Antidepressants
Depression is one of the most common medical conditions in the United States, with a lifetime prevalence of 16.2%.1 Whereas many treatment strategies are used in the management of depressed patients, pharmacotherapy remains a cornerstone of modern practice. Modern antidepressant therapy hinges on the 50-year-old monoamine hypothesis, which suggests that depressive symptoms are mediated through an imbalance of the dopaminergic, noradrenergic, and serotonergic systems.1,2 As a result, numerous antidepressant classes have emerged in an attempt to increase synaptic monoamine concentrations.
In the early 1950s, isoniazid and iproniazid were introduced for the treatment of tuberculosis. Shortly after, it was noted that these patients had improved mood, which was attributed to the ability of iproniazid to inhibit monoamine oxidase. Iproniazid, a derivative of isoniazid, subsequently became the first drug marketed specifically as an antidepressant.3 This led to the advent of other monoamine oxidase inhibitors (MAOIs). In 1956, the antidepressant effect of imipramine, a tricyclic agent, was recognized, and it was marketed the following year.4 The MAOIs and tricyclic antidepressants (TCAs) became the mainstay for treatment of depression for several decades until the advent of the safer selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs).
Antidepressant overdose is highly lethal. It accounts for only 3.7% of human exposures reported to U.S. Poison Control Centers but is involved in 10.5% of all poisoning fatalities.5
Monoamine Oxidase Inhibitors
Monoamine oxidase (MAO) is located on the outer mitochondrial membrane and is responsible for breakdown of cytoplasmic catecholamines. Type A (MAO-A) primarily deaminates serotonin and norepinephrine; type B (MAO-B) primarily deaminates phenylethylamine.6 Tyramine and dopamine are metabolized equally by both isoenzymes.3 Whereas most tissues contain both isozymes, MAO-A is primarily found in the placenta, sympathetic nerve terminals, and intestinal mucosa; MAO-B is found primarily in platelets and the basal ganglia.3
Although MAOIs have fallen out of favor for treatment of depression, their use in treatment of Parkinson’s disease is increasing. Drugs that selectively inhibit MAO-B disproportionately increase dopamine concentrations in the striatum.7 Selegiline is an irreversible MAO-B inhibitor used in the treatment of Parkinson’s disease. Although it is marketed as a selective inhibitor of MAO-B, at high doses its receptor specificity is lost, and it can cause inhibition of MAO-A as well.8 Rasagiline is also an irreversible inhibitor of MAO-B but appears to be more potent than selegiline.6 Furthermore, unlike selegiline, which is metabolized to L-methamphetamine, rasagiline is not metabolized to an amphetamine derivative.6
Clinical Features
Patients who take nonselective MAOIs in therapeutic doses are at risk for food-drug interactions. Tyramine is an indirectly acting sympathomimetic amine that is present in aged cheeses, red wine, smoked or pickled and aged meats, and other foods. Usually, tyramine is metabolized in the gut and liver by monoamine oxidase, rarely causing systemic effects. When MAO-A is inhibited, tyramine is absorbed systemically and enters presynaptic vesicles, ultimately causing release of norepinephrine and serotonin into the synapse, leading to a hypertensive crisis.9 This tyramine syndrome, which can occur within minutes to hours of ingestion of foods with high tyramine content, is characterized by headache, hypertension, flushing, and diaphoresis. This syndrome can occur up to 3 weeks after discontinuation of a nonselective MAOI. Although it is theoretically possible, this syndrome is rare with therapeutic use of MAO-B inhibitors.10
Diagnostic Strategies
Symptomatic patients presenting after an MAOI overdose should have an electrocardiogram with measurement of serum glucose and electrolytes if they are obtunded (see Chapter 147 for general management).
Management
Patients presenting with a tyramine reaction may have spontaneous resolution of symptoms during 6 hours. Severe hypertension higher than 200 mm Hg systolic with symptoms may be treated with phentolamine or nitroprusside. Patients with persistent severe headache and hypertension should have a head CT scan to look for intracranial hemorrhage. Patients with chest pain should be evaluated for myocardial infarction (see Chapter 26).
Treatment of suspected serotonin syndrome is supportive (see later section).
