[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Forward Genetics, Reverse Genetics, and Transgenesis
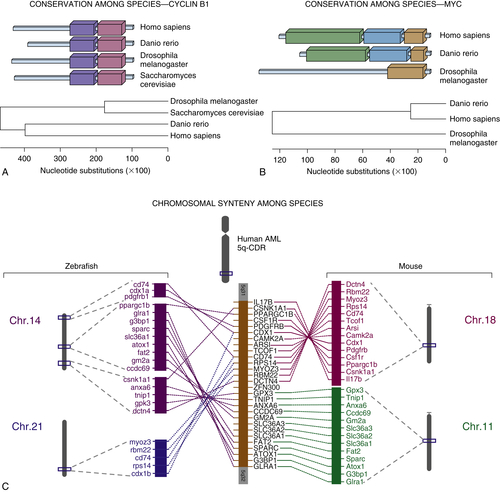
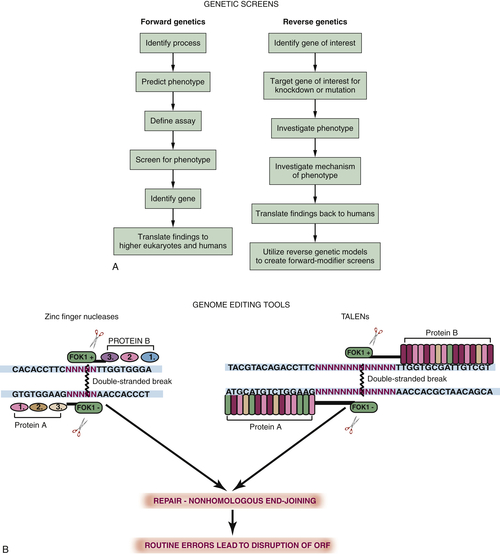
Drug Screens
Conditional Models
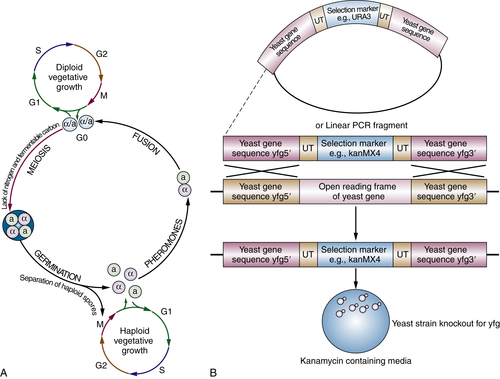
Yeast
Genetic Tools and Functional Genomics
The Cell Cycle
CDC2 and CDC28
Ploidy, Genome Instability, and Cancer
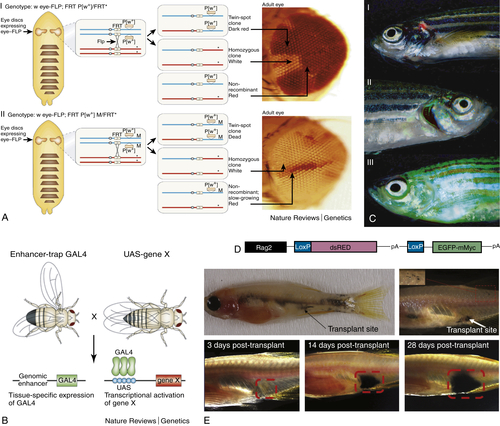
Flies
Why Use a Fly?
Genetic Tools
Malignant Neoplastic Tumor Suppressors in Drosophila: scribble and Others
Archipelago
Fish
Leukemia Models and Therapy Using Transgenic Zebrafish
Zebrafish Screen for Genomic Instability Mutants
Table 8-1
Internet Resources
http://zfin.org
http://flymove.unimuenster.de/Organogenesis/ImagDiscs/OrgDiscpage.html?http&&&flymove.uni-muenster.de/Organogenesis/ImagDiscs/OrgDiscTxt.html
http://www.neuro.uoregon.edu/k12/zfk12.html
http://www.dnaftb.org/dnaftb
http://zifit.partners.org/ZiFiT
https://boglab.plp.iastate.edu
http://www.sanger.ac.uk/Projects/D_rerio/zmp
Conclusion
1. Drosophila, the golden bug, emerges as a tool for human genetics . Nat Rev Genet . 2005 ; 6 : 9 – 23 .
2. Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2 . Nature . 1987 ; 327 : 31 – 35 .
3. A yeast BH3-only protein mediates the mitochondrial pathway of apoptosis . EMBO J . 2011 ; 30 : 2779 – 2792 .
4. BIM and other BCL-2 family proteins exhibit cross-species conservation of function between zebrafish and mammals . Cell Death Differ . 2008 ; 15 : 1063 – 1072 .
5. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics . Dev Cell . 2011 ; 21 ( 1 ) : 92 – 101 .
