Antimicrobial agents
After reading this chapter, the reader will be able to:
1. Define terms that pertain to antimicrobial agents
3. Describe the process involved in bacterial susceptibility testing
4. Discuss possible outcomes of antimicrobial combinations
5. List the various classes of the penicillins
6. List the various classes of the cephalosporins
7. Recognize similarities between members of the macrolides
8. Recognize similarities between members of the quinolones
9. List four mechanisms of action of antibacterials
10. List five commonly used antimycobacterials
11. Describe the commonly used azole antifungals and how they differ in spectrum of activity
12. Discuss similarities between members of the echinocandins
The antimicrobial properties of fermented beverages, moldy soybean curd, and spices were first described by the Chinese more than 2500 years ago.1 However, dissemination and acceptance of such knowledge did not take root until the early 1900s. The discovery of microscopic “little animals” by van Leeuwenhoek paved the way for Koch to validate that these “little animals,” or germs, caused disease. Before Koch’s work, almost no one believed that germs caused human disease. Despite the discovery of this important relationship, it would take numerous paradigm shifts and incredible luck before the discovery and usage of antimicrobials transpired.2
The sulfanilamides, azo compounds used in the German dye industry, were the first class of agents discovered to have antibacterial activity. These compounds were initially unsuccessful and required multiple modifications to reduce their unpleasant side effects. In 1928, Fleming discovered the first antibiotic, which he named penicillin. Fleming, similar to other scientists of the time, did not realize the utility of penicillin for systemic infections, however. A 1940 publication entitled “Penicillin as a Chemotherapeutic Agent” by Chain and colleagues3 set into motion what we now refer to as the “golden age” of antimicrobial chemotherapy.
The realization that living organisms may produce compounds that kill microbes is a relatively new concept. Since this realization, more than 30 classes of compounds have been identified from natural sources or created synthetically to treat infections resulting from bacteria, fungi, protozoa, and viruses. Techniques to identify organisms and to determine their susceptibility have also evolved over the years and are vital to the choice of the proper antimicrobial agent. In addition, other factors, such as the host, antimicrobial pharmacodynamics, antimicrobial combinations, and methods of monitoring therapy, are important parameters that need consideration before selecting an antimicrobial agent. This chapter focuses on these basic principles of antimicrobial therapy, provides a synopsis of mechanism of action and adverse effects, and emphasizes the clinical use of the various antimicrobial classes for the treatment of respiratory illnesses.1
Principles of antimicrobial therapy
Several factors require careful consideration before choosing a particular antimicrobial agent. Identification of the organism or organisms responsible for the infection is the first step toward treatment. Once the organism is isolated, antimicrobial susceptibility is determined according to standardized methods that can be replicated between laboratories. The susceptibility pattern of the organism narrows the choice of potential agents. Consideration must be given to host factors such as age, pregnancy, organ function, and site of infection. In addition, drug factors, such as available dosage forms, ease of administration, pharmacokinetics, potential adverse events, and cost, influence the choice of a specific agent.4
Identification of pathogen
In many clinical cases, the exact identity of the infecting organism is unknown. As a result, patients are treated empirically with an antimicrobial agent active against the organism or organisms that are most likely causing the infection. For example, 30% to 40% of patients with community-acquired pneumonia (CAP) fail to expectorate sputum, which prevents identification of a specific pathogen. However, research has shown, that the most common pathogens responsible for CAP include Streptococcus pneumoniae; Haemophilus influenzae; and atypical (intracellular) organisms such as Mycoplasma pneumoniae, Chlamydia pneumoniae, and Legionella pneumophila. As a result, empiric therapy for CAP involves antimicrobials active against this spectrum of organisms. Conversely, identification of an organism from culture material does not always indicate an infection. For example, hospitalized patients often have growth of gram-negative bacilli (rods) in sputum samples. However, these organisms may represent colonization only, and not hospital-acquired (nosocomial) pneumonia. Common pathogens and treatment of specific respiratory infections are listed in Table 14-1.5
TABLE 14-1
Common Pathogens and Treatment of Respiratory Infections in Adults*
| RESPIRATORY INFECTION | COMMON PATHOGENS | POTENTIAL ANTIBIOTIC REGIMENS |
| Sinusitis | ||
| Acute (community acquired) | Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis | Amoxicillin-clavulanate or cefuroxime axetil or respiratory quinolone (levofloxacin or moxifloxacin or gemifloxacin) or macrolide/ketolide or TMP-SMX |
| Acute (hospital acquired) | Pseudomonas aeruginosa, Acinetobacter spp., Staphylococcus aureus | Ceftazidime or cefepime or aztreonam or a carbapenem and vancomycin |
| Chronic | Bacteroides spp., Peptostreptococcus spp., Fusobacterium spp. | Antibiotics are usually unsuccessful; sinus drainage may be required |
| Bronchitis | ||
| Acute | Mycoplasma pneumoniae, Chlamydia pneumoniae, Bordetella pertussis | Antibiotics are usually not indicated; however, doxycycline or macrolide may be considered |
| Exacerbation of chronic bronchitis | S. pneumoniae, H. influenzae, M. catarrhalis | Value of antibiotics is controversial; doxycycline or macrolide may be considered |
| Pneumonia | ||
| Community acquired | S. pneumoniae, H. influenzae, M. catarrhalis, M. pneumoniae, C. pneumoniae, Legionella pneumophila | Azithromycin, clarithromycin, telithromycin, or respiratory quinolone, or doxycycline or β-lactam (ceftriaxone, cefuroxime, amoxicillin-clavulanate) and a macrolide |
| Hospital acquired (nonneutropenic patient) | S. pneumoniae, P. aeruginosa | Cefepime, ceftazidime, or aztreonam or a carbapenem or piperacillin-tazobactam ± an aminoglycoside or ciprofloxacin ± vancomycin |
| Hospital acquired (neutropenic patient) | As listed for nonneutropenic patients and fungi such as Aspergillus spp., Pneumocystis carinii (especially if HIV positive) | Cefepime, a carbapenem, ceftazidime, or piperacillin-tazobactam + an aminoglycoside or ciprofloxacin ± vancomycin ± amphotericin B ± TMP-SMX |
| Aspiration suspected | S. pneumoniae, Bacteroides fragilis, Peptostreptococcus spp., Fusobacterium spp. | Amoxicillin-clavulanate, ampicillin-sulbactam, piperacillin-tazobactam, clindamycin ± quinolone |
| Patient with cystic fibrosis | S. aureus, P. aeruginosa, Burkholderia cepacia | Aminoglycoside + piperacillin-tazobactam or ceftazidime, cefepime ± TMP-SMX (B. cepacia) |
| Empyema | ||
| Streptococcus milleri, B. fragilis, Enterobacteriaceae, Mycobacterium tuberculosis | Piperacillin-tazobactam or third-generation cephalosporin + clindamycin† |
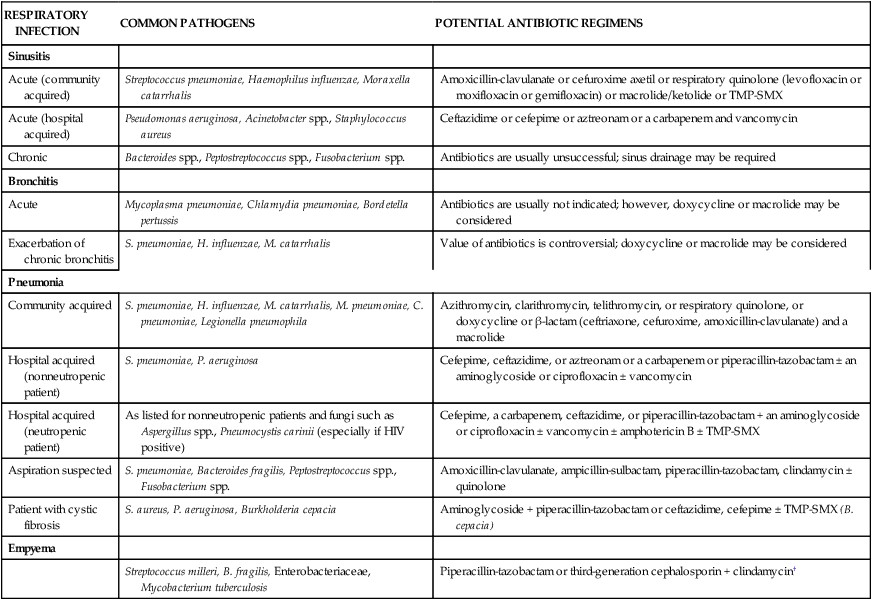
HIV, Human immunodeficiency virus; TMP-SMX, trimethoprim-sulfamethoxazole.
*The potential treatments listed here are not listed in order of superiority. Choice of antimicrobials depends on the individual susceptibility pattern of the suspected organisms within the specific institution.