Tricyclic Antidepressants
In the 1950s, imipramine became the first tricyclic antidepressant (TCA) used for the treatment of depression. Until the introduction of the SSRIs, TCAs remained the primary agents for treatment of depression. The therapeutic benefit of TCAs results from monoamine reuptake inhibition.11 Whereas use of TCAs for treatment of depression has waned, use for other conditions, including nocturnal enuresis, attention-deficit/hyperactivity syndrome, trigeminal neuralgia, and migraines, has increased.
Clinical Features
Cyclic antidepressant toxicity can result from overdose of a TCA or drug-drug interactions. Overdose is more commonly associated with life-threatening toxicity, but toxic effects can also occur when a TCA is combined with drugs that impair its metabolism through cytochrome P450. Tertiary amine TCAs, such as amitriptyline, imipramine, and clomipramine, are substrates of CYP2C19 and CYP1A2. Doxepin is also a substrate for CYP2D6. Drug-induced inhibition of these enzymes as well as genetic polymorphisms can decrease metabolism of these drugs, resulting in unexpectedly high serum concentrations and clinical toxicity.12 Conversely, inhibition of CYP2D6 and other P450 enzymes by these TCAs can also lead to increased serum concentrations of other drugs metabolized by the same enzymes. Because desipramine and nortriptyline are only weak CYP2D6 inhibitors, they cause fewer drug-drug interactions. Another drug interaction that occurs with TCAs is the serotonin syndrome, which occasionally results when a TCA is combined with another serotonergic drug, such as MAOI or SSRI.
After an overdose of a TCA, symptoms typically begin within 1 to 2 hours. With smaller ingested amounts, symptoms may be minimal and resolve quickly; patients who take large amounts may deteriorate rapidly soon after ingestion. All seriously poisoned patients have symptoms within 6 hours of an overdose. Early TCA poisoning is characterized primarily by anticholinergic effects. Patients typically present with tachycardia, flushed and dry skin, mydriasis, and altered level of consciousness. They may be alert and confused, severely agitated, mute, hallucinating, or even deeply comatose. Speech is often rapid and mumbling in character. Urinary retention is common. Seizures may occur and are likely to be multifactorial, resulting from increased synaptic monoamines, sodium channel inhibition, and possibly γ-aminobutyric acid (GABA) receptor antagonism. Early hypertension is common from the anticholinergic effects of the TCA and excess norepinephrine in the synapse from blockade of norepinephrine reuptake, but hypotension may also be due to alpha-receptor antagonism and also norepinephrine depletion. Later myocardial depression resulting from severe sodium channel antagonism may also lead to hypotension and bradycardia.13,14 Significant sodium channel blockade is associated with widening of the QRS interval. TCAs also block potassium efflux, which leads to a prolonged QT interval.15 With severe poisoning, the combined effects of the TCA on various receptors and ion channels lead to depressed level of consciousness, seizures, hypotension, and wide-complex cardiac arrhythmias.
Diagnostic Strategies
After overdose, the electrocardiogram can yield prognostic information. Early anticholinergic effects cause sinus tachycardia, which occurs virtually uniformly before other effects. Whereas the serum tricyclic concentrations are not particularly beneficial in predicting adverse events, the electrocardiogram is prognostic. A QRS duration longer than 100 milliseconds is predictive of seizures, whereas a QRS duration longer than 160 milliseconds is predictive of ventricular dysrhythmias.16 Additional findings on the electrocardiogram include a rightward shift of the terminal 40 milliseconds of the QRS complex seen as an R wave in aVR longer than 3 milliseconds.17 QT prolongation is less important clinically than the QRS duration. Urine drug of abuse screens commonly test for the presence of TCAs, but a positive test result suggests only use of a TCA or another xenobiotic that cross-reacts with the screen. Serum tricyclic levels do not correlate with severity of illness.16 General management (see Chapter 147) suggests other supportive care measures.
Management
Patients with sinus tachycardia alone do not need specific treatment but should be monitored to detect QRS widening early. Early hypertension should not be treated. Hypotensive patients should first receive fluid resuscitation with an isotonic crystalloid.13 Patients who remain hypotensive should be treated with direct-acting vasopressors, such as norepinephrine and epinephrine.13,14 There are some data that epinephrine may be superior to norepinephrine in this setting.
