Chapter 3 Antibodies
• Circulating antibodies (also called immunoglobulins) are soluble glycoproteins that recognize and bind antigens, specifically. They are present in serum, tissue fluids or on cell membranes. Their purpose is to help eliminate microorganisms bearing those antigens. Antibodies also function as membrane-bound antigen receptors on B cells, and play key roles in B cell differentiation.
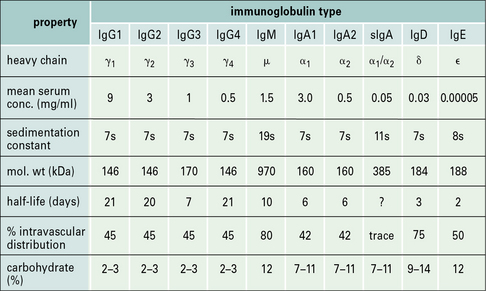
• There are five classes of antibody in mammals – IgG, IgA, IgM, IgD, and IgE. In humans, four subclasses of IgG and two of IgA are also defined. Thus, collectively, there are nine isotypes: IgM, IgA1, IgA2, IgG1, IgG2, IgG3, IgG4, IgD, and IgE.
• Antibodies have a basic structure of four polypeptide chains – two identical light chains and two identical heavy chains. The N- terminal ~110 amino acid residues of the light and heavy chains are highly variable in sequence; referred to as the variable regions VL and VH, respectively. The unique sequence of a VL/VH pair forms the specific antigen-binding site or paratope. The C-terminal regions of the light and heavy chains form the constant regions (CL and CH, respectively), which determine the effector functions of an antibody.
• Antigen-binding sites of antibodies are specific for the three-dimensional shape (conformation) of their target — the antigenic determinant or epitope.
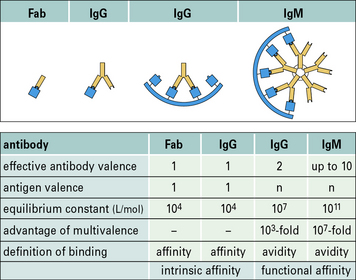
• Antibody affinity is a measure of the strength of the interaction between an antibody combining site (paratope) and its epitope. The avidity (or functional affinity) of an antibody depends on its number of binding sites (two for IgG) and its ability to engage multiple epitopes on the antigen – the more epitopes it binds, the greater the avidity.
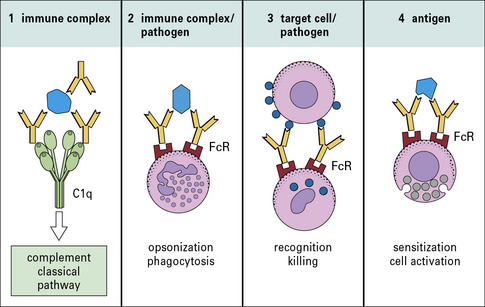
• Receptors for antibody heavy chain constant regions (Fc receptors) are expressed by mononuclear cells, neutrophils, natural killer cells, eosinophils, basophils and mast cells. They interact with the Fc regions of different isotypes of antibody and promote activities such as phagocytosis, tumor cell killing and mast cell degranulation.
• A vast repertoire of antigen-binding sites is achieved by random selection and recombination of a limited number of V, D and J gene segments that encode the variable (V) regions (domains). This process is known as V(D)J recombination and generates the primary antibody repertoire.
• Repeated rounds of somatic hypermutation and selection act on the primary repertoire to generate a secondary repertoire of antibodies with higher specificity and affinity for the stimulating antigen.
• Class switching combines rearranged VDJ genes with different heavy chain constant region genes so that the same antigen receptor can activate a variety of effector functions.
Antibodies recognize and bind antigens
Structural and functional diversity are characteristic features of these molecules.
Antibodies function as membrane-bound antigen receptors on B cells and soluble circulating antibodies
Antibodies are glycoproteins expressed as:
• membrane-bound receptors on the surface of B cells; or
• soluble molecules (secreted from plasma cells) present in serum and tissue fluids.
Contact between the B cell receptor on a particular B cell and the antigen it recognizes results in B cell activation and differentiation to generate a clone of plasma cells, which secrete large amounts of antibody. Each clone secretes only one type of antibody, with a unique specificity. The secreted antibody has the same binding specificity as the original B cell receptor (Fig. 3.1).
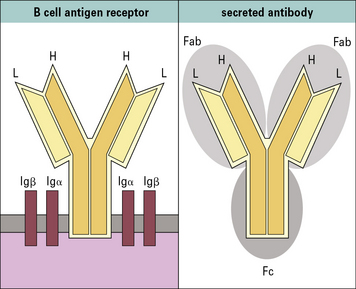
Antibodies are a family of glycoproteins
All antibody isotypes except IgD are bifunctional
Antibodies are bifunctional molecules. They:
• recognize and bind antigen; and
• promote the killing and/or removal of the immune complex formed through the activation of effector mechanisms.
• receptors expressed on host tissues (e.g. FcγRI on phagocytic cells); and
• the first component (C1q) of the complement system to initiate the classical pathway complement cascade (Fig. 3.2).
Antibody class and subclass is determined by the structure of the heavy chain
The basic structure of each antibody molecule is a unit consisting of:
Different antibody isotypes activate different effector systems
The relative proportions of IgA1 and IgA2 vary between serum and external secretion, where IgA is present in a secretory form (see Fig. 3.3).
Antibodies have a basic four chain structure
The basic four chain structure and folding of antibody molecules is illustrated for IgG1 (Fig. 3.4).
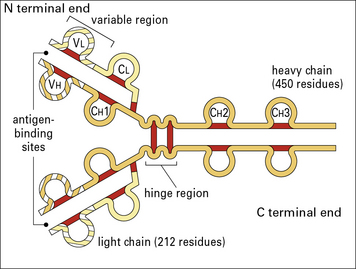
Fig. 3.4 The basic structure of IgG1
The N terminal end of IgG1 is characterized by sequence variability (V) in both the heavy and light chains, referred to as the VH and VL regions, respectively. The rest of the molecule has a relatively constant (C) structure. The constant portion of the light chain is termed the CL region. The constant portion of the heavy chain is further divided into three structurally discrete regions: CH1, CH2, and CH3. These globular regions, which are stabilized by intrachain disulfide bonds, are referred to as ‘domains’. The sites at which the antibody binds antigen are located in the variable domains. The hinge region is a segment of heavy chain between the CH1 and CH2 domains. Flexibility in this area permits the two antigen-binding sites to operate independently. There is close pairing of the domains except in the CH2 region (see Fig. 3.8). Carbohydrate moieties are attached to the CH2 domains.
