Chapter 80 Antiarrhythmic Drugs
Reorientation of drug therapy has also been necessary in the wake of the knowledge that certain antiarrhythmic agents, while suppressing supraventricular and ventricular arrhythmias, may increase mortality by their associated proarrhythmic effects. This was demonstrated in the Cardiac Arrhythmia Suppression Trial.1 The findings have important clinical implications with respect to the choice of antiarrhythmic agents in the short-term, long-term, and prophylactic control of arrhythmias. The issue is critical in the choice of intravenous therapy of ventricular arrhythmias not only in hospitalized patients but also in those developing out-of-hospital VT or VF, especially in the choice of agents as an integral part of the current Advanced Cardiovascular Life Support (ACLS) guidelines.2
Classification of Antiarrhythmic Drugs Revisited
Interest in the mechanisms of action of pharmacologic agents has provided the basis for their classification since the 1970s.3–9 The original intent was to classify the mechanisms of action of antiarrhythmic drugs and not the drugs themselves. Such an approach permitted grouping of drugs by their dominant action with the inevitable realization that many antiarrhythmic drugs in the clinic exerted single actions (e.g., β-blockers) or a varying spectrum of actions (sotalol and amiodarone) with their defined overall clinical effects both beneficial as well as deleterious.
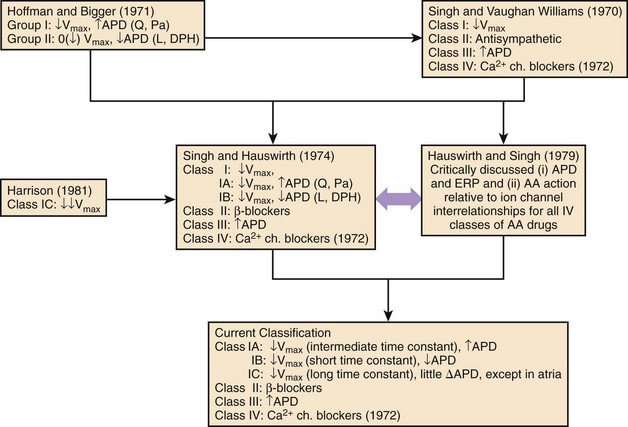
The attempts at classification of the manner in which antiarrhythmic agents might work beneficially in the control of disorders of rhythm; this follows closely in the wake of a series of experimental studies of a number of structurally disparate compounds having certain discrete electrophysiological actions.3–5,8,9 These compounds were local anesthetics or sodium channel blockers, β-adrenoceptor blockers, drugs that were also found to have anti-fibrillatory actions in the context of their property to selectively prolong cardiac repolarization, and those that were found to selectively block calcium channels at the membrane level. The premise of the initial classification was that each of the mechanisms described exerted, although in differing ways, an anti-fibrillatory effect in a standardized experimental model of VF.5 The evolution of the steps in the classification of antiarrhythmic mechanisms of drugs that are now widely used stems from the original framework, which has subsequently been modified slightly, the most recent being by Nattel and Singh.3–5,8,10 The final step completed in 1979 is shown in Figure 80-1.11 The need for the classification of antiarrhythmic drug mechanisms was conceived almost simultaneously and independently on both sides of the Atlantic. It will be noted that the Hoffman and Bigger attempt at classifying antiarrhythmic drugs placed compounds such as quinidine and procainamide, which had been known to slow conduction as well as to prolong repolarization in one category (type 1), and lidocaine and diphenylhydantoin (DPH), which also inhibited conduction but shortened repolarization into another category (type 2).12 Because propranolol in high doses exhibited potent conduction blocking properties, it was included among type 1 antiarrhythmic compounds.
On the basis of the findings in the experimental laboratory, it was suggested that type 1 compounds terminated or prevented arrhythmias by converting uni-directional block into a bi-directional one in an arrhythmia circuit, whereas the type 2 agents acted by eliminating uni-directional block by increasing conduction velocity. Subsequently, as an integral part of a series of studies on which the conventional antiarrhythmic classification has been based, it was found that this observation might have occurred as a consequence of the use of potassium ion (K+) concentration of 2.7 mEq/L in the perfusion media.9 The retesting of the effects of lidocaine and DPH in media containing physiological levels of potassium ions clearly established that both the compounds had their predominant actions as inhibitors of sodium (Na) channel–mediated conduction. It also became evident that in clinically significant concentrations, propranolol exerted no local anesthetic actions.8
In contrast, in the comprehensive classification scheme suggested by Singh, and Singh and Vaughan Williams, all drugs that exerted local anesthetic actions in the nerve as well as in the myocardial membrane (including lidocaine, quinidine, procainamide, disopyramide, and DPH among numerous others) were thought to act via class I actions; and propranolol and other β-adrenoceptor–blocking compounds that exhibited sympatholytic actions were characterized as acting via class II antiarrhythmic actions.3,5,7
When it was found that compounds such as sotalol (a β-blocker) and amiodarone, which prolonged repolarization, exerted unequivocal anti-fibrillatory actions not accounted for by class I or class II actions, the possibility of a class III action was suggested.3 Subsequently, on the basis of the electropharmacologic effects of the drug verapamil, a fourth class of antiarrhythmic actions was added by Singh and Vaughan Williams in 1972.7 Hauswirth and Singh subdivided class I compounds into those that suppressed sodium channels in all cardiac fast-channel tissues, as typified by quinidine and procainamide (class IA agents), and those that inhibited sodium channels only in diseased or depolarized tissues (class IB: lidocaine and DPH).11 Another difference between these two subclasses was that in the case of the 1A agents, repolarization was also prolonged, whereas in the case of 1B agents, repolarization was abbreviated. Harrison completed the subclassification by assigning the potent class I agents flecainide and encainide into a group designated as class IC.13 Experimental studies by Campbell validated the separation of subclasses of sodium channel blockers on the biophysical basis; he found that class IA agents had sodium channel–blocking kinetics between those of class IB agents (very fast) and IC compounds (very slow).14
This classification of antiarrhythmic drugs is still widely used in clinical practice. It has been the basis for the development of newer antiarrhythmic compounds. It was recognized early that a direct extrapolation of the experimental data to the clinical setting in this regard might not be readily possible. However, it was considered that for advances in understanding drug action, it was desirable to explore the effects of various classes of antiarrhythmic drugs in myocardial cells and membranes in terms of ion channels and related parameters, on the one hand, and relate them to their clinical effects, on the other hand.8,11 This approach was first suggested in detail by Hauswirth and Singh in 1979, and it re-emerged subsequently in the form of the Sicilian Gambit in 1991, incorporating an increasing understanding of membrane currents with the introduction of the patch clamp technique.11,15
Amiodarone and sotalol—two unique compounds—the dominant electrophysiological property of which is the prolongation of repolarization, formed the basis for the class III action.3,4,7,16–18 Amiodarone and sotalol have provided the background for the synthesis and characterization of simpler compounds (such as dofetilide and azimilide) as well as the impetus to develop other agents with similar properties but safer electropharmacologic profiles. The main properties of the major agents are discussed in this chapter relative to their roles in the control of supraventricular and ventricular tachyarrhythmias. Recently approved compounds for AF treatment, such as dronedarone and vernakalant, are also discussed. Certain electrophysiological properties of antiarrhythmic compounds are of much importance in the clinical area. They will be emphasized at the outset.
Heart Rate Dependency of the Action of Antiarrhythmic Agents
The electrophysiological property that also appears to be of major clinical interest is the rate-dependent effect of the compounds on the action potential duration (APD) and refractoriness.19 Rate dependency refers to a different effect at varying heart rates, with reverse-rate dependence (less effect at higher rates) being of great clinical importance. The differences in this parameter among the various agents may be of therapeutic relevance. This issue is of particular relevance in the case of their role in the control of AF. The drugs that exert the classic reverse-rate dependency (dofetilide, quinidine, and d,l-sotalol) appear to exhibit a similar ceiling of efficacy for maintaining the stability of sinus rhythm and a similar propensity for inducing torsades de pointes (TdP). Their effect on prolonging the APD and the effective refractory period (ERP) in the atrial muscle decline as the stimulation frequency increases. All such compounds are powerful blockers of the delayed rectifier current (IKr), and they are likely to be more effective in terminating AFL than AF when acutely administered. They are moderately effective in preventing recurrences of AF and AFL in the paroxysmal and persistent forms of these arrhythmias.
In contrast, compounds such as azimilide and possibly ambasilide, which may also inhibit the slow component of the delayed rectifier current (IKs), exhibit a different pattern of rate dependency of action with respect to the APD and associated refractoriness. Under the action of the drugs, a parallel increase occurs in the APD and the ERP, as shown in Figure 80-2. Whether this is clinically significant may require a direct comparison of these compounds with those of other so-called class III compounds such as dofetilide. In the case of azimilide, the available data suggest a low incidence of TdP, a neutral effect on mortality in high-risk patients with previous myocardial infarction (MI) and at least moderate efficacy in maintaining the stability of sinus rhythm in patients with AF.20,21 Large pivotal placebo-controlled trials with respect to azimilide are in progress.
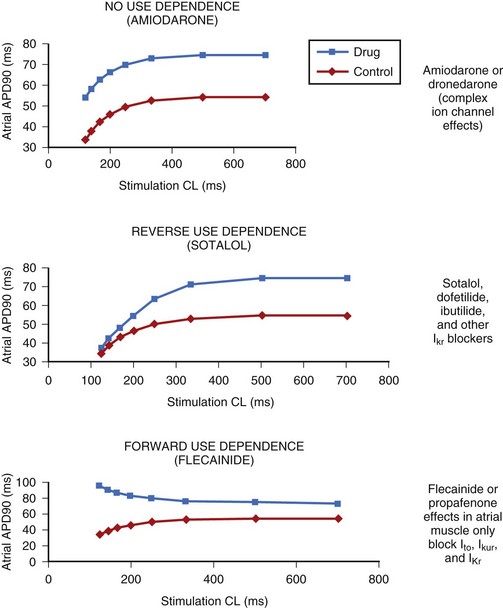
Of particular interest, the action of amiodarone and that of dronedarone, with respect to rate-dependent effects, are similar, as they are associated with a parallel shift (i.e., ERP increases in a parallel fashion over a wide range of frequencies), as noted in the case of azimilide and ambasilide.10,22 In the case of amiodarone evidence for the effectiveness of the drug for maintaining the stability of sinus rhythm is growing. Its potency (see later) appears the highest among all class III antiarrhythmic compounds, and it is associated with the lowest incidence of TdP in the context of the longest Q-T interval that amiodarone may produce during the course of long-term treatment. The precise reason for this combination of drug effects remains unclear, but the observations are clearly of theoretical importance for the purposes of developing compounds in the future. Perhaps of great importance also is the observation that in the case of flecainide (presumably also propafenone) is the nature of their effects on the ERP and the APD as a function in atrial tissue. Wang and coworkers found that in a variety of mammalian atria, flecainide (and presumably propafenone acts in a similar manner) had the property of prolonging the APD and the ERP as a function of rate.23 For example, greater effects were seen as the stimulation rates were increased (Figure 80-3) (see the discussion on atrial-selective drugs below). The available data suggest that the rate-related effects of antiarrhythmic drugs may vary in differing cardiac tissues, which is possibly a reflection of the differential action of drugs on the ion channels that they may inhibit to varying extents.
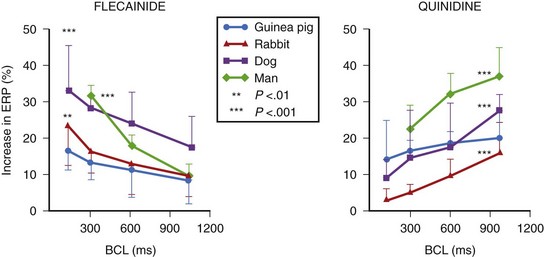
FIGURE 80-3 Rate-dependent effects of flecainide and quinidine on effective refractory period (ERP) in mammalian atria. The data contrast the forward use dependence of flecainide with the reverse use dependence of quinidine (see Figure 80-2).
(From Wang Z, Pelletier LC, Talajic M, Nattel S: Effects of flecainide and quinidine on human atrial potentials: Role of rate dependence and comparison with guinea pig, rabbit and dog tissues, Circulation 82:274–283, 1990.)
Class I Antiarrhythmic Compounds
Conversely, lidocaine and its oral congeners, mexiletine and tocainide, and DPH (class IB agents), in fact, shorten repolarization and hence refractoriness, in addition to slowing conduction. The actions of class I agents are usually more intense in diseased tissues that are partially depolarized, such as in myocardial ischemia. Here, their proarrhythmic effects in settings of ischemia or left ventricular dysfunction are often much greater and may prove fatal—as in the case of flecainide, encainide, and propafenone (class IC compounds), which are contraindicated in patients with significant ventricular dysfunction. As a class of antiarrhythmic drugs, the use of sodium channel blockers is declining because none of the agents has the potential to increase survival by controlling cardiac arrhythmias. If they are administered to patients with potentially serious cardiac disease, the risk of mortality may increase.24 Their main usefulness is in the conversion of AF to sinus rhythm and in maintaining the stability of sinus rhythm in patients in whom the arrhythmia occurs with normal or near-normal ventricular function. Relevant aspects of individual agents are presented below.
Quinidine
Quinidine has the dual electrophysiological properties of prolonging repolarization and slowing conduction by blocking inward sodium current, thereby slowing conduction and by blocking a variety of outward potassium currents; the effects of the drug on myocardial ion channels are compared with those of other antiarrhythmic drugs in Table 80-1. The drug is available in injectable and oral forms, but the injectable formulation is now rarely used. Quinidine may increase heart rate and facilitate AV nodal conduction by its vagolytic actions and increase ventricular response in AF. The major electrophysiological effect of clinical utility relative to the drug’s antiarrhythmic actions is the prolongation of the APD and the ERP (a class III action); the effects on both are attenuated as the heart rate increases. The drug has been used in patients with impaired ventricular function, but it may depress myocardial contractility. The elimination half life of quinidine is 8 to 9 hours, its metabolism being largely by hepatic hydroxylation. The usual oral dose of the drug is 1.2 to 1.6 g/day in 8 to 12 hourly divided doses relative to the preparation in use.
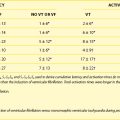
Table 80-1 Effects of IKr Inhibition on Effectiveness in Maintaining Sinus Rhythm in AF Relative to the Development of TdP and Total Mortality with and Without Blocking Other Ionic Currents
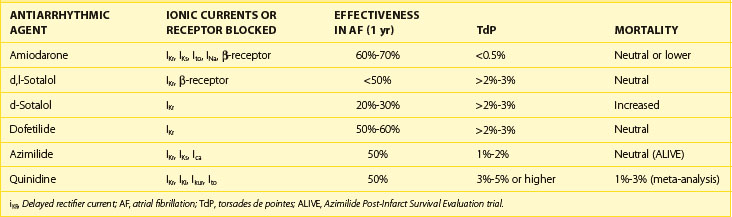
The most common adverse effects of quinidine include diarrhea, nausea, and vomiting, which occur in one third of all patients. Quinidine can also cause cinchonism (headache, dizziness, and tinnitus), and quinidine can cause syncope, a syndrome characterized by lightheadedness and fainting. The major shortcomings of quinidine are severe diarrhea, frequently caused by the drug, and TdP, which develops in 2% to 5% in association with a prolonged Q-T interval. Rarely, TdP may be fatal. In a detailed meta-analysis of the trials in AF involving quinidine, an increase in mortality has been reported.25 For these reasons, when considered in conjunction with the limited ceiling of effectiveness of the drug in ventricular arrhythmias as well as in supraventricular arrhythmias, the clinical utility of quinidine is limited. Its continued use can be justified only marginally.
Disopyramide
As in the case of quinidine and procainamide, disopyramide as a class IA antiarrhythmic agent has limited clinical use. Its electropharmacologic profile is similar to that of quinidine, but it has a more potent anticholinergic action and it produces a lesser degree of gastrointestinal disturbance. The elimination half life of the drug is about 8 hours, but slow-release formulations have been introduced. The effects of the drug in arrhythmia control have not been extensively studied except for the suppression of premature ventricular contractions (PVCs). Disopyramide has been used largely in the oral form at a dose of 100 to 200 mg q6h, but it is now rarely used for the control of ventricular tachyarrhythmias. Like quinidine, it does prolong the Q-T interval and may produce a variable incidence of TdP; of particular importance, in patients with impaired ventricular function and in those with heart failure, it is likely to severely aggravate cardiac decompensation by its potent negative inotropic actions. However, such a property may be of therapeutic value in the syndrome of idiopathic hypertrophic cardiomyopathy (HCM), a setting in which the drug produces a greater negative inotropic effect than do β-blockers with which disopyramide can be combined. Although not approved in the United States, disopyramide may have a significant anti-fibrillatory effect in AF in two settings where its role in controlling this arrhythmia may be unique—in patients with HCM who develop AF and in patients whose AF is entirely caused by excessive vagal tone.26 In the latter setting, the drug’s anticholinergic actions may be a specific therapy, which might be strikingly effective. It should be emphasized, however, that the drug’s anticholinergic actions may lead to serious adverse reactions in the form of urinary retention, worsening of glaucoma or myasthenia gravis, and severe constipation.
