[level-membership-for-pathology-category]
Chapter 9 Immunology and immunopathology
DEFENCE AGAINST INFECTION



The immune system evolved as a defence against infectious diseases. Individuals with deficient immune responses, if untreated, succumb to infections in early life. There is, therefore, a selective evolutionary pressure for an efficient immune system. Specific immunity is called into play only when micro-organisms bypass non-specific or innate mechanisms.
Non-specific defences
Many non-specific mechanisms prevent invasion of the body by micro-organisms:
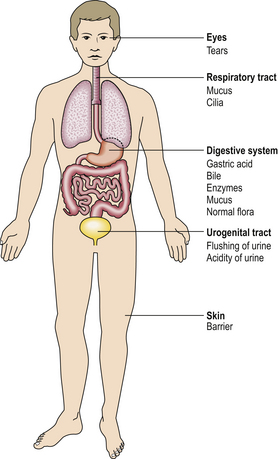
Innate immunity
The innate immune system is activated by pattern recognition receptors on dendritic cells recognising conserved polysaccharide molecular patterns on microbes. Key components include:
Specific immunity
The immune system has four essential features:
A specific or adaptive immune response consists of two parts: a specific response to the particular antigen and a non-specific augmentation of the effect of that response. For the specific response there is a quicker and larger response the second time that a particular antigen is encountered; memory of the initial specific immune response provides the efficiency.
The immune system has to recognise all pathogens, past and future, and must have considerable diversity of response. This diversity is partly genetic (germline encoded) and partly generated by somatic mutation during maturation of the immune system.
Immune responses, both innate and adaptive, have two phases: first the recognition phase, involving antigen-presenting cells and T-lymphocytes (see Key molecules), in which the antigen is recognised as foreign; and second the effector phase, in which antibodies and effector T-lymphocytes eliminate the antigen, often by recruiting innate mechanisms such as complement or macrophage activation.
KEY MOLECULES
 Antigens are substances able to provoke an immune response and react with the products of that response
Antigens are substances able to provoke an immune response and react with the products of that response Antibodies are immunoglobulin molecules produced by plasma cells. Antigen-binding properties reside in the Fab fragments, while effector functions lie in the Fc fragment
Antibodies are immunoglobulin molecules produced by plasma cells. Antigen-binding properties reside in the Fab fragments, while effector functions lie in the Fc fragment
 Major histocompatibility complex (MHC) antigens are of two main types—class I and class II. They play a fundamental role in the normal immune response by presenting antigenic peptides to T-cells
Major histocompatibility complex (MHC) antigens are of two main types—class I and class II. They play a fundamental role in the normal immune response by presenting antigenic peptides to T-cells Helper T-cells recognise antigen in association with MHC class II molecules, while cytotoxic T-cells recognise antigen associated with MHC class I
Helper T-cells recognise antigen in association with MHC class II molecules, while cytotoxic T-cells recognise antigen associated with MHC class I T-cell receptors will recognise antigen only as part of a complex of antigenic peptide and the MHC molecule—a process termed MHC restriction
T-cell receptors will recognise antigen only as part of a complex of antigenic peptide and the MHC molecule—a process termed MHC restriction

Antigens
Antigens are substances able to provoke an immune response and react with the immune products. They react both with the T-cell recognition receptor and with antibody. An antigenic molecule may have several antigenic determinants (epitopes); each epitope can bind with an individual antibody, and a single antigenic molecule can therefore provoke many antibody molecules with different binding sites. Some low molecular weight molecules, called haptens, are unable to provoke an immune response themselves, although they can react with existing antibodies. Such substances need to be coupled to a carrier molecule in order to have sufficient epitopes to be antigenic. For some chemicals, such as drugs, the carrier may be a host protein—called an auto-antigen. The tertiary structure, as well as the amino acid sequence, is important in determining antigenicity.
Antigens are conventionally divided into thymus-dependent and thymus-independent antigens. Thymus-dependent antigens require T-cell participation to provoke the production of antibodies; most proteins are examples. Thymus-independent antigens require no T-cell co-operation for antibody production; they directly stimulate specific B-lymphocytes by cross-linking antigen receptors on the B-cell surface but provoke poor immunological memory. Such antigens include bacterial cell wall polysaccharides.
Factors other than the intrinsic properties of the antigen also influence the quality of the immune response. These include:
Substances that improve a host’s immune response to a separate antigen are called adjuvants; these are routinely used in immunisation programmes in childhood.