• the light chain has two domains and an intrachain disulfide bond in each of the VL and CL domains;
• the heavy chain has four domains and an intrachain disulfide in each of the VH, CH1, CH2, and CH3 domains.
Each disulfide bond encloses a peptide loop of 60–70 amino acid residues.
Antibodies are prototypes of the immunoglobulin superfamily
Such molecules are said to belong to the immunoglobulin supergene family (IgSF).

Light chains are of two types
These are isotypes, being present in all individuals.
• the sequence of the ~110 N terminal residues was seen to be unique for each antibody protein analysed;
• the C terminal sequence (~110 residues) was constant for a given isotype (κ or λ) and allotype.
Thus, the light chain variable (VL) and constant (CL) regions were defined (Fig. 3.w1).
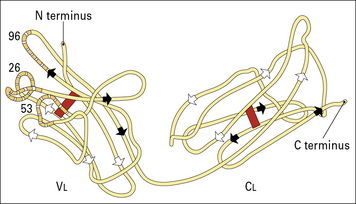
Fig. 3.w1 Basic folding in the light chain
The immunoglobulin domains in the light chain share a basic folding pattern with several straight segments of polypeptide chain lying parallel to the long axis of the domain. Light chains have two domains – one constant and one variable. Within each domain, the polypeptide chain is arranged in two layers, running in opposite directions, with many hydrophobic amino acid side chains between the layers. One of the layers has four segments (arrowed white), the other has three (arrowed black); both are linked by a single disulfide bond (red). Folding of the VL domains causes the hypervariable regions (see Fig. 3.6) to become exposed in three separate, but closely disposed, loops. One numbered residue from each hypervariable region is identified.
Hypervariable regions of VH and VL domains form the antigen-combining site
Within the variable regions of both heavy and light chains, some polypeptide segments show exceptional variability and are termed hypervariable regions. These segments are located around amino acid positions 30, 50, and 95 (Fig. 3.w2) and are referred to as Hv1, Hv2, and Hv3 or Lv1, Lv2, and Lv3, respectively.
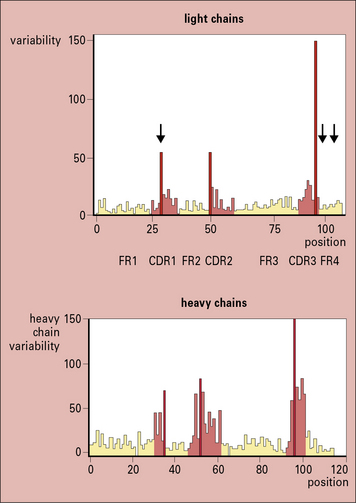
Fig. 3.w2 Amino acid variablility in the variable regions of immunoglobulins
(Courtesy of Professor EA Kabat.)
The intervening peptide segments are called framework regions (FRs) and determine the fold that ensures the CDRs are in proximity to each other (see Fig. 3.w2).
The overall structure of an antibody depends on its class and subclass
X-ray crystallography has provided structural data on complete IgG molecules (Fig. 3.5). Mobility around the hinge region of IgG allows for the generation of the Y- and T-shaped structures visualized by electron microscopy.
Assembled IgM molecules have a ‘star’ conformation
IgM is present in human serum as a pentamer of the basic four-chain structure ( see Fig. 3.w3). Each heavy chain is comprised of a VH and four CH domains. One advantage of this pentameric structure is that it provides 10 identical binding sites, which can dramatically increase the avidity with which IgM binds its cognate antigen. Given that serum IgM commonly functions to eliminate bacteria containing low affinity, polysaccharide antigens, the increased avidity provided by the pentameric structure provides an important functional advantage.
see Fig. 3.w3). Each heavy chain is comprised of a VH and four CH domains. One advantage of this pentameric structure is that it provides 10 identical binding sites, which can dramatically increase the avidity with which IgM binds its cognate antigen. Given that serum IgM commonly functions to eliminate bacteria containing low affinity, polysaccharide antigens, the increased avidity provided by the pentameric structure provides an important functional advantage.
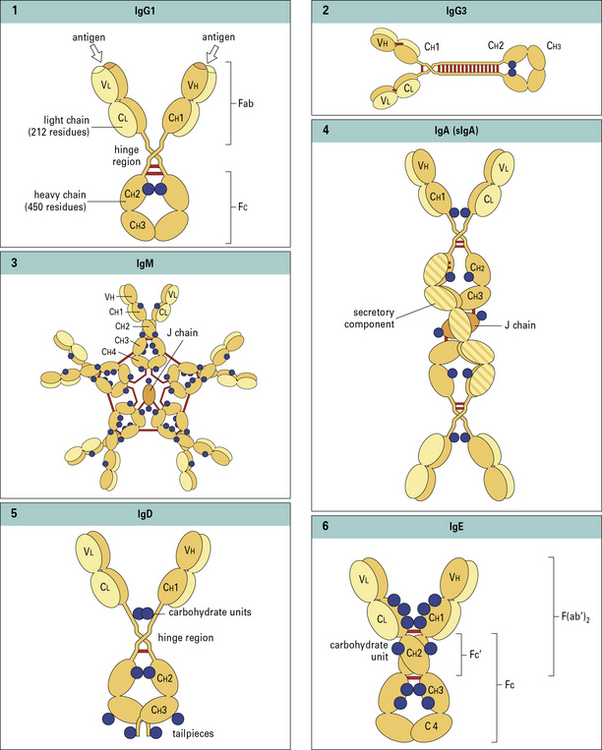
J chain is synthesized within plasma cells, has a mass of ~15 kDa and folds to form an immunoglobulin domain. Each heavy chain bears four N-linked oligosaccharide moieties, however, the oligosaccharides are not integral to the protein structure in the same way as in IgG-Fc. Oligosaccharides present on IgM activate the complement cascade via binding to the mannose binding lectin (see Chapter 4).