Lidocaine, Mexiletine, and Tocainide
The properties and the roles of intravenous lidocaine and its two orally active congeners, mexiletine and tocainide, as class IB compounds, will be discussed only briefly because their roles have declined considerably in recent years and they are likely to be only of historic interest. Despite the suppression of ventricular arrhythmias that class IB agents produce, data on their adverse effects on mortality continue to accrue.27,29 Lidocaine, a class I antiarrhythmic, is available as an injection. Lidocaine action is characterized by a fast, on-off block of sodium ion channels in both the activated and inactivated states, with a preference for inactivated state sodium channels. It also shortens the duration of the APD (hence the Q-T interval), and refractoriness, and has been shown in the experimental setting to elevate the ventricular defibrillation threshold. Thus, the drug and its oral congeners (also available as intravenous formulations) may act largely by slowing conduction rather than prolonging refractoriness. However, their actions in terminating and preventing arrhythmias may also stem from the conversion of unidirectional block to a bidirectional block in the arrhythmia re-entrant circuit although the proof for this in the clinical setting is lacking. Lidocaine is metabolized rapidly in the liver, and it is administered generally as an initial bolus of 100 to 200 mg, followed by 2 to 4 mg/min for varying durations of time (usually 24 to 36 hours) aiming at serum levels of 1.4 to 5 µg/mL.
Lidocaine gained prominence as a prophylactic agent in the early days of the inception of coronary care units and subsequently as the first-line agent in the control of VT/VF in all acute care units, including the emergency department, and in the resuscitation of those developing out-of-hospital cardiac arrest. The use of lidocaine and, indeed, other antiarrhythmic drugs in these settings was not based on data from controlled clinical trials. It is of interest to note that in a direct blinded comparison, intravenous lidocaine converted 18% of VT to sinus rhythm compared with 69% conversion with intravenous sotalol.30 The Cardiac Arrhythmias Suppression Trial on the drugs encainide, flecainide, and moricizine indicated a dichotomy between arrhythmia suppression and mortality.1 Abundant data now suggest that intravenous lidocaine and mexiletine are similarly ineffective in the setting of postmyocardial infarction and raise the issue that they might also be similar in the control of destabilizing VT or VF as well as in the survivors of out-of-hospital cardiac arrest.27–29 It is not surprising that in line with the data, the most recent AHA/ACC/ACLS (American Heart Association/American College of Cardiology/Advanced Cardiac Life Support) Guidelines have relegated the use of lidocaine as being “indeterminate.”2 This issue will be discussed in depth later in this chapter when the effects of intravenous lidocaine and intravenous amiodarone will be discussed relative to the broader implications of the choice antiarrhythmic agents in the control of life-threatening ventricular arrhythmias in various clinical settings.
Class 1c Agents
This class of antiarrhythmic agents developed on the basis of the belief that the suppression of ambient arrhythmias—whether PVCs, sustained VT, or nonsustained VT—by antiarrhythmic drugs should result in prevention of arrhythmic death and in the prolongation of survival. The fact that such a hypothesis was not vindicated has had a number of therapeutic consequences: (1) near-complete or complete suppression of PVCs in the patient with previous MI is associated with an increased mortality rate in those with cardiac disease and indicates that this class of drugs cannot be used with impunity even for the control of symptoms caused by arrhythmias; (2) increased observed mortality induced by class IC agents may be a property of class I agents in general and is also found in the case of lidocaine; and (3) data do not exclude the possibility for using such agents for controlling arrhythmia symptoms or for restoring and maintaining sinus rhythm in AF or AFL in patients without structural heart disease. This is especially so in the case of flecainide and propafenone. Moricizine is often included in the category of class IC agents, but its properties are difficult to classify, and it may not have any advantages over flecainide or propafenone. These agents slow conduction velocity profoundly but have little, if any, effect on refractoriness in ventricular tissues; and theoretically, they eliminate re-entry by slowing conduction to a point where the impulse is extinguished and cannot propagate further. This may be the basis of their effectiveness in markedly reducing ventricular ectopy. Their actions in atrial tissues differ markedly compared with those in the ventricle. They increase the ERP in the atria as a function of increases in heart rate, an effect that is likely to be the basis of their effectiveness in restoring and maintaining sinus rhythm.23 This is their major clinical utility as antiarrhythmic drugs.
Flecainide
Flecainide is a powerful blocker of sodium channels in virtually all cardiac tissues, but it does not significantly inhibit the pacemaker current or the calcium channels. It has effects on K+ channels but without a major effect on repolarization, except in the atria, where repolarization is prolonged and the effective period is lengthened. The drug has a significant negative inotropic effect especially at higher doses. The plasma half-life of the drug is 13 to 19 hours; two thirds of it is metabolized in the liver. The usual dose of flecainide is 100 to 300 mg, given twice daily. The intravenous formulation is available but is not used routinely. One clinical utility of flecainide is in the control of intractable symptoms caused by PVCs in patients with no significant cardiac disease. Perhaps its greatest value is in the restoration and prevention of recurrences of AF but only in patients without structural heart disease because of its serious proarrhythmic reactions in patients with cardiac disease.31 In the prophylactic control of AF or AFL, the drug should be combined with an AV nodal blocking drug (β-blockers or calcium channel blockers) to avoid the development of facilitated conduction across the AV node as the atrial rate slows under the action of the drug. In recent years, single oral doses (200 to 300 mg) of the drug have been successfully used for the acute termination of paroxysms of AF (“pill in the pocket” approach).
Propafenone
Propafenone is also a powerful class IC agent with an electrophysiological activity profile similar to that of flecainide; it also has a mild degree of β-blocking action, which does not appear to be clinically relevant.32 Its actions in the atria may also be similar to those of flecainide, and the drug does not prolong repolarization in ventricular tissues. The drug is rapidly absorbed with the bioavailability of 50%, its elimination half-life being 2 to 10 hours in normal subjects and 12 to 32 hours in poor metabolizers. Orally, propafenone is administered as 150 to 300 mg three times daily, and a longer acting formulation has been introduced. The drug can also be administered intravenously and as single oral doses (300 or 600 mg) for the acute conversion of paroxysms of atrial AF. As in the case of flecainide, propafenone should not be used in patients with significant cardiac disease, and its two main indications are for the suppression of resistant symptomatic PVCs and for the restoration and maintenance of sinus rhythm in the case of AF in patients with structurally normal hearts. Its efficacy rivals that of flecainide, although direct comparisons have not been made. As in the case of flecainide, propafenone should be combined with an AV nodal blocking drug to reduce the possibility of accelerated conduction with the slowing of atrial rate induced by the drug.
β-Adrenergic Blockers as Antiarrhythmic and Anti-fibrillatory Compounds
β-Blockers as a class of drugs exert distinctive antiarrhythmic and anti-fibrillatory effects because of their properties of consistently alleviating symptoms and prolonging survival in a wide subset of patients. The electrophysiological and anti-fibrillatory effects are most striking in the clinical context of most intense sympathetic stimulation. They prevent the development of VF in a variety of experimental animal models. The beneficial effects in this setting could not be accounted for by any known electrophysiological mechanisms. However, in the clinic, the antiarrhythmic potential of β-blockade was greatly overshadowed by its anti-ischemic actions for which this class of drugs was synthesized. Thus, the recognition of the fact that blunting the effects of catecholamines on the heart might be a potent anti-fibrillatory mechanism was slow in coming. It has been known for many years that increased activity of the sympathetic nervous system might induce cardiovascular morbidity and mortality through a variety of mechanisms. For example, given the appropriate pathologic substrate, increased sympathetic activity may be associated with sudden arrhythmic death.33–35 This is now known to be particularly striking in the case of patients developing MI with or without heart failure.
The intrinsic effects of β-blockers may be modified to varying extents by the associated pharmacologic properties that individual agents may have. For example, in the case of propranolol at high concentrations the sodium channel is significantly inhibited. Some agents (e.g., acebutolol, atenolol, bisoprolol, carvedilol, and metoprolol) are relatively cardioselective for blocking β1-adrenoceptors, and others are nonselective (propranolol, nadolol, timolol, and sotalol) with respect to β1– and β2-adrenoceptors. However, evidence that these varying degrees of selectivity of action significantly alter the antiarrhythmic and anti-fibrillatory actions of β-blockers in patients is scarce. However, little doubt exists that the presence of marked agonist actions (e.g., in pindolol) is associated with often significant increases rather than decreases in heart rate that largely offset the antiarrhythmic and anti-fibrillatory actions of a given agent.36
Electropharmacologic Properties and Anti-fibrillatory Actions
The precise effects of β-antagonists on ionic currents in differing myocardial cells in terms of depolarization and repolarization are difficult to characterize.37 It should be recognized that for a given agent within the broad class of these compounds, the effects may stem from their intrinsic properties of blocking β-receptors as well as from their associated pharmacologic actions. The dominant effect, however, is from β-receptor blockade, which has minimal effects on calcium channels (ICa-L) or various potassium channels (IK). It is known that sympathomimetic amines exert a stimulant effect on the pacemaker current (If), which is markedly inhibited by β-blockers. Indeed, the blocking effect of the pacemaker current by β-blockers is the most readily defined pharmacodynamic property of this class of drugs and one that correlates well with the beneficial effects on mortality in patients with cardiac disease. The effects of β-blockade on the nodal tissues are, therefore, significant with the slowing of the heart rate by effects in the sinoatrial (SA) node and by prolonging refractoriness in the AV node, which may be of clinical utility in terminating SVTs and slowing the ventricular response in the setting of AFL and AF.
Antiarrhythmic Actions of β-Blockers
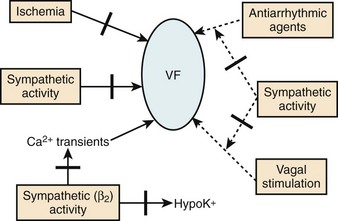
Perhaps, the antiarrhythmic and anti-fibrillatory actions of β-blockers are best characterized in terms of their property of attenuating the deleterious effects of excess catecholamines. The electrophysiological consequences of sympathetic hyperactivity have been extensively documented in numerous experimental and clinical studies.33–35 In the experimental setting, they have included (1) shortening of the ventricular APD, and hence the ERP; (2) augmenting ventricular conduction; (3) increasing ventricular automaticity; (4) reducing vagal tone; (5) decreasing VF threshold; and (6) the reversal or attenuation of the effects of antiarrhythmic drugs being administered in the expectation of preventing arrhythmic deaths. Conversely, it is known that the depletion of the adrenergic transmitters to the heart increases VF threshold; and in experimental models in which VF could be induced reproducibly, the arrhythmia is preventable by sympathetic blockade.33 Aspects of the anti-fibrillatory effects of β-adrenergic blocking drugs are shown in Figure 80-4.36 In fact, the appreciation of such an anti-fibrillatory effect was the basis for classifying it as a class II antiarrhythmic action.3–8
β-Blockade
Impact on Sudden Cardiac Death and Total Mortality in Survivors of Myocardial Infarction and in Patients with Heart Failure
β-Adrenergic blocking drugs are effective in reducing mortality in many subsets of patients with manifest arrhythmias and in those at high risk of dying from arrhythmic deaths.37–42 For example, they reduce death rates in patients with congenital long Q-T syndrome (LQTS), in survivors of cardiac arrest, and in selected cases of VT, although the data supporting these conclusions have not always been from controlled clinical trials.39–43 However, they are in line with the compelling data from randomized, placebo-controlled β-blocking trials, in which consistent and significant decreases in mortality have occurred, especially in survivors of acute MI (AMI) and in those with congestive heart failure.44–51 These trials have usually been of adequate sample size and of acceptable protocol design with features that include double-blind comparison in placebo-controlled studies. They have shown that these agents, as a class, not only bring about a beneficial change in total mortality but also on sudden, presumably arrhythmic death (Table 80-2). The overall clinical utility and versatility of this class of antiadrenergic compounds are well documented as indicated in Table 80-3.36
Table 80-2 Principal Uses of β-Blockers to Reduce Life-Threatening Ventricular Arrhythmias and Control Postoperative Atrial Fibrillation After Cardiac Surgery
| INDICATIONS | COMMENTS |
|---|---|
| Cardiac arrest survivors | No controlled clinical trials but uncontrolled data compelling |
| Adjunctive therapy to ICDs | |
| Controlled data still to be obtained, but support for VT/VF strong from large uncontrolled database | |
| Congenital long QT syndrome | Compelling but uncontrolled data used a single drug modality, now being replaced with ICDs, with which they often need to be combined |
| MI survivors | Most consistent antiarrhythmic agents increase survival in this setting, reduce total mortality, sudden death, and re-infarction rates |
| Overwhelmingly positive data from controlled clinical trials | |
| Congestive cardiac failure | Reduce total mortality and sudden death in CHF of ischemic and nonischemic origins: role well established |
| Prevention of AF | Prophylactic administration reduces the incidence of AF in postoperative cardiac patient undergoing cardiac surgery |
AF, Atrial fibrillation; ICDs, implantable cardioverter-defibrillators; MI, myocardial infarction; VT/VF, ventricular tachycardia/ventricular fibrillation.
Table 80-3 Actions of Intravenous Amiodarone Compared with Long-Term Amiodarone
| actions | INTRAVENOUS AMIODARONE | long-term AMIODARONE |
|---|---|---|
| Repolarization (Q-T interval) prolongation (atria and ventricles) | + | ++++ |
| Conduction velocity (atria and ventricles reduced) | ++ | ++ (as a function of rate) |
| Sinus rate reduced | + | +++ |
| AV nodal conduction slowed | + | ++ |
| AV nodal refractoriness increased | ++ | +++ |
| Atrial refractoriness increased | + | +++ |
| Ventricular refractoriness increased | + | +++ |
| Noncompetitive α- and β-blocking activities | + | ++ |
AV, Atrioventricular.
β-Blockers, as a class, consistently reduce the incidence of SCD and total mortality in the survivors of AMI and in patients with cardiac failure of ischemic and nonischemic origins. In the case of patients with MI, beneficial effects on SCD and total mortality have been documented at the time of diagnosis out of the hospital, during hospital stay, and following discharge from the hospital. In the latter case, SCD and total mortality were reduced from 18% to 39% during the first year. It is presumed that the benefit in survival is from the prevention of VF. It is noteworthy that with β-blockers, mortality is reduced over the first 10 days when the drugs are given in an initial intravenous dose followed by oral therapy as well as when they are given in conjunction with thrombolytic therapy during the early stages of AMI. Two other features of the response to β-blockers in this setting should be emphasized. First, unlike trials such as CAST, β-blocker trials have not been arrhythmia suppression trials, but the degree of benefit on survival correlated linearly with the degree of heart rate reduction, in the case of MI as well as with cardiac failure.1 Second, evidence from two post-MI trials showed that total mortality reduction was greater when amiodarone was administered in combination with β-blockers versus when it was used alone in the drug treatment limb.52–54
As summarized recently, β-blockers, given in controlled and graduated dose regimens, have been shown to variably but significantly reduce mortality in subsets of patients with advanced congestive cardiac failure.5,55–61 The outcomes of three recent trials with three different β-blockers should be emphasized (Table 80-4).56,57,60
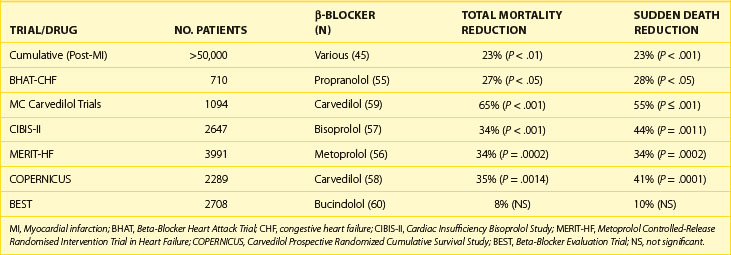
In the trial involving the use of metoprolol in 3991 patients with largely New York Heart Association class II to IV heart failure (LVEF <40%), randomized to long-acting metoprolol (n = 1990) in graduated doses of up to 200 mg/day or placebo (n = 2001), the primary endpoint was all-cause mortality, analyzed by intention to treat. The mean follow-up was 1 year. During the period of follow-up, 147 deaths (7.2%) occurred on metoprolol versus 217 deaths (11.0%) on placebo (P < .00009). A statistically significant reduction in SCD on metoprolol was seen compared with placebo.56
Similar data have been reported from a double-blind, multi-center study on the drug bisoprolol in 2647 patients randomized to a β-blocker (n = 1327) or placebo (n = 1320). The patients were all in classes III and IV with left ventricular ejection fraction (LVEF) of 35% or lower and all were taking diuretics and angiotensin-converting enzyme (ACE) inhibitors as in the case of Metoprolol Controlled-Release Randomised Intervention Trial in Heart Failure (MERIT-HF). All-cause mortality was 11.8% (156 deaths) versus 17.3% (17.3%). This difference was significant (P < .0001). The drug was effective in reducing SCD significantly. Cardiovascular deaths on bisoprolol were significantly fewer, fewer patients were admitted to the hospital, and the difference for the combined endpoints was also significant. The numbers of permanent treatment withdrawals were similar for bisoprolol and placebo. In the case of carvedilol, a number of relatively smaller individual trials have been conducted, but the cumulative data have been sufficiently compelling for making it the first β-blocker to be approved for use in congestive heart failure in the United States.60 The composite data from all the trials of β-blockers performed to date support the routine use of this mode of prophylactic therapy in all patients in whom β-blockers are not contraindicated.
Drugs Acting by Prolonging Repolarization
Of the currently available antiarrhythmic compounds, β-blockers, sotalol, amiodarone, ibutilide, and dofetilide are likely to remain the prototypes of the drugs of the future. Amiodarone and sotalol are of the greatest interest.62–67 They prolong repolarization and refractoriness in atria and ventricles while blocking sympathetic stimulation, although by differing mechanisms. Their propensities to decrease heart rate and to modulate the AV node function, and thereby slow ventricular response in AF and AFL, have additional value for the control of many arrhythmias. Thus, they have the greatest clinical utility for controlling ventricular as well as supraventricular arrhythmias. Their respective roles are further bolstered by controlled trials showing superiority over class I agents in patients with VT or VF.68,69 The perception that in spite being powerful sympathetic antagonists, sotalol and amiodarone acted dominantly by prolonging the APD led to the search for pure compounds devoid of other associated properties having simpler adverse effect profiles.70–73 Intravenous amiodarone has recently been studied extensively in clinical trials in destabilizing VT or VF in the hospital and in shock-resistant VF following out-of-hospital cardiac arrest.2,74–78
Current data suggest that compounds that induce an isolated or “pure” prolongation of the APD accompanied by a proportional lengthening of myocardial refractoriness may be anti-fibrillatory.37 Under certain circumstances, they have advantages over complex compounds such as sotalol or amiodarone. This has been the case with dofetilide, ibutilide, and azimilide.79 The properties of tedisamil, which is also under development, are somewhat more complex.37 Such compounds may be the result of the block of a single repolarizing ionic current (e.g., dofetilide) or multiple currents (ibutilide, tedisamil, or azimilide). Thus, for the present, it would appear that two subclasses of such agents might be appropriate to develop—those that are either safer and more effective chemical derivatives of sotalol and amiodarone or those that are relatively pure prolongers of repolarization and refractoriness. All such agents may need to produce a homogeneous lengthening of repolarization by selectively inhibiting multiple or single ionic channels in the myocardium with a minimal proclivity to cause TdP.