†See Table 14-7 for antimycobacterial regimens.
Susceptibility testing and resistance
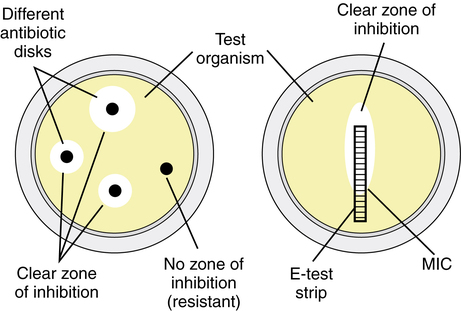
Once an organism is isolated, susceptibility test results can usually be obtained within 24 hours. Several methods are commonly used to determine the susceptibility of isolated pathogens. The Kirby-Bauer disk diffusion test involves the use of antibiotic-impregnated disks that are placed on an agar plate heavily inoculated (105 colony-forming units [cfu]/mL) with the isolated bacteria. If the organism is susceptible to the antibiotic, a clear zone of inhibition (no growth of the organism) develops around the disk. The degree of susceptibility or resistance of the organism depends on the diameter of this circular zone of inhibition; that is, a larger diameter indicates greater sensitivity. Another disk diffusion test is the elliptical test, or E-test. The E-test strip is placed on an agar plate heavily inoculated with the isolated organism. The strip creates an antimicrobial gradient, which results in a clear elliptical zone of inhibition. This method allows the determination of the minimal inhibitory concentration (MIC). MIC is defined as the least concentration of antimicrobial that prevents visible growth. The Kirby-Bauer and E-test methods are illustrated in Figure 14-1.
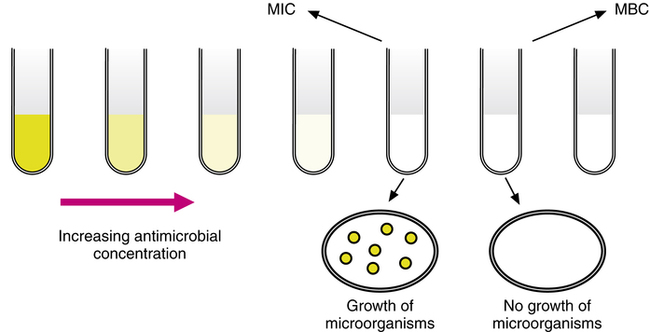
Other methods include inoculation of the organism into serial dilutions of an antimicrobial in agar or, more commonly, in broth culture media (Figure 14-2). Automated systems such as Vitek (bioMérieux, Durham, N.C.) and MicroScan (Dade Behring, West Sacramento, Calif.) take advantage of broth microdilution methods to provide efficient and rapid susceptibility results. When susceptibility testing is performed in broth media, a small sample can be removed from the test tubes or microwells with no growth and used to inoculate agar plates. The lowest concentration of antimicrobial agent that prevents growth of the organism on the agar plate after a 24-hour incubation is termed the minimal bactericidal concentration (MBC). Drugs that inhibit the growth of bacteria but do not kill them are termed bacteriostatic. A bactericidal drug is one that kills the bacteria. Examples of bacteriostatic and bactericidal drugs are listed in Box 14-1.
Susceptibility testing is a crucial part of antimicrobial therapy because the empiric regimen may fail when used to treat infections with resistant organisms. Microorganisms, similar to all living things, have genetic variability that affects their susceptibility to antimicrobials. Selective pressure from extensive clinical and agricultural use of antibiotics is thought to play a primary role in the emergence of resistant bacteria. Mechanisms of bacterial resistance include the production of enzymes that degrade or modify antibiotics, alteration of bacterial cell walls or membranes, upregulation of antimicrobial efflux pumps, and alteration of the site of antimicrobial action. Table 14-2 lists important emerging resistant bacteria.4

Host factors
The safety and efficacy of an antimicrobial agent vary, based on the population of patients being treated.5 For example, bone marrow transplant recipients with an active infection may not improve despite use of the ideal antimicrobial agent because of their impaired immune function. Similarly, other immunocompromised hosts, such as patients with acquired immunodeficiency syndrome (AIDS), recipients of cancer chemotherapy or steroids, and solid organ transplant recipients, are also at risk of failing to improve on antimicrobial therapy. Other factors such as the altered pharmacokinetics of an antimicrobial can affect response to therapy. For example, the absorption of certain antimicrobials such as itraconazole (an antifungal agent) is increased in the presence of gastric acid; others, such as penicillin G, are degraded in the presence of acid. The pH of the stomach varies with age; older patients tend to have achlorhydria, and young children tend to have a higher gastric pH. As a result, these two populations may have enhanced absorption of penicillin and decreased absorption of itraconazole relative to the rest of the population.
Antimicrobials concentrate in varying degrees within organ systems and can influence the outcome of therapy. Clindamycin achieves excellent bone concentrations and is very useful for treatment of osteomyelitis resulting from susceptible organisms. Similarly, drugs such as the aminoglycosides, most fluoroquinolones, and penicillins achieve very high concentrations in the urine and are useful for the treatment of urinary tract infections (UTIs). Conversely, certain drugs, although active against the organism in vitro, cannot achieve adequate concentrations at the site of infection. For example, aminoglycosides cannot penetrate the blood-brain barrier to treat meningitis adequately in adults. The blood-brain barrier represents tight junctions between the epithelial cells of the capillary wall that prevent drugs from entering the central nervous system.4
Pharmacodynamics
Pharmacodynamics refers to the science of understanding the optimal effect of a drug as a function of its concentration and the in vitro activity (MIC) against an organism. The pharmacodynamic properties of an antimicrobial are measured in vitro by using time-kill studies. These studies measure the rate and extent of microorganism killing when exposed to varying concentrations of antimicrobials. If the microbial kill rate increases proportionally with drug concentration, the antimicrobial is said to have a concentration-dependent effect. If the microbial kill rate is influenced by the time of drug concentration above the MIC, the antimicrobial is defined as time-dependent (or concentration-independent). Another pharmacodynamic phenomenon exhibited by antimicrobials is known as the postantibiotic effect (PAE). The PAE refers to the sustained suppression of bacterial growth even after the concentration of the antibiotic declines below detectable levels. The length of the PAE varies by the type of organism and the drug. Generally, time-dependent drugs, such as β-lactams, have short PAEs, whereas concentration-dependent drugs, such as aminoglycosides, metronidazole, and quinolones, have longer PAEs. Agents with a short PAE should be dosed frequently, and longer dosing intervals should be used for antimicrobials having a long PAE. These pharmacodynamic properties have been shown in vitro and in numerous animal studies. Clinical trials validating these principles are ongoing, and practical guidelines to incorporate pharmacodynamics in clinical practice are under study.4
Antimicrobial combinations
Empiric regimens must often cover a broad spectrum of organisms, which occasionally requires the use of two or more classes of antimicrobials. Ideally, the regimen should be narrowed after the specific organism has been isolated and susceptibilities are determined. Certain infections are polymicrobial, and in certain settings the use of antimicrobial combinations is justified. When antimicrobials are used in combination, it is important to know whether these agents act synergistically or are antagonistic. Synergy is shown in vitro when the combined effect of two antimicrobials is greater than their added effect (i.e., 1 + 1 > 2). Antagonism occurs when the effect of the combined drug is lower than the effect expected from either agent alone (i.e., 1 + 1 < 1). Antagonism may result in an unfavorable response, and such drug combinations should be avoided. A classic example of antagonism was the use of tetracycline and penicillin in children with pneumococcal meningitis. The mortality associated with the use of combination therapy was three times higher than the use of penicillin alone. Conversely, synergistic combinations have played a vital role in the treatment of resistant Pseudomonas infections in patients with cystic fibrosis. These patients have recurrent bouts of pseudomonal pneumonia and are often colonized with resistant species. Certain synergistic combinations of β-lactams and aminoglycosides have been shown to curb the development of resistance and to improve outcomes.4
Monitoring response to therapy
The use of antimicrobials can be associated with significant toxicities. The agent amphotericin B, which is used to treat fungal infections such as pulmonary aspergillosis, can cause significant renal dysfunction. Similarly, other agents can have adverse effects on the liver, gastrointestinal tract, neuromuscular system, hematologic system, heart, and lungs. The incidence of these adverse events varies among agents and is often reversible. Careful monitoring of patients receiving antimicrobials can prevent serious and potentially life-threatening adverse events.4
Antibiotics
Penicillins
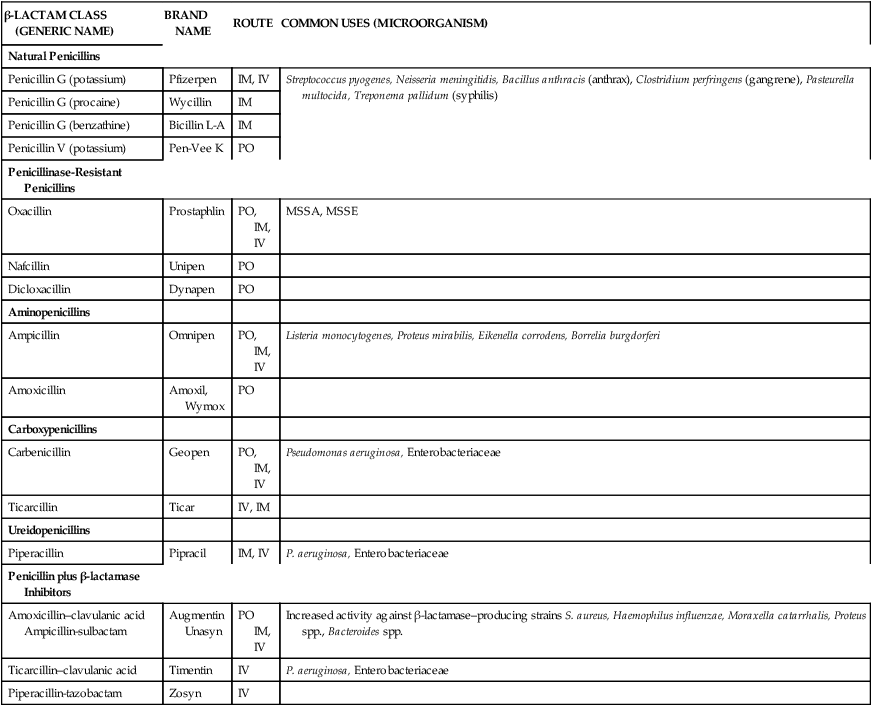
The discovery of penicillin in 1928 by Fleming ultimately led to the creation of a broad class of antibiotics commonly referred to as β-lactams. β-lactam antibiotics include the penicillins, cephalosporins, monobactams, and carbapenems.6 The main constituent of these antibiotics is the β-lactam ring structure. Chemical manipulation of β-lactam side chains led to the development of new agents with enhanced spectra of antimicrobial activity compared with penicillin. Specific side-chain modifications of penicillin have resulted in a broad class that includes the natural penicillins, aminopenicillins, penicillinase-resistant penicillins, carboxypenicillins, and ureidopenicillins (Table 14-3). Penicillins have also been combined with β-lactamase inhibitors to overcome a common mechanism of bacterial resistance.
