Chapter 55 Anoxic-Ischemic Encephalopathy
When the heart stops and cerebral blood flow is interrupted, patients lose consciousness and may remain comatose after resumption of circulation. Such a global injury to the brain is understandably profound, and more than 70% of patients die or remain comatose 24 hours after cardiopulmonary resuscitation (CPR) (Rogove et al., 1995; Zandbergen et al., 2006). Anoxic-ischemic injury—albeit less well defined and less clearly understood—may also occur in patients with respiratory arrest or severe hypoxemia (e.g., asphyxia) and in shock. Success of intervention in these conditions may be predicated on early correction of hypoxemia and hypovolemia. The time interval until correction may be less important than the initial severity of the abnormality.
Approximately 100,000 patients a year in the United States are admitted to intensive care units with anoxic-ischemic brain injury after CPR (Peberdy et al., 2003). Although the pathophysiology of brain injury caused by cardiac arrest is well understood, less is known about neuroprotection. There is, however, some hope that induced moderate hypothermia could not only affect survival but also improve neurological outcome if patients are immediately subjected to cooling (Broccard, 2006). This chapter critically evaluates the current knowledge of anoxic-ischemic brain injury after cardiac and respiratory arrest. Studies have reported tools for predicting outcomes, and guidelines for prediction of poor outcome have been developed by the American Academy of Neurology (Wijdicks et al., 2006). The accuracy of these predictors after the use of therapeutic hypothermia is a subject of ongoing investigations. Delayed postanoxic deterioration caused by carbon monoxide intoxication is discussed in Chapter 58.
Pathophysiological Concepts
One of the more vital questions for scientists and clinicians is whether there is a specific period during which these patients can be effectively treated. Is the damage to the brain permanent and present at ictus, or are there processes at work that could potentially be influenced and modulated? Several clinical facts are important. First, with cardiac arrest, whether due to asystole or ventricular fibrillation, there is no measurable flow to the brain. Moreover, even with standard CPR techniques, only one-third of the pre-arrest cerebral blood flow can be attained (Maramattom et al., 2005). In addition, the shockable rhythms (ventricular tachycardia and ventricular fibrillation) have a better outcome than “nonshockable” rhythms such as asystole and bradyarrhythmias, reflected by restoration of adequate cerebral blood flow when ejection fraction of the ventricle improves (Callans, 2004). Secondly, there might be a critical time period after which CPR may fail to restore neuronal function. This time interval is poorly defined, but we know that the neuronal oxygen stores are depleted within 20 seconds of cardiac arrest, and cerebral necrosis occurs as a result of ischemia. There is some uncertainty about whether hypoxemia alone could produce necrosis and, although it can cause damage (preferentially in the striatum), necrosis is rarely seen even in patients with arterial Po2 values less than 20 mm Hg.
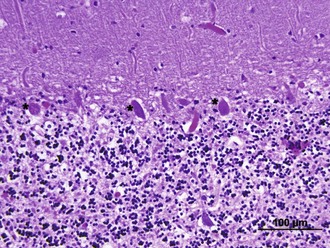
After the brain has been injured by anoxic-ischemic insult, several biochemical mechanisms may become operative (Fig. 55.1). Selective neuronal vulnerability to this type of injury involves areas in the hippocampus, the CA-1 sector, the thalami, the neocortex, and the Purkinje cells (Fig. 55.2). Necrosis of the cortex involves layers three, four, and five and is pathologically known as laminar necrosis. The vulnerability of these areas may be explained by the presence of receptors for excitatory neurotransmitters or the high metabolic demands of these neurons. An important question is whether necrosis or apoptosis occurs. The cell death cascade that involves several modulatory and degradation signals has been documented in global cerebral ischemia, but whether manipulation of these processes is effective remains unclear (Ogawa, 2007). A caspase inhibitor did not affect neurological outcome after 6 minutes of cardiopulmonary arrest in rats (Teschendorf et al., 2001).