Amiodarone as a Versatile and Complex Class III Agent
Amiodarone was synthesized as an antianginal drug in 1962, but attention was not drawn to the compound for its unique properties until 1970 when Singh and Vaughan Williams showed the properties of the drug to prolong cardiac repolarization, with anti-fibrillatory properties and the potential for slowing heart rate because of its nonspecific antiadrenergic actions.4 Since these early studies, it has become increasingly evident that amiodarone is an exceedingly complex antiarrhythmic compound (Figure 80-5).
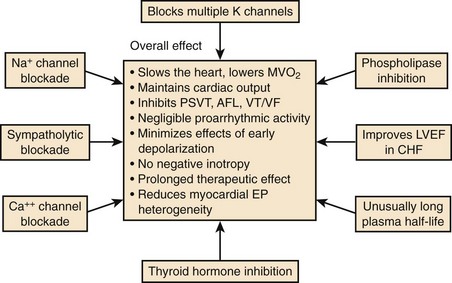
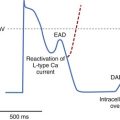
The electrophysiological effects of amiodarone administered acutely and after a period of long-term drug therapy differ markedly (see Table 80-3), both associated with distinct and potent antiarrhythmic actions.36 The electropharmacologic effects of the compound are multi-faceted.80 The most striking long-term property is the ability of the drug to increase the APD in atrial and ventricular tissues following long-term drug administration. Little or no effects occur in Purkinje fibers and M cells, a phenomenon that leads to the reduction in QT dispersion.81–83 The drug is also unique in that its effect on repolarization is not influenced by heart rate.84,85 In isolated myocardial fibers, it eliminates the tendency for the development of early afterdepolarization (EAD) produced in Purkinje fibers and in M cells. The ionic channels that amiodarone blocks after long-term drug administration are not fully defined, but it has been shown to block Ito and IK as well as INa and Ica.64 It abolishes EADs induced in isolated cardiac tissue.84 In humans, despite producing marked slowing of the heart rate and considerable increases in the Q-T or QTc interval, the drug has a very low propensity for producing TdP.84 Amiodarone rarely exhibits proarrhythmic actions typical of class I agents, although it is a fairly potent sodium channel blocker, especially at high heart rates. The salient pharmacodynamic features of amiodarone are summarized in Figure 80-5.
Amiodarone has a broad-spectrum antiarrhythmic effect with a varied adverse effect profile, which is generally dose and duration dependent. Included in Table 80-5 are the main potential clinical indications that have been accepted. Intravenous amiodarone is effective in the control of hemodynamically significant and refractory VT or VF (to lidocaine and procainamide), with a potency at least as high or higher than that of intravenous bretylium.74,75 The drug exerts a powerful suppressant effect on PVCs and nonsustained ventricular tachycardia (NSVT) and provides control in 60% to 80% of recurrent VT or VF when conventional drugs have failed during continuous oral therapy.86 In only a small number of patients does the drug prevent inducibility of VT or VF, there being little or no systematic relationship between the prevention of inducibility of VT or VF and the long-term clinical outcome.87
Table 80-5 Major Uses of Amiodarone in the Control of Ventricular and Supraventricular Arrhythmias
| INDICATIONS | COMMENTS |
|---|---|
| Cardiac arrest survivors | Superior to class I agents (guided therapy); tolerated in VT/VF and in all levels of LVEF and CHF; low incidence of torsades de pointes Increasingly perceived as the most potent antiarrhythmic agent, especially in the setting of low LVEF but being increasingly replaced with ICDs for primary therapy in many subsets of patients with VT/VF May increase VT defibrillation threshold; clinical significance unclear |
| Adjunctive therapy to ICDs | Controlled data still to be obtained but support for VT/VF is strong from large uncontrolled database; preferred agent in patients with significantly impaired LVEF and CHF |
| Conversion of VT to SR by IV regimen | Precise efficacy not known |
| Intravenous amiodarone in destabilizing refractory VT/VF | Superior to lidocaine and procainamide; efficacy equal to that of bretylium but with less hypotension; increases survival to hospitalization in patients with out-of-hospital cardiac arrest (ARREST and ALIVE trials) First-line antiarrhythmic agent in cardiac arrest resuscitation |
| Post-MI survivors | Numerous uncontrolled positive mortality studies; two recent placebo-controlled studies—EMIAT and CAMIAT—significant reduction in arrhythmic deaths, trend toward total mortality reduction in one of the studies, augmented effect, if combined with β-blockers Meta-analysis of all data consistent with reduction in total mortality and in sudden death while on drug |
| Congestive cardiac | Two controlled trials, one blinded placebo-controlled (CHF STAT) and one blinded (GESICA) indicate a spectrum of failure effects from reduction in total mortality (GESICA) and neutral (CHF STAT) with a trend in selective favorable effects in nonischemic cardiomyopathy Amiodarone increased LVEF |
| ATRIAL FIBRILLATION | |
| Acute conversion | Variable efficacy but systematic placebo-controlled study against a comparator agent not available |
| Maintenance of SR | Up to 60% or greater at 12 months after restoration of SR; maintains ventricular on relapse to atrial fibrillation on oral drug (controlled trials in progress) |
| Effect on AF prevention in postoperative cardiac surgery | Preoperative administration of oral drug reduces incidence of postoperative AF; IV drug given postoperatively may shorten hospital stay by reducing AF. |
AF, Atrial fibrillation; CHF, congestive heart failure; ICD, implantable cardioverter-defibrillator; IV, intravenous; LVEF, left ventricular ejection fraction; MI, myocardial infarction; SR, sinus rhythm; VT/VF, ventricular tachycardia and ventricular fibrillation; ARREST, Amiodarone in Out-of-Hospital Resuscitation of Refractory Sustained Ventricular Tachyarrhythmias trial; ALIVE, Azimilide Post-Infarct Survival Evaluation trial; EMIAT, European Myocardial Infarct Amiodarone Trial; CAMIAT, Canadian Amiodarone Myocardial Infarction Arrhythmia Trial; CHF STAT, Congestive Heart Failure Survival Trial of Antiarrhythmic Therapy; GESICA, Grupo de Estudio de la Sobrevida en la Insuficienca Cardiaca en Argentina.
The properties of amiodarone during long-term administration permit predictable control of recurrent paroxysmal SVTs, slowing of the ventricular response in AF and AFL, and maintaining the stability of sinus rhythm after chemical or electrical conversion.88 Amiodarone is the most potent drug for maintaining the stability of sinus rhythm in patients converted from AF. The drug is currently being compared with placebo and sotalol in a number of major multi-center studies.
Amiodarone is effective in converting AF to sinus rhythm both in valve disease as well as in patients with nonrheumatic AF.88,89 With respect to AF occurring after cardiac surgery, the drug has been studied in three different trials.90–92 Hohnloser and colleagues studied amiodarone in a randomized, double-blind, placebo-controlled study in 77 patients after coronary artery bypass surgery.90 They gave a 300-mg bolus of intravenous amiodarone followed by 1200 mg daily every 24 hours for 2 days and 900 mg daily for the next 2 days. At the end of the study period, the overall incidence of AF in the placebo group (n = 38) was 21%; in the amiodarone group (n = 39) it was 5% (P < .05).
More recently, Guarnieri and his associates have provided further supportive data from a larger double-blind study, in which they randomized 300 patients to placebo (n = 142) and to intravenous amiodarone (n = 158); 2 g of amiodarone were given over 2 days (1 g/day) through a central venous line started shortly after the completion of open heart surgery.91 In the placebo group, 67 (47%) of 142 patients developed AF compared with 56 (35%) of 158 in the amiodarone limb (P < .039). The time to the onset of AF in the amiodarone limb was significantly longer than in the placebo (2.9 vs. 2.2 days; P < .006).
Daoud and colleagues recently reported the first of the two studies on the effects of orally administered amiodarone in preventing postoperative AF in patients undergoing heart surgery.92 Their study was performed in 124 patients randomized to placebo (n = 60) and to amiodarone (n = 64) for a minimum of 7 days preoperatively (600 mg/day), followed by 200 mg/day until discharge from the hospital. Postoperative AF developed in 32 (53%) of the 60 patients in the placebo group and in 16 (25%) of the 64 patients in the group given amiodarone. Patients in the amiodarone group were hospitalized for significantly fewer days compared with those on placebo.
The unique pharmacodynamic profile of amiodarone holds much interest as the prototype of complex compounds that might be necessary to develop for anti-fibrillatory actions in atria and ventricles, provided the drug’s adverse effect profile can be improved.93–95 Clearly, along with sotalol, amiodarone has now emerged as one of the two leading antiarrhythmic drugs currently in use for the control of life-threatening ventricular tachyarrhythmias. However, their therapeutic roles in this setting need to be placed in perspective relative to the expanding indications in the use of implantable cardioverter-defibrillators (ICDs) to prevent arrhythmic deaths in patients with significant structural heart disease.95
Intravenous Amiodarone
Intravenous amiodarone has now emerged as the most powerful antiarrhythmic drug in the treatment of destabilizing VT or VF in hospitalized patients and as an antiarrhythmic agent for the resuscitation of patients developing cardiac arrest out of the hospital.74–77 It has also been shown to convert recent-onset AF to sinus rhythm, although it is not used widely for this indication because of slow onset of action. However, the intravenous drug is used prophylactically for the prevention of AF and AFL in patients who have undergone cardiac surgery.90,91 The major utility of intravenous amiodarone now is in the acute control of destabilizing VT or VF in hospitalized patients and in the resuscitation and continued control of pulseless VT or VF in patients surviving out-of-hospital cardiac arrest.74–77
A number of studies were performed in the early 1990s in destabilizing VT or VF, including those from electrical storm in patients resistant to lidocaine or procainamide. In particular, two amiodarone dose-response studies (125, 500, and 1000 or 2000 mg per 24 hours) with the drug, and one comparative trial with bretylium (2500 mg/24 hours), provided the initial data that intravenous amiodarone may exert a superior effect in controlling VT or VF (including electrical storm) in patients who were refractory to lidocaine or procainamide. In a direct comparison with intravenous bretylium in 305 patients in the randomized double-blind study, amiodarone showed a strong trend for a greater efficacy in reducing VT or VF events, but the difference did not reach statistical significance. However, the data provided the rationale for the exploration of the role of intravenous amiodarone in this setting. Two blinded randomized studies conducted in patients with persistent or recurrent pulseless VT or VF in patients developing out-of-hospital cardiac arrest have now been reported.76,77 In the Amiodarone in Out-of-Hospital Resuscitation of Refractory Sustained Ventricular Tachyarrhythmias (ARREST) trial, 504 patients experiencing out-of-hospital cardiac arrest with recurrent or persistent pulseless VT or VF were enrolled in a prospective randomized double-blind study in which the effects of intravenous amiodarone (300 mg) were compared with those of placebo against the background of an ACLS regimen, which included lidocaine in both limbs of the trial. The data revealed that amiodarone-treated patients were more likely to survive to reach the hospital compared with those in the placebo group (44% vs. 34%, P = .03). The difference was larger among the 221 patients in whom the arrest was witnessed (P = .008), possibly because of the shorter interval from the occurrence of the arrest and the administration of drug therapy.
ARREST did not directly compare the efficacy of amiodarone to that of lidocaine because the latter was an integral part of both treatment limbs. However, superiority of amiodarone demonstrated in ARREST was considered compelling enough to be acknowledged in the “Guidelines 2000 for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care.”2 The evidence (“fair to good evidence”) was considered to be in favor of amiodarone as a class IIB indication; the drug was considered “acceptable, safe and useful,” and the evidence with respect to lidocaine was deemed “indeterminate” because the “research quantity and quality fell short of supporting a final class decision.”
The Azimilide Post-Infarct Survival Evaluation (ALIVE) trial was undertaken by Dorian and associates to compare the effects of lidocaine and amiodarone in shock-resistant patients with VF with out-of-hospital cardiac arrest.77 In ALIVE, 347 patients were enrolled if they were resistant to three shocks, intravenous epinephrine, and further shock or if they had recurrent VF after initial successful defibrillation. The randomization was in a double-blinded manner so that the drugs were given either as intravenous amiodarone plus lidocaine placebo or as lidocaine plus amiodarone placebo. The primary endpoint was the percentage of patients who survived to reach the hospital. On amiodarone, 22.8% of 188 patients reached the hospital compared with 12% of 167 patients on lidocaine. The difference was highly statistically significant (P = .009; odds ratio, 2.17; 95% confidence interval, 1.21 to 3.83). The comparative data from the ALIVE trial are summarized in Figure 80-6.
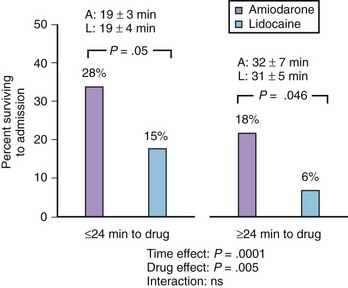
The cumulative clinical investigative data on intravenous lidocaine and intravenous amiodarone in the setting of cardiac resuscitation and in the control of ventricular tachyarrhythmias in the hospital and out of the hospital, when interpreted in light of the outcomes of the ARREST and ALIVE trials, permit a number of conclusions. The most compelling is the issue that intravenous lidocaine is inferior to intravenous amiodarone in increasing the numbers of cardiac arrest survivors reaching hospital admission. Lidocaine does not appear to have a role in the control of life-threatening ventricular tachyarrhythmias, so its continued use in this setting is no longer justifiable.30 Current data support the premise that intravenous amiodarone should now supersede intravenous lidocaine as the first-line antiarrhythmic drug in the resuscitation of patients with shock-resistant VT or VF.30,77
Sotalol as an Antiarrhythmic Drug
The overall action of sotalol is dominated by its dual propensity for competitively blocking β-adrenoceptors without selectivity for β1– or β2-adrenoceptors and by the property for prolonging the myocardial APD. Sotalol is a racemate of D- and L-isomers, both of which have equal class III activity, only the L-isomer having significant β-adrenoceptor-blocking activity.96,97
Electrophysiological Actions
Sotalol is a nonselective β-adrenoceptor-blocking drug with little or no sodium channel–blocking actions without intrinsic sympathomimetic activity. In 1970, Singh and Vaughan Williams found that sotalol markedly prolonged the APD in isolated atrial and ventricular multi-cellular preparations.3 This was classified as a class III antiarrhythmic mechanism. Sotalol slows the sinus node frequency by depressing phase 4 depolarization. Sotalol increases the APD primarily by blocking the time-dependent outward delayed rectifier potassium current (Ik). The effect of increased frequency of stimulation leading to decreases in the APD has been termed reverse-rate dependency.19 The drug prolongs the Q-T and QTc intervals without significant effects on QRS or P-R intervals, with no effect on the His-Purkinje (H-V interval), or ventricular (Q-R interval) conduction velocity.98
Pharmacokinetics of Sotalol and Optimal Dosing
Because the drug is fully bioavailable, not metabolized, and not bound to plasma proteins, fluctuations in serum concentration are small, the plasma half life is long (10 to 15 hours), and plasma levels are linearly related to dose. The usual starting dose is 80 mg bid (rarely 40 mg bid), with stepwise increases (120 mg bid, 160 mg bid, rarely higher) for optimal effect. Because sotalol is largely excreted by the kidneys in an unchanged form, the plasma concentrations of the drug vary linearly with creatinine clearance. Dose adjustment is necessary in proportion to the degree of renal dysfunction present. For a creatinine clearance greater than 60 mL/min, 12-hour dosing intervals are appropriate. The interval may be increased to 12 to 24 hours when the creatinine clearance is 30 to 69 mL/min and 36 to 48 hours for patients with the clearance of 10 to 30 mL/min. However, it may be clinically prudent not to use the drug when creatinine clearance is less than 60 mL/min. Emerging data suggest that a close monitoring of the Q-T interval relative to the dose may be effective in preventing the development of TdP. Numerous pharmacodynamic interactions with numerous cardioactive compounds are likely. This is especially likely in the case of other antiadrenergic drugs (verapamil, diltiazem, and amiodarone) and other QT-prolonging agents (class III agents, including amiodarone, tricyclic anti-depressants, and phenothiazines). Coadministration with erythromycin and other QT-prolonging drugs should also be avoided. Intravenously (not approved by the U.S. Food and Drug Administration), it is given in a dosage of 0.2 to 1.5 mg/kg of dl-sotalol over 2 to 3 minutes under electrocardiographic and hemodynamic control. Although doses of up to 960 mg/day had been used in the past, especially in investigative protocols, it is now clear that higher doses are rarely necessary and may be associated with a greater incidence of TdP, heart failure (NYHA classes III and IV), or severe bradycardia.66,67 Although in rare patients, 80 mg/day may be effective, the range of total daily doses of 240, 360, and 480 mg administered in two equally divided doses allows flexibility of optimal dosing in most patients. In a recent study in AF, 240 mg (as 120 mg bid) was found to be the optimal daily dosage in terms of efficacy and safety.