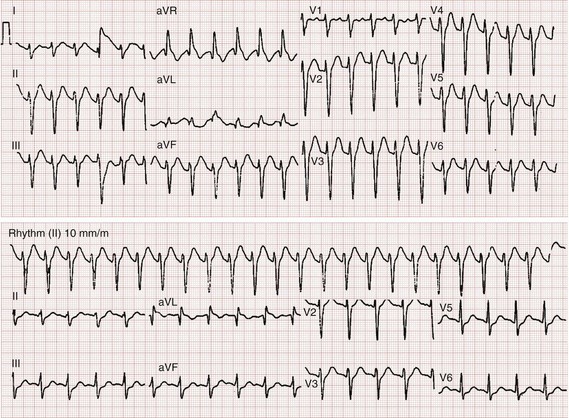
Hypertonic sodium bicarbonate should not be given prophylactically and should be given only to treat specific evidence of sodium channel blockade, such as a wide QRS and ventricular dysrhythmias. Recommendations vary about how to administer this therapy. A conservative approach is to administer a bolus of 1 to 2 mEq/kg hypertonic sodium bicarbonate if the QRS interval exceeds 100 milliseconds. This dose may be repeated in a few minutes if the QRS does not narrow. A sodium bicarbonate infusion can be used to maintain a pH between 7.50 and 7.55.13 This can be done by administration of 5% dextrose in water with 150 mEq of sodium bicarbonate and 40 mEq of potassium added to each liter of fluid at a rate of twice maintenance starting at 35 mEq/hr and titrating to pH. Alternatively, infusions of 1 mEq sodium bicarbonate per milliliter of fluid may be used if volume overload is a concern. Additional boluses of sodium bicarbonate may be necessary if the QRS widens or if adequate alkalinization is not achieved with infusion alone. Figure 151-1 demonstrates a 12-lead electrocardiogram from a patient poisoned with a TCA before and after sodium bicarbonate therapy. If ventricular dysrhythmias persist despite maximal alkalinization (pH > 7.55), 200 mL of 3% hypertonic saline (in an adult) can be used.18 Class IA and IC antidysrhythmics can worsen cardiac toxicity and should be avoided.13 Seizures are treated with lorazepam or diazepam and with phenobarbital if they are refractory to benzodiazepines. Because seizure leads to acidosis and worsens the cardiac status, seizing patients who do not respond quickly should be rapidly paralyzed and intubated if necessary to prevent increasing acidosis.
Intravenous lipid emulsion (ILE) therapy has gained interest recently for reversal of toxicity caused by lipophilic drugs, including TCAs. Although the exact mechanism of ILE is not known, it probably involves redistribution of a lipophilic drug from the tissue receptors back into the vascular compartment in the context of a large bolus of concentrated lipid solution, the so-called lipid sink phenomenon.19,20 Other mechanisms also are possible. Because of the potential for iatrogenic harm, its use is currently reserved for life-threatening toxicity that remains refractory to sodium bicarbonate. There are several different dosing strategies. One approach is to give 1.5 mL/kg of a 20% lipid solution during 2 to 3 minutes. This bolus can be repeated once in 5 minutes if there is no clinical improvement. If clinical improvement does occur, the bolus should be followed by an infusion of 0.25 mL/kg/min for 15 to 30 minutes.21
Disposition
If no sinus tachycardia, decreased level of consciousness, or seizures have developed within 6 hours of an overdose, it is unlikely that toxicity will occur,22 and the patient can be medically cleared from the emergency department for psychiatric disposition, if needed. Patients with signs of cyclic antidepressant toxicity should be admitted to an intensive care unit.
Selective Serotonin Reuptake Inhibitors
SSRIs have a wide therapeutic index.23 Although they are safer in overdose than MAOIs and TCAs, they do have therapeutic limitations, such as the long delay until onset of antidepressant effect.24 The SSRIs undergo hepatic metabolism. There is considerable variability in their half-life; however, paroxetine has one of the shortest half-lives (17 hours) compared with fluoxetine, which has one of the longest half-lives (53 hours for parent drug, 240 hours for active metabolite).25
Clinical Features
Overdose of SSRIs is usually well tolerated and rarely fatal, with ingestions of up to 30 times the daily dose associated with few or no symptoms. Gastrointestinal upset and mild CNS depression can occur with large overdoses. Coma and seizures are rare, with incidences of approximately 2.4 and 1.9%, respectively.23 The incidence of serotonin syndrome after SSRI overdose is variable, up to 14%,23 but most other series report a much lower incidence.
