[level-membership-for-basic-science-category]
Chapter 13 Antibacterial drugs
Classification
Inhibition of cell wall synthesis
β-lactams
the structure of which contains a β-lactam ring. The major subdivisions are:
• Penicillins, whose official names usually include, or end in, ‘cillin’.
• Cephalosporins and cephamycins, which are recognised by the inclusion of ‘cef ’ or ‘ceph’ in their official names. In the UK recently all these names have been standardised to begin with ‘cef’.
Other subcategories of β-lactams include:
Other inhibitors of cell wall synthesis include vancomycin and teicoplanin.
Inhibition of cell wall synthesis
β-lactams
Penicillins
Mode of action
Penicillins act by inhibiting the enzymes (penicillin binding proteins, PBPs) involved in the cross-linking of the peptidoglycan layer of the cell wall, which is weakened, and this leads to osmotic rupture. Penicillins are thus bactericidal and are ineffective against resting organisms which are not making new cell wall. The main defence of bacteria against penicillins is to produce enzymes, β-lactamases, which hydrolyse the β-lactam ring. Other mechanisms that have been described include modifications to PBPs to render them unable to bind β-lactams, reduced permeability of the outer cell membrane of Gram-negative bacteria, and pumps in the outer membrane which remove β-lactam molecules. Some particularly resistant bacteria may possess several mechanisms that act in concert. The remarkable safety and high therapeutic index of the penicillins is due to the fact that human cells, while bounded by a cell membrane, lack a cell wall. They exhibit time-dependent bacterial killing (see p.164).
Narrow-spectrum penicillins
Benzylpenicillin
Benzylpenicillin (t½ 0.5 h) (penicillin G) has to be given with spaced doses that have to be large to maintain a therapeutic concentration, but the large therapeutic ratio of penicillin allows the resulting fluctuations to be tolerable.1 Benzylpenicillin is eliminated by the kidney, with about 80% being actively secreted by the renal tubule and this can be blocked by probenecid.
Antistaphylococcal penicillins
Examples of agents stable to staphylococcal β-lactamases include:
• Flucloxacillin (t½ 1 h) is better absorbed and so gives higher blood concentrations than does cloxacillin. It may cause cholestatic jaundice, particularly when used for more than 2 weeks or given to patients > 55 years.
• Cloxacillin (t½ 0.5 h) has been withdrawn from the market in some countries, including the UK.
• Methicillin and oxacillin: their use is now confined to laboratory sensitivity tests. Identification of methicillin-resistant Staphylococcus aureus (MRSA) in patients indicates the organisms are resistant to all β-lactam antibiotics and often to other antibacterial drugs.
Cephalosporins
Mode of action
is that of the β-lactams, i.e. cephalosporins impair bacterial cell wall synthesis and hence are bactericidal. They exhibit time-dependent bacterial killing (see p. 164).
Pharmacokinetics
Usually, cephalosporins are excreted unchanged in the urine, but some, including cefotaxime, form a desacetyl metabolite which possesses some antibacterial activity. Many are actively secreted by the renal tubule, a process which can be blocked with probenecid. As a rule, the dose of cephalosporins should be reduced in patients with poor renal function. Cephalosporins in general have a t½ of 1–4 h although there are exceptions (e.g. ceftriaxone, t½ 8 h). Wide distribution in the body allows treatment of infection at most sites, including bone, soft tissue, muscle and (in some cases) CSF. Data on individual cephalosporins appear in Table 13.1.