In electron micrographs the assembled IgM molecule is seen to have a ‘star’ conformation with a densely packed central region and radiating arms (Fig. 3.6); however, electron micrographs of IgM antibodies binding to poliovirus show molecules adopting a ‘staple’ or ‘crab-like’ configuration (see Fig. 3.6), which suggests that flexion readily occurs between the CH2 and CH3 domains, though this region is not structurally homologous to the IgG hinge. Distortion of this region, referred to as dislocation, results in the ‘staple’ configuration of IgM required to activate complement.

Secretory IgA is a complex of IgA, J chain and secretory component
The IgA1 and IgA2 subclasses differ substantially in the structure of their hinge regions:
IgA is the predominant antibody isotype in external secretions but is present as a complex secretory form. IgA is secreted by gut localized plasma cells as a dimer in which the heavy chain ‘tailpiece’ is covalently bound to a J chain, through a disulphide bond (see  Fig. 3.w3).
Fig. 3.w3).
Electron micrographs of IgA dimers show double Y-shaped structures, suggesting that the monomeric subunits are linked end-to-end through the C terminal Cα3 regions (Fig. 3.7).

The dimeric form of IgA binds a poly-Ig receptor (Fig. 3.8) expressed on the basolateral surface of epithelial cells. The complex is internalized, transported to the apical surface where the poly-Ig receptor is cleaved to yield the secretory component (SC) that is released still bound to the IgA dimer. The released secretory form of IgA is relatively resistant to cleavage by enzymes in the gut and is comprised of:
 see
see 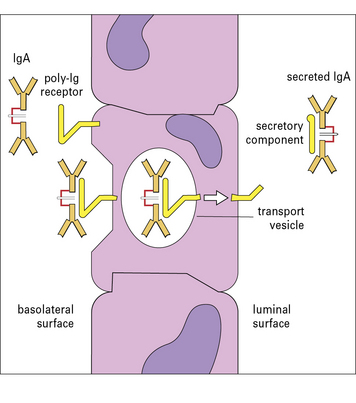
Serum IgD has antigen specificity but not effector functions
Serum IgD accounts for less than 1% of the total serum immunoglobulin. Although serum IgD has been shown to have specific antigen binding activity no effector functions have been identified. Each heavy chain is comprised of a VH domain  and three CH domains with an extended hinge region (see Figs. 3.w3). IgD also functions as an antigen specific receptor on B cells, together with IgM, and as such exhibits the same diversity of antigen specificity.
and three CH domains with an extended hinge region (see Figs. 3.w3). IgD also functions as an antigen specific receptor on B cells, together with IgM, and as such exhibits the same diversity of antigen specificity.
The heavy chain of IgE is comprised of four constant region domains
Each heavy chain is comprised of a VH and four CH domains and bears six N-linked oligosaccharides ( see Figs. 3.w3). An N-linked oligosaccharide, present in the CH3 domain, and equivalent to CH2 in IgG, influences binding to FcεRII but not FcεRI.
see Figs. 3.w3). An N-linked oligosaccharide, present in the CH3 domain, and equivalent to CH2 in IgG, influences binding to FcεRII but not FcεRI.
Antibody structural variation
Antibodies show structural variation of three different types – isotypic, allotypic, and idiotypic. The human immunoglobulin isotypes are products of defined immunoglobulin genes encoding the constant regions of heavy and light chains, and the allotypes are polymorphic variants of these genes. The idiotype of an antibody molecule results from antigenic uniqueness reflecting the structural uniqueness of the VH and VL regions (Fig. 3.w4).
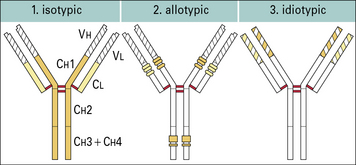
Antibody isotypes are the products of genes present within the genome of all healthy members of a species
Allotypes result from genetic variation at a locus within the species
• definition of the heavy chain class, G;
• followed by the subclass, G3;
Idiotypes result from antigenic uniqueness
• may be specific for individual B cells or plasma cell products (antibodies) (private idiotypes);
• are sometimes shared between different B cell clones and or monoclonal antibodies (public, cross-reacting, or recurrent idiotypes).
Idiotypy (from the Greek ‘idios’, meaning ‘private’) originally referred to the antigenic uniqueness of an individual antibody molecule as recognized by antisera raised in rabbits and mice, by immunization of an individual with a single antibody molecule raised in another member of the same species and allotype. For human IgG proteins heterologous antisera are raised, in rabbits or mice, and absorbed with polyclonal IgG, to absorb cross-reactive antibody. In the modern era monoclonal anti-idiotype antibodies would be generated. They are important reagents when generating assays for quantitative and qualitative analysis of an antibody therapeutic (see Method box 3.1).
Method box 3.1 Recombinant antibodies for human therapy
Q. What problems could you envisage with the use of mouse antibodies to treat diseases in humans?
In response, scientists then produced:
• chimeric antibodies, in which the mouse V domains were linked to human antibody C domains;
• humanized antibodies, for which the CDRs of the mouse V region were transferred to a human V region framework sequence;
• fully human antibodies, from phage display libraries of expressed human antibody genes or transgenic mice expressing human antibody genes.
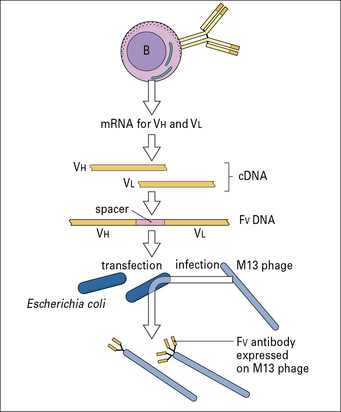
Antigen–antibody interactions
The conformations of the epitope and the paratope are complementary
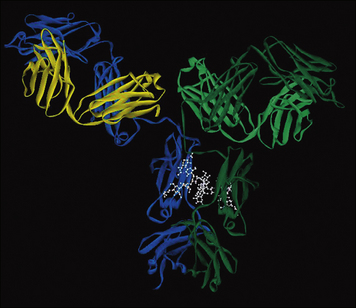
An examination of the interaction between the Fab fragment of the mouse D1.3 monoclonal antibody and hen egg white lysozyme (HEL) reveals the complementary surfaces of the epitope and the antibody’s combining site (paratope); comprised of 17 amino acid residues of the antibody and 16 residues of the lysozyme molecule (Fig. 3.9). All hypervariable regions of the heavy and light chains contribute, though the third hypervariable region in the heavy chain appears to be most important.