Control of Cardiac Arrhythmias with Sotalol
The main clinical indications for the use of sotalol in supraventricular and ventricular arrhythmias are shown in Table 80-6. The spectrum of sotalol effects in arrhythmias is wider than that of conventional β-blockers on the one hand and wider than that for the pure class III agents on the other hand.67 Sotalol has a modest suppressant effect on PVCs; it is generally not used for this indication. It exerts a variable effect in suppressing inducible VT or VF, but this approach is no longer used in the control of VT or VF. The two main uses of the drug are in the control of cardiac arrhythmias: (1) as an adjunctive therapy with the ICD for reducing the number of shocks and (2) for the maintenance of sinus rhythm in patients with AF restored to sinus rhythm either chemically or by DC cardioversion. Both indications have been substantiated in controlled clinical trials.99–101
Table 80-6 Major Uses of Sotalol in the Control of Ventricular and Supraventricular Arrhythmias
| INDICATIONS | COMMENTS |
|---|---|
| Cardiac arrest survivors | Superior to class I agents (guided therapy); VT/VF may be comparable with amiodarone (no significant direct comparison but incidence of TdP higher |
| Now being increasingly replaced with ICDs for primary therapy in many subsets of patients | |
| Adjunctive therapy to ICDs | Controlled data still to be obtained but support for VT/VF is strong from large uncontrolled database |
| Conversion of VT to SR by IV regimen | Superior to lidocaine; use not approved in the United States |
| MI survivors | Positive in one study with an 18% reduction in total mortality but not statistically significant |
| Congestive cardiac failure | No controlled studies performed |
| ATRIAL FIBRILLATION | |
| Acute conversion | Variable efficacy but systematic placebo-controlled study or against a comparator agent not available |
| Maintenance of SR | Up to 50% at 12 months after restoration of SR; maintains ventricular rate on relapse to atrial fibrillation |
| Results of placebo-controlled trials imminent | |
| Prevention of postoperative sequelae | Up to 50% reduction when drug given orally before period of cardiac surgery and after cardiac surgery |
The role of sotalol in supraventricular arrhythmias, other than atrial fibrillation, is not discussed in this review.
ICD, Implantable cardioverter-defibrillator; IV, intravenous; MI, myocardial infarction; SR, sinus rhythm; TdP, torsades de pointes; VT/VF, ventricular tachycardia and ventricular fibrillation.
Experience has indicated that concomitant anti-arrhythmic therapy with an ICD is an integral part of therapy of recurrent VT or VF.101 Sotalol may slow the VT rate and aid termination by anti-tachycardia pacing; it may decrease the defibrillation threshold and facilitate termination at lower energy levels and reduce the number of shocks by suppressing episodes of supraventricular (including AF) and ventricular tachyarrhythmias. The overall effects of sotalol (as is the case with any other pharmacologic agent now being introduced) in supraventricular arrhythmias should be viewed in relation to the changes that have occurred in the electrode catheter ablation of paroxysmal supraventricular tachycardias (PSVTs) with or without accessory tracts in the heart and many cases of AFL.98 Because these newer approaches carry an extremely high frequency of success with prospects for cure, prophylactic drug therapy of PSVTs is likely to be used much less often than previously in areas of the world where ablative techniques are readily available.
The predictive accuracy of the acute response for the long-term effects of the drug remains unclear, however. The anti-fibrillatory effects of sotalol as a result of its class III action are likely to make it more effective in maintaining sinus rhythm in patients with AF and AFL after cardioversion.65–67 A well-defined electrophysiological rationale exists for the potential usefulness of sotalol for the acute conversion of AFL and AF to sinus rhythm and for maintaining the stability of sinus rhythm during long-term drug prophylaxis.66,67 Conversion rates have been variable, but a slowing of the ventricular rates has been consistent, as might be expected for a β-blocker. The conversion rate may be up to 40% to 50% of selected patients, which compares favorably with intravenous procainamide and possibly with intravenously administered newer class III agents (ibutilide or dofetilide) or class IC agents (e.g., flecainide). The maintenance of sinus rhythm after conversion is potentially of greater practical importance. Sotalol is as effective as propafenone or quinidine in maintaining sinus rhythm in patients with AF but appears to be less effective than amiodarone in direct comparisons.93,94,102–104
Of particular interest are two placebo-controlled trials that dealt with stability of sinus rhythm under the action of dl-sotalol. Benditt and colleagues conducted a double-blinded placebo-controlled trial in which they compared the efficacy, safety, and dose-response relationship of three doses of d,l-sotalol (80, 120, and 160 mg twice daily) for the maintenance of sinus rhythm in 253 patients converted to sinus rhythm.99 Compared with the placebo (27 days), the time to recurrence was significantly prolonged by two doses of the drug—226 days on 120-mg dose (P = .001) and 175 days on 160-mg dose (P = .012). No deaths or cases of TdP sustained VT or VF occurred. A number of controlled studies have indicated the efficacy of sotalol in preventing the occurrence of AF and AFL developing in patients undergoing cardiac surgery.105,106 Although no cases of TdP occurred, it is known from studies in patients receiving the drug for other arrhythmias that the incidence of TdP varies with dose and can be higher than 5% at the highest doses of the drug sometimes used.107
Bretylium Tosylate
Developed as an antihypertensive agent, bretylium gained prominence for many decades as an intravenous anti-fibrillatory agent for the control of destabilizing VT or VF, especially in the resuscitation of patients experiencing out-of-hospital cardiac arrest. Its introduction into therapeutics was based largely on studies in animal models and comparative studies with lidocaine in the acute control of VT or VF.108,109 No superiority over lidocaine has been demonstrated. Bretylium was recently compared with intravenous amiodarone in a multi-center study of patients with VT or VF who were refractory to or intolerant of lidocaine and procainamide. Bretylium showed equivalent efficacy, measured as time to recurrence and survival but caused more adverse effects (e.g., hypotension) compared with amiodarone.75 Bretylium drug is no longer included in the list of antiarrhythmic drugs in the latest ACLS Guidelines in the control of destabilizing VT or VF developing in or out of the hospital and is no longer available.2 Thus, as an antiarrhythmic drug, along with lidocaine, it is likely to become only of historic interest.30
“Pure” Class III Antiarrhythmic Drugs
Compounds such as dofetilide, ibutilide, and azimilide among others are examples of pure class III agents.71–73 Isolated or pure prolongation of the APD accompanied by a proportional lengthening of myocardial refractoriness is now accepted as anti-fibrillatory therapy.16–18 Under certain circumstances, drugs with such effects may have advantages over complex compounds such as sotalol or amiodarone.
Ibutilide, a Prototype of Intravenous Class III Agents
Ibutilide was the first of the “pure” class III agents approved by the FDA for the acute termination of AFL and AF.110–117 It does this by prolonging the atrial action potential and refractoriness.72,110
Electropharmacology and Pharmacokinetics
The class III action of the drug stems from its property of blocking the rapid component of the delayed rectifier current (IKr); at much lower concentrations the drug also activates a slow inward sodium current, which is not blocked by IKr blockers.72,110 In isolated atrial and ventricular muscle preparations, ibutilide increases the APD and the ERP, effects on both of which are attenuated at increasing rates. In humans, the drug produces concentration-related increases in the Q-T and the QTc intervals.111,112 Ibutilide, like most other class III compounds, is likely to produce anti-fibrillatory as well as profibrillatory actions.72
Ibutilide exerts little or no effect on sinus rates or AV nodal conduction in healthy volunteers and patients with a minimal influence on the P-R or the QRS intervals.114 Also, it does not lower blood pressure or worsen heart failure because it does not exert a negative inotropic effect in the atria or the ventricles.72 It exhibits no significant interaction with the autonomic nervous system. The property that is critical for its therapeutic utility is the acute prolongation of the atrial ERP leading to rapid termination of AF and AFL and the lowering of the energy for atrial defibrillation when these arrhythmias are not amenable solely to electrical conversion.72,112
Ibutilide is only available as an intravenous formulation, as the drug is rapidly metabolized because of a strong first-pass effect in the liver. For this reason, the oral formulation is unlikely to be effective. The elimination half life is variable (2 to 12 hours; mean, 6 hours). The drug is extensively metabolized and excreted largely by the kidney but intravenous dosing does not require dose adjustment relative to changes in renal and hepatic functions. Coadministration of ibutilide with digoxin, calcium channel blockers, or β-blockers exerts effects in the pharmacokinetics, safety, or the efficacy of the drug for the conversion of AF or AFL. Similarly, patient age and gender or the nature of the arrhythmia may have an effect on the drug’s pharmacokinetics.72
Ibutilide in Conversion of Atrial Fibrillation and Atrial Flutter
The effectiveness of ibutilide in converting AF and AFL to sinus rhythm when given intravenously has been established in two pivotal controlled studies.111,112 For example, intravenous ibutilide was studied in 200 patients who were hemodynamically stable, free of heart failure and angina but who had AF and AFL with a duration of between 3 hours and 90 days. They were randomized in equal numbers to placebo and four doses of the drug (0.005, 0.010, 0.015, and 0.025 mg/kg given over 10 minutes). The conversion rate on placebo was 3%. In those for the four ascending doses of ibutilide, the corresponding rates of conversions were 12%, 33%, 45%, and 46%, respectively.110 The overall rate of conversion in the case of AF was 29% and 38% for AFL. The mean time to conversion was 19 minutes, with conversion being accompanied by the prolongation of the Q-T and QTc intervals on the electrocardiogram (ECG); six patients in the study developed polymorphic VT (3.6%).
The second study involved 266 patients who had AF or AFL, with a duration of 3 hours to 45 days.112 They were randomized to be given up to two 10-minute infusions of ibutilide (1 and 0.5 mg, or 1 and 1 mg) or placebo. Most patients had a history of cardiac disease with an enlarged left atrium. The conversion rate was 2% on placebo and 47% on ibutilide (63% in AFL and 31% in AF). The mean time to conversion was 27 minutes, with an incidence of TdP of 8.3% (12.5% in AFL, 6.2% in AF), with1.7% being sustained requiring DC cardioversion. These experiences with ibutilide have been confirmed in subsequent clinical experience.
The major issues regarding the use of pure class III agents in the acute conversion of AF and AFL have been discussed critically by Singh and Roden.116,117 The conversion rates exceed 30% in the case of AF and approximately 50% in the case of AFL of recent onset. The associated rate of TdP developing during the conversion has been as low as 2% to 3% in the case of AF and as high as 8% to 12% in AFL. These data provide the basis for the use of intravenous ibutilide in the acute conversion of AF and AFL.118 To date, this is the only intravenous compound that has been formally approved in recent years by the FDA for the standardized chemical conversion of AF and AFL; meaningful comparisons with other drug regimens, oral or intravenous, have not been established. It is of much interest that the conversion of AF and AFL by intravenous ibutilide in patients treated long term with amiodarone has recently been reported. In 70 such patients, the conversion of AF by ibutilide was 54% in AFL and 39% in AF, with one instance of nonsustained TdP (1.4%) in the entire study. Data suggest that despite the combination of amiodarone and ibutilide producing a further increase in the Q-T interval, amiodarone appeared to decrease the incidence of TdP usually associated with intravenous ibutilide.
Facilitation of Trans-thoracic Cardioversion with Ibutilide Pretreatment
Pure class III agents predictably lower the threshold for defibrillation in atria and ventricles. Recently, Oral and colleagues reported that patients with AF who have not been electrically cardioverted may be successfully cardioverted if they are pretreated with intravenous ibutilide.114 They randomized 100 such patients who had AF for a mean duration of 117 days to undergo trans-thoracic cardioversion, with or without pretreatment with 1 mg of ibutilide. The protocol for conversion was a step-up process involving sequential shocks of 50, 100, 200, 300, and 360 J. If the trans-thoracic shock failed to cardiovert in the absence of ibutilide, the drug was given, and the shock protocol was repeated. Cardioversion to sinus rhythm in the group who had not been given ibutilide was 36 (76%) of 50 patients, but all 50 patients pretreated with ibutilide could be cardioverted (100%; P < .001). It was of interest that all 14 patients who did not cardiovert with the initial direct-current cardioversion could be cardioverted following ibutilide pretreatment. The results reported by Oral and coworkers are striking and need to be confirmed, as the approach will undoubtedly have major implications for clinical practice, especially in conjunction with biphasic cardioversion shocks.114
Adverse Effects
The safety profile of intravenous ibutilide on the basis of experience with the drug in 586 patients involved in phase II and III clinical trials has been reported.120 Noncardiac adverse effects were indistinguishable from those of placebo, as were hypotension, conduction block, or bradycardia. The most significant adverse reaction was the overall incidence of 4.3% in the case of TdP, with 1.7% requiring cardioversion. In almost all patients, the onset of the arrhythmia was within 40 minutes of the commencement of the drug infusion.
Use of Dofetilide in Maintaining the Stability of Sinus Rhythm in Atrial Fibrillation
Dofetilide (Tikosyn) is the prototype of pure or simple class III antiarrhythmic agent. Its oral formulation was approved by the FDA for the maintenance of sinus rhythm in patients with paroxysmal and persistent AF and AFL. Like ibutilide, the drug is effective in terminating recent-onset AF and AFL, although this indication has not yet been approved for routine clinical use.121
Pharmacodynamics and Pharmacokinetics
The drug is a highly selective agent that delays repolarization in the atria, ventricles, and Purkinje fibers by IKr blockade. Such an Ikr block is the sole identifiable action of the drug in cardiac muscle, although it is likely that M-cell APD may be prolonged by the reduction of IKs. The compound exerts no effects on sodium and calcium channels and, as such, has minimal effects on conduction velocity. It has no significant effect on the antagonizing autonomic transmitters.70,122 Thus, it does not alter the P-R interval or the QRS duration of the surface ECG.30 Its sole measurable electrophysiological action is the lengthening of cardiac repolarization as reflected in the Q-T or QTc interval, an effect that correlates directly with the lengthening of the ERP. The drug has no effect on myocardial contractility or on systemic hemodynamics. It does not depress systemic blood pressure. Therefore, the drug can be used in the control of AF in patients with heart failure. The drug is excreted largely via the kidneys, so dose adjustment is necessary in patients with impaired renal function.
The impact of the drug on mortality in the patient with previous MI has been found to be neutral in patients with left ventricular dysfunction with or without cardiac failure.71 However, these studies have shown that the safety of the compound is critically dependent on the use of appropriate doses of the drug relative to renal function, therapy initiation in the hospital, and subsequent monitoring of the patient by following the changes in the Q-T and QTc intervals with adjustment of drug dose.
Effectiveness of Dofetilide in Atrial Fibrillation and Atrial Flutter
Two blinded placebo-controlled pivotal studies have provided the pivotal data on the effectiveness of the compound. The first trial, European and Australian Multicenter Evaluative Research on Atrial Fibrillation of Dofetilide (EMERALD), the effects of three doses of dofetilide (25, 250, and 500 µg bid) were compared with placebo over a 12-month period in 534 patients with AF or AFL durations of between 1 week and 2 years since onset.123 The conversion rate to sinus rhythm at the highest dose was 29% compared with 1% in placebo (P = .001). Those not converting on the drug were restored to sinus rhythm by electrical conversion; in all, 427 patients who did convert by either means were followed up for arrhythmia recurrence. At the highest dose, 66% remained in sinus rhythm compared with 26% on placebo (P = .001) at the end of the first year. The median time to relapse of AF or AFL at the two higher doses of the drug was greater than 365 days compared with 34 days for placebo.
The data from the EMERALD and SAFIRE-D studies indicate, therefore, that dofetilide in a defined group of patients with AF or AFL is a useful anti-fibrillatory agent for restoring and maintaining sinus rhythm.123,124 Supportive data have also been reported from the Danish Trial in Acute Myocardial Infarction on Dofetilide (DIAMOND).71 The major aspect of this study focused on mortality rate in two groups of patients after MI—those with ventricular dysfunction and those with overt congestive heart failure. The study included 1518 patients, who were randomized to dofetilide or placebo. After a median follow-up of 18 months, no impact on mortality was observed. In the patients (n = 506) who had AF or developed AF during the course of the study, dofetilide was effective in converting AF to sinus rhythm (12% vs. 2% on placebo); once the sinus rhythm was restored in these groups, either chemically or electrically, the 1-year maintenance of sinus rhythm was 79% in the group taking dofetilide versus 42% in that on placebo. These data are consistent with the effects of the drug given intravenously to patients with AF or AFL; in 91 patients (75 with AF and 16 with AFL), Falk and colleagues found a conversion rate of 31% in AF patients given 8 µg/kg of dofetilide intravenously in a double-blinded study; the conversion rate was 12.5% on 4 µg/kg, there being no conversions on placebo.121 In AFL, the conversion rate was 54%. However, the major significance of dofetilide in clinical therapeutics is likely to be the maintenance of sinus rhythm in patients with paroxysmal or persistent AF as indicated by the outcomes of placebo-controlled clinical trials discussed above. Indication for its use in the prophylactic therapy of AF is likely to be the major niche for dofetilide in patients with or without heart failure.125
Calcium Channel Blockers, Adenosine, and Digoxin
The electropharmacologic properties and the clinical utility of structurally disparate compounds will be considered together because the bulk of their actions relative to their antiarrhythmic effects involves the inhibition of the AV node to varying extents. Transient complete block of anterograde conduction at the AV node by these agents has been used for the termination of re-entrant supraventricular tachycardias either for therapeutic purposes or for differentiating narrow-QRS tachycardia on the surface electrocardiogram.38 The blocking actions of verapamil, diltiazem, and adenosine induce a prompt and predictable conversion of PSVT in 80% to 100% of cases of the arrhythmia. The conversion rate is achieved by intravenous therapy with 3 to 5 mg (in children) to 10 to 15 mg (in adults) with verapamil, 17 to 25 mg of diltiazem, and 6 to 12 mg of adenosine. Adenosine is now the most frequently used compound in this setting because of near complete efficacy and ultrashort elimination half life accounting for the nature of the drug’s transient adverse effects. However, in some clinical settings, the use of the drug may not be appropriate. Adenosine is preferred in patients with depressed ventricular function and who have recently received β-blockers and in neonates. Alternatively, for termination of PSVT, verapamil may be preferable in patients on drugs known to interfere with the actions of adenosine or its metabolism or in patients with bronchospasm. In patients in whom the diagnosis of PSVT is suspected but not certain, it might be preferable to use verapamil or diltiazem because either drug will not produce sustained hypotension.
