Chapter 11 Carcinogenesis and neoplasia
GENERAL CHARACTERISTICS OF NEOPLASMS (TUMOURS)




Definitions
The word tumour means literally an abnormal swelling. However, in modern medicine, the word has a much more specific meaning. A tumour (neoplasm) is a lesion resulting from the autonomous or relatively autonomous abnormal growth of cells that persists after the initiating stimulus has been removed.
Tumours result from the neoplastic transformation of any nucleated cell in the body; the transformed cells are called neoplastic cells. By transformation involving a series of genetic alterations (e.g. mutations), cells escape permanently from normal growth regulatory mechanisms. The neoplastic cells in tumours designated malignant possess additional potentially lethal abnormal characteristics enabling them to invade and to metastasise, or spread, to other tissues.
Neoplastic cells grow to form abnormal swellings (except for leukaemias), but note that swellings or organ enlargement can also result from inflammation, cysts, hypertrophy or hyperplasia.
The term neoplasm (new growth) is synonymous with the medical meaning of the word tumour and is often used in preference because it is less ambiguous and not quite so alarming when overheard by patients. Cancer is a word used more in the public arena than in medicine; it has emotive connotations and generally refers to a malignant neoplasm.
Incidence of tumours
Malignant neoplasms—those that invade and spread and are therefore of greater clinical importance—develop in approximately 25% of the human population. The risk increases with age, but tumours can occur even in infancy (Fig. 11.1). The mortality rate is high, despite modern therapy, so that cancer accounts for about one-fifth of all deaths in developed countries. However, the mortality rate varies considerably between specific tumour types.
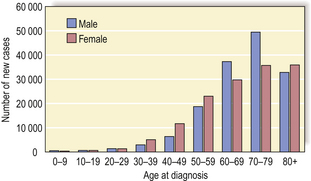
Fig. 11.1 Age and cancer incidence in England (2004). Cancer occurs at all ages but is most common over the age of 50 years.
(Based on data from the Office for National Statistics)
The relative incidence by diagnosis of various common types of cancer is shown in Fig. 11.2. Lung cancer is the most frequent malignant neoplasm in the UK and USA, and its importance is compounded by the extremely poor prognosis. In other countries other cancers are more common, and these differences often provide important aetiological clues.
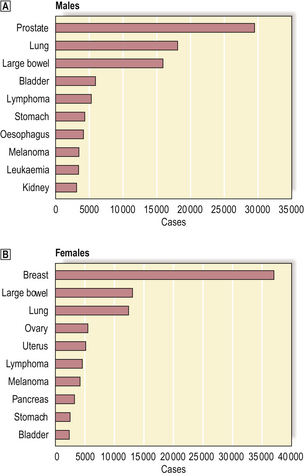
Fig. 11.2 Incidence of the ten most frequent cancers in the UK (2004).  Top ten cancers in males.
Top ten cancers in males.  Top ten cancers in females. The data exclude non-melanoma skin cancers (e.g. basal cell carcinoma) because, although common, they are so rarely fatal.
Top ten cancers in females. The data exclude non-melanoma skin cancers (e.g. basal cell carcinoma) because, although common, they are so rarely fatal.
(Data published by Cancer Research UK, 2007)
For various reasons most epidemiological data on cancer incidence probably underestimate the true incidence. Not all tumours become clinically evident and, unless a thorough autopsy is performed, may never be detected. For example, autopsy surveys have revealed a higher than expected incidence of occult carcinoma of the prostate in elderly men, although these often minute lesions are probably of little clinical consequence. Cancer incidence may also be underestimated due to a failure of detection or diagnosis in countries and communities with poor health care.
Structure of tumours
Solid tumours consist of neoplastic cells and stroma (see below and Fig. 11.3). The neoplastic cells reproduce to a variable extent the growth pattern and synthetic activity of the parent cell of origin. Depending on their functional resemblance to the parent tissue, they continue to synthesise and secrete cell products such as collagen, mucin or keratin; these often accumulate within the tumour where they are recognisable histologically. Other cell products may be secreted into the blood where they can be used clinically to monitor tumour growth and the effects of therapy (p. 234).
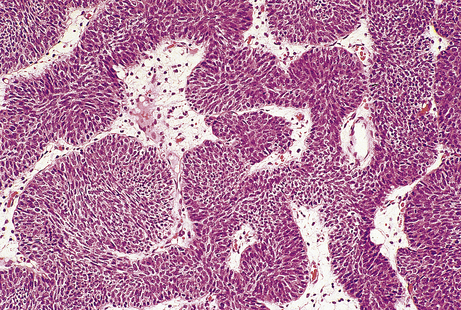
Fig. 11.3 Tumour cells and stroma. Histology of an epithelial neoplasm showing the darkly staining tumour cells embedded in a paler connective tissue stroma.
Stroma
The neoplastic cells are embedded in and supported by a connective tissue framework called the stroma (from the Greek word meaning a mattress), which provides mechanical support and nutrition to the neoplastic cells. The process of stroma formation, called a desmoplastic reaction when it is particularly fibrous, is due to induction of connective tissue proliferation by growth factors in the immediate tumour environment.
The stroma always contains blood vessels which perfuse the tumour (Fig. 11.4). The growth of a tumour is dependent upon its ability to induce blood vessels to perfuse it, for unless it becomes permeated by a vascular supply its growth will be limited by the ability of nutrients to diffuse into it, and the tumour cells will cease growing when the nodule has attained a diameter of no more than 1–2mm (Fig. 11.5). Angiogenesis in tumours is induced by factors such as vascular endothelial growth factor (VEGF). This action is opposed by factors such as angiostatin and endostatin which have potential in cancer therapy.
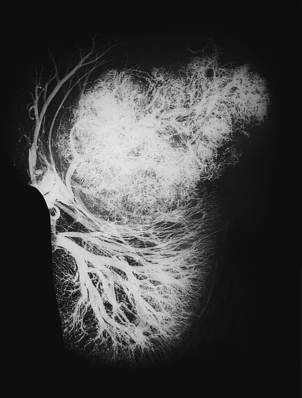
Fig. 11.4 Vascular stroma. Increased vascularity of a malignant tumour in a kidney, revealed by radiology after intra-arterial injection of X-ray contrast fluid.
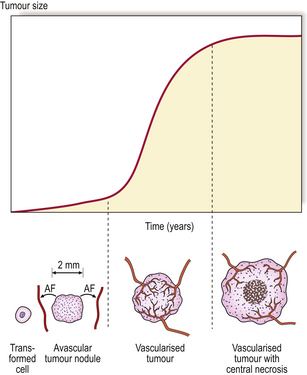
Fig. 11.5 Tumour angiogenesis. Neoplastic transformation of a single cell results in the growth of a tumour nodule, limited by the ability of nutrients to diffuse into it, to a diameter of 1–2 mm. Production of angiogenic factors (AF) stimulates the proliferation and ingrowth of blood vessels, enabling tumour growth to be supported by perfusion. Eventually, the tumour outgrows its blood supply, and areas of necrosis appear, resulting in slower growth.
Fibroblasts and the matrix they secrete give some mechanical support to the tumour cells and may in addition have nutritive properties. Stromal myofibroblasts are often abundant, particularly in carcinomas of the breast; their contractility is responsible for the puckering and retraction of adjacent structures.
The stroma often contains a lymphocytic infiltrate of variable density. This may reflect a host immune reaction to the tumour (Ch. 9), a hypothesis supported by the observation that patients whose tumours are densely infiltrated by lymphocytes tend to have a better prognosis.
Tumour shape and correlation with behaviour
The gross appearance of a tumour on a surface (e.g. gastrointestinal mucosa) may be described as sessile, polypoid, papillary, fungating, ulcerated or annular (Fig. 11.6). The behaviour of a tumour (i.e. whether it is benign or malignant) can often be deduced from its gross appearance: polypoid tumours are generally benign, i.e. unlikely to spread beyond the tissue of origin (Fig. 11.7); ulceration is more commonly associated with aggressive behaviour because invasion is the defining feature of malignancy (Fig. 11.8).
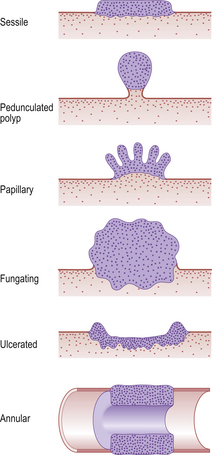
Fig. 11.6 Tumour shapes. Sessile, polypoid and papillary tumours are usually benign. Fungating, ulcerated or annular tumours are more likely to be malignant. Annular tumours encircling a tubular structure (e.g. intestine) are common in the large bowel, where they often cause intestinal obstruction.
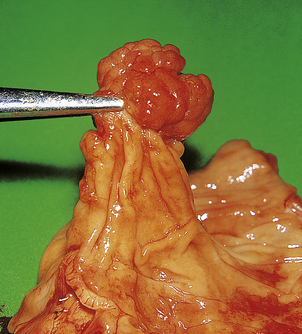
Fig. 11.7 Adenomatous polyp of the colon. This common lesion has a clearly visible stalk. Although benign, these lesions are precursors of adenocarcinoma of the large bowel. When seen endoscopically, adenomatous polyps can be removed by snaring the stalk.
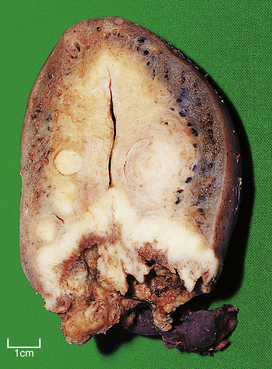
Fig. 11.8 Squamous cell carcinoma of the cervix. This uterus is invaded by a carcinoma arising in and destroying the cervix. The small round tumours in the myometrium are benign leiomyomas (‘fibroids’).
Ulcerated tumours can often be distinguished from non-neoplastic ulcers, such as peptic ulcers in the stomach, because the former tend to have heaped-up or rolled edges.
The shape of connective tissue neoplasms can be misleading. Although circumscription by a clearly defined border is one of the characteristics of benign epithelial tumours, some malignant connective tissue tumours are also well circumscribed.
Tumours are usually firmer than the surrounding tissue, causing a palpable lump in accessible sites such as the breasts. Extremely hard tumours are often referred to as ‘scirrhous’. Softer lesions are sometimes called ‘medullary’; they occur in the thyroid and breast.
The cut surfaces of malignant tumours are often variegated due to areas of necrosis and degeneration, but some, such as lymphomas and seminomas, appear uniformly bland.
Tumour histology
Neoplasms differ histologically from their corresponding normal tissue by various features; these are useful in diagnosis and include:
These features are often seen to their greatest degree in malignant neoplasms (Fig. 11.9).
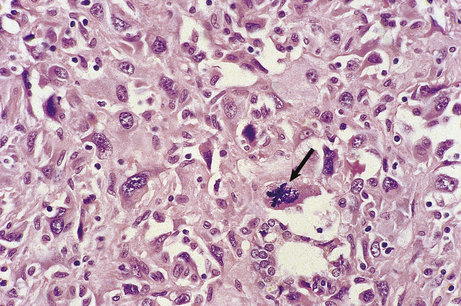
CLASSIFICATION OF TUMOURS



Tumours are classified according to their behaviour and histogenesis (cell of origin).
Behavioural classification
The behavioural classification divides tumours into:
The principal pathological criteria for classifying a tumour as benign or malignant are summarised in Table 11.1. Some tumours, such as some ovarian tumours, defy precise behavioural classification, because their histology is intermediate between that associated with benign and malignant tumours; these are often referred to as ‘borderline’ tumours.
Table 11.1 Principal characteristics of benign and malignant tumours
| Feature | Benign | Malignant |
|---|---|---|
| Growth rate | Slow | Relatively rapid |
| Mitoses | Infrequent | Frequent and often atypical |
| Histological resemblance to normal tissue | Good | Variable, often poor |
| Nuclear morphology | Often normal | Usually hyperchromatic, irregular outline, multiple nucleoli and pleomorphic |
| Invasion | No | Yes |
| Metastases | Never | Frequent |
| Border | Often circumscribed or encapsulated | Often poorly defined or irregular |
| Necrosis | Rare | Common |
| Ulceration | Rare | Common on skin or mucosal surfaces |
| Direction of growth on skin or mucosal surfaces | Often exophytic | Often endophytic |
Benign tumours



Benign tumours remain localised. They are slowly growing lesions that do not invade the surrounding tissues or spread to other sites in the body. They are often enveloped by a thin layer of compressed connective tissue (i.e. encapsulated).
When a benign tumour arises in an epithelial or mucosal surface, the tumour grows away from the surface, because it cannot invade, often forming a polyp which may be either pedunculated (stalked) or sessile; this non-invasive outward direction of growth creates an exophytic lesion (Fig. 11.10). Histologically, benign tumours closely resemble the parent cell or tissue.
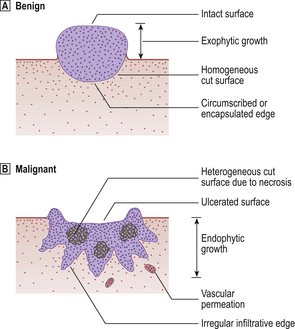
Fig. 11.10 Benign and malignant tumours growing on surfaces (e.g. skin, bowel wall), showing the principal differences in their gross appearances.
Although benign tumours are, by definition, confined to their site of origin, they may cause clinical problems due to:
Malignant tumours



Malignant tumours are, by definition, invasive. They are typically rapidly growing and poorly circumscribed. Histologically, they resemble the parent cell or tissue to a lesser extent than do benign tumours. Malignant tumours encroach on and destroy the adjacent tissues (Fig. 11.10), enabling the neoplastic cells to penetrate the walls of blood vessels and lymphatic channels and thereby disseminate to other sites. This important process is called metastasis and the resulting secondary tumours are called metastases. Patients with widespread metastases are often said to have carcinomatosis.
Not all tumours categorised as malignant exhibit metastatic behaviour. For example, basal cell carcinoma of the skin (rodent ulcer) rarely forms metastases, yet is regarded as malignant because it is highly invasive and destructive.
Malignant tumours on epithelial or mucosal surfaces may form a protrusion in the early stages, but eventually invade the underlying tissue; this invasive inward direction of growth gives rise to an endophytic tumour. Ulceration is common.
Malignant tumours in solid organs tend to be poorly circumscribed, often with strands of neoplastic tissue penetrating adjacent normal structures. The resemblance of the cut surface of these lesions to a crab (Latin: cancer) gives the disease its popular name. Malignant tumours often show central necrosis because of inadequate vascular perfusion.
The considerable morbidity and mortality associated with malignant tumours may be due to:
Histogenetic classification




