody production, and severe inflammatory destruction of multiple organs. Individuals deficient in AIRE gene expression develop autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED).
Primary alterations in the activity of T and/or B cells, cytokine imbalances, or defective immunoregulatory circuits may also contribute to the emergence of autoimmunity. Diminished production of tumor necrosis factor (TNF) and interleukin (IL) 10 has been reported to be associated with the development of autoimmunity. Overproduction or therapeutic administration of type 1 interferon has also been associated with autoimmunity. Overexpression of costimulatory molecules on T cells similarly can lead to autoantibody production.
Autoimmunity may also result from an abnormality of immunoregulatory mechanisms. Observations made in both human autoimmune disease and animal models suggest that defects in the generation and expression of regulatory T cell (Treg) activity may allow the production of autoimmunity. It has recently been appreciated that the IPEX (immunodysregulation, polyendocrinopathy, enteropathy X-linked) syndrome results from the failure to express the FOXP3 gene, which encodes a molecule critical in the differentiation of Tregs. Administration of normal Tregs or of factors derived from them can prevent the development of autoimmune disease in rodent models of autoimmunity, and allogeneic stem cell transplantation ameliorates human IPEX. Abnormalities in the function of Tregs have been noted in a number of human autoimmune diseases, including rheumatoid arthritis and SLE, although it remains uncertain whether these functional abnormalities are causative or are secondary to inflammation. One of the mechanisms by which Tregs control immune/inflammatory responses is by the production of the cytokine IL-10. In this regard, children with a deficiency in the expression of IL-10 or the IL-10 receptor develop inflammatory bowel disease that mimics Crohn’s disease and that can be cured by allogeneic stem cell transplantation. Finally, recent data indicate that B cells may also exert regulatory function, largely through the production of IL-10. Deficiency of IL-10-producing regulatory B cells can prolong the course of multiple sclerosis in an animal model, and such cells are thought to be functionally diminished in human SLE.
It should be apparent that no single mechanism can explain all the varied manifestations of autoimmunity or autoimmune disease. Furthermore, genetic evaluation has shown that convergence of a number of abnormalities is often required for the induction of an autoimmune disease. Additional factors that appear to be important determinants in the induction of autoimmunity include age, sex (many autoimmune diseases are far more common in women), exposure to infectious agents, and environmental contacts. How all of these disparate factors affect the capacity to develop self-reactivity is currently being investigated intensively.
GENETIC CONSIDERATIONS
![]() Evidence in humans that there are susceptibility genes for autoimmunity comes from family studies and especially from studies of twins. Studies in type 1 diabetes mellitus, rheumatoid arthritis, multiple sclerosis, and SLE have shown that ~15–30% of pairs of monozygotic twins show disease concordance, whereas the figure is <5% for dizygotic twins. The occurrence of different autoimmune diseases within the same family has suggested that certain susceptibility genes may predispose to a variety of autoimmune diseases. Genome-wide association studies have begun to identify polymorphisms in individual genes that are associated with specific autoimmune diseases. More than 50 genetic polymorphisms associated with one or more autoimmune diseases have been identified to date. It is notable that some genes are associated with multiple autoimmune diseases, whereas others are specifically associated with only one autoimmune condition. Moreover, recent genetic evidence suggests that clusters of genetic risk factors can commonly be found in groups of autoimmune diseases. Four general clusters have been identified: one group of 6 genetic polymorphisms most frequently associated with Crohn’s disease, psoriasis, and multiple sclerosis; a second cluster of 8 polymorphisms most strongly associated with celiac disease, rheumatoid arthritis, and SLE; a third cluster of 7 polymorphisms most strongly associated with type 1 diabetes, multiple sclerosis, and rheumatoid arthritis; and a fourth cluster of more than 12 polymorphisms most strongly associated with type 1 diabetes, rheumatoid arthritis, celiac disease, Crohn’s disease, and SLE. These results imply that autoimmune diseases with widely different clinical presentations and patterns of organ involvement could involve similar immunopathogenic pathways. For example, the same allele of the gene encoding PTPN22 is associated with multiple autoimmune diseases. Its product is a phosphatase expressed by a variety of hematopoietic cells that downregulates antigen receptor–mediated stimulation of T and B cells. The risk allele is associated with type 1 diabetes mellitus, rheumatoid arthritis, and SLE in some populations. The explanation for the association of this polymorphism with autoimmune disease is uncertain, but it is likely that it diminishes antigen receptor signaling during lymphocyte development, permitting escape of autoreactive clones or decreased activation-induced apoptosis of autoantigen-reactive lymphocytes in the periphery. In recent years, genome-wide association studies have demonstrated a variety of other genes that are involved in human autoimmune diseases. Most genes individually confer a relatively low risk for autoimmune diseases and are found in normal individuals. No gene has been identified that is essential for autoimmune diseases. In addition to this evidence from humans, certain inbred mouse strains reproducibly develop specific spontaneous or experimentally induced autoimmune diseases, whereas others do not. These findings have led to an extensive search for genes that determine susceptibility to autoimmune disease and for genes that might be protective.
Evidence in humans that there are susceptibility genes for autoimmunity comes from family studies and especially from studies of twins. Studies in type 1 diabetes mellitus, rheumatoid arthritis, multiple sclerosis, and SLE have shown that ~15–30% of pairs of monozygotic twins show disease concordance, whereas the figure is <5% for dizygotic twins. The occurrence of different autoimmune diseases within the same family has suggested that certain susceptibility genes may predispose to a variety of autoimmune diseases. Genome-wide association studies have begun to identify polymorphisms in individual genes that are associated with specific autoimmune diseases. More than 50 genetic polymorphisms associated with one or more autoimmune diseases have been identified to date. It is notable that some genes are associated with multiple autoimmune diseases, whereas others are specifically associated with only one autoimmune condition. Moreover, recent genetic evidence suggests that clusters of genetic risk factors can commonly be found in groups of autoimmune diseases. Four general clusters have been identified: one group of 6 genetic polymorphisms most frequently associated with Crohn’s disease, psoriasis, and multiple sclerosis; a second cluster of 8 polymorphisms most strongly associated with celiac disease, rheumatoid arthritis, and SLE; a third cluster of 7 polymorphisms most strongly associated with type 1 diabetes, multiple sclerosis, and rheumatoid arthritis; and a fourth cluster of more than 12 polymorphisms most strongly associated with type 1 diabetes, rheumatoid arthritis, celiac disease, Crohn’s disease, and SLE. These results imply that autoimmune diseases with widely different clinical presentations and patterns of organ involvement could involve similar immunopathogenic pathways. For example, the same allele of the gene encoding PTPN22 is associated with multiple autoimmune diseases. Its product is a phosphatase expressed by a variety of hematopoietic cells that downregulates antigen receptor–mediated stimulation of T and B cells. The risk allele is associated with type 1 diabetes mellitus, rheumatoid arthritis, and SLE in some populations. The explanation for the association of this polymorphism with autoimmune disease is uncertain, but it is likely that it diminishes antigen receptor signaling during lymphocyte development, permitting escape of autoreactive clones or decreased activation-induced apoptosis of autoantigen-reactive lymphocytes in the periphery. In recent years, genome-wide association studies have demonstrated a variety of other genes that are involved in human autoimmune diseases. Most genes individually confer a relatively low risk for autoimmune diseases and are found in normal individuals. No gene has been identified that is essential for autoimmune diseases. In addition to this evidence from humans, certain inbred mouse strains reproducibly develop specific spontaneous or experimentally induced autoimmune diseases, whereas others do not. These findings have led to an extensive search for genes that determine susceptibility to autoimmune disease and for genes that might be protective.
The strongest consistent association for susceptibility to autoimmune disease is with particular MHC alleles. It has been suggested that the association of MHC genotype with autoimmune disease relates to differences in the ability of different allelic variations of MHC molecules to present autoantigenic peptides to autoreactive T cells. An alternative hypothesis involves the role of MHC alleles in shaping the T cell receptor repertoire during T cell ontogeny in the thymus. In addition, specific MHC gene products may themselves be the source of peptides that can be recognized by T cells. Cross-reactivity between such MHC peptides and peptides derived from proteins produced by common microbes may trigger autoimmunity by molecular mimicry. However, MHC genotype alone does not determine the development of autoimmunity. Identical twins are far more likely to develop the same autoimmune disease than MHC-identical nontwin siblings; this observation suggests that genetic factors other than the MHC affect disease susceptibility. Studies of the genetics of type 1 diabetes mellitus, SLE, rheumatoid arthritis, and multiple sclerosis in humans and mice have identified several independently segregating disease susceptibility loci in addition to the MHC. Genes that encode molecules of the innate immune response are also involved in autoimmunity. In humans, inherited homozygous deficiency of the early proteins of the classic pathway of complement (C1q, C4, or C2) as well as genes involved in the type 1 interferon pathway are very strongly associated with the development of SLE.
IMMUNOPATHOGENIC MECHANISMS IN AUTOIMMUNE DISEASES
The mechanisms of tissue injury in autoimmune diseases can be divided into antibody-mediated and cell-mediated processes. Representative examples are listed in Table 377e-3.
|
MECHANISMS OF TISSUE DAMAGE IN AUTOIMMUNE DISEASE |
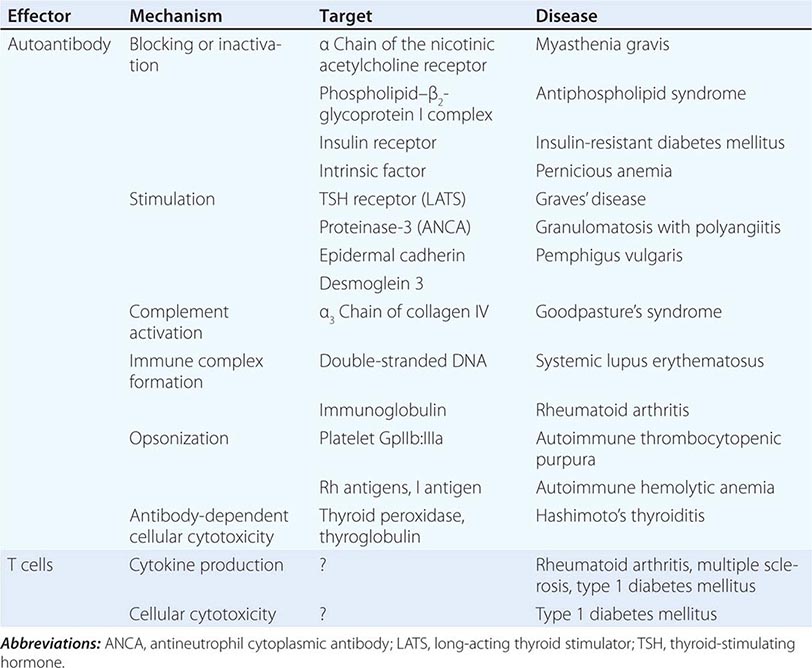
The pathogenicity of autoantibodies can be mediated through several mechanisms, including opsonization of soluble factors or cells, activation of an inflammatory cascade via the complement system, and interference with the physiologic function of soluble molecules or cells.
In autoimmune thrombocytopenic purpura, opsonization of platelets targets them for elimination by phagocytes. Likewise, in autoimmune hemolytic anemia, binding of immunoglobulin to red cell membranes leads to phagocytosis and lysis of the opsonized cell. Goodpasture’s syndrome, a disease characterized by lung hemorrhage and severe glomerulonephritis, represents an example of antibody binding leading to local activation of complement and neutrophil accumulation and activation. The autoantibody in this disease binds to the α3 chain of type IV collagen in the basement membrane. In SLE, activation of the complement cascade at sites of immunoglobulin deposition in renal glomeruli is considered to be a major mechanism of renal damage. Moreover, the DNA- and RNA-containing immune complexes in SLE activate TLR9 and TLR7, respectively, in dendritic cells and promote a proinflammatory, immunogenic milieu conducive to amplification of the autoimmune response.
