CHAPTER 97 ANTIBACTERIAL THERAPY: THE OLD, THE NEW, AND THE FUTURE
Infections remain the leading cause of death in hospitalized patients, and antimicrobial therapy is a mainstay of treatment. However, widespread overuse and misuse of antibiotics have led to an alarming increase in multiple-drug-resistant (MDR) pathogens. New agents may allow shorter courses of therapy and prophylaxis, which are desirable for cost control and control of microbial flora. Moreover, antibiotics are second only to analgesic agents in the number of adverse drug reactions.
PRINCIPLES OF PHARMACOKINETICS
Half-life refers to the amount of time required for the drug concentration to reduce by half, and thus is a hybrid of consider ations of both clearance and volume of distribution. Half-life is useful to estimate when a steady-state drug concentration will be achieved. If a “loading dose” is not administered intravenously, thereby creating instantaneously a desired drug concentration to be maintained throughout therapy, four to five half-lives must elapse to achieve a steady state. Changes in dosage and changes in half-life owing to disease state (e.g., renal failure) must be accounted for. Interpretation of drug concentration data is difficult if the patient is not at a steady state, especially so in critical illness characterized by fluctuating organ function and volume of distribution.
EMPIRIC ANTIBIOTIC THERAPY
Must antibiotics be started immediately? If the presumed infection is not destabilizing, this decision also depends on the overall status of the patient and should take into consideration such host factors as age, debility, renal and hepatic function, and immunosuppression. Culture yields are highest before antibiotics are administered, which for certain types of specimens (e.g., blood, cerebrospinal fluid) can be crucial. However, for many infections (e.g., bacteremia, intraabdominal infection, pneumonia) early appropriate therapy improves outcome.
CHOICE OF ANTIBIOTIC
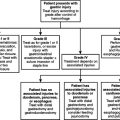
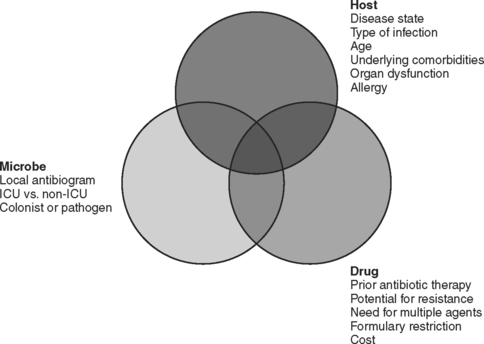
The choice of which antibiotic to prescribe is made based on several interrelated factors. Paramount is activity against identified pathogens, presuming that a distinction between infecting and colonizing organisms can be made and that narrow-spectrum coverage is always most desirable. Knowledge of antimicrobial resistance patterns, nationally and especially in one’s own institution and unit, is essential. Also important is an assumption regarding likely pathogens, which is paramount in cases where empiric therapy is necessary. Estimation of likely pathogens depends on the disease process believed responsible, whether the infection is community- or hospital-acquired, whether MDR organisms are present, and proximity to other infected patients. Also important are patient-specific factors, including age, debility, immunosuppression, intrinsic organ function, prior allergy or another adverse reaction, and recent antibiotic therapy. Institutional factors that may play a role include the existence of guidelines or practice parameters that may specify a particular therapy, or the availability of specific agents as defined by inclusion on the formulary or restriction by antibiotic control programs (Figure 1).
Development of Bacterial Resistance
Cephalosporin resistance among Gram-negative bacilli can be the result of induction of chromosomal β-lactamases after exposure to the antibiotic. The extended-spectrum cephalosporins are rendered ineffective when bacteria such as enteric Gram-negative bacilli mutate to constitutively produce a β-lactamase that is normally an inducible enzyme. Although resistance to cephalosporins can occur by several mechanisms, the appearance of chromosomally mediated β-lactamases has been identified as a consequence of the use of third-generation cephalosporins. Resistance rates decline when use is restricted. The induction of an extended-spectrum β-lactamase (ESBL) in Klebsiella by ceftazidime was first reported approximately 20 years ago, but more than 200 mutations have now been described in several species of Gram-negative bacteria. The mutant bacteria develop resistance rapidly not only to all cephalosporins but to entire other classes of β-lactam antibiotics. It is therefore justifiable to restrict the use of ceftazidime, especially in institutions grappling with an ESBL-producing bacterium. The carbapenems generally retain useful microbicidal activity against ESBL-producing strains. Increasingly, Pseudomonas aeruginosa produces beta-lactamases of the ampC type.