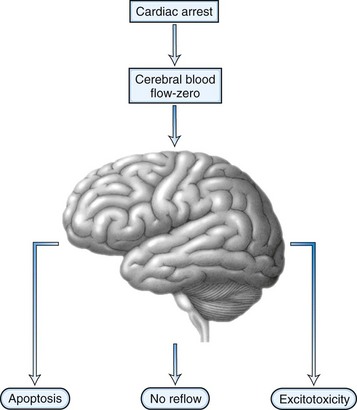
Another mechanism of damage is excitatory brain injury. Glutamate efflux due to ischemic injury increases intracellular calcium concentration, which results in neuronal injury. The excess release of calcium leads to other processes that include activation of catabolic enzymes and endonucleases. Glutamate excitotoxicity has remained the major hypothesis to explain the neuronal injury and was made more probable after the documentation of neuroprotection with N-methyl-d-aspartate (NMDA) or α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonists. Research interest has pointed toward a phenomenon called no reflow. This concept is based on the premise that after resumption of circulation, there are major microcirculatory reperfusion deficits. Coagulation may occur within these reperfusion zones, with intravascular fibrin formation and microthrombosis; this concept is currently an incentive for a reperfusion trial using recombinant tissue-type plasminogen activator (tPA). Also, the use of hypertonic hyperoncotic solutions improved these perfusion deficits. Despite our understanding of the pathophysiology of anoxic-ischemic injury based on careful animal experiments, the clinical reality is discouraging. Clinical trials using barbiturates or calcium channel antagonists have been unsuccessful (Maramattom and Wijdicks, 2005). Induced hypothermia, which inhibits apoptosis and reduces free radical formation and excitatory neurotransmitters, might be the only current beneficial intervention (Bernard et al., 2002; Group THACAS, 2002). Patients who are comatose after CPR unfortunately often have a devastating outcome. Improvement of outcome might come from very early intervention and administration of neuroprotective agents at the onset of resuscitation, rather than when a patient enters the hospital.
Clinical Examination
Early awakening after CPR, clinical signs that indicate localization of the pain stimuli, or following simple commands are indicators of a good outcome. However, the current literature provides no criteria on which a good outcome can be based. Most studies have specifically concentrated on the examination of the patient, assuming a poor outcome. An estimated 25% of patients remain comatose in the first 24 hours. The mortality rate in patients who have been resuscitated following cardiopulmonary arrest approaches 80% to 90% when they have not awakened within the first 24 hours (Zandbergen et al., 2006).
Clinical neurological examination follows a standard procedure, with examination of the brainstem reflexes, motor response to pain, specific attention to myoclonus, and spontaneous or elicited eye movement abnormalities. Because the brainstem is far more resilient to anoxic-ischemic injury than the cortex, brainstem reflexes, including the pupillary reflex to light, are often normal. Absent pupil responses can be caused by a high dose of intravenous atropine used during resuscitation, although a pupil response can often still be found when examined under the magnifying glass. Fixed, dilated pupils presenting 6 hours after resuscitation are a sign of poor prognosis, but this is rarely present in isolation and is usually an indication that the brainstem has also been involved in the anoxic-ischemic injury. Corneal reflexes have been absent in about a third of patients, but they often reappear soon. Far more important is the presence of eye movement abnormalities (Wijdicks, 2002). Sustained upward gaze is indicative of a significant global bihemispheric injury that may include the thalamus. In some patients, downward gaze can be elicited using rapid head shaking or attempting to elicit a vestibular ocular response (Johkura et al., 2004). Other eye abnormalities, including ping-pong gaze or periodic lateral gaze deviations, have not been specifically examined for their prognostic value (Diesing and Wijdicks, 2004). Continuous blinking is often a common finding in comatose patients, although its anatomical substrate is unknown. An important clinical sign is myoclonus status epilepticus, defined as continuous jerking that involves the facial muscles, limbs, and abdominal muscles (Thomke et al., 2005; Young et al., 2005). These jerks can often be elicited by touch or hand clap and may also involve the diaphragm, which complicates ventilation. Myoclonus status epilepticus is an agonal phenomenon indicating a very poor prognosis. A high percentage of these patients have a burst suppression pattern on electroencephalogram (EEG) and computed tomography (CT), indicating either cerebral infarction or cerebral edema. Myoclonus status epilepticus must be distinguished from myoclonus due to intoxication or hepatic encephalopathy and from generalized tonic-clonic seizures. Convulsive status epilepticus is uncommon, as is nonconvulsive status. The motor response to pain should be classified and described as absence to pain, extensor response, pathological flexion response, or no motor response.