Autoantibodies can also interfere with normal physiologic functions of cells or soluble factors. Autoantibodies to hormone receptors can lead to stimulation of cells or to inhibition of cell function through interference with receptor signaling. For example, long-acting thyroid stimulators—autoantibodies that bind to the receptor for thyroid-stimulating hormone (TSH)—are present in Graves’ disease and function as agonists, causing the thyroid to respond as if there were an excess of TSH. Alternatively, antibodies to the insulin receptor can cause insulin-resistant diabetes mellitus through receptor blockade. In myasthenia gravis, autoantibodies to the acetylcholine receptor can be detected in 85–90% of patients and are responsible for muscle weakness. The exact location of the antigenic epitope, the valence and affinity of the antibody, and perhaps other characteristics determine whether activation or blockade results from antibody binding.
Antiphospholipid antibodies are associated with thromboembolic events in primary and secondary antiphospholipid syndrome and have also been associated with fetal wastage. The major antibody is directed to the phospholipid–β2-glycoprotein I complex and appears to exert a procoagulant effect. In pemphigus vulgaris, autoantibodies bind to desmoglein 3, a component of the epidermal cell desmosome, and play a role in the induction of the disease. These antibodies exert their pathologic effect by disrupting cell–cell junctions through stimulation of the production of epithelial proteases, with consequent blister formation. Cytoplasmic antineutrophil cytoplasmic antibody (c-ANCA), found in granulomatosis with polyangiitis, is an antibody to an intracellular antigen, the 29-kDa serine protease (proteinase-3). In vitro experiments have shown that IgG anti-c-ANCA causes cellular activation and degranulation of primed neutrophils.
It is important to note that autoantibodies of a given specificity may cause disease only in genetically susceptible hosts, as has been shown in experimental models of myasthenia gravis, SLE, rheumatic fever, and rheumatoid arthritis. Furthermore, once organ damage is initiated, new inflammatory cascades are initiated that can sustain and amplify the autoimmune process. Finally, some autoantibodies seem to be markers for disease but have, as yet, no known pathogenic potential.
AUTOIMMUNE DISEASES
Manifestations of autoimmunity are found in a large number of pathologic conditions. However, their presence does not necessarily imply that the pathologic process is an autoimmune disease. A number of attempts to establish formal criteria for the classification of diseases as autoimmune have been made, but none is universally accepted. One set of criteria is shown in Table 377e-4; however, this scheme should be viewed merely as a guide in consideration of the problem.
|
HUMAN AUTOIMMUNE DISEASE: PRESUMPTIVE EVIDENCE FOR IMMUNOLOGIC PATHOGENESIS |
To classify a disease as autoimmune, it is necessary to demonstrate that the immune response to a self-antigen causes the observed pathology. Initially, the detection of antibodies to the affected tissue in the serum of patients suffering from various diseases was taken as evidence that these diseases had an autoimmune basis. However, such autoantibodies are also found when tissue damage is caused by trauma or infection and in these cases are secondary to tissue damage. Thus, autoimmunity must be shown to be pathogenic before a disease is categorized as autoimmune.
To confirm autoantibody pathogenicity, it may be possible to transfer disease to experimental animals by the administration of autoantibodies from a patient, with the subsequent development of pathology in the recipient similar to that seen in the patient. This scenario has been documented, for example, in Graves’ disease. Some autoimmune diseases can be transferred from mother to fetus and are observed in the newborn babies of diseased mothers. The symptoms of the disease in the newborn usually disappear as the levels of maternal antibody decrease. An exception, however, is congenital heart block, in which damage to the developing conducting system of the heart follows in utero transfer of anti-Ro antibody from the mother to the fetus. This antibody transfer can result in a permanent developmental defect in the heart.
In most situations, the critical factors that determine when the development of autoimmunity results in autoimmune disease have not been delineated. The relationship of autoimmunity to the development of autoimmune disease may be associated with the fine specificity of the antibodies or T cells or their specific effector capabilities. In many circumstances, a mechanistic understanding of the pathogenic potential of autoantibodies has not been established. In some autoimmune diseases, biased production of cytokines by helper T (TH) cells may play a role in pathogenesis. In this regard, T cells can differentiate into specialized effector cells that predominantly produce interferon γ (TH1), IL-4 (TH2), or IL-17 (TH17) or that provide help to B cells (T follicular helper, TFH) (Chap. 372e). TH1 cells facilitate macrophage activation and classic cell-mediated immunity, whereas TH2 cells are thought to have regulatory functions and are involved in the resolution of normal immune responses as well as in the development of responses to a variety of parasites. TH17 cells produce a number of inflammatory cytokines, including IL-17 and IL-22, and seem to be prominently involved in host resistance to certain fungal infections. TFH cells help B cells by constitutively producing IL-21. In a number of autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis, type 1 diabetes mellitus, and Crohn’s disease, there appears to be biased differentiation of TH1 and TH17 cells, with resultant organ damage. Studies suggest an accentuated differentiation of TH17 cells associated with animal models of inflammatory arthritis, whereas increased differentiation of TFH cells has been associated with animal models of SLE.
ORGAN-SPECIFIC VERSUS SYSTEMIC AUTOIMMUNE DISEASES
The spectrum of autoimmune diseases ranges from conditions specifically affecting a single organ to systemic disorders that involve many organs (Table 377e-5). Hashimoto’s autoimmune thyroiditis is an example of an organ-specific autoimmune disease (Chap. 405). In this disorder, a specific lesion in the thyroid is associated with infiltration of mononuclear cells and damage to follicular cells. Antibody to thyroid constituents can be demonstrated in nearly all cases. Other organ- or tissue-specific autoimmune disorders include pemphigus vulgaris, autoimmune hemolytic anemia, idiopathic thrombocytopenic purpura, Goodpasture’s syndrome, myasthenia gravis, and sympathetic ophthalmia. One important feature of some organ-specific autoimmune diseases is the tendency for overlap, such that an individual with one specific syndrome is more likely to develop a second syndrome. For example, there is a high incidence of pernicious anemia in individuals with autoimmune thyroiditis. More striking is the tendency for individuals with an organ-specific autoimmune disease to develop multiple other manifestations of autoimmunity without the development of associated organ pathology. Thus, as many as 50% of individuals with pernicious anemia have non-cross-reacting antibodies to thyroid constituents, whereas patients with myasthenia gravis may develop antinuclear antibodies, antithyroid antibodies, rheumatoid factor, antilymphocyte antibodies, and polyclonal hypergammaglobulinemia. Part of the explanation may relate to the genetic elements shared by individuals with these different diseases.
|
DISEASES ON THE AUTOIMMUNE SPECTRUM |
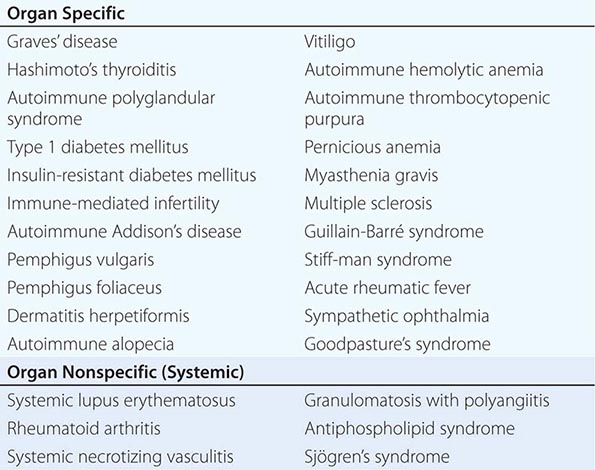
Systemic autoimmune diseases differ from organ-specific diseases in that pathologic lesions are found in multiple diverse organs and tissues. The hallmark of these conditions is the demonstration of associated relevant autoimmune manifestations that are likely to have an etiologic role in organ pathology. SLE represents the prototype of these disorders because of its abundant autoimmune manifestations. SLE is a disease of protean manifestations that characteristically involves the kidneys, joints, skin, serosal surfaces, blood vessels, and central nervous system (Chap. 378). The disease is associated with a vast array of autoantibodies whose production appears to be a part of a generalized hyperreactivity of the humoral immune system. Other features of SLE include generalized B cell hyperresponsiveness and polyclonal hypergammaglobulinemia. Current evidence suggests that both hypo- and hyperresponsiveness to antigen can lead to survival and activation of autoreactive B cells in SLE. The autoantibodies in SLE are thought to arise as part of an accentuated T cell–dependent B cell response since most pathogenic anti-DNA autoantibodies exhibit evidence of extensive somatic hypermutation.
378 Systemic Lupus Erythematosus
DEFINITION AND PREVALENCE
Systemic lupus erythematosus (SLE) is an autoimmune disease in which organs and cells undergo damage initially mediated by tissue-binding autoantibodies and immune complexes. In most patients, autoantibodies are present for a few years before the first clinical symptom appears. Ninety percent of patients are women of child-bearing years; people of all genders, ages, and ethnic groups are susceptible. Prevalence of SLE in the United States is 20 to 150 per 100,000 women depending on race and gender; highest prevalence is in African-American and Afro-Caribbean women, and lowest prevalence is in white men.
PATHOGENESIS AND ETIOLOGY
The proposed pathogenic mechanisms of SLE are illustrated in Fig. 378-1. Interactions between susceptibility genes and environmental factors result in abnormal immune responses, which vary between different patients. Those responses may include (1) activation of innate immunity (dendritic cells, monocyte/macrophages) by CpG DNA, DNA in immune complexes, viral DNA or RNA, and RNA in RNA/protein self-antigens; (2) lowered activation thresholds and abnormal activation pathways in adaptive immunity cells (mature T and B lymphocytes); (3) ineffective regulatory CD4+ and CD8+ T cells, B cells, and myeloid-derived suppressor cells; and (4) reduced clearance of immune complexes and apoptotic cells. Self-antigens (nucleosomal DNA/protein; RNA/protein in Sm, Ro, and La; phospholipids) are recognized by the immune system in surface blebs of apoptotic cells; thus autoantigens, autoantibodies, and immune complexes persist for prolonged periods of time, allowing inflammation and disease to develop. Immune cell activation is accompanied by increased secretion of proinflammatory type 1 and 2 interferons (IFNs), tumor necrosis factor α (TNF-α), interleukin (IL) 17 and B cell-maturation/survival cytokines B lymphocyte stimulator (BLyS/BAFF), and IL-10. Upregulation of genes induced by IFNs is a genetic “signature” in peripheral blood cells of 50–60% of SLE patients. Decreased production of other cytokines also contributes to SLE: lupus T and natural killer (NK) cells fail to produce enough IL-2 and transforming growth factor beta (TGF-β) to induce and sustain regulatory CD4+ and CD8+ T cells. The result of these abnormalities is sustained production of autoantibodies (referred to in Fig. 378-1 and described in Table 378-1) and immune complexes; pathogenic subsets bind target tissues, with activation of complement, leading to release of cytokines, chemokines, vasoactive peptides, oxidants, and proteolytic enzymes. This results in activation of multiple tissue cells (endothelial cells, tissue-fixed macrophages, mesangial cells, podocytes, renal tubular epithelial cells) and influx into target tissues of T and B cells, monocyte/macrophages, and dendritic cells. In the setting of chronic inflammation, accumulation of growth factors and products of chronic oxidation contribute to irreversible tissue damage, including fibrosis/sclerosis in glomeruli, arteries, lungs, and other tissues.