Calcium Channel Blockers as Antiarrhythmic Drugs
Verapamil and diltiazem have no significant electrophysiological effects on atrial, ventricular, or His-Purkinje fiber refractoriness or conduction. However, they may shorten the APD in atria and uncommonly induce AF.38 They slow the phase 4 depolarization in the SA and AV nodes, with slowing of conduction mediated by the block of L-type calcium channels. Their major effect is in the AV node, where they reduce conduction and prolong the effective and functional refractory periods in the anterograde as well as retrograde directions. The major depressant effects of verapamil and diltiazem on the AV node are also used in three other specific settings: (1) In the prevention of the recurrences of episodes of PSVT, they can be combined with digoxin or β-blockers, but this use of the drugs is declining with the increasing preference for cure with radiofrequency (RF) ablation of the arrhythmia; (2) they are used for rapid acute control of ventricular response in the case of AFL and fibrillation; and (3) they bring about chronic modulation of the ventricular rate in these arrhythmias when rate control is deemed to be the preferred approach in treatment. In this context, calcium channel blockers can be combined with varying doses of digoxin, β-blockers, or both.126 Limited data are available to support the possible value of verapamil in the acute control of multi-focal atrial tachycardia, but its efficacy in the long-term prophylaxis of the arrhythmia remains uncertain.38 Recent data suggest the interesting possibility that the use of calcium-lowering drugs, such as verapamil given during AF, may reduce the recurrence of AF after electrical cardioversion.127,128
Because the electrophysiological effects of calcium channel blockers are minimal in the ventricular muscle, they are unlikely to be potent antiarrhythmic agents in most types of ventricular tachyarrhythmias.128,129 For the same reasons, these compounds do not appear to induce proarrhythmic reactions and have not been shown to adversely affect mortality, although this may possibly occur by virtue of the negative inotropic actions in patients with advanced levels of heart failure. The role of calcium channel blockers in the treatment of ventricular arrhythmias is limited—as might be expected from the nature of their actions in ventricular muscle. They are poor suppressants of premature ventricular premature beats and nonsustained or sustained VT. It is possible that in some subsets of patients with ischemic heart disease, calcium channel blockers may prevent VT or VF by the anti-ischemic actions, but such a possibility has not been tested in relevant clinical models.
In addition to their use in supraventricular tachyarrhythmias, including for rate control in AFL and AF, at least two relatively uncommon forms of VT respond to calcium channel blockers.126,128,129 Such arrhythmias occur in the context of what appears to be a structurally normal heart. The first is the syndrome of the left ventricular septal VT. Such a VT occurs largely in males, and electrocardiographically, it has a right bundle branch block (RBBB) pattern, with left axis deviation; it can be induced by rapid atrial pacing or by programmed electrical stimulation. It can be terminated by intravenous verapamil but not by adenosine. The arrhythmia can be controlled by oral calcium channel blockers, especially verapamil, but the primary mode of treatment is by RF ablation. The other idiopathic VT that may respond to calcium channel blockers (also to β-blockers) is the right ventricular outflow tract (RVOT) tachycardia. It has the left bundle branch block (LBBB) pattern on the ECG, with a vertical axis, and occurs more frequently in females. The arrhythmia is not readily induced by programmed electrical stimulation but can be induced by exercise or by isoproterenol infusion. The arrhythmia is terminated predictably and promptly to intravenous verapamil or adenosine. Again, the primary mode of therapy is catheter ablation, but it is also controlled by β-blockade or calcium channel blockers and may respond to β-blockers.
Verapamil
The electrophysiological properties of this compound formed the basis for the so-called class IV antiarrhythmic actions.7 When verapamil is administered intravenously (5 to 20 mg over 2 minutes), the peak effects on the AV node occur in 10 to 15 minutes, the effects lasting for 6 hours. After oral administration, the drug acts in hours, with a peak effect occurring at 3 hours, with an elimination half life of 3 to 7 hours, but the effects last much longer as a function of duration of drug administration. For sustained oral therapy for modulating the ventricular response, the usual dose range is 80 to 120 mg three times daily or four times daily. Alternatively, single oral doses of the long-acting preparations (240 to 480 mg/day) may be used. The major cardiovascular adverse effect of the drug relates to excessive depressant action on the AV node. Drug-drug interactions are with digoxin and amiodarone.
Adenosine
The electrophysiological actions of adenosine are mediated via a receptor-effector mechanism that includes the A1 receptor and a guanine nucleotide–binding G protein. The primary direct actions of adenosine are the activation of an outward potassium current (known as IK,ACh or IKado) present in atria and the SA and AV nodes but absent in ventricles. In the AV node, the drug depresses the nodal action potential; and in the sinus node, the sinus rate slows with shifts of the pacemaker and hyperpolarization. As mentioned earlier, the major therapeutic effect of adenosine is the consistent and predictable termination of all forms of PSVT in which the antegrade limb of the arrhythmia is in the AV node. It should be emphasized that adenosine does shorten the APD in atria, making them susceptible to the initiation of AF, which in the case of patients with the WPW syndrome, may be potentially dangerous. Adenosine does not have a major role in the control of arrhythmia generated in ventricles. The role of the drug in the acute termination and the diagnosis of supraventricular tachyarrhythmias is now well established.130,131
Digoxin
Classifying digoxin as an antiarrhythmic agent has always been controversial. The major effects of the cardiac glycoside are mediated by the drug’s central and peripheral actions to increase vagal activity.132 Such an action has two electrophysiological effects: in atria, the atrial refractory period (conducive to the development of AF) is shortened, and on the AV node, the vagal effect leads to delay in conduction and an increase in the ERP, which slows the ventricular response in AF and AFL, a slowing that may lead to conversion of these arrhythmias to sinus rhythm. Such a conversion does not appear to be caused by the direct anti-fibrillatory actions of the drug. This may also be the mechanism of conversion of recent-onset AF and AFL by β-blockers as well by calcium channel blockers. The effects of digoxin on the myocardium stems from the drug’s propensity to inhibit sodium-potassium adenosine triphosphatase (ATPase), with an increase in the intracellular concentration of calcium by the modulation of the calcium channels and the inhibition of sodium-calcium exchange. This may be the basis for the drug’s known, although weak, positive inotropic effects in the ventricular myocardium. The effect of digoxin on the ventricular myocardium is in therapeutic concentrations (0.8 to 2.0 ng/mL). However, at higher doses, it may produce electrocardiographic changes; in toxic doses, it may induce premature atrial and ventricular arrhythmias such as AT with block, premature atrial and ventricular contractions, and bi-directional VT. However, in a large clinical trial that included patients with heart failure, it had a significant adverse effect on total mortality.
Recently Approved Antiarrhythmic Drugs
Dronedarone
In 2009, dronedarone was approved by the FDA for sinus rhythm maintenance in patients with a history of AF or AFL with ejection fractions greater than 35%, on the basis of the results of several randomized clinical trials. This compound is the noniodinated derivative of amiodarone, a compound that was created to reduce the adverse effect profile of amiodarone without the loss of its complex electrophysiological and pharmacologic profile.22,133–134 Besides the deletion of the iodine in the benzene ring, a methane sulfonamide group has been included in the benzofuran ring, and the ethyl groups in the side chain have been replaced by butyls in dronedarone. Such structural changes have led to the shortening of the elimination half life to 20 to 30 hours, no effect on thyroid hormone metabolism, and the propensity to block M2 receptors so that, despite a significant noncompetitive antiadrenergic action, a somewhat lower heart rate-reducing effect compared with amiodarone is achieved.
Dronedarone is a multi-channel blocker that also inhibits atrial-selective currents (IKur and IK,ACh) and possesses antiadrenergic properties (Table 80-7).135–139 In other respects, experimental and preliminary clinical studies have shown that the electropharmacologic effects of dronedarone and amiodarone are similar. For example, in acute electrophysiological studies, in vitro dronedarone shortened the APD in cardiac muscle but, like amiodarone, induced significant prolongation of the APD and the ERP in the atrial as well as ventricular myocardium and in the AV node, with the depression of the phase 4 depolarization in the SA node following 1 week of drug administration. The effects of dronedarone on Purkinje fibers as well as in M cells have been reported to be similar to those in the case of amiodarone, suggesting that the drug is likely to exhibit a negligible incidence of TdP, although the clinical experience with the drug has not been extensive. Thus, dronedarone prolongs atrial and ventricular APD and reduces heart rate, with low risk of ventricular TdP arrhythmias.140 Amiodarone suppresses APD abbreviation during experimental AF, and it has, therefore, been assumed that prevention of ion channel remodeling (suppression of ICa,L reduction) may contribute to amiodarone’s superior efficacy in AF.141 Inhibition of two-pore-domain TASK-1 leak channels, which are preferentially expressed in atria, may also contribute to amiodarone’s anti-AF efficacy.142 It is not known whether suppression of ICa,L remodelling and inhibition of TASK-1 channels contribute to the antiarrhythmic effects of dronedarone.
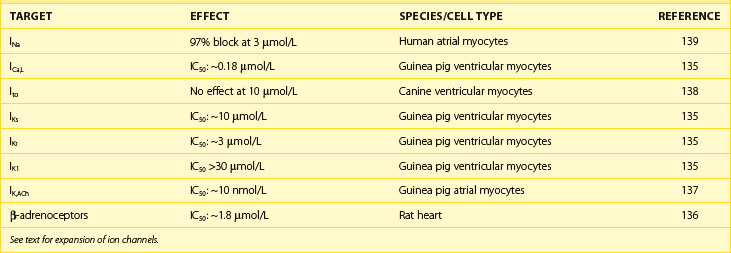
Table 80-8 summarizes the prospective randomized clinical trials with dronedarone.143–148 In the European Trial in Atrial Fibrillation Patients Receiving Dronedarone for the Maintenance of Sinus Rhythm (EURIDIS) and Atrial Fibrillation or Flutter Patients for the Maintenance of Sinus Rhythm (ADONIS) trials, the median times to recurrence of paroxysmal and persistent AF (primary endpoint) were significantly longer, and ventricular rates during AF recurrence were lower for dronedarone compared with placebo.144 Post hoc analyses of both trials demonstrated that dronedarone significantly reduced the combined endpoint of hospitalization or death. The Antiarrhythmic Trial with Dronedarone in Moderate-to-Severe CHF Evaluating Morbidity Decrease (ANDROMEDA) trial investigated patients with systolic left ventricular deterioration but was prematurely discontinued because of increased mortality from heart failure worsening in the dronedarone droup.146 Therefore dronedarone is contraindicated in class III and IV heart-failure.
In the Placebo-Controlled, Double-Blind, Parallel Arm Trial to Assess the Efficacy of Dronedarone 400 mg bid for the Prevention of Cardiovascular Hospitalization or Death from Any Cause in Patients with Atrial Fibrillation/Atrial Flutter (ATHENA) trial, dronedarone significantly reduced the primary study endpoint (all-cause mortality and cardiovascular hospitalization) of AF patients by 24% and the total hospitalization burden by 28%.147 A post hoc analysis on non-prespecified secondary endpoints also showed that dronedarone reduced the risk of stroke (ischemic or hemorrhagic) in patients with AF or AFL adequately treated by standard therapy, including anti-thrombotics in 34%.147 Dronedarone prolonged the median time to first AF or AFL recurrence (dronedarone, 737 days vs. placebo, 498 days) and reduced the likelihood of permanent AF or AFL.149 The overall efficacy for AF suppression was approximately 50%.147 Heart rate during AF or AFL was lower with dronedarone than with placebo.149 Thus, dronedarone exerts both rhythm and rate control effects in patients with AF or AFL and may reduce the risk of hospitalization for and death from cardiovascular diseases. because of these issues the recently published European guidelines for AF management have suggested dronedarone as first-line drug option for patients with nonpersistent AF.150 Although the mechanism of stroke reduction still needs to be determined, it is possible, but not proven, that dronedarone itself may reduce the stroke risk in AF, an effect that has not previously been observed with antiarrhythmic drugs. However, because stroke was not a prespecified endpoint resulting from post hoc subanalyses of ATHENA only, this assumption can be regarded as hypothesis-generating at best. It is possible that prevention of thromboembolism is attributable to reduction in AF burden, reductions in blood pressure, and the potential anti-ischemic properties of dronedarone. Further prospective studies are required to definitively prove the efficacy of dronedarone to reduce stroke incidence in AF patients. Although the mechanism of stroke reduction is unknown, the ATHENA results suggest the possibility that dronedarone itself reduces stroke risk in AF, an effect that has not previously been observed with antiarrhythmic drugs.
However, in the head-to-head Randomized, Double-Blind Trial to Evaluate the Efficacy and Safety of Dronedarone (400 mg bid) Versus Amiodarone (600 mg qd for 28 Days, then 200 mg qd Thereafter) for at least 6 months for the Maintenance of Sinus Rhythm in Patients with AF (DIONYSOS) trial, amiodarone was shown to be superior to dronedarone for the maintenance of sinus rhythm in patients with persistent AF.148 These preliminary results show that AF recurrence during 7-month follow-up after electrical cardioversion was greater with dronedarone (63.5%) than with amiodarone (42.0%), whereas drug discontinuation was more frequent with amiodarone (13.3%) than with dronedarone (10.4%). These data point to a higher tolerability but less efficacy for dronedarone compared with amiodarone. Of note, amiodarone was associated with a trend toward greater all-cause mortality and more adverse effects leading to more frequent drug discontinuation. These results, together with those from ANDROMEDA, underscore the need for head-to-head mortality trials and stroke-prevention studies to more precisely define the therapeutic value of dronedarone in the treatment of AF.
The reason for the inferior efficacy of dronedarone compared with amiodarone is unclear and likely involves multiple factors. Greater cardiac accumulation of amiodarone than of dronedarone and the greater efficacy of N-desethylamiodarone (the major amiodarone metabolite) compared with that of N-debutyldronedarone are potential but unproven candidate mechanisms.151 It is possible that amiodarone interactions with thyroid hormones, dependent on the iodide moieties that are absent in dronedarone, are important for its antiarrhythmic efficacy. Regardless of the reasons, dronedarone has much lower anti-fibrillatory efficacy, and, as can be estimated from an indirect treatment-effect comparison, the fact that switching patients from amiodarone to dronedarone increases the risk of AF recurrence fivefold to eightfold should be kept in mind.
Clinical trials with dronedarone have provided no evidence of thyroid dysfunction or pulmonary toxicity. The most frequently reported adverse events for dronedarone versus placebo in ATHENA were gastrointestinal disorders (26% vs. 22%), skin disorders (10% vs. 8%), and increased blood creatinine (4.7% vs. 1%) because of the inhibition of its renal tubular secretion, which is not considered of clinical relevance. Of note, in EURIDIS, ADONIS and ATHENA, patients with class III heart failure, class IV heart failure, or both were excluded.144,147,149 The increased incidence of hospitalization for worsening congestive heart failure or death in the dronedarone group of the discontinued ANDROMEDA trial raises safety concerns for patients with CHF and moderate to severe left ventricular dysfunction.146 In DIONYSOS, the dronedarone group experienced more diarrhea, vomiting, and nausea but less cardiac adverse effects (bradycardia or Q-T interval prolongation), than amiodarone. Despite its improved safety profile, recent cases of significant liver toxicity related to dronedarone have been reported (www.fda.gov/Drugs/DrugSafety/ucm240011.htm); considering the structural similarities between dronedarone and amiodarone, this is not unexpected and requires routine liver function tests. The overall incidence and significance of dronedarone-related hepatic toxicity are currently unknown, and prospectively collected data in larger patient cohorts are needed to further evaluate the safety profile of dronedarone in different patient subpopulations. A recent study showed adverse outcomes in patient with permanent atrial fibrillation (Permanent Atrial Fibrillation Outcome Study Using Dronedarone on Top of Standard Therapy [PALLAS], personal communication, 2011).