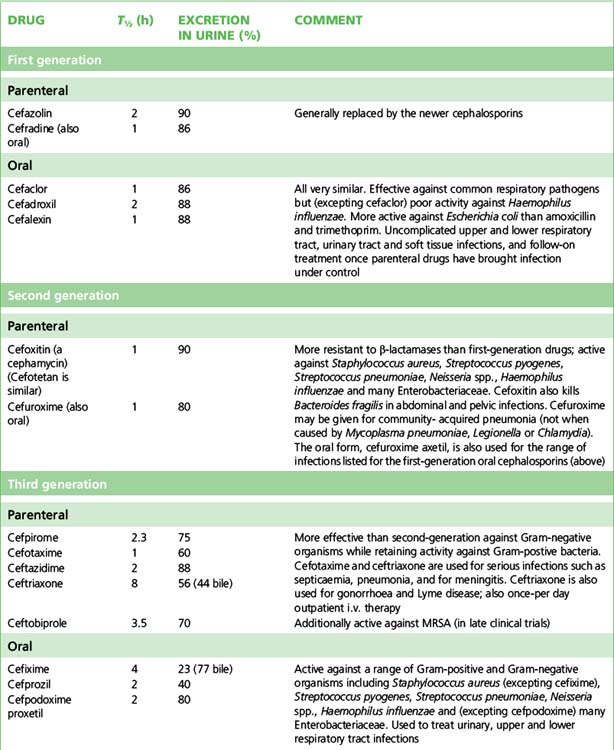
Classification and uses
The cephalosporins are conventionally categorised by ‘generations’ sharing broadly similar antibacterial and pharmacokinetic properties; newer agents have rendered this classification less precise but it retains sufficient usefulness to be presented in Table 13.1.
Adverse effects
Cephalosporins are well tolerated. The most usual unwanted effects are allergic reactions of the penicillin type, and gastrointestinal upset. Overall the rate of cephalosporin skin reactions such as urticarial rashes and pruritis lies between 1% and 3%. There is cross-allergy between penicillins and cephalosporins involving up to 10% of patients; if a patient has had a severe or immediate allergic reaction or if serum or skin testing for penicillin allergy is positive (see p. 176), then a cephalosporin should not be used. Pain may be experienced at the sites of i.v. or i.m. injection. If cephalosporins are continued for more than 2 weeks, reversible thrombocytopenia, haemolytic anaemia, neutropenia, interstitial nephritis or abnormal liver function tests may occur. The broad spectrum of activity of the third generation cephalosporins may predispose to opportunist infection with resistant bacteria or Candida albicans and to Clostridium difficile diarrhoea. In the UK, reduction of broad-spectrum cephalosporin use is one component of the bundle of measures aimed to reduce the incidence of Clostridium difficile-associated diarrhoea. Ceftriaxone achieves high concentrations in bile and, as the calcium salt, may precipitate to cause symptoms resembling cholelithiasis (biliary pseudolithiasis).
Other inhibitors of cell wall synthesis and membrane function
Vancomycin
Daptomycin
(t½ 9 h) is a recently released lipopeptide antibiotic, naturally produced by the bacterium Streptomyces roseosporus which was first isolated from a soil sample from Mount Ararat in Turkey.3 It has activity against virtually all Gram-positive bacteria, including penicillin-resistant Streptococcus pneumoniae and MRSA, regardless of vancomycin resistance phenotype. It is unable to cross the Gram-negative outer membrane, rendering these bacteria resistant.
Inhibition of protein synthesis
Aminoglycosides
Mode of action
The aminoglycosides act inside the cell by binding to the ribosomes in such a way that incorrect amino acid sequences are entered into peptide chains. Aminoglycosides are bactericidal and exhibit concentration-dependent bacterial killing (see p. 164).
Pharmacokinetics
Current practice is to administer aminoglycosides as a single daily dose rather than as twice- or thrice-daily doses. Algorithms are available to guide such dosing according to patients’ weight and renal function, and in this case only trough concentrations need to be assayed. Lean body-weight should be used because aminoglycosides distribute poorly in adipose tissue. Single daily dose therapy is probably less oto- and nephrotoxic than divided-dose regimens, and appears to be as effective. The immediate high plasma concentrations that result from single daily dosing are advantageous, e.g. for acutely ill septicaemic patients, as aminoglycosides exhibit concentration-dependent killing (see p. 164).
Antibacterial activity
Aminoglycosides are in general active against staphylococci and aerobic Gram-negative organisms including almost all the Enterobacteriaceae. Bacterial resistance to aminoglycosides is an increasing but patchily distributed problem, notably by acquisition of plasmids (see p. 169) which carry genes coding for the formation of drug-destroying enzymes. Gentamicin resistance is rare in community-acquired pathogens in many hospitals in the UK.