Citalopram overdose deserves special mention because of a higher rate of QTc prolongation and seizures compared with other SSRIs.26–28 Furthermore, onset of QTc prolongation may be delayed for up to 13 hours.29 Despite being the active enantiomer of citalopram, escitalopram appears to be less toxic than citalopram, with a lower incidence of seizure and QTc prolongation.26
Therapeutic administration of SSRIs may be associated with the syndrome of inappropriate secretion of antidiuretic hormone (SIADH).30,31 Most cases of hyponatremia develop within 1 month and frequently within the first 2 weeks.31 The overall incidence is not clear; however, in one small study, 12.5% of elderly patients taking an SSRI had SIADH, with an additional 12.5% of patients having mild asymptomatic hyponatremia.
Diagnostic Strategies
Diagnosis of SSRI toxicity is often dependent on obtaining a history of overdose. Clinical features of toxicity are similar to those seen after overdose of many other toxicants, and urine drug of abuse screens do not detect SSRIs. An electrocardiogram can assess for conduction disturbances, especially QT prolongation. See Chapter 147 for other general management strategy. Specific SSRI levels are not performed by most hospital laboratories and do not influence management. They may help confirm overdose retrospectively.
Disposition
Patients who overdose with an SSRI who are asymptomatic after 6 hours of monitoring are unlikely to have toxicity. Some authors advocate for 13 hours of observation29 after the ingestion of more than 1000 mg of citalopram or escitalopram. Symptomatic patients should be admitted to a monitored care setting.
Serotonin-Norepinephrine Reuptake Inhibitors
Duloxetine, venlafaxine, desvenlafaxine, and milnacipran are collectively referred to as serotonin-norepinephrine reuptake inhibitors (SNRIs). Desvenlafaxine, venlafaxine, and duloxetine are marketed in the United States for treatment of depression. Milnacipran, although it is used as an antidepressant in Europe, is currently approved in the United States only for treatment of fibromyalgia. In addition to the treatment of depression, duloxetine is commonly used in the management of diabetic neuropathy, fibromyalgia, and urinary stress incontinence.32 Venlafaxine and its active metabolite desvenlafaxine are both available medicinally. The SNRIs may also produce dose-dependent inhibition of sodium channels.33
Clinical Features
Duloxetine overdose produces CNS depression.34 Most fatal overdoses of duloxetine described thus far have involved coingestants.32 Thus, ascribing causality to duloxetine in these fatal cases is difficult.32
Venlafaxine and desvenlafaxine overdoses can result in neurologic, muscular, and cardiovascular toxicity.35 Common findings include CNS depression, seizures, and tachycardia. Ventricular arrhythmias and QRS prolongation on the electrocardiogram may also occur.33,36,37 After venlafaxine overdose, rhabdomyolysis can develop independently or as a result of seizures.35,38 Both serotonin syndrome and takotsubo cardiomyopathy occur with venlafaxine overdose.39
Experience with milnacipran overdose is limited. Expected toxic effects include CNS depression and tachycardia. Acute, reversible cardiomyopathy,40 serotonin syndrome, and at least one fatal intoxication have been described after overdose.40–42
Diagnostic Strategies
Diagnosis of SNRI ingestion depends on history before onset of symptoms. Specific drug levels are not rapidly available and do not aid management. An electrocardiogram can detect QRS or QT interval prolongation. SNRIs are not detected by urine drug of abuse screens, but venlafaxine is associated with a false-positive phencyclidine screen.43,44
Miscellaneous Antidepressants
Bupropion is a monocyclic antidepressant that is structurally related to amphetamine.45 In addition to its use as an antidepressant, it is used to assist in smoking cessation.46,47 Its primary mechanism of action is inhibition of dopamine and norepinephrine reuptake, but it also acts as a noncompetitive inhibitor of nicotinic acetylcholine receptors.47
Seizures are a dose-dependent phenomenon and can occur with therapeutic dosing or overdose of bupropion.48 At doses of 300 to 450 mg/day, the incidence of seizures is approximately 0.4%. Doses of 450 to 600 mg/day, however, are associated with a tenfold increased incidence of seizures.48 Among sustained-release preparations, doses less than 450 mg/day are associated with a seizure incidence of 0.1%.48
Sinus tachycardia, tonic-clonic seizures, and agitation are common after overdose.45,49,50 Intraventricular conduction delay is rare.45 Because of the risk of delayed seizures, patients with intentional overdoses of extended-release bupropion should be admitted for 24 hours of observation.51
