CHAPTER 10 Angiogenesis in Meningiomas
INTRODUCTION
To obtain nutrients, tumor cells initially rely on passive diffusion from their microbioenvironment.1 In 1947, Algire2 suggested that the formation of new blood vessels is required for progressive tumor growth. Folkman3 proved this theory in 1971 and demonstrated that solid tumors were absolutely dependent on angiogenesis for growth beyond a diameter of 2 mm. Angiogenesis research will probably change the face of medicine in the next decades, with more than 500 million people worldwide predicted to benefit from pro- or anti-angiogenesis treatments.4
Meningiomas are no exceptions to these fundamental rules of angiogenesis.5 Meningiomas derive their blood supply predominantly from meningeal vessels originating in the external carotid circulation. An additional supply from cerebral–pial vessels is present in approximately 60% of patients.6 Meningiomas are of variable vascularity, ranging from sparsely vascularized to highly vascular angiomatous meningiomas.7–9 They further display variable degrees of peritumoral brain edema, ranging from absent to life-threatening conditions.10 In contrast to gliomas, there is no scientifically proven association between edema and histologic grade in meningiomas.11 Nevertheless, extent of angiogenic activity during the meningioma formation and progression is certainly important in the surgical and endovascular management and recurrence of these tumors, regardless of their anatomic location.
BASICS OF VASCULOGENESIS AND ANGIOGENESIS IN NORMAL AND PATHOLOGIC CONDITIONS
The first vascular network in the embryo is established by the process of vasculogenesis at the time when somites begin to form. In the mouse the first blood islands arise by in situ differentiation from the extraembryonic mesoderm around E7.0 to 7.5. T’hey are located between the two germ layers and form the inner layer of the yolk sac.12–15 The extraembryonic yolk sac circulation, the primitive heart, and the primary vascular plexus inside the embryo, as the dorsal aortae, and the vitelline veins, thus form by the process of vasculogenesis. While this first vascular plexus is still emerging, early signs of its remodeling, especially inside the embryo, are visible. Network remodeling is defined as rearrangement of number or location of vascular segments to establish a functional adaptation without measurable network expansion.16–18 Vascular fusion reduces the number of segments and gives rise to larger vessels. At other places larger vessels disappear and/or are remodeled into a network of smaller ones subsequently increasing the number of segments.19,20 These processes lead to transformation of the uniform primary plexus into the secondary one that is more complexly structured. Further expansion of the primary and secondary vascular plexus occurs by the process of angiogenesis. Angiogenesis refers to blood vessel formation from preexisting ones.20 It implies two distinct mechanisms: endothelial sprouting and intussusceptive microvascular growth (IMG). Angiogenesis establishes the circulation in so far avascular regions. Concerning the formation of vascular networks in the forming organs, some are established by vasculogenesis, for example, lung and spleen, while others, such as the brain, are derived from angiogenesis.
Vasculogenesis
The first descriptions of the in situ formation of blood vessels in living animals, some of them derived from manipulations of the early chicken embryo and in vivo microscopic observations, date back to the 19th century.21,22 Sabin23 contributed the most detailed analysis of the process of vasculogenesis and clearly separated it by definition from angiogenesis.
Mesodermal cells in the extraembryonic yolk sac that are termed hemangioblasts give rise to the blood islands. These cells proliferate and differentiate to form the precursors of the endothelial cells of the vessel wall, the angioblasts, and the precursors of the hematopoietic cells located in the lumen. Fusion of the blood islands results in the primary vascular plexus.24,25 Inside the embryo, cells of the proximal lateral mesoderm assemble symmetrically at the lateral sides of the embryo to form the “preendocardial tubes”.24 The latter fuse at the anterior intestinal portal. The fused region forms the endocardium of the heart. Closely related to heart formation is the formation of the two ventral and dorsal aortae. These vessels develop by assembly of angioblasts to form four major channels. The two dorsal aortae fuse later in development to give rise to one single vessel. The vitelline or omphalomesenteric arteries arise from the distal parts of the dorsal aortae. They fuse with the yolk sac vessels similar to the two vitelline veins that form by splitting of the sinus venosus area of the developing heart. Mesodermal cells of the allantois give rise to the umbilical vessels. This demonstrates that formation of the large intraembryonic vessels is very early depending on the morphogenetic steps of vessel fusion and splitting that cause further growth and remodeling by the process of angiogenesis. Angioblasts can also migrate inside the embryo to form vascular plexus at distant locations. Chick-quail grafting experiments suggest that two types of intraembryonic hemangioblasts exist; one derived from the splanchnopleuric mesoderm can produce hematopoietic cells, unlike the other that originates from the somatopleuric mesoderm.26 Vascularization of organs derived form mesoderm and endoderm (as the lung and spleen) occurs primarily by vasculogenesis.25
Vasculogenesis is subsequently induced by the endoderm and mesoderm.25–28 Vascular endothelial growth factor (VEGF) is expressed in the extraembryonic visceral endoderm and in the extraembryonic mesoderm when the first blood islands emerge. At day 8.5, when the intraembryonic vascular plexus begins to form, surrounding endodermal cells exhibit strong VEGF expression, while the expression is moderate in mesodermal derived cells. Endothelial cells at this stage express the VEGF-A receptor flk-l (VEGFR-2, KDR) in a paracrine manner12,27 and flk-l upregulation is even induced by VEGF.29 Subsequently, gene targeting studies showed that flk-1−/− mice lacked vasculogenesis and failed to develop blood islands throughout the embryo and the yolk sac based on a lack of differentiation of mesodermal cells to form angioblasts. The embryos died between E8.5 and 9.5.30 The second tyrosine kinase receptor for VEGF, flt-t (VEGFR-1), is simultaneously expressed in endothelial cells during early embryonic development.31 Flt-1−/− mice also died by E8.0/E9.0 and lacked a proper organization of the blood islands.
lnactivation of a single VEGF allele in mice caused death of the embryos between El1 and El2. The VEGF+/− embryos exhibited a number of developmental anomalies, among them malformations of the heart, rudimentary dorsal aortae, and a reduced number of nucleated red blood cells in the yolk sac. This indicates that a threshold level of VEGF is required to maintain angioblast differentiation.32 Homozygous VEGF-A–deficient embryos that were completely derived from embryonic stem (ES) cells (generated by aggregation of homozygous VEGF-A–deficient ES cells with tetraploid embryos) died in mid-gestation (E9.5) based on even more severe cardiovascular defects. These results taken together indicate that the embryonic VEGF expression requires a precise regulation. In addition, other growth factors, such as transforming growth factor β (TGF-β), have also been linked to yolk sac hematopoiesis and vasculogenesis.12,16
Angiogenesis
Angiogenesis, the formation of blood vessels from preexisting ones, consists of two distinct processes: sprouting of endothelial cells and splitting of vessel lumens by IMG. Organs derived from the ectoderrn–mesoderm, such as the brain and neuroectoderm, are vascularized by angiogenesis.16
The sprouting mode of angiogenesis
One of the first descriptions of the sprouting process dates back to Galen of Pergamon (c. A.D. 130–200), who compared the developing embryo to a plant that grows along with the branching umbilical veins.33 More recently, a precise concept consistent with sprout formation has been elaborated using a variety of models. The most important ones are the chicken chorioallantoic membrane (CAM) and the corneal pocket.34,35 The sprouting process consists of several consecutive steps that have been described by Ausprunk and Folkman35 and many other authors:36,37
(Note that steps 1 and 11 are specific for tumoral, such as meningioma, conditions.)
The intussusceptive mode of angiogenesis
IMG has been detected in a variety of tissues and organs, in embryonic as well as adult angiogenesis.17,38,39 In addition, its existence has been proven by the demonstration of different mechanisms of its cellular implementation by in vivo video microscopy using the chicken CAM and tumor xenografts as model systems. These mechanisms were confirmed by the analysis of sequential serial sections using light- and electron microscopy.17,18
The following steps are common to all mechanisms of IMG analyzed so far:16–18,38,39
ln the adult situation, as in tissue repair or tumor angiogenesis, periendothelial cells of the vessel wall (fibroblast-like cells, pericytes, and smooth muscle cells) are involved in IMG.38,39 Fibroblast-like cells that expressed endothelial-specific markers such as platelet–endothelial cell adhesion molecule (PECAM), flt-l, and tie-2 were detected in tissue repair and tumors.