|
AUTOANTIBODIES IN SYSTEMIC LUPUS ERYTHEMATOSUS (SLE) |
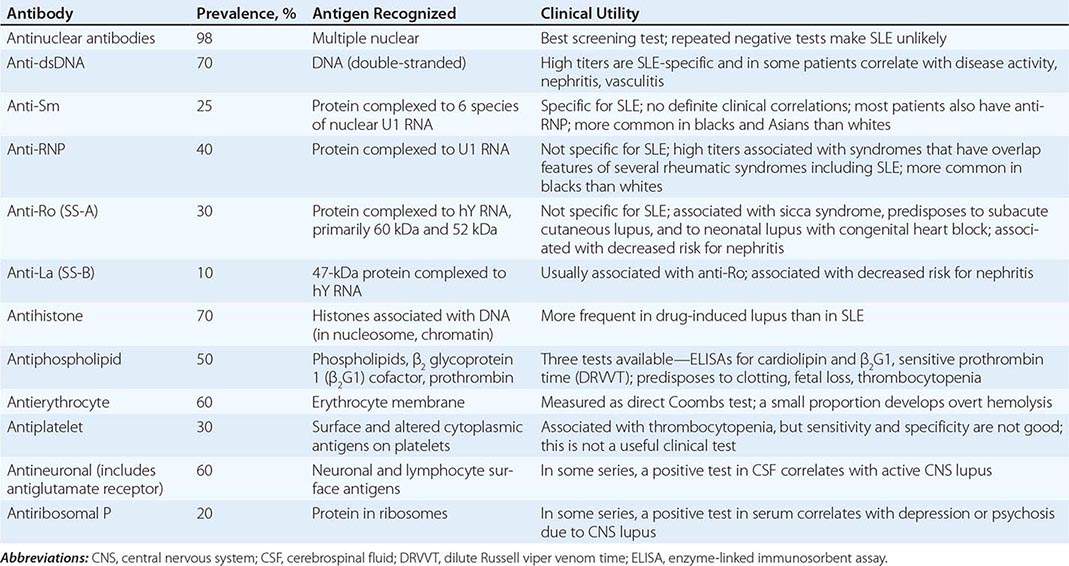
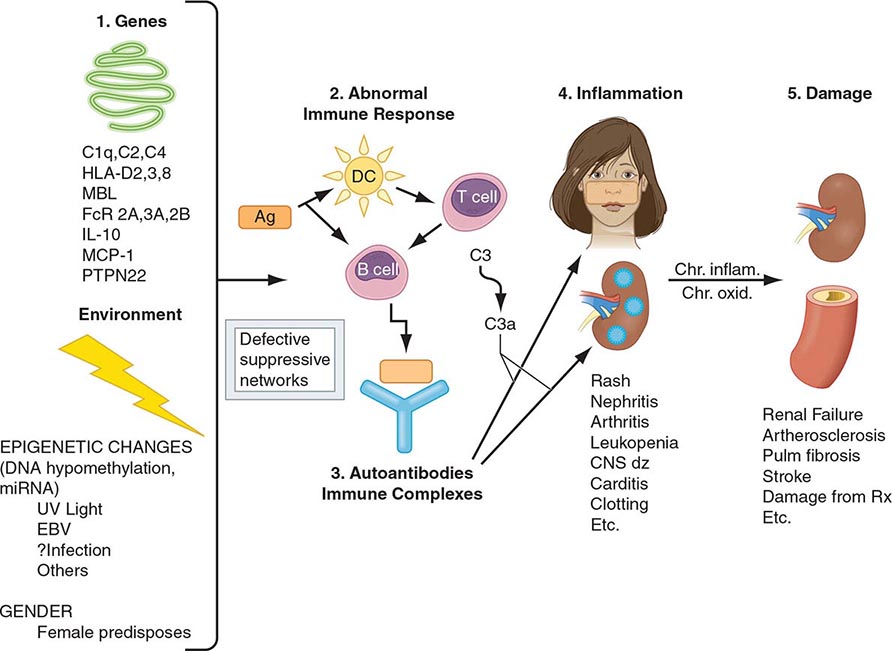
FIGURE 378-1 Pathogenesis of systemic lupus erythematosus (SLE). Genes confirmed in more than one genome-wide association analysis in northern European whites (several confirmed in Asians as well) as increasing susceptibility to SLE or lupus nephritis are listed (reviewed in SG Guerra et al: Arthritis Res Ther 14:211, 2012). Gene-environment interactions (reviewed in KH Costenbader et al: Autoimmune Rev 11:604, 2012) result in abnormal immune responses that generate pathogenic autoantibodies and immune complexes that deposit in tissue, activate complement, cause inflammation, and over time lead to irreversible organ damage (reviewed in GC Tsokos: N Engl J Med 365:2110, 2011; and BH Hahn, in DJ Wallace, BH Hahn [eds]: Dubois’ Lupus Erythematosus and Related Syndromes, 8th ed. New York, Elsevier, 2013). Ag, antigen; C1q, complement system; C3, complement component; CNS, central nervous system; DC, dendritic cell; EBV, Epstein-Barr virus; HLA, human leukocyte antigen; FcR, immunoglobulin Fc-binding receptor; IL, interleukin; MCP, monocyte chemotactic protein; PTPN, phosphotyrosine phosphatase; UV, ultraviolet.
SLE is a multigenic disease. Rare single-gene defects confer high hazard ratios (HRs) for SLE (5 to 25), including homozygous deficiencies of early components of complement (C1q,r,s; C2; C4) and a mutation in TREX1 on the × chromosome. In most genetically susceptible individuals, normal alleles of multiple genes each contribute a small amount to abnormal immune/inflammation/tissue damage responses; if enough predisposing variations are present, disease results. Approximately 45 predisposing genes (examples listed in Fig. 378-1) have been identified in recent genome-wide association studies in different racial groups. Individually, they confer an HR for SLE of 1.5–3 and account for approximately 18% of disease susceptibility, suggesting that environmental exposures and epigenetics play major roles. Predisposing, antigen-presenting human leukocyte antigen (HLA) molecules are most commonly found, in multiple ethnic groups (HLA DRB1 *0301 and *1501, as well as multiple genes across the major histocompatibility complex (MHC) 120-gene region). Other genetic factors in whites include innate immunity pathway gene polymorphisms, especially associated with IFN-α (STAT4, IRF5, IRAK1, TNFAIP3, PTPN22), genes in lymphocyte signaling pathways (PTPN22, PDCD-1, Ox40L, BANK-1, LYN, BLK), genes that affect clearance of apoptotic cells or immune complexes (C1q, FCRGIIA, FCRGIIIA, CRP, ITGAM), genes that influence neutrophil adherence (ITGAM), and genes that influence DNA repair (TREX-1). Some polymorphisms influence clinical manifestations; such as single nucleotide polymorphisms (SNPs) of STAT4 that associate with severe disease, anti-DNA, nephritis, and antiphospholipid syndrome, and an allele of FCGRIIA encoding a receptor that binds immune complexes poorly and predisposes to nephritis. Some gene effects are in promoter regions (e.g., IL-10), and others are conferred by copy numbers (e.g., C4A). In addition to genome-encoded susceptibility and protective genes, the influence of certain microRNAs (miRNAs) on gene transcription, as well as posttranscriptional epigenetic modification of DNA (which is hypomethylated in T cells of SLE patients), probably play major roles in disease susceptibility.
Some gene polymorphisms contribute to several autoimmune diseases, such as STAT4 and CTLA4. All of these gene polymorphisms/transcription/epigenetic combinations influence immune responses to the external and internal environment; when such responses are too high and/or too prolonged and/or inadequately regulated, autoimmune disease results.
Female sex is permissive for SLE with evidence for hormone effects, genes on the × chromosome, and epigenetic differences between genders playing a role. Females of many mammalian species make higher antibody responses than males. Women exposed to estrogen-containing oral contraceptives or hormone replacement have an increased risk of developing SLE (1.2- to 2-fold). Estradiol binds to receptors on T and B lymphocytes, increasing activation and survival of those cells, thus favoring prolonged immune responses. Genes on the × chromosome that influence SLE, such as TREX-1, may play a role in gender predisposition, possibly because some genes on the second × in females are not silent. People with XXY karyotype (Klinefelter’s syndrome) have a significantly increased risk for SLE.
Several environmental stimuli may influence SLE (Fig. 378-1). Exposure to ultraviolet light causes flares of SLE in approximately 70% of patients, possibly by increasing apoptosis in skin cells or by altering DNA and intracellular proteins to make them antigenic. Some infections induce normal immune responses that involve certain T and B cells that recognize self-antigens; such cells are not appropriately regulated, and autoantibody production occurs. Most SLE patients have autoantibodies for 3 years or more before the first symptoms of disease, suggesting that regulation controls the degree of autoimmunity for years before quantities and qualities of autoantibodies and pathogenic B and T cells cause clinical disease. Epstein-Barr virus (EBV) may be one infectious agent that can trigger SLE in susceptible individuals. Children and adults with SLE are more likely to be infected by EBV than age-, sex-, and ethnicity-matched controls. EBV contains amino acid sequences that mimic sequences on human spliceosomes (RNA/protein antigens) often recognized by autoantibodies in people with SLE. Current tobacco smoking increases risk for SLE (odds ratio [OR] 1.5). Prolonged occupational exposure to silica (e.g., inhalation of soap powder dust or soil in farming activities) increases risk (OR 4.3) in African-American women. Thus, interplay between genetic susceptibility, environment, gender, and abnormal immune responses results in autoimmunity (Chap. 377e).
PATHOLOGY
In SLE, biopsies of affected skin show deposition of Ig at the dermal-epidermal junction (DEJ), injury to basal keratinocytes, and inflammation dominated by T lymphocytes in the DEJ and around blood vessels and dermal appendages. Clinically unaffected skin may also show Ig deposition at the DEJ.
In renal biopsies, the pattern and severity of injury are important in diagnosis and in selecting the best therapy. Most recent clinical studies of lupus nephritis have used the International Society of Nephrology (ISN) and the Renal Pathology Society (RPS) classification (Table 378-2). In the ISN/RPS classification, the addition of “a” for active and “c” for chronic changes gives physicians information regarding the potential reversibility of disease. The system focuses on glomerular disease, although the presence of tubular interstitial and vascular disease is important to clinical outcomes. In general, class III and IV disease, as well as class V accompanied by III or IV disease, should be treated with aggressive immunosuppression if possible, because there is a high risk for end-stage renal disease (ESRD) if patients are untreated or undertreated. In contrast, treatment for lupus nephritis is not recommended in patients with class I or II disease or with extensive irreversible changes. In the recent Systemic Lupus International Collaborating Clinic (SLICC) criteria for classification of SLE, a diagnosis can be established on the basis of renal histology without meeting additional criteria (Table 378-3).
|
CLASSIFICATION OF LUPUS NEPHRITIS (INTERNATIONAL SOCIETY OF NEPHROLOGY AND RENAL PATHOLOGY SOCIETY) |
Note: Indicate and grade (mild, moderate, severe) tubular atrophy, interstitial inflammation and fibrosis, and severity of arteriosclerosis or other vascular lesions.
Source: JJ Weening et al: Kidney Int 65:521, 2004. Reprinted by permission from Macmillan Publishers Ltd., Copyright 2004.
|
SYSTEMIC LUPUS INTERNATIONAL COLLABORATING CLINIC CRITERIA FOR CLASSIFICATION OF SYSTEMIC LUPUS ERYTHEMATOSUS |
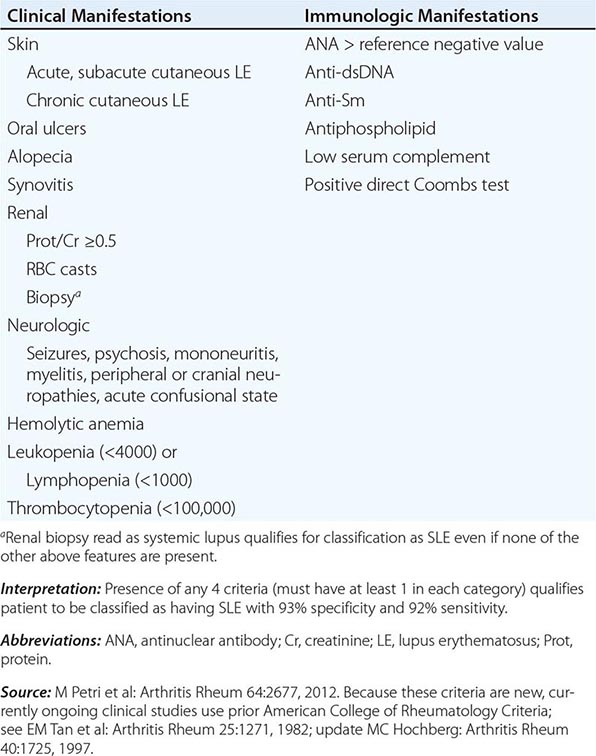
Histologic abnormalities in blood vessels may also determine therapy. Patterns of vasculitis are not specific for SLE but may indicate active disease: leukocytoclastic vasculitis is most common (Chap. 385).
