145 Anticholinergics
 Key Points
Key Points• Antimuscarinic poisoning syndrome is a more appropriate description than anticholinergic overdose because only the muscarinic receptors, not the nicotinic acetylcholine receptors, are involved. In this chapter the term anticholinergic will be used for consistency and is specifically meant to indicate antimuscarinic.
• Anticholinergic agents antagonize the neurotransmitter acetylcholine at both central and peripheral muscarinic acetylcholine receptors, which leads to altered mental status, mydriasis, tachycardia, urinary retention, ileus, and dry, flushed skin.
• The diagnosis of anticholinergic syndrome is largely clinical and should include physical examination, fingerstick serum glucose measurement, and an electrocardiogram.
• Anticholinergic syndrome is a key clinical finding leading to the diagnosis of poisoning by tricyclic antidepressants (a subset of antimuscarinic agents).
• Basic treatment involves supportive care of the vital signs, activated charcoal, benzodiazepines for agitation, sodium bicarbonate for a QRS complex longer than 100 msec or wide-complex tachycardia, and physostigmine for consequential central and peripheral anticholinergic (antimuscarinic) manifestations, if appropriate.
Epidemiology
Because anticholinergic toxicologic syndrome (toxidrome) is common, recognition of its associated signs and symptoms is a necessary clinical skill. It occurs following exposure to many seemingly unrelated agents, many of which are available without prescription or used in patients with a propensity toward self-harm (Box 145.1). For example, according to data from the American Association of Poison Control Centers, 25,788 single exposures to diphenhydramine alone occurred in 2008, with 201 major outcomes and 3 deaths.1
Pathophysiology and Pharmacology
Muscarinic acetylcholine receptors are linked to G proteins to execute their postreceptor effects. They are found primarily in the CNS (most abundantly in the brain). They are also present at effector organs innervated by postganglionic parasympathetic neurons. Stimulation of these end-organs, either pharmacologically or through enhanced neuronal output, results in miosis, lacrimation, salivation, bronchospasm, bronchorrhea, bradydysrhythmia, urination, and increased gastrointestinal motility (Table 145.1). Finally, muscarinic receptors are located in sweat glands innervated by postsynaptic sympathetic neurons and cause diaphoresis when stimulated.
Table 145.1 Pathophysiology of Anticholinergic (Antimuscarinic Poisoning Syndrome) Symptoms
| ANTICHOLINERGIC EFFECT | SYMPTOMS |
|---|---|
| Central inhibition of muscarinic acetylcholine receptors | Confusion, disorientation, psychomotor agitation, ataxia, myoclonus, tremor, picking movements, abnormal speech, visual and auditory hallucinations, psychosis, seizures, cardiovascular collapse, coma |
| Inhibition of postsynaptic sympathetic muscarinic acetylcholine receptors in the sweat glands, as well as vasodilation of peripheral blood vessels | Dry, flushed skin |
| Inhibition of postsynaptic parasympathetic muscarinic acetylcholine receptors in the: | |
| Salivary glands | Dry mucous membranes |
| Eye | Paralysis of the sphincter muscle of the iris and the ciliary muscle of the lens resulting in mydriasis, cycloplegia, and blurred vision |
| Heart (vagus nerve) | Tachycardia |
| Bladder | Urinary retention and overflow incontinence |
| Bowel | Adynamic ileus |
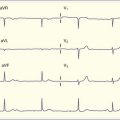
Tricyclic antidepressants are a unique subset of antimuscarinic agents that deserve special attention. Their antidepressant effect is achieved pharmacologically through blockade of the reuptake of norepinephrine, dopamine, and serotonin in the CNS. Additionally, tricyclic antidepressants interact with other channels and receptors and cause considerably more profound clinical toxicity in overdose than occurs with most other agents that exhibit anticholinergic effects. Adverse effects of a tricyclic antidepressant overdose include competitive inhibition at both central and peripheral muscarinic acetylcholine receptors (antimuscarinic poisoning syndrome); histamine receptor antagonism (sedation); sodium channel blockade in the myocardium (widening of the QRS complex or wide-complex dysrhythmias, atrioventricular block, QT prolongation, and rightward shift of the terminal 40-msec QRS axis on an electrocardiogram [ECG], as well as negative inotropy leading to hypotension); α-adrenergic receptor antagonism on vascular smooth muscle (vasodilation leading to hypotension); and although the mechanism of this effect is unclear, γ-aminobutyric acid (GABA) antagonism (seizures). In the right clinical setting, anticholinergic syndrome is a key clinical finding leading to the diagnosis of tricyclic antidepressant poisoning (Table 145.2).