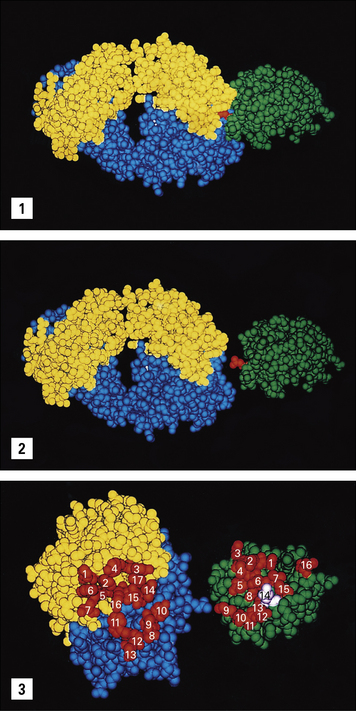
Fig. 3.9 The Fab–lysozyme complex
(Reprinted with permission from Poljak RJ, Science 1986;233:747–753. Copyright 1986 AAAS and reprinted with permission from Garcia KC et al, Science 1996;274:209–219. Copyright 1996 AAAS.)
Such structural studies are essential when engineering antibody molecules (e.g. when ‘humanizing’ a mouse antibody to generate an antibody therapeutic, see  Method box 3.1).
Method box 3.1).
Antibody affinity is a measure of the strength of interaction between a paratope and its epitope
Antibodies form multiple non-covalent bonds with antigen
Thus interacting groups must be in intimate contact before these attractive forces come into play.
For a paratope to combine with its epitope (see Fig. 3.9) the interacting sites must be complementary in shape, charge distribution, and hydrophobicity, and in terms of donor and acceptor groups, capable of forming hydrogen bonds.
The great specificity of the antigen–antibody interaction is exploited in a number of widely used assays (see  Method box 3.2).
Method box 3.2).
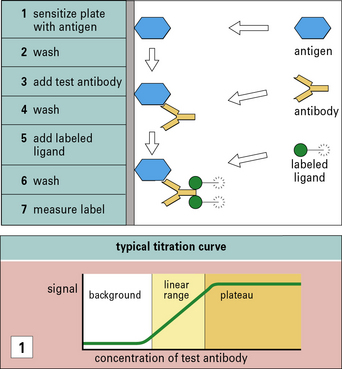
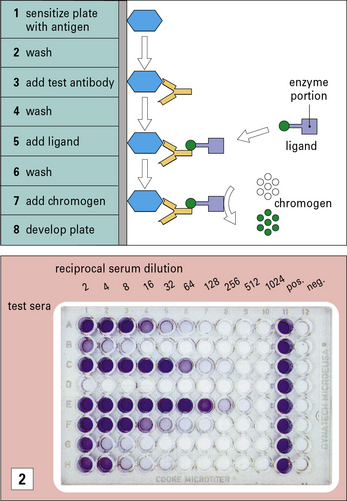
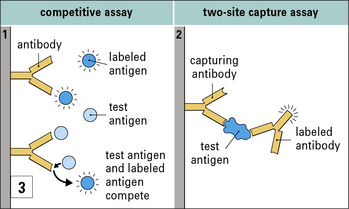
Antigen–antibody interactions are reversible
The affinity of an antibody is the sum of the attractive and repulsive forces resulting from binding between the paratope of a monovalent Fab fragment and its epitope. This interaction will be reversible, so at equilibrium the Law of Mass Action can be applied and an equilibrium constant, K (the association constant), can be determined (Fig. 3.10). In practice, due to the divalency of antibody and multiple epitopes expressed on an antigen, large three dimensional complexes may be formed that do not readily dissociate; when a paratope–epitope interaction is broken both the antibody and antigen remain in proximity due to other paratope–epitope interactions, thus reassociation is favored.
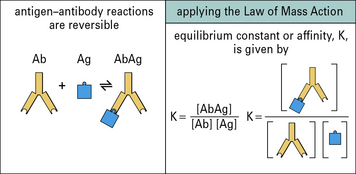
Avidity is likely to be more relevant than affinity
The avidity of an antibody for its antigen is dependent on the affinities of the individual antigen-combining sites for the epitopes on the antigen. Avidity will be greater than the sum of these affinities if both antibody-binding sites bind to the antigen because all antigen–antibody bonds would have to be broken simultaneously for the complex to dissociate (Fig. 3.11).
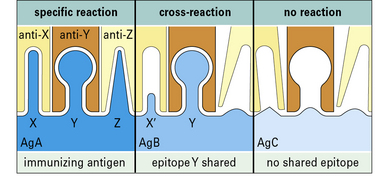
Cross-reactive antibodies recognize more than one antigen
Antigen–antibody reactions can show a high level of specificity, but can also be cross-reactive, binding to a structurally related but different antigen (Fig. 3.12). Thus, monoclonal antibodies to hen egg lysozyme (HEL) may also bind structurally homologous duck egg lysozyme (DEL). A polyclonal antiserum to HEL will contain populations of antibodies specific for HEL and others that cross-react with DEL.
Antibodies recognize the conformation of antigenic determinants
Analysis of antibodies to protein antigens reveals that specificity may be for epitopes:
• consisting of a single contiguous stretch of amino acids (a continuous epitope);
• dependent on the native conformation of the antigen and formed from two or more stretches of sequence separated in the primary structure (discontinuous or conformational epitopes).
Q. How might these differing characteristics of antigen be relevant when producing antibodies for immunological assays?
A. Antibodies specific for discontinuous epitopes may not bind denatured antigen, for example on immunoblots (see  Method box 3.3), whereas antibodies to continuous epitopes may bind denatured antigen.
Method box 3.3), whereas antibodies to continuous epitopes may bind denatured antigen.
Method box 3.3 Assays to characterize antigens
These techniques may be useful for comparing the abundance of antigens, but are less well suited to the quantitation of antigens than the immunoassays described in Method box 3.2.