Histogenesis—the specific cell of origin of an individual tumour—is determined by histopathological examination and specifies the tumour type. This is then incorporated in the name given to the tumour (e.g. squamous cell carcinoma).
Histogenetic classification includes numerous subdivisions, but the major categories of origin are:
Although some general differences exist between the main groups of malignant tumours (Table 11.2), individual lesions have to be categorised more precisely both in clinical practice and for epidemiological purposes. It is inadequate to label the patient’s tumour as merely having an epithelial or connective tissue origin; efforts must be made to determine the precise cell type. The classification of individual tumours is vitally important. Thorough histological examination of the tumour, sometimes using special techniques like genetic analysis and immunocytochemistry, detects subtle features that betray its provenance.
Table 11.2 Principal characteristics of carcinomas and sarcomas
| Feature | Carcinoma | Sarcoma |
|---|---|---|
| Origin | Epithelium | Connective tissues |
| Behaviour | Malignant | Malignant |
| Frequency | Common | Relatively rare |
| Preferred route of metastasis | Lymph | Blood |
| In situ phase | Yes | No |
| Age group | Usually over 50 years | Usually below 50 years |
Histological grade (degree of differentiation)
The extent to which the tumour resembles histologically its cell or tissue of origin determines the tumour grade (Fig. 11.11) or degree of differentiation. Benign tumours are not usually further classified in this way because they nearly always closely resemble their parent tissue and grading the degree of differentiation offers no further clinical benefit in terms of choosing the most appropriate treatment. However, the degree of differentiation of malignant tumours is clinically useful both because it correlates strongly with patient survival (prognosis), and because it often indicates the most appropriate treatment. Thus, malignant tumours are usually graded either as well, moderately or poorly differentiated, or numerically, often by strict criteria, as grade 1, grade 2 or grade 3.
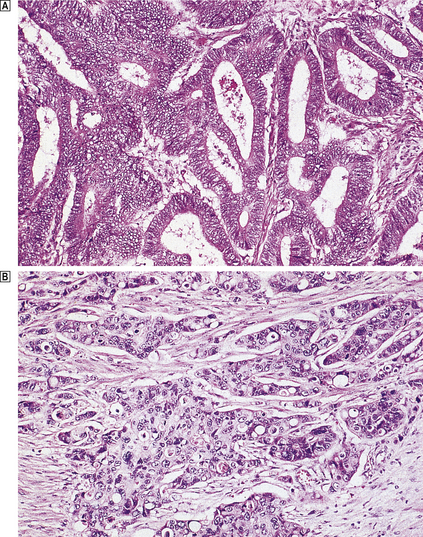
Fig. 11.11 Histological grading of differentiation.  Well-differentiated adenocarcinoma of the colon characterised by glandular structures similar to those in normal mucosa.
Well-differentiated adenocarcinoma of the colon characterised by glandular structures similar to those in normal mucosa.  Poorly differentiated adenocarcinoma of the colon characterised by a more solid growth pattern with little evidence of gland formation.
Poorly differentiated adenocarcinoma of the colon characterised by a more solid growth pattern with little evidence of gland formation.
A well-differentiated tumour more closely resembles the parent tissue than does a poorly differentiated tumour, while moderately differentiated tumours are intermediate between these two extremes. Poorly differentiated tumours are more aggressive than well-differentiated tumours.
A few tumours are so poorly differentiated that they lack easily recognisable histogenetic features. There may even be great difficulty in deciding whether they are carcinomas or lymphomas, for example, although immunocytochemistry and genetic analysis often enable a distinction to be made. Tumours defying precise histogenetic classification are often referred to as ‘anaplastic’, or by some purely descriptive term such as ‘spindle cell’ or ‘small round cell’ tumour. Fortunately, advances in diagnostic histopathology have resulted in considerably fewer unclassifiable tumours and these descriptive terms are rapidly becoming obsolete.
NOMENCLATURE OF TUMOURS





Tumours justify separate names because, although they are all manifestations of the same disease process, each separately named tumour has its own characteristics in terms of cause, appearance and behaviour. Accurate diagnosis and naming of tumours is essential so that patients can be optimally treated. A tumour that defies accurate classification is designated anaplastic; such tumours are always malignant.
The specific name of an individual tumour invariably ends in the suffix ‘-oma’. However, relics of this suffix’s former wider usage remain, as in ‘granuloma’ (an inflammatory aggregate of epithelioid macrophages), ‘tuberculoma’ (the large fibrocaseating lesion of tuberculosis), ‘atheroma’ (lipid-rich intimal deposits in arteries), and ‘mycetoma’ (a fungal mass populating a lung cavity) and ‘haematoma’ (mass of coagulated blood); these are not neoplasms.
There are exceptions to the rules of nomenclature that follow and these are a potential source of misunderstanding. For example, the words ‘melanoma’ and ‘lymphoma’ are both commonly used to refer to malignant tumours of melanocytes and lymphoid cells respectively, even though, from the rules of tumour nomenclature, these terms can be mistakenly interpreted as meaning benign lesions. To avoid confusion, which could be clinically disastrous, their names are often preceded by the word ‘malignant’. Similarly, a ‘myeloma’ is a malignant neoplasm of plasma cells.
The suffix for neoplastic disorders of blood cells is ‘-aemia’, as in leukaemia; but again, exceptions exist. For example, anaemia is not a neoplastic disorder.
Detailed descriptions of individual tumours are, in most instances, included in the relevant systematic chapters. Examples of tumour nomenclature are given below and, for reference, in Table 11.3.
Table 11.3 Examples of tumour nomenclature
| Type | Benign | Malignant |
|---|---|---|
| Epithelial | ||
| Squamous cell | Squamous cell papilloma | Squamous cell carcinoma |
| Transitional | Transitional cell papilloma | Transitional cell carcinoma |
| Basal cell | Basal cell papilloma | Basal cell carcinoma |
| Glandular | Adenoma (e.g. thyroid adenoma) | Adenocarcinoma (e.g. adenocarcinoma of breast) |
| Mesenchymal | ||
| Smooth muscle | Leiomyoma | Leiomyosarcoma |
| Striated muscle | Rhabdomyoma | Rhabdomyosarcoma |
| Adipose tissue | Lipoma | Liposarcoma |
| Blood vessels | Angioma | Angiosarcoma |
| Bone | Osteoma | Osteosarcoma |
| Cartilage | Chondroma | Chondrosarcoma |
| Mesothelium | Benign mesothelioma | Malignant mesothelioma |
| Synovium | Synovioma | Synovial sarcoma |
Epithelial tumours
Epithelial tumours are named histogenetically according to their specific epithelial type and behaviourally as benign or malignant.
Benign epithelial tumours
Benign epithelial tumours are either:
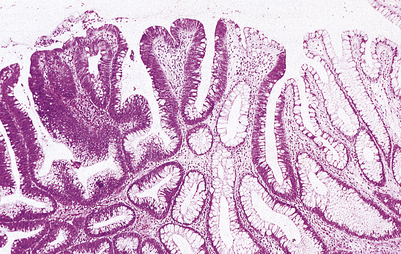
A papilloma is a benign tumour of non-glandular or non-secretory epithelium, such as transitional or stratified squamous epithelium (Fig. 11.12). An adenoma is a benign tumour of glandular or secretory epithelium (Fig. 11.13). The name of a papilloma or adenoma is incomplete unless prefixed by the name of the specific epithelial cell type or glandular origin; examples include squamous cell papilloma, transitional cell papilloma, colonic adenoma and thyroid adenoma.
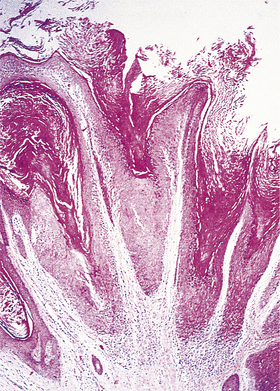
Fig. 11.12 Histology of a benign tumour of non-secretory epithelium: squamous cell papilloma. The tumour is non-invasive and grows outwards from the skin surface (i.e. it is exophytic). The tumour cells closely resemble those of the normal epidermis. This benign tumour is commonly caused by a human papillomavirus.
Malignant epithelial tumours
Malignant tumours of epithelium are always called carcinomas. Carcinomas of non-glandular epithelium are always prefixed by the name of the epithelial cell type; examples include squamous cell carcinoma and transitional cell carcinoma. Malignant tumours of glandular epithelium are always designated adenocarcinomas, coupled with the name of the tissue of origin; examples include adenocarcinoma of the breast, adenocarcinoma of the prostate and adenocarcinoma of the stomach.
Carcinomas should be further categorised according to their degree of differentiation: their resemblance to the tissue of origin.
Carcinoma in situ
The term carcinoma in situ refers to an epithelial neoplasm exhibiting all the cellular features associated with malignancy, but which has not yet invaded through the epithelial basement membrane separating it from potential routes of metastasis—blood vessels and lymphatics (Fig. 11.14). Complete excision at this very early stage will guarantee a cure. Detection of carcinomas at the in situ stage, or of their precursor lesions, is the aim of population screening programmes for cervical, breast and some other carcinomas. The phase of in situ growth may last for several years before invasion commences.
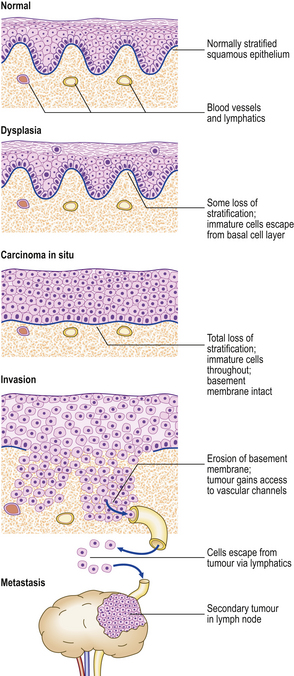
Fig. 11.14 Evolution of an invasive squamous cell carcinomafrom the precursor lesions of dysplasia and carcinoma in situ (usually grouped together as intra-epithelial neoplasia). Note that the tumour cells cannot reach routes of metastasis such as blood vessels and lymphatics until the basement membrane has been breached.
Carcinoma in situ may be preceded by a phase of dysplasia, in which the epithelium shows disordered maturation short of frank neoplasia. Some dysplastic lesions are almost certainly reversible. As there are other applications of the word ‘dysplasia’, as well as some difficulty in reliably distinguishing between carcinoma in situ and dysplasia in biopsies, the term is now less favoured. The term ‘intra-epithelial neoplasia’, as in cervical intra-epithelial neoplasia (CIN), is used increasingly to encompass both carcinoma in situ and the precursor lesions formerly known as dysplasia.
Connective tissue and other mesenchymal tumours
Tumours of connective and other mesenchymal tissues are, like epithelial tumours, named according to their cell of origin and their behavioural classification.
Benign connective tissue and mesenchymal tumours
Benign mesenchymal tumours are named after the cell or tissue of origin suffixed by ‘-oma’, as follows:
Malignant connective tissue and mesenchymal tumours
Malignant tumours of mesenchyme are always designated sarcomas, prefixed by the name that describes the cell or tissue of origin. Examples include:
As with carcinomas, sarcomas can be further categorised according to their grade or degree of differentiation (Fig. 11.15).
Eponymously named tumours
Some tumours have inherited the name of the person who first recognised or described the lesion. Examples include:
Miscellaneous tumours
Most tumours can be categorised according to the scheme of nomenclature already described. There are, however, important exceptions.
Teratomas
A teratoma is a neoplasm formed of cells representing all three germ cell layers: ectoderm, mesoderm and endoderm. In their benign form, these cellular types are often easily recognised; the tumour may contain teeth and hair, and, on histology, respiratory epithelium, cartilage, muscle, neural tissue, etc. In their malignant form, these representatives of ectoderm, mesoderm and endoderm will be less easily identifiable.
Teratomas are of germ cell origin. They occur most often in the gonads, where germ cells are abundant. Although all cells in the body contain the same genetic information, arguably in germ cells this information is in the least repressed state and is therefore capable of programming such divergent lines of differentiation. Supporting evidence for a germ cell origin for teratomas comes from karyotypic analysis of their sex chromosome content. Teratomas in the female are always XX, whereas only 50% of those in the male are XX and the remainder XY; this correlates with the sex chromosome distribution in the germ cells of the two sexes.
Ovarian teratomas are almost always benign and cystic; in the testis, they are almost always malignant and relatively solid. As germ cells in the embryo originate at a site remote from the developing gonads, teratomas arise occasionally elsewhere in the body, usually in the midline, possibly from germ cells that have been arrested in their migration. These extragonadal sites for teratomas include the mediastinum and sacro-coccygeal region.
Embryonal tumours: the ‘blastomas’
Some types of tumour occur almost exclusively in the very young, usually in those below 5 years of age, and bear a histological resemblance to the embryonic form of the organ in which they arise. Examples include:
Mixed tumours
Mixed tumours show a characteristic combination of cell types. The best example is the mixed parotid tumour (pleomorphic salivary adenoma); this consists of glands embedded in a cartilaginous or mucinous matrix derived from the myoepithelial cells of the gland. Another common mixed tumour is the fibroadenoma of the breast, a lobular tumour consisting of epithelium-lined glands or clefts in a loose fibrous tissue matrix.
The occurrence of mixed tumours in an individual organ can sometimes be predicted from its embryology. This is illustrated by the Müllerian tract tumours that occur in the female genital tract; these often contain a mixture of carcinomatous and sarcomatous elements reflecting the intrinsic capacity of the tissue for divergent differentiation.
A tumour may also have a mixed appearance because of metaplasia within it. For example, transitional cell carcinomas of the bladder sometimes have foci of glandular or squamous differentiation.
Carcinosarcomas combine the appearances of carcinoma and sarcoma in one tumour, as a result of either collision of adjacent carcinoma and sarcoma or divergent carcinomatous and sarcomatous differentiation from one original transformed cell.
Neuroendocrine tumours
Neuroendocrine tumours are derived from peptide hormone-secreting cells scattered diffusely in various epithelial tissues. These cells are sometimes referred to as APUD cells; this acronym signifies their biochemical properties (amine content and/or precursor uptake and decarboxylation). (For this reason, the tumours have been called ‘apudomas’.)
The name of those neuroendocrine tumours producing a specific peptide hormone is usually derived from the name of the hormone, together with the suffix ‘-oma’. For example, the insulin-producing tumour originating from the beta-cells of the islets of Langerhans is called an insulinoma. There are exceptions: for example, the calcitonin-producing tumour of the thyroid gland is called a ‘medullary carcinoma of the thyroid gland’ because it was described as a specific entity before calcitonin had been discovered.
Neuroendocrine tumours of the gut and respiratory tract that do not produce any known peptide hormone are called carcinoid tumours. The appendix is the commonest site, but, here, these tumours are usually an incidental finding of little clinical significance. Carcinoids arising elsewhere (the small bowel is the next commonest site) often metastasise to mesenteric lymph nodes and the liver. Extensive metastases lead to the carcinoid syndrome (tachycardia, sweating, skin flushing, anxiety and diarrhoea) due to excessive production of 5-hydroxytryptamine and prostaglandins.
Many neuroendocrine tumours are functionally active, and clinical syndromes often result from excessive secretion of their products (Table 11.4).
Table 11.4 Some neuroendocrine tumours and their associated clinical syndromes
| Tumour | Clinical syndrome |
|---|---|
| Insulinoma | Episodes of hypoglycaemia |
| Gastrinoma | Extensive peptic ulceration of the upper gut (Zollinger–Ellison syndrome) |
| Phaeochromocytoma | Paroxysmal hypertension |
| Carcinoid | If metastases are present, flushing, palpitations and pulmonary valve stenosis |
These neoplasms often pursue an indolent course, growing relatively slowly and metastasising late. Their behaviour cannot always be predicted from their histological features.
Some individuals inherit a familial predisposition to develop neuroendocrine tumours; they have a multiple endocrine neoplasia (MEN) syndrome (Ch. 17).
Hamartomas
A hamartoma is a tumour-like lesion, the growth of which is co-ordinated with the individual; it lacks the autonomy of a true neoplasm. Hamartomas are always benign and usually consist of two or more mature cell types normally found in the organ in which the lesion arises. A common example occurs in the lung, where a hamartoma typically consists of a mixture of cartilage and bronchial-type epithelium (the so-called ‘adenochondroma’; Ch. 14). Pigmented naevi or ‘moles’ (Ch. 24) may also be considered as hamartomatous lesions. Their clinical importance is:
Cysts
A cyst is a fluid-filled space lined by epithelium. Cysts are not necessarily tumours or neoplasms but, because they may have local effects similar to those produced by true tumours and some tumours are typically cystic, it is pertinent to consider them here. Common types of cyst are:
The only type of cyst whose aetiology merits its inclusion within this chapter is the neoplastic cyst. This is seen most commonly in the ovary, where it may be either a benign cystic teratoma, filled with sebaceous material, or a cystadenoma or cystadenocarcinoma, each of which may be filled with either serous fluid or mucus depending on the secretory properties of the lining epithelium.
BIOLOGY OF TUMOUR CELLS