Lymph node biopsies are usually performed to rule out infection or malignancies. In SLE, they show nonspecific diffuse chronic inflammation.
DIAGNOSIS
The diagnosis of SLE is based on characteristic clinical features and autoantibodies. Current criteria for classification are listed in Table 378-3, and an algorithm for diagnosis and initial therapy is shown in Fig. 378-2. The criteria are intended for confirming the diagnosis of SLE in patients included in studies; the author uses them in individual patients for estimating the probability that a disease is SLE. Any combination of four or more criteria, with at least one in the clinical and one in the immunologic category, well documented at any time during an individual’s history, makes it likely that the patient has SLE. (Specificity and sensitivity are ~93% and ~92%, respectively.) In many patients, criteria accrue over time. Antinuclear antibodies (ANA) are positive in >98% of patients during the course of disease; repeated negative tests by immunofluorescent methods suggest that the diagnosis is not SLE, unless other autoantibodies are present (Fig. 378-2). High-titer IgG antibodies to double-stranded DNA and antibodies to the Sm antigen are both specific for SLE and, therefore, favor the diagnosis in the presence of compatible clinical manifestations. The presence in an individual of multiple autoantibodies without clinical symptoms should not be considered diagnostic for SLE, although such persons are at increased risk.
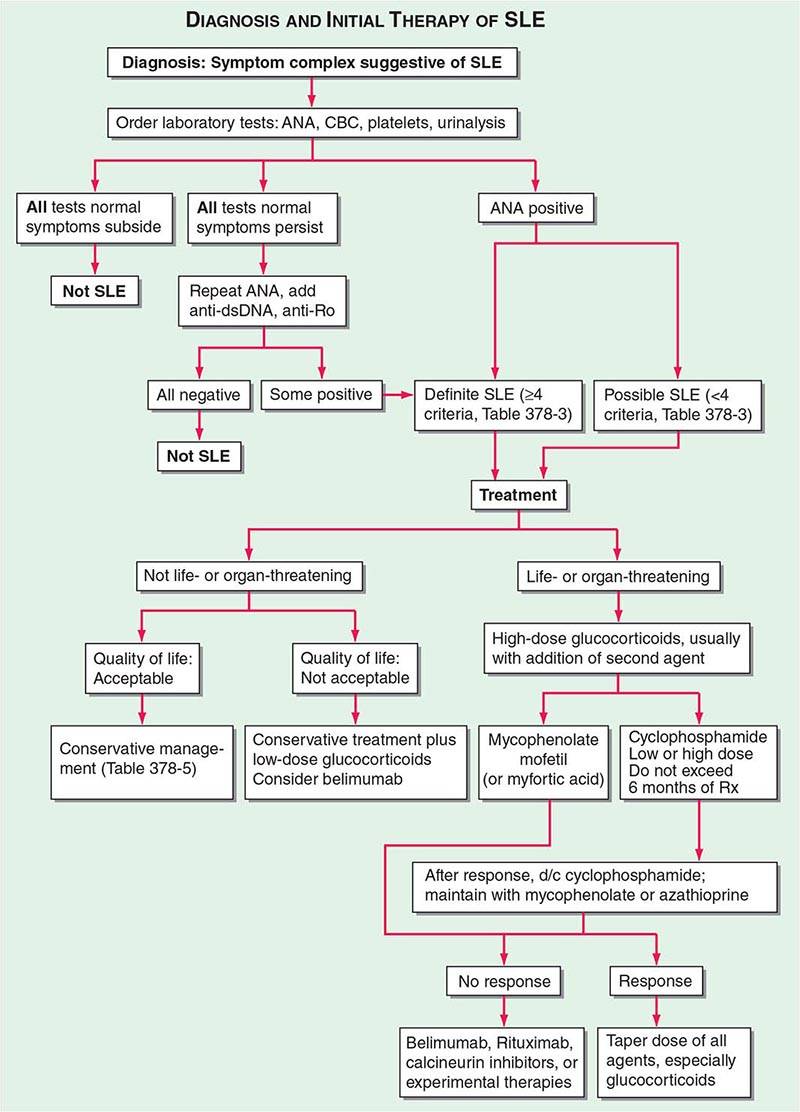
FIGURE 378-2 Algorithm for diagnosis and initial therapy of systemic lupus erythematosus (SLE). For guidelines on management of lupus and lupus nephritis, see BH Hahn et al: Arthritis Care Res (Hoboken) 64:797, 2012; GK Bertsias et al: Ann Rheum Dis 71:1771, 2012; and G Bertsias et al: Ann Rheum Dis 67:195, 2008. For details on mycophenolate and cyclophosphamide induction and maintenance therapies, see L Henderson et al: Cochrane Database Syst Rev 12:CD002922, 2012; Z Touma et al: J Rheumatol 38:69, 2011; EM Ginzler et al: Arthritis Rheum 62:211, 2010; FA Houssiau et al: Ann Rheum Dis 69:61, 2010; and MA Dooley et al: N Engl J Med 365:1886, 2011. For belimumab in treatment, see BH Hahn: N Eng J Med 368:1528, 2013. For rituximab, see L Lightstone: Lupus 22:390, 2013; and BH Rovin et al: Arthritis Rheum 64:1215, 2012. ANA, antinuclear antibodies; CBC, complete blood count.
INTERPRETATION OF CLINICAL MANIFESTATIONS
When a diagnosis of SLE is made, it is important to establish the severity and potential reversibility of the illness and to estimate the possible consequences of various therapeutic interventions. In the following paragraphs, descriptions of some disease manifestations begin with relatively mild problems and progress to those more life-threatening.
OVERVIEW AND SYSTEMIC MANIFESTATIONS
At its onset, SLE may involve one or several organ systems; over time, additional manifestations may occur (Tables 378-3 and 378-4). Most of the autoantibodies characteristic of each person are present at the time clinical manifestations appear (Tables 378-1 and 378-3). Severity of SLE varies from mild and intermittent to severe and fulminant. Approximately 85% of patients have either continuing active disease (while being treated) or one or more flares of active disease annually. Permanent complete remissions (absence of symptoms with no treatment) are rare. Systemic symptoms, particularly fatigue and myalgias/arthralgias, are present most of the time. Severe systemic illness requiring glucocorticoid therapy can occur with fever, prostration, weight loss, and anemia with or without other organ-targeted manifestations.
|
CLINICAL MANIFESTATIONS OF SLE AND PREVALENCE OVER THE ENTIRE COURSE OF DISEASEa |
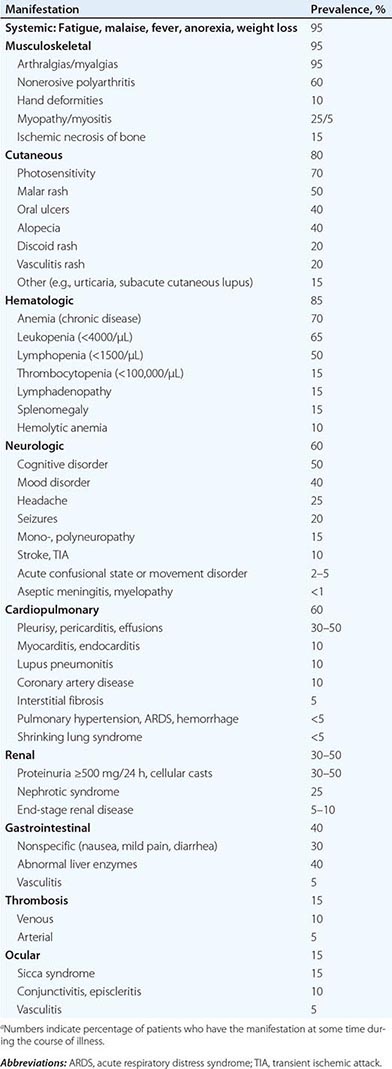
MUSCULOSKELETAL MANIFESTATIONS
Most people with SLE have intermittent polyarthritis, varying from mild to disabling, characterized by soft tissue swelling and tenderness in joints and/or tendons, most commonly in hands, wrists, and knees. Joint deformities (hands and feet) develop in only 10%. Erosions on joint x-rays are rare but can be identified by ultrasound in almost half of patients. Some individuals have rheumatoid-like arthritis with erosions and fulfill criteria for both RA and SLE (“rhupus”); they may be coded as having both diseases. If pain persists in a single joint, such as knee, shoulder, or hip, a diagnosis of ischemic necrosis of bone should be considered, particularly if there are no other manifestations of active SLE because its prevalence is increased in SLE, especially in patients treated with systemic glucocorticoids. Myositis with clinical muscle weakness, elevated creatine kinase levels, positive magnetic resonance imaging (MRI) scan, and muscle necrosis and inflammation on biopsy can occur, although most patients have myalgias without frank myositis. Glucocorticoid therapies (commonly) and antimalarial therapies (rarely) can cause muscle weakness; these adverse effects must be distinguished from active inflammatory disease.
CUTANEOUS MANIFESTATIONS
Lupus dermatitis can be classified as acute, subacute, or chronic, and there are many different types of lesions encompassed within these groups. Discoid lupus erythematosus (DLE) is the most common chronic dermatitis in lupus; lesions are roughly circular with slightly raised, scaly hyperpigmented erythematous rims and depigmented, atrophic centers in which all dermal appendages are permanently destroyed. Lesions can be disfiguring, particularly on the face and scalp. Treatment consists primarily of topical or locally injected glucocorticoids and systemic antimalarials. Only 5% of people with DLE have SLE (although half have positive ANA); however, among individuals with SLE, as many as 20% have DLE. The most common acute SLE rash is a photosensitive, slightly raised erythema, occasionally scaly, on the face (particularly the cheeks and nose—the “butterfly” rash), ears, chin, V region of the neck and chest, upper back, and extensor surfaces of the arms. Worsening of this rash often accompanies flare of systemic disease. Subacute cutaneous lupus erythematosus (SCLE) consists of scaly red patches similar to psoriasis, or circular flat red-rimmed lesions. Patients with these manifestations are exquisitely photosensitive; most have antibodies to Ro (SS-A). Other SLE rashes include recurring urticaria, lichen planus-like dermatitis, bullae, and panniculitis (“lupus profundus”). Rashes can be minor or severe; they may be the major disease manifestation. Small ulcerations on the oral or nasal mucosa are common in SLE; the lesions resemble aphthous ulcers.