Uses
• Gram-negative bacillary infection, particularly septicaemia, renal, pelvic and abdominal sepsis. Gentamicin remains the drug of choice, but tobramycin may be preferred for Pseudomonas aeruginosa. Amikacin has the widest antibacterial spectrum of the aminoglycosides but is best reserved for infection caused by gentamicin-resistant organisms. If local resistance rates are low, an aminoglycoside may be included in the initial best-guess regimen for treatment of serious septicaemia. A potentially less toxic antibiotic may be substituted when culture results are known (48–72 h), and toxicity is very rare after such a short course.
• Bacterial endocarditis. An aminoglycoside, usually gentamicin, usually comprises part of the antimicrobial combination for enterococcal, streptococcal or staphylococcal infection of the heart valves.
• Other infections: tuberculosis, tularaemia, plague, brucellosis.
• Topical uses. Neomycin and framycetin, too toxic for systemic use, are effective for topical treatment of infections of the conjunctiva or external ear. Tobramycin is given by inhalation for therapy of infective exacerbations of cystic fibrosis: sufficient systemic absorption may occur to recommend assay of serum concentrations in such patients.
Adverse effects
• Ototoxicity. Both vestibular (especially with gentamicin and streptomycin) and auditory (amikacin, neomycin) damage may occur, causing hearing loss, vertigo, nystagmus and tinnitus which may be permanent (see above). Tinnitus may give warning of auditory nerve damage. Early signs of vestibular toxicity include motion-related headache, dizziness or nausea. Serious ototoxicity can occur with topical application, including ear drops. At least five mutations in the mitochondrial gene encoding 12 S rRNA have been found that predispose patients to irreversible aminoglycoside hearing loss, and the possibility of screening individuals before commencing therapy is being investigated. Anti-free radical agents such as salicylate may reduce aminoglycoside toxicity.
• Nephrotoxicity. Dose-related changes, which are usually reversible, occur in renal tubular cells, where aminoglycosides accumulate. Low blood pressure, loop diuretics and advanced age are recognised as added risk factors.
• Neuromuscular blockade. Aminoglycosides may impair neuromuscular transmission and aggravate (or reveal) myasthenia gravis, or cause a transient myasthenic syndrome in patients whose neuromuscular transmission is normal.
• Other reactions include rashes and haematological abnormalities, including marrow depression, haemolytic anaemia and bleeding due to antagonism of factor V.
For gentamicin and tobramycin, oto- and nephrotoxicity are increased if peak concentrations exceed 12–14 mg/L consistently, or troughs exceed 2 mg/L. For amikacin the corresponding concentrations are 32–34 mg/L and 10 mg/L.
Tetracyclines
Uses
Tetracyclines are active against nearly all Gram-positive and Gram-negative pathogenic bacteria, but increasing bacterial resistance and low innate activity limit the clinical use of most members of the class. Although 4-quinolone usage has replaced them especially in the developed world, they remain drugs of first choice for infection with chlamydiae (psittacosis, trachoma, pelvic inflammatory disease, lymphogranuloma venereum), mycoplasma (pneumonia), rickettsiae (Q fever, typhus), Bartonella spp., and borreliae (Lyme disease, relapsing fever) (for use in acne, see p.273). Doxycycline is used in therapeutic and prophylactic regimens for malaria (see p. 230) and is active against amoebae and a variety of other protozoa. Their most common other uses are as second-line therapy of minor skin and soft tissue infections especially in β-lactam allergic patients; surprisingly, many MRSA strains currently remain susceptible to tetracyclines in the UK.
Macrolides
Erythromycin
Erythromycin (t½ 2–4 h) binds to bacterial ribosomes and interferes with protein synthesis; it is bacteriostatic and exhibits time-dependent killing (see p. 164). It is effective against Gram-positive organisms because these accumulate the drug more efficiently, and its antibacterial spectrum is similar to that of penicillin.