TABLE 14-3
Classification and Clinical Uses of Penicillins
| β-LACTAM CLASS (GENERIC NAME) | BRAND NAME | ROUTE | COMMON USES (MICROORGANISM) |
| Natural Penicillins | |||
| Penicillin G (potassium) | Pfizerpen | IM, IV | Streptococcus pyogenes, Neisseria meningitidis, Bacillus anthracis (anthrax), Clostridium perfringens (gangrene), Pasteurella multocida, Treponema pallidum (syphilis) |
| Penicillin G (procaine) | Wycillin | IM | |
| Penicillin G (benzathine) | Bicillin L-A | IM | |
| Penicillin V (potassium) | Pen-Vee K | PO | |
| Penicillinase-Resistant Penicillins | |||
| Oxacillin | Prostaphlin | PO, IM, IV | MSSA, MSSE |
| Nafcillin | Unipen | PO | |
| Dicloxacillin | Dynapen | PO | |
| Aminopenicillins | |||
| Ampicillin | Omnipen | PO, IM, IV | Listeria monocytogenes, Proteus mirabilis, Eikenella corrodens, Borrelia burgdorferi |
| Amoxicillin | Amoxil, Wymox | PO | |
| Carboxypenicillins | |||
| Carbenicillin | Geopen | PO, IM, IV | Pseudomonas aeruginosa, Enterobacteriaceae |
| Ticarcillin | Ticar | IV, IM | |
| Ureidopenicillins | |||
| Piperacillin | Pipracil | IM, IV | P. aeruginosa, Enterobacteriaceae |
| Penicillin plus β-lactamase Inhibitors | |||
| Amoxicillin–clavulanic acid Ampicillin-sulbactam |
Augmentin Unasyn |
PO IM, IV |
Increased activity against β-lactamase–producing strains S. aureus, Haemophilus influenzae, Moraxella catarrhalis, Proteus spp., Bacteroides spp. |
| Ticarcillin–clavulanic acid | Timentin | IV | P. aeruginosa, Enterobacteriaceae |
| Piperacillin-tazobactam | Zosyn | IV |
The penicillins generally are widely distributed throughout the body and are associated with relatively low levels of toxicity. Most penicillins are acid-labile (destroyed in the stomach) and therefore are poorly absorbed after oral administration. Most agents in this class are not metabolized but are excreted unchanged in the urine. Therefore, most penicillins require reductions in dosage for patients with renal dysfunction.7
Mode of action
Penicillins exert their pharmacologic activity by inhibiting cell wall synthesis. Penicillins bind to enzymes (penicillin-binding proteins) located within the cell wall and prevent cross-linking of the peptidoglycan structure necessary for cell wall development. In addition, penicillins activate an endogenous autolytic system within bacteria, which subsequently leads to cell lysis and death. Penicillins are bactericidal, exhibit time-dependent killing, and can act synergistically with aminoglycosides against some bacteria (i.e., Pseudomonas aeruginosa and enterococci).8
Clinical uses
Natural penicillins.
Benzylpenicillin (penicillin G) is the parent compound of this class. This agent may be administered by the parenteral (intravenous [IV]) or intramuscular (IM) route. Penicillin G procaine is used for intramuscular dosing only and provides a less painful alternative when sustained serum concentrations are required. Phenoxymethyl penicillin (penicillin V) resists degradation by gastric acid and can be administered orally (PO). Natural penicillins are effective primarily against gram-positive bacteria and anaerobes. Penicillin G is the drug of choice for the treatment of primary and secondary syphilis (Treponema pallidum) and pharyngitis caused by group A streptococci (Streptococcus pyogenes). Because of the increasing frequency of resistance in Staphylococcus aureus, S. pneumoniae, and Neisseria gonorrhoeae, penicillin G should no longer be considered the agent of choice for infections caused by these organisms.7
Penicillinase-resistant penicillins.
In an attempt to overcome the emergence of penicillinase-producing (β-lactamase–producing) staphylococci, semisynthetic penicillinase-resistant antibiotics were developed. These agents are commonly referred to as antistaphylococcal agents because of their excellent activity against S. aureus. Methicillin was the first agent in this class of antibiotics, followed by oxacillin, nafcillin, cloxacillin, and dicloxacillin. Chemical modification of penicillin by the addition of an acyl side chain prevents hydrolysis of the agents in the presence of penicillinase. This class has activity against gram-positive cocci (staphylococci and streptococci) and is routinely used in skin and soft tissue infections. These agents are ineffective in the treatment of infections caused by gram-negative organisms or anaerobes. Until the 1980s, these antibiotics were the mainstay of treatment against staphylococci. However, the emergence of methicillin-resistant staphylococci has greatly reduced the clinical effectiveness of these agents.7
Aminopenicillins.
Aminopenicillins were the first penicillin class developed that were considered clinically active against some gram-negative bacteria (Escherichia coli, H. influenzae). Ampicillin and amoxicillin are the primary antibiotics in this class. In contrast to penicillin, ampicillin and amoxicillin are stable in gastric acid and therefore are suitable for oral administration. Both ampicillin and amoxicillin are frequently used in infections with susceptible organisms of the respiratory (S. pneumoniae, H. influenzae) and urinary (E. coli) tracts.7
Carboxypenicillins.
With the emergence of more resistant gram-negative bacilli, penicillins with increased gram-negative activity were needed. Carbenicillin was the first penicillin to have activity against Pseudomonas aeruginosa. It was also active against most members of the family Enterobacteriaceae, including E. coli, Enterobacter, Proteus, Morganella, and Serratia. Subsequent modification of carbenicillin resulted in ticarcillin, which has even greater in vitro activity against P. aeruginosa and members of Enterobacteriaceae (including Klebsiella). Neither of these agents is considered to have appreciable activity against gram-positive organisms (staphylococci or streptococci). These agents are administered primarily in the intravenous form because high serum concentrations cannot be achieved with the oral formulations.7
Ureidopenicillins.
Although carbenicillin and ticarcillin provided increased gram-negative coverage, antimicrobial agents with enhanced antipseudomonal activity were still needed. The ureidopenicillins were developed to fill this need. Piperacillin is a penicillin antibiotic with enhanced gram-negative activity (especially against P. aeruginosa) and with fewer adverse reactions than carboxypenicillins. Ureidopenicillins also exhibit activity against streptococci and enterococci and many anaerobes. Piperacillin is the primary agent of this class and has been efficacious in the treatment of pneumonia, bacteremia, UTIs, osteomyelitis, and soft tissue infections.7
β-lactam and β-lactamase inhibitor combinations.