RENAL MANIFESTATIONS
Nephritis is usually the most serious manifestation of SLE, particularly because nephritis and infection are the leading causes of mortality in the first decade of disease. Because nephritis is asymptomatic in most lupus patients, urinalysis should be ordered in any person suspected of having SLE. The classification of lupus nephritis is primarily histologic (see “Pathology,” above, and Table 378-2). Renal biopsy is recommended for every SLE patient with any clinical evidence of nephritis; results are used to plan current and near-future therapies. Patients with dangerous proliferative forms of glomerular damage (ISN III and IV) usually have microscopic hematuria and proteinuria (>500 mg per 24 h); approximately one-half develop nephrotic syndrome, and most develop hypertension. If diffuse proliferative glomerulonephritis (DPGN) is inadequately treated, virtually all patients develop ESRD within 2 years of diagnosis. Therefore, aggressive immunosuppression is indicated (usually systemic glucocorticoids plus a cytotoxic drug), unless damage is irreversible (Fig. 378-2, Table 378-5). African Americans are more likely to develop ESRD than are whites, even with the most current therapies. Overall in the United States, ~20% of individuals with lupus DPGN die or develop ESRD within 10 years of diagnosis. Such individuals require aggressive control of SLE and of the complications of renal disease and of therapy. Approximately 20% of SLE patients with proteinuria (usually nephrotic) have membranous glomerular changes without proliferative changes on renal biopsy. Their outcome is better than for those with DPGN, but patients with class V and nephrotic range proteinuria should be treated in the same way as those with classes III or IV proliferative disease. Lupus nephritis tends to be an ongoing disease, with flares requiring re-treatment or increased treatment over many years. For most people with lupus nephritis, accelerated atherosclerosis becomes important after several years of disease; attention must be given to control of systemic inflammation, blood pressure, hyperlipidemia, and hyperglycemia.
|
MEDICATIONS FOR THE MANAGEMENT OF SLE |
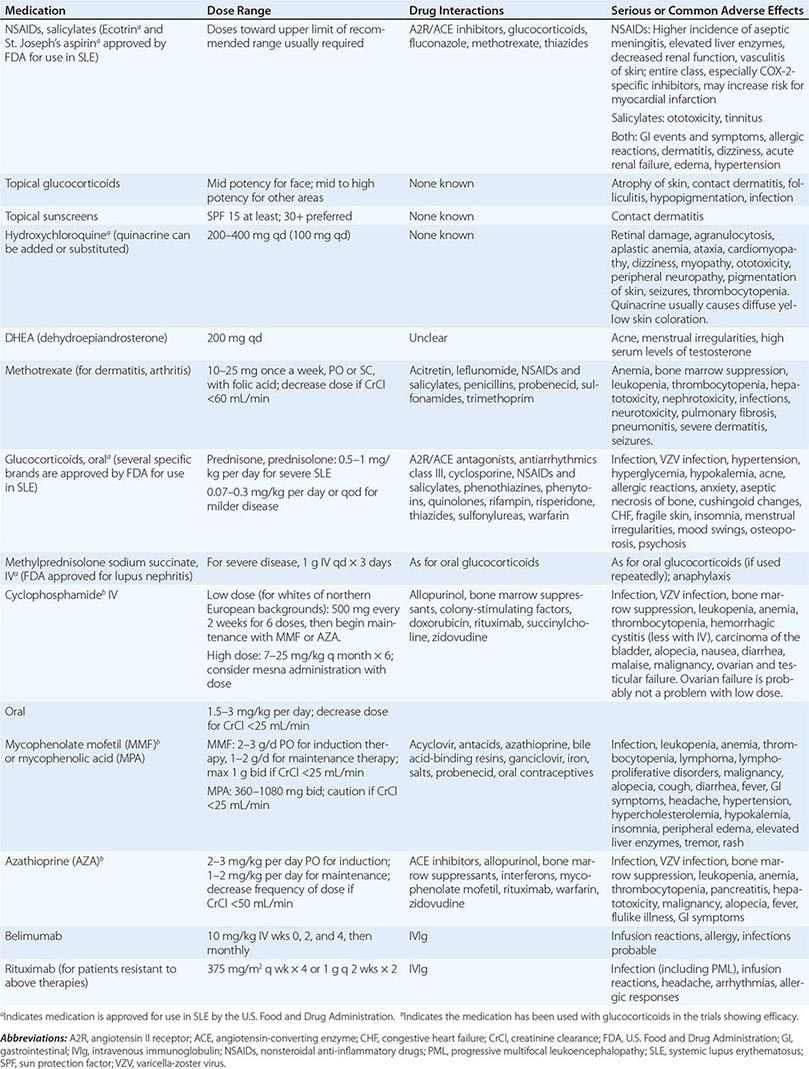
NERVOUS SYSTEM MANIFESTATIONS
There are many central nervous system (CNS) and peripheral nervous system manifestations of SLE; in some patients, these are the major cause of morbidity and mortality. It is useful to approach this diagnostically by asking first whether the symptoms result from SLE or another condition (such as infection in immunosuppressed individuals or side effects of therapies). If symptoms are related to SLE, it should be determined whether they are caused by a diffuse process (requiring immunosuppression) or vascular occlusive disease (requiring anticoagulation). The most common manifestation of diffuse CNS lupus is cognitive dysfunction, including difficulties with memory and reasoning. Headaches are also common. When excruciating, they often indicate SLE flare; when milder, they are difficult to distinguish from migraine or tension headaches. Seizures of any type may be caused by lupus; treatment often requires both antiseizure and immunosuppressive therapies. Psychosis can be the dominant manifestation of SLE; it must be distinguished from glucocorticoid-induced psychosis. The latter usually occurs in the first weeks of glucocorticoid therapy, at daily doses of ≥40 mg of prednisone or equivalent; psychosis resolves over several days after glucocorticoids are decreased or stopped. Myelopathy is not rare and is often disabling; rapid initiation of immunosuppressive therapy starting with high-dose glucocorticoids is standard of care.
VASCULAR OCCLUSIONS
The prevalence of transient ischemic attacks, strokes, and myocardial infarctions is increased in patients with SLE. These vascular events are increased in, but not exclusive to, SLE patients with antibodies to phospholipids (antiphospholipid antibodies), which are associated with hypercoagulability and acute thrombotic events (Chap. 379). Chronic SLE with or without antiphospholipid antibodies is associated with accelerated atherosclerosis. Ischemia in the brain can be caused by focal occlusion (either noninflammatory or associated with vasculitis) or by embolization from carotid artery plaque or from fibrinous vegetations of Libman-Sacks endocarditis. Appropriate tests for antiphospholipid antibodies (see below) and for sources of emboli should be ordered in such patients to estimate the need for, intensity of, and duration of anti-inflammatory and/or anticoagulant therapies. In SLE, myocardial infarctions are primarily manifestations of accelerated atherosclerosis. The increased risk for vascular events is three- to tenfold overall, and is highest in women <49 years old. Characteristics associated with increased risk for atherosclerosis include older age, hypertension, dyslipidemia, dysfunctional proinflammatory high-density lipoproteins, repeated high scores for disease activity, high cumulative or daily doses of glucocorticoids, and high levels of homocysteine. When it is most likely that an event results from clotting, long-term anticoagulation is the therapy of choice. Two processes can occur at once—vasculitis plus bland vascular occlusions—in which case it is appropriate to treat with anticoagulation plus immunosuppression. Statin therapies reduce levels of low-density lipoproteins (LDL) in SLE patients; reduction of cardiac events by statins has been shown in SLE patients with renal transplants but not in other SLE cohorts to date.
PULMONARY MANIFESTATIONS
The most common pulmonary manifestation of SLE is pleuritis with or without pleural effusion. This manifestation, when mild, may respond to treatment with nonsteroidal anti-inflammatory drugs (NSAIDs); when more severe, patients require a brief course of glucocorticoid therapy. Pulmonary infiltrates also occur as a manifestation of active SLE and are difficult to distinguish from infection on imaging studies. Life-threatening pulmonary manifestations include interstitial inflammation leading to fibrosis, shrinking lung syndrome, and intraalveolar hemorrhage; all of these probably require early aggressive immunosuppressive therapy as well as supportive care.
CARDIAC MANIFESTATIONS
Pericarditis is the most frequent cardiac manifestation; it usually responds to anti-inflammatory therapy and infrequently leads to tamponade. More serious cardiac manifestations are myocarditis and fibrinous endocarditis of Libman-Sacks. The endocardial involvement can lead to valvular insufficiencies, most commonly of the mitral or aortic valves, or to embolic events. It has not been proven that glucocorticoid or other immunosuppressive therapies lead to improvement of lupus myocarditis or endocarditis, but it is usual practice to administer a trial of high-dose steroids along with appropriate supportive therapy for heart failure, arrhythmia, or embolic events. As discussed above, patients with SLE are at increased risk for myocardial infarction, usually due to accelerated atherosclerosis, which probably results from immune attack, chronic inflammation, and/or chronic oxidative damage to arteries.
HEMATOLOGIC MANIFESTATIONS
The most frequent hematologic manifestation of SLE is anemia, usually normochromic normocytic, reflecting chronic illness. Hemolysis can be rapid in onset and severe, requiring high-dose glucocorticoid therapy, which is effective in most patients. Leukopenia is also common and almost always consists of lymphopenia, not granulocytopenia; lymphopenia rarely predisposes to infections and by itself usually does not require therapy. Thrombocytopenia may be a recurring problem. If platelet counts are >40,000/μL and abnormal bleeding is absent, therapy may not be required. High-dose glucocorticoid therapy (e.g., 1 mg/kg per day of prednisone or equivalent) is usually effective for the first few episodes of severe thrombocytopenia. Recurring or prolonged hemolytic anemia or thrombocytopenia, or disease requiring an unacceptably high dose of daily glucocorticoids, should be treated with an additional strategy (see “Management of Systemic Lupus Erythematosus” below).
GASTROINTESTINAL MANIFESTATIONS
Nausea, sometimes with vomiting, and diarrhea can be manifestations of an SLE flare, as can diffuse abdominal pain probably caused by autoimmune peritonitis and/or intestinal vasculitis. Increases in serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are common when SLE is active. These manifestations usually improve promptly during systemic glucocorticoid therapy. Vasculitis involving the intestine may be life-threatening; perforations, ischemia, bleeding, and sepsis are frequent complications. Aggressive immunosuppressive therapy with high-dose glucocorticoids is recommended for short-term control; evidence of recurrence is an indication for additional therapies.
OCULAR MANIFESTATIONS
Sicca syndrome (Sjögren’s syndrome; Chap. 383) and nonspecific conjunctivitis are common in SLE and rarely threaten vision. In contrast, retinal vasculitis and optic neuritis are serious manifestations: blindness can develop over days to weeks. Aggressive immunosuppression is recommended, although there are no controlled trials to prove effectiveness. Complications of systemic and intraorbital glucocorticoid therapy include cataracts (common) and glaucoma.
LABORATORY TESTS
Laboratory tests serve (1) to establish or rule out the diagnosis; (2) to follow the course of disease, particularly to suggest that a flare is occurring or organ damage is developing; and (3) to identify adverse effects of therapies.
TESTS FOR AUTOANTIBODIES (TABLES 378-1 AND 378-3)
Diagnostically, the most important autoantibodies to detect are ANA because the test is positive in >95% of patients, usually at the onset of symptoms. A few patients develop ANA within 1 year of symptom onset; repeated testing may thus be useful. ANA tests using immunofluorescent methods are more reliable than enzyme-linked immunosorbent assays (ELISAs) and/or bead assays, which have less specificity. ANA-negative lupus exists but is rare in adults and is usually associated with other autoantibodies (anti-Ro or anti-DNA). High-titer IgG antibodies to double-stranded DNA (dsDNA) (but not to single-stranded DNA) are specific for SLE. ELISA and immunofluorescent reactions of sera with the dsDNA in the flagellate Crithidia luciliae have ~60% sensitivity for SLE; identification of high-avidity anti-dsDNA in the Farr assay is not as sensitive but may correlate better with risk for nephritis. Titers of anti-dsDNA vary over time. In some patients, increases in quantities of anti-dsDNA herald a flare, particularly of nephritis or vasculitis, especially when associated with declining levels of C3 or C4 complement. Antibodies to Sm are also specific for SLE and assist in diagnosis; anti-Sm antibodies do not usually correlate with disease activity or clinical manifestations. Antiphospholipid antibodies are not specific for SLE, but their presence fulfills one classification criterion, and they identify patients at increased risk for venous or arterial clotting, thrombocytopenia, and fetal loss. There are three widely accepted tests that measure different antibodies (anticardiolipin, anti-β2-glycoprotein, and the lupus anticoagulant). ELISA is used for anticardiolipin and anti-β2-glycoprotein (both internationally standardized with good reproducibility); a sensitive phospholipid-based activated prothrombin time such as the dilute Russell venom viper test is used to identify the lupus anticoagulant. The higher the titers of IgG anticardiolipin (>40 IU is considered high), and the greater the number of different antiphospholipid antibodies that are detected, the greater is the risk for a clinical episode of clotting. Quantities of antiphospholipid antibodies may vary markedly over time; repeated testing is justified if clinical manifestations of the antiphospholipid syndrome (APS) appear (Chap. 379). To classify a patient as having APS, with or without SLE, by international criteria requires the presence of one or more clotting episodes and/or repeated fetal losses plus at least two positive tests for antiphospholipid antibodies, at least 12 weeks apart; however, many patients with APS do not meet these stringent criteria, which are intended for inclusion of patients into studies.