The outcomes for patients in coma range from death, including brain death to persistent vegetative state (see Chapters 5 and 32A), to awakening with disabilities ranging from the minimally conscious state (see Chapter 5) to complete recovery (see Chapter 48).
Awakening from coma can be protracted and prolonged, although the vast majority of patients awaken within the first 48 hours. In our series of patients, 94 of 101 patients with postanoxic ischemic injury awoke within 3 days after cardiac arrest and induced hypothermia did not seem to directly influence this (Fugate et al., 2011). However, awakening can occur even 3 months after onset, although rarely without a severe deficit such as an amnesic syndrome or other neurological findings (Table 55.1).
Table 55.1 Clinical Syndromes after Postanoxic-Ischemic Encephalopathy
| Clinical Syndrome | Mechanism | Outcome |
|---|---|---|
| “Man-in-the-barrel” syndrome | Bilateral watershed infarcts | Uncertain, may improve substantially |
| Parkinsonism | Infarcts in the striatum | Improvement possible |
| Action myoclonus | Cerebellar infarcts | In awake patients, could improve with medication |
The neurological examination can be confounded by an additional systemic injury associated with CPR. Several patients may have an associated acute renal failure or liver injury. In addition, medications may have been administered to counter pain or to facilitate mechanical ventilation. Often patients have been treated with fentanyl and lorazepam, both of which have long elimination half-lives (Table 55.2). The use of therapeutic hypothermia may further contribute, as hepatic metabolism and renal clearance are decreased, which may cause an enhanced and prolonged effect of medications (Polderman, 2009).
| Agent | Elimination Half-Life (Hours) |
|---|---|
| Morphine | 1.5-4 |
| Fentanyl | 2-5 |
| Alfentanil | 1.5-3.5 |
| Midazolam | 1-4 |
| Lorazepam | 10-20 |
| Propofol | 2 |
Laboratory Tests
An evidence-based review of all laboratory tests found that many have insufficient prognosticating value (Wijdicks et al., 2006). Widely used laboratory tests are EEG, CT or magnetic resonance imaging (MRI), evoked potentials, and serum biomarkers. Most frequently used is an EEG, which is classified using grades of severity. The so-called malignant patterns include burst suppression, alpha coma with periodic sharp waves and spikes, and isoelectric or markedly suppressed EEG. The last pattern often indicates loss of all brainstem function, and the patient should be reexamined to document the clinical diagnosis of brain death. However, marked suppression has been described in patients with intact lower brainstem function. With the increasing use of therapeutic hypothermia (which requires sedation and neuromuscular blockade), there has been interest in EEG monitoring during cooling (Abend et al., 2009; Al Thenayan et al., 2010; Legriel et al., 2009; Rundgren et al., 2006). While longer monitoring will almost certainly increase detection of epileptiform activity, there remains no evidence that earlier detection and treatment of seizures in this setting alters outcome.
Also of interest are somatosensory evoked potentials (SSEP) (Madl and Holzer, 2004). SSEPs are not influenced by drugs, temperature, or acute metabolic derangements and are more accurate for prognostication (Chen et al., 1996). SSEP requires stimulation of the median nerve that then results in a potential at the brachial plexus, cervical spinal cord, and finally bilateral cortex potentials (N20). For SSEP to be reliable, the cervical spine potential has to be recognized, and this could be of potential concern in patients with a severe anoxic-ischemic injury involving the cervical spinal cord.
There has been interest in the use of biomechanical markers in prognostication. Most studies have examined serum neuron-specific enolase (NSE) and S100 in comatose survivors. NSE is a gamma isomer of enolase that is located in neurons, and S100 (Bottiger et al., 2001; Tiainen et al., 2003; Wang et al., 2004) is a calcium-binding astroglial protein. In addition, earlier studies have looked at creatinine kinase brain isoenzyme (CKBB). The usefulness of these biomarkers is questionable. None of these studies are automated, it may take time to obtain the test, and standardization may not be optimal.