Contrary to past claims and an enduring hope, there is no therapeutically exploitable feature unique to neoplastic cells other than the general property of relative or absolute growth autonomy. Many of the other features have normal counterparts: mitotic activity is a feature also of regenerating cells; placental trophoblast is invasive; and the nucleated cells of the blood and lymph wander freely around the body, settling in other sites.
The autonomy of neoplastic cells is often relative rather than absolute. For example, approximately two-thirds of breast carcinomas contain oestrogen receptors; these tumours are better differentiated than receptor-negative breast carcinomas and they have a better prognosis. Furthermore, if women with oestrogen receptor-positive breast carcinomas are given tamoxifen (a drug that blocks the receptor) they survive longer than women with receptor-positive tumours who have not been treated in this way.
One of the many difficulties in studying tumours is their genetic instability, leading to the formation of many clones with divergent properties within one tumour. This is often reflected in the histology which may show a heterogeneous growth pattern, some areas appearing better differentiated than others. Clinically, this instability and consequent cellular heterogeneity is important because thereby some tumours resist chemotherapy; consequently, many chemotherapy regimes involve a combination of agents administered simultaneously or sequentially.
Cellular immortalisation
Cells that have undergone neoplastic transformation appear immortal, especially when studied in cell cultures. Whereas normal untransformed cells have a limited life-span, neoplastic cells have a prolonged or indefinite life-span. This is enabled by:
DNA of tumour cells
Tumour cells have abnormal nuclear DNA. The total amount of DNA per cell commonly exceeds that of the normal diploid (2N) population. This is evident in histological sections as nuclear hyperchromasia. The amount of DNA may increase in exact multiples of the diploid state (polyploidy) such as tetraploid (4N) and octaploid (8N); alternatively there may be aneuploidy—the presence of inexact multiples of DNA per cell.
Aneuploidy and polyploidy are associated with increased tumour aggressiveness and are recognisable in histological sections as variations in nuclear size and staining (pleomorphism).
At a chromosomal level these abnormalities of DNA are associated with the presence of additional chromosomes and often with chromosomal translocations. Some of these karyotypic abnormalities have a regular association with specific tumours; the best known and one of the most consistent is the association of the Philadelphia chromosome with chronic myeloid leukaemia.
Genetic abnormalities are being discovered with increasing frequency in tumours. Some of these may be relatively late events, epiphenomena with no central role in the cancer process. However, others are of fundamental importance, appearing at an early stage in the development of the tumour. Abnormalities of oncogenes and tumour suppressor genes are of considerable interest in this regard because of their central involvement in carcinogenesis (p. 246).
Mitotic and apoptotic activity
Malignant tumours frequently exhibit more mitotic activity than the corresponding normal cell population. In histological sections, mitoses are abundant, and mitotic figures are often grossly abnormal, showing tripolar and other bizarre arrangements. Cellular proliferation can be estimated by mitosis counting, DNA measurements and determination of the frequency of expression of cell cycle-associated proteins (e.g. Ki-67 antigen). Prognostic information can be derived from these estimations: higher frequencies of cellular proliferation are associated with a worse prognosis.
However, assessment of the growth characteristics of a tumour must involve also an appraisal of the rate of cell loss, through either ischaemic necrosis or apoptotic cell death. Although tumours often contain abundant apoptotic bodies, a common biological defect of neoplastic cells is abrogation of the cell death mechanisms. In some lymphomas, for example, this is mediated by abnormal expression of bcl-2, an apoptosis-inhibiting gene.
Metabolic abnormalities
Although tumour cells show a tendency towards anaerobic glycolysis, there are no metabolic abnormalities entirely specific to the neoplastic process. The known metabolic abnormalities of tumour cells are simply discordant with the normal physiological state of the tissue or host.
The surface of tumour cells is abnormal. Tumour cells are less cohesive. In many neoplasms, poor cellular cohesion is due to a reduction in specialised intercellular junctions such as desmosomes. This loss of adhesiveness enables malignant tumour cells to spread through tissues and detach themselves to populate distant organs.
Tumour cells may retain the capacity to synthesise and secrete products characteristic of the normal cell type from which they are derived, often doing so in an excessive and uncontrolled manner. In addition, tumours often show evidence of gene derepression. All somatic cells contain the same genetic information, but only a small proportion of the genome is transcribed into RNA and translated into protein in any normal cell. Most genes are repressed, and only those required for the function of the particular cell are selectively expressed. However, in many tumour cells, some genes become derepressed, resulting in the inappropriate synthesis of unexpected substances.
Tumour products
The major types of tumour product are:
Some tumour products are useful as markers for diagnosis or follow-up (Table 11.5). They can be detected in histological sections or their concentrations measured in the blood. Rising blood levels suggest the presence of tumour; falling levels indicate a sustained response to therapy (Fig. 11.16).
Table 11.5 Tumours secreting markers used in diagnosis or follow-up
| Tumour | Marker | Comment |
|---|---|---|
| Myeloma |
Hepatocellular carcinomaAlpha-fetoprotein (AFP)Also associated with testicular teratomaGastrointestinal adenocarcinomasCarcinoembryonic antigen (CEA)False positives occur in some non-neoplastic conditionsNeuroendocrine tumoursPeptide hormones (e.g. insulin, gastrin)Excessive hormone production may have clinical effectsPhaeochromocytomaVanillyl mandelic acid (VMA)Metabolite of catecholamines in urineCarcinoid5-Hydroxyindole-acetic acid (5-HIAA)Metabolite of 5-hydroxytryptamine (5-HT) in urineChoriocarcinomaHuman chorionic gonadotrophin (hCG)In blood or urineMalignant teratoma
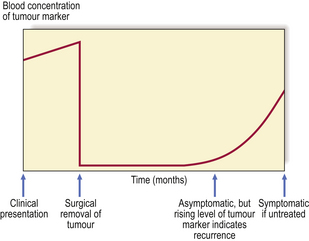
Fig. 11.16 Use of tumour markers to monitor clinical progress. Abnormally high levels of the marker can be used to detect tumours before they become symptomatic, either by screening a population at risk or, as in the example shown here, by regular monitoring to detect early recurrences. The events shown here could take place over a total period of 12 months.
CARCINOGENESIS
Carcinogenesis is the process that results in the transformation of normal cells to neoplastic cells by causing permanent genetic alterations.
Neoplasms arise from single cells that have become transformed by cumulative mutational events. Because of this presumed single-cell origin, neoplasms are said to be clonal proliferations; this distinguishes them from non-neoplastic masses which are typically polyclonal. Spontaneous mutations during normal DNA replication are probably common, but many are rectified by repair mechanisms. The probability of neoplastic transformation increases with the number of cell divisions experienced by a cell; this may explain why the incidence of cancer increases with age. Exposure to carcinogens increases the probability of specific mutational events.
A carcinogen is an environmental agent participating in the causation of tumours. Such agents are said to be carcinogenic (cancer causing) or oncogenic (tumour causing). The ultimate site of action of all carcinogens is the DNA in which genes are encoded. Carcinogens are therefore also mutagenic. Very often more than one carcinogen is necessary for the complete neoplastic transformation of a cell, and there is good evidence that the process occurs in several discrete steps; this is the multistep hypothesis.
Once established, neoplastic behaviour does not require the continued presence of the carcinogen. It is rather a ‘hit-and-run’ situation and evidence of the specific causative agent(s) is not usually found in the eventual tumours. Exceptions include some suspected carcinogenic viruses, genetic material of which persists in the resulting tumours, and some insoluble substances, such as asbestos, which cannot be eliminated from the tissues. The ‘hit-and-run’ character of carcinogenesis is one of several reasons why carcinogens have proved so elusive.
Recent research has considerably improved our knowledge and understanding of the molecular basis of carcinogenesis. Genetic alterations are absolutely fundamental to the carcinogenic process. The central role of these genetic abnormalities will be considered in detail after the different classes of carcinogen have been described.
Identification of carcinogens