ANTIBIOTIC SPECTRUM OF ACTIVITY
Penicillins
The carboxypenicillins (ticarcillin and carbenicillin) and ureidopenicillins (azlocillin, mezlocillin, and piperacillin; sometimes referred to as acylampicillins) have enhanced activity against Gram-negative bacteria and some activity against P. aeruginosa. Ureidopenicillins have greater intrinsic activity against Pseudomonas, but with the advent of β-lactamase inhibitor combination drugs none of these agents is used widely anymore. Beta-lactamase inhibitors (sulbactam, tazobactam, and clavulanic acid) result in enzymatic inactivation and enhanced effectiveness of the antibacterial agent. The effectiveness of these drugs as antibacterial agents is primarily a function of the inherent antibacterial properties of the parent compound (ampicillin < ticarcillin < piperacillin), and to a lesser extent of the effectiveness of the inhibitor (sulbactam ∼ clavulanic acid < tazobactam). The spectrum of activity varies as a result, and the treating clinician needs to be familiar with each of the drugs in this class.
Cell-Wall–Active Agents
Lipoglycopeptides
Both S. aureus and S. epidermidis are susceptible to vancomycin, although MICs for S. aureus are increasing and may require higher doses for therapeutic effect. Streptococcus pyogenes, group B streptococci, S. pneumoniae (including penicillin-resistant strains), and C. difficile are also susceptible. Listeria monocytogenes, anaerobic cocci, other clostridial species, and Actinomyces are usually susceptible. Most strains of E. faecalis are inhibited (but not killed) by concentrations attainable in serum, but E. faecalis is increasingly resistant to vancomycin. Resistant enterococci have emerged because of prolonged or indiscriminate use of vancomycin (Table 1), occasioned by the ubiquity of MRSA/MRSE. Both GISA and strains of S. aureus fully resistant to vancomycin are recognized, but so far only in association with prolonged (i.e., weeks to months) exposure to vancomycin.
Table 1 Situations in Which Use of Vancomycin Is Discouraged









Vancomycin usage is often inappropriate, and it is important for the public health that inappropriate usage should be curtailed. Bona fide indications include serious infections caused by MRSA/MRSE, Gram-positive infections in patients with serious penicillin allergy, and oral therapy (or by enema in patients with ileus) for C. difficile-related colitis in patients who have failed or are intolerant to metronidazole. Parenteral vancomycin is now usually administered in a dose of 15 mg/kg actual body weight q12h. The infusion must be performed over the course of at least 1 hour. The dose must be reduced in renal failure, and monitoring of serum concentrations may be helpful in that circumstance. New high-flux hemodialysis membranes dialyze vancomycin partially, and a 500-mg dose should be given after each dialysis.
PROTEIN SYNTHESIS INHIBITORS
Tetracyclines
Tetracyclines bind irreversibly to the 30S ribosomal subunit, but unlike aminoglycosides are bacteriostatic agents. Widespread resistance limits their utility in the hospital setting (with two exceptions), but they are still prescribed as oral agents. Short-acting oral tetracyclines include oxytetracycline and tetracycline HCl. Intermediate-acting oral agents of this class include demeclocycline, whereas those with a long half-life include the semisynthetic lipophilic congeners doxycycline and minocycline. Most pneumococci and H. influenzae are inhibited by achievable concentrations in serum. Thus, the tetracyclines may be used for management of sinusitis and acute exacerbations of chronic bronchitis. Gonococci and meningococci are quite susceptible. Unfortunately, penicillin-resistant gonococci tend also to be resistant to tetracycline. Outpatient urinary isolates of E. coli can be treated with tetracyclines, as can most infections caused by Vibrio spp. Most recently, doxycycline has been used with some success against VRE.