Vascular network remodeling
Remodeling is necessary to optimize the functional adaptation of the newly formed network.42,43 It implies addition of new vascular segments (vessels), but also the deletion of previous ones.16,17 Remodeling also includes the differential growth of segments, a process termed pruning.20 Pruning can occur by generation of new endothelial cells in the vessel wall. The size of segments is also increased by their fusion, and conversely the division of segments decreases their sizes. The number of segments is inversely affected by these two processes. Thus remodeling and pruning occur simultaneously.18
Vascular network remodeling also occurs as a response to divergent flow conditions. Numerous angiogenic molecules, for example, platelet-derived growth factor B (PDGF-B), and transcription factors are expressed by endothelial cells, whereas their receptors are up-regulated in perivascular cells in the vessel wall after the alteration of shear stress profiles.44 Endothelial cells in vitro that are exposed to large shear stress gradients respond with increased cell division and motility in the vicinity of flow separation.45 Network remodeling is also linked to maturation and stabilization of the vessel wall in larger vessels that is based on the recruitment and differentiation of mesenchymal cells or fibroblasts to form pericytes and smooth muscle cells.18,46
The molecular regulation of angiogenesis
In general, the molecular regulation of angiogenesis is the regulation of its distinct steps. Thus an angiogenic molecule promotes endothelial cell proliferation, migration, or tube formation while an inhibitor interferes with these steps. Concerning IMG, the ability to induce stretching and thinning of endothelial cells and movement of the endothelial layer is critical. Induction of cell membrane fusion to form transcellular holes in endothelial cells is another requisite as well as matrix proteolysis, collagen fibril synthesis, and recruitment of periendothelial cells. To date, a number of angiogenic growth factors have been identified. The most prominent ones are fibroblast growth factor 1 and 2 (FGF-1, acidic and FGF-2, basic), platelet-derived growth factor (PDGF), hepatocyte growth factor (HGF, scatter factor), VEGF-A, VEGF-B, VEGF-C, transforming growth factor α (TGF-) and interleukin 8 (IL-8).47 In addition, an important role in angiogenesis has been established for the tie/Angiopoietin and the Eph-B/ephrin-B system of tyrosine kinase receptors and their ligands.46,48,49
VEGF-A and its receptors (flk-l and flt-1)
Similar to its role in vasculogenesis, VEGF-A is also responsible for the regulation of angiogenesis. Many studies have demonstrated, mostly in vitro, that VEGF promotes migration and tube formation in endothelial cells. The VEGF family members have differing capabilities to promote these functions.50,51 VEGF+/− embryos die between day 11 and 12 from ubiquitous defects in their vasculature. These defects include abnormally large vessels in the yolk sac and inside the embryo accompanied by tissue necrosis, a lack of fusion of the vitelline veins with the yolk sac vessels, and failing growth of vessels from the perineural plexus into the neuroepithelium of the brain.32 This confirms the importance of VEGF for embryonic angiogenesis. Interestingly, high expression levels are detected in organs where vasculogenesis is the leading mechanism of vascularization, while lower levels are found in organs that receive their vascular supply by angiogenesis.12 Whereas the flk-1 receptor (VEGFR-2) is dominant during early vasculogenesis, flt-l (VEGFR-1) is prominent during remodeling of the primary vascular plexus and subsequent angiogenesis. Flt-l−/− mice thus exhibit large, dilated vessels in the yolk sac and throughout the embryo instead of a plexus of smaller ones.52
The Angiopoietin/tie system
The tie receptors form another family of receptor tyrosine kinases. They are important for the regulation of angiogenesis together with the ligands of the tie-2 receptor, the Angiopoietins. The tie tyrosine kinase receptors, like the VEGF receptors, form the only receptor families that are predominantly expressed on endothelial cells.53 Homozygous mutant mice deficient of the tie-l or tie-2 receptor undergo severe vascular malformations and die between days E13.5 and P0 (tie-1) and E9.5 and 10.5 (tie-2).54 This suggests that both receptors play a critical role in embryonic angiogenesis. The detailed analysis of tie-2 receptor deficient embryos revealed malformations of the heart derived from a lack of interaction between endocardium and myocardium. Abnormally large vessels were detected in the extraembryonic yolk sac circulation, and in many vascular networks throughout the embryo, for example, the perineural and intestinal vascular plexus.16,54 Ultrastructural analysis demonstrated a failure of endothelial cell stretching (interaction with their extracellular matrix) in numerous locations as well as a lack of recruitment of periendothelial cells (pericyte-, smooth muscle-, and myocardial cell precursors). This caused an aberrant morphology of tissue folds and tissue pillars that were not able to divide the embryonic vessels as they did in the control embryos with subsequent collapse of some vascular segments. In addition, the formation of vascular loops that formed in situ according to the principles of IMG was disturbed. Thus loop systems that invaded the neuroectoderm and the surrounding mesenchyme from the perineural plexus in the control embryo were diminished in number and rudimentary in structure in the tie-2 mutant.18 The only mechanism by which vessels could remodel in the tie-2 knockout embryo was based on a flat collapse of opposing vessel walls. This was frequently followed by thinning and cell membrane fusion of the opposing endothelial cells with formation of two transcellular holes. Invasion of these holes by matrix elements formed a new branching point and divided the vessel segment permanently into two parts. However, the collapsed area was much larger in diameter as compared to the region of insertion of tissue folds that divided vessel segments in the normal case. Thus the newly separated segments were much smaller and located more distant to each other as compared to wild-type segments. These malformations of the vascular network demonstrated that the embryo was very likely unable to perform a physiologic gas exchange. Necrotic cells detected throughout the embryonic tissues support this interpretation.18 Homozygous mice for a targeted mutation of Angiopoietin-1 (Ang-1), a ligand that specifically activates the tie-2 receptor, exhibited a corresponding phenotype.46 Angiopoietin-1 is expressed on periendothelial cells, suggesting a paracrine regulation.18 In addition, Ang-1 is chemotactic for endothelial cells, but does not cause endothelial cell proliferation or tube formation in vitro.55 These data taken together support the hypothesis that the Angiopoietin-1/tie-2 system promotes IMG rather than endothelial sprouting. Although Koblizek and colleagues56 detected endothelial sprouting induced by Ang-1 in a specialized in vitro system of angiogenesis, they confirmed the notion that Angiopoietin-1 was only weakly mitogenic for endothelial cells. Further, Ang-1 increases the survival of networks exposed to VEGF or acidic FGF and prevents apoptotic death triggered by growth factor withdrawal. In addition, transgenic overexpression of Ang-1 in the skin of adult mice results in larger, more numerous, and highly branched vessels as compared to controls.57 The view that Ang-1 cooperates with VEGF to ensure embryonic angiogenesis16,18,22 was confirmed in the cornea micropocket assay in the adult. In this system the combination of Ang-1 and VEGF resulted in an enhanced capillary density and an increased luminal diameter of the basal limbus artery as compared to the effect of VEGF alone.58
Angiopoietin-2 also binds specifically to the tie-2 receptor; however, it has no activating effect and consequently blocks Ang-1 activity. Thus mutant mice that overexpress the Ang-2 ligand exhibit a phenotype corresponding to the tie-2-or Ang-1–deficient mice.59 Interestingly in, situ hybridization during the ovarian an cycle in adult mice showed that Ang-2 is coexpressed with VEGF within a region of vessel growth. presumably promoting angiogenesis by endothelial sprouting. Ang-2 expressed in the absence of VEGF was related to an area of vessel regression.59 Corresponding data were obtained in the corneal micropocket assay where Ang-2 together with VEGF caused the formation of elongated vessels and “isolated sprouting cells located at the tip of developing capillaries.”58
The tie-1 receptor tyrosine kinase was originally associated with a survival function for endothelial cells and a stabilization effect on the vessel wall. Mutant mice deficient in the tie-1 receptor exhibited holes in vessel walls and endothelial cells that appeared to be necrotic.54 A more recent investigation of these mice demonstrated significantly increased numbers of blood vessels throughout all tissues and organs investigated. Ultrastructural analysis revealed that many endothelial cells were in a “hyperactive state” compared to others that matched those in the control animals. These hyperactive endothelial cells were extremely stretched with numerous cellular filopodia that projected into the vessel lumen and were even connected to the opposite vessel wall. These cells also showed frequent intra- and transcellular holes at places not related to tissue pillar separation. These data taken together support the hypothesis that the tie-1 receptor could be an inhibitor of angiogenesis that would inverse the functions of the tie-2/Ang-1 system. It likely also influences the process of cell membrane fusion that is necessary for all mechanisms of IMG.18 The recent analysis of double mutant embryos that lack both tie-1 and tie-2 receptors revealed a phenotype largely corresponding to the one of tie-2–deficient mice with an increased severity and an earlier onset of the mutant characteristics.