Superantigen is the term given to foreign proteins that simultaneously activate large numbers of T-lymphocytes carrying a particular T-cell receptor V-beta gene (see T-cell receptors). Widespread T-cell activation results in florid cytokine release, as exemplified by toxic shock syndrome induced by certain streptococcal toxins.
Antibody
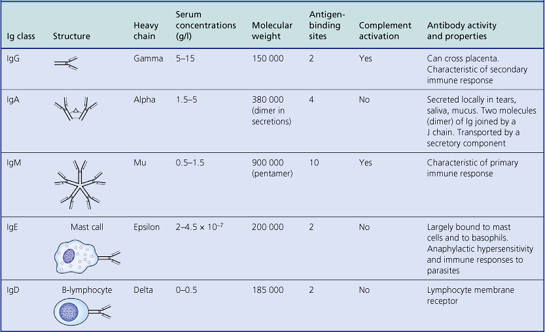
Humoral immunity is dependent on the production of antibodies and their actions. All antibodies belong to the immunoglobulin class of proteins and are produced by plasma cells, themselves derived from B-lymphocytes. The basic structure of an immunoglobulin molecule is shown in Figure 9.2. It has a four-chain structure: two identical heavy (H) chains (molecular weight 50 kD) and two identical light (L) chains (mol wt 25 kD). There are two alternative types of light chain, known as kappa and lambda; an antibody molecule has either two kappa or two lambda light chains, never one of each. In contrast, there are five types of heavy chain, each with important functional differences (Table 9.1). The heavy chains determine the class (isotype) of the antibody and the physiological function of the antibody molecule. Once the antigen-binding site has reacted with its antigen, the molecule undergoes a change in the conformation of its heavy chains in order to take part in effector functions (Table 9.1).
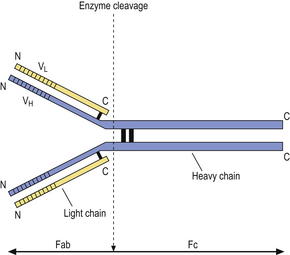
Fig. 9.2 Basic structure of an immunoglobulin molecule. The two identical light chains and two identical heavy chains are held together by disulphide bonds. (Fab, fragment antigen binding; Fc, fragment crystallisable; VL, variable domain of a light chain; VH, variable domain of a heavy chain.)
The amino (N) terminal regions of the heavy and light chains include the antigen-binding sites. The amino acid sequences of these N- terminal domains vary between different antibody molecules of the same isotype and are known as variable (V) regions. Most of these differences reside in three hypervariable areas of the molecule, each only 6–10 amino acid residues long. In the folded molecules, these hypervariable regions in each chain come together, with their counterparts on the other pair of heavy and light chains, to form the antigen-binding site. The structure of this part of the antibody molecule is unique to that molecule and is known as the idiotypic determinant. In any individual, about 106–107 different antibody molecules could be made up by 103 different heavy chain variable regions associating with 103 different light chain variable regions. Somatic mutation during multiple divisions of B-lymphocytes generates further diversity of around 1014 antibody specificities.
IgM is the oldest class of immunoglobulin in evolutionary terms. It is a large molecule consisting of five basic units held together by a joining (J) chain; it penetrates poorly into tissues on account of its large size (Table 9.1). The major physiological role of IgM is intravascular neutralisation of organisms (especially viruses) aided by its 10 antigen-binding sites. IgM also has multiple complement-binding sites; this results in excellent complement activation and lysis of the organism or removal of the antigen–antibody–complement complexes by complement receptors on phagocytic cells. It is the first class of antibody to be formed in response to an initial encounter with an antigen (primary immune response).
IgG is a smaller immunoglobulin which penetrates tissues easily. It is the most abundant immunoglobulin in the plasma and extracellular fluid. It is the only immunoglobulin that crosses the placenta to provide immune protection to the neonate; this is an active process involving specific placental receptors for the Fc portion of the IgG molecule. Polymorphs and macrophages also have surface receptors for the Fc fragment of IgG; thus binding of IgG to particulate antigen promotes adhesion of these cells and subsequent phagocytosis of the antigen.
There are four subclasses of IgG: IgG1 and IgG3 activate complement efficiently and are responsible for clearing most protein antigens; IgG2 and IgG4 react predominantly with carbohydrate antigens (in adults).
IgA is sometimes referred to as ‘mucosal antiseptic paint’. It is secreted locally by plasma cells in the intestinal and respiratory mucosa and is an important constituent of breast milk. It consists of two basic units (a dimer) linked by a ‘joining’ or J chain. The addition of a ‘secretory component’ prevents digestion of the immunoglobulin molecule by enzymes present in intestinal or bronchial secretions. Secretory component is a fragment of the polymeric immunoglobulin receptor synthesised by epithelial cells and transports secretory IgA from the mucosa into the lumen of the gut or bronchi.