Chloramphenicol
Chloramphenicol is a bacteriostatic agent that binds to the 50S ribosomal subunit. The drug has limited activity against the Enterobacteriaceae but remains effective against Salmonella/Shigella spp., including S. typhimurium. Chloramphenicol retains useful activity against most anaerobic organisms except for C. difficile. A resurgence in the use of chloramphenicol was occasioned by the emergence of VRE, but newer agents have supplanted that usage. Chloramphenical penetrates well into cerebrospinal fluid, and receives occasional usage for meningitis—especially when caused by H. influenzae. The bone marrow toxicity of chloramphenicol is feared, but rare in actuality. Reversible dose-related bone marrow toxicity is more common than aplastic anemia, which occurs in only about 1/25,000 courses of therapy. It is one of only a few antibiotics that require a dosage reduction in liver disease (Table 2) but not in renal insufficiency.
Table 2 Antimicrobials Requiring Dosage Reduction in Hepatic Disease
The Macrolide-Lincosamide-Streptogramin Family
Clindamycin
The lincosamide antibiotics in clinical use include lincomycin and clindamycin, but lincomycin is no longer widely available. Clindamycin also binds to the 50S ribosome and has good antianaerobic activity (although B. fragilis resistance is increasing), but in contrast to chloramphenicol it is devoid of activity against Gram-negative organisms while possessing reasonably good activity against Gram-positive cocci. Clindamycin is used occasionally for anaerobic infections, and it is a preferred choice to vancomycin for prophylaxis of clean surgical cases in penicillin-allergic patients (where the primary concern is the prevention of Gram-positive surgical site infections). Because clindamycin inhibits production of exotoxins in vitro, it has been advocated in preference to penicillin as first-line therapy of invasive infections caused by S. pyogenes. The toxicity of clindamycin is far less than that of chloramphenicol, but its use has been associated with the development of antibiotic-associated colitis due to overgrowth of C. difficile.
DRUGS THAT DISRUPT NUCLEIC ACIDS
Rifampin
In addition to antituberculosis chemotherapy, rifampin is used for meningococcal meningitis prophylaxis of close contacts, synergistic therapy of MSSA endocarditis (this is controversial because of questions about antagonism and a propensity to develop resistance), the staphylococcal carrier state (including MRSA), chronic staphylococcal arthritis or osteomyelitis, synergistic therapy of Legionnaire’s disease, brucellosis, and staphylococcal prosthetic device infections. Synergistic therapy with rifampin and vancomycin is controversial for MRSA endocarditis, and there are no data to support synergistic therapy for other MRSA infections.
Rifampin is a potent inducer of the hepatic microsomal enzyme system. Reduced oral bioavailability and decreased serum half-life occurs for a number of drugs, including barbiturates, benzodiazepines, calcium channel blockers, chloramphenicol, cyclosporine, digitalis, estrogens, fluconazole, haloperidol, histamine H2-antagonists, metoprolol, phenytoin, prednisone, propranolol, quinidine, theophylline, and warfarin (Table 3).
| Potentiated Effect of Oral Anticoagulants |
ANTIBIOTIC TOXICITIES
Beta-Lactam Allergy
Complement-independent toxicity may result from binding to neutrophil or macrophage cell membranes. Examples include leukopenia, thrombocytopenia, hemolytic anemia, and interstitial nephritis. Immune complex (Arthus) reactions occur when circulating antigenantibody (IgG, IgM) complexes fix complement and lodge in various tissue sites, causing serum-sickness–like reactions and possibly drug fever. The onset of these reactions is usually 7–14 days after therapy has begun, even if drug has already been stopped. In cell-mediated hypersensitivity, β-lactam antigen-specific T-cell receptors bind the antigen—causing cytokine release and lymphocyte proliferation. Contact dermatitis is the usual manifestation. Certain reactions do not fall under these classifications, including pruritis, maculopapular reactions, erythema multiforme, erythema nodosum, photosensitivity, and exfoliative dermatitis.