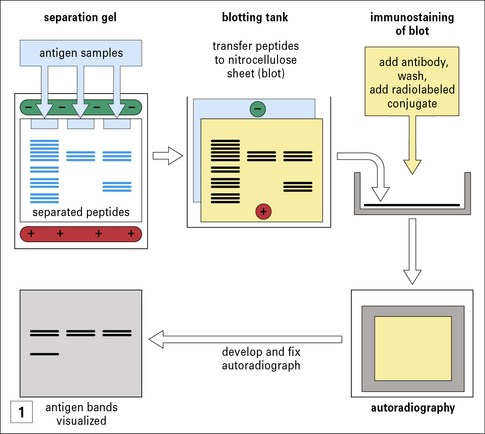
In immunoblotting, denatured antigen samples are separated in an analytical gel (e.g. an SDS polyacrylamide gel or an isoelectric focusing gel). The resolved molecules are transferred electrophoretically, in a blotting tank, to a nitrocellulose membrane. The blot is then exposed to antibody, washed and further exposed to a radiolabeled conjugate (second antibody) to detect antibody bound to antigen. The principle is similar to that of a radioimmunoassay (RIA, see Method box 3.2) or enzyme-linked immunosorbent assay (ELISA, see Method box 3.2). After washing again, the blot is placed in contact with X-ray film in a cassette. The autoradiograph is developed and the antigen bands that have bound the antibody are visible. This immunoblotting technique can be modified for use with a chemiluminescent label or an enzyme-coupled conjugate (as in ELISA), where the bound material can be detected by treatment with a chromogen, which deposits an insoluble reagent directly onto the blot.
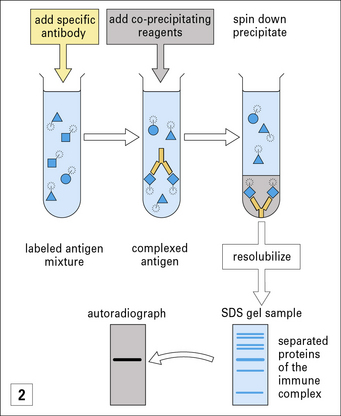
In immunoprecipitation the antigens being tested are labeled with 125I, and antibody is added that binds only to its specific antigen. The complexes are precipitated by the addition of co-precipitating agents, such as anti-immunoglobulin antibodies or staphylococcal protein A. The insoluble complexes are spun down and washed to remove any unbound labeled antigens. The precipitate is then resolubilized (e.g. in SDS), and the components are separated on analytical gels. After running, the fixed gels are autoradiographed, to show the position of the specific labeled antigen. Frequently the antigens are derived from the surface of radiolabeled cells, which are solubilized with detergents before the immunoprecipitation. It is also possible to label the antigens with biotin, and detect them at the end chromatographically using streptavidin (binds biotin) coupled to an enzyme such as peroxidase (see ELISA technique in Method box 3.2).
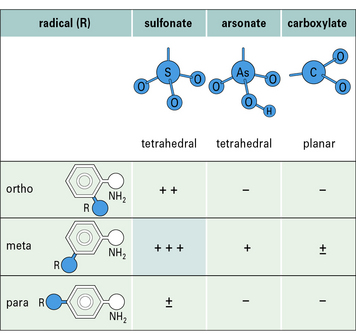
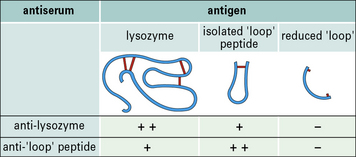
Antibodies are capable of expressing remarkable specificity and are able to distinguish small differences in the shape and chemical properties (e.g. charge, hydrophobicity) of epitopes. Small changes in the epitope, such as the position of a single chemical group, can therefore abolish binding (Figs. 3.13 and 3.14).
Antibody effector functions
Antibodies are bifunctional because they both:
• specifically bind antigen to form large complexes thus limiting the spread of pathogens in vivo; and
• the immune complexes formed elicit host responses to facilitate their removal and destruction.
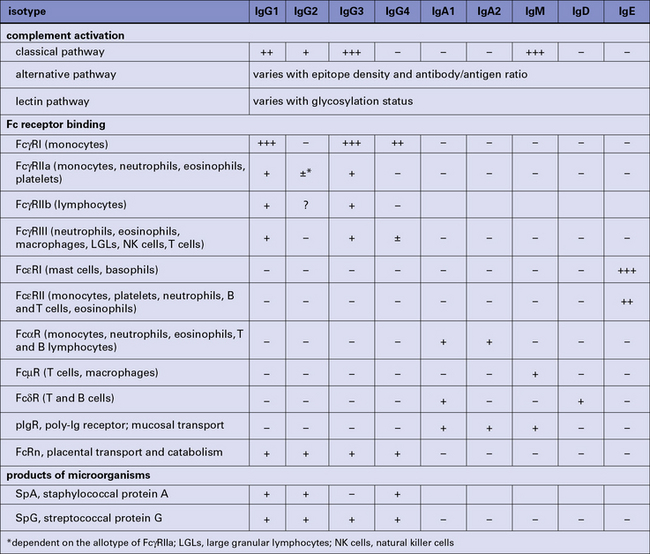
In antibody–antigen complexes the antibody molecules are essentially aggregated such that the multiple Fc regions are able to engage, cross link and activate ligands or receptors (e.g. FcγR and C1q) (Fig. 3.15). Antibody is said to be an opsonin; it opsonizes the antigen (bacterium, virus); to opsonise means to make the antigen more ‘tasty’ or ‘attractive’ targets for phagocytic cells, i.e. they ingest or eat the complexes.
IgM predominates in the primary immune response
Once bound to its target, IgM is a potent activator of the classical pathway of complement.
Although the pentameric IgM antibody molecule consists of five Fcμ regions, it does not activate the classical pathway of complement in its uncomplexed form. However, when bound to an antigen with repeating identical epitopes, it forms a ‘staple’ structure (see Fig. 3.6), undergoing a conformational change referred to as dislocation, and the multiple Fcμ presented in this form are able to initiate the classical complement cascade.
IgG is the predominant isotype of secondary immune responses
The IgG subclasses also interact with a complex array of cellular Fc receptors (FcλR) expressed on various cell types (see Fig. 3.15 and below). IgG equilibrates between the intravascular and extravascular pools, so providing comprehensive systemic protection.
In humans, the newborn infant is not immunologically competent and the fetus is protected by passive IgG antibody selectively transported across the placenta (Fig. 3.16). Transport is mediated by the neonatal Fc receptor (FcRn) – all IgG subclasses are transported but the cord/maternal blood ratios differ, being approximately 1.2 for IgG1 and approximately 0.8 for IgG2.