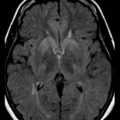
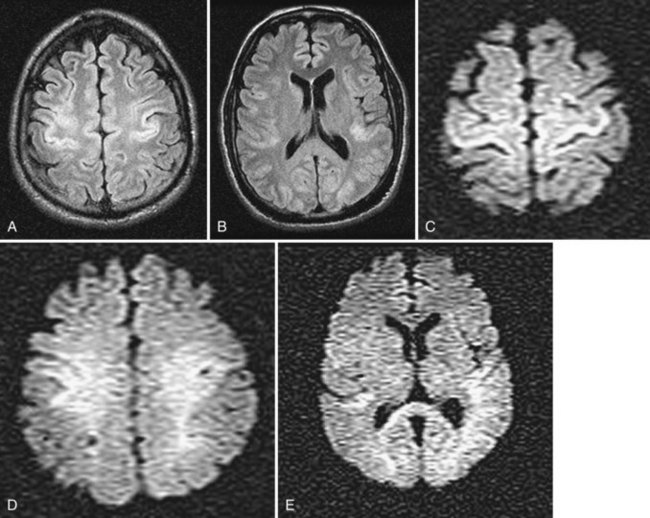
Neuroimaging, particularly MRI, has been of interest. MRI could document a significant injury, and earlier studies suggest that cortical abnormalities are important in predicting awakening (Wijdicks et al., 2001). Conventional MRI can be normal, with abnormalities only seen on diffusion-weighted MRI (Fig. 55.3). Whether MRI is useful in prognostication is unclear. Studies have been performed in a small selected population of patients, and the transport of patients (who often require vasopressor pumps) may be difficult. Many patients are not sufficiently stable to move to the MR suite. CT studies are often normal but may show early brain edema and significant cerebral edema in 3 to 4 days in the more extreme cases. The disappearance of the gray/white junction is associated with failure to awaken.
Management Options
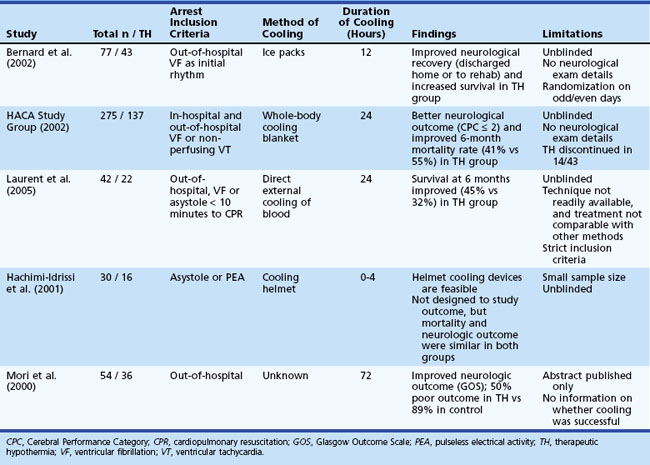
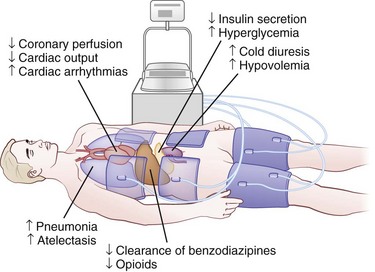
The practice of induced hypothermia in postcardiac arrest management among intensive care units has increased in recent years. Induced hypothermia, a complex undertaking, is now standard therapy for those with out-of-hospital ventricular fibrillation cardiac arrests. Two early pivotal trials found improved survival, but details on the neurological condition of the patients are unclear because the neurological examination in both trials was insufficient (Bottiger et al., 2001; Maramattom et al., 2005). More recently, a Cochrane systematic review and meta-analysis to assess its efficacy was conducted (Arrich et al., 2009). Four randomized controlled trials and one abstract encompassing a total of 481 patients were included. The results indicated that induced hypothermia improves survival and neurological outcome after cardiac arrest (Table 55.3). A cooling protocol for out-of-hospital cardiopulmonary assessment is shown in Box 55.1. This requires reduction in core temperature with ice packs, rapid infusion of cold intravenous fluids, and the use of external cooling devices or endovascular cooling systems (Holzer et al., 2006). Hypothermia is initiated within 2 to 3 hours to reduce core temperatures to 32°C to 34°C and is maintained for 24 hours, followed by gradual rewarming. Sedation and neuromuscular blockade are needed to control shivering. Major potential systemic complications include pneumonia, cardiac arrhythmias, pancreatitis, and hyperglycemia (Fig. 55.4) but are sometimes difficult to directly attribute to hypothermia, as these patients are systemically critically ill. The benefit of hypothermia has not been clearly established in patients after in-hospital CPR or in those with initial cardiac rhythms other than ventricular fibrillation.