Trazodone
Trazodone is an atypical antidepressant with a mechanism of action that includes antagonism of the 5-hydroxytryptamine type 2A (5-HT2A) receptor and alpha1 receptor. Its use as an antidepressant has been somewhat limited by adverse effects, such as orthostatic hypotension, priapism, and sedation.52
Serotonin Syndrome
Serotonin syndrome is a potentially lethal condition resulting from excess serotonin accumulation in the synaptic cleft. This syndrome occurs after an isolated overdose of an SSRI, but it is more commonly a result of drug-drug interactions, especially with drug combinations that raise synaptic serotonin concentrations by different mechanisms.53 Agonism of the 5-HT2A receptor appears to be largely responsible for this condition in humans.53–55
Whereas numerous xenobiotics have been implicated in causing serotonin syndrome, some of the most common are the SSRIs, SNRIs, TCAs, MAOIs, dextromethorphan, amphetamines, and designer amphetamines, including methylenedioxymethamphetamine (“ecstasy”), cocaine, meperidine, lithium, tramadol, buspirone, lysergic acid diethylamide (LSD), and linezolid53,56–60 (Table 151-1). Serotonin syndrome is more likely to develop when drugs from different classes are combined, resulting in increased serotonin in the synaptic cleft from different mechanisms (e.g., increased release and impaired uptake).
Table 151-1
Xenobiotics Commonly Implicated in Serotonin Syndrome
Analgesics: tramadol, meperidine, pentazocine
Drugs of abuse: cocaine, amphetamine derivatives (e.g., methylenedioxymethamphetamine), lysergic acid diethylamide (LSD)
Monoamine oxidase inhibitors (e.g., isocarboxazid, linezolid, phenelzine, moclobemide, selegiline)
Miscellaneous: dextromethorphan, lithium, metoclopramide, St. John’s wort
Selective serotonin reuptake inhibitors (e.g., milnacipran, venlafaxine)
Serotonin-norepinephrine reuptake inhibitors (e.g., citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, sertraline)
Tricyclic antidepressants (e.g., amitriptyline, clomipramine, desipramine, doxepin, imipramine, nortriptyline)
Clinical Features
Serotonin syndrome is described as a triad of mental status changes, autonomic instability, and increased neuromuscular activity, but the condition exists along a spectrum; some patients have only mild tremor and diarrhea, whereas others exhibit life-threatening manifestations.53 Clinical features may include tremor, akathisia, gastrointestinal illness, clonus (inducible or spontaneous), rigidity, fever, seizures, and autonomic instability. The clonus is typically more pronounced in the lower extremities than in the upper extremities.
Diagnostic Strategies
The Sternbach criteria were developed in 1991 and became the first widely used diagnostic algorithm.61 Additional criteria, including the Hunter criteria62 and the Boyer and Shannon criteria,53 have been developed. The Hunter criteria (Table 151-2) appear to be more sensitive than the Sternbach criteria, with fewer false positives.62
Table 151-2
The Hunter Criteria for Serotonin Syndrome62
In the setting of exposure to a known serotonergic agent, serotonin syndrome can be diagnosed by the presence of any of the following:
Management
Management is supportive, with removal of the offending agents being paramount. Mild cases may require only discontinuation of the offending agent and low-dose benzodiazepines for rigidity. More severe cases may require intravenous fluid resuscitation and large doses of benzodiazepines or other sedative-hypnotic agents to gain control of symptoms. A trial of cyproheptadine, a 5-HT2A antagonist, is an appropriate adjunctive therapy for more severe cases,53,63 but there are no randomized controlled trials demonstrating improved benefit with cyproheptadine over supportive care and benzodiazepines alone.64 Patients with hyperthermia that does not respond promptly to sedation with benzodiazepines should receive a nondepolarizing neuromuscular blocking agent after endotracheal intubation.
Discontinuation Syndromes
After the abrupt discontinuation of certain antidepressants, patients can experience a withdrawal, or discontinuation, syndrome. Unlike potentially life-threatening GABA withdrawal from ethanol or benzodiazepines, the discontinuation syndrome from antidepressants is rarely life-threatening but can result in significant discomfort. One notable exception involves neonates born to mothers using TCAs65 or SSRIs, who can have serious, potentially life-threatening withdrawal. Antidepressant discontinuation syndrome does not always develop, but when it does, it typically starts within the first 3 days after therapy is stopped.66 This syndrome is difficult to distinguish from recurrence of the underlying depression, which has overlap of some symptoms.