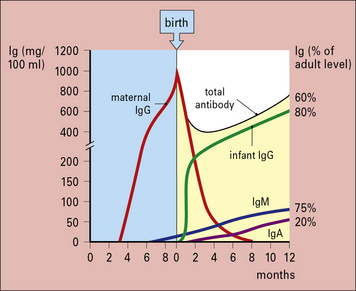
Serum IgA is produced during a secondary immune response
IgE may have evolved to protect against helminth parasites infecting the gut
Fc receptors
The three types of Fc receptor for IgG are FcγRI, FcγRII, and FcγRIII
Three types of cell surface receptor for IgG (FcγR) are defined in humans:
Each receptor is characterized by a glycoprotein α chain that has an extracellular domain, homologous with immunoglobulin domains, that binds to antibody (Fig. 3.17) – that is, they belong to the immunoglobulin superfamily, as do receptors for IgA (FcαR) and IgE (FcεRI).
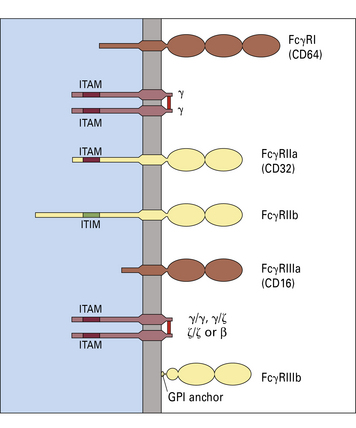
Phosphorylation of the ITAM motif triggers activities such as:
In contrast, phosphorylation of ITIM blocks cellular activation.
FcγRI is involved in phagocytosis of immune complexes and mediator release
FcγRII is expressed in two forms
FcγRIII is expressed as FcγRIIIa and FcγRIIIb
FcγRIIIa is a transmembrane protein (like FcγRI, FcγRIIa, and FcγRIIb), whereas FcγRIIIb is GPI (glycosyl phosphatidyl inositol) anchored (see Fig. 3.17).
• have a moderate affinity for monomeric IgG; and
• may be associated with γ/ξ and/or β chains bearing ITAM motifs.
FcγRIIIa is expressed on monocytes, macrophages, NK cells, and a fraction of T cells.
Polymorphism in FcγRIIIa and FcγRIIIb may affect disease susceptibility
IgG Fc interaction sites for several ligands have been identified
The interactions between maternal IgG and the MHC class I molecule-like FcRn expressed on the intestinal epithelium of the neonatal rat have now been studied at high resolution (Fig. 3.18) and are believed to mimic closely the binding of the human placental counterpart, hFcRn, with maternal IgG. Titration of IgG histidine residues in the binding site for FcRn may explain its:
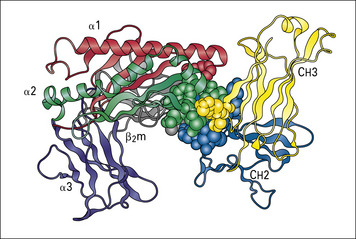
Fig. 3.18 Neonatal rat intestinal Fc receptor FcRn
(Reproduced from Ravetch JV, Margulies DH. New tricks for old molecules. Nature 1994;372:323–324.)
IgM–antigen complexes are very efficient activators of the classical complement system, but the mechanism by which IgM binds C1q appears different from that of IgG. The conformational change from a ‘star’ to a ‘staple’ conformation upon binding to multivalent antigen is thought to unveil a ring of occult C1q-binding sites that are not accessible in the star-shaped configuration (see Fig. 3.6).

The two types of Fc receptor for IgE are FcεRI and FcεRII
Two types of Fc receptor for IgE (FcεR) are defined in humans (Fig. 3.19):
• the high-affinity FcεRI which is expressed on mast cells and basophils and is the ‘classical’ IgE receptor; and
• the low-affinity FcεRII (CD23), which is expressed on leukocytes and lymphocytes.
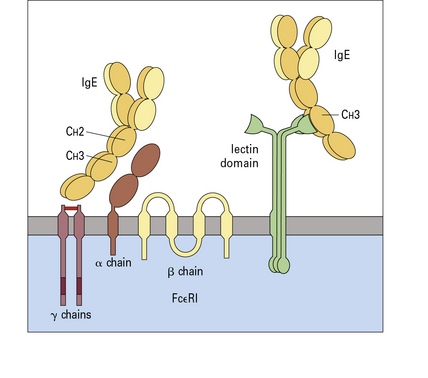
Cross-linking of IgE bound to FcεRI results in histamine release
The high-affinity receptor (FcεRI) is present on the surface of mast cells and basophils as a complex with a β (33 kDa) and two γ (99 kDa) chains to form the αβγ2 receptor unit (see Fig. 3.19).
FcεRII is a type 2 transmembrane molecule
FcεRII is the low-affinity (CD23) receptor and is a type 2 transmembrane molecule (i.e. one in which the C termini of the polypeptides are extracellular, see Fig. 3.19). The two forms of human CD23 are:
Development of the antibody repertoire by gene recombination
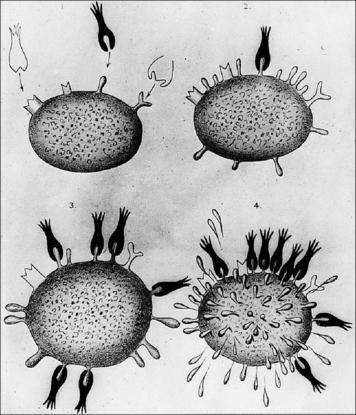
Ehrlich’s side-chain hypothesis, put forward at the beginning of the 20th century (Fig. 3.20), proposed antigen-induced selection. His model is close to our present view of clonal selection, except that he placed receptors of several different specificities on the same cell and did not address the question of how the diverse receptors were generated.
Heavy chain gene recombination precedes light chain recombination
• an open reading frame is transcribed to nuclear RNA;
• the introns are ‘spliced’ out of nuclear RNA to yield a continuous messenger RNA (mRNA);
The primary antibody repertoire is:
• generated through recombination of germline gene segments, with selection to eliminate B cells producing anti-self antibodies;
• present in the B cell population and expressed as membrane-bound IgM and IgD acting as antigen receptors.
Antibody genes undergo rearrangement during B cell development
Rearrangement at the heavy chain VH locus precedes rearrangement at light chain loci
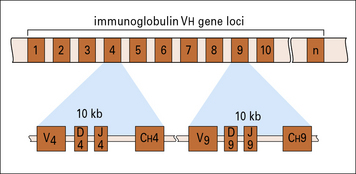
The germline human heavy chain locus, on chromosome 14 (Fig. 3.21), contains a library of 38 to 46 functional VH gene segments that encode the N-terminal 95 residues of the VH region.