Table 145.2 Pathophysiology of Tricyclic Antidepressants and Associated Symptoms
| EFFECT | SYMPTOMS |
|---|---|
| Blockade of reuptake of norepinephrine, dopamine, and serotonin in the central nervous system | Mood elevation |
| Competitive inhibition at both central and peripheral muscarinic acetylcholine receptors | Antimuscarinic poisoning syndrome |
| Histamine receptor antagonism | Sedation |
| Sodium channel blockade in the myocardium | QRS complex widening or wide-complex dysrhythmias, atrioventricular block, QT prolongation and rightward shift of the terminal 40-msec QRS axis on the electrocardiogram, as well as negative inotropy leading to hypotension |
| α-Adrenergic receptor antagonism on vascular smooth muscle | Vasodilation leading to hypotension |
| γ-Aminobutyric acid antagonism | Seizures |
Presenting Signs and Symptoms
See Tables 145.1 and 145.2, which detail the pathophysiology associated with the signs and symptoms of antimuscarinic poisoning syndrome and tricyclic antidepressants.
Differential Diagnosis and Medical Decision Making
Diagnostic Testing
 Facts and Formulas
Facts and Formulas
A QRS duration shorter than 100 msec predicts that no serious clinical toxicity will occur, a QRS duration longer than 100 msec is associated with a 30% incidence of seizures, and a QRS duration longer than 160 msec is associated with a 50% likelihood of the development of ventricular dysrhythmia.2 Additionally, an R wave in lead aVR measuring 3 mm or greater3 or a terminal 40-msec right axis deviation between 130 and 270 degrees4 is a predictor of tricyclic antidepressant–induced toxicity.
 Red Flags
Red Flags
Cautions for Physicians
Physostigmine is contraindicated in patients after a tricyclic antidepressant overdose with a QRS interval longer than 100 msec because it may cause cardiac arrhythmias and profound hypotension.
Flumazenil should not generally be administered to patients with anticholinergic (antimuscarinic) toxicity because it may cause seizures or other complications.
Failure to become cholinergic (bradycardia, bronchorrhea, diaphoretic, drooling) after the administration of physostigmine is essentially diagnostic of anticholinergic toxicity.
Physostigmine will prolong the action of drugs metabolized by cholinesterases, such as succinylcholine. A nondepolarizing agent should be used instead.
Treatment
Most patients with anticholinergic (antimuscarinic) toxicity can be treated adequately with general supportive care of the airway, breathing, and circulation, followed by frequent reassessment and close observation (Fig. 145.1). To avoid the risk for aspiration, activated charcoal (1 g/kg) is recommended only for patients who are capable of spontaneously drinking and protecting their own airway. It may also be administered cautiously via a nasogastric tube to patients who are endotracheally intubated. Because anticholinergic (antimuscarinic) agents may slow gastrointestinal transit, activated charcoal may be useful several hours after ingestion.5 The specific role of activated charcoal in most of these poisonings has not been studied.
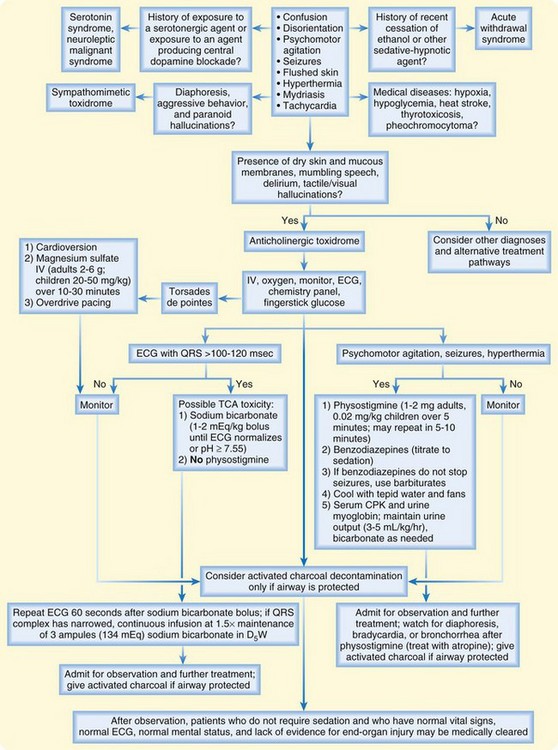
The initial therapy for cardiovascular toxicity secondary to sodium channel blockade is hypertonic sodium bicarbonate in 1- to 2-mEq/kg boluses.6 Treatment with sodium bicarbonate is indicated for patients with a QRS complex longer than 100 to 120 msec or a wide-complex or ventricular tachycardia until the abnormality is reversed or serum pH reaches 7.55. The ECG should be repeated within 60 seconds after a bolus of sodium bicarbonate to check for narrowing of the QRS complex. If narrowing has occurred, a continuous infusion at 1.5 times the maintenance intravenous fluid rate (three ampules [132 mEq] of sodium bicarbonate in 1 L of 5% dextrose in water [D5W]) should be administered. Profound cardiovascular toxicity may require more aggressive interventions, such as the initiation of vasopressors or use of an intraaortic balloon pump.