Uses
Dose
is 250 mg 6-hourly or twice this in serious infection and four times for legionnaires’ disease.
Clindamycin,
Clindamycin is used for staphylococcal bone and joint infections, dental infections and serious intra-abdominal sepsis (in the last, it is usually combined with an agent active against Gram-negative pathogens such as gentamicin). Because of its ability to inhibit production of bacterial protein toxins, it is the antibiotic of choice for serious invasive Streptococcus pyogenes infections (although surgical resection of affected tissue plays a prime role) and it is also an alternative to linezolid for treatment of Panton-Valentine leukocidin-producing strains of Staphylococcus aureus (see p. 210). It is a second choice in combination for some Toxoplasma infections (see p. 236). Topical preparations are used for therapy of severe acne and non-sexually transmitted infection of the genital tract in women.
The most serious adverse effect is antibiotic-associated (pseudomembranous) colitis (see p. 170); clindamycin should be stopped if any diarrhoea occurs.
Other inhibitors of protein synthesis
Resistance to antimicrobials: linezolid, quinupristin-dalfopristin and fosfomycin
No antibiotic should be used recklessly, however difficult it appears to be to select for resistance in vitro. On the other hand, the attitude that ‘All new antibiotics should be locked away’ risks stifling innovation whilst denying life-saving treatments … Debates on the use of new anti-Gram-positive agents are sure to intensify … and it is vital that they take place on a basis of science not knee-jerk restrictions or over-zealous marketing.4
These agents are inactive against most Gram-negative bacteria.
Inhibition of nucleic acid synthesis
Sulfonamides and sulfonamide combinations
Systemic use
Sulfonamide-trimethoprim combination
• Prevention and treatment of pneumonia due to Pneumocystis carinii infection in immunosuppressed patients (for therapy, high doses of 120 mg/kg/day in 2–4 divided doses are used and therapeutic drug monitoring is essential).
• Prevention and treatment of toxoplasmosis, and treatment of nocardiasis and Stenotrophomonas maltophilia infection (90–120 mg/kg/day dosing).
Quinolones
(4-quinolones, fluoroquinolones)
The first widely used quinolone, nalidixic acid, was discovered serendipitously as a by-product of chloroquine synthesis. It is effective for urinary tract infections because it is concentrated in the urine, but it has little systemic activity. Fluorination of the quinolone structure was subsequently found to produce compounds that were up to 60 times more active than nalidixic acid and killed a wider range of organisms. These newer ‘4-quinolones’ act principally by inhibiting bacterial (but not human) DNA gyrase (topoisomerase II and IV), thus preventing the supercoiling of DNA, a process that is necessary for compacting chromosomes in the bacterial cell; they are bactericidal and exhibit concentration-dependent bacterial killing (see p. 164). In general quinolones are extremely active against Gram-negative organisms and most have useful activity against Pseudomonas aeruginosa, mycobacteria and Legionella pneumophila. Most are less active against Gram-positive organisms (resistance commonly emerges) and anaerobes. Resistance typically arises via mutation of the target enzymes and these are coded on mobile plasmids; efflux pumps may also contribute. Quinolone resistance rates of a wide range of Gram-negative bacteria have risen alarmingly worldwide during the past 10 years, and clinical cross-resistance across all members of the group is common.
Adverse effects
include gastrointestinal upset and allergic reactions (rash, pruritus, arthralgia, photosensitivity and anaphylaxis). High rates of quinolone usage in hospitals have been associated with outbreaks of diarrhoea caused by Clostridium difficile, so reduced use is one component of the bundles of recommended control measures (see p. 170). CNS effects may develop with dizziness, headache and confusion. Convulsions have occurred during treatment (avoid or use with caution where there is a history of epilepsy or concurrent use of NSAIDs, which potentiate this effect). Reversible arthropathy has developed in weight-bearing joints in immature animals exposed to quinolones. Quinolones should be used only for serious infections and then with caution in children and adolescents; however, ciprofloxacin is licensed for treatment of Pseudomonas aeruginosa lung infection in children over 5 years of age with cystic fibrosis. Rupture of tendons, notably the Achilles, has occurred, more commonly in the elderly and those taking corticosteroids concurrently. Levofloxacin and ofloxacin are less likely than ciprofloxacin to cause corneal precipitates during topical therapy to the eye and are preferred for this as much as for their enhanced anti-Gram-positive activity.