Most tumours are thought to result from an environmental cause, although there is increasing evidence, in some tumours, of an inherited risk. It has been estimated that approximately 85% of the cancer risk is due to environmental agents.
Ethics prohibit the testing of suspected carcinogens in humans, so much of our knowledge of carcinogenesis in humans is derived from indirect or circumstantial evidence. Identification is hampered both by the complexity of the human environment, which makes it difficult to isolate a single causative factor from the many possible candidates, and by the very long time interval between exposure to a carcinogen and the appearance of signs and symptoms leading to the diagnosis of the tumour; this latent interval may be two or three decades.
Carcinogens may be identified from:
Epidemiological evidence
Some types of cancer are more common in certain countries, regions or communities within them (Fig. 11.17). Epidemiology has proved to be a fruitful source of information about the causes of tumours. Tumour incidence is more important than mortality data in this regard, because only a proportion of tumours prove fatal and the precise causes of death may not be well documented. It is thus essential to survey populations thoroughly for tumour incidence; in countries with well-developed health services, investigators can usually rely on diagnostic records and cancer registries, but elsewhere it may be necessary to visit and examine the population under study. Variations in tumour incidence may genuinely be due to environmental factors, but the data must first be standardised to eliminate the effect of, for example, any differences in the age distribution. The long latency between exposure to a carcinogen and the appearance of the tumour makes it necessary to consider also the effect of population movement. This effect can be used to distinguish between racial (hereditary) and environmental factors in determining cancer incidence in migrants.
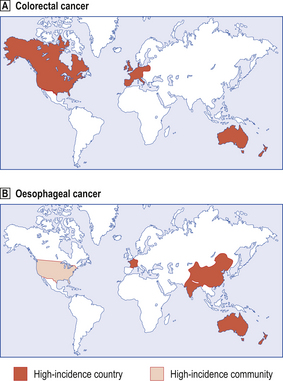
Fig. 11.17 World map showing countries in which there is a relatively high incidence of specific types of cancer.  Colorectal cancer.
Colorectal cancer.  Oesophageal cancer. Low-incidence countries may conceal high-incidence regions or communities; for example, oesophageal carcinoma is more common among blacks in the USA (lighter shaded area). Note that colorectal cancer is much commoner in countries whose inhabitants eat a more refined diet. Dietary associations with oesophageal cancer are less well defined.
Oesophageal cancer. Low-incidence countries may conceal high-incidence regions or communities; for example, oesophageal carcinoma is more common among blacks in the USA (lighter shaded area). Note that colorectal cancer is much commoner in countries whose inhabitants eat a more refined diet. Dietary associations with oesophageal cancer are less well defined.
Having found a high tumour incidence in a population, comparisons of lifestyle, diet and occupational risks with those of a low tumour-incidence control population often leads to specific causative associations being identified.
The following examples illustrate how carcinogens can be identified in this way.
Hepatocellular carcinoma
In countries such as the UK and the USA, hepatocellular carcinoma is a relatively uncommon tumour and, when it does occur, it usually arises in a cirrhotic liver. However, the worldwide incidence of hepatocellular carcinoma is high, and in some countries it is the most common tumour (Ch. 16). Epidemiology reveals two factors that may be involved in the high prevalence in endemic areas: mycotoxins and hepatitis viruses B and C.
The incidence of hepatocellular carcinoma in different regions of Uganda is associated with the frequency with which food samples in those regions were found to be contaminated with aflatoxins. Aflatoxins are mycotoxins produced by the fungus Aspergillus flavus, and are a highly carcinogenic group of compounds. The fungus grows on food stored in humid conditions in the high-incidence areas. However, the situation is not clear-cut, because of the prevalence of hepatitis B virus in the area. There is a high incidence of point mutations of specific codons in p53, a tumour suppressor gene, in hepatocellular carcinomas associated epidemiologically with aflatoxins.
There is a strong correlation between the incidence of hepatitis B and C virus infection and hepatocellular carcinoma in many countries. Suspicion that hepatitis B virus may be oncogenic (tumour-causing) is reinforced by the discovery, in such cases, of a copy of the viral genome incorporated within the genome of the liver cancer cells.
Oesophageal carcinoma
The very high incidence of oesophageal carcinoma in China and in the Caspian littoral region of Iran has been intensively studied by epidemiologists. Several factors have been implicated, including the dyes used in carpet making, nitrates in the soil, abrasives in the diet, and opium dross, but no single aetiological factor has yet emerged from these studies.
One of the most informative natural experiments on the causation of oesophageal carcinoma took place in China when a reservoir was built in the Linhsien area. People living in this district were known to have a very high incidence of oesophageal carcinoma as, remarkably, did their domestic chickens. A dietary factor was suspected because the chickens were usually fed with their owners’ waste food. The reservoir displaced these people to Fanhsien, where they continued to have a high incidence of oesophageal carcinoma. They left their chickens behind and re-stocked with a local strain in which oesophageal carcinoma was uncommon. However, fed with the table-scraps and waste food from their new owners, the local strain subsequently developed a high incidence of oesophageal carcinoma. A dietary factor was clearly implicated.
Occupational and behavioural risks
Certain types of cancer are, or have been, more common in people engaged in specific activities. The discovery in a given community of a link between a higher risk of developing a cancer and the person’s occupation helps to separate general environmental causes of carcinogenesis from those that are probably specific to an individual person or group in the community.
Scrotal carcinoma
Percival Pott is credited with the first observation, in 1775, linking a particular tumour with a specific occupation. He noticed a high incidence of carcinoma of the scrotal skin in men who were or had been chimney sweeps, and postulated that the soot was responsible. It was not until 150 years later that the specific carcinogen, a polycyclic aromatic hydrocarbon, was identified.
Lung carcinoma
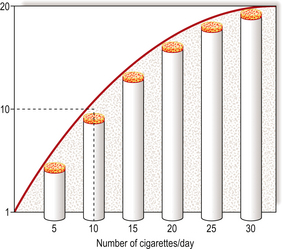
Lung carcinoma is a major public health problem in many countries. In the UK, more then 30000 deaths are attributed to this cause annually; the actual incidence is only marginally higher because this form of cancer has an extremely poor prognosis. The unarguable association with cigarette smoking was established by meticulous epidemiological research. The problem, a common one for epidemiologists, was that people who smoke are commonly exposed to many other possible risks: they tend to live in cities, inhale atmospheric pollutants from motor vehicles, domestic fires and industry, be fond of alcoholic drinks, etc. However, careful analysis of environmental factors showed that cigarette smoking correlated most strongly with the incidence of lung carcinoma. There is an almost linear dose–response relationship between the number of cigarettes smoked daily and the risk of developing lung cancer (Fig. 11.18). Furthermore, the incidence of lung carcinoma declined in those groups of people, such as British male doctors, whose tobacco consumption fell substantially.
Carcinoma of the cervix
The observation that carcinoma of the cervix is commonest amongst prostitutes and an extreme rarity in celibate nuns suggested that the disease may be due to a venereally transmitted agent. The risk of carcinoma of the cervix is strongly associated with sexual intercourse, in particular with the number of partners and thus the risk of exposure to a possible carcinogenic agent conveyed by the male. Specific genotypes of human papillomavirus (HPV) are now proven to cause cancer of the cervix. Indeed, the evidence is so compelling that HPV immunisation is being introduced with the specific aim of reducing the incidence of this disease. HPV is an essential causative agent for squamous carcinoma of the cervix. Smoking is an aetiological co-factor.
Bladder carcinoma
In the 1890s, epidemiologists noted a higher than expected incidence of bladder cancer among men employed in the aniline dye and rubber industries. Further analysis led to the identification of beta-naphthylamine as the causative agent. Although stringent precautions are now taken to minimise the risk, people working in these industries are regularly screened for bladder cancer by cytological examination of their urine and, if bladder cancer occurs, the patient is entitled to compensation on the assumption that the disease has been acquired occupationally.
Direct evidence
It is fortunately a rare event for someone to be knowingly exposed to a single agent that causes cancer. Sadly, such happenings are often the adverse result of diagnostic or therapeutic medical intervention, but we must not overlook the opportunity to learn much from them.
Thorotrast
Thorotrast was a colloidal suspension of thorium dioxide widely used in many countries during 1930–1950 as a contrast medium in diagnostic radiology. When it was first introduced for medical use, two potentially hazardous properties of Thorotrast were known: thorium dioxide is naturally radioactive, emitting alpha-radiation and possessing an extremely long half-life of 1.39 × 1010 years; the colloidal suspension is rapidly and irreversibly taken up by the body’s phagocytic cells, such as those lining the vascular sinusoids in the liver and the spleen. Despite these potential hazards, Thorotrast proved such a good contrast medium, particularly for angiography, that its popularity was assured. However, in 1947 the first report was published of a patient who developed angiosarcoma of the liver after Thorotrast administration. As other cases were recognised, Thorotrast was withdrawn from use. Nevertheless, this unfortunate episode has given us an opportunity to learn about and confirm the carcinogenic effects of alpha-radiation.
Thyroid carcinoma and radiation in children
The thyroid gland is vulnerable to the carcinogenic effects of external irradiation and of the radioactive isotopes of iodine (the latter are concentrated by the thyroid gland in the synthesis of thyroid hormone). For example, in April 1986 a nuclear reactor exploded at Chernobyl in Ukraine, releasing a large quantity of radioactive material into the atmosphere. The material released included radioactive iodine. After a 4-year latent interval, there has been a dramatic increase in the local incidence of thyroid carcinoma in children (Fig. 11.19). To minimise this risk, non-radioactive iodine is usually given to people immediately after any accidental exposure to radioactive iodine to compete with the latter for uptake by the thyroid gland.
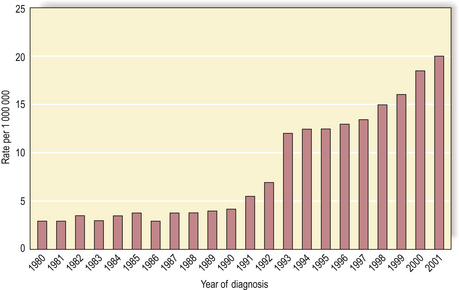
Fig. 11.19 Rising incidence of thyroid carcinoma in children (0–14 years) in Belarus. Thyroid carcinoma in children is relatively uncommon, but since the nuclear reactor explosion in 1986 at Chernobyl there has been a significantly increased local incidence.
(Based on data in Mahoney MC, Lawvere S, Falkner KL et al 2004 Thyroid cancer incidence trends in Belarus. International Journal of Epidemiology 33: 1025–1033.)
Experimental testing
Carcinogens are not united by any common physical or chemical properties; it is therefore considered necessary to screen all new drugs, food additives and potential environmental pollutants in non-human systems before they are introduced for human use. Three types of test system for carcinogenic or mutagenic activity are employed:
None of these is perfect; animals and isolated cell cultures often metabolise the agent being tested in a way that differs from normal human metabolic pathways, and mutagenicity in bacterial DNA may not correspond to carcinogenicity. In addition, the dynamics of these test systems are very different from that of clinical cancer; cancer in humans is a chronic process often lasting decades, whereas tests for carcinogenic activity in experimental systems usually seek more immediate effects. Nevertheless, despite these limitations, it is still appropriate to investigate possible carcinogens in this way.
Known or suspected carcinogens
The main classes of carcinogenic agent are:
As a result of direct testing for mutagenicity, or from accidental exposures or epidemiological evidence, many known or strongly suspected carcinogens have been identified (Fig. 11.20). In many countries, legislation prohibits or restricts the use of proven carcinogens.
Chemical carcinogens



Many chemical carcinogens have now been identified. The main categories are shown in Table 11.6.
Table 11.6 Examples of proven or suspected chemical carcinogens and the tumours with which they are associated
| Chemical | Tumour | Comments |
|---|---|---|
| Polycyclic aromatic hydrocarbons (e.g. 3,4-benzpyrene) | Lung cancerSkin cancer | Strong link with smokingFollowing repeated exposure to mineral oils |
| Aromatic amines (e.g. beta-naphthylamine) | Bladder cancer | In rubber and dye workers |
| Nitrosamines | Gut cancers | Proven in animals |
| Azo dyes (e.g. 2-acetylaminofluorene) | Bladder and liver cancer | Proven in animals |
| Alkylating agents (e.g. cyclophosphamide) | Leukaemia | Small risk in humans |
| Other organic chemicals (e.g. vinyl chloride) | Liver angiosarcoma | Used in PVC manufacture |
The carcinogenic risk cannot be predicted from the structural formula alone; even apparently closely related compounds can have different effects.
Some agents act directly, requiring no metabolic conversion. Others (procarcinogens) require metabolic conversion into active carcinogens (ultimate carcinogens) (Fig. 11.21). If the enzyme required for conversion is ubiquitous within tissues, tumours will occur at the site of contact or entry; for example, polycyclic aromatic hydrocarbons induce skin tumours if painted on to the skin, or lung cancer if inhaled in tobacco smoke. Other agents require metabolic conversion by enzymes confined to certain organs, and thus often induce tumours remote from the site of entry; for example, aromatic amines require hydroxylation in the liver before expressing their carcinogenic effects. In a few instances the carcinogen is synthesised in the body from ingredients in the diet; thus, carcinogenic nitrosamines are synthesised by gut bacteria utilising dietary nitrates and nitrites.
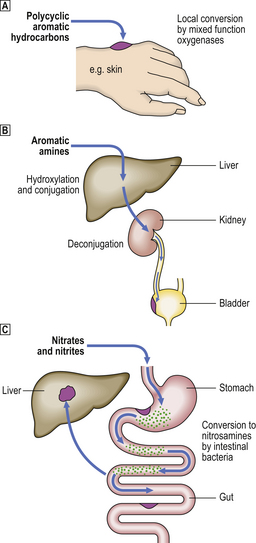
Fig. 11.21 Summary of some metabolic pathways for conversion of chemical procarcinogens into the active ultimate carcinogens.  Polycyclic aromatic hydrocarbons.
Polycyclic aromatic hydrocarbons.  Aromatic amines.
Aromatic amines.  Nitrates and nitrites. (See text for details.)
Nitrates and nitrites. (See text for details.)
Polycyclic aromatic hydrocarbons
Polycyclic aromatic hydrocarbons were the first chemical carcinogens to be intensively studied. In 1917, Yamagiwa and Itchikawa in Japan showed that skin tumours could be induced in rabbits by painting their skin with tar. Tar was a suspected carcinogen because of the high incidence of skin cancer among tar-workers, particularly on the hands, which were frequently in contact with it. In the 1930s in London, Cook and Kennaway fractionated tar and attributed the carcinogenic effect to the polycyclic aromatic hydrocarbons. Like many chemicals implicated in the development of cancer, these are procarcinogens, requiring metabolic conversion by hydroxylation to form ultimate carcinogens. In this case, the carcinogenic effect is invariably at the site of contact because the hydroxylating enzymes (e.g. aryl carbohydrate hydroxylase) are ubiquitous in human tissues and readily induced in susceptible individuals. However, if the substance is absorbed into the body, this may lead to a risk of cancer at sites remote from the point of initial contact; there is, for example, an increased incidence of bladder cancer in tobacco smokers.
The tumour most commonly associated with exposure to polycyclic aromatic hydrocarbons is carcinoma of the lung. This tumour is much more common in smokers than in non-smokers and the risk to an individual or group parallels the quantity of tobacco consumed. Tobacco smoke contains many candidates for carcinogenic activity; the most important is probably 3,4-benzpyrene. Tobacco is also chewed in some countries, and there it is associated with a risk of carcinoma of the mouth.
Aromatic amines
The high incidence of bladder carcinoma in workers in the dye and rubber industries has now been attributed to beta-naphthylamine. Unlike the polycyclic aromatic hydrocarbons, this substance has no local carcinogenic effect. It requires conversion by hydroxylation in the liver into the active carcinogenic metabolite, 1-hydroxy-2-naphthylamine. However, the carcinogenic effect is masked immediately by conjugation with glucuronic acid in the liver. Bladder cancer results because the conjugated metabolite is excreted in the urine and deconjugated in the urinary tract by the enzyme glucuronidase, thus exposing the urothelium to the active carcinogen.
Nitrosamines
While ultimate proof of a causal relationship with human cancers is lacking, there is epidemiological evidence linking carcinomas of the gastrointestinal tract to the ingestion of nitrosamines and to dietary nitrates and nitrites. Nitrates are used widely as fertilisers, and are eventually washed by the rain into rivers and underground water tables where they can contaminate drinking water. In addition, both nitrates and nitrites have been used as food additives. Although these radicals are not in themselves carcinogenic, they are readily metabolised by commensal bacteria within the gut and converted to carcinogenic nitrosamines by combination with secondary amines and amides. Direct proof of a major role in carcinogenesis in humans is still awaited, but these substances are potent carcinogens in laboratory animals and it is unlikely that humans would be exempt from this effect.
Azo dyes
The carcinogenic potential of azo dyes, derivatives of aromatic amines, was recognised at an early stage and their use has thus been severely restricted. In laboratory animals, dimethylaminoazobenzene—otherwise known as ‘butter yellow’ because it was once used to impart an appetising yellow colour to margarine—causes liver cancer.
Alkylating agents
Many categories of chemical carcinogen, including polycyclic hydrocarbons, have alkylation as the ultimate common pathway, so it is not surprising that alkylating agents themselves can be carcinogenic. Alkylating agents bind directly to DNA, the ultimate site of action of all carcinogens. Nitrogen mustard is a well-known example, but these agents are not otherwise widely implicated as a major cause of human cancer.
Oncogenic viruses






Viruses were first implicated as carcinogenic agents through the experiments of Rous (in 1911) and Shope (in 1932), who studied fowl sarcomas and rabbit skin tumours, respectively. They showed that it was possible to transmit the tumours from one animal to another, in the manner of an infectious disease; tumours could be induced by injecting a cell-free filtrate of each tumour. The only possible transmissible agent was considered to be a virus, because the pores of the filter were too fine to permit the passage of bacteria or whole tumour cells. The study of oncogenic retroviruses in laboratory animals has had a seminal effect on our understanding of the molecular basis of tumour development and has led to the discovery of oncogenes (Fig. 11.22 and Table 11.12, p. 251).
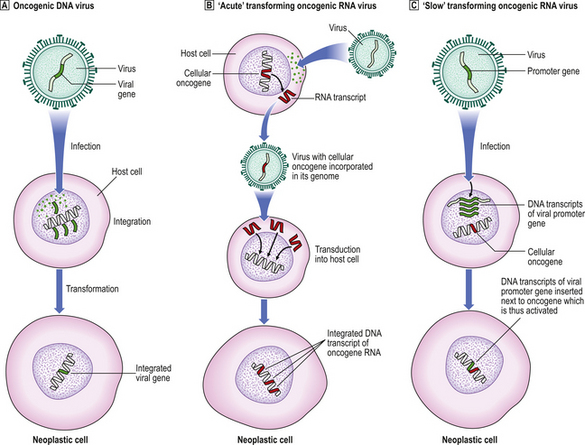
Fig. 11.22 Simplified mechanisms of integration of oncogenic viral genes, or DNA transcripts, into the host cell DNA.  Oncogenic DNA virus: the viral genome is integrated into host cell DNA; neoplastic transformation is a postulated consequence.
Oncogenic DNA virus: the viral genome is integrated into host cell DNA; neoplastic transformation is a postulated consequence.  ‘Acute’ transforming oncogenic RNA virus: transduction into the host cell of an RNA transcript of a cellular oncogene picked up from another cell.
‘Acute’ transforming oncogenic RNA virus: transduction into the host cell of an RNA transcript of a cellular oncogene picked up from another cell.  ‘Slow’ transforming oncogenic RNA virus: insertion of a viral promoter gene next to a cellular oncogene. In both
‘Slow’ transforming oncogenic RNA virus: insertion of a viral promoter gene next to a cellular oncogene. In both  and
and  , DNA transcripts are made from the RNA conveyed by the virus using the enzyme reverse transcriptase and, in contrast to cellular oncogenes, they lack introns.
, DNA transcripts are made from the RNA conveyed by the virus using the enzyme reverse transcriptase and, in contrast to cellular oncogenes, they lack introns.
Many human tumours are now known to be associated with viruses (Table 11.7).
Table 11.7 Viruses implicated in human tumours
| Virus | Tumour | Comments |
|---|---|---|
| Human papillomavirus |
Epstein–Barr virus
Hepatitis B and C virusesHepatocellular carcinomaStrong associationHuman herpes virus-8
Human T-cell lymphotropic virus-1Adult T-cell leukaemia/lymphomaEndemic in Southern Japan and Caribbean basin
Human tumours for which a viral aetiology has been proposed or proven include:
Human papillomavirus
Human papillomavirus (HPV), of which there are many subtypes, causes the common wart (squamous cell papilloma). This lesion occurs most commonly on the hand, a frequent site of physical contact enabling transmission between individuals, and the virus is abundant within the abnormal cells of the lesion. Anogenital warts are also due to HPV, raising the possibility that other genital epithelial neoplasms may also be attributable to this cause. Evidence is also accumulating to suggest involvement of HPV in squamous neoplasia of the upper respiratory and digestive tracts.
Epidemiological and laboratory evidence reveals HPV as an essential cause of cancer of the cervix; this is discussed in more detail in Chapter 19.
Epstein–Barr virus
The Epstein–Barr (EB) virus was discovered first in cell cultures from Burkitt’s lymphoma, a B-cell lymphoma endemic in certain regions of Africa and occurring only sporadically elsewhere. Early hopes that EB virus was the sole cause of Burkitt’s lymphoma were dashed when it was discovered, following the accidental infection of a laboratory worker, that infection by the virus on its own causes infectious mononucleosis, a common, benign lymphoproliferative disorder which remits spontaneously in most cases. Clearly a co-factor is involved in the pathogenesis of Burkitt’s lymphoma; epidemiological evidence suggests that this is malaria.
EB virus is also implicated in the causation of nasopharyngeal carcinoma in the Far East, where there is a relatively high incidence of this tumour.
Radiant energy