The ephrin-B/Eph-B system
Recently another receptor tyrosine kinase family, the Eph-B receptors and their ligands, the ephrins, have been shown to promote embryonic and tumorigenic angiogenesis.48,49 lnterestingly, the ephrins are not soluble ligands, but they are membrane attached and their binding to the receptor requires cell–cell contacts.48 A bidirectional signaling between ligand and receptor is suggested.60 The ephrin-B2 ligand was located strictly on arterial endothelial cells, while the corresponding Eph-B4 receptor marked only venous endothelial cells. Analysis of ephrin-B2 knockout mice showed vascular defects comparable to those of the Ang-1 or tie-2 receptor-deficient mouse, such as the persistence of the primary uniform vascular plexus.48 From these studies it appeared that arterial and venous sites are at least partly genetically determined before the typical physiologic characteristics have developed. While in the control yolk sac arterial and venous vessels were alternating the ephrin-B2–deficient mice showed a strict separation of arterial and venous sides. Similar to the situation in the Ang-1/tie-2 knockout mice, periendothelial cells were missing or they remained distant to the endothelial layer.The branches of the anterior cardinal vein were also affected and lacked signs of remodeling.48 In addition, in another investigation a more widespread expression of the Eph-B receptors has been suggested. The expression of Eph-B receptors and the ephrin-B2 ligand on periendothelial cells was demonstrated and a possible role of the Eph-B/ephrin-B system in remodeling of the primary vascular plexus confirmed.49 To investigate whether the function of endothelial ephrin-B2 can be compensated by ephrin-B2 that is expressed on perivascular cells, transgenic mice were generated in which ephrin-B2 is specifically deleted in the endothelium and endocardium of the developing vasculature and heart. The resulting phenotype corresponded to the one of the ephrin-B2 null mutants, indicating that periendothelial ephrin-B2 expression is not sufficient to compensate for the endothelial and endocardial ephrin-B2 loss.61
lnteractions of VEGFs, angiopoietins, and ephrins
Taken together these data show that VEGF might be one of the leading gowth factors involved in the regulation of vasculogenesis. The Angiopoietin/tie system is activated later and cooperates with VEGF during the period of angiogenesis. While VEGF promotes endothelial cell migration, proliferation, and tube formation, its effect on intussusception still is not known. Yet the flt-1−/−and the VEGF+/− phenotypes resemble those of the Ang-l /tie-2 knockout. Whether this could be derived from a disturbance of the cellular mechanisms of IMG needs further investigation. Ang-1 and its tie-2 receptor are clearly involved in the regulation of IMG, as has been established in two detailed studies.18,46 Concerning the role of Ang-2 and its cooperaion w ith VEGI, the interpretation of the present data has suggested that endothelial sprouting be promoted. This is based on the conclusion that Ang-2, because it interferes with the effect of Ang-1 and is coexpressed with VEGF in areas of vessel growth, must enhance the established function of VEGF, which is to promote endothelial sprouting.59 However, this implies that VEGF is not effective in the mechanisms of IMG. Further, because the vessels that developed during the ovarian cycle reflect the morphology of loops rather than of single blind-ending sprouts that are not perfused, it can be imagined that in situ loop formation by IMG could be a relevant mechanism. These questions will of course need careful investigation in the future by analysis of serial sections at the histologic and ultrastructural level.
Interestingly, the Ang/tie system and the ephrin-B/Eph-B system have similar roles in angiogenesis. The Angiopoietins might transport signals from mesenchymal cells to endothelial cells through a larger intercellular space to ensure their “communication and connection.” This facilitates varying mechanisms of IMG that require the involvement of periendothelial cells to guarantee the formation of stable tissue folds and ITSs for vascular network formation, growth, and remodeling. During IMG the endothelial layer actually retreats toward the mesenchyme to form tissue folds and loops. The ephrin-B/Eph-B system could exert a similar effect. In addition, it allows for “communication” between endothelial cells of opposite sides of the circulation, subsequently establishing direct cellular connections between the arterial and venous sides. These arterial and venous endothelial cells come into contact during IMG within the capillary areas of the circulation that lack supporting cells.39,62
Examples of other regulators of angiogenesis
Many of the junctional complexes between endothelial cells and endothelial and periendothelial cells that not only connect these cells, but also allow for their communication, are of the gap junction type. Transgenic mice deficient in the gap junction protein connexin 45 (Cx45) exhibited striking abnormalities in angiogenesis and died between E9.5 and E10.5. While vasculogenesis appeared to be unaffected, the subsequent remodeling of the primitive vascular plexus to form vessels of different sizes, especially in the yolk sac, did not occur. ln addition, smooth muscle cells were absent from most major arteries throughout the embryo.61 These defects emphasize the importance of the communication between the endothelial and periendothelial cell layers as it is especially required for IMG.
Matrix metalloproteinases (MMPs) are essential for extracellular matrix (ECM) remodeling by proteolytic cleavage of growth factors, degradation of the ECM to promote cellular migration, and regulated receptor cleavage to terminate migratory signaling.63 Recently, RECK, a membrane-anchored glycoprotein, that inhibits three MMPs (MMP-9, MMP-2, and MTI-MMP) and is widely expressed in mesenchymal and vascular smooth muscle cells, was found to be essential for mouse development. RECK−/− mice died around E10.5. RECK deficiency caused increased proteolysis of collagen and defects in the basal lamina with subsequent failure of remodeling of the primitive vascular plexus to form a mature one. In contrast, RECK overexpression impaired tumor angiogenesis, likely because ECM remodeling was compromised.64 This indicates that a tight balance between degradation and synthesis of collagen fibers is required for normal vascular remodeling (collagen fibers are an important component of ITS and pillar cores).
The Notch gene family encodes large transmembrane receptors that are involved in intercellular signaling. Notch1 and Notch4 are expressed in endothelial cells in the embryonic vasculature.65 Notchl −/− and Notchl/Notch4 double-mutant embryos displayed severe defects in angiogenesis, as indicated by the persistence of the primitive vascular plexus in the yolk sac, disorganized intersomitic vessels, a collapse of the dorsal aortae and the anterior cardinal veins.66 These defects are similar to the ones observed in tie-2-, Angiopoietin−/−, and ephrin B2/EphB4–deficient embryos.