6. Zebrafish hox clusters and vertebrate genome evolution . Science . 1998 ; 282 : 1711 – 1714 .
7. Liu TX, Zhou Y, Kanki JP, et al. Evolutionary conservation of zebrafish linkage group 14 with frequently deleted regions of human chromosome 5 in myeloid malignancies. i2002 Apr 30; 99(9): 6136–6141.
8. Fast homozygosity mapping and identification of a zebrafish ENU-induced mutation by whole-genome sequencing . PLoS One . 2012 ; 7 ( 4 ) : e34671 .
9. Efficient mapping and cloning of mutations in zebrafish by low-coverage whole-genome sequencing . Genetics . 2012 Mar ; 190 ( 3 ) : 1017 – 1024 .
10. Targeted gene disruption in somatic zebrafish cells using engineered TALENs . Nat Biotechnol . 2011 Aug ; 29 ( 8 ) : 697 – 698 .
11. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA) . Nat Methods . 2011 Jan ; 8 ( 1 ) : 67 – 69 .
12. Cre/lox-regulated transgenic zebrafish model with conditional myc-induced T cell acute lymphoblastic leukemia . Proc Natl Acad Sci U S A . April 26, 2005 ; 102 ( 17 ) : 6068 – 6073 2005 .
13. Myc-Induced T cell leukemia in transgenic zebrafish . Science 2003 . February 7, 2003 ; 299 ( 5608 ) : 887 – 890 .
14. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis . Cancer Cell . 2012 Mar 20 ; 21 ( 3 ) : 362 – 373 .
15. Cancer therapeutics in yeast . Cancer Cell . 2002 Oct ; 2 ( 4 ) : 267 – 273 .
16. DHODH modulates transcriptional elongation in the neural crest and melanoma . Nature . 2011 Mar 24 ; 471 ( 7339 ) : 518 – 522 .
17. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology . Nature . 2012 Jun 7 ; 486 ( 7401 ) : 80 – 84 .
18. Using Drosophila melanogaster to map human cancer pathways . Nat Rev Cancer . 2005 Aug ; 5 ( 8 ) : 626 – 639 .
19. A unique and universal molecular barcode array . Nat Methods . 2006 ; 3 ( 8 ) : 601 – 603 .
20. Global synthetic-lethality analysis and yeast functional profiling . Trends Genet . 2006 ; 22 ( 1 ) : 56 – 63 .
21. Revealing hidden relationships among yeast genes involved in chromosome segregation using systematic synthetic lethal and synthetic dosage lethal screens . Cell Cycle . 2006 Mar ; 5 ( 6 ) : 592 – 595 .
22. Global mapping of the yeast genetic interaction network . Science . 2004 ; 303 : 808 – 813 .
23. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast . Cell . 2006 ; 126 ( 3 ) : 611 – 625 .
24. Proteomics to study genes and genomes . Nature . 2000 ; 405 ( 6788 ) : 837 – 846 .
25. Global landscape of protein complexes in the yeast . Saccharomyces cerevisiae. Nature . 2006 ; 440 ( 7084 ) : 637 – 643 .
26. Proteome survey reveals modularity of the yeast cell machinery . Nature . 2006 ; 440 ( 7084 ) : 631 – 636 .
27. The Josef Steiner Lecture: CDKs and cell-cycle control in fission yeast: relevance to other eukaryotes and cancer . Int J Cancer . 1997 ; 71 : 707 – 708 .
28. Synchronization of haploid yeast cell cycles, a prelude to conjugation . Exp Cell Res . 1973 ; 76 ( 1 ) : 111 – 117 .
29. Saccharomyces cerevisiae: a diffusible sex factor . Science . 1970 ; 168 : 1472 – 1473 .
30. Nobel Lecture. Yeast and cancer . Biosci Rep . 2002 ; 22 : 373 – 394 .
31. Living with or without cyclins and cyclin-dependent kinases . Genes Dev . 2004 ; 18 : 2699 – 2711 .
32. Recycling the cell cycle: cyclins revisited . Cell . 2004 ; 116 ( 2 ) : 221 – 234 .
33. The Bloom syndrome helicase is a substrate of the mitotic Cdc2 kinase . Cell Cycle . 2006 Aug ; 5 ( 15 ) : 1681 – 1686 .
34. Mutator suppression and escape from replication error-induced extinction in yeast . PLoS Genet . 2011 Oct ; 7 ( 10 ) : e1002282 .
35. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers . N Engl J Med . 2009 Jul 9 ; 361 ( 2 ) : 123 – 134 .
36. Thicker than blood: conserved mechanisms in Drosophila and vertebrate hematopoiesis . Dev Cell . 2003 ; 5 ( 5 ) : 673 – 690 .
37. Drosophila in cancer research: an expanding role . Trends Genet . 2000 ; 16 ( 1 ) : 33 – 39 .
38. A genetic screen for modifiers of the lats tumor suppressor gene identifies C-terminal Src kinase as a regulator of cell proliferation in . Drosophila. Oncogene . 2003 Sep 25 ; 22 ( 41 ) : 6436 – 6444 .