Certain bacteria have the ability to produce enzymes (β-lactamases) that destroy the activity of penicillins by disrupting the β-lactam structure. β-lactamase inhibitors were developed to overcome this form of resistance. Consequently, combination β-lactam and β-lactamase inhibitors have a large spectrum of activity, making them particularly useful in polymicrobial infections. At present, three β-lactamase inhibitors are approved for combination with penicillins: clavulanic acid, sulbactam, and tazobactam. β-lactamase inhibitors enhance the activity of β-lactams against β-lactamase–producing strains of S. aureus, Moraxella catarrhalis, E. coli, H. influenzae, Klebsiella species, and Bacteroides species.7
Adverse reactions and precautions
The most common adverse reaction to penicillins is hypersensitivity. Approximately 3% to 10% of the population are allergic to penicillin. Reactions vary in severity from a mild rash to life-threatening anaphylaxis. Patients allergic to a penicillin could be potentially allergic to all classes of β-lactams (cephalosporins, carbapenems). In addition to allergic reactions, hematologic reactions such as thrombocytopenia and increased bleeding times have been reported. Hematologic disturbances are thought to be greater with carboxypenicillins than with ureidopenicillins. Gastrointestinal disturbances (nausea, vomiting, and diarrhea) are more common with oral dosage forms of penicillins, especially ampicillin. Interstitial nephritis has occurred most commonly with methicillin (not commercially available) but may occur with other penicillins as well. Central nervous system toxicities (i.e., seizures) have been reported with penicillins. Patients with an underlying seizure disorder and patients with renal insufficiency are at greatest risk for developing this complication.7
Cephalosporins
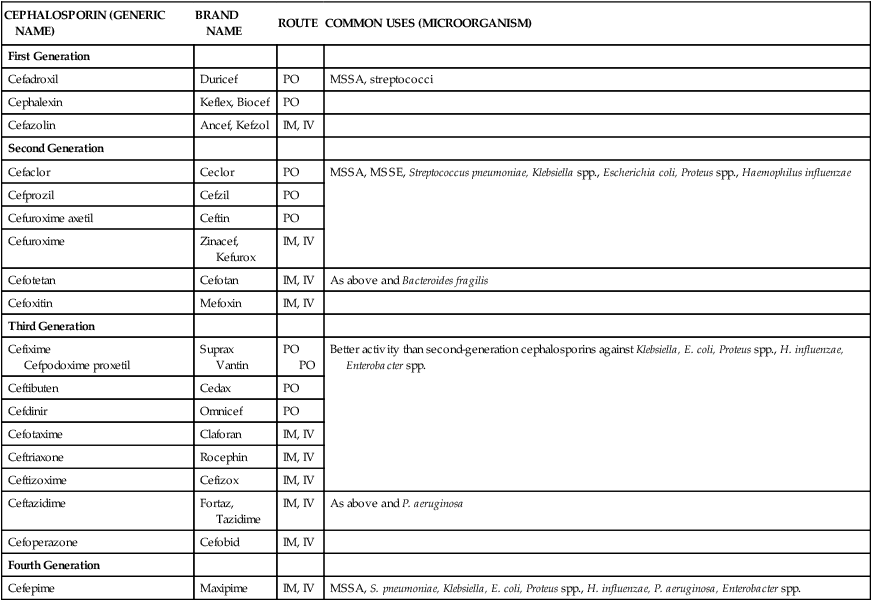
The cephalosporins include a large group of antimicrobials that are structurally related to the penicillins. Discovered in the 1940s as a microbial by-product of the fungus Cephalosporium acremonium, this class is now widely used in clinical practice. Similar to the penicillins, this class exhibits bactericidal activity, is distributed throughout the body, and produces relatively few adverse effects. Cephalosporins are used for various clinical indications and are available in oral and intravenous formulations (Table 14-4). Agents from this class have been loosely grouped into “generations” based on their spectrum of activities. At present there are four generations (classes) of cephalosporins.
TABLE 14-4
Classification and Clinical Uses of Cephalosporins
| CEPHALOSPORIN (GENERIC NAME) | BRAND NAME | ROUTE | COMMON USES (MICROORGANISM) |
| First Generation | |||
| Cefadroxil | Duricef | PO | MSSA, streptococci |
| Cephalexin | Keflex, Biocef | PO | |
| Cefazolin | Ancef, Kefzol | IM, IV | |
| Second Generation | |||
| Cefaclor | Ceclor | PO | MSSA, MSSE, Streptococcus pneumoniae, Klebsiella spp., Escherichia coli, Proteus spp., Haemophilus influenzae |
| Cefprozil | Cefzil | PO | |
| Cefuroxime axetil | Ceftin | PO | |
| Cefuroxime | Zinacef, Kefurox | IM, IV | |
| Cefotetan | Cefotan | IM, IV | As above and Bacteroides fragilis |
| Cefoxitin | Mefoxin | IM, IV | |
| Third Generation | |||
| Cefixime Cefpodoxime proxetil |
Suprax Vantin |
PO PO |
Better activity than second-generation cephalosporins against Klebsiella, E. coli, Proteus spp., H. influenzae, Enterobacter spp. |
| Ceftibuten | Cedax | PO | |
| Cefdinir | Omnicef | PO | |
| Cefotaxime | Claforan | IM, IV | |
| Ceftriaxone | Rocephin | IM, IV | |
| Ceftizoxime | Cefizox | IM, IV | |
| Ceftazidime | Fortaz, Tazidime | IM, IV | As above and P. aeruginosa |
| Cefoperazone | Cefobid | IM, IV | |
| Fourth Generation | |||
| Cefepime | Maxipime | IM, IV | MSSA, S. pneumoniae, Klebsiella, E. coli, Proteus spp., H. influenzae, P. aeruginosa, Enterobacter spp. |
Clinical uses
As a class, cephalosporins are active against a wide variety of organisms. Because of their broad spectrum of activity and low level of toxicity, these agents are commonly used for a wide variety of infections. The spectrum of activity differs for each cephalosporin generation. All cephalosporins are ineffective against enterococci.9
Adverse reactions and precautions
Similar to penicillins, the cephalosporins as a group are well tolerated. Hypersensitivity reactions occur in 1% to 3% of patients, with cross-reactivity of cephalosporins in patients with penicillin allergy ranging from 5% to 15%. Generally, patients with a penicillin allergy (limited to a rash) may be challenged with a cephalosporin. Cephalosporin use is contraindicated, however, in patients with a history of anaphylaxis to β-lactams.10 Desensitization should be performed if no therapeutic alternative exists for the use of a cephalosporin. Oral cephalosporins have been associated with minor gastrointestinal complaints such as nausea, vomiting, and diarrhea. Hypoprothrombinemia has been reported, especially with agents (cefotetan and cefoperazone) with a methylthiotetrazole (MTT) side chain. The MTT side chain may also induce a disulfiram-like reaction in patients who concurrently ingest alcohol (disulfiram inhibits the metabolism of alcohol). The symptoms of this uncomfortable reaction include flushing, nausea, thirst, palpitations, chest pain, vertigo, and death in some cases. Most cephalosporins are eliminated through the kidneys and require dosage adjustment in the presence of renal insufficiency.11
Carbapenems
The carbapenems are the newest class of β-lactam antibiotics. At present, four carbapenems—imipenem-cilastatin, meropenem, doripenem, and ertapenem—are available for use in the United States. Cilastatin is used to inhibit the metabolism of imipenem within the kidney to prolong the half-life of this agent. Carbapenems are broad-spectrum antibiotics, displaying activity against a wide variety of gram-positive, gram-negative, and anaerobic bacteria.12
Clinical uses
Imipenem, meropenem, and doripenem all are active against P. aeruginosa, multidrug-resistant (MDR) gram-negative bacilli, and most anaerobes. Ertapenem differs from other carbapenems in that it possesses no activity against P. aeruginosa.13 All four carbapenems have activity against gram-positive organisms such as MSSA and Streptococcus species, including pneumococci (S. pneumoniae). Imipenem, meropenem, and doripenem have been used clinically for empiric treatment of bacteremia and sepsis, community-acquired and nosocomial pneumonia, skin and skin structure infections, complicated UTIs, intraabdominal infections, obstetric and gynecologic infections, osteomyelitis, and infections in patients with cancer and neutropenia. Because ertapenem is ineffective against P. aeruginosa, it should not be used in the treatment of nosocomial pneumonia, neutropenic fever, or any other infection in which P. aeruginosa is a likely pathogen.13 Because of their excellent in vitro activity and broad spectrum of coverage, these agents are often reserved to treat infections that are caused by bacteria resistant to most other agents.