Azoles
• Metronidazole and tinidazole (antibacterial and antiprotozoal) which are described here.
• Fluconazole, itraconazole, clotrimazole, econazole, ketoconazole, isoconazole and miconazole which are described under Antifungal drugs (p. 223).
• Albendazole, mebendazole and thiabendazole which are described under Anthelminthic drugs (p. 236).
Metronidazole
Uses
Metronidazole’s clinical indications are:
• Treatment of sepsis to which anaerobic organisms, e.g. Bacteroides spp. and anaerobic cocci, are contributing, including post-surgical infection, intra-abdominal infection and septicaemia, osteomyelitis and abscesses of brain or lung.
• Antibiotic-associated pseudomembraneous colitis (caused by Clostridium difficile).
• Trichomoniasis of the urogenital tract in both sexes.
• Amoebiasis (Entamoeba histolytica), including both intestinal and extra-intestinal infection.
• Giardiasis (Giardia lamblia).
• Acute ulcerative gingivitis and dental infections (Fusobacterium spp. and other oral anaerobic flora).
• Anaerobic vaginosis (Gardnerella vaginalis and vaginal anaerobes).
Adverse effects
A disulfiram-like effect (see p. 147) occurs with alcohol because metronidazole inhibits alcohol and aldehyde dehydrogenase; patients should be warned appropriately.
Minor antimicrobials
Mupirocin
is primarily active by inhibition of tRNA synthetase in Gram-positive organisms, including those commonly associated with skin infections. It is available as an ointment for use, e.g. in folliculitis and impetigo, and to eradicate Staphylococcus aureus site, e.g. in carriers of resistant staphylococci and to clear nasal carriage before surgery (see p. 168). It is also effective at reducing rates of staphylococcal peritonitis in patients receiving chronic ambulatory peritoneal dialysis, and for this indication it needs only to be applied to the nares for a few days per month. Re-colonisation after eradication of carriage occurs quite swiftly, with 5–30% being positive around 4 days after treatment, and 85–100% after a month.
Alvarez-Elcoro S., Enzler M.J. The macrolides: erythromycin, clarithromycin, and azithromycin. Mayo Clin. Proc.. 1999;74:613–634.
Andriole V.T. The quinolones: past present and future. Clin. Infect. Dis.. 2005;41:S113–S119.
Anon. Quinupristin + dalfopristin for infections. Drug Ther. Bull. 2002;40:15–16.
Anon. Moxifloxacin. Drug Ther. Bull. 2004;42:61–62.
Anon. What role for tigecycline in infections? Drug Ther. Bull. 2008;46:62–64.
Chambers H.F. Methicillin resistance in staphylococci: molecular and biochemical basis and clinical implications. Clin. Microbiol. Rev.. 1997;10:781–791.
Cunha B.A. New uses for older antibiotics: nitrofurantoin, amikacin, colistin, polymyxin B, doxycycline, and minocycline revisited. Med. Clin. North Am.. 2006;90:1089–1107.
Diekema D.J., Jones R.N. Oxazolidine antibiotics. Lancet. 2001;358:1975–1982.
Drawz S.M., Bonomo R.A. Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev.. 2010;23:160–201.
Falagas M.E., Kastoris A.C., Kapaskelis A.M., et al. Fosfomycin for the treatment of multidrug-resistant, including extended-spectrum beta-lactamase producing, enterobacteriaceae infections: a systematic review. Lancet Infect. Dis.. 2010;10:43–50.
Fisman D.N., Kaye K.M. Once-daily dosing of aminoglycoside antibiotics. Infect. Dis. Clin. North Am.. 2000;14:475.