Vernakalant
In 2010, vernakalant was approved by the European Commission for the rapid conversion of recent-onset AF to sinus rhythm. The atrial-selective effects of vernakalant were initially explored in a vagotonic canine model of AF.152 The clinical relevance of its atrial selectivity has been confirmed in human electrophysiology studies demonstrating significantly greater atrial than ventricular ERP prolongation.153 Vernakalant affects a variety of cardiac ion channels (Figure 80-7 and Table 80-9), but its atrial-selective action most likely results from inhibition of ion currents exclusively present in atria (IKur and IK,ACh) and from a selective inhibition of atrial sodium channels.154,155 The sodium channel–blocking potency of vernakalant, particularly during AF, is likely underestimated because of the experimental conditions needed to record sodium currents and because the vernakalant-induced sodium channel block is substantially enhanced at the very rapid rates of the fibrillating atrium (see below).154–155
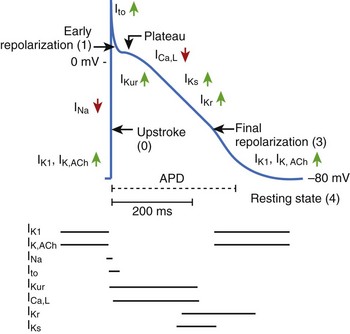
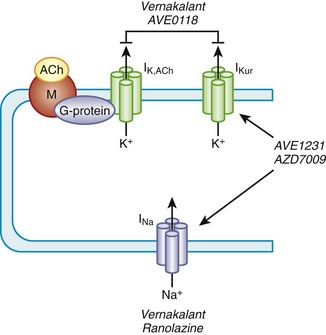
Table 80-9 Efficacy of Dronedarone in Randomized Clinical Trials in Patients with Atrial Fibrillation or Heart Failure
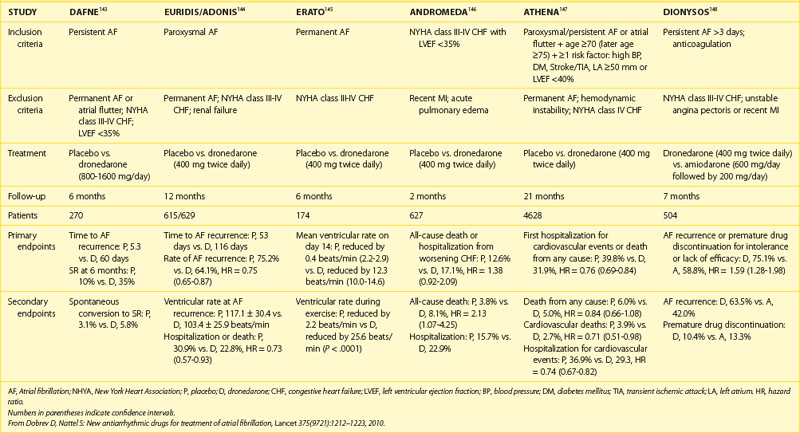
The biophysical properties of atrial sodium channels differ from those of ventricles. For instance, the sodium current has larger amplitude in atria and inactivates at a more negative half-inactivation potential, causing less available current at any given membrane potential.156 In addition, the resting membrane potential is more depolarized in atrial cardiomyocytes than in ventricular cardiomyocytes, leaving a larger fraction of sodium channels in their inactivated state.156 Therefore, application of activated-state sodium channel blockers such as vernakalant suppress sodium channel–dependent parameters (e.g., action potential upstroke velocity, conduction velocity, and diastolic threshold of activation) predominantly in the atria. In addition, the very rapid atrial activation during AF reduces the duration of the diastolic interval in atria, which decreases the dissociation rate of the blocker from the resting sodium channel in atria, whereas the long diastolic interval in ventricles allows enough time for drug dissociation from the channel. As a consequence, the combination of a predominant open-channel block and an inhibitory effect on late sodium current produces rate-dependent depression of sodium channel parameters and atrial excitability.156 Thus, vernakalant’s atrial anti-fibrillatory activity, but the absence of pro-fibrillatory effects at ventricles, is most likely caused by a combination of rapidly unbinding sodium channel blocking action with atrial-selective inhibition of IKur and IK,ACh.154–157
At faster rates, amiodarone and dronedarone, both of which are inactivated-state sodium channel blockers with rapid dissociation kinetics, have a greater potential to depress INa properties in atria than in ventricles.158 Recently, the combination of the two mechanistically different sodium channel blockers—ranolazine, an activated-state sodium channel blocker like vernakalant, and dronedarone, an inactivated-state sodium channel blocker—was highly effective for AF termination and prevention, with the combination being much more effective than expected from the mathematical sum of the two separate efficacy values.159 Although similar beneficial efficacy effects could also result from the combination of vernakalant and dronedarone, this hypothesis requires direct experimental proof.
Vernakalant’s main use is as an intravenous drug for rapid AF termination. Several studies have shown substantial efficacy, approximately 50% (compared with nearly 0% for placebo), with very few adverse effects (Table 80-10).160–163 Efficacy is high for recent-onset AF (<48 hours), whereas longstanding AF is resistant to vernakalant. Vernakalant is useless in patients with AFL, perhaps because of insufficient IKr inhibition. The higher efficacy of class I antiarrhythmic drugs such as vernakalant to cardiovert recent-onset AF versus persistent AF might result from the lower degree of atrial remodeling in recent-onset AF.164 Because of the more complex pathophysiology in longstanding persistent AF with progressive structural changes in both atria (structural remodeling), AF stabilization in such patients depend more on structural, rather than ion channel, remodeling. Complex interaction patterns between cardiomyocytes and fibroblasts may lead to re-entrant circuits that depend to a lesser extent on functional re-entry determinants such as the properties of sodium currents.
Table 80-10 Efficacy of Vernakalant in Randomized Clinical Trials in Patients with Atrial Fibrillation
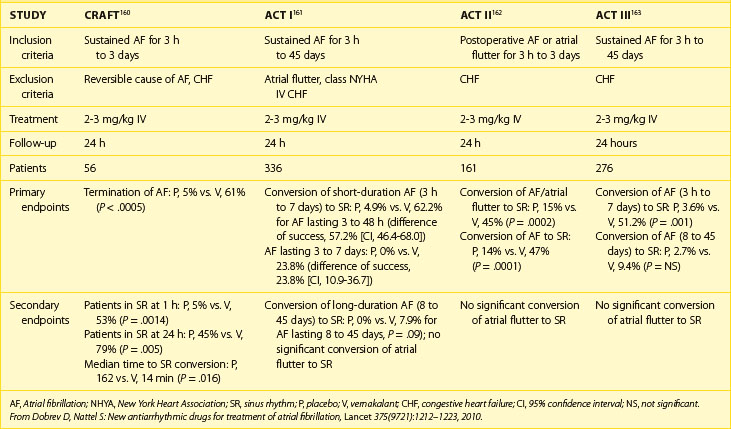
The efficacy of vernakalant for acute AF termination appears comparable with other available compounds, although head-to-head comparative studies have not been performed yet.165 The Phase III Superiority Study of Vernakalant vs. Amiodarone in Subjects with Recent Onset Atrial Fibrillation (AVRO) trial compared intravenous vernakalant versus amiodarone in recent-onset AF, but because of slow (up to 24 hours) onset of action, amiodarone is less suitable for acute AF cardioversion and thus is not the right comparator when assessing the short-term (90 minutes) success rate of pharmacologic cardioversion.166,167 Head-to-head trials with intravenous flecainide are needed to validate the superiority of vernakalant for acute AF cardioversion in different patient populations. In addition, the AF conversion rate with vernakalant is substantially lower in congestive heart failure (26.9%) than in hemodynamically stable patients (54.1%), clearly suggesting its reduced efficacy in patients with structural heart disease.161,162,166 Intravenous ibutilide and dofetilide are approved for acute AF cardioversion, but they have lower efficacy and higher risk for proarrhythmia because of their inhibitory effects on Ikr, which are not shared by vernakalant.154,168
The most common adverse effects of vernakalant are sneezing and dysgeusia related to sodium channel blockade of the central nervous system. Adverse hemodynamic effects such as hypotonia may occur, particularly in the setting of pre-existing ventricular dysfunction, but are rarely observed. Because of its short plasma half life, oral administration of vernakalant as pure salt is clinically not practical, but a slow-release preparation is currently in development.169 The value and safety of the oral agent for sinus rhythm maintenance remain to be established.
Newer Antiarrhythmic Drugs Under Development
Azimilide
The structure of this compound does not include the methane sulfonamide group present in sotalol, dofetilide, or ibutilide. Azimilide, although not a benzofuran, in some respects, resembles amiodarone, which, like azimilide, maintains a class III effect at high stimulation frequencies.170–173 Azimilide is likely to exhibit a lower incidence of TdP compared with the other pure class III agents (d-sotalol, dofetilide, and ibutilide). The terminal half life of azimilide is 4 to 5 days, and it can be administered once daily. It is eliminated by hepatic metabolism.174 It takes approximately 2 weeks for the drug to achieve steady state if no loading regimen is administered. No clinically significant pharmacokinetic interactions have been observed between azimilide and warfarin or digoxin.
The compound prolongs the myocardial APD by predominantly blocking the slow component of the delayed rectifier current (IKs) with presumably a somewhat smaller effect on the rapid component (IKr). Such a property may not be associated with reverse use and rate dependency of action on repolarization. Thus, azimilide is likely to be less “torsadogenic” compared with other specific IKr blockers. The effect of the drug on the M cells in the mid-myocardial region of ventricular tissue is not fully defined, but in other tissues, the drug prolongs the cardiac APD and ERP. Like other pure class III agents, the drug does not slow conduction across the AV node. In isolated human atrial and ventricular myocytes, azimilide produces a concentration-dependent inhibition of both IKs and Ikr. In intact animal models, azimilide has been shown to suppress both atrial and ventricular arrhythmias and has the potential to prevent SCD following coronary artery occlusion.37 Of particular interest, the drug has been found to be unusually and consistently effective in terminating AF and AFL in various canine models.
As indicated previously, in the currently changing therapeutic landscape of arrhythmia control, azimilide may have particular value in the acute conversion of AFL and AF with a potential for maintaining sinus rhythm after pharmacologic or electrical conversion of these arrhythmias.63 The overall efficacy of the drug is likely to rival that of dofetilide (see Table 80-2), and as in the case of other pure class III agents, it is likely to be of value in the reduction of the number of shocks in patients with ICDs for VT or VF. On theoretical grounds, the drug is of much interest as the first example of a class III agent that blocks both Iks and IKr. Thus, its effects in patients with recent MI to improve survival by reducing the risk for sudden arrhythmic deaths, as shown in a controlled study, will be of much interest and significance.
It should be emphasized that as in the case of other pure class III agents, the greatest usefulness of the drug is likely to be in the prophylactic therapy of AF. A number of studies have determined the effect of 35 to 125 mg/day of azimilide on the time to first recurrence of a symptomatic atrial arrhythmia. Analyses of combined data for 100 mg doses and data for the 125 mg dose have shown statistically significant differences from placebo. A dose-dependent response has been observed, with placebo patients having an 83% greater recurrence rate versus patients treated with 125 mg of azimilide. To date, the most effective dose of azimilide in preventing recurrences of AF is 125 mg/day. For example, Pritchett and colleagues found that in symptomatic patients, the mean recurrence time for AF was 17 days for placebo, 22 days for 50 mg dose, 41days for 100 mg dose (all nonsignificant), but 130 days when the dose was 125 mg/day (P = .002).175
Key References
Black SC, Butterfield JL, Lucchesi BR. Protection against programmed electrical stimulation-induced ventricular tachycardia and sudden cardiac death by NE-10064, a class III antiarrhythmic drug. J Cardiovasc Pharmacol. 1993;22:810-818.
Burashnikov A, Belardinelli L, Antzelevitch C. Acute dronedarone is inferior to amiodarone in terminating and preventing atrial fibrillation in canine atria. Heart Rhythm. 2010;7(9):1273-1279.
Burashnikov A, Di Diego JM, Zygmunt AC, et al. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: Differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116(13):1449-1457.
Camm AJ, Capucci A, Hohnloser SH, et al. A randomized active-controlled study comparing the efficacy and safety of vernakalant to amiodarone in recent-onset atrial fibrillation. J Am Coll Cardiol. 2011;57(3):313-321.
Camm AJ, Kirchhof P, Lip GY, et al. Guidelines for the management of atrial fibrillation: The Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Eur Heart J. 2010;31(19):2369-2429.
Chatelain P, Meysmans L, Matteazzi JR, et al. Interaction of the antiaarhythmic agents SR 33589 and amiodarone with the beta-adrenoceptor and adenylate cyclase in rat heart. Br J Pharmacol. 1995;116(3):1949-1956.
Comtois P, Sakabe M, Vigmond EJ, et al. Mechanisms of atrial fibrillation termination by rapidly unbinding Na+ channel blockers: Insights from mathematical models and experimental correlates. Am J Physiol Heart Circ Physiol. 2008;295(4):H1489-H1504.
Corey AE, Al-Khalidi H, Brezovic C, et al. Azimilide pharmacokinetics and pharmacodynamics upon multiple oral dosing. Clin Pharmacol Ther. 1997;61:205-212.
Davy JM, Herold M, Hoglund C, et al. Dronedarone for the control of ventricular rate in permanent atrial fibrillation: The Efficacy and safety of dRonedArone for the cOntrol of ventricular rate during atrial fibrillation (ERATO) study. Am Heart J. 156(3), 2008. 527 e1–e9
Dorian P, Pinter A, Mangat I, et al. The effect of vernakalant (RSD1235), an investigational antiarrhythmic agent, on atrial electrophysiology in humans. J Cardiovasc Pharmacol. 2007;50(1):35-40.
Ehrlich JR, Nattel S. Novel approaches for pharmacological management of atrial fibrillation. Drugs. 2009;69(7):757-774.
Fedida D. Vernakalant (RSD1235): A novel, atrial-selective antifibrillatory agent. Expert Opin Investig Drugs. 2007;16(4):519-532.
Fedida D, Orth PM, Chen JY, et al. The mechanism of atrial antiarrhythmic action of RSD1235. J Cardiovasc Electrophysiol. 2005;16(11):1227-1238.
Fermini B, Jurkiewicz NK, Jow B. Use dependent effect of the class III antiarrhythmic agent NE-10064 (azimilide) on cardiac repolarization block or delayed rectifier potassium and L-type calcium currents. J Cardiovasc Pharmacol. 1995;26:259-267.
Galve E, Rius T, Ballester R, et al. Intravenous amiodarone in treatment of recent-onset atrial fibrillation: Results of a randomized, controlled study. J Am Coll Cardiol. 1996;27(5):1079-1082.
Gautier P, Guillemare E, Marion A, et al. Electrophysiologic characterization of dronedarone in guinea pig ventricular cells. J Cardiovasc Pharmacol. 2003;41(2):191-202.
Gierten J, Ficker E, Bloehs R, et al. The human cardiac K2P3.1 (TASK-1) potassium leak channel is a molecular target for the class III antiarrhythmic drug amiodarone. Naunyn Schmiedebergs Arch Pharmacol. 2010;381(3):261-270.
Guillemare E, Martion A, Nisato D, et al. Acute effects of dronedarone and amiodarone on IK1, I Kr, and IKs in guinea pig ventricular myocytes. Fund Clin Pharmacol. 1999;13:389-395.
Guillemare E, Marion A, Nisato D, Gautier P. Inhibitory effects of dronedarone on muscarinic K+ current in guinea pig atrial cells. J Cardiovasc Pharmacol. 2000;36(6):802-805.
Hohnloser SH, Crijns HJ, van Eickels M, et al. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med. 2009;360(7):668-678.
Kathofer S, Thomas D, Karle CA. The novel antiarrhythmic drug dronedarone: Comparison with amiodarone. Cardiovasc Drug Rev. 2005;23(3):217-230.
Kober L, Torp-Pedersen C, McMurray JJ, et al. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med. 2008;358(25):2678-2687.
Kowey PR, Dorian P, Mitchell LB, et al. Vernakalant hydrochloride for the rapid conversion of atrial fibrillation after cardiac surgery: A randomized, double-blind, placebo-controlled trial. Circ Arrhythm Electrophysiol. 2009;2(6):652-659.
Lalevee N, Nargeot J, Barrere-Lemaire S, et al. Effects of amiodarone and dronedarone on voltage-dependent sodium current in human cardiomyocytes. J Cardiovasc Electrophysiol. 2003;14(8):885-890.
Le Heuzey JY, De Ferrari GM, Radzik D, et al. A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: The DIONYSOS study. J Cardiovasc Electrophysiol. 2010;21(6):597-605.
Naccarelli GV, Wolbrette DL, Samii S, et al. Vernakalant—a promising therapy for conversion of recent-onset atrial fibrillation. Expert Opin Investig Drugs. 2008;17(5):805-810.
Nattel S, De Blasio E, Wang W-Q, Beatch GN. RSD1235: A novel antiarrhythmic agent with a unique electrophysiological profile that terminates AF in dogs. Eur Heart J. 2001;22(Suppl):448.
Page RL, Connolly SJ, Crijns HJ, et al. Rhythm- and Rate-Controlling Effects of Dronedarone in Patients with Atrial Fibrillation (from the ATHENA Trial). Am J Cardiol. 2011;107(7):1019-1022.
Pratt CM, Roy D, Juul-Moller S, et al. Efficacy and tolerance of RSD1235 in the treatment of atrial fibrillation or atrial flutter: Results of a phase III, randomized, placebo-controlled, multicenter trial. Am J Cardiol. 2010;106(9):1277-1283.
Pritchett E, Page P, Connelly S, et al. Azimilide treatment in atrial fibrillation [abstract]. Circulation. 1999;98(Suppl):1633.
Restivo M, Hegazy M, El-Hamamy M. Antiarrhythmic efficacy of azimilide dihydrochloride on functional circus movement atrial flutter in the canine right atrial enlargement model. PACE. 1996;19:664.
Roy D, Pratt CM, Torp-Pedersen C, et al. Vernakalant hydrochloride for rapid conversion of atrial fibrillation: A phase 3, randomized, placebo-controlled trial. Circulation. 2008;117(12):1518-1525.
Roy D, Rowe BH, Stiell IG, et al. A randomized, controlled trial of RSD1235, a novel anti-arrhythmic agent, in the treatment of recent onset atrial fibrillation. J Am Coll Cardiol. 2004;44(12):2355-2361.
Salata JJ, Brooks RR. Pharmacology of azimilide dihydrochloride (NE-10064), a class III antiarrhythmic agent. Cardiovas Drug Rev. 1997;15:137-156.
Schilling R. Cardioversion of atrial fibrillation and the use of antiarrhythmic drugs. Heart. 2010;96:333-338.
Shinagawa K, Shiroshita-Takeshita A, Schram G, Nattel S. Effects of antiarrhythmic drugs on fibrillation in the remodeled atrium: Insights into the mechanism of the superior efficacy of amiodarone. Circulation. 2003;107(10):1440-1446.
Sicouri S, Burashnikov A, Belardinelli L, Antzelevitch C. Synergistic electrophysiologic and antiarrhythmic effects of the combination of ranolazine and chronic amiodarone in canine atria. Circ Arrhythm Electrophysiol. 2010;3(1):88-95.
Singh BN, Connolly SJ, Crijns HJ, et al. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med. 2007;357(10):987-999.