Aminoglycosides
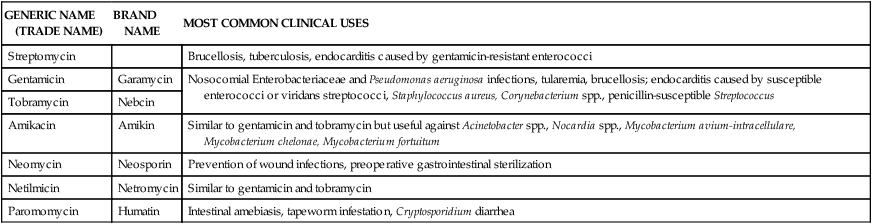
Streptomycin was discovered in 1943 and was the first antimicrobial agent available to treat tuberculosis. Numerous other aminoglycosides have been developed since and include gentamicin, tobramycin, netilmicin, and amikacin. These agents are used for gram-negative infections, including infections caused by P. aeruginosa. These antimicrobials have poor gastrointestinal absorption and require parenteral administration.15 Table 14-5 lists aminoglycosides and their clinical uses.
TABLE 14-5
Clinical Uses of Aminoglycosides*
| GENERIC NAME (TRADE NAME) | BRAND NAME | MOST COMMON CLINICAL USES |
| Streptomycin | Brucellosis, tuberculosis, endocarditis caused by gentamicin-resistant enterococci | |
| Gentamicin | Garamycin | Nosocomial Enterobacteriaceae and Pseudomonas aeruginosa infections, tularemia, brucellosis; endocarditis caused by susceptible enterococci or viridans streptococci, Staphylococcus aureus, Corynebacterium spp., penicillin-susceptible Streptococcus |
| Tobramycin | Nebcin | |
| Amikacin | Amikin | Similar to gentamicin and tobramycin but useful against Acinetobacter spp., Nocardia spp., Mycobacterium avium-intracellulare, Mycobacterium chelonae, Mycobacterium fortuitum |
| Neomycin | Neosporin | Prevention of wound infections, preoperative gastrointestinal sterilization |
| Netilmicin | Netromycin | Similar to gentamicin and tobramycin |
| Paromomycin | Humatin | Intestinal amebiasis, tapeworm infestation, Cryptosporidium diarrhea |
*The aminoglycosides (most commonly gentamicin and tobramycin) are used to treat infections caused by gram-negative bacteria.
Mode of action
Aminoglycosides bind irreversibly to the 30S bacterial ribosome and inhibit the translation of ribonucleic acid (RNA) into proteins. Aminoglycosides also competitively displace cations that link lipopolysaccharides in the outer cell wall of gram-negative bacteria. This destabilization of the cell wall results in increased cell permeability and lysis. Aminoglycosides are bactericidal agents and exhibit concentration-dependent killing. They are often synergistic when used in combination with β-lactam antibiotics.16
Clinical uses
Gentamicin, tobramycin, and amikacin all have been used for nosocomial gram-negative infections, such as ventilator-associated pneumonias. However, aminoglycosides do not achieve high concentrations in bronchial secretions when administered systemically; this is thought to be particularly problematic for patients infected with resistant gram-negative organisms. As a result, aminoglycosides (particularly tobramycin and now amikacin) have been administered by inhalation to control P. aeruginosa infections in patients with cystic fibrosis (see Chapter 13). Amikacin is currently more expensive and is generally reserved for organisms resistant to other aminoglycosides. Aminoglycosides are used synergistically with β-lactams when treating endocarditis caused by Streptococcus species and Enterococcus species. Aminoglycosides are also used extensively to treat intraabdominal infections. Streptomycin is used in combination with other antitubercular antimicrobials, especially for MDR tuberculosis.
Adverse reactions and precautions
The primary toxicities associated with the use of aminoglycosides are nephrotoxicity and ototoxicity. Nephrotoxicity usually develops after at least 5 to 7 days of therapy and occurs more commonly in patients with hypotension, liver disease, advanced age, and coadministration of other nephrotoxic agents. Ototoxicity (both cochlear toxicity and vestibular toxicity) may be irreversible because significant damage must occur before it can be detected. The most common symptoms associated with the development of cochlear toxicity include tinnitus (ringing in the ears); vestibular toxicity manifests as dizziness and nausea. Another serious but rare toxicity is neuromuscular blockade associated with peritoneal irrigation and rapid high-dose aminoglycoside use. Underlying conditions such as myasthenia gravis or concomitant use of neuromuscular blockers may potentiate this side effect, requiring supportive measures such as intubation and possible ventilation support.17
Tetracyclines
Tetracyclines are broad-spectrum antibiotics with activity against gram-positive and gram-negative microorganisms and many rickettsiae, chlamydiae, mycoplasmas, spirochetes, protozoa, and mycobacteria. The most commonly used agent in this class is doxycycline because it can be administered twice daily and is relatively inexpensive. Other available tetracyclines include minocycline and tetracycline. The agents are available in oral and parenteral formulations.18
Tigecycline
Tigecycline, a glycylcycline antibiotic, is similar in structure to tetracycline antibiotics. Because of substitution of a central four-ring carbocyclic nucleus, tigecycline has a greater spectrum of activity than tetracyclines. Tigecycline is active against most gram-positive bacteria, including S. pneumoniae, vancomycin-susceptible enterococci, vancomycin-resistant enterococci, coagulase-negative staphylococci, MSSA, and methicillin-resistant S. aureus (MRSA). Tigecycline also has activity against most gram-negative bacteria, including H. influenzae, M. catarrhalis, most members of the Enterobacteriaceae family (including extended-spectrum β-lactamase [ESBL]–producing organisms), and Acinetobacter species. It is also active against many anaerobic bacteria. However, tigecycline is not active against P. aeruginosa and Proteus species.19
Adverse reactions and precautions
During clinical trials, the most common side effects observed with the use of tigecycline were gastrointestinal and included nausea, vomiting, diarrhea, and abdominal pain. Other frequently reported adverse effects were headache, thrombocytopenia, and elevations of liver enzymes. Because tigecycline is a structural derivative of minocycline, it is appropriate to monitor for side effects associated with other tetracycline antibiotics, such as phototoxicity and dental disorders.20 Tigecycline should not be used in individuals with hypersensitivity reactions to tetracycline antibiotics.
Macrolides
Erythromycin was introduced in 1952 and was the first agent of this class to be used for infections with atypical organisms and for infections in patients intolerant to penicillin G. Early work with erythromycin involved production of various salt derivatives to improve its gastrointestinal tolerability and absorption. In the last decade, clarithromycin and azithromycin (an azalide) have been introduced. Macrolides exhibit activity against gram-positive bacteria (streptococci, MSSA), gram-negative bacteria (H. influenzae, M. catarrhalis), and atypical bacteria (mycoplasmas, rickettsiae, Legionella, and Chlamydia). Clarithromycin and azithromycin are also active against Mycobacterium avium and other mycobacterial species.21
Adverse reactions and precautions
Clarithromycin and azithromycin are generally better tolerated than erythromycin. The most common adverse reactions of macrolides include gastrointestinal complaints such as nausea, vomiting, abdominal cramps, and diarrhea (30% of patients taking erythromycin). The use of intravenous erythromycin is associated with thrombophlebitis. Ventricular tachycardia and Q–T interval prolongation have been reported with the use of the macrolides. Erythromycin and clarithromycin are potent inhibitors of the hepatic drug metabolism system known as the cytochrome P450 (CYP) system. As a result, erythromycin and clarithromycin can increase the systemic concentrations of drugs metabolized through the CYP system. For drugs with narrow therapeutic indices, such as theophylline, warfarin, and triazolam, this interaction can lead to potential life-threatening complications. Abnormalities in liver function, tinnitus, dizziness, and reversible hearing loss also have been associated with the use of macrolides.22
Telithromycin
Telithromycin is the first member of the ketolide group of antimicrobials, which are structurally related to the macrolide antibiotics. It has good antimicrobial activity against respiratory pathogens typically responsible for CAP, such as S. pneumoniae, H. influenzae, M. pneumoniae, C. pneumoniae, L. pneumophila, and M. catarrhalis. Telithromycin has improved acid stability compared with erythromycin and therefore has good oral absorption.23
Clinical uses
At present, telithromycin is indicated for the treatment of acute exacerbation of chronic bronchitis (AECB), acute bacterial sinusitis, and CAP. For AECB and acute bacterial sinusitis, a 5-day course of telithromycin was shown to have comparable efficacy to comparator agents (amoxicillin-clavulanate, cefuroxime, or clarithromycin). Longer courses of telithromycin (7 to 10 days) are required for treatment of CAP. It is not currently approved for the treatment of streptococcal pharyngitis; however, it has been shown to be equally as effective as clarithromycin or penicillin V. Telithromycin may serve as an alternative agent for patients at risk of infection with penicillin-resistant or macrolide-resistant S. pneumoniae.23 It is also an alternative treatment in patients with penicillin allergies. Telithromycin is available only in oral formulation and is typically dosed as 800 mg daily.