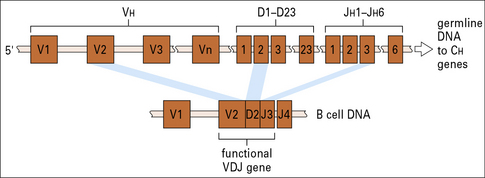
The C terminal residues of the VH region are encoded within 23 DH and six JH gene segments (see Fig. 3.21).
The first event is recombination between a JH gene segment and DH segments, followed by recombination with a VH gene segment
• are highly variable, both in the number of codons and in the nucleotide sequence;
• may be read in three possible reading frames without generating stop codons; and
Productive recombination between DH and JH gene segments signals recombination of this DJ sequence to a VH gene segment, forming a contiguous DNA sequence encoding the entire VH protein sequence (see Fig. 3.21).
Rearrangement results in a VK gene segment becoming contiguous with a JK gene segment
The germline human κ light chain locus on chromosome 2 (Fig. 3.22) contains a library of 31–35 functional Vκ gene segments that encode the N terminal 95 residues of the Vκ region.
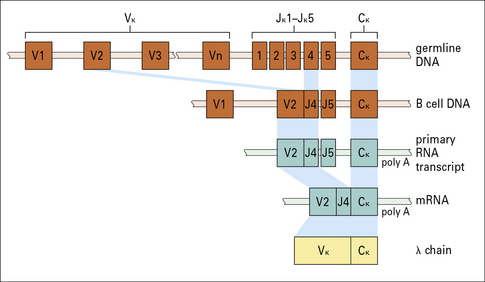
The C terminal residues of the Vκ region are encoded within five Jκ gene segments (see Fig. 3.22).
Recombination results in a Vλ gene segment becoming contiguous with a functional Jλ gene segment
The germline human λ light chain locus (Fig. 3.23) on chromosome 22 contains a library of 29–33 functional Vλ gene segments that encode the N terminal 95 residues of the Vλ region.
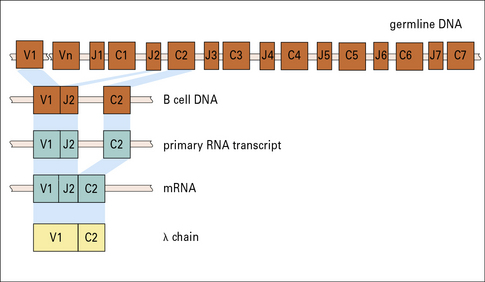
There are 7–11 Jλ gene segments with each linked to a Cλ gene sequence (see Fig. 3.23) – the number of JλCλ sequences depends on the haplotype.
Recombination involves recognition of signal sequences by the V(D)J recombinase
Each V, D, and J segment is flanked by recombination signal sequences (RSS):
• a signal sequence found downstream (3′) of VH,VL, and DH gene segments consists of a heptamer CACAGTG or its analog, followed by a spacer of non-conserved sequence (12 or 23 bases), and then a nonamer ACAAAAACC or its analog (Fig. 3.24);
• immediately upstream (5′) of a germline JL, DH, and JH segment is a corresponding signal sequence of a nonamer and a heptamer, again separated by an unconserved sequence (12 or 23 bases).
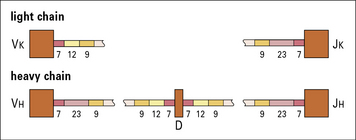
Fig. 3.24 Recombination sequences in immunoglobulin genes
The recombination sequences in the light chain genes (top) and heavy chain genes (bottom) consist of heptamers (7), 12 or 23 unconserved bases, and nonamers (9). The sequences of heptamers and nonamers are complementary and the nonamers act as signals for the recombination activating genes to form a synapsis between the adjoining exons. Similar recombination sequences are present in the T cell receptor V, D, and J gene segments (see Chapter 5).
The 12 and 23 base spacers correspond to either one or two turns of the DNA helix (see Fig. 3.24).
• a RAG-1–RAG-2 complex recognizes the RSS, bringing a 12-RSS and a 23-RSS together into a synaptic complex (Fig. 3.25);
• the RAG proteins initiate cleavage by introducing a nick in the area bordering the 5′ end of the signal heptamer and the coding region;
• the RAG proteins then convert this nick into a double-strand break, generating a hairpin at the coding end and a blunt cut at the signal sequence, resulting in a blunt signal end;
• the hairpinned coding end must be opened before the joining step, and usually undergoes further processing (the addition or deletion of nucleotides), resulting in an imprecise junction. The loss or addition of nucleotides during coding joint formation (termed ‘junctional diversity’) creates additional diversity that is not encoded by the V, D, or J segments, and also contributes to the generation of nonproductive joints, since some of the modification create stop codons.
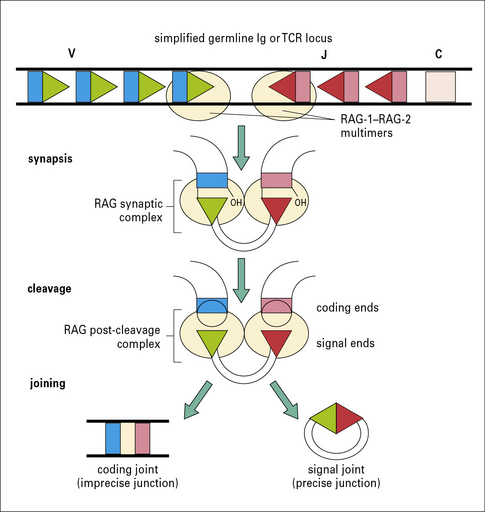
Signal ends, in contrast, are usually joined precisely to form circular signal joints that have no known immunological function and are lost from the cell (see Fig. 3.25).
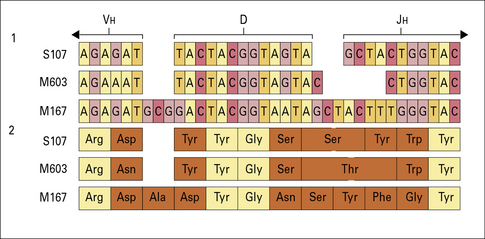
Additional diversification is provided by the enzyme terminal deoxynucleotidyl transferase, which may add random nucleotides to the exposed cut ends of the DNA. Nucleotides may therefore be inserted between DH and JH, and between VH and DH, without need of a template (Figs 3.26 and 3.27).