Slavik RS, Tisdale JE, Borzak S. Pharmacologic conversion of atrial fibrillation: A systematic review of available evidence. Prog Cardiovasc Dis. 2001;44(2):121-152.
Sun W, Sarma JSM, Singh BN. Electrophysiologic effects of dronedarone (SR33589), on non-diodinate benzfuran derivative, in the rabbit heart: Comparison with amiodarone. Circulation. 1999;100:2276-2283.
Talajic M, DeRoode MR, Nattel S. Comparative electrophysiologic effects of intravenous amiodarone and desethylamiodarone in dogs: Evidence for clinically relevant activity of the metabolite. Circulation. 1987;75(1):265-271.
Touboul P, Brugada J, Capuccl A, et al. Dronedarone for prevention of atrial fibrillation: A dose-ranging study. Eur Heart J. 2003;24(16):1481-1487.
Varró A, Takács J, Németh M, et al. Electrophysiological effects of dronedarone (SR 33589), a noniodinated amiodarone derivative in the canine heart: comparison with amiodarone. Br J Pharmacol. 2001;133(5):625-634.
1 The Cardiac Arrhythmia Suppression Trial II Investigators. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary Report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:227-233. 406–412
2 Guidelines 2000 for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. An international consensus on science. 6. Advanced cardiovascular life support. 5. Pharmacology 1: Agents for arrhythmias. Circulation. 2000;102(Suppl 1):1-112-1-128.
3 Singh BN, Vaughan Williams EM. A third class of anti-arrhythmic action: Effects on atrial and ventricular intracellular potentials, and other pharmacological actions on cardiac muscle, of MJ 1999 and AH 3474. Br J Pharmacol. 1970;39:675-687.
4 Singh BN, Vaughan Williams. The effect of amiodarone, a new antianginal drug, cardiac muscle. Br J Pharmacol. 1970;39:657-667.
5 Singh BN. Pharmacological actions of certain cardiac drugs and hormones: Focus on antiarrhythmic mechanisms [D. Phil. Thesis, 1971; Hertford College & University of Oxford. UK]. Mt Kisco, NY: Futura; 1991.
6 Vaughan Williams EM. Classification of antiarrhythmic drugs. In: Sandoe E, Flenstedt-Johnson E, Olesen KH, editors. Symposium on cardiac arrhythmias. Sodertalje, Sweden: AB Astra, 1970.
7 Singh BN, Vaughan Williams EM. A fourth class of anti-dysrhythmic action? Effect of verapamil on ouabain toxicity, on atrial and ventricular intracellular potentials, and on other features of cardiac function. Cardiovasc Res. 1972;6:109-119.
8 Singh BN, Hauswirth O. Comparative mechanisms of action of antiarrhythmic drugs. Am Heart J. 1974;87:367-382.
9 Singh BN, Williams EM. Effect of altering potassium concentration on the action of lidocaine and diphenylhydantoin on rabbit atrial and ventricular muscle. Circ Res. 1971;29:286-295.
10 Nattel S, Singh BN. Evolution, mechanisms, and classification of antiarrhythmic drugs: Focus on class III actions. Am J Cardiol. 1999;84:11-19.
11 Hauswirth O, Singh BN. Ionic mechanisms in heart muscle in relationship to the genesis and the pharmacological control of cardiac arrhythmias. Pharmacol Rev. 1979;30:5-63.
12 Hoffman BF, Bigger JTJr. Antiarrhythmic drugs. In DiPalma JR, editor: Drill’s Pharmacology in medicine, ed 4, New York: McGraw-Hill, 1971.
13 Harrison DC. Is there a rational basis for the modified classification of antiarrhythmic drugs? In: Morganroth J, Moore EN, editors. Cardiac arrhythmias: New therapeutic drugs and devices. Boston, MA: Martinus Nijhoff, 1985.
14 Campbell TJ. Kinetics of onset of rate-dependent effects of class I antiarrhythmic drugs are important in determining their effects on refractoriness in guinea-pig ventricle, and provide a theoretical basis for their subclassification. Cardiovasc Res. 1983;17:344-352.
15 The Task Force of the Working Group on Arrhythmias of the European Society of Cardiology. The Sicilian Gambit: A new approach to the classification of antiarrhythmic drugs based on their actions on arrhythmogenic mechanisms. Circulation. 1991;84:1831-1851.
16 Singh BN. The coming of age of class III antiarrhythmic principle: Retrospective and future trends. Am J Cardiol. 1996;78(Suppl 4a):17-27.
17 Lazzara R. From first class to third class: Recent upheaval in antiarrhythmic therapy—lessons from clinical trials. Am J Cardiol. 1996;78:28-33.
18 Singh BN. Expanding indications for the use of class III antiarrhythmic agents in patients at high risk for sudden death. J Cardiovasc Electrophysiol. 1995;6:887-900.
19 Hondeghem LM, Snyders DJ. Class III anti-arrhythmic agents have a lot of potential but a long way to go: Reduced effectiveness and dangers of reverse use dependence. Circulation. 1990;81:686.
20 Camm AJ, Karam R, Pratt CM. The azimilide post-infarct survival evaluation (ALIVE) trial. Am Cardiol. 1998;81:35D-39D.
21 Pritchett E, Page R, Connolly S, Marcello S. Azimilide treatment of atrial fibrillation. Circulation. 1998;98(17):633. 1
22 Sun W, Sarma JSM, Singh BN. Chronic and acute effects of dronedarone in the action potential of rabbit atrial muscle preparations: Comparison with amiodarone. J Cardiovasc Pharmacol. 2002;39:677-684.
23 Wang Z, Pelletier LC, Talajic M, Nattel S. Effects of flecainide and quinidine on human atrial potentials: Role of rate dependence and comparison with guinea pig, rabbit and dog tissues. Circulation. 1990;82:274-283.
24 Teo KK, Yusuf S, Furberg CD. Effects of prophylactic antiarrhythmic drug therapy in acute myocardial infarction: An overview of results from randomized controlled trials. JAMA. 1993;270:1589-1595.
25 Coplen SE, Antman EM, Berlin JA. Efficacy and safety of quinidine therapy for maintenance of sinus rhythm after cardioversion: A meta-analysis of randomized control trials. Circulation. 1990;82:1106-1116.
26 Kirk MM, Gold MR. Disopyramide phosphate. Card Electrophysiol Rev. 2000;4:269-271.
27 MacMahon S, Collins R, Perot R, et al. Effects of prophylactic lidocaine in suspected acute myocardial infarction: An overview of results from randomized, controlled trials. JAMA. 1988;260:1910-1916.
28 Impact Research Group. International mexiletine and placebo antiarrhythmic coronary trial: Report on arrhythmias and other findings. J Am Coll Cardiol. 1984;4:1148-1156.
29 Singh BN. Routine prophylactic lidocaine administration in acute myocardial infarction: An idea whose time is all but gone? Circulation. 1992;86:1033-1035.
30 Ho DS, Zecchin RP, Richards DA, et al. Double-blind trial of lignocaine versus sotalol for acute termination of spontaneous sustained ventricular tachycardia. Lancet. 1994;344:18-23.
31 Ruffy R. Flecainide—2000 update. Card Electrophysiol Rev. 2000;4:277-279.
32 Valderrabano M, Singh BN. Electrophysiologic and antiarrhythmic effects of propafenone: Focus on atrial fibrillation. J Cardiovasc Pharmacol Ther. 1999;4:183-198.
33 Schwartz PJ, La Rovere MT, Vanoli E. Autonomic nervous system and sudden cardiac death: Experimental basis and clinical observations for post-myocardial infarction risk stratification. Circulation. 1992;85(1 Suppl):I77-I87.
34 Meredith IT, Broughton A, Jennings GL, Esler MD. Evidence of a selective increase in cardiac sympathetic activity in patients with sustained ventricular arrhythmias. N Engl J Med. 1991;325:618-624.
35 Reiter MJ, Reiffel JA. Importance of beta- blockade in the therapy of serious ventricular arrhythmias. Am J Cardiol. 1998;82:91-105.
36 Reiter MJ. Antiarrhythmic impact of anti-ischemic, antifailure and other cardiovascular strategies. Card Electrophysiol Rev. 2000;4:194-205.
37 Singh BN. Current antiarrhythmic drugs: An overview of mechanisms of action and potential clinical utility. J Cardiovasc Electrophysiol. 1999;10:283-301.
38 Singh BN, Sarma JSM. Beta-blockers and calcium-channel blockers as antiarrhythmic drugs. In: Zipes DP, Jalife J, editors. Cardiac electrophysiology. From cell to the bedside. Philadelphia: WB Saunders, 2000.
39 Schwartz PJ. The idiopathic long QT interval syndrome: Progress and questions. Am Heart J. 1985;109:399-405.
40 Tan HL, Hou CJ, Lauer MR, Sung RJ. Electrophysiologic mechanisms of the long QT interval syndromes and torsades de pointes. Ann Int Med. 1995;122(9):701-714.
41 Wiesfeld AC, Crijns HJ, Tuininga YS, Lie KI. Beta adrenergic blockade in the treatment of sustained ventricular tachycardia or ventricular fibrillation. Pacing Clin Electrophysiol. 1996;19:1026-1035.
42 Steinbeck G, Andersen D, Bach P, et al. A comparison of electrophysiologically guided antiarrhythmic drug therapy with beta-blocker therapy to patients with symptomatic sustained tachyarrhythmias. N Engl J Med. 1992;327:987-993.
43 Hallstrom AP, Cobb LA, Yu BH, et al. An antiarrhythmic drug experience in 941 patients resuscitated from an initial cardiac arrest between 1970 and 1985. Am J Cardiol. 1991;68:1025-1031.
44 Yusuf S, Wittes J, Friedman L. Overview of results of randomized clinical trials in heart disease. I. Treatments following myocardial infarction. JAMA. 1988;260:2088-2093.
45 Norwegian Multicenter Study Group. Timolol-induced reduction in mortality and reinfarction in patients surviving acute myocardial infarction. N Engl J Med. 1981;304:801-807.
46 Beta-Blocker Heart Attack Trial Research Group. A randomized trial of propranolol in patients with acute myocardial infarction. I. Mortality results. JAMA. 1982;247:1707-1714.
47 Quattlebaum TG, Varghese J, Neill CA, Donahoo JS. Sudden death among postoperative patients with tetralogy of Fallot: A follow-up study of 243 patients for an average of twelve years. Circulation. 1976;54:289.
48 Marin-Garcia J, Moller JH. Sudden death after operative repair of tetralogy of Fallot. Br Heart J. 1977;39:1380.
49 Kendall MJ, Lynch KP, Hjalmarson A, Kjekshus J. Beta-blockers and sudden cardiac death. Ann Int Med. 1995;123:358-367.
50 Hjalmarson A. Effects of beta blockade on sudden cardiac death during acute myocardial infarction and the postinfarction period. Am J Cardiol. 1997;80(9B):35J-39J.
51 Gottlieb SS, McCarter RJ, Vogel RA. Effect of beta-blockade on mortality among high-risk and low-risk patients after myocardial infarction. N Engl J Med. 1998;339:489-495.
52 Julian DG, Camm AJ, Franglin G, et al. For the European Myocardial Infarct Amiodarone Trial Investigators. Randomized trial of effect of amiodarone on mortality in patients with left- ventricular dysfunction after recent myocardial infarction: EMIAT. Lancet. 1997;349:667-673.
53 Cairns JA, Connolly SJ, Roberts RJ, Gent M. Randomized trial of outcome after myocardial infarction in patients with frequent or repetitive ventricular premature depolarizations: CAMIAT, for the Canadian Amiodarone Myocardial Infarction Arrhythmia Trial Investigators. Lancet. 1997;394:675-682.
54 Boutitie F, Boissel JP, Connolly SJ, et al. and the EMIAT and CAMIAT Investigators: Amiodarone interaction with β-blockers: Analysis of the merged EMIAT and CAMIAT databases. Circulation. 1999;99:2268-2275.
55 Teerlink JR, Bassie BM. Beta-adrenergic blocker mortality trials in congestive heart failure. Am J Cardiol. 1999;8:94R-102R.
56 Chadda K, Goldstein S, Byington R, Curb JD. Effect of propranolol after acute myocardial infarction in patients with congestive heart failure. Circulation. 1986;73:503-513.
57 MERIT-HEFT Study Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL randomized intervention trial in congestive heart Failure (MERIT-HF). Lancet. 1999;353:2001-2007.
58 CIBIS II Investigators and Committees. The cardiac insufficiency bisoprolol study CIBIS II. Lancet. 1999;353:9-13.
59 Packer M, Bristow MR, Cohn JN, et al. For the US Carvedilol Heart Failure Study group: The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. N Engl J Med. 1996;334:1349-1360.
60 Packer M, et al. The Carvedilol, Prospective Randomized Cumulative Survival Study Group. N Engl J Med. 2001;344:1651-1658.
61 The Best Steering Committee Design of the Beta Blocker Evaluation Trial (BEST). Am J Cardiol. 1995;75:122-132. 75I
62 Singh BN. The coming of age of the class III antiarrhythmic principle: Retrospective and future trends. Am J Cardiol. 1996;78(Suppl 4A):17-27.
63 Singh BN. Antiarrhythmic drugs: A re-orientation in light of recent developments in the control of disorders of rhythm. Am J Cardiol. 1998;81(6A):3D-13D.
64 Kodama I, Kamiya K, Toyama J. Cellular electropharmacology of amiodarone. Cardiovasc Res. 1997;35:13-29.
65 Singh BN. Expanding indications for the use of class III antiarrhythmic agents in patients at high risk for sudden death. J Cardiovasc Electrophysiol. 1995;6:887-900.
66 Singh BN. Antiarrhythmic action of dl-sotalol in ventricular and supraventricular arrhythmias. J Cardiovasc Pharmacol. 1992;2:590.
67 Singh BN. Sotalol: Current status and expanding indications. J Cardiovasc Pharmacol Ther. 1999;4:59-65.
68 The CASCADE Investigators. The Cascade Study—randomized anti-arrhythmic drug therapy in survivors of cardiac arrest in Seattle. Am J Cardiol. 1993;72:280-287.
69 Mason JW, the ERVEM Investigators. A randomized comparison of electrophysiologic study to electrocardiographic monitoring for prediction of antiarrhythmic drug efficacy in patients with ventricular tachyarrhythmias. N Engl J Med. 1993;329:445-451.
70 Singh BN, Ahmed R. Class III antiarrhythmic drugs. Curr Opin Cardiol. 1994;9:12-22.
71 Torp-Pedersen C, Moller M, Bloch-Thomsen PE, et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction, for the Danish Investigations of Arrhythmia and Mortality on Dofetilide Study group. N Engl J Med. 1999;341:857-865.
72 Murray KT. Ibutilide. Circulation. 1998;97:493-497.
73 Nattel S, Liu L, St George D. Effects of the novel antiarrhythmic agent azimilide on experimental atrial fibrillation and atrial electrophysiologic properties. Cardiovasc Res. 1998;37:627-635.
74 Scheinman MM, Levine JH, Cannom DS, et al. Intravenous Amiodarone Multicenter Investigative Group: Dose ranging study of intravenous amiodarone in patients with life-threatening ventricular tachyarrhythmias. Circulation. 1995;92:326.
75 Kowey PR, Levine JH, Herre JM, et al. Intravenous Amiodarone Multicenter Investigative Group: Randomized, double-blind comparison of intravenous amiodarone and bretylium in the treatment of patients with recurrent hemodynamically destabilizing ventricular tachycardia or fibrillation. Circulation. 1995;92:3255-3263.
76 Kudenchuk PJ, Cobb LA, Copass MK, et al. Amiodarone for resuscitation after out-of-hospital cardiac arrest due to ventricular fibrillation. N Engl J Med. 1999;341:871.
77 Dorian P, Cass D, Schwartz, et al. Amiodarone as compared with lidocaine for shock-resistant ventricular fibrillation. N Engl J Med. 2002;346:884-890.
78 Singh BN. Initial antiarrhythmic drug therapy during resuscitation from sudden cardiac death: A time for a fundamental change in strategy? J Cardiovasc Pharmacol Ther. 2000;5:3-9.
79 Singh BN. What niche will newer class III antiarrhythmic drugs occupy? Curr Cardiol Rep. 2001;3:314-323.
80 Singh BN. Antiarrhythmic actions of amiodarone: A profile of a paradoxical agent. Am J Cardiol. 1996;78:41-53.
81 Papp JG, Nemeth M, Krassoi I, et al. Differential electro- physiologic effects of chronically administered amiodarone on canine Purkinje fibers versus ventricular muscle. J Pharmacol Exp Ther. 1996;1:187-196.
82 Sicouri S, Moro S, Litovsky S, et al. Chronic amiodarone reduces transmural dispersion of repolarization in the canine heart. J Cardiovasc Electrophysiol. 1997;8:1269-1279.
83 Cui G, Sen L, Sager PT, et al. Effects of amiodarone, sematilide and sotalol on QT dispersion. Am J Cardiol. 1995;75:465-469.
84 Hohnloser SH, Singh BN. Proarrhythmia with class III antiarrhythmic drugs: Definition, electrophysiologic mechanisms, incidence, predisposing factors, and clinical implications. J Cardiovasc Electrophysiol. 1995;6:920-936.
85 Sager PT, Uppal P, Follmer CT, et al. The frequency-dependent electrophysiologic effects of amiodarone in humans. Circulation. 1993;88:1063-1068.
86 Connolly SJ. Evidence based-analysis of amiodarone efficacy and safety. Circulation. 1999;100:2025-2034.
87 Nasir N, Swarna US, Boahene KA, et al. Therapy of sustained ventricular arrhythmias with amiodarone: Prediction of efficacy with serial electrophysiologic studies. J Cardiovasc Pharmacol Ther. 1996;1:123-133.