Metronidazole Toxicity
Metronidazole is generally well tolerated. Minor adverse reactions include gastrointestinal upset and metallic taste, which sometimes necessitate stopping the drug. Discolored urine, rash, urticaria, urethral or vaginal burning, gynecomastia, and reversible neutropenia have also been noted. Rare but serious adverse neurologic reactions include seizures, encephalopathy, ataxia, and peripheral neuropathy. Other rare but potentially serious reactions include disulfiram-like reactions in the presence of alcohol, potentiation of warfarin effect (see Table 2), C. difficile-associated disease (despite its therapeutic efficacy), and acute pancreatitis. Suggestions of mutagenicity from in vitro studies have not been borne out clinically, but the drug crosses the placenta readily and should be used in pregnancy only when necessary.
AVOIDING TOXICITY
Adjustment of Antibiotic Therapy in Hepatic Insufficiency
Liver disease has the greatest effect on those drugs that undergo extensive oxidative metabolism. With such a multiplicity of factors involved, it is difficult to predict the effect of disease on drug disposition in individual patients. There is no useful clinically available test of liver function that can be used as a guide to dosage, such as glomerular filtration rate in the case of renal failure. A general rule is that dosage reduction should be up to 25% of the usual dose if hepatic metabolism is 40% or less and renal function is normal, the drug is given acutely, and has a large therapeutic index (see Table 2). Greater dosage reductions (up to 50%) are advisable if the drug is administered chronically, there is a narrow therapeutic index, protein binding is significantly reduced, or the drug is excreted renally and renal function is severely impaired. In circumstances where renally excreted therapeutic substitutes exist for patients with liver disease, such drugs should be used.
Adjustment of Antibiotic Therapy in Renal Insufficiency
Drug elimination by the kidneys depends on the GFR, tubular secretion, and reabsorption. Renal dysfunction may alter any or all of these parameters, which in turn may be influenced by nonrenal organ dysfunction. Different types of renal disease, or acute versus chronic renal failure, may result in different drug clearance rates among patients with the same GFR. The management of antibiotics in renal failure must be individualized because most antibiotics are excreted via the kidneys. Relatively precise estimates of renal function are especially important in patients with impaired renal function who have not yet come to dialysis because the clearance of many drugs by dialysis actually makes management easier.
Factors influencing drug clearance by hemofiltration include molecular size, aqueous solubility, plasma protein binding, equilibration kinetics between plasma and tissue, and the apparent volume of distribution. Generally, drugs that have a molecular weight greater than 500 daltons are less efficiently dialyzed by standard dialysis membranes. However, the new high-flux polysulfone membranes can clear efficiently molecules up to 5 kD (the molecular weight of vancomycin is 1.486 kD) (Table 4).
Table 4 Dosing of Selected Parenteral Antibiotics Applied After Dialysis
| Antibiotic | Dose |
|---|---|
| Amikacin | 2.5–3.75 mg/kg |
| Ampicillin | 1 g |
| Azlocillin | 3 g |
| Aztreonam | 0.125 g |
| Cefamandole | 0.5–1 g |
| Cefepime | 0.5 g |
| Cefoxitin | 1 g |
| Ceftazidime | 1 g |
| Ceftizoxime | 1–3 g |
| Cefuroxime | 0.75 g |
| Chloramphenicol | 1 g |
| Gentamicin | 1.0–1.7 mg/kg |
| Imipenem/cilastatin | 0.25–0.5 g |
| Meropenem | 0.5 g |
| Mezlocillin | 2–3 g |
| Netilmicin | 2 mg/kg |
| Piperacillin | 2 g |
| Piperacillin/tazobactam | 2.25 g |
| Ticarcillin | 3 g |
| Ticarcillin/clavulanic acid | 3.1 g |
| Tobramycin | 1.0–1.7 mg/kg |
| Trimethoprim/sulfamethoxazole | 5 mg/kg trimethoprim |
| Vancomycin | 0.5 g if using polysulfone dialysis membrane; otherwise no supplement |
Cefaclor, cefoperazone, ceftriaxone, chloramphenicol, clindamycin, cloxacillin and dicloxacillin, doxycycline, erythromycin, linezolid, methicillin/nafcillin/oxacillin, metronidazole, rifampin, and tigecycline do not require dosage reductions in renal failure. Many penicillins and cephalosporins require a dosage reduction only when severe renal insufficiency (variously defined as a creatinine clearance <30–50 ml/min) exists (Table 5). Tetracyclines other than doxycycline and tigecycline are contraindicated in renal failure.