Antidepressant discontinuation syndrome was first described in the 1950s with imipramine, but it has been described with all major classes of antidepressants.66 Withdrawal from SSRIs involves both physical and psychological symptoms, most commonly nausea, lethargy, headache, and dizziness. The symptoms can be divided into six general categories: dysequilibrium (e.g., dizziness, ataxia), sleep disturbances, gastrointestinal symptoms, affective symptoms (e.g., irritability, anxiety), sensory symptoms (e.g., electric shock–like sensation, paresthesias), and general somatic symptoms (e.g., headache, tremor, anorexia, diaphoresis).66 The syndrome is more common after discontinuation of drugs with shorter half-lives (e.g., paroxetine) than of drugs with longer half-lives (e.g., fluoxetine). TCA withdrawal is similar to SSRI withdrawal, although sensory abnormalities and equilibrium disturbances are rare with TCA discontinuation. Non–life-threatening arrhythmias are rare after discontinuation of the TCAs.
References
1. Bostwick, JM. A generalist’s guide to treating patients with depression with an emphasis on using side effects to tailor antidepressant therapy. Mayo Clin Proc. 2010;85:538–550.
2. Lee, S, Jeong, J, Kwak, Y, Park, SK. Depression research: Where are we now? Mol Brain. 2010;10:8.
3. Youdim, MB, Weinstock, M. Therapeutic applications of selective and non-selective inhibitors of monoamine oxidase A and B that do not cause significant tyramine potentiation. Neurotoxicology. 2004;25:243–250.
4. Lopez-Munoz, F, Alamo, C. Monoaminergic neurotransmission: The history of the discovery of antidepressants from 1950s until today. Curr Pharm Des. 2009;15:1563–1586.
5. Bronstein, AC, et al. 2010 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 28th Annual Report. Clin Toxicol (Phila). 2011;49:910–941.
6. Chen, JJ, Ly, AV. Rasagiline: A second-generation monoamine oxidase type B inhibitor for the treatment of Parkinson’s disease. Am J Health Syst Pharm. 2006;63:915–928.
7. Youdim, MB, Bakhle, YS. Monoamine oxidase: Isoforms and inhibitors in Parkinson’s disease and depressive illness. Br J Pharmacol. 2006;147:S287–S296.
8. Gerlach, M, Youdim, MB, Reiderer, P. Pharmacology of selegiline. Neurology. 1996;47(6 Suppl 3):S137–S145.
9. Stahl, SM, Felker, A. Monoamine oxidase inhibitors: A modern guide to an unrequited class of antidepressants. CNS Spectr. 2008;13:855–870.
10. LeWitt, PA. MAO-B inhibitor know-how: Back to the pharm. Neurology. 2009;72:1352–1357.
11. Preskorn, SH. CNS drug development. Part I: The early period of CNS drugs. J Psychiatr Pract. 2010;16:344–349.
12. Gillman, PK. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol. 2007;151:737–748.
13. Kerr, GW, McGuffie, AC, Wilkie, S. Tricyclic antidepressant overdose: A review. Emerg Med J. 2001;18:236–241.
14. Bradberry, SM, Thanacoody, HK, Watt, BE, Thomas, SH, Vale, JA. Management of the cardiovascular complications of tricyclic antidepressant poisoning: Role of sodium bicarbonate. Toxicol Rev. 2005;24:195–204.
15. Alvarez, PA, Pahissa, J. QT alterations in psychopharmacology: Proven candidates and suspects. Curr Drug Saf. 2010;5:97–104.
16. Boehnert, MT, Lovejoy, FHJr. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an acute overdose of tricyclic antidepressants. N Engl J Med. 1985;313:474–479.
17. Liebelt, EL, Francis, PD, Woolf, AD. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med. 1995;26:195–201.
18. McCabe, JL, Cobaugh, DJ, Menegazzi, JJ, Fata, J. Experimental tricyclic antidepressant toxicity: A randomized, controlled comparison of hypertonic saline solution, sodium bicarbonate, and hyperventilation. Ann Emerg Med. 1998;32:329–333.