Somatic hypermutation in antibody genes
The secondary response is characterized by the appearance of germinal centers, within the spleen, bone marrow and lymph glands, within which the recombined DNA encoding variable light and heavy chain sequences undergoes repeated rounds of random mutation (somatic hypermutation) to generate B cells expressing structurally distinct receptors (Fig. 3.28). Survival and expansion of these B cells requires that their receptors engage antigen presented to them by follicular dendritic cells in the lymphoid tissues:
• a majority will acquire a deleterious mutation and, in the absence of a survival signal, die;
• a minority bind antigen with increased affinity and compete with those of lower affinity (i.e. antibodies characteristic of the primary response).
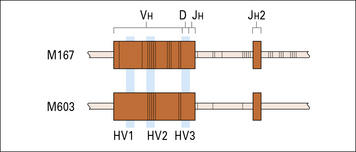
This process, called affinity maturation, is dependent on both T cells and germinal centers.
Athymic mice lacking T cells do not form germinal centers and show no affinity maturation.
Diversity is generated at several different levels
Antibody diversity therefore arises at several levels:
• there are multiple V genes recombining with D and J segments;
• recombinational inaccuracies and N-nucleotide addition;
• gene conversion (in some species – see  Fig. 3.w6);
Fig. 3.w6);
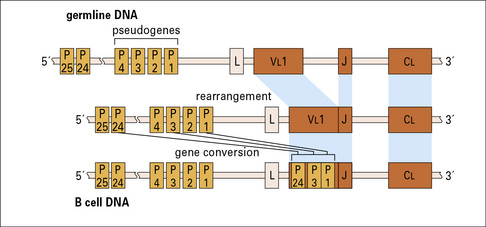

Different species have different strategies for generating diversity
• sharks rely on a large number of antibody genes and do not use somatic recombination (Fig. 3.w5);
• chickens have a very small number of antibody genes that undergo a very high level of gene conversion (Fig. 3.w6).
Critical thinking: The specificity of antibodies (see p. 433 for explanations)
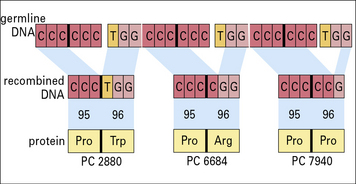
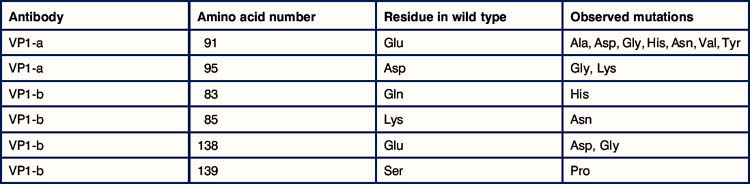
1. When virus is propagated in the presence of neutralizing antiviral antiserum it is found that mutated forms of the virus develop. Mutations are detected in VP1, VP2, or VP3, but never in VP4. Why should this be so? The most effective neutralizing antibodies are directed against the protein VP1 – this is termed an immunodominant antigen. Two different monoclonal antibodies against VP1 were developed and used to induce mutated forms of the virus. When the sequences of the mutated variants were compared with the original virus, it was found that only certain amino acid residues became mutated (see table below).
2. What can you tell about the epitopes that are recognized by the two different monoclonal antibodies?
3. When the binding of the antibody VP1-a is measured against the different mutant viruses, it is found that it binds with high affinity to the variant with glycine (Gly) at position 138, with low affinity to the variant with Gly at position 95, and does not bind to the variant with lysine (Lys) at position 95. How can you explain these observations?
Arnold J.N., Wormald M.R., Sim R.B., et al. The Impact of Glycosylation on the Biological Function and Structure of Human Immunoglobulins. Annu Rev Immunol. 2007;25:21–50.
Gould H.J., Sutton B.J. IgE in allergy and asthma today. Nat Rev Immunol. 2008;8:205–217.
Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov. 2009;8:226–234.
Jefferis R., Lefranc M.-P. Human immunoglobulin allotypes: possible implications for immunogenicity. MAbs. 2009;1:332–338.
Litman G.W., Rast J.P., Fugmann S.D. The origins of vertebrate adaptive immunity. Nat Rev Immunol. 2010;10:543–553.
Monteiro R.C. Role of IgA and IgA Fc receptors in inflammation. J Clin Immunol. 2010;30:1–9.
Neuberger M.S. Antibody diversification by somatic mutation: from Burnet onwards. Immunol Cell Biol. 2008;86:124–132.
Reichert J.M. Antibody-based therapeutics to watch in 2011. MAbs. 2011;11:3.
Schroeder H.W., Jr., Cavacini L. Structure and function of immunoglobulins. J Allergy Clin Immunol. 2010;125:S41–S52.
Shukla A.A., Thömmes J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. 2010;28:253–261.
Woof J.M., Burton D. Human antibody-Fc receptor interactions illuminated by crystal structures. Nat Rev Immunol. 2004;4:89–99.
Woof JM, Kerr MA. The function of immunoglobulin A in immunity. J Pathol 208:270–282
IMGT, the International ImMunoGeneTics Information System®. http://www.imgt.org/. An integrated knowledge resource for the immunoglobulins (IG), T cell receptors (TR), major histocompatibility complex (MHC), immunoglobulin superfamily and related proteins of the immune system of human and other vertebrate species
Mike Clark’s Immunoglobulin Structure/Function Home Page. http://www.path.cam.ac.uk/~mrc7/mikeimages.html. Provides a wealth of information, of his own generation and through access to many other related sites
National Center for Biotechnology Information (NCBI). http://www.ncbi.nlm.nih.gov/. Established in 1988 as a national resource for molecular biology information, NCBI creates public databases, conducts research in computational biology, develops software tools for analyzing genome data, and disseminates biomedical information – all for the better understanding of molecular processes affecting human health and disease
CD antigens. http://www.uniprot.org/docs/cdist.txt. Human cell differentiation molecules: CD nomenclature and list of entries
Summary of Antibody Structures in the Protein Databank. http://acrmwww.biochem.ucl.ac.uk/abs/sacs/index.html.