Integrins, a family of heterodimeric transmembrane proteins comprising at least 16 α and 8 β units, which form cell adhesion receptors to the ECM, play an important role in vascular morphogenesis. The role of integrins appears to be complex. While integrin ligation supports specific cell–ECM interactions for cell adhesion and migration, it also induces a wide range of intracellular signaling events, many of which are also activated after growth factor receptor binding.67,68 For example, the pro-angiogenic function of basic fibroblast growth factor (FGF-2) seems to be dependent on integrin avB3 ligation.67 Recent progress in the identification of the transcriptional regulation of vascular development has demonstrated that the homebox gene Hox D3 is induced in endothelial cells as a response to FGF-2 treatment.69 Correspondingly the overexpression of Hox D3 causes integrin avB3 expression.70 Important roles for the EPAS/hypoxia-induclble factor 2a and for the oncogenic LIM-only transcription factor Lmo2 concerning the remodeling of the primitive vascular plexus during angiogenesis could be determined by analyses of mutant mice.71
Angiogenesis in the Pathologic State
Cellular mechanisms
Angiogenesis is induced in many pathologic conditions such as wound healing, chronic inflammation, atherosclerosis, and tumors. Blood vessel growth in tumors has been extensively investigated, as it was shown that tumors are angiogenesis dependent.3,72 Further, it was demonstrated that tumors switch to an angiogenic phenotype by starting to release angiogenic growth factors.73 So far, tumor angiogenesis, along with angiogenesis in other pathologic states, is thought to be implemented by the process of normal angiogenesis, that is, by endothelial sprouting.36 What is the difference between physiologic angiogenesis and angiogenesis in tumors? The switch to the angiogenic phenotype and the recent discovery of potent anti-angiogenic agents as thrombospondin,74,75 angiostatin,76 endostatin,77 and vasostatin78 implicate that a net balance of promoters and inhibitors regulates blood vessel growth. This balance can shift toward a pathologic situation, typically detected in tumors, in which negative regulators are decreased while positive ones prevail.79–81 This would explain why tumor angiogenesis is an unlimited process. However, what is the nature of the imbalance between promoters and inhibitors? Is there a structural correlate to the unbalanced vessel growth or are blood vessels just more frequently formed in tumors? The pathologic parameters of the tumor circulation suggest that blood vessel growth in tumors is a disturbed process.82 The comparison between healing wounds and growing to the composition of their stroma revealed many similarities and differences of both systems and led to the characterization of tumors as “wounds that do not heal.”83,84 The similarity of gene expression profiles of endothelial cells derived from tumors and from physiologic angiogenesis in the adult as compared to endothelial cells of normal tissue suggests, however, that endothelial cells are not the primary source of differences between the more physiologic implementation of angiogenesis in tissue repair as compared to tumor angiogenesis.85
Patan and colleagues,17 by using in vivo video microscopy, demonstrated that intussusceptive microvascular growth is an alternative mechanism of tumor angiogenesis. In these studies it is shown that angiogenesis in tumors follows the normal principles of intussusceptive growth, but also consists of pathologic differences. The formation of tissue pillars and ITSs occurs more frequently and includes sequences of contradictory steps (formation of segments followed by their occlusion) that are implemented in much shorter periods of time as compared to the situation in the embryo.16,17
In the tissue surrounding the tumor or distant to the wound suture in wound healing, venules and smaller veins expanded the previously existing vascular network by the process of segmentation. Segmentation started with intraluminal fibrin deposits that became ensheathed by migrating endothelial cells to form tiny folds. These folds were stabilized by collagen fibrils and connected in the center of the vessel lumen in a spoke-wheel–like pattern. ITSs frequently separated from them. Thus segmentation was also based on IMG and was even detected in large arteries.39 In tissue repair, the growth of the preexistent network established connections to the newly formed loop systems that were not detected in the tumors. In the latter, but not in wound healing, segmentation also consisted of pathologic variations that caused formation of blind ending tubes.38,39 Thus the wound, unlike the tumor, forms a network of perfectly connected vessels that facilitates perfusion and reoxygenation. Hypoxia-induced angiogenesis subsequently ceases in wound healing. In the tumors, the pathologic structure of the vascular network facilitates abnormal circulatory conditions and likely perpetuates hypoxia-driven angiogenesis that could also mediate the dominance of positive regulators over negative ones. These data demonstrate that the pathophysiologic conditions of the tumor circulation are based on an abnormal vascular network structure that is derived from pathologic mechanisms of vessel formation and growth.38,39 The latter could likely be the consequence of tumor cell invasion of blood vessel walls and the subsequent replacement of endothelial cells by periendothelial tumor cells to form mosaic vessels.86
A body of literature has recently demonstrated evidence for the involvement of circulating endothelial precursor cells (CEPs) derived from the bone marrow in adult neovascularization.87–89 Especially in ischemic tissues, CEPs are thought to contribute to angiogenesis as an alternative to endothelial cell proliferation. In angiogenic-defective, tumor-resistant Id-mutant mice, tumor vascularization could be restored after transplantation of wild-type bone marrow or VEGF-mobilized stem cells. The newly formed tumor blood vessels were largely composed of donor CEPs.90
Molecular regulation
The literature on the role of growth factors that induce and promote angiogenesis in tumors and in other pathologic conditions is increasing on daily. The VEGFs and the FGFs are among the most extensively characterized ones. VEGF was even originally detected in tumors and characterized as vascular permeability factor (VPF) based on its ability to increase vascular permeability.91 Hypoxia is prevalent in tumors, and VEGF as well as FGF expression is up-regulated under hypoxic conditions.92,93 Thus it is not surprising that members of both growth factor families are expressed by many tumors in vivo and in vitro.47,91,94,95 Further, interfering with the VEGF pathway in tumors by application of neutralizing antibodies96 or by retrovirus-mediated expression of a dominant negative flk-1 mutant suppresses the growth of many tumor cell lines in vivo.97,98 Similarly, high local expression of sflt-l inhibits tumor growth and metastases.99,100 The Angiopoietins and tie receptors have also been implicated in angiogenesis in the pathologic state. It has been demonstrated that targeting the tie-2 receptor in tumors using a soluble tie-2 receptor delivered by an adenoviral vector reduces tumor growth and metastases.101 In addition, it was shown in melanomas that the VEGF receptor pathway and the tie-2 pathway are essential for melanoma growth. However, because the inhibition of one pathway could not be compensated by the other, it was suggested that both systems are independent mediators.102 During early tumor development, Ang-2 is detected in host vessels that are co-opted by tumor cells and subsequently regress. At a later stage, Ang-2 is coexpressed with VEGF’ at the tumor margin where angiogenesis is dominant.103 The theory of early host vessel co-option has been challenged by data that demonstrated signs of IMG (tissue pillars and ITSs) in host vessels that were surrounded by tumor cells 3 to 4 days after tumor implantation. These vessels appeared to form an initial anchor point for tumor cells, the majority of the latter was still migrating in the area of implantation.39 Host vessel growth toward extremely small tumor aggregates (<l mm3) with the simultaneous expression of VEGF, VEGFR-2 and Ang-2 has been demonstrated. Surrounding host blood vessels were invaded by tumor cells rather than being co-opted.104 All these data are derived from the study of tumor xenografts in which cells of already highly metastatic tumors (that produce angiogenic factors) are injected into the so far healthy animal. Thus the early events of tumor development in the xenograft might more likely correspond to the physiology of metastatic growth. It can be imagined that all described phenomena coexist. More relevant for the investigation of early tumor physiology is the analysis of spontaneous developing tumors in transgenic mouse models that reveal an induction of angiogenesis in premalignant lesions.105 Angiogenesis depends on vascular endothelial growth factor (VEGF) for initiation and platelet-derived growth factor (PDGF) for maintenance of blood vessels.106 Sun and colleagues107 designed a novel molecule, GFB-204, that binds PDGF and VEGF, blocks binding of PDGF and VEGF to their receptors and subsequently inhibits PDGFR and Flk-1 tyrosine phosphorylation. GFB-204 is selective for PDGF and VEGF and does not inhibit EGF, IGF-1, and FGF stimulation of Erk1/2, Akt, and STAT3. GFB-204 inhibits endothelial cell migration and capillary network formation in vitro. Treatment of mice with GFB-204 suppresses human tumor growth and angiogenesis.
The concept of angiogenesis-dependent tumor growth has been complicated by the demonstration that tumor cells deficient in the p53 tumor suppressor gene (which is inactivated in most human cancers) display a diminished rate of apoptosis under hypoxia, which might render them independent of vascular supply and less responsive to anti-angiogenic strategies.108 Interestingly, the tie-1 receptor is expressed during hypoxia and as a response to VEGF in vitro.109 Tie-1 was also detected in large vessels close to the wound suture on day 3 in wound healing and on day 7 in the small vessels of the neovasculature that course throughout the wound.109 Flt-1 showed corresponding expression pattern in respect to time and location.109 These findings match the onset of formation of loops in the wall of large veins and their further remodeling.38,39 Recent studies demonstrated the expression of Ang-1 by pericytes during cutaneous wound healing, comparable to the embryonic situation.110 Concerning angiogenesis in ischemic tissues, bFGF and VEGF-A with its two tyrosine kinase receptors are increased in myocytes and macrophages of the ischemic myocardium.111–113 Today the first clinical trials using VEGF and FGF to induce neovascularization in ischemic tissues are in progress.47,114,115
It thus appears that the most important regulators of embryonic angiogenesis play very similar roles in angiogenesis in the adult under pathologic conditions. However, their precise function in respect to the mechanisms of physiologic and pathologic angiogenesis has still to be determined. Further, the detection of potent angiogenic inhibitors in tumors that interfere with tumor growth and metastasis74–76,78–81,116–118 enhances the chances that the imbalance between promoters and inhibitors of angiogenesis will be further characterized and anti-angiogenic therapy of human tumors and other pathologic conditions will be successful in the future.
MENINGIOMA ANGIOGENESIS
Meningiomas are vascular tumors and display the fundamental rules of angiogenesis.5,119–122 Therefore, inhibition of angiogenesis is a promising field of research, especially for the treatment of recurrent meningiomas.123,124 Meningiomas derive their blood supply predominantly from meningeal vessels originating in the external carotid circulation. An additional supply from cerebral–pial vessels is present in approximately 60% of patients.6 Meningiomas are of variable vascularity, ranging from sparsely vascularized to highly vascular angiomatous meningiomas.7–9 They further display variable degrees of peritumoral brain edema, ranging from absent to life-threatening conditions.10 In contrast to gliomas, there is no scientifically proven association between edema and histologic grade in meningiomas.11 Nevertheless, the extent of angiogenic activity during the meningioma formation and progression is certainly important in the surgical and endovascular management and recurrence of these tumors, regardless of their anatomic location.