Quinolones (fluoroquinolones)
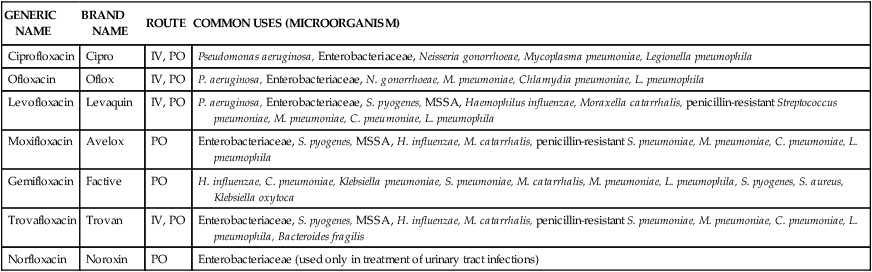
The quinolones, also known as fluoroquinolones (Table 14-6), are a semisynthetic group of antimicrobials structurally related to nalidixic acid (quinolone), one of the by-products of chloroquine synthesis. They are widely distributed into most body fluids and tissues (achieving high respiratory tract concentrations). Quinolones are eliminated primarily through the kidneys and achieve high concentrations in the urine. Agents from this class have variable activity against gram-negative bacteria, gram-positive bacteria, anaerobes, atypical bacteria, and mycobacteria.24 Ciprofloxacin, levofloxacin, and moxifloxacin are the most commonly used fluoroquinolones in the United States. Gatifloxacin was removed from the market in 2006 because of the high incidence of diabetes and other blood glucose irregularities.
TABLE 14-6
Classification and Clinical Uses of Quinolones
| GENERIC NAME | BRAND NAME | ROUTE | COMMON USES (MICROORGANISM) |
| Ciprofloxacin | Cipro | IV, PO | Pseudomonas aeruginosa, Enterobacteriaceae, Neisseria gonorrhoeae, Mycoplasma pneumoniae, Legionella pneumophila |
| Ofloxacin | Oflox | IV, PO | P. aeruginosa, Enterobacteriaceae, N. gonorrhoeae, M. pneumoniae, Chlamydia pneumoniae, L. pneumophila |
| Levofloxacin | Levaquin | IV, PO | P. aeruginosa, Enterobacteriaceae, S. pyogenes, MSSA, Haemophilus influenzae, Moraxella catarrhalis, penicillin-resistant Streptococcus pneumoniae, M. pneumoniae, C. pneumoniae, L. pneumophila |
| Moxifloxacin | Avelox | PO | Enterobacteriaceae, S. pyogenes, MSSA, H. influenzae, M. catarrhalis, penicillin-resistant S. pneumoniae, M. pneumoniae, C. pneumoniae, L. pneumophila |
| Gemifloxacin | Factive | PO | H. influenzae, C. pneumoniae, Klebsiella pneumoniae, S. pneumoniae, M. catarrhalis, M. pneumoniae, L. pneumophila, S. pyogenes, S. aureus, Klebsiella oxytoca |
| Trovafloxacin | Trovan | IV, PO | Enterobacteriaceae, S. pyogenes, MSSA, H. influenzae, M. catarrhalis, penicillin-resistant S. pneumoniae, M. pneumoniae, C. pneumoniae, L. pneumophila, Bacteroides fragilis |
| Norfloxacin | Noroxin | PO | Enterobacteriaceae (used only in treatment of urinary tract infections) |
IV, Intravenous; MSSA, methicillin-susceptible Staphylococcus aureus; PO, oral.
Other antibiotics
Chloramphenicol
Chloramphenicol has been available for use in the United States since 1949. This agent has a broad spectrum of activity against gram-positive, gram-negative, and anaerobic bacteria. Chloramphenicol distributes well into various tissues including the brain. Use of this antibiotic has declined with the availability of less toxic agents.25
Colistin (colistimethate)
Colistin, a member of the polymyxin family, was used in the early 1960s for serious gram-negative infections, including infections caused by P. aeruginosa. It was approved in 1968 by the U.S. Food and Drug Administration (FDA) but was later abandoned for drugs with similar gram-negative efficacy and more favorable side-effect profiles.26 However, there has been a resurgence in the use of colistin, because of the emergence of MDR gram-negative bacteria including P. aeruginosa and Acinetobacter species.
Clinical uses.
Colistin has a broad range of activity against most gram-negative bacteria, including MDR P. aeruginosa and Acinetobacter species. Use of colistin is often reserved for severe systemic infections, including ventilator-associated pneumonias. Proteus and Neisseria species are generally resistant to colistin, along with most anaerobes and gram-positive bacteria. Colistin is administered in an inactive form (colistimethate) intravenously, intramuscularly, or by nebulization. After administration, inactive colistimethate is converted in vivo to the active form colistin.26 Nebulized colistin is used often in patients with cystic fibrosis who harbor MDR P. aeruginosa and Acinetobacter.
Daptomycin
Mode of action.
The exact mechanism of daptomycin has not been completely elucidated; however, it is believed to occur by irreversible binding to the cytoplasmic membrane of bacterial cells and subsequent disruption of the membrane potential. This disruption apparently causes leakage of intracellular ions, leading to rapid cell death.27
Clinical uses.
Daptomycin is currently approved for use in complicated skin and skin structure infections caused by susceptible gram-positive bacteria and S. aureus bacteremia. It has excellent activity against resistant staphylococci and enterococci, including vancomycin-resistant strains, although it is not approved for this use. In a phase 3 trial involving daptomycin for the treatment of CAP, daptomycin was inferior to the comparator agent (ceftriaxone), especially in patients with more serious infections. This poor response in pulmonary infections has been attributed to inactivation of daptomycin by pulmonary surfactants.27 Consequently, daptomycin is not indicated for use in the treatment of pneumonia. Daptomycin is typically dosed at 4 to 6 mg/kg every 24 hours. It is available only as an intravenous infusion.
Trimethoprim-sulfamethoxazole
Sulfamethoxazole belongs to the class of antibiotics known as the sulfonamides. Trimethoprim is a pyrimidine found to potentiate the activity of sulfamethoxazole. The combination of trimethoprim and sulfamethoxazole (TMP-SMX; Bactrim) was introduced in 1968 and has since gained a place in the treatment of numerous infections. This combination is active against gram-positive bacteria (streptococci, MSSA, MRSA) and gram-negative bacteria (H. influenzae, Burkholderia cepacia, Stenotrophomonas maltophilia). In addition, it is active against Pneumocystis jiroveci (formerly known as Pneumocystis carinii).28
Metronidazole
Metronidazole is a nitroimidazole that was used initially for its antiprotozoal effects against pathogens such as Trichomonas vaginalis, Giardia lamblia, and Entamoeba histolytica. Its anaerobic properties were discovered after an observation that acute ulcerative gingivitis improved in patients being treated for trichomonal vaginitis.25
Nitrofurantoin
Multiple nitrofuran compounds have been synthesized since their discovery in the early 1940s. The most widely used agent of this class has been nitrofurantoin. This antibacterial does not achieve therapeutic concentrations in body tissues other than the kidney. As a result, it is used only for UTIs.30
Quinupristin and dalfopristin
Quinupristin and dalfopristin are streptogramins that act synergistically when used together (as in the product Synercid). These agents are active against gram-positive bacteria and are used primarily to treat infections caused by vancomycin-resistant Enterococcus faecium (VREF).31
Linezolid
The antibiotic linezolid belongs to a novel class of antibiotics known as the oxazolidinones. Linezolid, similar to quinupristin-dalfopristin, is active against gram-positive bacteria and is approved for the treatment of severe life-threatening VREF infections. In contrast to vancomycin and quinupristin-dalfopristin, linezolid is available as an oral formulation that is completely absorbed from the gastrointestinal tract.32
Antimycobacterials
Treatment consists of multiple antibiotic regimens for 6 to 12 months in duration. Single-agent regimens should never be used for treatment because the likelihood of developing resistance is high. Treatment failures often result from poor patient compliance and from resistance to antibiotics. Drugs used in the treatment of tuberculosis can be categorized as either first-line or second-line agents depending on their efficacy and side-effect profiles. Initial therapy generally involves a combination of isoniazid, pyrazinamide, rifampin, and ethambutol. Table 14-7 summarizes clinically used antimycobacterial agents, doses, routes of administration, and side effects. Addition or subtraction of agents from this regimen is usually based on culture and sensitivity data, along with patient response to treatment. Guidelines for the treatment of active pulmonary tuberculosis are provided in Table 14-833,34 or may be viewed directly at http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5211a1.htm.