88 Skoulargis J, Rothlisberger C, Skudicky D, et al. Effectiveness of amiodarone and electrical cardioversion for chronic rheumatic atrial fibrillation after mitral valve surgery. Am J Cardiol. 1993;72:423-427.
89 Chun SH, Sager PT, Stevenson WG, et al. Long-term efficacy of amiodarone for the maintenance of sinus rhythm in patients with refractory atrial fibrillation or flutter. Am J Cardiol. 1995;76:47-50.
90 Hohnloser SH, Meinertz T, Dummbacher T, et al. Electro cardiographic and antiarrhythmic effects of intravenous amiodarone: Results of prospective, placebo-controlled study. Am Heart J. 1991;121:89-90.
91 Guarnieri T. Intravenous amiodarone reduces CABG hospitalization: The ARCH Trial, Presentation at the NASPE Annual Scientific Meeting, San Diego, May 1998.
92 Daoud EG, Strickenberger AS, Man C, et al. Preoperative amiodarone as prophylaxis against atrial fibrillation after heart surgery. N Engl J Med. 1997;337:1785-1791.
93 Roy D, Talajic M, Dorian P, et al. Amiodarone to prevent recurrence of atrial fibrillation, for the Canadian Trial of Atrial Fibrillation Investigators. N Engl J Med. 2000;342:913-918.
94 Kochiadakis GE, Igoumenidis NE, Marketou ME, et al. Low-dose amiodarone versus sotalol for suppression of recurrent symptomatic atrial fibrillation. Am J Cardiol. 1998;81:995-1005.
95 Singh BN. A Symposium: Approaches to controlling cardiac arrhythmias: Focus on amiodarone—the last 15 years. Am J Cardiol. 1999;84(Supp 9A):1R-174R.
96 Kato R, Yabek L, Ikeda N, et al. Electrophysiologic effects of dextro- and levo-isomers of sotalol in isolated cardiac muscle and their in vivo pharmacokinetics. J Am Coll Cardiol. 1986;7:116-126.
97 Antonaccio MJ, Gomoll AW. Pharmacology, pharmacodynamics and pharmacokinetics of sotalol. Am J Cardiol. 1990;65:12A-20A.
98 Nademanee K, Feld G, Hendrickson JA, et al. Electrophysiologic and antiarrhythmic effects of sotalol in patients with life- threatening ventricular tachyarrhythmias. Circulation. 1985;72:555-564.
99 Benditt DG, Williams JH, Jin J, et al. Maintenance of sinus rhythm with oral dl-sotalol therapy in patients with symptomatic atrial fibrillation and flutter: A dose-response study, for the dl-Sotalol Atrial Fibrillation/Flutter Study Group. Am J Cardiol. 1999;84:270-277.
100 Pacifico A, Hohnloser S, Williams JH, et al, for the dl-sotalol implantable cardioverter-defibrillator study group. N Engl J Med. 1999;340:1855-1862.
101 Movsowitz C, Marchlinski FE. Interactions between implantable cardioverter-defibrillators and class III antiarrhythmic drugs. Am J Cardiol. 1998;82:411.
102 Singh BN, Deedwania P, Nademanee K, et al. Sotalol: A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use. Drugs. 1987;34:311-330.
103 Reimold SC, Cantillon CO, Priedman PL, et al. Propafenone versus sotalol for suppression of recurrent symptomatic atrial fibrillation. Am J Cardiol. 1993;71:558.
104 Juul-Moller S, Edvardsson N, Ahlberg NR. Sotalol versus quinidine for the maintenance of sinus rhythm after direct current conversion of atrial fibrillation. Circulation. 1990;82:1932.
105 Suttorp MJ, Kignma JH, Peels HOJ. Effectiveness of sotalol in preventing supraventricular tachyarrhythmias shortly after coronary artery bypass grafting. Am J Cardiol. 1991;68:1163.
106 Singh BN, Lopez B, Sarma JSM. Significance and prevention of atrial fibrillation occurring after surgery: A time for fundamental change in strategy? J Cardiovasc Pharmacol Therapeut. 1998;3:259.
107 Kehoe R, Zheutlin T, Dunnington C, et al. Safety and efficacy of sotalol in patients with drug refractory sustained ventricular tachyarrhythmias. Am J Cardiol. 1990;65:58A.
108 Haynes RE, Chinn RL, Copass MK, et al. Comparison of bretylium tosylate and lidocaine in management of out-of-hospital ventricular fibrillation: A randomized clinical trial. Am J Cardiol. 1981;48:353.
109 Olson DW, Thompson BM, Darin JC, et al. A randomized comparison study of bretylium tosylate and lidocaine in resuscitation of patients from out-of-hospital ventricular fibrillation in a paramedic system. Ann Emerg Med. 1984;13:807-811.
110 Lee KS, Gibson JK. Unique ionic mechanism of action of ibutilide on freshly isolated heart cells. Circulation. 1995;92:2755-2757.
111 Ellenbogen KA, Sambler BS, Wood MA, et al. Efficacy of intravenous ibutilide for rapid termination of atrial fibrillation and flutter: A dose-response study. J Am Coll Cardiol. 1996;28:130-136.
112 Stambler BS, Wood MA, Ellenbogen KA, et al. Efficacy of safety of repeated intravenous doses of ibutilide for rapid conversion of atrial flutter or fibrillation. Circulation. 1996;94:1613-1621.
113 Ellenbogen KA, Clemo HF, Stambler BS, et al. Efficacy of ibutilide termination of atrial fibrillation and flutter. Am J Cardiol. 1996;78:42-45.
114 Oral H, Souza HJ, Michaud GF, et al. Facilitating transthoracic cardioversion of atrial fibrillation with ibutilide treatment. N Engl J Med. 1999;340:1849-1854.
115 Nacarelli GV, Lee KS, Gibson JK, et al. Electrophysiology and pharmacology of ibutilide. Am J Cardiol. 1996;78:12-16.
116 Singh BN. Acute conversion of atrial flutter and fibrillation: Direct current cardioversion versus intravenously administered pure class III agents. J Amer Coll Cardiol. 1997;29:391-393.
117 Roden DM. Ibutilide and the treatment of atrial arrhythmias. A new drug—almost unheralded—is now available to US physicians. Circulation. 1996;94:1499-1502.
118 Stambler BS. Update on intravenous ibutilide. Cardiac Electrophysiol Rev. 2000;4:243-247.
119 Reference deleted in proofs.
120 Kowey PR, Vanderlugt JT, Luderer JR. Safety and risk/benefit analysis of ibutilide for acute conversion of atrial flutter and fibrillation. Am J Cardiol. 1996;78:46-52.
121 Falk RH, Pollak A, Singh SN, Friedrich T. Intravenous dofetilide, a class III antiarrhythmic agent, for the termination of sustained atrial fibrillation or flutter. J Am Coll Cardiol. 1997;29:385-390.
122 Carmeliet E. Voltage- and time-dependent block of the delayed rectifier K+ current in cardiac myocytes by dofetilide. J Pharmacol Ther. 1992;262:809-815.
123 Data on file in Pfizer database, New York, New York, 2001.
124 Singh SN, Zoble RG, Yellen L, et al. Efficacy and safety of oral dofetilide in converting to and maintaining sinus rhythm in patients with chronic atrial fibrillation or flutter. The Symptomatic Atrial Fibrillation Investigative Research on Dofetilide (SAFIRE-D) study, for the Dofetilide Atrial Fibrillation Investigators. Circulation. 2000;102:2383-2390.
125 Doshi S, Singh BN. Pure class III antiarrhythmic drugs: Focus on dofetilide. J Cardiovasc Pharmacol Ther. 2000;5:237-247.
126 Farshi R, Kistner D, Sarma JS, et al. Ventricular rate control in chronic atrial fibrillation during daily and programmed exercise: A crossover open-label study of five drug regimens. J Am Coll Cardiol. 1999;33:304-310.
127 Tieleman RG, Van Gelder IC, Crijn HJ, et al. Early recurrences of atrial fibrillation after electrical conversion: A result of fibrillation-induced electrical remodeling of the atria? J Am Coll Cardiol. 1998;31:167-173.
128 Wyse DG. Calcium channel blockers. Cardiac Electrophysiol Rev. 2000;4:308-311.
129 Lee SH, Chen SA, Tai CT, et al. Electropharmacologic characteristics and radiofreqency catheter ablation of sustained ventricular tachycardia in patients without structural heart disease. Cardiology. 1996;87:33-41.
130 Wilbur SL, Marchlinski FE. Adenosine as an antiarrhythmic agent. Am J Cardiol. 1997;12A:30-37.
131 Conti JB, Belardinelli LC, Curtis AB. Usefulness of adenosine in the diagnosis of tachyarrhythmias. Am J Cardiol. 1995;75:952-955.
132 Smith TW. Digitalis: Mechanisms of action and clinical use. N Engl J Med. 1988;318:358-365.
133 Sun W, Sarma JSM, Singh BN. Electrophysiologic effects of dronedarone (SR33589), on non-diodinate benzfuran derivative, in the rabbit heart: Comparison with amiodarone. Circulation. 1999;100:2276-2283.
134 Guillemare E, Martion A, Nisato D, et al. Acute effects of dronedarone and amiodarone on IK1, I Kr, and IKs in guinea pig ventricular myocytes. Fund Clin Pharmacol. 1999;13:389-395.
135 Gautier P, Guillemare E, Marion A, et al. Electrophysiologic characterization of dronedarone in guinea pig ventricular cells. J Cardiovasc Pharmacol. 2003;41(2):191-202.
136 Chatelain P, Meysmans L, Matteazzi JR, et al. Interaction of the antiaarhythmic agents SR 33589 and amiodarone with the beta-adrenoceptor and adenylate cyclase in rat heart. Br J Pharmacol. 1995;116(3):1949-1956.
137 Guillemare E, Marion A, Nisato D, Gautier P. Inhibitory effects of dronedarone on muscarinic K+ current in guinea pig atrial cells. J Cardiovasc Pharmacol. 2000;36(6):802-805.
138 Varró A, Takács J, Németh M, et al. Electrophysiological effects of dronedarone (SR 33589), a noniodinated amiodarone derivative in the canine heart: Comparison with amiodarone. Br J Pharmacol. 2001;133(5):625-634.
139 Lalevee N, Nargeot J, Barrere-Lemaire S, et al. Effects of amiodarone and dronedarone on voltage-dependent sodium current in human cardiomyocytes. J Cardiovasc Electrophysiol. 2003;14(8):885-890.
140 Kathofer S, Thomas D, Karle CA. The novel antiarrhythmic drug dronedarone: Comparison with amiodarone. Cardiovasc Drug Rev. 2005;23(3):217-230.
141 Shinagawa K, Shiroshita-Takeshita A, Schram G, Nattel S. Effects of antiarrhythmic drugs on fibrillation in the remodeled atrium: Insights into the mechanism of the superior efficacy of amiodarone. Circulation. 2003;107(10):1440-1446.
142 Gierten J, Ficker E, Bloehs R, et al. The human cardiac K2P3.1 (TASK-1) potassium leak channel is a molecular target for the class III antiarrhythmic drug amiodarone. Naunyn Schmiedebergs Arch Pharmacol. 2010;381(3):261-270.
143 Touboul P, Brugada J, Capucci A, et al. Dronedarone for prevention of atrial fibrillation: A dose-ranging study. Eur Heart J. 2003;24(16):1481-1487.
144 Singh BN, Connolly SJ, Crijns HJ, et al. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med. 2007;357(10):987-999.
145 Davy JM, Herold M, Hoglund C, et al. Dronedarone for the control of ventricular rate in permanent atrial fibrillation: The Efficacy and safety of dRonedArone for the cOntrol of ventricular rate during atrial fibrillation (ERATO) study. Am Heart J. 156(3), 2008. 527 e1–e9
146 Kober L, Torp-Pedersen C, McMurray JJ, et al. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med. 2008;358(25):2678-2687.
147 Hohnloser SH, Crijns HJ, van Eickels M, et al. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med. 2009;360(7):668-678.
148 Le Heuzey JY, De Ferrari GM, Radzik D, et al. A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: The DIONYSOS study. J Cardiovasc Electrophysiol. 2010;21(6):597-605.
149 Page RL, Connolly SJ, Crijns HJ, et al. Rhythm- and rate-controlling effects of dronedarone in patients with atrial fibrillation (from the ATHENA Trial. Am J Cardiol. 2011;107(7):1019-1022.
150 Camm AJ, Kirchhof P, Lip GY, et al. Guidelines for the management of atrial fibrillation: The Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Eur Heart J. 2010;31(19):2369-2429.
151 Talajic M, DeRoode MR, Nattel S. Comparative electrophysiologic effects of intravenous amiodarone and desethylamiodarone in dogs: Evidence for clinically relevant activity of the metabolite. Circulation. 1987;75(1):265-271.
152 Nattel S, De Blasio E, Wang W-Q, Beatch GN. RSD1235: A novel antiarrhythmic agent with a unique electrophysiological profile that terminates AF in dogs. Eur Heart J. 2001;22(supplement):448.
153 Dorian P, Pinter A, Mangat I, et al. The effect of vernakalant (RSD1235), an investigational antiarrhythmic agent, on atrial electrophysiology in humans. J Cardiovasc Pharmacol. 2007;50(1):35-40.
154 Fedida D, Orth PM, Chen JY, et al. The mechanism of atrial antiarrhythmic action of RSD1235. J Cardiovasc Electrophysiol. 2005;16(11):1227-1238.
155 Fedida D. Vernakalant (RSD1235): A novel, atrial-selective antifibrillatory agent. Expert Opin Investig Drugs. 2007;16(4):519-532.
156 Burashnikov A, Di Diego JM, Zygmunt AC, et al. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: Differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116(13):1449-1457.
157 Comtois P, Sakabe M, Vigmond EJ, et al. Mechanisms of atrial fibrillation termination by rapidly unbinding Na+ channel blockers: Insights from mathematical models and experimental correlates. Am J Physiol Heart Circ Physiol. 2008;295(4):H1489-H1504.
158 Burashnikov A, Belardinelli L, Antzelevitch C. Acute dronedarone is inferior to amiodarone in terminating and preventing atrial fibrillation in canine atria. Heart Rhythm. 2010;7(9):1273-1279.
159 Sicouri S, Burashnikov A, Belardinelli L, Antzelevitch C. Synergistic electrophysiologic and antiarrhythmic effects of the combination of ranolazine and chronic amiodarone in canine atria. Circ Arrhythm Electrophysiol. 2010;3(1):88-95.
160 Roy D, Rowe BH, Stiell IG, et al. A randomized, controlled trial of RSD1235, a novel anti-arrhythmic agent, in the treatment of recent onset atrial fibrillation. J Am Coll Cardiol. 2004;44(12):2355-2361.
161 Roy D, Pratt CM, Torp-Pedersen C, et al. Vernakalant hydrochloride for rapid conversion of atrial fibrillation: A phase 3, randomized, placebo-controlled trial. Circulation. 2008;117(12):1518-1525.
162 Kowey PR, Dorian P, Mitchell LB, et al. Vernakalant hydrochloride for the rapid conversion of atrial fibrillation after cardiac surgery: A randomized, double-blind, placebo-controlled trial. Circ Arrhythm Electrophysiol. 2009;2(6):652-659.
163 Pratt CM, Roy D, Juul-Moller S, et al. Efficacy and tolerance of RSD1235 in the treatment of atrial fibrillation or atrial flutter: results of a phase iii, randomized, placebo-controlled, multicenter trial. Am J Cardiol. 2010;106(9):1277-1283.
164 Ehrlich JR, Nattel S. Novel approaches for pharmacological management of atrial fibrillation. Drugs. 2009;69(7):757-774.
165 Slavik RS, Tisdale JE, Borzak S. Pharmacologic conversion of atrial fibrillation: A systematic review of available evidence. Prog Cardiovasc Dis. 2001;44(2):121-152.
166 Camm AJ, Capucci A, Hohnloser SH, et al. A randomized active-controlled study comparing the efficacy and safety of vernakalant to amiodarone in recent-onset atrial fibrillation. J Am Coll Cardiol. 2011;57(3):313-321.
167 Galve E, Rius T, Ballester R, et al. Intravenous amiodarone in treatment of recent-onset atrial fibrillation: Results of a randomized, controlled study. J Am Coll Cardiol. 1996;27(5):1079-1082.
168 Schilling R. Cardioversion of atrial fibrillation and the use of antiarrhythmic drugs. Heart. 2010;96:333-338.
169 Naccarelli GV, Wolbrette DL, Samii S, et al. Vernakalant—a promising therapy for conversion of recent-onset atrial fibrillation. Expert Opin Investig Drugs. 2008;17(5):805-810.
170 Fermini B, Jurkiewicz NK, Jow B. Use dependent effect of the class III antiarrhythmic agent NE-10064 (azimilide) on cardiac repolarization block or delayed rectifier potassium and L-type calcium currents. J Cardiovasc Pharmacol. 1995;26:259-267.
171 Salata JJ, Brooks RR. Pharmacology of azimilide dihydrochloride (NE-10064), a class III antiarrhythmic agent. Cardiovas Drug Rev. 1997;15:137-156.
172 Black SC, Butterfield JL, Lucchesi BR. Protection against programmed electrical stimulation-induced ventricular tachycardia and sudden cardiac death by NE-10064, a class III antiarrhythmic drug. J Cardiovasc Pharmacol. 1993;22:810-818.
173 Restivo M, Hegazy M, El-Hamamy M. Antiarrhythmic efficacy of azimilide dihydrochloride on functional circus movement atrial flutter in the canine right atrial enlargement model. PACE. 1996;19:664.
174 Corey AE, Al-Khalidi H, Brezovic C, et al. Azimilide pharmacokinetics and pharmacodynamics upon multiple oral dosing. Clin Pharmacol Ther. 1997;61:205-212.
175 Pritchett E, Page P, Connelly S, et al. Azimilide treatment in atrial fibrillation [abstract]. Circulation. 1999;98(Suppl):1633.