In the last 10 years, investigators studying angiogenic mechanisms in meningiomas documented expression of the angiogenic factors fibroblast growth factor-2, VEGF, and PDGF.11,125–127 Reports have also noted VEGF expression in association with expression of its cognate receptors in endothelial cells, thus supporting a paracrine mechanism of neoangiogenesis in meningiomas.128,129 However, growth factors are not the only regulators of angiogenesis. A network of interactions among these cytokines, matrix proteins, and vascular cells is key to the initiation and regulation of angiogenic events. For example, tenascin is one of the matrix proteins in the angiogenesis-promoting microenvironment and may play an important role in meningioma vascularization.5 Endothelial cells in vitro attach to the tenascin substrate, where they become elongated and form interconnecting processes. Such changes do not occur when endothelial cells are grown on other matrix proteins, such as fibronectin, collagen, or laminin.40,41,68 This suggests that tenascin may promote the proliferation and motility of these cells. Research has also shown that attachment of endothelial cells to tenascin is mediated by annexin and integrins, including alpha v beta 3 integrin, which is required for angiogenesis. Bello and colleagues130 proposed that alpha v beta 3 integrin in meningiomas promotes endothelial cell adhesion and migration through interaction with vitronectin and tenascin. In vivo and in situ studies of endothelial cells in human brain microvessels have also demonstrated that vascular cells are involved in neovascular proliferation and that they contribute to the deposition of tenascin. A correlation was shown between tenascin expression and VEGF expression in meningioma. This relationship suggests that tenascin is a nonspecific angiogenic matrix molecule that may play a role in angiogenesis in these tumors.5
The “edema” that surrounds meningiomas has been studied by a number of authors (see Chapter 9).6,119–122 Age and sex of the patient, tumor size, location, aggressiveness, vascularity, invasiveness, and the compression of vascular structures do not appear to correlate with peritumoral edema. However, angioblastic subtype, presence of progesterone receptors, and proliferation index in meningioma are all correlated with edema, which may reflect a breakdown of the blood–brain barrier as well as secretion of material by the tumor itself.131 Hirashima and colleagues132 identified PDGF as important in the development of peritumoral edema. Data presented by Goldman and colleagues133 and Yoshioka and colleagues134 suggest that VEGF may be the main factor in meningioma-associated edema. In 16 meningiomas analyzed by Kalkanis and colleagues135 and 18 analyzed by Provias and colleagues,10 the tumors with surrounding edema contained significantly higher levels of VEGF mRNA than those with no associated edema. A study of 15 meningiomas by Quindlen and Bucher136 showed a correlation between volume of peritumoral edema and tissue plasminogen activator content. The results of these investigations suggest that meningiomas with peritumoral edema exhibit greater proliferative activity and produce higher amounts of vasotrophic and vasoactive factors. In a study of 135 patients, Mantle and colleagues137 identified peritumoral edema as a potential radiologic marker of brain invasion and suggested that invaded brain tissue is a possible source of residual cells in tumor recurrence after gross total resection.
Vascular endothelial growth factor-A (VEGF-A), which has also been termed vascular permeability factor, is considered a key regulator of angiogenesis and edema formation. The VEGF-A mRNA is expressed by meningioma cells.10,133 Several studies demonstrated that VEGF-A levels in meningiomas are associated with the extent of peritumoral edema.6,10,133 Moreover, meningiomas with striking VEGF-A expression usually appeared to receive some blood supply through cerebral–pial arteries, and both VEGF-A expression as well as cerebral–pial blood supply were associated with the extent of brain edema.6
Two small studies suggested that VEGF-A mRNA expression may correlate with meningioma vascularity.10,138 However, when determining VEGF-A protein levels in a relatively large number of 69 meningiomas, no association with microvessel density could be confirmed.11 Moreover, and in contrast to gliomas, there was no association between microvessel density and histological grade. Nevertheless, VEGF-A levels were increased 10-fold in anaplastic meningiomas and 2-fold in atypical meningiomas compared to benign ones. VEGF-A contained in protein extracts of human meningiomas induced capillary-like tube formation and migration of endothelial cells in vitro, indicating biological activity in this context.11 Another study reported a correlation between VEGF-A protein expression and recurrence of benign meningiomas.139 Taken together, these findings suggest that VEGF-A is probably involved in vascular remodeling and angiogenesis in meningiomas, which, however, does not result in a net increase in vessel number with increasing histologic grade. Supposing that the more malignant meningiomas have a higher oxygen and metabolic demand, VEGF-A might facilitate adaptation by modulating vascular permeability. Hypoxia leading to tumor necrosis is observed only in atypical and anaplastic meningiomas. Therefore, other pathogenic mechanism may be involved in the up-regulation of VEGF in meningiomas. Overexpression of EGF and PDGF has been documented in meningiomas and postulated to contribute to their growth.140 This raises the possibility that these growth-promoting factors not only act as mitogen for tumor cells but also induce neovasculanzation by up-regulating VEGF.
Meningioma-growth has been reported to be steroid dependent.141 Further, progesterone receptor were found to be expressed in high levels in meningiomas.142 From a cellular perspective, there may be a meaningful relationship between VEGF up-regulation and progesterone levels in meningiomas. Findings in the rat female reproductive system indicate that VEGF mRNA expression is hormonally regulated in steroid-producing and steroid-responsive cells.143 These findings may suggest that female hormone receptor stimulation might increase VEGF expression in meningiomas.
It was shown that meningiomas express P1GF and VEGF-B.144,145 Weindel and colleagues146 also reported expression of P1GF in some meningiomas. Both VEGF- related molecules bind with high affinity to VEGFR-1 and are not up-regulated to hypoxia.147 Although the biologic significance of these observations remain to be elucidated, it is tempting to speculate that meningiomas may use distinct repertoires of VEGF and VEGF-related molecules to stimulate angiogenesis and induce edema formation. This would explain the heterogeneous expression of VEGF observed in these tumors. Pistolesi and colleagues148,149 speculated that the microvessel pattern could underlie a higher metabolic demand, probably due to a rapid growth with a consequent worse clinical behaviour of the tumor. In this sense, the vascular pattern may be used as a prognostic factor, in order to focus attention mostly on the grade I meningiomas that have a higher likelihood of either recurrence or development of perilesional edema. The pattern of vasculature itself seems to be dependent on the types of VEGF isoforms: the grade II and III meningiomas (that presented numerous microvessels) expressed the soluble isoforms 121 and 165, while isoform 189 was more frequently detected in grade I meningiomas.