There is little free IgD or IgE in serum or normal body fluids. These two classes mainly act as cell receptors. IgD is synthesised by antigen-sensitive B-lymphocytes and acts as a cell surface receptor for antigen. IgE is produced by plasma cells but taken up by specific IgE receptors on mast cells and basophils. IgE probably evolved as a way of expelling intestinal parasites via mast cell degranulation.
T-cell receptors
Like B-cells, each T-cell is committed to a given antigen, which it recognises by one of two types of T-cell receptor (TCR). T-cells have either alpha/beta TCR (a heterodimer of alpha and beta chains) or gamma/delta TCR (a heterodimer of gamma and delta chains). Alpha/beta TCRs predominate in adults, although 10% of T-cells in epithelial structures are of the gamma/delta TCR type. Each type of TCR is associated with several transmembrane proteins which make up the cluster differentiation 3 (CD3) molecule (Fig. 9.3) to form the CD3–TCR complex responsible for taking the antigen recognition signal inside the cell (transduction). The CD3 antigen is widely used as a marker of mature T-cells in diagnostic and investigative pathology.
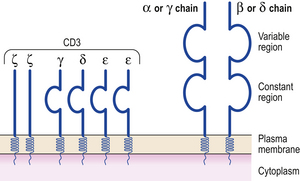
Fig. 9.3 The structure of the T-cell receptor (TCR). The variable regions of the alpha (α) and beta (β) chains make up the T idiotype. The TCR is closely associated on the cell surface with the CD3 molecule.
The TCR complex recognises small processed antigen peptides in the context of major histocompatibility complex (MHC) class I and II antigens (see below), depending on the type of T-cell. Helper T-cells recognise MHC class II molecules in association with foreign antigen and use the CD4 molecule to enhance binding and intracellular signalling. Cytotoxic T-cells recognise antigen associated with MHC class I molecules and use CD8 molecules for increased binding and signalling. However, recognition of processed antigen alone is not enough to activate T-cells. Additional signals through soluble interleukins are needed; some of these are generated during ‘antigen processing’.
Major histocompatibility complex antigens
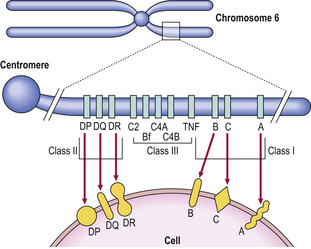
Histocompatibility antigens were so named because of the vigorous reactions they provoked during mismatched organ transplantation. However, these antigens play a fundamental role in the normal immune response by presenting antigenic peptides to T-cells. Human major histocompatibility complex (MHC) antigens are also known as human leukocyte antigens(HLAs). MHC antigens are cell surface glycoproteins of two basic types: class I and class II (Fig. 9.4). They exhibit extensive genetic polymorphism with multiple alleles at each locus. As a result, genetic variability between individuals is very great and most unrelated individuals possess different HLA molecules. This means that it is very difficult to obtain perfect HLA matches between unrelated persons for transplantation.
The antigen-specific receptor of an individual T-cell (TCR) will only recognise antigen as part of a complex of antigenic peptide and that individual’s MHC. This process of dual recognition of peptide and MHC molecule is known as MHC restriction because the MHC molecule restricts the ability of the T-cell to recognise antigen. T-cells from one person will not co-operate with antigen-presenting cells from a person of different HLA type.
MHC class I antigens are subdivided into three groups: A, B and C. Each group is controlled by a different gene locus within the major histocompatibility complex on chromosome 6 (Fig. 9.4). The products of the genes at all three loci are chemically similar. MHC class I antigens (Fig. 9.5) are made up of a heavy chain (alpha) controlled by a gene in the relevant MHC locus, associated with a smaller chain called beta-2-microglobulin, controlled by a gene on chromosome 15. The differences between individual MHC class I antigens are due to variations in the alpha chains; the beta-2-microglobulin component is constant. The detailed structure of class I antigens was determined by X-ray crystallography. This shows that small antigenic peptides are tightly bound to a groove in the surface alpha chains.