Table 55.3 Summary of Studies on the Practice of Induced Hypothermia in Postcardiac Arrest Management Among Intensive Care Units
Operate the Arctic Sun cooling device
Place bladder catheter to monitor temperature
Systematic Approach to Prediction of Prognosis
The assessment of prognosis has been summarized in a published algorithm commissioned by the Quality Standard Subcommittee of the American Academy of Neurology (Fig. 55.5) (Wijdicks et al., 2006). This extensive literature review found that the circumstances surrounding CPR were not predictive of outcome. Several clinical features were highly predictive. The presence of myoclonus status epilepticus within the first 24 hours in patients with circulatory arrest, absence of pupillary responses within day 1 to 3 after cardiopulmonary arrest, absence of corneal reflex within day 1 to 3, and absent or extensor motor responses after day 3 are all associated with invariably poor outcome. Eye movement abnormalities were insufficiently predictive, but clinical studies in these patients have not focused on the prediction of specific eye motor abnormalities. These guidelines were based on studies done prior to the routine use of induced hypothermia, and predictors are being reevaluated for their reliability after cooling. The reliability of the motor response to predict poor outcome as early as day 3 after the use of hypothermia has been questioned (Al Thenayan et al., 2008; Rossetti et al., 2010). However, it remains unknown to what extent sedative medications used in this setting are a confounding factor (Fugate et al., 2010), and this subject deserves further study.
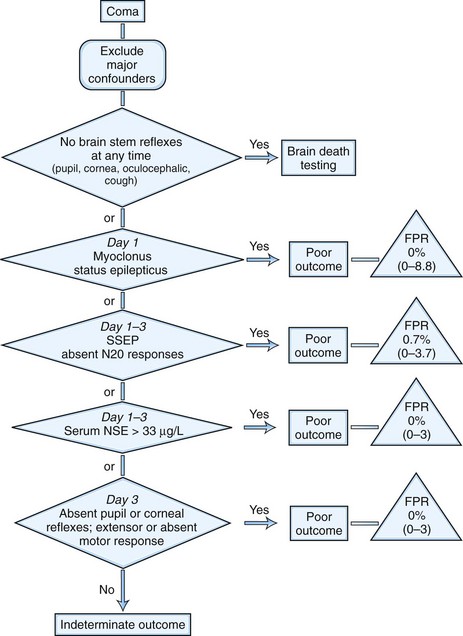
Burst suppression or generalized epileptiform discharges on EEG predicted poor outcome, but the prognostic criteria were insufficiently accurate. Improvement has been noted in patients who displayed an alpha coma pattern and even in patients with markedly suppressed EEGs (10-20 mV). The malignant EEG patterns showed a false-positive ratio of 3% (95% confidence interval, 0.9%–11%). More recently it has been suggested that the presence of EEG background reactivity may be of prognostic significance (Rossetti et al., 2010; Al Thenayan et al., 2010). SSEP measured after 24 hours and up to 3 days after CPR was highly predictive of poor outcome; the bilateral absence of the N20 component of SSEP with median nerve stimulation predicted poor outcome (Wijdicks et al., 2006). Preliminary evidence suggests that absent N20 responses during mild hypothermia after resuscitation maintains accuracy in predicting a poor neurological outcome (Bouwes et al., 2009).