19. Jamaty, C, et al. Lipid emulsions in the treatment of acute poisoning: A systematic review of human and animal studies. Clin Toxicol (Phila). 2010;48:1–27.
20. Harvey, M, Cave, G. Intralipid outperforms sodium bicarbonate in a rabbit model of clomipramine toxicity. Ann Emerg Med. 2007;49:178–185.
21. American College of Medical Toxicology. Interim guidance for the use of lipid resuscitation therapy. http://www.acmt.net/_Library/Membership_Documents/Lipid_resuscitation_G_4F9E49_-_for_member_comment.pdf, 2010.
22. Woolf, AD, et al. Tricyclic antidepressant poisoning: An evidence-based consensus guideline for out-of-hospital management. Clin Toxicol (Phila). 2007;45:203–233.
23. Isbister, GK, Bowe, SJ, Dawson, A, Whyte, IM. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol. 2004;42:277–285.
24. Olver, JS, Burrows, GD, Norman, TR. Third-generation antidepressants: Do they offer advantages over the SSRIs? CNS Drugs. 2001;15:941–954.
25. O’Donnell, JM, Shelton, RC. Drug therapy of depression and anxiety disorders. In: Brunton LL, Chabner BA, Knollman BC, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 12th ed. New York: McGraw-Hill; 2011:397–415.
26. Hayes, BD, Klein-Schwartz, W, Clark, RF, Muller, AA, Miloradovich, JE. Comparison of toxicity of acute overdoses with citalopram and escitalopram. J Emerg Med. 2010;39:44–48.
27. Jimmink, A, Caminada, K, Hunfeld, NG, Touw, DJ. Clinical toxicology of citalopram after acute intoxication with the sole drug or in combination with other drugs: Overview of 26 cases. Ther Drug Monit. 2008;30:365–371.
28. Catalano, G, Catalano, MC, Epstein, MA, Tsambiras, PE. QTc interval prolongation associated with citalopram overdose: A case report and literature review. Clin Neuropharmacol. 2001;24:158–162.
29. Isbister, GK, Friberg, LE, Duffell, SB. Application of pharmacokinetic-pharmacodynamic modeling in management of QT abnormalities after citalopram overdose. Intensive Care Med. 2006;32:1060–1065.
30. Jacob, S, Spinler, SA. Hyponatremia associated with selective serotonin-reuptake inhibitors in older adults. Ann Pharmacother. 2006;40:1618–1622.
31. Kirby, D, Ames, D. Hyponatremia and selective serotonin re-uptake inhibitors in elderly patients. Int J Geriatr Psychiatry. 2001;16:484–493.
32. Vey, EL, Kovelman, I. Adverse events, toxicity, and post-mortem data on duloxetine: Case reports and literature survey. J Forensic Leg Med. 2010;17:175–185.
33. Bosse, GM, Spiller, HA, Collins, AM. A fatal case of venlafaxine overdose. J Med Toxicol. 2008;4:18–20.
34. Kruithof, MK, Bruins, NA, van Roon, EN. Coma after overdose with duloxetine. Ann Pharmacother. 2011;45:e5.
35. Pascale, P, Oddo, M, Pacher, P, Augsburger, M, Liaudet, L. Severe rhabdomyolysis following venlafaxine overdose. Ther Drug Monit. 2005;27:562–564.
36. Isbister, GK. Electrocardiogram changes and arrhythmias in venlafaxine overdose. Br J Clin Pharmacol. 2009;67:572–576.
37. Howell, C, Wilson, AD, Waring, WS. Cardiovascular toxicity due to venlafaxine poisoning in adults: A review of 235 consecutive cases. Br J Clin Pharmacol. 2007;64:192–197.
38. Wilson, AD, Howell, C, Waring, WS. Venlafaxine ingestion is associated with rhabdomyolysis in adults: A case series. J Toxicol Sci. 2007;32:97–101.
39. Christoph, M, et al. Broken heart syndrome: Tako Tsubo cardiomyopathy associated with an overdose of the serotonin-norepinephrine reuptake inhibitor venlafaxine. Eur Neuropsychomarmacol. 2010;20:594–597.
40. Levine, M, Truitt, CA, O’Connor, AD. Acute cardiomyopathy complicating a milnacipran overdose. J Med Toxicol. 2011;7:312–316.