Antidotal Therapy—Physostigmine
In the setting of a clear diagnosis of anticholinergic toxicity, physostigmine should be administered. It is beneficial in the treatment of agitation and delirium and also shortens the time to recovery after agitation. This agent should not be given once a benzodiazepine has been administered because the end point of clear mental status has been lost.7
Physostigmine is indicated for patients with central (and perhaps peripheral) anticholinergic manifestations. It is contraindicated in patients with a QRS interval longer than 100 msec if the history suggests overdose of tricyclic antidepressant or other cardiotoxic agents. The latter contraindication is based on two case reports of patients with tricyclic antidepressant overdose in whom asystole developed after the administration of physostigmine. The cause of the asystole was theorized to be physostigmine-induced bradycardia that resulted in cardiac conduction defects and decreased cardiac output in the presence of tricyclic antidepressant–induced sodium channel blockade.8 Other contraindications to the use of physostigmine are bronchospastic disease, peripheral vascular disease, intestinal or bladder obstruction, intraventricular conduction defects, and atrioventricular block.
Physostigmine is administered intravenously over a 5-minute period, 1 to 2 mg in adults and 0.02 mg/kg (maximum of 0.5 mg) in children. Its onset of action occurs within minutes.9 This initial dose can be repeated in 5 to 10 minutes if an adequate response is not achieved and muscarinic effects are not noted. Failure of the patient to become cholinergic after the administration of physostigmine is essentially diagnostic of anticholinergic toxicity. Although the total effective dose of physostigmine depends on the individual, as well as the dose and duration of action of the anticholinergic (antimuscarinic) agent, 4 mg is usually a sufficient dose for most patients.10 The half-life of physostigmine is 16 minutes, and its usual duration of action exceeds 1 hour.11
Follow-Up, Next Steps in Care, and Patient Education
 Documentation
Documentation Patient Teaching Tips
Patient Teaching Tips
The regional poison control center can be contacted by telephone at 1-800-222-1222.
A dose of medication other than what was prescribed by the patient’s physician should never be taken unless it has been approved by the ordering physician.
A second medication or herbal supplement should never be added to a previously taken medication without approval from the physician.
Boehnert MT, Lovejoy FH. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an overdose of tricyclic antidepressants. N Engl J Med. 1985;313:474–479.
Niemann JT, Bessen HA, Rothstein RJ, et al. Electrocardiographic criteria for tricyclic antidepressant cardiotoxicity. Am J Cardiol. 1986;57:1154–1159.
Pentel P, Peterson CD. Asystole complicating physostigmine treatment of tricyclic antidepressant overdose. Ann Emerg Med. 1980;9:588–590.
1 Bronstein AC, Spyker DA, Cantilena LR, Jr., et al. 2008 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 26th Annual Report. Clin Toxicol. 2009;47:911–1084.
2 Boehnert MT, Lovejoy FH. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an overdose of tricyclic antidepressants. N Engl J Med. 1985;313:474–479.
3 Leibelt EL. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant overdose. Ann Emerg Med. 1995;26:195–201.
4 Niemann JT, Bessen HA, Rothstein RJ, et al. Electrocardiographic criteria for tricyclic antidepressant cardiotoxicity. Am J Cardiol. 1986;57:1154–1159.
5 Green R, Sitar DS, Tenenbein M. Effect of anticholinergic drugs on the efficacy of activated charcoal. Clin Toxicol. 2004;42:267–272.
6 Sharma AN, Hexdall AH, Chang EK, et al. Diphenhydramine-induced wide complex dysrhythmia responds to treatment with sodium bicarbonate. Am J Emerg Med. 2003;21:212–215.
7 Burns MJ, Linden CH, Graudins A, et al. A comparison of physostigmine and benzodiazepines for the treatment of anticholinergic poisoning. Ann Emerg Med. 2000;35:374–381.
8 Pentle P, Peterson CD. Asystole complicating physostigmine treatment of tricyclic antidepressant overdose. Ann Emerg Med. 1980;9:588–590.
9 Holzgrate RE, Vondrell JJ, Mintz SM. Reversal of postoperative reactions to scopolamine with physostigmine. Anesth Analg. 1973;52:921–925.
10 Forrer GR, Miller JJ. Atropine coma—a somatic therapy in psychiatry. Am J Psychiatry. 1958;115:455–458.
11 Asthana S, Greig NH, Hegedus L, et al. Clinical pharmacokinetics of physostigmine in patients with Alzheimer’s disease. Clin Pharmacol Ther. 1995;58:299–309.