An additional autoantibody test with predictive value (not used for diagnosis) detects anti-Ro/SS-A, which indicates increased risk for neonatal lupus, sicca syndrome, and SCLE. Women with child-bearing potential and SLE should be screened for antiphospholipid antibodies and anti-Ro, because both antibodies have the potential to cause fetal harm.
STANDARD TESTS FOR DIAGNOSIS
Screening tests for complete blood count, platelet count, and urinalysis may detect abnormalities that contribute to the diagnosis and influence management decisions.
TESTS FOR FOLLOWING DISEASE COURSE
It is useful to follow tests that indicate the status of organ involvement known to be present during SLE flares. These might include urinalysis for hematuria and proteinuria, hemoglobin levels, platelet counts, and serum levels of creatinine or albumin. There is great interest in identification of additional markers of disease activity. Candidates include levels of anti-DNA and anti-C1q antibodies, several components of complement (C3 is most widely available), activated complement products (including those that bind to the C4d receptor on erythrocytes), IFN-inducible gene expression in peripheral blood cells, serum levels of BLyS (B lymphocyte stimulator, also called BAFF), and urinary levels of TNF-like weak inducer of apoptosis (TWEAK), neutrophil gelatinase-associated lipocalin (NGAL), or monocyte chemotactic protein 1 (MCP-1). None is uniformly agreed upon as a reliable indicator of flare or of response to therapeutic interventions. It is likely that a panel of multiple proteins will be developed to predict both impending flare and response to recently instituted therapies. For now, the physician should determine for each patient whether certain laboratory test changes predict flare. If so, altering therapy in response to these changes may be advisable (30 mg of prednisone daily for 2 weeks has been shown to prevent flares in patients with rising anti-DNA plus falling complement). In addition, given the increased prevalence of atherosclerosis in SLE, it is advisable to follow the recommendations of the National Cholesterol Education Program for testing and treatment, including scoring of SLE as an independent risk factor, similar to diabetes mellitus.
MANAGEMENT OF SYSTEMIC LUPUS ERYTHEMATOSUS
There is no cure for SLE, and complete sustained remissions are rare. Therefore, the physician should plan to induce remissions of acute flares and then maintain improvements with strategies that suppress symptoms to an acceptable level and prevent organ damage. Usually patients will endure some adverse effects of medications. Therapeutic choices depend on (1) whether disease manifestations are life-threatening or likely to cause organ damage, justifying aggressive therapies; (2) whether manifestations are potentially reversible; and (3) the best approaches to preventing complications of disease and its treatments. Therapies, doses, and adverse effects are listed in Table 378-5.
CONSERVATIVE THERAPIES FOR MANAGEMENT OF NON-LIFE-THREATENING DISEASE
Among patients with fatigue, pain, and autoantibodies indicative of SLE, but without major organ involvement, management can be directed to suppression of symptoms. Analgesics and antimalarials are mainstays. NSAIDs are useful analgesics/anti-inflammatories, particularly for arthritis/arthralgias. However, two major issues indicate caution in using NSAIDs. First, SLE patients compared with the general population are at increased risk for NSAID-induced aseptic meningitis, elevated serum transaminases, hypertension, and renal dysfunction. Second, all NSAIDs, particularly those that inhibit cyclooxygenase-2 specifically, may increase risk for myocardial infarction. Acetaminophen to control pain may be a good strategy, but NSAIDs are more effective in some patients. The relative hazards of NSAIDs compared with low-dose glucocorticoid therapy have not been established. Antimalarials (hydroxychloroquine, chloroquine, and quinacrine) often reduce dermatitis, arthritis, and fatigue. A randomized, placebo-controlled, prospective trial has shown that withdrawal of hydroxychloroquine results in increased numbers of disease flares; hydroxychloroquine also reduces accrual of tissue damage, including renal damage, over time. Because of potential retinal toxicity, patients receiving antimalarials should undergo ophthalmologic examinations annually. A placebo-controlled prospective trial suggests that administration of dehydroepiandrosterone may reduce disease activity. If quality of life is inadequate despite these conservative measures, treatment with low doses of systemic glucocorticoids may be necessary. The clinician may also consider treatment with belimumab (anti-BLyS) in these patients, although published clinical trials enrolled patients who had failed to respond to conservative therapies. Lupus dermatitis should be managed with topical sunscreens, antimalarials, topical glucocorticoids, and/or tacrolimus, and if severe or unresponsive, systemic glucocorticoids with or without mycophenolate mofetil.
LIFE-THREATENING SLE: PROLIFERATIVE FORMS OF LUPUS NEPHRITIS
Guidelines for management of lupus nephritis have been published recently by the American College of Rheumatology and the European League Against Rheumatism (encompassed and referenced in Fig. 378-2 and Table 378-5). The mainstay of treatment for any inflammatory life-threatening or organ-threatening manifestations of SLE is systemic glucocorticoids (0.5–1 mg/kg per day PO or 500–1000 mg of methylprednisolone sodium succinate IV daily for 3 days followed by 0.5–1 mg/kg of daily prednisone or equivalent). Evidence that glucocorticoid therapy is life-saving comes from retrospective studies from the predialysis era; survival was significantly better in people with DPGN treated with high-dose daily glucocorticoids (40–60 mg of prednisone daily for 4–6 months) versus lower doses. Currently, high doses are recommended for much shorter periods; recent trials of interventions for severe SLE use 4–6 weeks of 0.5–1 mg/kg per day of prednisone or equivalent. Thereafter, doses are tapered as rapidly as the clinical situation permits, usually to a maintenance dose ranging from 5 to 10 mg of prednisone or equivalent per day. Most patients with an episode of severe SLE require many years of maintenance therapy with low-dose glucocorticoids, which can be increased to prevent or treat disease flares. Frequent attempts to gradually reduce the glucocorticoid requirement are recommended because virtually everyone develops important adverse effects (Table 378-5). High-quality clinical studies regarding initiating therapy for severe, active SLE with IV pulses of high-dose glucocorticoids are not available. Most recent clinical trials in lupus nephritis have initiated therapy with high-dose IV glucocorticoid pulses (500–1000 mg daily for 3–5 days). This approach must be tempered by safety considerations, such as the presence of conditions adversely affected by glucocorticoids (e.g., infection, hyperglycemia, hypertension, osteoporosis).
Cytotoxic/immunosuppressive agents added to glucocorticoids are recommended to treat serious SLE. Almost all prospective controlled trials in SLE involving cytotoxic agents have been conducted in combination with glucocorticoids in patients with lupus nephritis. Therefore, the following recommendations apply to treatment of nephritis. Either cyclophosphamide (an alkylating agent) or mycophenolate mofetil (a relatively lymphocyte-specific inhibitor of inosine monophosphatase and therefore of purine synthesis) is an acceptable choice for induction of improvement in severely ill patients; azathioprine (a purine analogue and cycle-specific antimetabolite) may be effective but is slower to influence response and associated with more flares. In patients whose renal biopsies show ISN grade III or IV disease, early treatment with combinations of glucocorticoids and cyclophosphamide reduces progression to ESRD and death. Shorter-term studies with glucocorticoids plus mycophenolate mofetil (prospective randomized trials of 6 months, follow-up studies of 36 months) show that this regimen is similar to cyclophosphamide in achieving improvement. Comparisons are complicated by effects of race, since higher proportions of African Americans (and other non-Asian, nonwhite races) respond to mycophenolate than to cyclophosphamide, whereas similar proportions of whites and Asians respond to each drug. Regarding toxicity, diarrhea is more common with mycophenolate mofetil; amenorrhea, leukopenia, and nausea are more common with cyclophosphamide. Importantly, rates of severe infections and death are similar in meta-analyses. Two different regimens of IV cyclophosphamide are available. For white patients with northern European backgrounds, low doses of cyclophosphamide (500 mg every 2 weeks for six total doses, followed by azathioprine or mycophenolate maintenance) are as effective as standard high doses, with less toxicity. Ten-year follow-up has shown no differences between the high-dose and low-dose groups (death or ESRD in 9–20% of patients in each group). The majority of the European patients were white; it is not clear whether the data apply to U.S. populations. High-dose cyclophosphamide (500–1000 mg/m2 body surface area given monthly IV for 6 months, followed by azathioprine or mycophenolate maintenance) is an acceptable approach for patients with severe nephritis (e.g., multiple cellular crescents and/or fibrinoid necrosis on renal biopsy, or rapidly progressive glomerulonephritis). Cyclophosphamide and mycophenolate responses begin 3–16 weeks after treatment is initiated, whereas glucocorticoid responses may begin within 24 h.
For maintenance therapy, mycophenolate and azathioprine probably are similar in efficacy and toxicity; both are safer than cyclophosphamide. In a recently published multicenter study, mycophenolate was superior to azathioprine in maintaining renal function and survival in patients who responded to induction therapy with either cyclophosphamide or mycophenolate. The incidence of ovarian failure, a common effect of high-dose cyclophosphamide therapy (but probably not of low-dose therapy), can be reduced by treatment with a gonadotropin-releasing hormone agonist (e.g., leuprolide 3.75 mg intramuscularly) prior to each monthly cyclophosphamide dose. Patients with high serum creatinine levels (e.g., ≥265 μmol/L [≥3.0 mg/dL]) many months in duration and high chronicity scores on renal biopsy are not likely to respond to any of these therapies. In general, it may be better to induce improvement in an African-American or Hispanic patient with proliferative glomerulonephritis with mycophenolate mofetil (2–3 g daily) rather than cyclophosphamide, with the option to switch if no evidence of response is detectable after 3–6 months of treatment. For whites and Asians, induction with either mycophenolate mofetil or cyclophosphamide is acceptable. Cyclophosphamide may be discontinued when it is clear that a patient is improving. The number of SLE flares is reduced by maintenance therapy with mycophenolate mofetil (1.5–2 g daily) or azathioprine (1–2.5 mg/kg per day). Both cyclophosphamide and mycophenolate mofetil are potentially teratogenic; patients should be off either medication for at least 3 months before attempting to conceive. Azathioprine can be used if necessary to control active SLE in patients who are pregnant. If azathioprine is used either for induction or maintenance therapy, patients may be prescreened for homozygous deficiency of the TMPT enzyme (which is required to metabolize the 6-mercaptopurine product of azathioprine) because they are at higher risk for bone marrow suppression.
Good improvement occurs in ~80% of lupus nephritis patients receiving either cyclophosphamide or mycophenolate at 1–2 years of follow-up. However, in some studies, at least 50% of these individuals have flares of nephritis over the next 5 years, and re-treatment is required; such individuals are more likely to progress to ESRD. Long-term outcome of lupus nephritis to most interventions is better in whites than in African Americans. Methotrexate (a folinic acid antagonist) may have a role in the treatment of arthritis and dermatitis but probably not in nephritis or other life-threatening disease. Small controlled trials (in Asia) of leflunomide, a relatively lymphocyte-specific pyrimidine antagonist licensed for use in rheumatoid arthritis, have suggested it can suppress disease activity in some SLE patients. Cyclosporine and tacrolimus, which inhibit production of IL-2 and T lymphocyte functions, have not been studied in prospective controlled trials in SLE in the United States; several studies in Asia have shown they are effective in lupus nephritis. Because they have potential nephrotoxicity but little bone marrow toxicity, the author uses them for periods of a few months in patients with steroid-resistant cytopenias of SLE or in steroid-resistant patients who have developed bone marrow suppression from standard cytotoxic agents.