In studies done prior to the routine use of hypothermia, only NSE predicted outcome, but CKBB or S100 were not reliable indicators. Therapeutic hypothermia may have an effect on the metabolism and clearance of these biomarkers, further clouding their prognostic value. Results of recent studies are conflicting, with some finding that NSE levels maintain prognostic accuracy after hypothermia (Oksanen et al., 2009; Rundgren et al., 2009) while others finding the prognostic value to be reduced (Fugate et al., 2010; Steffen et al., 2010).
Neuroimaging may become an important confirmatory test, and in particular, the use of diffusion-weighted MRI holds promise for prognostication (Wijman et al., 2009). Nonetheless, currently there are insufficient data to guide prognosis using neuroimaging.
Abend N.S., Topjian A., Ichord R., et al. Electroencephalographic monitoring during hypothermia after pediatric cardiac arrest. Neurology. 2009;72:1931-1940.
Al Thenayan E., Savard M., Sharpe M., et al. Predictors of poor neurologic outcome after induced mild hypothermia following cardiac arrest. Neurology. 2008;71:1535-1537.
Al Thenayan E., Savard M., Sharpe M., et al. Electroencephalogram for prognosis after cardiac arrest. J Crit Care. 2010;25:300-304.
Arrich, J., Holzer, M., Herkner, H., et al., 2009 Hypothermia for neuroprotection in adults after cardiopulmonary resuscitation. Cochrane Database Syst Rev. 2009;4:CD004128.
Bernard S.A., Gray T.W., Buist M.D., et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557-563.
Bottiger B.W., Mobes S., Glatzer R., et al. Astroglial protein S-100 is an early and sensitive marker of hypoxic brain damage and outcome after cardiac arrest in humans. Circulation. 2001;103:2694-2698.
Bouwes A., Binnekade J.M., Zandstra D.F., et al. Somatosensory evoked potentials during mild hypothermia after cardiopulmonary resuscitation. Neurology. 2009;73:1457-1461.
Broccard A. Therapeutic hypothermia for anoxic brain injury following cardiac arrest: a “cool” transition toward cardiopulmonary cerebral resuscitation. Crit Care Med. 2006;34:2008-2009.
Callans D.J. Out-of-hospital cardiac arrest—the solution is shocking. N Engl J Med. 2004;351:632-634.
Chen R., Bolton C.F., Young B. Prediction of outcome in patients with anoxic coma: a clinical and electrophysiologic study. Crit Care Med. 1996;24:672-678.
Diesing T.S., Wijdicks E.F. Ping-pong gaze in coma may not indicate persistent hemispheric damage. Neurology. 2004;63:1537-1538.
Fugate J.E., Wijdicks E.F., Mandrekar J., et al. Predictors of neurologic outcome in hypothermia after cardiac arrest. Ann Neurol. 2010;68:907-914.
Fugate, J.E., Wijdicks, E.F., White, R.D., et al., 2011. Does therapeutic hypothermia affect time to awakening in cardiac arrest survivors? Neurology (In press).
Group THACAS. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549-556.
Hachimi-Idrissi S., Corne L., Ebinger G., et al. Mild hypothermia induced by a helmet device: a clinical feasibility study. Resuscitation. 2001;51(3):275-281.
Holzer M., Mullner M., Sterz F., et al. Efficacy and safety of endovascular cooling after cardiac arrest. Cohort study and bayesian approach. Stroke. 2006;37:1792-1797.
Johkura K., Komiyama A., Kuroiwa Y. Vertical conjugate eye deviation in post-resuscitation coma. Ann Neurol. 2004;56:878-881.
Laurent I., Adrie C., Vinsonneau C., et al. High-volume hemofiltration after out-of-hospital cardiac arrest: a randomized study. J Am Coll Cardiol. 2005;46(3):432-437.
Legriel S., Bruneel F., Sediri H., et al. Early EEG monitoring for detecting postanoxic status epilepticus during therapeutic hypothermia: a pilot study. Neurocrit Care. 2009;11:338-344.
Madl C., Holzer M. Brain function after resuscitation from cardiac arrest. Curr Opin Crit Care. 2004;10:213-217.