Ultraviolet light
Skin cancer is more common on parts of the body regularly exposed to sunlight, and ultraviolet light (UVL) is now considered to be a major causal factor, UVB more so than UVA. Skin cancer is less common in people with naturally pigmented skin, as the melanin has a protective effect; it is more common in fair-skinned people, particularly those who get sunburnt easily, living in sunny climates (e.g. Australia).
Most types of skin cancer are associated with UVL exposure, but the risk is particularly high for malignant melanoma and basal cell carcinoma (‘rodent ulcer’). This risk is greatly increased in patients with xeroderma pigmentosum, a rare congenital deficiency of DNA repair enzymes, in whom numerous skin cancers occur due to unrepaired damage to the DNA of the skin cells induced by UVL.
Ionising radiation
The carcinogenic effects of radiation are long-term and must be distinguished from the more immediate, dose-related, acute effects such as skin erythema and, more seriously, bone marrow aplasia (Ch. 6).
Evidence that relatively high doses of ionising radiation are carcinogenic is indisputable. The carcinogenic effect of low levels of radiation continues to be a matter of great public concern because of the debate over the safety of nuclear power sources. Exposure to some ionising radiation from cosmic and other natural sources (background radiation) is inescapable; however, linear extrapolation of the low-dose risk from the quantifiable carcinogenic risk from higher levels of radiation is generally conceded to exaggerate the problem.
An increased incidence of cancer following exposure to ionising radiation has been witnessed since the earliest work with radioactive materials. Before protective measures were introduced there was a well-recognised increased incidence of leukaemia in radiology workers, and of skin cancer in those who regularly placed their hands in X-ray beams. The therapeutic use of radiation, often without adequate justification (e.g. radiation of the thymus gland in children with miscellaneous ailments; Ch. 6), has resulted in cancers. Radiation from military sources, such as in Hiroshima and Nagasaki in 1945, resulted in a high incidence of certain tumours in survivors. Industrial exposure to radiation includes the risk of carcinoma of the lung associated with the mining of radioactive uranium. There has been a dramatic increase in the incidence of thyroid cancer in children near Chernobyl in Ukraine, the site of a nuclear accident in 1986 (p. 237).
Some tissues are more vulnerable than others to the carcinogenic effects of ionising radiation, and specific risks are associated with particular radioactive elements if they are concentrated in specific tissues, for example radioactive iodine concentrated in the thyroid gland. Tissues that appear particularly sensitive to the carcinogenic effects of ionising radiation include thyroid, breast, bone and haemopoietic tissue.
Hormones
It is somewhat surprising that substances occurring naturally in the body and indispensable for normal bodily functions should be implicated as at least co-factors in carcinogenesis. For example, exogenous oestrogens can be shown experimentally to promote the formation of mammary and endometrial carcinomas; the association between breast carcinoma and oral contraceptives containing oestrogens is weak. Androgenic and anabolic steroids are known to induce hepatocellular tumours in humans, and oestrogenic steroids may make pre-existing lesions (e.g. adenomas and focal nodular hyperplasia) abnormally vascular, thus causing otherwise asymptomatic lesions to present clinically.
Bacteria, fungi and parasites
Cancer may also result from infection with other living organisms (e.g. bacteria, parasites) or from the ingestion of food contaminated with the metabolic products of other organisms (e.g. mycotoxins).
Bacteria
Helicobacter pylori, a major cause of gastritis and peptic ulceration, is now strongly implicated in the pathogenesis of gastric lymphomas. Initially, the lesions are dependent on the continuing presence of H. pylori (the lymphoma regresses if the bacteria are eradicated), but eventually the lymphoma becomes fully autonomous.
Fungi
Mycotoxins are toxic substances produced by fungi. Those having the greatest relevance in human carcinogenesis are the aflatoxins produced by Aspergillus flavus. Aflatoxins, particularly aflatoxin B1, are among the most potent carcinogens and have been specifically linked to the high incidence of hepatocellular carcinoma in certain parts of Africa (Ch. 16).
Parasites
There is good evidence, both epidemiological and direct, to implicate Schistosoma haematobium, Opisthorchis viverrini and Clonorchis sinensis in the causation of human cancer. In such cases, there is a high incidence of the tumour in infested areas, and the parasites can often be found actually within or in the immediate vicinity of the tumour.
Schistosoma haematobium is strongly implicated in the high incidence of bladder carcinoma, usually of squamous cell type, in areas where infestation with the parasite is rife, such as Egypt. The ova of the parasite can often be found in or close to the tumour.
The liver flukes Clonorchis sinensis and Opisthorchis viverrini dwell in the bile ducts, where they induce an inflammatory reaction, epithelial hyperplasia and sometimes eventually adenocarcinoma of the bile ducts (cholangiocarcinoma). There is a high incidence of this tumour in parts of the Far East and other fluke-infested areas.
Miscellaneous carcinogens
In contrast to radiation, chemicals and viruses, which ultimately damage or bind to DNA, there are some miscellaneous carcinogens whose mechanism of action is not well understood, despite their proven association with cancer.
Asbestos
Inhalation of asbestos fibres results in various lesions: asbestosis, pleural plaques, mesothelioma and carcinoma of the lung (Fig. 11.23). Of the two neoplastic consequences, the association with mesothelioma is the more specific because this tumour is exceptionally rare in the absence of asbestos exposure. The pleura is the most frequent site for mesothelioma, but the association with asbestos is just as strong for peritoneal mesothelioma. There is also an association with carcinoma of the lung, which is enhanced by cigarette smoking.
Host factors in carcinogenesis
In addition to the extrinsic or environmental factors in carcinogenesis, there are also several important host factors that influence the cancer risk. These are:
Race
The precise role of race in determining an individual’s risk of developing specific types of cancer is complicated by the fact that racial differences often coincide with differences in place of residence, diet and habit. While in some instances the link is obvious—for example, skin cancer is uncommon in blacks because the melanin in their skin protects them from the carcinogenic effects of ultraviolet sunlight—apparent racial differences are often explicable in terms of habit or cultural practices. Thus, oral cancer is relatively common in India and South-East Asia; but this is not associated directly with race, rather with tobacco or betel chewing and the remarkable habit of ‘reverse smoking’ in which the burning end of the cigarette is habitually placed in the mouth!
The relative contributions of race and environment to the incidence of cancer can be deduced from comparing the incidence in racial groups that have migrated to other countries. For example, cancer of the stomach is relatively uncommon in Africa, but the incidence in North American blacks of African descent approximates to the higher risk in the white population.
Diet
Dietary factors may be linked to cancer risk because:
There is a positive correlation between high dietary fat and the risk of breast and colorectal cancer; alcohol appears to be a risk factor for breast and oesophageal cancer; and there is experimental evidence to suggest that a low protein diet has a protective effect against certain chemical carcinogens by reducing the levels of mixed-function oxygenases in the liver. Dietary fibre appears to be protective for colorectal cancer by promoting more rapid intestinal transit; any carcinogens in the bowel contents therefore remain in contact with the mucosa for a shorter time.
Constitutional factors
Inherited predisposition
Some individuals inherit an increased risk of developing certain tumours (Table 11.8). There is, for example, an inherited predisposition to breast cancer; a woman whose mother and one sister have developed breast cancer has a c. 50% probability of developing it herself. Genes responsible for this inherited risk (BRCA1 on chromosome 17 and BRCA2 on chromosome 13) have been identified. Sometimes the inherited risk is well defined, as in the condition xeroderma pigmentosum, a deficiency of DNA repair enzymes. Polyposis coli is an autosomal dominant inherited predisposition to develop multiple adenomatous polyps of the large bowel; consequently there is an increased risk of carcinoma of the colon and rectum arising in these polyps. Retinoblastoma, a malignant tumour of the eye in children, is familial and often bilateral in approximately one-third of cases; in these patients there is usually an abnormality of the RB1 gene on chromosome 13.
Table 11.8 Examples of inherited cancer risks
| Inherited disorder | Tumour(s) | Comment |
|---|---|---|
| Multiple endocrine neoplasia (MEN) syndromes | Endocrine tumours, e.g. phaeochromocytoma, medullary carcinoma of the thyroid, parathyroid adenoma | Several types (MEN I, II, etc.) attributed to RET gene on chromosome 10 and others on chromosome 11 |
| Xeroderma pigmentosum | Skin cancers, e.g. basal cell carcinoma, melanoma | Deficiency of DNA repair enzymes |
| Familial polyposis coli | Colorectal carcinoma | Preceded by numerous adenomatous polyps; autosomal dominant APC gene on chromosome 5 |
| Hereditary non-polyposis colorectal cancer | Colorectal carcinoma | Mutated genes (MLH1, MSH2) involved in DNA repair |
| von Hippel–Lindau syndrome | Cerebellar haemangioblastoma, phaeochromocytoma, hypernephroma | Autosomal dominant inheritance associated with VHL gene on chromosome 3 |
| Li–Fraumeni syndrome | Breast carcinoma, soft-tissue sarcomas | Autosomal dominant inheritance associated with abnormalities on chromosomes 13 (RB1 gene), 11 and 17 (p53 gene) |
| Retinoblastoma | Retinoblastoma (frequently bilateral) | Inherited allelic loss of one inhibitory RB1 gene on chromosome 13 |
| Familial breast carcinoma |
Attributed to mutated BRCA1 gene on chromosome 17
Age
The incidence of cancer increases with age. There are several possible explanations: the cumulative risk of exposure to carcinogens with increasing age; the long latent interval between exposure to the initiating carcinogenic agent and the clinical appearance of the resulting tumour means that there is inevitably a tendency for most tumours to begin to appear only after a few decades of life have elapsed; accumulating genetic lesions (mutations) may render the ageing cell more sensitive to carcinogenic effects. Finally, it may be that incipient tumours developing in young individuals are recognised and eliminated by some innate defence system, such as natural killer cells, and that this protective effect is lost with age.
Gender
Breast cancer is at least 200 times commoner in women than in men. This is probably due to the greater mammary epithelial volume and to the promoting effects of oestrogens in females. It is more common in women who are nulliparous or who have not breast-fed their children, and those who have experienced an early menarche and/or late menopause. Endocrine factors are undoubtedly important.
Associations with gender occur in other cancers, but these are more often due to, for example, smoking habits than to hormonal factors.
Premalignant lesions and conditions
A premalignant lesion is an identifiable local abnormality associated with an increased risk of a malignant tumour developing at that site. Examples include adenomatous polyps of the colon and rectum, and epithelial dysplasias in various sites, notably the cervix. Studies of these lesions reinforce the multistep theory of carcinogenesis (Fig. 11.24); it may be that these lesions represent the growth of partially transformed cells which have not yet achieved full neoplastic status.
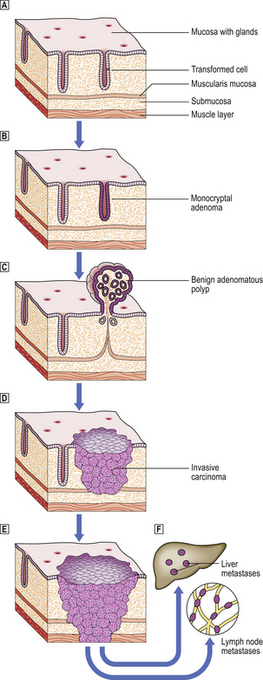
Fig. 11.24 Carcinoma of the large bowel as a model of tumour progression.  A single epithelial cell within a mucosal gland becomes transformed into a tumour cell by carcinogenic events.
A single epithelial cell within a mucosal gland becomes transformed into a tumour cell by carcinogenic events.  The abnormal cell proliferates to produce a clone of cells populating one gland.
The abnormal cell proliferates to produce a clone of cells populating one gland.  Further proliferation results in the formation of a benign, non-invasive polyp (adenoma) protruding from the mucosal surface.
Further proliferation results in the formation of a benign, non-invasive polyp (adenoma) protruding from the mucosal surface.  The transformed cells become invasive as a result of further genetic changes; the lesion is now regarded as malignant (carcinoma).
The transformed cells become invasive as a result of further genetic changes; the lesion is now regarded as malignant (carcinoma).  The malignant cells invade blood vessels and lymphatics, and are carried to the liver and lymph nodes, respectively, to form secondary tumours (metastases)
The malignant cells invade blood vessels and lymphatics, and are carried to the liver and lymph nodes, respectively, to form secondary tumours (metastases)  .
.
A premalignant condition is one that is associated with an increased risk of malignant tumours. In chronic ulcerative colitis, for example, there is an increased risk of colorectal cancer and this can be predicted by seeking the premalignant lesion (in this case dysplasia) in rectal biopsies. Sometimes congenital abnormalities predispose to cancer; the undescended testis is, for example, more prone to neoplasms than the normally located organ.
If patients are found to have premalignant lesions and conditions they can be followed up carefully, and tumours detected at an early stage when they are more amenable to potentially curative treatment (Table 11.9). This is the principle of the population screening programmes for carcinoma of the cervix.
Table 11.9 Examples of premalignant lesions and conditions
| Lesion/condition | Cancer risk |
|---|---|
| Premalignant lesion | |
| Adenomatous polyp of colorectum | Colorectal adenocarcinoma |
| Cervical epithelial dysplasia | Carcinoma of the cervix |
| Mammary ductal epithelial hyperplasia | Carcinoma of the breast |
| Premalignant condition | |
| Hepatic cirrhosis | Hepatocellular carcinoma |
| Xeroderma pigmentosum | Skin cancer |
| Ulcerative colitis |
Transplacental carcinogenesis
In the 1940s some pregnant women with threatened miscarriages were treated with diethylstilbestrol, a synthetic oestrogenic compound, in an attempt to avert the fetus from being aborted. The female progeny of those pregnancies that went successfully to full term were later discovered to have a high incidence of vaginal adenocarcinoma, an otherwise rare tumour, in early adult life. This is an example of transplacental carcinogenesis; the carcinogen, presumably diethylstilbestrol, was administered to the mother, but the carcinogenic effect was exhibited only in the child resulting from the pregnancy, when she reached young adulthood.
CELLULAR AND MOLECULAR EVENTS IN CARCINOGENESIS