Several other growth factors, including VEGF-B, placenta growth factor, scatter factor/hepatocyte growth factor, and fibroblast growth factor-2 have also been analyzed in meningiomas. However, no clear correlation between either of these factors and meningioma angiogenesis or malignancy grade has yet been established. Moreover, expression of several other growth factors and their receptors, including epidermal growth factor, platelet-derived growth factor, and insulin-like growth factor and others has been analyzed in meningiomas.120,150
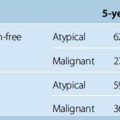
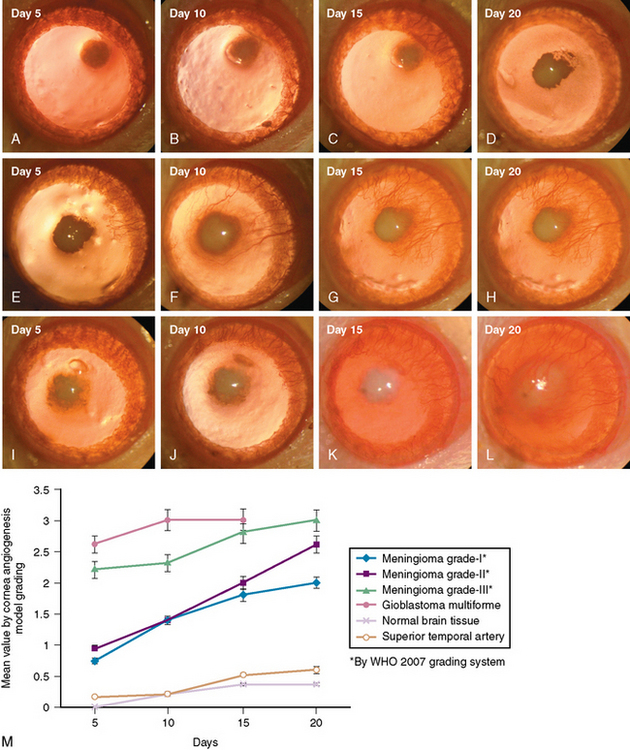
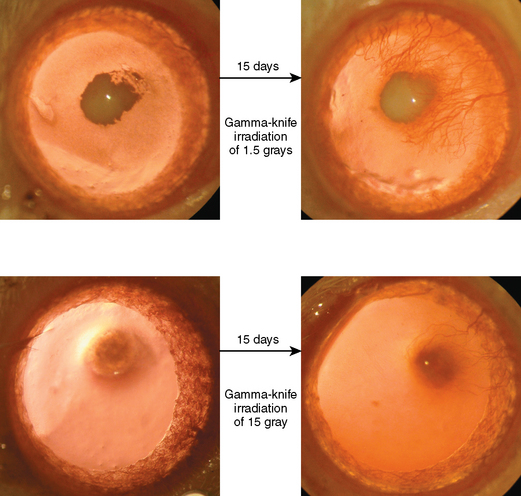
Our laboratory recent data using cornea angiogenesis model provides in vivo information that histologic grading in meningiomas is definitely determines its angiogenetic activity (Fig. 10-1).34 Figure 10-2 displays that angiogenic activity of malignant meningiomas can be compared only with the angiogenic activity of glioblastoma multiforme, which has the highest potential among neoplasms regarding neovascularization. Angiogenic activity of recurrent meningiomas is currently being tested in our laboratory. One other clinically important laboratory finding is the gamma-knife inhibition of angiogenesis. Our recent data showed that focused irradiation of gamma-knife radiosurgery dose dependently inhibits neovascularization induced by tissue of arteriovenous malformation in the cornea angiogenesis model.151 Similarly, preliminary data provide proof that stereotactic gamma-knife irradiation inhibits neovascularization around meningioma tissue embedded in the rat cornea (see Fig. 10-2).
[1] Dvorak H.F. Angiogenesis: update 2005. J Thromb Haemost. 2005;3:1835-1842.
[2] Algire G. The Biology of Melanomas. New York: New York Academy of Sciences Press, 1947.
[3] Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182-1186.
[4] Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932-936.
[5] Kilic T., et al. Tenascin in meningioma: expression is correlated with anaplasia, vascular endothelial growth factor expression, and peritumoral edema but not with tumor border shape. Neurosurgery. 2002;51:183-192. discussion 192–3
[6] Bitzer M., et al. Angiogenesis and brain oedema in intracranial meningiomas: influence of vascular endothelial growth factor. Acta Neurochir (Wien). 1998;140:333-340.
[7] Pamir M.N., Ozduman K., Belirgen M., Kilic T., Ozek M.M. Outcome determinants of pterional surgery for tuberculum sellae meningiomas. Acta Neurochir (Wien). 2005;147:1121-1130. discussion 1130
[8] Pamir M.N., Kilic T., Bayrakli F., Peker S. Changing treatment strategy of cavernous sinus meningiomas: experience of a single institution. Surg Neurol. 2005;64(Suppl. 2):S58-66.
[9] Pamir M.N., Kilic T., Ozduman K., Ture U. Experience of a single institution treating foramen magnum meningiomas. J Clin Neurosci. 2004;11:863-867.
[10] Provias J., et al. Meningiomas: role of vascular endothelial growth factor/vascular permeability factor in angiogenesis and peritumoral edema. Neurosurgery. 1997;40:1016-1026.
[11] Lamszus K., et al. Vascular endothelial growth factor, hepatocyte growth factor/scatter factor, basic fibroblast growth factor, and placenta growth factor in human meningiomas and their relation to angiogenesis and malignancy. Neurosurgery. 2000;46:938-947. discussion 947–8
[12] Miquerol L., Gertsenstein M., Harpal K., Rossant J., Nagy A. Multiple developmental roles of VEGF suggested by a LacZ-tagged allele. Dev Biol. 1999;212:307-322.
[13] Poole T.J., Coffin J.D. Vasculogenesis and angiogenesis: two distinct morphogenetic mechanisms establish embryonic vascular pattern. J Exp Zool. 1989;251:224-231.
[14] Cockerill G.W., Gamble J.R., Vadas M.A. Angiogenesis: models and modulators. Int Rev Cytol. 1995;159:113-160.
[15] Coffin J.D., Harrison J., Schwartz S., Heimark R. Angioblast differentiation and morphogenesis of the vascular endothelium in the mouse embryo. Dev Biol. 1991;148:51-62.
[16] Patan S., Haenni B., Burri P.H. Implementation of intussusceptive microvascular growth in the chicken chorioallantoic membrane (CAM). Microvasc Res. 1997;53:33-52.
[17] Patan S., Munn L.L., Jain R.K. Intussusceptive microvascular growth in a human colon adenocarcinoma xenograft: a novel mechanism of tumor angiogenesis. Microvasc Res. 1996;51:260-272.
[18] Patan S. TIE1 and TIE2 receptor tyrosine kinases inversely regulate embryonic angiogenesis by the mechanism of intussusceptive microvascular growth. Microvasc Res. 1998;56:1-21.
[19] Risau W. Angiogenesis and endothelial cell function. Arzneimittelforschung. 1994;44:416-417.
[20] Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671-674.
[21] Reagan F. Vascularization phenomenain fragments of embryonic bodiescompletely isolated from yolk-sac blastoderm. Anat Rec. 1915;9:329-341.
[22] His W. Untersuchungen über die erste Anlage des Wirbelthierliebes. Leibzig, 1868.
[23] Sabin F. Studies on the origin of blood-vessels and of red blood-corpuscles as seen in the living blastoderm of chicks during the second day of incubation. Contributions to Embryology. 1920.
[24] Risau W., Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73-91.
[25] Pardanaud L., Yassine F., Dieterlen-Lievre F. Relationship between vasculogenesis, angiogenesis and haemopoiesis during avian ontogeny. Development. 1989;105:473-485.
[26] Pardanaud L., et al. Two distinct endothelial lineages in ontogeny, one of them related to hemopoiesis. Development. 1996;122:1363-1371.
[27] Flamme I., Breier G., Risau W. Vascular endothelial growth factor (VEGF) and VEGF receptor 2 (flk-1) are expressed during vasculogenesis and vascular differentiation in the quail embryo. Dev Biol. 1995;169:699-712.
[28] Flamme I. Is extraembryonic angiogenesis in the chick embryo controlled by the endoderm? A morphology study. Anat Embryol (Berl). 1989;180:259-272.
[29] Kremer C., Breier G., Risau W., Plate K.H. Up-regulation of flk-1/vascular endothelial growth factor receptor 2 by its ligand in a cerebral slice culture system. Cancer Res. 1997;57:3852-3859.
[30] Shalaby F., et al. Failure of blood-island formation and vasculogenesis in Flk-1–deficient mice. Nature. 1995;376:62-66.
[31] Breier G., Clauss M., Risau W. Coordinate expression of vascular endothelial growth factor receptor-1 (flt-1) and its ligand suggests a paracrine regulation of murine vascular development. Dev Dyn. 1995;204:228-239.
[32] Ferrara N., et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439-442.
[33] Harris C. The Heart and Vascular System in Ancient Greek medicine. Oxford: Clarendon Press, 1973.
[34] Konya D., et al. Testing the angiogenic potential of cerebrovascular malformations by use of a rat cornea model: usefulness and novel assessment of changes over time. Neurosurgery. 2005;56:1339-1345. discussion 1345–6
[35] Ausprunk D.H., Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc Res. 1977;14:53-65.
[36] Folkman J. How is blood vessel growth regulated in normal and neoplastic tissue? G.H.A. Clowes memorial Award lecture. Cancer Res. 1986;46:467-473.
[37] Folkman J. Tumor angiogenesis. Adv Cancer Res. 1985;43:175-203.
[38] Patan S., et al. Vascular morphogenesis and remodeling in a human tumor xenograft: blood vessel formation and growth after ovariectomy and tumor implantation. Circ Res. 2001;89:732-739.
[39] Patan S., et al. Vascular morphogenesis and remodeling in a model of tissue repair: blood vessel formation and growth in the ovarian pedicle after ovariectomy. Circ Res. 2001;89:723-731.