41. Yacoub, HA, Johnson, WG, Souayah, N. Serotonin syndrome after administration of milnacipran for fibromyalgia. Neurology. 2010;74:699–700.
42. Fanton, L, et al. Fatal intoxication with milnacipran. J Forensic Leg Med. 2008;15:388–390.
43. Santos, PM, et al. False positive phencyclidine results caused by venlafaxine. Am J Psychiatry. 2007;164:349.
44. Bond, GR, Steele, PE, Uges, DR. Massive venlafaxine overdose resulted in a false positive Abbott AxSYM urine immunoassay for phencyclidine. J Toxicol Clin Toxicol. 2003;41:999–1002.
45. Curry, SC, Kashani, JS, LoVecchio, F, Holubek, W. Intraventricular conduction delay after bupropion overdose. J Emerg Med. 2005;29:299–305.
46. McNeil, JJ, Piccenna, L, Ioannides-Demos, LL. Smoking cessation—recent advances. Cardiovasc Drugs Ther. 2010;24:359–367.
47. Damaj, MI, et al. Enantioselective effects of hydroxyl metabolites of bupropion on behavior and on function of monoamine transporters and nicotinic receptors. Mol Pharmacol. 2004;66:675–682.
48. Dhillon, S, Yang, LP, Curran, MP. Bupropion: A review of its use in the management of major depressive disorder. Drugs. 2008;68:653–689.
49. Belson, MG, Kelley, TR. Bupropion exposures: Clinical manifestations and medical outcome. J Emerg Med. 2002;23:223–230.
50. Shepherd, G, Velez, LI, Keyes, DC. Intentional bupropion overdoses. J Emerg Med. 2004;27:147–151.
51. Starr, P, et al. Incidence and onset of delayed seizures after overdoses of extended-release bupropion. Am J Emerg Med. 2009;27:911–915.
52. Mendelson, WB. A review of the evidence for the efficacy and safety of trazodone in insomnia. J Clin Psychiatry. 2005;66:469–476.
53. Boyer, EW, Shannon, M. The serotonin syndrome. N Engl J Med. 2005;352:1112–1120.
54. Nisijima, K, Shioda, K, Yoshino, T, Takano, K, Kato, S. Memantine, an NMDA antagonist, prevents the development of hyperthermia in an animal model for serotonin syndrome. Pharmacopyschiatry. 2004;37:57–62.
55. Sun-Edelstein, C, Tepper, SJ, Shapiro, RE. Drug-induced serotonin syndrome: A review. Expert Opin Drug Saf. 2008;7:587–596.
56. Quinn, DK, Stern, TA. Linezolid and serotonin syndrome. Prim Care Companion J Clin Psychiatry. 2009;11:353–356.
57. Schwartz, AR, Pizon, AF, Brooks, DE. Dextromethorphan-induced serotonin syndrome. Clin Toxicol (Phila). 2008;46:771–773.
58. Adan-Manes, J, Novalbos, J, Lopez-Rodriguez, R, Ayuso-Mateos, JL, Abad-Santos, F. Lithium and venlafaxine interaction: A case of serotonin syndrome. J Clin Pharm Ther. 2006;31:397–400.
59. Devlin, RJ, Henry, JA. Clinical review: Major consequences of illicit drug consumption. Crit Care. 2008;12:202.
60. Bijl, D. The serotonin syndrome. Neth J Med. 2004;62:309–313.
61. Sternbach, H. The serotonin syndrome. Am J Psychiatry. 1991;148:705–713.
62. Dunkley, EJ, Isbister, GK, Sibbritt, D, Dawson, AH, Whyte, IM. The Hunter Serotonin Toxicity Criteria: Simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003;96:635–642.
63. Rusyniak, DE, Sprague, JE. Toxin-induced hyperthermic syndromes. Med Clin North Am. 2005;89:1277–1296.
64. Graudins, A, Stearman, A, Chan, B. Treatment of the serotonin syndrome with cyproheptadine. J Emerg Med. 1998;16:615–619.
65. Cowe, L, Lloyd, DJ, Dawling, S. Neonatal convulsion caused by withdrawal from maternal clomipramine. Br Med J (Clin Res Ed). 1982;284:1837–1838.
66. Haddad, PM. Antidepressant discontinuation syndromes: Clinical relevance, prevention and management. Drug Saf. 2001;24:183–197.