Use of biologics directed against B cells for active SLE is under intense study. Use of anti-CD20 (rituximab), particularly in patients with SLE who are resistant to the more standard combination therapies discussed above, is controversial. Several open trials have shown efficacy in a majority of such patients, both for nephritis and for extrarenal lupus. However, recent prospective placebo-controlled randomized trials, one in renal and one in nonrenal SLE, did not show a difference between anti-CD20 and placebo when added to standard combination therapies. In contrast, recent trials of standard therapy plus belimumab (anti-BLyS, which binds soluble BLyS/BAFF, which is required for maturation of naïve and transitional B cells to plasma cells and memory B cells) showed improvement in 51% of SLE patients compared to 36% of those on placebo; these differences were statistically significant. The U.S. Food and Drug Administration (FDA) has approved belimumab for treatment of seropositive patients with SLE who have failed standard treatments. The belimumab trial did not include patients with active nephritis or CNS disease. Post hoc analyses have shown that the SLE patient most likely to respond to belimumab has fairly robust clinical activity (a Systemic Lupus Erythematosus Disease Activity Index [SLEDAI] score of ≥10), positive anti-DNA, and low serum complement. SLEDAI is a widely used measure of SLE disease activity; scores >3 reflect clinically active disease. At this time, it is useful to add belimumab to the therapeutic armamentarium in SLE, and it is clear that some patients benefit. However, its role in management of lupus nephritis is not yet known.
SPECIAL CONDITIONS IN SLE THAT MAY REQUIRE ADDITIONAL OR DIFFERENT THERAPIES
Crescentic Lupus Nephritis The presence of cellular or fibrotic crescents in glomeruli with proliferative glomerulonephritis indicates a worse prognosis than in patients without this feature. There are no large prospective multinational controlled trials showing efficacy of cyclophosphamide, mycophenolate, cyclosporine, or tacrolimus in such cases. Most authorities currently recommend that high-dose cyclophosphamide is the induction therapy of choice, in addition to high-dose glucocorticoids. One prospective trial from China showed superiority of mycophenolate to cyclophosphamide.
Membranous Lupus Nephritis Most SLE patients with membranous (INS-V) nephritis also have proliferative changes and should be treated for proliferative disease. However, some have pure membranous changes. Treatment for this group is less well defined. Some authorities do not recommend immunosuppression unless proteinuria is in the nephrotic range (although treatment with angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers is recommended). In those patients, recent prospective controlled trials suggest that alternate-day glucocorticoids plus cyclophosphamide or mycophenolate mofetil or cyclosporine are all effective in the majority of patients in reducing proteinuria. It is more controversial whether they preserve renal function over the long term.
Pregnancy and Lupus Fertility rates for men and women with SLE are probably normal. However, rate of fetal loss is increased (approximately two- to threefold) in women with SLE. Fetal demise is higher in mothers with high disease activity, antiphospholipid antibodies, and/or active nephritis. Suppression of disease activity can be achieved by administration of systemic glucocorticoids. A placental enzyme, 11-β-dehydrogenase 2, deactivates glucocorticoids; it is more effective in deactivating prednisone and prednisolone than the fluorinated glucocorticoids dexamethasone and betamethasone. Glucocorticoids are listed by the FDA as pregnancy category A (no evidence of teratogenicity in human studies); cyclosporine, tacrolimus, and rituximab are listed as category C (may be teratogenic in animals but no good evidence in humans); azathioprine, hydroxychloroquine, mycophenolate mofetil, and cyclophosphamide are category D (there is evidence of teratogenicity in humans, but benefits might outweigh risks in certain situations); and methotrexate is category × (risks outweigh benefits). Therefore, active SLE in pregnant women should be controlled with hydroxychloroquine and, if necessary, prednisone/prednisolone at the lowest effective doses for the shortest time required. Azathioprine may be added if these treatments do not suppress disease activity. Adverse effects of prenatal glucocorticoid exposure (primarily betamethasone) on offspring may include low birth weight, developmental abnormalities in the CNS, and predilection toward adult metabolic syndrome. It is likely that each of these glucocorticoids and immunosuppressive medications gets into breast milk, at least in low levels; patients should consider not breastfeeding if they need therapy for SLE. In SLE patients with antiphospholipid antibodies (on at least two occasions) and prior fetal losses, treatment with heparin (usually low-molecular-weight) plus low-dose aspirin has been shown in prospective controlled trials to increase significantly the proportion of live births; however, a recent prospective trial showed no differences in fetal outcomes in women taking aspirin compared to those taking aspirin plus low-molecular-weight heparin. An additional potential problem for the fetus is the presence of antibodies to Ro, sometimes associated with neonatal lupus consisting of rash and congenital heart block with or without cardiomyopathy. The cardiac manifestations can be life-threatening; therefore the presence of anti-Ro requires vigilant monitoring of fetal heart rates with prompt intervention (delivery if possible) if distress occurs. Recent evidence shows that hydroxychloroquine treatment of an anti-Ro-positive mother whose infant develops congenital heart block significantly reduces the chance that subsequent fetuses will develop heart block. There is some evidence that dexamethasone treatment of a mother in whom first- or second-degree heart block is detected in utero may sometimes prevent progression of heart block. Women with SLE usually tolerate pregnancy without disease flares. However, a small proportion develops severe flares requiring aggressive glucocorticoid therapy or early delivery. Poor maternal outcomes are highest in women with active nephritis or irreversible organ damage in kidneys, brain, or heart.
Lupus and Antiphospholipid Syndrome (APS) Patients with SLE who have venous or arterial clotting and/or repeated fetal losses and at least two positive tests for antiphospholipid antibodies have APS and should be managed with long-term anticoagulation (Chap. 379). A target international normalized ratio (INR) of 2.0–2.5 is recommended for patients with one episode of venous clotting; an INR of 3.0–3.5 is recommended for patients with recurring clots or arterial clotting, particularly in the CNS. Recommendations are based on both retrospective and prospective studies of posttreatment clotting events and adverse effects from anticoagulation.
Microvascular Thrombotic Crisis (Thrombotic Thrombocytopenic Purpura, Hemolytic-Uremic Syndrome) This syndrome of hemolysis, thrombocytopenia, and microvascular thrombosis in kidneys, brain, and other tissues carries a high mortality rate and occurs most commonly in young individuals with lupus nephritis. The most useful laboratory tests are identification of schistocytes on peripheral blood smears, elevated serum levels of lactate dehydrogenase, and antibodies to ADAMS13. Plasma exchange or extensive plasmapheresis is usually life-saving; most authorities recommend concomitant glucocorticoid therapy; there is no evidence that cytotoxic drugs are effective.
Lupus Dermatitis Patients with any form of lupus dermatitis should minimize exposure to ultraviolet light, using appropriate clothing and sunscreens with a sun protection factor of at least 30. Topical glucocorticoids and antimalarials (such as hydroxychloroquine) are effective in reducing lesion severity in most patients and are relatively safe. Systemic treatment with retinoic acid is a useful strategy in patients with inadequate improvement on topical glucocorticoids and antimalarials; adverse effects are potentially severe (particularly fetal abnormalities), and there are stringent reporting requirements for its use in the United States. Extensive, pruritic, bullous, or ulcerating dermatitides usually improve promptly after institution of systemic glucocorticoids; tapering may be accompanied by flare of lesions, thus necessitating use of a second medication such as hydroxychloroquine, retinoids, or cytotoxic medications such as methotrexate, azathioprine, or mycophenolate mofetil. In therapy-resistant lupus dermatitis there are reports of success with topical tacrolimus (caution must be exerted because of the possible increased risk for malignancies) or with systemic dapsone or thalidomide (the extreme danger of fetal deformities from thalidomide requires permission from and supervision by the supplier).
PREVENTIVE THERAPIES
Prevention of complications of SLE and its therapy include providing appropriate vaccinations (the administration of influenza and pneumococcal vaccines has been studied in patients with SLE; flare rates are similar to those receiving placebo) and suppressing recurrent urinary tract infections. Vaccination with attenuated live viruses is generally discouraged in patients who are immunosuppressed. Strategies to prevent osteoporosis should be initiated in most patients likely to require long-term glucocorticoid therapy and/or with other predisposing factors. Postmenopausal women can be protected from steroid-induced osteoporosis with either bisphosphonates or denosumab. Safety of long-term use of these strategies in premenopausal women is not well established. Control of hypertension and appropriate prevention strategies for atherosclerosis, including monitoring and treatment of dyslipidemias, management of hyperglycemia, and management of obesity, are recommended.
EXPERIMENTAL THERAPIES
Studies of highly targeted experimental therapies for SLE are in progress. They include targeting (1) activated B lymphocytes with anti-CD22 or TACI-Ig, (2) inhibition of IFN-α, (3) inhibition of B/T cell second signal coactivation with CTLA-Ig, (4) inhibition of innate immune activation via TLR7 or TLR7 and 9, (5) induction of regulatory T cells with peptides from immunoglobulins or autoantigens; (6) suppression of T cells, B cells, and monocyte/macrophages with laquinimod; and (7) inhibition of lymphocyte activation by blockade of Jak/Stat. A few studies have used vigorous untargeted immunosuppression with high-dose cyclophosphamide plus anti-T cell strategies, with rescue by transplantation of autologous hematopoietic stem cells for the treatment of severe and refractory SLE. One U.S. report showed an estimated mortality rate over 5 years of 15% and sustained remission in 50%. It is hoped that in the next edition of this text, we will be able to recommend more effective and less toxic approaches to treatment of SLE based on some of these strategies.
PATIENT OUTCOMES, PROGNOSIS, AND SURVIVAL
![]() Survival in patients with SLE in the United States, Canada, Europe, and China is approximately 95% at 5 years, 90% at 10 years, and 78% at 20 years. In the United States, African Americans and Hispanic Americans with a mestizo heritage have a worse prognosis than whites, whereas Africans in Africa and Hispanic Americans with a Puerto Rican origin do not. The relative importance of gene mixtures and environmental differences accounting for ethnic differences is not known. Poor prognosis (~50% mortality in 10 years) in most series is associated with (at the time of diagnosis) high serum creatinine levels (>124 μmol/L [>1.4 mg/dL]), hypertension, nephrotic syndrome (24-h urine protein excretion >2.6 g), anemia (hemoglobin <124 g/L [<12.4 g/dL]), hypoalbuminemia, hypocomplementemia, antiphospholipid antibodies, male sex, ethnicity (African American, Hispanic with mestizo heritage), and low socioeconomic status. Data regarding outcomes in SLE patients with renal transplants show mixed results: some series show a twofold increase in graft rejection compared to patients with other causes of ESRD, whereas others show no differences. Overall patient survival is comparable (85% at 2 years). Lupus nephritis occurs in approximately 10% of transplanted kidneys. Disability in patients with SLE is common due primarily to chronic fatigue, arthritis, and pain, as well as renal disease. As many as 25% of patients may experience remissions, sometimes for a few years, but these are rarely permanent. The leading causes of death in the first decade of disease are systemic disease activity, renal failure, and infections; subsequently, thromboembolic events become increasingly frequent causes of mortality.
Survival in patients with SLE in the United States, Canada, Europe, and China is approximately 95% at 5 years, 90% at 10 years, and 78% at 20 years. In the United States, African Americans and Hispanic Americans with a mestizo heritage have a worse prognosis than whites, whereas Africans in Africa and Hispanic Americans with a Puerto Rican origin do not. The relative importance of gene mixtures and environmental differences accounting for ethnic differences is not known. Poor prognosis (~50% mortality in 10 years) in most series is associated with (at the time of diagnosis) high serum creatinine levels (>124 μmol/L [>1.4 mg/dL]), hypertension, nephrotic syndrome (24-h urine protein excretion >2.6 g), anemia (hemoglobin <124 g/L [<12.4 g/dL]), hypoalbuminemia, hypocomplementemia, antiphospholipid antibodies, male sex, ethnicity (African American, Hispanic with mestizo heritage), and low socioeconomic status. Data regarding outcomes in SLE patients with renal transplants show mixed results: some series show a twofold increase in graft rejection compared to patients with other causes of ESRD, whereas others show no differences. Overall patient survival is comparable (85% at 2 years). Lupus nephritis occurs in approximately 10% of transplanted kidneys. Disability in patients with SLE is common due primarily to chronic fatigue, arthritis, and pain, as well as renal disease. As many as 25% of patients may experience remissions, sometimes for a few years, but these are rarely permanent. The leading causes of death in the first decade of disease are systemic disease activity, renal failure, and infections; subsequently, thromboembolic events become increasingly frequent causes of mortality.