[40] Kilic T., et al. Expression of structural proteins and angiogenic factors in normal arterial and unruptured and ruptured aneurysm walls. Neurosurgery. 2005;57:997-1007. discussion 997–1007
[41] Kilic T., et al. Expression of structural proteins and angiogenic factors in cerebrovascular anomalies. Neurosurgery. 2000;46:1179-1191. discussion 1191–2
[42] Harrigan M.R. Angiogenic factors in the central nervous system. Neurosurgery. 2003;53:639-660. discussion 660–1
[43] Zadeh G., Guha A. Angiogenesis in nervous system disorders. Neurosurgery. 2003;53:1362-1376.
[44] Sumpio B.E., et al. Regulation of PDGF-B in endothelial cells exposed to cyclic strain. Arterioscler Thromb Vasc Biol. 1998;18:349-355.
[45] Nagel T., Resnick N., Dewey C.F.Jr, Gimbrone M.A.Jr. Vascular endothelial cells respond to spatial gradients in fluid shear stress by enhanced activation of transcription factors. Arterioscler Thromb Vasc Biol. 1999;19:1825-1834.
[46] Suri C., et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171-1180.
[47] Ware J.A., Simons M. Angiogenesis in ischemic heart disease. Nat Med. 1997;3:158-164.
[48] Wang H.U., Chen Z.F., Anderson D.J. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93:741-753.
[49] Adams R.H., et al. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999;13:295-306.
[50] Keyt B.A., et al. Identification of vascular endothelial growth factor determinants for binding KDR and FLT-1 receptors. Generation of receptor-selective VEGF variants by site-directed mutagenesis. J Biol Chem. 1996;271:5638-5646.
[51] Carmeliet P., et al. Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat Med. 1999;5:495-502.
[52] Shibuya M. Vascular endothelial growth factor (VEGF)-Receptor2: its biological functions, major signaling pathway, and specific ligand VEGF-E. Endothelium. 2006;13:63-69.
[53] Sato T.N., Qin Y., Kozak C.A., Audus K.L. Tie-1 and tie-2 define another class of putative receptor tyrosine kinase genes expressed in early embryonic vascular system. Proc Natl Acad Sci USA. 1993;90:9355-9358.
[54] Sato T.N., et al. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376:70-74.
[55] Witzenbichler B., Maisonpierre P.C., Jones P., Yancopoulos G.D., Isner J.M. Chemotactic properties of angiopoietin-1 and -2, ligands for the endothelial-specific receptor tyrosine kinase Tie2. J Biol Chem. 1998;273:18514-18521.
[56] Koblizek T.I., Weiss C., Yancopoulos G.D., Deutsch U., Risau W. Angiopoietin-1 induces sprouting angiogenesis in vitro. Curr Biol. 1998;8:529-532.
[57] Suri C., et al. Increased vascularization in mice overexpressing angiopoietin-1. Science. 1998;282:468-471.
[58] Asahara T., et al. Tie2 receptor ligands, angiopoietin-1 and angiopoietin-2, modulate VEGF-induced postnatal neovascularization. Circ Res. 1998;83:233-240.
[59] Maisonpierre P.C., et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55-60.
[60] Davis S., et al. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science. 1994;266:816-819.
[61] Gerety S.S., Anderson D.J. Cardiovascular ephrinB2 function is essential for embryonic angiogenesis. Development. 2002;129:1397-1410.
[62] Patan S. Vasculogenesis and angiogenesis as mechanisms of vascular network formation, growth and remodeling. J Neurooncol. 2000;50:1-15.
[63] Chang C., Werb Z. The many faces of metalloproteases: cell growth, invasion, angiogenesis and metastasis. Trends Cell Biol. 2001;11:S37-43.
[64] Oh J., et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107:789-800.
[65] Del Amo F.F., et al. Expression pattern of Motch, a mouse homolog of Drosophila Notch, suggests an important role in early postimplantation mouse development. Development. 1992;115:737-744.
[66] Krebs L.T., et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343-1352.
[67] Eliceiri B.P. Integrin and growth factor receptor crosstalk. Circ Res. 2001;89:1104-1110.
[68] Seker A., et al. Expression of integrins in cerebral arteriovenous and cavernous malformations. Neurosurgery. 2006;58:159-168. discussion 159–68
[69] Oettgen P. Transcriptional regulation of vascular development. Circ Res. 2001;89:380-388.
[70] Boudreau N., Andrews C., Srebrow A., Ravanpay A., Cheresh D.A. Induction of the angiogenic phenotype by Hox D3. J Cell Biol. 1997;139:257-264.
[71] Yamada Y., Pannell R., Forster A., Rabbitts T.H. The oncogenic LIM-only transcription factor Lmo2 regulates angiogenesis but not vasculogenesis in mice. Proc Natl Acad Sci USA. 2000;97:320-324.
[72] Greenblatt M., Shubi P. Tumor angiogenesis: transfilter diffusion studies in the hamster by the transparent chamber technique. J Natl Cancer Inst. 1968;41:111-124.
[73] Folkman J., Watson K., Ingber D., Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 1989;339:58-61.
[74] Good D.J., et al. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci USA. 1990;87:6624-6628.
[75] DiPietro L.A. Thrombospondin as a regulator of angiogenesis. EXS. 1997;79:295-314.
[76] O’Reilly M.S., et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79:315-328.
[77] O’Reilly M.S. Angiostatin: an endogenous inhibitor of angiogenesis and of tumor growth. EXS. 1997;79:273-294.
[78] Pike S.E., et al. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med. 1998;188:2349-2356.
[79] Rastinejad F., Polverini P.J., Bouck N.P. Regulation of the activity of a new inhibitor of angiogenesis by a cancer suppressor gene. Cell. 1989;56:345-355.
[80] Bouck N. Tumor angiogenesis: the role of oncogenes and tumor suppressor genes. Cancer Cells. 1990;2:179-185.
[81] Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27-31.
[82] Jain R.K. Determinants of tumor blood flow: a review. Cancer Res. 1988;48:2641-2658.
[83] Dvorak H.F. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650-1659.
[84] Nagy J.A., et al. Pathogenesis of ascites tumor growth: angiogenesis, vascular remodeling, and stroma formation in the peritoneal lining. Cancer Res. 1995;55:376-385.
[85] St Croix B., et al. Genes expressed in human tumor endothelium. Science. 2000;289:1197-1202.
[86] Hammersen F., Endrich B., Messmer K. The fine structure of tumor blood vessels. I. Participation of non-endothelial cells in tumor angiogenesis. Int J Microcirc Clin Exp. 1985;4:31-43.
[87] Asahara T., et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221-228.
[88] Asahara T., et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964-967.
[89] Luttun A., Carmeliet G., Carmeliet P. Vascular progenitors: from biology to treatment. Trends Cardiovasc Med. 2002;12:88-96.
[90] Lyden D., et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194-1201.
[91] Folkman J. Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N Engl J Med. 1995;333:1757-1763.
[92] Shweiki D., Itin A., Soffer D., Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843-845.
[93] Kuwabara K., et al. Hypoxia-mediated induction of acidic/basic fibroblast growth factor and platelet-derived growth factor in mononuclear phagocytes stimulates growth of hypoxic endothelial cells. Proc Natl Acad Sci U S A. 1995;92:4606-4610.
[94] Ferrara N. Role of vascular endothelial growth factor in the regulation of angiogenesis. Kidney Int. 1999;56:794-814.
[95] Dvorak H.F., Nagy J.A., Feng D., Brown L.F., Dvorak A.M. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol. 1999;237:97-132.
[96] Kim K.J., et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841-844.
[97] Millauer B., et al. Dominant-negative inhibition of Flk-1 suppresses the growth of many tumor types in vivo. Cancer Res. 1996;56:1615-1620.
[98] Millauer B., Shawver L.K., Plate K.H., Risau W., Ullrich A. Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature. 1994;367:576-579.
[99] Kong H.L., et al. Regional suppression of tumor growth by in vivo transfer of a cDNA encoding a secreted form of the extracellular domain of the flt-1 vascular endothelial growth factor receptor. Hum Gene Ther. 1998;9:823-833.
[100] Goldman C.K., et al. Paracrine expression of a native soluble vascular endothelial growth factor receptor inhibits tumor growth, metastasis, and mortality rate. Proc Natl Acad Sci USA. 1998;95:8795-8800.
[101] Lin P., et al. Antiangiogenic gene therapy targeting the endothelium-specific receptor tyrosine kinase Tie2. Proc Natl Acad Sci USA. 1998;95:8829-8834.
[102] Siemeister G., et al. Two independent mechanisms essential for tumor angiogenesis: inhibition of human melanoma xenograft growth by interfering with either the vascular endothelial growth factor receptor pathway or the Tie-2 pathway. Cancer Res. 1999;59:3185-3191.