Having considered the various types of carcinogen, we can now turn our attention to the way in which these agents actually transform normal cells into neoplastic cells, capable of autonomous growth and, in malignant neoplasms, of invasion and metastasis.
Experimental observations
Evidence for a multistep theory of carcinogenesis is derived from observations on the experimental induction of tumours in laboratory animals and from the sequential genetic alterations in the development of human tumours.
Latency
Part of the reason for the long latent interval between exposure to a carcinogen and clinical recognition of the tumour is the fact that tumours result from the clonal proliferation of single cells; it takes an appreciable time for this transformed single cell to grow into a nodule of cells large enough to cause signs and symptoms. However, another important factor is that, with the possible exceptions of ionising radiation and of some fast-transforming oncogenic retroviruses, the change from a normal cell into a growing and potentially fatal neoplasm is thought to entail more than one genetic event.
Initiation, promotion and progression
Experimental carcinogenesis has revealed two major steps— initiation and promotion—in the transformation of cells from normal to neoplastic and a further step—progression— resulting in the malignant phenotype.
A frequently cited example of this sequence is the effect of successive applications of methylcholanthrene and croton oil on mouse skin. A single application of methylcholanthrene results in a visible tumour only if it is followed by repeated painting of the site with non-carcinogenic croton oil. Methylcholanthrene is the initiator inducing lesions in the DNA of the target cell, and croton oil promotes the growth of the initiated cell; further mutational events then cause the lesion to progress to malignancy (Fig. 11.25).
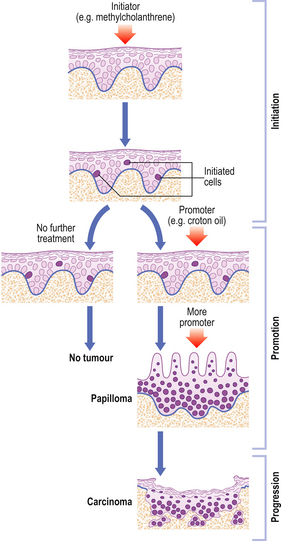
Fig. 11.25 Initiation, promotion and progression, as illustrated by the multiple steps involved in experimental chemical carcinogenesis in the epidermis. Latency is represented by the time interval between exposure to the initiating agent and the growth of a detectable neoplasm. (See text for details.)
Such experiments cannot, of course, be performed in humans, but there are many malignant tumours that develop from observable precursor lesions such as epithelial dysplasia or benign adenomas; for example, the adenoma–carcinoma sequence is well characterised in the large bowel.
This sequential development of neoplasms is associated with genetic abnormalities which drive the uncontrolled proliferation of tumour cells and, in malignant tumours, their progression to invasive behaviour.
Genetic abnormalities in tumours




Chromosomal abnormalities
The simplest technique for examining the genome of cells is chromosomal (karyotypic) analysis. This involves culturing the cells in the presence of colchicine, which blocks formation of the mitotic spindle and arrests mitosis in metaphase. On exposure to a hypotonic medium, the osmotic shock causes the cells to explode and spill their chromosomes onto the surface of a glass slide where they can be stained, counted and examined in detail. Unfortunately, at this relatively crude level of analysis in molecular terms, very few recurring patterns of chromosomal abnormality have been found in tumours. Abnormalities such as additional chromosomes and translocation of part of one chromosome to another are very common, but few are constant even among a single tumour type (Table 11.10). A notable exception is the Philadelphia chromosome; this 9;11 translocation resulting in the bcr–abl fusion gene is one of the most consistent chromosomal abnormalities yet discovered, and is commonly found in chronic myeloid (granulocytic) leukaemia. More recently, however, chromosomes have been studied by in situ hybridisation, a technique that enables determination of the number and location of specific DNA sequences (Fig. 11.26).
Table 11.10 Examples of non-random chromosomal abnormalities in neoplastic diseases
| Neoplasm | Chromosomal abnormality | Comment |
|---|---|---|
| Burkitt’s lymphoma | Translocation of c-myc oncogene from chromosome 8 to an immunoglobulin gene locus on chromosome 14 | Results in expression of c-myc gene |
| Chronic myeloid leukaemia | Translocation involving chromosomes 9 and 22 (Philadelphia chromosome) | Results in fusion of c-abl and bcr genes; bcr-abl protein has tyrosine kinase activity |
| Follicle centre cell lymphoma | Translocation involving chromosomes 14 and 18 | Results in expression of bcl-2 gene inhibiting apoptosis |
| Ewing’s tumour Peripheral neuroectodermal tumour | Translocation involving chromosomes 11 and 22 | Distinguishes these tumours from neuroblastoma, which they may resemble histologically |
Fig. 11.26 Fluorescent in situ hybridisation (FISH) of nuclei from an adult germ cell tumour of testis. This reveals, as green dots, extra copies of the short arm of chromosome 12 (12p). The fewer red dots mark the normally represented long arm of chromosome 12 (12q). The extra copies of 12p are in the form of isochromosomes (i(12p)), a useful diagnostic marker of adult germ cell tumours.
(Courtesy of Jill Elliott, Sheffield Regional Cytogenetics Service, Sheffield Children’s Hospital.)
Genetic mechanisms in carcinogenesis
Genetic alterations are the root cause of neoplastic cellular behaviour. Research has revealed that a minimum of three genetic alterations are needed to transform a normal cell into a neoplastic cell (Fig. 11.27):
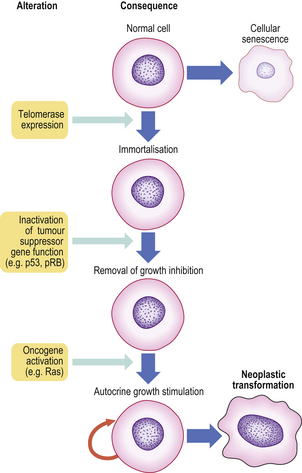
Fig. 11.27 Essential steps in neoplastic transformation. Three key genetic events are the minimum needed to convert a normal human cell into a neoplastic cell. Telomerase expression prevents telomeric shortening with each cell division and thus thwarts cellular senescence. Inactivation of tumour suppressor gene function in the immortalised cells removes inhibition of growth control. Oncogene activation sets up autocrine growth stimulation; the cell now produces a growth factor for which it already has a receptor or expresses a receptor for a growth factor it normally produces. The cell is now fully transformed.
(Based on observations by Hahn WC, Counter CM, Lundberg AS et al 1999 Creation of human tumor cells with defined genetic elements. Nature 400: 464–468)
Telomerase expression confers immortalisation on the cells. Cells lacking telomerase (most cells in the body) have only a limited replicative ability. Our chromosomal telomeres shorten as we age. Eventually the telomeres become so short with each cell division that replication cannot start, and cellular senescence and death ensues.
Tumour suppressor gene inactivation and abnormal oncogene expression work in concert (Fig. 11.28) to drive cells from their normal state of regulated growth to the dysregulated and uncontrolled growth that characterises neoplastic cells.