TABLE 14-7
Dose, Route, and Side-Effect Profile of Commonly Used Antimycobacterials
| ANTIMYCOBACTERIAL | ADULT DOSE | ROUTE | SIDE EFFECTS |
| Isoniazid | 5 mg/kg/day; maximum 300 mg/day | PO, IM | Hepatotoxicity (symptoms include nausea, loss of appetite, abdominal pain), peripheral neuritis, rash, fever, anemia |
| Rifampin | 10 mg/kg/day; maximum 600 mg/day | PO, IV | Hepatotoxicity, flulike symptoms, discolorations of body secretions to an orange color |
| Rifabutin | 300 mg/day | PO | Hepatotoxicity, flulike symptoms, discolorations of body secretions to an orange color |
| Pyrazinamide | 15-30 mg/kg/day; maximum 2000 mg/day | PO | Hepatotoxicity, arthralgia, hyperuricemia |
| Ethambutol | 15-25 mg/kg/day; maximum 2500 mg/day | PO | Optic neuritis (greater incidence in patients receiving >15 mg/kg/day) |
| Streptomycin | 15 mg/kg/day; maximum 1000 mg/day | IM | Ototoxicity (high-frequency hearing loss, vertigo), nephrotoxicity |
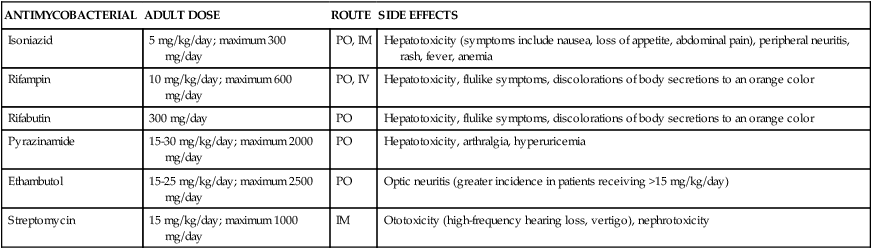
TABLE 14-8
Guidelines for Treatment of Active Pulmonary Tuberculosis
| CLINICAL SCENARIO | TREATMENT REGIMEN |

Data from American Thoracic Society, CDC, Infectious Diseases Society of America: Treatment of tuberculosis, MMWR Recomm Rep 52(RR-11):1-77, 2003.
Antifungals
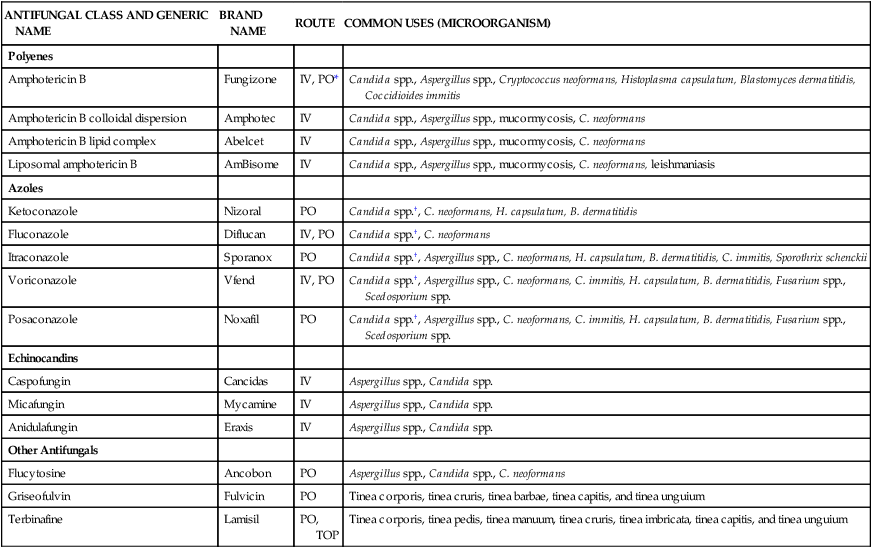
The incidence of fungal infections has increased dramatically. Candida species are now the fourth most commonly isolated bloodstream pathogens. Candidemia has a mortality rate of 40%. The number of patients immunocompromised as a result of AIDS, cancer chemotherapy, and organ transplantation has been increasing. These patient populations have diminished cell-mediated immunity and are predisposed to numerous fungal pathogens that vary in incidence geographically. The treatment of choice for most fungal infections has been the polyene amphotericin B. The high incidence of nephrotoxicity associated with this agent served as the impetus for the development of the azoles. Ketoconazole was the first agent of this class, but it has largely been replaced by the triazoles fluconazole and itraconazole. Newer triazoles with improved activity against molds are being developed. In addition, a new class of antifungals known as the echinocandins is now available. Systemically used antifungals, including route and clinical uses, are summarized in Table 14-9.35–37
TABLE 14-9
Classification of Systemically Used Antifungals
| ANTIFUNGAL CLASS AND GENERIC NAME | BRAND NAME | ROUTE | COMMON USES (MICROORGANISM) |
| Polyenes | |||
| Amphotericin B | Fungizone | IV, PO* | Candida spp., Aspergillus spp., Cryptococcus neoformans, Histoplasma capsulatum, Blastomyces dermatitidis, Coccidioides immitis |
| Amphotericin B colloidal dispersion | Amphotec | IV | Candida spp., Aspergillus spp., mucormycosis, C. neoformans |
| Amphotericin B lipid complex | Abelcet | IV | Candida spp., Aspergillus spp., mucormycosis, C. neoformans |
| Liposomal amphotericin B | AmBisome | IV | Candida spp., Aspergillus spp., mucormycosis, C. neoformans, leishmaniasis |
| Azoles | |||
| Ketoconazole | Nizoral | PO | Candida spp.†, C. neoformans, H. capsulatum, B. dermatitidis |
| Fluconazole | Diflucan | IV, PO | Candida spp.†, C. neoformans |
| Itraconazole | Sporanox | PO | Candida spp.†, Aspergillus spp., C. neoformans, H. capsulatum, B. dermatitidis, C. immitis, Sporothrix schenckii |
| Voriconazole | Vfend | IV, PO | Candida spp.†, Aspergillus spp., C. neoformans, C. immitis, H. capsulatum, B. dermatitidis, Fusarium spp., Scedosporium spp. |
| Posaconazole | Noxafil | PO | Candida spp.†, Aspergillus spp., C. neoformans, C. immitis, H. capsulatum, B. dermatitidis, Fusarium spp., Scedosporium spp. |
| Echinocandins | |||
| Caspofungin | Cancidas | IV | Aspergillus spp., Candida spp. |
| Micafungin | Mycamine | IV | Aspergillus spp., Candida spp. |
| Anidulafungin | Eraxis | IV | Aspergillus spp., Candida spp. |
| Other Antifungals | |||
| Flucytosine | Ancobon | PO | Aspergillus spp., Candida spp., C. neoformans |
| Griseofulvin | Fulvicin | PO | Tinea corporis, tinea cruris, tinea barbae, tinea capitis, and tinea unguium |
| Terbinafine | Lamisil | PO, TOP | Tinea corporis, tinea pedis, tinea manuum, tinea cruris, tinea imbricata, tinea capitis, and tinea unguium |
IV, Intravenous; PO, oral; TOP, topical.
*The oral form of amphotericin B is not absorbed through the gastrointestinal tract.
Polyenes
Polyenes include amphotericin B and nystatin. Amphotericin B has been available for more than 50 years and remains the drug of choice for most systemic fungal infections. More recently, amphotericin B has been formulated into lipid-based products. These lipid-based products alter the distribution of amphotericin B, resulting in a higher uptake of the agent into the reticuloendothelial system (liver, spleen, lymphatics) relative to the kidneys.38 The net effect of this shift in distribution has been shown to reduce the incidence of nephrotoxicity. Nystatin is used only topically for oral and intertriginous (between skin surfaces) candidiasis. A lipid-based formulation of nystatin is currently under investigation.