[103] Holash J., et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994-1998.
[104] Vajkoczy P., et al. Microtumor growth initiates angiogenic sprouting with simultaneous expression of VEGF, VEGF receptor-2, and angiopoietin-2. J Clin Invest. 2002;109:777-785.
[105] Naumov G.N., Akslen L.A., Folkman J. Role of Angiogenesis in Human Tumor Dormancy: Animal models of the Angiogenic Switch. Cell Cycle. 5, 2006.
[106] Kilic T., et al. Intracranial inhibition of platelet-derived growth factor-mediated glioblastoma cell growth by an orally active kinase inhibitor of the 2–phenylaminopyrimidine class. Cancer Res. 2000;60:5143-5150.
[107] Sun J., et al. Inhibiting angiogenesis and tumorigenesis by a synthetic molecule that blocks binding of both VEGF and PDGF to their receptors. Oncogene. 2005;24:4701-4709.
[108] Yu J.L., Rak J.W., Coomber B.L., Hicklin D.J., Kerbel R.S. Effect of p53 status on tumor response to antiangiogenic therapy. Science. 2002;295:1526-1528.
[109] McCarthy M.J., Crowther M., Bell P.R., Brindle N.P. The endothelial receptor tyrosine kinase tie-1 is upregulated by hypoxia and vascular endothelial growth factor. FEBS Lett. 1998;423:334-338.
[110] Sundberg C., Kowanetz M., Brown L.F., Detmar M., Dvorak H.F. Stable expression of angiopoietin-1 and other markers by cultured pericytes: phenotypic similarities to a subpopulation of cells in maturing vessels during later stages of angiogenesis in vivo. Lab Invest. 2002;82:387-401.
[111] Banai S., et al. Upregulation of vascular endothelial growth factor expression induced by myocardial ischaemia: implications for coronary angiogenesis. Cardiovasc Res. 1994;28:1176-1179.
[112] Arras M., et al. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J Clin Invest. 1998;101:40-50.
[113] Li J., et al. VEGF, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. Am J Physiol. 1996;270:H1803-H1811.
[114] Baumgartner I., et al. Constitutive expression of phVEGF165 after intramuscular gene transfer promotes collateral vessel development in patients with critical limb ischemia. Circulation. 1998;97:1114-1123.
[115] Laham R.J., Simons M., Sellke F. Gene transfer for angiogenesis in coronary artery disease. Annu Rev Med. 2001;52:485-502.
[116] O’Reilly M.S., et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277-285.
[117] Boehm T., Folkman J., Browder T., O’Reilly M.S. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404-407.
[118] O’Reilly M.S., Pirie-Shepherd S., Lane W.S., Folkman J. Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science. 1999;285:1926-1928.
[119] McCutcheon I.E. The biology of meningiomas. J Neurooncol. 1996;29:207-216.
[120] Sanson M., Cornu P. Biology of meningiomas. Acta Neurochir (Wien). 2000;142:493-505.
[121] Smith D.A., Cahill D.W. The biology of meningiomas. Neurosurg Clin N Am. 1994;5:201-215.
[122] Whittle I.R., Smith C., Navoo P., Collie D. Meningiomas. Lancet. 2004;363:1535-1543.
[123] Folkman J., Browder T., Palmblad J. Angiogenesis research: guidelines for translation to clinical application. Thromb Haemost. 2001;86:23-33.
[124] Folkman J. Angiogenesis inhibitors: a new class of drugs. Cancer Biol Ther. 2003;2:S127-S133.
[125] Black P., Carroll R., Zhang J. The molecular biology of hormone and growth factor receptors in meningiomas. Acta Neurochir Suppl (Wien). 1996;65:50-53.
[126] Lamszus K. Meningioma pathology, genetics, and biology. J Neuropathol Exp Neurol. 2004;63:275-286.
[127] Lamszus K., Heese O., Westphal M. Angiogenesis-related growth factors in brain tumors. Cancer Treat Res. 2004;117:169-190.
[128] Zagzag D., et al. Tenascin-C expression by angiogenic vessels in human astrocytomas and by human brain endothelial cells in vitro. Cancer Res. 1996;56:182-189.
[129] Zagzag D., et al. Tenascin expression in astrocytomas correlates with angiogenesis. Cancer Res. 1995;55:907-914.
[130] Bello L., et al. Alpha(v)beta3 and alpha(v)beta5 integrin expression in meningiomas. Neurosurgery. 2000;47:1185-1195.
[131] Black P.M. Meningiomas. Neurosurgery. 1993;32:643-657.
[132] Hirashima Y., et al. Platelet-activating factor and edema surrounding meningiomas. J Neurosurg. 1998;88:304-307.
[133] Goldman C.K., et al. Brain edema in meningiomas is associated with increased vascular endothelial growth factor expression. Neurosurgery. 1997;40:1269-1277.
[134] Yoshioka H., et al. Peritumoral brain edema associated with meningioma: influence of vascular endothelial growth factor expression and vascular blood supply. Cancer. 1999;85:936-944.
[135] Kalkanis S.N., Carroll R.S., Zhang J., Zamani A.A., Black P.M. Correlation of vascular endothelial growth factor messenger RNA expression with peritumoral vasogenic cerebral edema in meningiomas. J Neurosurg. 1996;85:1095-1101.
[136] Quindlen E.A., Bucher A.P. Correlation of tumor plasminogen activator with peritumoral cerebral edema. A CT and biochemical study. J Neurosurg. 1987;66:729-733.
[137] Mantle R.E., Lach B., Delgado M.R., Baeesa S., Belanger G. Predicting the probability of meningioma recurrence based on the quantity of peritumoral brain edema on computerized tomography scanning. J Neurosurg. 1999;91:375-383.
[138] Samoto K., et al. Expression of vascular endothelial growth factor and its possible relation with neovascularization in human brain tumors. Cancer Res. 1995;55:1189-1193.
[139] Yamasaki F., et al. Recurrence of meningiomas. Cancer. 2000;89:1102-1110.
[140] Carroll R.S., et al. Expression and activation of epidermal growth factor receptors in meningiomas. J Neurosurg. 1997;87:315-323.
[141] Speirs V., Boyle-Walsh E., Fraser W.D. Constitutive co-expression of estrogen and progesterone receptor mRNA in human meningiomas by RT-PCR and response of in vitro cell cultures to steroid hormones. Int J Cancer. 1997;72:714-719.
[142] Maxwell M., Galanopoulos T., Neville-Golden J., Antoniades H.N. Expression of androgen and progesterone receptors in primary human meningiomas. J Neurosurg. 1993;78:456-462.
[143] Shweiki D., Itin A., Neufeld G., Gitay-Goren H., Keshet E. Patterns of expression of vascular endothelial growth factor (VEGF) and VEGF receptors in mice suggest a role in hormonally regulated angiogenesis. J Clin Invest. 1993;91:2235-2243.
[144] Donnini S., Machein M.R., Plate K.H., Weich H.A. Expression and localization of placenta growth factor and PlGF receptors in human meningiomas. J Pathol. 1999;189:66-71.
[145] Maxwell M., Galanopoulos T., Hedley-Whyte E.T., Black P.M., Antoniades H.N. Human meningiomas co-express platelet-derived growth factor (PDGF) and PDGF-receptor genes and their protein products. Int J Cancer. 1990;46:16-21.
[146] Weindel K., Moringlane J.R., Marme D., Weich H.A. Detection and quantification of vascular endothelial growth factor/vascular permeability factor in brain tumor tissue and cyst fluid: the key to angiogenesis? Neurosurgery. 1994;35:439-448. discussion 448–9
[147] Enholm B., et al. Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene. 1997;14:2475-2483.
[148] Pistoles S., et al. Angiogenesis in intracranial meningiomas: immunohistochemical and molecular study. Neuropathol Appl Neurobiol. 2004;30:118-125.
[149] Peker S., Abacioglu U., Bayrakli F., Kilic T., Pamir M.N. Gamma knife radiosurgery for cavernous sinus plasmacytoma in a patient with breast cancer history. Surg Neurol. 2005;63:174-176.
[150] Black P., Carroll R., Zhang J. The molecular biology of hormone and growth factor receptors in meningiomas. Acta Neurochir Suppl. 1996;65:50-53.
[151] Kilic K., et al. Inhibition of angiogenesis induced by cerebral arteriovenous malformations using gamma knife irradiation. J Neurosurg. 2007;106:463-469.