39. Functional identification of Api5 as a suppressor of E2F-dependent apoptosis in vivo . PLoS Genet . 2006 Nov 17 ; 2 ( 11 ) : e196 .
40. A genetic screen for modifiers of E2F in . Drosophila melanogaster. Genetics . 1999 ; 153 : 275 – 287 .
41. Genetic mosaic techniques for studying Drosophila development . Development . 2003 ; 130 : 5065 – 5072 .
42. dMyc Transforms cells into super-competitors . Cell . 2004 ; 117 ( 1 ) : 117 – 129 .
43. Drosophila Myc regulates organ size by inducing cell competition . Cell . 2004 ; 117 ( 1 ) : 107 – 116 .
44. Drosophila myc regulates cellular growth during development . Cell . 1999 ; 98 ( 6 ) : 779 – 790 .
45. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors . Science . 2000 Jul 7 ; 289 ( 5476 ) : 113 – 116 .
46. Junctional recruitment of mammalian Scribble relies on E-cadherin engagement . Oncogene . 2005 Jun 23 ; 24 ( 27 ) : 4330 – 4339 .
47. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila . EMBO J . 2003 Nov 3 ; 22 ( 21 ) : 5769 – 5779 .
48. J.N.K.- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila . EMBO J . 2006 Nov 15 ; 25 ( 22 ) : 5294 – 5304 .
49. The art and design of genetic screens: . Drosophila melanogaster. Nat Rev Genet . 2002 Mar ; 3 ( 3 ) : 176 – 188 .
50. Analyzing the function of tumor suppressor genes using a Drosophila model . Methods Mol Biol . 2003 ; 223 : 349 – 382 .
51. HPV E6 specifically targets different cellular pools of its PDZ domain-containing tumour suppressor substrates for proteasome-mediated degradation . Oncogene . 2004 Oct 21 ; 23 ( 49 ) : 8033 – 8039 .
52. Analysis of the expression and localisation of a LAP protein, human scribble, in the normal and neoplastic epithelium of uterine cervix . Br J Cancer . 2004 Jan 12 ; 90 ( 1 ) : 194 – 199 .
53. Human discs large and scrib are localized at the same regions in colon mucosa and changes in their expression patterns are correlated with loss of tissue architecture during malignant progression . Int J Cancer . 2006 ; 119 ( 6 ) : 1285 – 1290 .
54. Archipelago regulates cyclin E levels in Drosophila and is mutated in human cancer cell lines . Nature . 2001 ; 413 ( 6853 ) : 311 – 316 .
55. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase . Science 2001 . October 5, 2001 ; 294 ( 5540 ) : 173 – 177 .
56. General Motors Cancer Research Prizewinner Laureates Lectures. Alfred P. Sloan, Jr. Prize. The formation of the embryonic axes in . Drosophila. Cancer . 1993 May 15 ; 71 ( 10 ) : 3189 – 3193 .
57. The identification of genes with unique and essential functions in the development of the zebrafish . Danio rerio . 1996 : 1 – 36 .
58. Large-scale mutagenesis in the zebrafish: in search of genes controlling development in a vertebrate . Curr Biol . 1994 ; 4 ( 3 ) : 189 – 202 .
59. Zebrafish as a cancer model system . Cancer Cell . 2002 ; 1 ( 3 ) : 229 – 231 .
60. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma . Curr Biol . 2005 ; 15 ( 3 ) : 249 – 254 .
61. Transgenes as screening tools to probe and manipulate the zebrafish genome . Dev Dynam . 2005 ; 234 ( 2 ) : 255 – 268 .
62. Transparent adult zebrafish as a tool for in vivo transplantation analysis . Cell Stem Cell . 2008 Feb 7 ; 2 ( 2 ) : 183 – 189 .
63. T-lymphoblastic lymphoma cells express high levels of BCL2, S1P1, and ICAM1, leading to a blockade of tumor cell intravasation . Cancer Cell . 2010 Oct 19 ; 18 ( 4 ) : 353 – 366 .
64. Zebrafish screen identifies novel compound with selective toxicity against leukemia . Blood . 2012 Jun 14 ; 119 ( 24 ) : 5621 – 5631 .
65. Attainment of minimal biological variability and measurements of genotoxicity: production of homozygous diploid zebra fish . Natl Cancer Inst Monogr . 1984 May ; 65 : 53 – 58 .
66. Zebrafish genomic instability mutants and cancer susceptibility . Genetics 2006 . October 1, 2006 ; 174 ( 2 ) : 585 – 600 .
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Forward Genetics, Reverse Genetics, and Transgenesis
Drug Screens
Conditional Models
Yeast
Genetic Tools and Functional Genomics
The Cell Cycle
CDC2 and CDC28
Ploidy, Genome Instability, and Cancer
Flies
Why Use a Fly?
Genetic Tools
Malignant Neoplastic Tumor Suppressors in Drosophila: scribble and Others
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