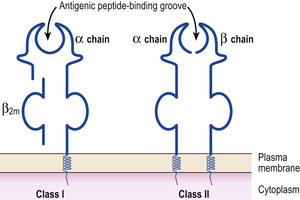
MHC class II antigens have a folded structure similar to class I antigens with the peptide-binding groove found between the alpha and beta chains (Fig. 9.5). Whereas class I molecules are expressed by most nucleated cells, expression of class II molecules is restricted to dendritic cells, B-lymphocytes, activated T-cells, macrophages, inflamed vascular endothelium and some epithelial cells. However, other cells (e.g. thyroid, pancreas, gut epithelium) can be induced to express class II molecules under the influence of interferon-gamma released during inflammation. In humans, there are three groups of class II antigen: the loci are known as HLA-DP, HLA-DQ and HLA-DR.
MHC class III antigens (Fig. 9.4) constitute early complement proteins C4 and C2. Other inflammatory proteins, e.g. tumour necrosis factor (TNF), are encoded in adjacent areas.
Accessory and co-stimulatory molecules
T-cell activation needs more than just binding between the T-cell receptor and the MHC class II molecule and processed antigen complex on the antigen-presenting cell.
Accessory and co-stimulatory molecules are needed for efficient binding and signalling (Fig. 9.6). Each accessory molecule has a corresponding protein, or ligand, to which it binds. The interaction between antigen-presenting cells and T-cells is strongly influenced by accessory molecules which function as co-stimulators; for example, CD80 and CD86 on the activated dendritic cell engage with their counter-receptors CD28 and CTLA-4 (CD152) on the T-cell surface (Fig. 9.6). A functional co-stimulatory pathway is essential for T-cell activation. In the absence of a co-stimulatory signal, interaction between the dendritic cell and T-cell leads to T-cell unresponsiveness (a state called anergy).
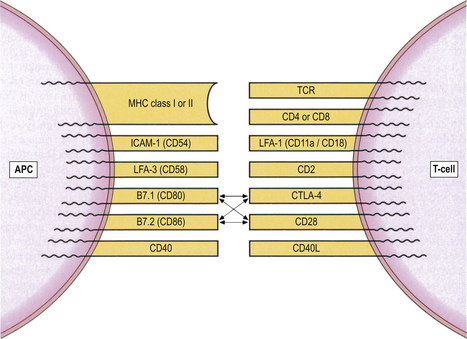
Fig. 9.6 Accessory and co-stimulatory molecules on T-lymphocytes and their ligands on antigen-presenting cells (APCs).
Adhesion molecules mediate cell-to-cell adhesion as well as adhesion between leukocytes and endothelial cells, and are grouped into two main families: integrins and selectins.
The migration of leukocytes to sites of inflammation depends on three key sequential steps mediated by adhesion molecules:
Integrins are subdivided into five families (beta-1 to beta-5 integrins) which mediate binding of lymphocytes and monocytes to the endothelial adhesion receptor called vascular cell adhesion molecule (VCAM-1). Defective expression of certain integrins is associated with a severe immunodeficiency characterised by marked neutrophil leukocytosis because neutrophils are unable to migrate from blood vessels into sites of infection.
The selectin family comprises three glycoproteins designated by the prefixes E- (endothelial), L- (leukocyte) or P- (platelet) to denote the cells on which they were first described. Selectins bind strongly to carbohydrate molecules on leukocytes and endothelial cells, and regulate the homing of these cells to sites of inflammation.
Cytokines
Cytokines are soluble mediators secreted by lymphocytes (lymphokines) or by macrophages/monocytes (monokines). They act as stimulatory or inhibitory signals between cells.
Cytokines that act between cells of the immune system are called interleukins; those that induce chemotaxis of leukocytes are called chemokines. All cytokines share common features:
Among the array of cytokines produced by macrophages and T-cells, interleukin-1 (IL-1) and interleukin-2 (IL-2) have a pivotal role in amplifying immune responses. IL-1 acts on a wide range of targets, including T- and B-cells (Table 9.2). In contrast, the effects of IL-2 are restricted largely to lymphocytes: it has a trophic effect on T-cells, IL-2 receptor-bearing B-cells and natural killer (NK) cells. The considerable overlap between individual cytokines and interleukins is summarised in Table 9.3.