Azoles
Systemically used azoles include ketoconazole, fluconazole, itraconazole, voriconazole, and posaconazole. Ketoconazole was the first oral agent available to treat systemic fungal infections. This agent is poorly absorbed from the gastrointestinal tract (when an acidic environment is not present) and has potential for substantial toxicity. As a result, ketoconazole has largely been replaced by the newer triazoles fluconazole, itraconazole, and voriconazole. Fluconazole is available in oral and intravenous formulations, is widely distributed into the tissues, and is relatively nontoxic. Compared with ketoconazole, it has a narrow spectrum of activity. Itraconazole has an enhanced spectrum of activity compared with fluconazole but slightly less than that of voriconazole. Voriconazole is also available in oral and intravenous forms. Posaconazole was approved more recently for prophylaxis of invasive aspergillosis and disseminated candidiasis in severely immunocompromised hosts and for the treatment of refractory oropharyngeal candidiasis. Use of posaconazole is intended for patients failing or refractory to other therapies and has been documented to be active against Zygomycetes.37
Adverse reactions and precautions
Fluconazole is well tolerated and has minimal side effects. Common adverse effects with ketoconazole, itraconazole, and voriconazole are anorexia, nausea, and vomiting. In addition, transaminase and bilirubin elevations have been reported. Impotence, decreased libido, and gynecomastia are also known to occur with ketoconazole and are attributed to its inhibition of sex steroid synthesis, and consequently it is used clinically for certain endocrine disorders. Voriconazole can cause visual disturbances such as photopsia and chromatopsia, which can occur in 30% of patients.36,39 Ketoconazole, itraconazole, and voriconazole are metabolized in the liver and are potent inhibitors of the CYP 3A4 system, so each has a significant potential for drug interactions. Conversely, fluconazole does not undergo significant hepatic metabolism and has the lowest drug interaction potential of the triazoles. Fluconazole is, however, eliminated unchanged in the urine and so dosage adjustment is required in patients with impaired renal function. Renal function is also important when administering the intravenous formulations of itraconazole and voriconazole. These drugs should be avoided in patients with creatinine clearance values less than 30 mL/min to prevent the accumulation of cyclodextrin, an intravenous solubilizing agent. Cyclodextrin is also used to formulate oral itraconazole solution and has been cited as the cause of the high incidence of adverse gastrointestinal intolerance relative to itraconazole capsules, which do not contain this agent.
Echinocandins
Caspofungin was the first echinocandin to receive FDA approval in early 2001. Micafungin and anidulafungin received FDA approval in 2004 and 2006, respectively. These agents have similar pharmacokinetic and pharmacodynamic properties. They have poor gastrointestinal absorption, and all require parenteral administration.40
Adverse reactions and precautions
Echinocandins seem to be well tolerated, although most of the safety data are for caspofungin and micafungin. In most studies, common adverse reactions were infusion-related and included fever, rash, flushing, and thrombophlebitis. Infusion-related reactions were approximately 10% for caspofungin and slightly less for micafungin. Anidulafungin has produced infusion-related reactions in 15% of patients. Nausea and vomiting were also frequently reported for all three agents. Elevations in liver transaminase (AST and ALT) levels and hyperbilirubinemia were noted with echinocandins, but these were mild and reversible on drug discontinuation. Echinocandins are not metabolized through the CYP system, reducing the potential for drug interactions.41 All echinocandins can increase the area under the curve (AUC) of cyclosporine when coadministered, with caspofungin creating the most significant changes in AUC. The mechanism of this interaction is unknown. Concurrent use of cyclosporine and echinocandins may warrant careful monitoring.
Griseofulvin and terbinafine
Griseofulvin was one of the first antifungals discovered; however, only dermatophytes (skin fungi) are susceptible to this agent. The absorption of this agent is greatly increased when taken with a high-fat meal. Terbinafine, which was developed more recently, has shown more potent activity against dermatophytes. Terbinafine is a highly lipophilic allylamine that concentrates in the stratum corneum, sebum, and hair follicles. This property makes it an excellent agent for cutaneous dermatophytosis (e.g., ringworm infection, athlete’s foot) and onychomycosis (fungal infection in nails).35
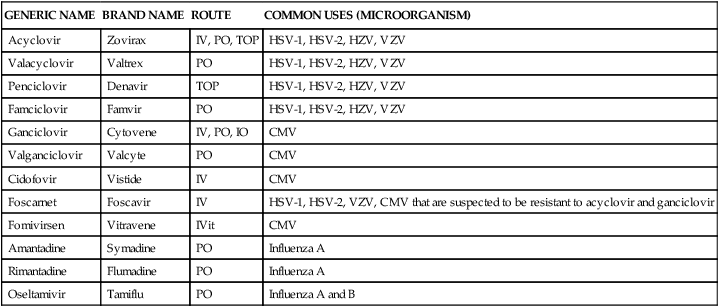
Antiviral agents
Several agents are available for treating viral infections (Table 14-10). All of these agents act by inhibiting steps involved in viral replication; none of the agents inhibit nonreplicating viruses. Antivirals (excluding antiretrovirals) mimic nucleosides and inhibit DNA synthesis. Agents used to treat HIV are not discussed here.42
TABLE 14-10
| GENERIC NAME | BRAND NAME | ROUTE | COMMON USES (MICROORGANISM) |
| Acyclovir | Zovirax | IV, PO, TOP | HSV-1, HSV-2, HZV, VZV |
| Valacyclovir | Valtrex | PO | HSV-1, HSV-2, HZV, VZV |
| Penciclovir | Denavir | TOP | HSV-1, HSV-2, HZV, VZV |
| Famciclovir | Famvir | PO | HSV-1, HSV-2, HZV, VZV |
| Ganciclovir | Cytovene | IV, PO, IO | CMV |
| Valganciclovir | Valcyte | PO | CMV |
| Cidofovir | Vistide | IV | CMV |
| Foscarnet | Foscavir | IV | HSV-1, HSV-2, VZV, CMV that are suspected to be resistant to acyclovir and ganciclovir |
| Fomivirsen | Vitravene | IVit | CMV |
| Amantadine | Symadine | PO | Influenza A |
| Rimantadine | Flumadine | PO | Influenza A |
| Oseltamivir | Tamiflu | PO | Influenza A and B |
Acyclovir and valacyclovir
Acyclovir is available in intravenous, oral, and topical formulations. Oral acyclovir is not readily absorbed and requires frequent daily dosing. Valacyclovir, a prodrug of acyclovir, was developed to improve gastrointestinal absorption of acyclovir. Valacyclovir is available only in oral formulation.42
Penciclovir and famciclovir
Penciclovir and famciclovir are similar in structure and activity to acyclovir. Famciclovir, the prodrug of penciclovir, is converted to its active form (penciclovir) in the gastrointestinal tract. Famciclovir is available in oral formulation, and penciclovir is available only in a 1% topical cream. Penciclovir and famciclovir seem to have greater in vitro activity against HSV and VZV than acyclovir.42
Ganciclovir and valganciclovir
Ganciclovir is a guanine nucleoside analogue with a mechanism of action similar to acyclovir. Valganciclovir is a newly approved prodrug of ganciclovir that improves ganciclovir absorption. Ganciclovir has a higher affinity for DNA transferase than acyclovir, which increases the intracellular half-life of the drug and allows less frequent dosing. Valganciclovir is available only in oral formulation; ganciclovir is available in oral, intravenous, and intraocular (eye implant) formulations.42
Cidofovir
Cidofovir is an acyclic phosphonate nucleoside analogue that has potent antiviral activity against a wide variety of viruses. In contrast to the guanine nucleoside analogues, cidofovir has enhanced activity against HSV, EBV, VZV, and CMV. Cidofovir is available only in intravenous formulation.42
Foscarnet
Foscarnet is a pyrophosphonate nucleoside analogue that has potent antiviral activity against HSV, EBV, VZV, and CMV. In addition, foscarnet has shown activity against hepatitis B and influenza viruses. It is poorly absorbed, and it is available only in intravenous formulation.42
Amantadine and rimantadine
Amantadine and rimantadine are closely related antiviral agents with activity against influenza A only. Amantadine has also been used in the treatment of Parkinson disease. Both agents are well absorbed from the gastrointestinal tract and are suitable for oral administration.42
Oseltamivir
Oseltamivir belongs to a new class of antivirals known as neuraminidase inhibitors. Oseltamivir is a prodrug that is converted to its active form (oseltamivir carboxylate) after it is absorbed. It is available only as an oral formulation.42
Adverse reactions and precautions
 RESPIRATORY CARE ASSESSMENT OF ANTIBIOTIC THERAPY
RESPIRATORY CARE ASSESSMENT OF ANTIBIOTIC THERAPY SELF-ASSESSMENT QUESTIONS
SELF-ASSESSMENT QUESTIONS
1. What is the difference between bacteriostatic and bactericidal antimicrobial agents?
2. Describe the difference between antimicrobial agents that act in a concentration-dependent manner and agents that act in a time-dependent manner.
3. Describe at least three parameters that may indicate antibiotic failure in a patient.
4. Why is combination antibiotic therapy useful? (Be specific.)
5. Describe the mechanism of action of penicillin antibiotics. Name at least two additional antibiotic classes with similar mechanisms of action.
6. Which β-lactam antibiotic is least likely to cause an allergic reaction in a patient with a penicillin allergy?
7. Name three antimicrobial agents that would be useful in the treatment of community-acquired pneumonia.
8. What is the antimicrobial agent of choice for treatment of Pneumocystis pneumonia (PCP)?
9. What agents are considered first-line therapy for treatment of pulmonary tuberculosis?
10. Which antimicrobial agents are useful for the treatment of nosocomial pneumonia caused by Pseudomonas aeruginosa?
 CLINICAL SCENARIO
CLINICAL SCENARIO