Maramattom B.V., Wijdicks E.F. Postresuscitation encephalopathy. Current views, management, and prognostication. Neurologist. 2005;11:234-243.
Mori K., Takeyama Y., Itoh Y., et al. A multivariate analysis of prognostic factors in survivors of out-of-hospital cardiac arrest with brain hypothermia. Crit Care Med.. 2000;28:A168.
Ogawa S., Kitao Y., Hori O. Ischemia-induced neuronal cell death and stress response. Antioxid Redox Signal. 2007;9:573-587.
Oksanen T., Tiainen M., Skrifyars M.B., et al. Predictive power of serum NSE and OHCA score regarding 6-month neurologic outcome after out-of-hospital ventricular fibrillation and therapeutic hypothermia. Resuscitation. 2009;80:165-170.
Peberdy M.A., Kaye W., Ornato J.P. Cardiopulmonary resuscitation of adults in the hospital: a report of 14,720 cardiac arrests from the National Registry of Cardiopulmonary Resuscitation. Resuscitation. 2003;58:297-308.
Polderman K.H. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. 2009;37:S186-S202.
Rogove H.J., Safar P., Sutton-Tyrrell K., et al. Old age does not negate good cerebral outcome after cardiopulmonary resuscitation: analyses from the brain resuscitation clinical trials. The Brain Resuscitation Clinical Trial I and II Study Groups. Crit Care Med. 1995;23:18-25.
Rossetti A.O., Oddo M., Logroscino G., et al. Prognostication after cardiac arrest and hypothermia. A prospective study. Ann Neurol. 2010;67:301-307.
Rundgren M., Karlsson T., Nielsen N., et al. Neuron specific enolase and S-100B as predictors of outcome after cardiac arrest and induced hypothermia. Resuscitation. 2009;80:784-789.
Rundgren M., Rosen I., Friberg H. Amplitude-integrated EEG (aEEG) predicts outcome after cardiac arrest and induced hypothermia. Intensive Care Med. 2006;32:836-842.
Steffen I.G., Hasper D., Ploner C.J., et al. Mild therapeutic hypothermia alters neuron specific enolase as an outcome predictor after resuscitation: 97 prospective hypothermia patients compared to 133 historical non-hypothermia patients. Crit Care. 2010;14:R69.
Teschendorf P., Popp E., Motsh J. Effective inhibition of caspases on neuronal degeneration and outcome following global cerebral ischemia due to cardiocirculatory arrest in rats. Anesthesiology. 2001;95:788. Abstract
Thomke F., Marx J.J., Sauer O., et al. Observations on comatose survivors of cardiopulmonary resuscitation with generalized myoclonus. BMJ Neurol. 2005;5:14.
Tiainen M., Roine R.O., Pettila V., et al. Serum neuron-specific enolase and S-100B protein in cardiac arrest patients treated with hypothermia. Stroke. 2003;34:2881-2886.
Wang J.T., Young G.B., Connolly J.F. Prognostic value of evoked responses and event-related brain potentials in coma. Can J Neurol Sci. 2004;31:438-450.
Wijdicks E.F. Neurologic Complications of Critical Illness, second ed. New York: Oxford University Press; 2002.
Wijdicks E.F., Campeau N.G., Miller G.M. MR imaging in comatose survivors of cardiac resuscitation. AJNR Am J Neuroradiol. 2001;22:1561-1565.
Wijdicks E.F., Hijdra A., Young G.B., et al. Practice parameter: prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence-based review). Neurology. 2006;67:203-210.
Wijman C.A., Mlynash M., Caulfield A.F., et al. Prognostic value of brain diffusion-weighted imaging after cardiac arrest. Ann Neurol. 2009;65:394-402.
Young G.B., Doig G.S., Ragazzoni A. Anoxic-ischemic encephalopathy; clinical and electrophysiological associations with outcome. Neurocrit Care. 2005;2:159-164.
Zandbergen E.G., Hijdra A., Koelman J.H. Prediction of poor outcome within the first three days of postanoxic coma. Neurology. 2006;66:62-68.