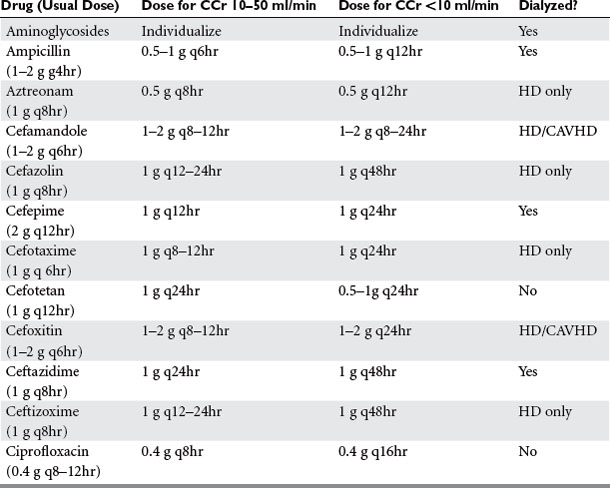
When adjusting therapy in renal failure, the dose can be reduced or the interval between doses can be prolonged. The initial dose should be the same regardless, in order to obtain adequate peak serum concentrations. It is preferred to maintain the dose and prolong the interval with aminoglycosides because of the importance of maintaining a high peak concentration. However, it makes sense to reduce dose but maintain the interval when administering β-lactam drugs (especially those with no PAE) in order to maintain a constant drug concentration. The need to dose patients during or after a renal replacement therapy treatment must be borne in mind. During continuous renal replacement therapy, the estimated creatinine clearance is 15 ml/minute in addition to the patient’s intrinsic clearance.
American Thoracic Society. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171:388-416.
GM Anstead & AD Owens Recent advances in the treatment of infections due to resistant Staphylococcus aureus Curr Opin Infect Dis 17 549–555
Bartlett JG, Perl TM. The new Clostridium difficile—what does it mean? N Engl J Med. 2005;343:2503-2505.
Benko AS, Cappelletty DM, Kruse JA, et al. Continuous infusion versus intermittent administration of ceftazidime in critically ill patients with suspected Gram-negative infections. Antimicrob Agents Chemother. 1996;40:691-695.
Bosso JA. The antimicrobial armamentarium: evaluating current and future treatment options. Pharmacotherapy. 2005;25:55S-62S.
Carlet J, Ben Ali A, Chalfine A. Epidemiology and control of antibiotic resistance in the intensive care unit. Curr Opin Infect Dis. 2004;17:309-316.
Chastre J, Wolff M, Fagon JY, et al. Comparison of 15 vs. 8 days of antibiotic therapy for ventilator-associated pneumonia in adults: a randomized trial. JAMA. 2003;290:2588-2598.
Clark NM, Hershberger E, Zervosc MJ, et al. Antimicrobial resistance among Gram-positive organisms in the intensive care unit. Curr Opin Crit Care. 2003;9:403-412.
Dellinger EP. Duration of antibiotic treatment in surgical infections of the abdomen. Undesired effects of antibiotics and future studies. Eur J Surg. 1996;576:29-31. (Suppl)
DiPiro JT, Edmiston CE, Bohnen JMA. Pharmacodynamics of antimicrobial therapy in surgery. Am J Surg. 1996;171:615-622.
Evans RS, Pestotnik SL, Classen DC, et al. A computer-assisted management program for antibiotics and other antiinfective agents. N Engl J Med. 1998;338:232-238.
Fry DE. The importance of antibiotic pharmacokinetics in critical illness. Am J Surg. 1996;172:20S-25S. (Suppl)
Garnacho-Montero J, Garcia-Garmendia JL, Barrero-Almodovar A, et al. Impact of adequate empirical antibiotic therapy on the outcome of patients admitted to the intensive care unit with sepsis. Crit Care Med. 2003;31:2742-2751.
Gold HS, Moellering RC. Antimicrobial drug resistance. N Engl J Med. 1996;335:1445-1453.