Sánchez García M., De la Torre M.A., Morales G., et al. Clinical outbreak of linezolid-resistant Staphylococcus aureus in an intensive care unit. J. Am. Med. Assoc.. 2010;303(22):2260–2264.
Gaynes R.P. Preserving the effectiveness of antibiotics. J. Am. Med. Assoc.. 2010;303(22):2293–2294.
Holgate S. Penicillin allergy: how to diagnose and when to treat. Br. Med. J.. 1988;296:1213.
Howden B.P., Davies J.K., Johnson P.D., et al. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanism, laboratory detection and clinical implications. Clin. Microbiol. Rev.. 2010;23:99–139.
Kelkar P.S., Li J.T.C. Cephalosporin allergy. N. Engl. J. Med.. 2001;345:804–809.
McLean-Tooke A., Aldridge C., Stroud C., et al. Practical management of antibiotic allergy in adults. J. Clin. Pathol. 2011;64(3):192–199. Available online at: http://jcp.bmj.com/content/early/2010/12/20/jcp.2010.077289.full.pdf (accessed October 2011)
Romano A., Viola M., Guéant-Rodriguez R.M., et al. Tolerability of meropenem in patients with IgE-mediated hypersensitivity to pencillins. Ann. Intern. Med.. 2007;146:266–269.
Rybak M., Lomaestro B., Rotschafer J.C., et al. Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am. J. Health Syst. Pharm. 2009;66:82–98. Available online at: http://www.ajhp.org/cgi/reprint/66/1/82 (accessed October 2011)
Torres M.J., Blanca M. The complex clinical picture of beta-lactam hypersensitivity: penicillins, cephalosporins, monobactams, carbapenems, and clavams. Med. Clin. North Am.. 2010;94:805–820.
Van Rijen M., Bonten M., Wenzel R., Kluytmans J. Mupirocin ointment for preventing Staphylococcus aureus infections in nasal carriers. Cochrane Database Syst. Rev. (4):2010. cd006216, 2008
1 Is it surprise at the answer that reduces most classes of students to silence when asked the trough:peak ratio for a drug given 6-hourly with a t½ of 0.5 h? (answer: 212 = 4096).
2 600 mg = 1 000 000 units, 1 mega-unit.
3 Eisenstein B I, Oleson F B Jr, Baltz R H 2010 Daptomycin: from the mountain to the clinic, with essential help from Francis Tally, MD. Clinical Infectious Diseases 50(Suppl. 1):S10–15.
4 Livermore D M 2000 Quinupristin/dalfopristin and linezolid: where, when, which and whether to use? Journal of Antimicrobial Chemotherapy 46:347–350.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 13 Antibacterial drugs
Classification
Inhibition of cell wall synthesis
β-lactams
the structure of which contains a β-lactam ring. The major subdivisions are:
• Penicillins, whose official names usually include, or end in, ‘cillin’.
• Cephalosporins and cephamycins, which are recognised by the inclusion of ‘cef ’ or ‘ceph’ in their official names. In the UK recently all these names have been standardised to begin with ‘cef’.
Other subcategories of β-lactams include:
Other inhibitors of cell wall synthesis include vancomycin and teicoplanin.
Inhibition of cell wall synthesis
β-lactams
Penicillins
Mode of action
Penicillins act by inhibiting the enzymes (penicillin binding proteins, PBPs) involved in the cross-linking of the peptidoglycan layer of the cell wall, which is weakened, and this leads to osmotic rupture. Penicillins are thus bactericidal and are ineffective against resting organisms which are not making new cell wall. The main defence of bacteria against penicillins is to produce enzymes, β-lactamases, which hydrolyse the β-lactam ring. Other mechanisms that have been described include modifications to PBPs to render them unable to bind β-lactams, reduced permeability of the outer cell membrane of Gram-negative bacteria, and pumps in the outer membrane which remove β-lactam molecules. Some particularly resistant bacteria may possess several mechanisms that act in concert. The remarkable safety and high therapeutic index of the penicillins is due to the fact that human cells, while bounded by a cell membrane, lack a cell wall. They exhibit time-dependent bacterial killing (see p.164).