Fig. 11.28 Oncogenes and tumour suppressor genes. Abnormal expression of oncogenes drives normal cells towards the neoplastic state. Loss of tumour suppressor gene function enables neoplastic transformation by permitting mutations.
Genomic instability
Maintenance of genomic integrity involves genes and their products (e.g. p53) involved in sensing and repairing DNA damage. Failure of these processes causes genomic instability, an important general mechanism enabling the specific genetic alterations associated with neoplastic transformation. Cells that have lost these mechanisms for preserving genomic integrity are said to have a mutator phenotype.
Genomic instability increases naturally with age and is itself postulated to be involved in the ageing process. However, there are also inherited conditions characterised by genomic instability that indicate two major levels at which preservation of DNA integrity may fail:
Tumour suppressor genes
Clues to the existence of inhibitory genes came from observations on the behaviour of transformed cells that were fused with untransformed cells; the resulting hybrid cells behaved like untransformed cells until specific chromosomes bearing the inhibitory genes were lost, causing the cells to revert to their transformed state.
The existence of these inhibitory genes was also postulated by Alfred Knudson in 1971. Using a statistical approach to familial cancer incidence he formulated a two-hit hypothesis. The first ‘hit’ is the inheritance of a defective (mutant) allele of a tumour suppressor gene, the other allele being normal (wild) and expressing sufficient suppressive effect. The second ‘hit’ is the mutational loss of function of the normal allele, thus now fully depriving the cell of the suppressive effect of that tumour suppressor gene.
‘Caretakers’ and ‘gatekeepers’
Tumour suppressor genes are categorised further according to their mechanism of action:
Examples and the tumour susceptibilities with which inherited abnormalities of these genes are associated are given in Table 11.11.
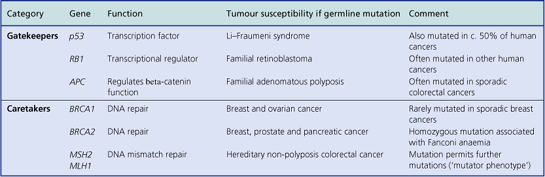
The RB gene was the first inhibitory gene to have been well characterised, and is associated with retinoblastomas. Retinoblastomas are malignant tumours derived from the retina; they occur almost exclusively in children. In some cases they are hereditary, occurring bilaterally and also in some of the patient’s siblings. In other cases they are sporadic, occurring unilaterally and without any familial associations. Individuals with hereditary retinoblastomas show a germline deletion on chromosome 13, corresponding to the known site of the RB1 gene. Therefore, only one further mutational loss of the paired gene in the target retinal cell is required for the tumour to develop. Sporadic retinoblastoma cases have a normal chromosome 13 and therefore require two mutational losses before the tumour can develop (Fig. 11.29).
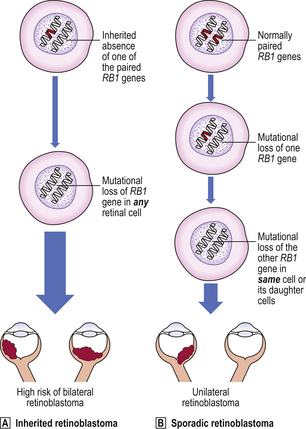
Fig. 11.29 Loss of tumour suppressor gene function and inherited retinoblastoma. Loss of functional tumour suppressor genes permits tumour development.  Individuals with an inherited risk of retinoblastoma are born with a predisposing germline mutation in one of the paired alleles of the RB1 suppressor gene; mutational loss of the remaining RB1 allele is required for retinoblastomas to develop.
Individuals with an inherited risk of retinoblastoma are born with a predisposing germline mutation in one of the paired alleles of the RB1 suppressor gene; mutational loss of the remaining RB1 allele is required for retinoblastomas to develop.  Normal individuals without an inherited germline mutation of the RB1 gene have a low incidence of retinoblastoma because acquired mutations in both alleles have to occur in the same cell or its daughters; sporadic retinoblastoma is, therefore, a rare event.
Normal individuals without an inherited germline mutation of the RB1 gene have a low incidence of retinoblastoma because acquired mutations in both alleles have to occur in the same cell or its daughters; sporadic retinoblastoma is, therefore, a rare event.
The tumour suppressor gene p53, situated on the short arm of chromosome 17, is the gene most frequently mutated and extensively studied in human cancer. The normal functions of p53 are to enable:
The p53 levels rise in cells that have sustained DNA damage, until either the damage is repaired or the cell undergoes apoptosis. This prevents propagation of possibly mutated genes. This important function of p53 results in it being called ‘the guardian of the genome’.p53 can lose its normal function by a variety of mechanisms:
These events have major implications. Cells with damaged DNA, possibly with mutated oncogenes, undergo mitotic replication rather than apoptotic death (Fig. 11.30). Also, cytotoxic chemotherapy against the tumour may be less effective if the cells fail to respond by apoptosis.
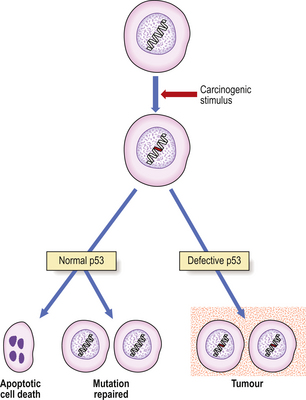
Fig. 11.30 Role of p53 in cells with damaged DNA. In the presence of normal p53, cells with a mutation resulting from a potentially carcinogenic stimulus are arrested in G1 of the cell cycle until either the mutation is repaired or, if the damage is severe, apoptosis occurs. If the p53 is defective, as a result of mutation or binding, the cells proceed to S phase and the mutation is propagated to daughter cells, possibly eventually leading to a tumour.
Inherited germline (present in all cells) mutations of p53 occur in the rare Li–Fraumeni syndrome. Affected individuals have an inherited predisposition to a wide range of tumours. At birth they are heterozygous for the defective gene (only very rarely are the maternal and paternal alleles both defective). Eventually, the normal allele is itself lost or mutated (loss of heterozygosity) in a variety of cells, thus enabling their neoplastic transformation.
Oncogenes
Oncogenes are genes driving the neoplastic behaviour of cells. Originally proposed as a hypothesis, oncogenes were discovered as a result of studies of oncogenic RNA retroviruses. These are RNA viruses that have the ability to transfer their genome, or parts of it, to the genome of the cells they infect. Normally the transfer of genomic information is in the opposite direction: DNA sequences are transcribed into RNA, which then determines the amino acid sequence of a peptide or protein. However, retroviruses contain an enzyme, reverse transcriptase, that enables the viral RNA to be transcribed into complementary DNA which is then incorporated into the infected cell’s genome. In the case of oncogenic retroviruses, these genes were called oncogenes.
The next major discovery was of the presence of DNA sequences almost identical to viral oncogenes (v-oncogenes) in the genome of normal cells (cellular or proto-oncogenes). Numerous oncogenes have now been identified. However, in normal cells these oncogenes are present at the frequency of only one copy per haploid genome, and their transcription is tightly controlled as required for cell growth and differentiation.
They are present in the genome of even the most primitive protozoa and metazoa; this high degree of evolutionary conservation implies a function indispensable to normal life. The result of much research now leads us to conclude that these cellular oncogenes are essential for normal cell and tissue growth and differentiation, particularly during embryogenesis and healing. But when they are aberrant or inappropriately expressed they result in the growth of a tumour.
Normal or partially transformed cell cultures can be fully transformed by the addition of DNA bearing oncogenes, a process known as transfection. Alternatively, oncogenic (or carcinogenic) retroviruses can transform cells by transferring oncogenes from another cell, a process known as transduction.
Oncogenes can be classified into five groups according to the function of the gene product (oncoprotein):
Abnormalities of oncogene expression are crucial for the growth and behaviour of tumour cells.
Abnormalities of oncogene expression in tumours
Oncogenes can have tumour-promoting effects by either:
Increased expression of oncogenes has been found in most tumours. The mechanisms are summarised in Figure 11.31. Increased expression may be detected by:
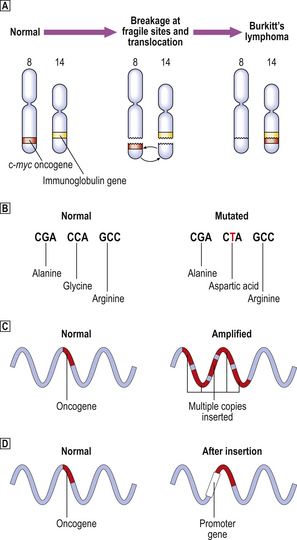
Fig. 11.31 Mechanisms of oncogene activation.  Translocation of an oncogene from an untranscribed site to a position adjacent to an actively transcribed gene; e.g. simplified chromosomal translocation in Burkitt’s lymphoma, in which the c-myc oncogene is often translocated from chromosome 8, its normal location, to chromosome 14, where it is placed adjacent to one of the immunoglobulin genes and is thus inappropriately transcribed.
Translocation of an oncogene from an untranscribed site to a position adjacent to an actively transcribed gene; e.g. simplified chromosomal translocation in Burkitt’s lymphoma, in which the c-myc oncogene is often translocated from chromosome 8, its normal location, to chromosome 14, where it is placed adjacent to one of the immunoglobulin genes and is thus inappropriately transcribed.  Point mutation (in this case in codon 12 of the ras oncogene), in which the substitution of a single base in the oncogene is translated into an amino acid substitution in the oncoprotein causing it to be hyperactive.
Point mutation (in this case in codon 12 of the ras oncogene), in which the substitution of a single base in the oncogene is translated into an amino acid substitution in the oncoprotein causing it to be hyperactive.  Amplification by the insertion of multiple copies of the oncogene (in this case, c-myc in neuroblastoma), resulting in cellular proliferation stimulated by excessive quantities of the oncoprotein.
Amplification by the insertion of multiple copies of the oncogene (in this case, c-myc in neuroblastoma), resulting in cellular proliferation stimulated by excessive quantities of the oncoprotein.  Increased oncogene expression by gene insertion (insertional mutagenesis) resulting in proximity of an oncogene to a promoter or enhancing gene; this is one mechanism of retroviral carcinogenesis.
Increased oncogene expression by gene insertion (insertional mutagenesis) resulting in proximity of an oncogene to a promoter or enhancing gene; this is one mechanism of retroviral carcinogenesis.
Increased numbers of oncogene copies may result from infection by a retrovirus which causes reverse transcription of its RNA and insertion of multiple copies of the resulting DNA into the DNA of the host cell genome. A more common occurrence in human tumours is gene amplification resulting in multiple copies, such as in the myc family of oncogenes in neuroblastoma; this can be recognised in chromosome preparations from tumour cells by the presence of homogeneously staining regions and double minute chromosomes.
Increased transcription can occur if a normally silent (i.e. not transcribed) oncogene is moved to another part of the genome where active transcription is occurring. This is often evident from the karyotype; part of one chromosome which is known to bear an oncogene may be translocated to another chromosome where a gene known to be actively transcribed is situated. Specific examples include:
Alternatively, the cellular oncogene may undergo a point mutation resulting in a gene product, such as a protein kinase, with increased or inappropriate activity.
Autocrine stimulation of neoplastic cell growth
Oncogene products play an important role in cellular growth and behaviour (Table 11.12). By their expression in inappropriate circumstances a cell can become autonomous, proliferating without the usual requirement for external signals (Fig. 11.32). For example, an oncogene product may be a receptor for a growth factor (Ch. 5) already normally produced by that cell; the cell then responds to stimulation by its own growth factor. Alternatively, the oncogene product may be a growth factor for which the cell already normally bears a specific receptor. In both cases the result is autocrine stimulation of growth.
Table 11.12 Examples of oncogenes and the function of their products
| Oncogene | Function of oncoprotein | Abbreviated from |
|---|---|---|
| abl | Protein-tyrosine kinase activity | Abelson mouse leukaemia |
| myc | Binds to DNA, directly stimulating synthesis | Myelocytomatosis |
| sis | Growth factor (platelet-derived growth factor) | Simian sarcoma |
| erbB | Receptor for epidermal growth factor | Avian erythroblastosis (also erbA) |
| ras | Acts on intracellular signalling (cyclic nucleotides) | Rat sarcoma |
Fig. 11.32 Self-stimulation of neoplastic cell proliferation mediated by oncoproteins. (A) Direct stimulation of DNA synthesis by an oncoprotein that binds to DNA, (B) Synthesis of receptors (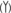 ) for a growth factor (red triangles) already present in the extracellular environment. (C) Synthesis of a growth factor (green circles), receptors for which (
) for a growth factor (red triangles) already present in the extracellular environment. (C) Synthesis of a growth factor (green circles), receptors for which ( ) are normally present on the cell. (D) Interference with intracellular signalling between the cell membrane and the nucleus.
) are normally present on the cell. (D) Interference with intracellular signalling between the cell membrane and the nucleus.
Other oncogene products act directly within the nucleus to stimulate mitosis or on intracytoplasmic second messengers, such as cyclic nucleotides, thus modulating intracellular signalling.
Interaction of carcinogens with oncogenes and tumour suppressor genes
The neoplastic behaviour of tumour cells persists after withdrawal of carcinogenic stimuli and this behaviour is passed on to subsequent cellular generations through mitotic divisions; it is therefore concluded that the lesion responsible for neoplastic behaviour is within the genome. This has led to a search for the final common pathway through which the very diverse range of known carcinogens acts—a search for the molecular lesion within the genome that is the end result of carcinogenesis.
Ultimately, the metabolism of chemical carcinogens results in the formation of DNA adducts, but the mere presence of adducts is insufficient for tumours to develop. Further molecular alterations, such as mutations, during DNA replication, and clonal expansion of the mutated cells are required before a tumour results. The formation of adducts can be reversed by virtue of their innate instability, or by DNA repair enzymes; their effect may also be minimised by dilution with new DNA through normal replication.
The selectivity of a carcinogenic metabolite for a particular nucleotide is thought to explain the site-specific mutations induced in oncogenes that can result in their abnormal expression. For example, several chemical carcinogens have been shown experimentally to result in single base substitutions in codons 12 and 61 of the ras oncogene, leading to the synthesis of a hyperactive mutant protein. Site-specific mutations of p53 are present in hepatocellular carcinomas associated with aflatoxin exposure.
The mutational effects of ionising radiation are probably random throughout the genome, but when they occur in oncogenes or tumour suppressor genes the cells harbouring the mutant genes have a selective growth advantage, eventually resulting in tumours.
The role of viruses in tumour induction can be attributed directly to the genetic material within them and its incorporation within the host cell.
Epigenetic control of tumour growth
Current evidence emphasises the genetic theory of carcinogenesis, that tumours result only from induced or congenital genetic abnormalities that dictate the aberrant behaviour of the cells. Experimental evidence favouring the epigenetic theory—that the behaviour of tumour cells results from the expression of deregulated or abnormally controlled non-mutated genes—does not necessarily imply that the primary carcinogenic event is epigenetic; it simply shows that, in some instances, the neoplastic behaviour of tumour cells can be influenced by epigenetic factors. Possible mechanisms include:
In some neoplasms at least, an epigenetic influence can be demonstrated. If the cells of a malignant teratoma are injected into an early mouse embryo, the neoplastic cells differentiate normally and no tumour develops. In other words, the otherwise autonomous growth and incompletely differentiated state of this particular malignant neoplasm can be corrected by an epigenetic influence.
BEHAVIOUR OF TUMOURS
The clinical effects of tumours are determined by the biological behaviour of the neoplastic cells within them. The most important property of malignant tumours is the ability to invade and metastasise.
Invasion and metastasis




Invasion and metastasis are responsible for most of the fatal consequences of tumours. They also dictate the most appropriate treatment. There is no point in simply removing the tumour itself. In most instances the tumour should be removed in continuity with a wide margin of apparently normal tissue, to ensure that the plane of resection is clear of the often ill-defined invasive edge of the tumour; the regional lymph nodes may also be resected. Incomplete local removal of a tumour may result in a local recurrence because the original plane of resection transected the invasive edge of the lesion.
Tumours should be manipulated with care during clinical examination or surgical removal, to minimise the risk of pumping tumour cells into blood and lymphatic channels. A ligature is therefore often tied around the vascular pedicle at an early stage in the surgical removal of a tumour.
In epithelial neoplasms, invasion and metastasis require the acquisition of motile and migratory properties normally associated with cells of mesenchymal lineage. This shift in behaviour is often referred to as epithelial–mesenchymal transition.
Invasion
The invasiveness of malignant neoplasms is determined by the properties of the neoplastic cells within them. Factors influencing tumour invasion are:
Cellular motility is abnormal in that the cells are not only more motile than their normal counterparts (which may not move at all), but also show loss of the normal mechanism that arrests or reverses normal cellular migration: contact inhibition of migration.
Proteinases and inhibitors
Matrix metalloproteinases are among the most important proteinases in neoplastic invasion. These enzymes are secreted by malignant neoplastic cells, enabling them to digest the surrounding connective tissue. There are three families:
These enzymes are counteracted by tissue inhibitors of metalloproteinases (TIMPs). The net effect is determined by the balance between metalloproteinases and their inhibitors. It may be possible to limit the invasiveness of tumour cells by artificially increasing the level of inhibitory activity.
Invasion often occurs along tissue planes offering less resistance to tumour growth, such as perineural spaces and, of course, vascular lumina. Other tissues are extremely resistant to neoplastic invasion, such as cartilage and the fibrocartilage of intervertebral discs.
Clinicopathological significance
Invasion is the single most important criterion of malignancy. Metastases are a consequence of invasion and, when detected clinically, are unequivocal markers of malignancy. In epithelial tumours, invasion is relatively easy to recognise because the basement membrane serves as a clear line of demarcation between the tissue boundaries (Fig. 11.14). In connective tissue tumours, invasion is less easy to recognise unless there is clear evidence of vascular or lymphatic permeation; other histological features, such as mitotic activity, are usually assessed for prognostic purposes.
Invasion within epithelium is known as pagetoid infiltration; it is named after Paget’s disease of the nipple, which is due to infiltration of the epidermis of the nipple by tumour cells from a ductal carcinoma in the underlying breast. This pattern of invasion can also occur with a few other epithelial malignancies.
Metastasis
Metastasis is the process whereby malignant tumours spread from their site of origin (the primary tumour) to form other tumours (secondary tumours) at distant sites. The total tumour burden resulting from this process can be very great indeed, and the total mass of the secondary tumours invariably exceeds that of the primary lesion; it is not uncommon at autopsy to find a liver weighing several kilograms more than normal, laden with metastases. The word carcinomatosis is used to denote extensive metastatic disease.
Sometimes, metastases can be the presenting clinical feature. Bone pain or fractures due to skeletal metastases can be the first manifestation of a clinically occult internal malignancy. Palpable lymph nodes, due to metastatic involvement, may appear before the signs and symptoms of the primary tumour.
The metastatic cascade
Neoplastic cells must successfully complete a cascade of events before forming a metastatic tumour (Fig. 11.33). Only a proportion of the neoplastic cells in a malignant tumour may have the full repertoire of properties necessary for completion of the cascade. Many tumours studied experimentally in animals consist of metastatic and non-metastatic clones, and metastatic tumours in humans often appear histologically less well differentiated than the primary lesion, suggesting that there is clonal evolution of the metastatic phenotype. There is experimental evidence for the inactivation of ‘anti-metastatic’ genes (‘metastogenes’), such as nm23, in neoplastic cells capable of metastasis, but their precise role in the metastatic cascade in human tumours is uncertain.
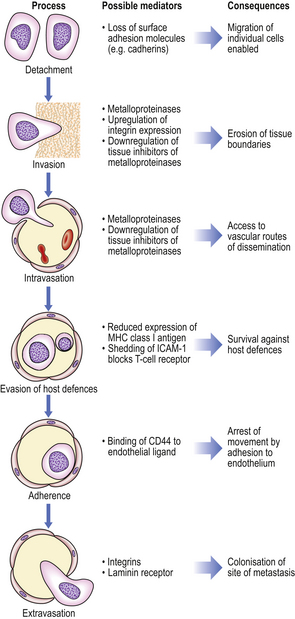
Fig. 11.33 Metastatic cascade. The spread of tumour cells from the site of origin, the primary tumour, to form secondary tumours in other locations requires completion of a logical sequence of events mediated by tumour–host interactions.
The sequential steps involved in the metastatic cascade are:
On reaching the site of metastasis there is a recapitulation of the events that were required for primary tumour growth. The tumour cells must proliferate and, if they are to grow to form a nodule larger than a few millimetres in diameter, the ingrowth of blood vessels must be elicited by angiogenic factors.
Alterations in cell adhesion molecules are important at several points in the metastatic cascade; these affect cell–cell and cell–substrate adhesion. Studies on experimental and human tumours show that reduced expression of cadherins, which are involved in adhesion between epithelial cells, correlates positively with invasive and metastatic behaviour. Increased expression of integrins appears to be important for the invasive migration of neoplastic cells into connective tissues.
Routes of metastasis
The routes of metastasis are (Fig. 11.34):
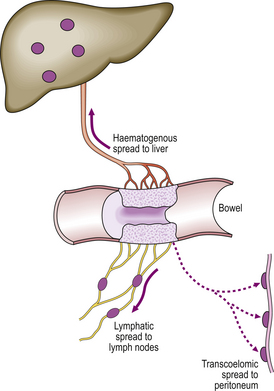
Carcinomas tend to prefer lymphatic spread, at least initially, while sarcomas prefer haematogenous spread. However, exceptions to these tendencies are common, and carcinomas often generate blood-borne metastases.
Haematogenous metastasis
Bone is a site favoured by haematogenous metastases from five carcinomas—lung, breast, kidney, thyroid and prostate. Other organs commonly involved by haematogenous metastases are lung, liver and brain (Fig. 11.35). The metastases are frequently multiple, whereas primary tumours arising in the affected organs are usually solitary. Curiously, tumours rarely metastasise to skeletal muscle or to the spleen, despite their lavish blood supply.
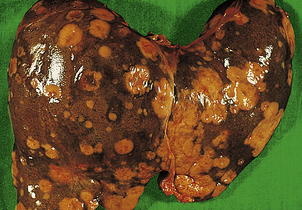
Lymphatic metastasis
Tumour cells reach the lymph node through the afferent lymphatic channel. The tumour cells settle and grow in the periphery of the node, gradually extending to replace it (Fig. 11.36).Lymph nodes involved by metastatic tumours are usually firmer and larger than normal. Groups of involved lymph nodes may be matted together by both tumour tissue and the connective tissue reaction to it. Lymph node metastases often interrupt lymphatic flow, thus causing oedema in the territory that they drain.
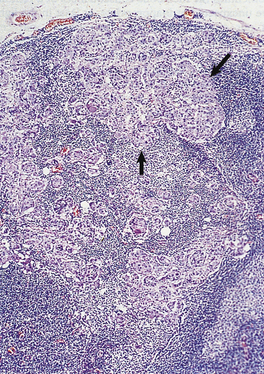
Fig. 11.36 Lymph node metastasis. The lymph node is partly replaced by a deposit of metastatic adenocarcinoma (arrowed) from a primary in the stomach.
Clinically, it is necessary to be cautious in interpreting the significance of enlarged lymph nodes draining tumours because the enlargement could simply be due to reactive changes.
Transcoelomic metastasis
The peritoneal, pleural and pericardial cavities are common sites of transcoelomic metastasis, which results in an effusion of fluid into the cavity. The fluid is rich in protein (i.e. it is an exudate) and may contain fibrin. The fluid also contains the neoplastic cells causing the effusion, and cytological examination of the aspirated fluid is very important in diagnosing the cause of effusions into body cavities. The tumour cells often grow as nodules on the mesothelial surface of the cavity.
Peritoneal effusions (ascites) may be due to involvement by any abdominal tumour, but primaries within the ovaries are particularly common. Pleural and pericardial effusions are common consequences of carcinomas of the breast and lung.
Clinical effects of tumours