DRUG-INDUCED LUPUS
This is a syndrome of positive ANA associated with symptoms such as fever, malaise, arthritis or intense arthralgias/myalgias, serositis, and/or rash. The syndrome appears during therapy with certain medications and biologic agents, is predominant in whites, has less female predilection than SLE, rarely involves kidneys or brain, is rarely associated with anti-dsDNA, is commonly associated with antibodies to histones, and usually resolves over several weeks after discontinuation of the offending medication. The list of substances that can induce lupus-like disease is long. Among the most frequent are the antiarrhythmics procainamide, disopyramide, and propafenone; the antihypertensive hydralazine; several angiotensin-converting enzyme inhibitors and beta blockers; the antithyroid propylthiouracil; the antipsychotics chlorpromazine and lithium; the anticonvulsants carbamazepine and phenytoin; the antibiotics isoniazid, minocycline, and nitrofurantoin (Macrodantin); the antirheumatic sulfasalazine; the diuretic hydrochlorothiazide; the antihyperlipidemics lovastatin and simvastatin; and IFNs and TNF inhibitors. ANA usually appears before symptoms; however, many of the medications mentioned above induce ANA in patients who never develop symptoms of drug-induced lupus. It is appropriate to test for ANA at the first hint of relevant symptoms and to use test results to help decide whether to withdraw the suspect agent.
379 |
Antiphospholipid Syndrome |
DEFINITIONS
Antiphospholipid syndrome (APS) is an autoantibody-mediated acquired thrombophilia characterized by recurrent arterial or venous thrombosis and/or pregnancy morbidity. The major autoantibodies detected in the patient’s sera are directed against phospholipid (PL)-binding plasma proteins, mainly against a 43-kDa plasma apolipoprotein known as β2 glycoprotein I (β2GPI) and prothrombin. The plasma concentration of β2GPI is 50–200 μg/mL. β2GPI consists of 326 amino acids arranged in five domains (I through V). Domain V forms a positively charged patch, suitable to interact with negatively charged PL. In plasma, β2GPI has a circular conformation with domain V binding to and concealing the B cell epitopes lying on domain I. Another group of antibodies termed lupus anticoagulant (LA) elongate clotting times in vitro; this elongation is not corrected by adding normal plasma to the detection system (Table 379-1). Patients with APS often possess antibodies recognizing Treponema pallidum PL/cholesterol complexes, which are detected as biologic false-positive serologic tests for syphilis (BFP-STS) and Venereal Disease Research Laboratory (VDRL) tests. APS may occur alone (primary) or in association with any other autoimmune disease (secondary). Catastrophic APS (CAPS) is defined as a rapidly progressive thromboembolic disease involving simultaneously three or more organs, organ systems, or tissues leading to corresponding functional defects.
|
CLASSIFICATION AND NOMENCLATURE OF ANTIPHOSPHOLIPID ANTIBODIES |
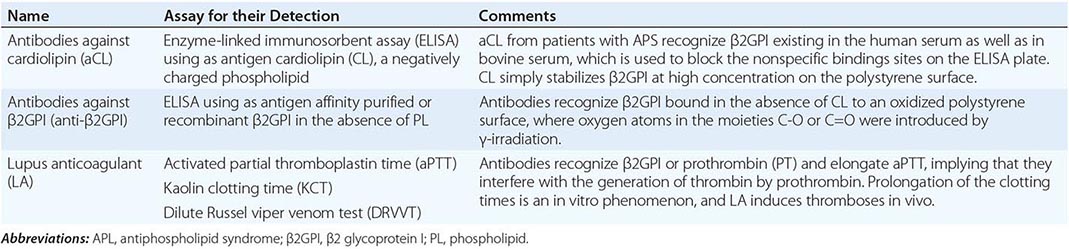
EPIDEMIOLOGY
Anti-PL (aPL)-binding plasma protein antibodies occur in 1–5% of the general population. Their prevalence increases with age; however, it is questionable whether they induce thrombotic events in elderly individuals. One-third of patients with systemic lupus erythematosus (SLE) (Chap. 378) possess these antibodies, whereas their prevalence in other autoimmune connective tissue disorders, such as systemic sclerosis (scleroderma), Sjögren’s syndrome, dermatomyositis, rheumatoid arthritis, and early undifferentiated connective tissue disease, ranges from 6 to 15%. One-third of aPL-positive individuals experience thrombotic events or pregnancy morbidity.
PATHOGENESIS
The trigger for the induction of antibodies to PL-binding proteins is not known. However infections, oxidative stress, major physical stresses such as surgery, and discontinuation of anticoagulant treatment may induce the exacerbation of the disease. Experimental data have shown that these phenomena are induced via (1) conformational changes of β2GPI either complexed with microbial antigens or dimerization through interaction with endothelial cell surface receptor annexin 2/TLR4, the platelet receptors apolipoprotein E receptor 2′ (apoER2′) and/or GPIb/IX/V receptor, and/or the chemokine platelet factor 4 (PF4); or (2) impaired defensive mechanisms such as reduced generation of endothelial nitric oxide synthase. Adherence of β2GPI to apoER2′, GPIb/IX/V receptor, and/or PF4 induces activation of endothelial cells, platelets, and monocytes. This process activates downstream pathways such as p38 mitogen-activated protein (p38 MAP) kinase and nuclear factor (NF)-κB, leading to the following events: secretion of proinflammatory cytokines, such as interleukin (IL) 1, IL-6, and IL-8; the expression of adhesion molecules; inhibition of cell-surface plasminogen activation; and expression of tissue factor. The above events change the phenotype of these cells to a prothrombotic form. In addition, anti-β2GPI antibodies induce fetal injury in mice through complement activation, as shown by the evidence that C4-deficient mice were protected from fetal injury.
CLINICAL MANIFESTATIONS AND LABORATORY FINDINGS
Clinical manifestations represent mainly a direct or indirect expression of venous or arterial thrombosis and/or pregnancy morbidity (Table 379-2). Clinical features associated with venous thrombosis are superficial and deep vein thrombosis, cerebral venous thrombosis, signs and symptoms of intracranial hypertension, retinal vein thrombosis, pulmonary emboli, pulmonary arterial hypertension, and Budd-Chiari syndrome. Livedo reticularis consists of a mottled reticular vascular pattern that appears as a lace-like, purplish discoloration of the skin. It is probably caused by swelling of the venules owing to obstruction of capillaries by thrombi. This clinical manifestation correlates with vascular lesions such as those in the central nervous system as well as aseptic bone necrosis. Arterial thrombosis is manifested as migraines, cognitive dysfunction, transient ischemic attacks, stroke, myocardial infarction, arterial thrombosis of upper and lower extremities, ischemic leg ulcers, digital gangrene, avascular necrosis of bone, retinal artery occlusion leading to painless transient vision loss, renal artery stenosis, and glomerular lesions, as well as infarcts of spleen, pancreas, and adrenals. Libman-Sacks endocarditis consists of very small vegetations, histologically characterized by organized platelet-fibrin microthrombi surrounded by growing fibroblasts and macrophages. Glomerular lesions are manifested with hypertension, mildly elevated serum creatinine levels, proteinuria, and mild hematuria. Histologically, these lesions are characterized in an acute phase by thrombotic microangiopathy involving glomerular capillaries, and in a chronic phase with fibrous intima hyperplasia, fibrous and/or fibrocellular occlusions of arterioles, and focal cortical atrophy (Table 379-2). Premature atherosclerosis has been recognized as a rare feature of APS. Coombs-positive hemolytic anemia and thrombocytopenia are laboratory findings associated with APS. Discontinuation of therapy, major surgery, infection, and trauma may trigger CAPS.
|
CLINICAL FEATURES OF ANTIPHOSPHOLIPID SYNDROME |
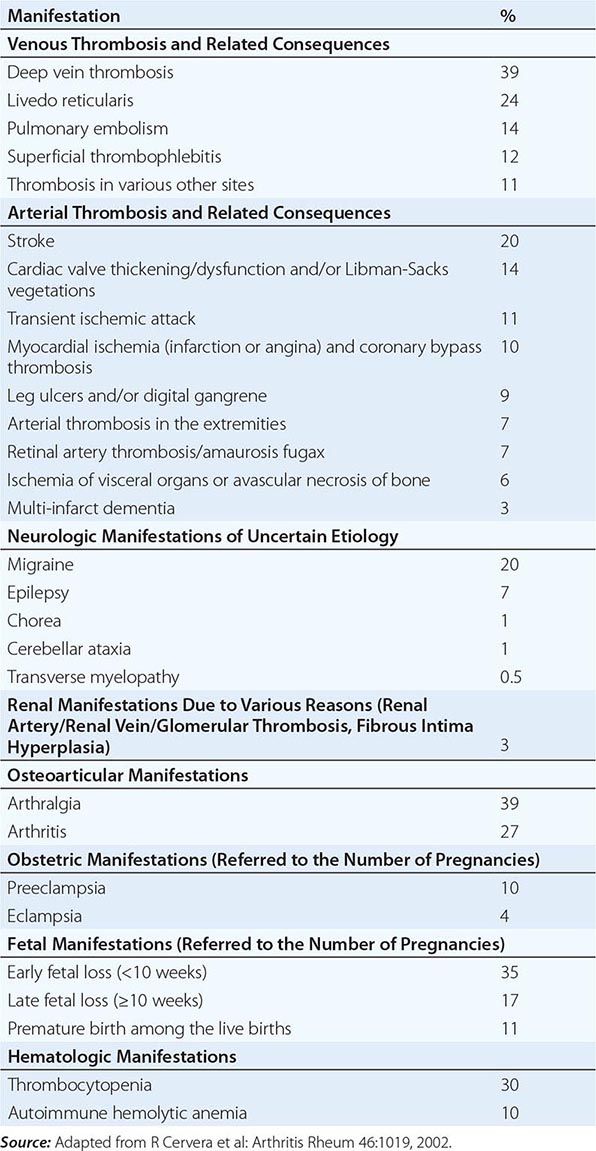
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
The diagnosis of APS should be seriously considered in cases of thrombosis, cerebral vascular accidents in individuals younger than 55 years of age, or pregnancy morbidity in the presence of livedo reticularis or thrombocytopenia. In these cases, aPL antibodies should be measured. The presence of at least one clinical and one laboratory criterion ensures the diagnosis even in the presence of other causes of thrombophilia. Clinical criteria include: (1) vascular thrombosis defined as one or more clinical episodes of arterial, venous, or small vessel thrombosis in any tissue or organ; and (2) pregnancy morbidity, defined as (a) one or more unexplained deaths of a morphologically normal fetus at or beyond the tenth week of gestation; (b) one or more premature births of a morphologically normal neonate before the thirty-fourth week of gestation because of eclampsia, severe preeclampsia, or placental insufficiency; or (c) three or more unexplained consecutive spontaneous abortions before the tenth week of gestation. Laboratory criteria include (1) LA, (2) anticardiolipin (aCL), and/or (3) anti-β2GPI antibodies, at intermediate or high titers on two occasions, 12 weeks apart.
Differential diagnosis is based on the exclusion of other inherited or acquired causes of thrombophilia (Chap. 141), Coombs-positive hemolytic anemia (Chap. 129), and thrombocytopenia (Chap. 140). Livedo reticularis with or without a painful ulceration on the lower extremities also may be a manifestation of disorders affecting (1) the vascular wall, such as polyarteritis nodosa, SLE, cryoglobulinemia, and lymphomas; or (2) the vascular lumen, such as myeloproliferative disorders, atherosclerosis, hypercholesterolemia, or other causes of thrombophilia.