Narrow-spectrum penicillins
Benzylpenicillin
Benzylpenicillin (t½ 0.5 h) (penicillin G) has to be given with spaced doses that have to be large to maintain a therapeutic concentration, but the large therapeutic ratio of penicillin allows the resulting fluctuations to be tolerable.1 Benzylpenicillin is eliminated by the kidney, with about 80% being actively secreted by the renal tubule and this can be blocked by probenecid.
Antistaphylococcal penicillins
Examples of agents stable to staphylococcal β-lactamases include:
• Flucloxacillin (t½ 1 h) is better absorbed and so gives higher blood concentrations than does cloxacillin. It may cause cholestatic jaundice, particularly when used for more than 2 weeks or given to patients > 55 years.
• Cloxacillin (t½ 0.5 h) has been withdrawn from the market in some countries, including the UK.
• Methicillin and oxacillin: their use is now confined to laboratory sensitivity tests. Identification of methicillin-resistant Staphylococcus aureus (MRSA) in patients indicates the organisms are resistant to all β-lactam antibiotics and often to other antibacterial drugs.
Cephalosporins
Mode of action
is that of the β-lactams, i.e. cephalosporins impair bacterial cell wall synthesis and hence are bactericidal. They exhibit time-dependent bacterial killing (see p. 164).
Pharmacokinetics
Usually, cephalosporins are excreted unchanged in the urine, but some, including cefotaxime, form a desacetyl metabolite which possesses some antibacterial activity. Many are actively secreted by the renal tubule, a process which can be blocked with probenecid. As a rule, the dose of cephalosporins should be reduced in patients with poor renal function. Cephalosporins in general have a t½ of 1–4 h although there are exceptions (e.g. ceftriaxone, t½ 8 h). Wide distribution in the body allows treatment of infection at most sites, including bone, soft tissue, muscle and (in some cases) CSF. Data on individual cephalosporins appear in Table 13.1.
Classification and uses
The cephalosporins are conventionally categorised by ‘generations’ sharing broadly similar antibacterial and pharmacokinetic properties; newer agents have rendered this classification less precise but it retains sufficient usefulness to be presented in Table 13.1.
Adverse effects
Cephalosporins are well tolerated. The most usual unwanted effects are allergic reactions of the penicillin type, and gastrointestinal upset. Overall the rate of cephalosporin skin reactions such as urticarial rashes and pruritis lies between 1% and 3%. There is cross-allergy between penicillins and cephalosporins involving up to 10% of patients; if a patient has had a severe or immediate allergic reaction or if serum or skin testing for penicillin allergy is positive (see p. 176), then a cephalosporin should not be used. Pain may be experienced at the sites of i.v. or i.m. injection. If cephalosporins are continued for more than 2 weeks, reversible thrombocytopenia, haemolytic anaemia, neutropenia, interstitial nephritis or abnormal liver function tests may occur. The broad spectrum of activity of the third generation cephalosporins may predispose to opportunist infection with resistant bacteria or Candida albicans and to Clostridium difficile diarrhoea. In the UK, reduction of broad-spectrum cephalosporin use is one component of the bundle of measures aimed to reduce the incidence of Clostridium difficile-associated diarrhoea. Ceftriaxone achieves high concentrations in bile and, as the calcium salt, may precipitate to cause symptoms resembling cholelithiasis (biliary pseudolithiasis).
Other inhibitors of cell wall synthesis and membrane function
Vancomycin
Daptomycin
(t½ 9 h) is a recently released lipopeptide antibiotic, naturally produced by the bacterium Streptomyces roseosporus which was first isolated from a soil sample from Mount Ararat in Turkey.3 It has activity against virtually all Gram-positive bacteria, including penicillin-resistant Streptococcus pneumoniae and MRSA, regardless of vancomycin resistance phenotype. It is unable to cross the Gram-negative outer membrane, rendering these bacteria resistant.
[/not-level-membership-for-basic-science-category]