The clinical effects of tumours are attributable to their location, their cell of origin and their behaviour. The effects may be local, or occur at some distance from the tumour.
Local effects
Tumours exert local effects through compression and displacement of adjacent tissues and, if malignant, through their destruction by actual invasion. These effects can be clinically inconsequential if the organ is large relative to the size of the tumour or if no vital structure is threatened. However, even benign tumours can have life-threatening effects on neighbouring structures; for example, a functionally inactive adenoma of the pituitary gland may obliterate the adjacent functioning pituitary tissue, such is the confined space in which the gland sits, resulting in hypopituitarism.
Malignant neoplasms obviously have more serious local effects because they invade and destroy local structures. This may be rapidly fatal if a vital structure is eroded, for example a pulmonary artery by a carcinoma of the lung. In the case of basal cell carcinoma of the skin (‘rodent ulcer’), its local effects are sufficient to justify the label ‘carcinoma’ because, although the tumour rarely metastasises, its invasiveness can be very disfiguring.
Malignant tumours on mucosal surfaces are often ulcerated. Blood can ooze from these lesions; this blood loss can beoccult in the case of gastrointestinal tumours and this is a very important cause of anaemia. Ulcerated surfaces also expose the patient to the risk of infection.
Metabolic effects
The metabolic effects of tumours can be subdivided into those specific to individual tumours and those common to many tumours.
Tumour-type specific effects
Well-differentiated endocrine tumours often retain the functional properties of the parent tissue. Since such tumours are relatively autonomous and because the total number of functioning cells often greatly exceeds that in the normal organ, clinical effects are common. For example:
Sometimes the metabolic consequences of a tumour are unexpected or inappropriate, at least in the light of our current knowledge; for example, small-cell carcinomas of the lung commonly secrete ACTH and ADH, although this rarely gives rise to clinically significant consequences.
Other specific tumour-associated phenomena have no metabolic consequences but are nevertheless probably mediated by humoral factors. The most common example is finger-clubbing and hypertrophic osteoarthropathy in patients with carcinoma of the lung.
Non-specific metabolic effects
Disseminated malignant tumours are commonly associated with profound weight loss despite apparently adequate nutrition. The catabolic clinical state of a cancer patient with severe weight loss and debility is known as cachexia and is thought to be mediated by tumour-derived humoral factors that interfere with protein metabolism. Cachexia can also occur quite early in the course of the disease, notably in patients with carcinoma of the lung. Weight loss can, of course, also be due to interference with nutrition because of, for example, oesophageal obstruction, severe pain or depressive illness.
Neuropathies and myopathies are associated with the presence of malignant neoplasms, particularly with carcinoma of the lung. A tendency to venous thrombosis is associated with mucus-producing adenocarcinomas, notably of the pancreas. Glomerular injury can result from deposition of immune complexes in which one of the ingredients is tumour antigen (Ch. 9).
Prognosis
Malignant tumours have a variable prognosis (Table 11.13). This is determined partly by the innate characteristics of the tumour cells (e.g. growth rate, invasiveness), and partly by the effectiveness of modern cancer therapy for individual types of tumour.
Table 11.13 Prognosis of some different types of solid malignant tumour, based on experience of responses to treatment in the UK
| Prognostic category | ||
|---|---|---|
| Good | Intermediate | Poor |
A good prognosis implies a greater than 80% 5-year survival; poor prognosis implies a less than 20% 5-year survival. Prognosis in individual cases is, of course, influenced by tumour grade and stage at presentation.
Prognostic indices
One of the major efforts in histopathology continues to be the search for features that more accurately predict the likely behaviour of individual tumours. It is insufficient merely to diagnose a tumour as malignant and to identify its origin. The patient’s treatment is guided by the most accurate determination of:
It is also important to determine whether the presenting lesion is a primary tumour or a metastasis. This can be difficult. There may be little point in performing radical surgery to remove a tumour if it is a metastasis, and the primary tumour and perhaps other metastases remain in the patient.
Tumour type
The tumour type is usually determined from the growth pattern of the tumour and its relationship to the surrounding structures from which a direct origin may be evident. Thus, a gland-forming neoplasm in the breast is most likely to be a primary adenocarcinoma of the breast, particularly if carcinoma cells are also present within the breast ducts near the tumour (ductal carcinoma in situ). A squamous cell carcinoma is often recognisable from the production of keratin, and it may be in continuity with adjacent squamous epithelium that may show carcinoma in situ.
Some types of tumour need to be subclassified because variants with differing behaviour exist. Malignant lymphomas, for example, are subclassified into Hodgkin’s and non-Hodgkin’s lymphoma, each of which is then further subclassified by detailed appraisal of the histology (Ch. 22).
Genetic analysis or immunohistology may be necessary to type tumours that do not have obvious differentiated features detectable on routine light microscopy.
Tumour grade
The grade of a tumour is an assessment of its degree of malignancy or aggressiveness. This can be inferred from its histology. The most important features contributing to the assessment of tumour grade are:
Grading systems have been devised for many types of tumour,and most involve an assessment of the above features. Tumours are often heterogeneous, and the grading should be performed on what appears to be the least differentiated area as this is likely to contain the most aggressive clone or clones of tumour cells.
Tumour stage
The stage of a tumour is the extent of spread. This is determined by histopathological examination of the resected tumour and by clinical assessment of the patient, often involving imaging techniques. Perhaps the best-known staging system is that devised in the 1930s by Cuthbert Dukes for colorectal carcinomas (Ch. 15):
The most generally applicable staging system is the TNM system (Fig. 11.37):
Fig. 11.37 Summary of TNM system for staging of tumours. This concept (tumour, nodes and distant metastases) is the basis of most tumour staging systems.
For example, a T1 breast carcinoma is equal to, or less than, 20mm in diameter; large numbers denote large tumours. N0 denotes no nodal metastases, N1 one or few nodal metastases, and N2 many nodal metastases. M0 denotes an absence of metastases, and M1 and greater denotes increasing extent of distant metastases.
For many tumours, the TNM status is used to derive a stage score. Typically, a stage 1 tumour is confined to the organ of origin and a stage 4 tumour has disseminated widely.
Tumour dormancy
After surgical removal, radiotherapy and/or chemotherapy there may be no clinically detectable tumour remaining in a patient. This does not necessarily mean that the tumour has been completely eradicated: minute deposits can evade detection by even the most sophisticated imaging techniques. These occult tumour foci can remain clinically dormant for perhaps several years before their regrowth causes signs and symptoms. For this reason, it is virtually impossible to speak of a cancer patient as being ‘cured’, and prognosis can be given only in terms of the probability of survival or the length of the disease-free interval. The prognostic information derived from tumour type, grade and stage is used to predict the patient’s chances of surviving, say, 5 years.
EARLY DETECTION OF CANCER BY SCREENING
Because of the dynamics of neoplastic progression and spread, early diagnosis is just as important as treatment in determining the outcome of the disease. The success of early diagnosis relies upon finding tumours at a curable stage before they have had a chance to spread from their site of origin. This is best achieved by screening asymptomatic people, concentrating on those at greatest risk, in the hope of detecting very early lesions (Fig. 11.38). In many countries there are active screening programmes for cervical and breast cancer; screening for colorectal cancer is also being introduced.
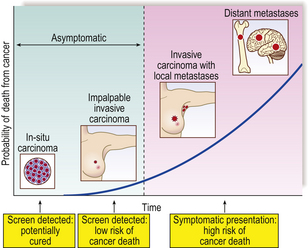
Fig. 11.38 Pathological basis of cancer screening. Using breast cancer as the example, detection at the pre-invasive stage of ductal carcinoma in situ confers a potential cure because there is no risk of metastases. Once the tumour has invaded and gained access to blood vessels and lymphatics, the prospect of cure progressively diminishes.
Cervical intra-epithelial neoplasia (CIN) can be detected by exfoliative cytology of the cervix. Cells are scraped from the cervix, deposited on to glass slides, stained, and then examined by an expert cytologist trained to detect subtle abnormalities. Breast cancer can be detected at an early stage by regular screening by mammography (X-ray imaging of the breast), followed by diagnosis of any abnormalities by fine-needle aspiration cytology or biopsy.
While early cancer detection is of proven benefit in individual cases, the overall population benefit may be less than anticipated. This is partly because some people are reluctant to be screened; those that do volunteer may not be from the socio-economic groups most at risk, particularly in the case of cancer of the cervix. Furthermore, early detection may not significantly affect the overall mortality from the screened cancer, but merely cause individuals premature anxiety about a disease that would not have become symptomatic for a few more years. Finally, it is not certain that all of the early abnormalities detected by screening would have progressed to more serious lesions within the otherwise natural lifetime of the individual concerned. Thus, the biases that must be allowed for in measuring the benefits of screening are:
However, despite these possible reasons for an exaggerated benefit, screening for early cancers and precursor lesions is yielding genuine reductions in cancer mortality.
Commonly confused conditions and entities relating to carcinogenesis and neoplasia
| Commonly confused | Distinction and explanation |
|---|---|
| Proto-oncogenes, cellular oncogenes and oncogenes | Proto-oncogenes and cellular oncogenes are normal unmutated genes with important functions in morphogenesis and in the growth and differentiation of normal cells. When these genes become mutated or abnormally expressed as part of the neoplastic process, they are referred to as oncogenes. |
| Gatekeepers and caretakers | These are two categories of tumour suppressor gene. Gatekeeper genes stop cells with mutated or damaged DNA from proceeding through the cell cycle and replicating the error. Caretaker genes repair damaged DNA. |
| Grade and stage | The grade of a tumour is its degree of histological resemblance to the parent tissue. The stage of a tumour is its anatomical extent of spread. |
| Histogenesis and differentiation | Histogenesis indicates the cell type from which a neoplasm has arisen; this can be often deduced from the morphological features of the neoplastic cells. Differentiation is the extent to which the neoplastic cells resemble the normal cell lineage from which they are assumed to have arisen. |
| Sarcoma and carcinoma | Both are malignant neoplasms. A sarcoma is of connective tissue (mesenchymal) origin. A carcinoma is of epithelial origin. |
| In situ carcinoma and intra-epithelial neoplasia | An in situ carcinoma has all the cytological features of a malignant epithelial neoplasm, but has not yet invaded through the basement membrane and, therefore, cannot have metastasised. Because epithelial dysplasia (disordered differentiation) may progress to in situ carcinoma and histopathologists may be unable reliably to distinguish the entities, they are merged together as intra-epithelial neoplasia and, for example, in the cervix categorised according to the severity of the abnormality (CIN1, CIN2 or CIN3). |
Alizadeh A.A., Ross D.T., Perou C.M., van de Rijn M.. Towards a novel classification of human malignancies based on gene expression patterns. Journal of Pathology. 2001;195:41-52.
Baak J.P.A., Hermsen M.A.J.A., Meijer G., et al. Genomics and proteomics in cancer. European Journal of Cancer. 2003;39:1199-1215.
Bicknell R., Lewis C.E., Ferrara N.. Tumour angiogenesis. Oxford: Oxford University Press; 1997.
Butel J.S.. Viral carcinogenesis: revelation of molecular mechanisms and etiology of human disease. Carcinogenesis. 2000;21:405-426.
De Wever O., Mareel M.. Role of tissue stroma in cancer cell invasion. Journal of Pathology. 2003;200:429-447.
Eccles S.A., Welch D.R.. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742-1757.
Fearon E.R.. Human cancer syndromes: clues to the origin and nature of cancer. Science. 1997;278:1043-1050.
Finkel T., Serrano M., Blasco M.A.. The common biology of cancer and ageing. Nature. 2007;448:767-774.
Fletcher C.D.M., editor. Diagnostic histopathology of tumours, 3rd edn, Edinburgh: Churchill Livingstone, 2007.
Haber D.A., Fearon E.R.. The promise of cancer genetics. Lancet. 1998;351(SII):1-8.
Hanahan D., Weinberg R.A.. The hallmarks of cancer. Cell. 2000;100:57-70.
Jass J.R.. Familial colorectal cancer: pathology and molecular characteristics. Lancet Oncology. 2000;1:220-226.
Kastan M.B., Bartek J.. Cell-cycle checkpoints and cancer. Nature. 2004;432:316-323.
Keith W.N., Evans T.R., Glasspool R.M.. Telomerase and cancer: time to move from a promising target to a clinical reality. Journal of Pathology. 2001;195:404-414.
Kinzler K.W., Vogelstein B.. Gatekeepers and caretakers. Nature. 1997;386:761-763.
Loeb L.A.. A mutator phenotype in cancer. Cancer Research. 2001;61:3230-3239.
Murphy G.. Matrix metalloproteinases in neoplastic progression: where are we now? Recent Advances in Histopathology. 2007;22:81-92.
Rabbitts T.H.. Chromosomal translocations in human cancer. Nature. 1994;372:143-149.
Stiewe T.. The p53 family in differentiation and tumorigenesis. Nature Reviews. Cancer. 2007;7:165-168.
Thompson E.W., Newgreen D.F.. Carcinoma invasion and metastasis: a role for epithelial–mesenchymal transition. Cancer Research. 2005;65:5991-5999.
Weitzman J.B., Yaniv M.. Rebuilding the road to cancer. Nature. 1999;400:401-402.
Yokota J.. Tumor progression and metastasis. Carcinogenesis. 2000;21:497-503.
Cancer Research UK website (a good source of key facts about different cancers, with statistical data): http://www.cancerresearchuk.org