Harbarth S, Ferriere K, Hugonnet S, et al. Epidemiology and prognostic determinants of bloodstream infections in surgical intensive care. Arch Surg. 2002;137:1353-1359.
Jones RN. Microbiological features of vancomycin in the 21st century: minimum inhibitory concentration creep, bactericidal/static activity, and applied breakpoints to predict clinical outcomes or detect resistant strains. Clin Infect Dis. 2005;42:S13-S24.
Kollef MH, Micek ST. Strategies to prevent antimicrobial resistance in the intensive care unit. Crit Care Med. 2005;33:1845-1853.
LeDell K, Muto CA, Jarvis WR, et al. SHEA guideline for preventing nosocomial transmission of multidrug-resistant strains of. Staphylococcus aureus and Enterococcus. Infect Control Hosp Epidemiol. 2003;24:639-641.
Livermore DM. Bacterial resistance: origins, epidemiology, and impact. Clin Infect Dis. 2003;36:S11-S23.
V Loo, L Poirier & MA Miller et al A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med 353 2442–2449
McDonald LC, Kilgore GE, Thompson A, et al. An epidemic, toxin genevariant strain of Clostridium difficile. N Engl J Med. 2005;353:2433-2441.
Naiemi NA, Duim B, Savelkoul PH, et al. Widespread transfer of resistance genes between bacterial species in an intensive care unit: implications for hospital epidemiology. J Clin Microbiol. 2005;43:4862-4864.
Naimi TS, LeDell KH, Como-Sabetti K, et al. Comparison of community- and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA. 2004;290:2976-2984.
Neuhauser MM, Weinstein RA, Rydman R, et al. Antibiotic resistance among Gram-negative bacilli in US intensive care units: implications for fluoro-quinolone use. JAMA. 2003;289:885-888.
Nseir S, Di Pompeo C, Soubrier S, et al. First-generation fluoroquinolone use and subsequent emergence of multiple drug-resistant bacteria in the intensive care unit. Crit Care Med. 2005;33(2):283-289.
Padmanabhan RA, Larosa SP, Tomecki KJ. What’s new in antibiotics? Dermatol Clin. 2005;23:301-312.
Paul M, Benuri-Silbiger I, Soares-Weiser K, et al. Beta-lactam monotherapy versus beta-lactam-aminoglycoside combination therapy for sepsis in immunocompetent patients: systematic review and meta-analysis of randomized trials. BMJ. 2004;328(7441):668.
Rello J, Ollendorf DA, Oster G, et al. Epidemiology and outcomes of ventilator-associated pneumonia in a large US database. Chest. 2002;122:2115-2121.
Raymond DP, Pelletier SJ, Crabtree TD, et al. Impact of a rotating empiric antibiotic schedule on infectious mortality in an intensive care unit. Crit Care Med. 2001;29:1101-1108.
Schentag JJ, Gilliland KK, Paladino JA. What have we learned from pharma-cokinetic and pharmacodynamic theories? Clin Infect Dis. 2001;32:S39-S46.
Schlaes DM, Gerding DN, John JFJr, et al. Society for Healthcare Epidemiology of America and Infectious Diseases Society of America Joint Committee on the Prevention of Antimicrobial Resistance: guidelines for the prevention of antimicrobial resistance in hospitals. Clin Infect Dis. 1997;25:584-599.
Sehulster L, Chinn RY, et al. Guidelines for environmental infection control in health-care facilities. Recommendations of CDC and the Healthcare Infection Control Practices Advisory Committee (HICPAC). MMWR Recomm Rep. 2003;6:1-42.
Shorr AF, Sherner JH, Jackson WL, et al. Invasive approaches to the diagnosis of ventilator-associated pneumonia: a meta-analysis. Crit Care Med. 2005;33:46-53.
Trouillet JL, Chastre J, Vuagnat A, et al. Ventilator-associated pneumonia caused by potentially drug-resistant bacteria. Am J Respir Crit Care Med. 1998;157:531-539.
Viviani M, Silvestri L, van Saene HK, et al. Surviving Sepsis Campaign Guidelines: selective decontamination of the digestive tract still neglected. Crit Care Med. 2005;33:462-463.