Table 9.2 Actions of interleukin-1
| Target cell | Effect |
|---|---|
| T-lymphocytes |
B-lymphocytes
Neutrophils
Proliferation/activationOsteoclastsReabsorption of boneHepatocytesAcute-phase protein synthesisHypothalamusProstaglandin-induced feverMuscleProstaglandin-induced proteolysis
Table 9.3 Cytokines and their actions
| Cytokine | Source | Action |
|---|---|---|
| IL-2 | T-cells | Proliferation and maturation of T-cells, induction of IL-2 receptors and activation of NK cells |
 |
Induction of MHC class II, FcR and IL-2R on B- and T-cells; induction of isotype switching in B-cells; facilitate IgE production (mainly IL-4) | |
| IL-6 | Macrophages, fibroblasts, T-cells, mast cells | Growth and differentiation of T-, B- and haemopoietic cells; acute phase response |
| IL-8 | Monocytes/macrophages, T-cells, fibroblasts, keratinocytes, endothelial cells, NK cells | Chemotaxis of neutrophils |
| IL-10 | T-cells, monocytes, keratinocytes | Inhibitory cytokine production |
| IL-12 | B-cells, monocytes/macrophages | Synergism with IL-2; regulates IFN-gamma production |
| IL-13 | T-cells | Actions overlap with IL-4 |
| GM-CSF | T-cells, macrophages, endothelial cells | Stimulates growth of polymorphs and mononuclear progenitors |
| IFN-gamma | T-cells, NK cells | Activation of macrophages, endothelial cells and NK cells; increases expression of MHC class I and II; inhibits IgE production |
| TNF | Macrophages, T-cells, B-cells | Promotion of inflammation; interferes with catabolism in muscle and fat |
STRUCTURAL ORGANISATION OF THE IMMUNE SYSTEM







T- and B-lymphocyte development
All lymphoid cells originate in the bone marrow although the nature of the uncommitted lymphoid stem cell remains unclear (Fig. 9.7). An understanding of the developmental pathway is important, not only to clarify the physiology of the normal immune response but also because some leukaemias and immunodeficiency states reflect maturation arrest of cells in their early stages of development. Lymphoid progenitors destined to become T-lymphocytes migrate from the bone marrow into the cortex of the thymus where further differentiation into mature T-cells occurs. Passage of T-cells from the thymic cortex to the medulla is associated with the acquisition of characteristic surface glycoprotein molecules so that medullary thymocytes resemble mature peripheral blood T-cells. T-cell development in the thymus is characterised by a process of positive selection whereby T-cells that recognise and bind with low affinity to fragments of self-antigen in association with self-MHC molecules proceed to full maturation. In contrast, T-cells that do not recognise self-MHC or that recognise and bind with high affinity to self-antigen are selected out—negative selection—and do not develop further. Negatively selected T-cells kill themselves by apoptosis, i.e. programmed cell death. This process is an important mechanism in preventing autoimmune disease. In summary, the thymus selects out the useful, neglects the useless and destroys the harmful, i.e. autoreactive T-cells.
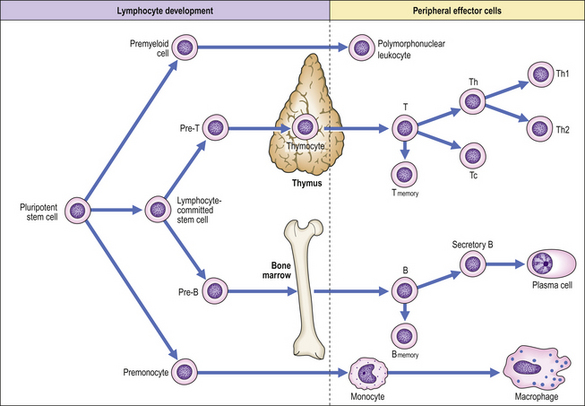
In contrast, B-cell development occurs in the bone marrow and depends on the secretion of cytokines by stromal cells.
Primary and secondary lymphoid organs
The thymus and the bone marrow are primary lymphoid organs. They contain cells undergoing a process of maturation from stem cells to antigen-sensitive but antigen-restricted cells. This process of maturation is independent of antigenic stimulation. In contrast, secondary lymphoid organs are those that contain antigen-reactive cells in the process of recirculating through the body. They include the lymph nodes, spleen and mucosa-associated lymphoid tissues. Antigenic stimulation changes the relative proportions of mature cell types in secondary tissues.
Peripheral T- and B-cells circulate in a characteristic pattern through the secondary lymphoid organs. Most of the recirculating cells are T-cells and the complete cycle takes about 24 hours; some B-cells, including long-lived memory B-cells, also recirculate. Lymphocyte circulation is strongly influenced by chemokine receptors on lymphocyte surfaces which act as homing agents. Adhesion molecules direct cells to their respective ligands on high endothelial venules of lymph nodes.
Lymph node architecture is well adapted to its function (Fig. 9.8). Lymphatic vessels draining the tissues penetrate the lymph node capsule and drain into the marginal sinus from which a branching network of sinuses passes through the cortex to the medulla and into the efferent lymphatic. This network provides a filtration system for antigens entering the node from peripheral tissue.
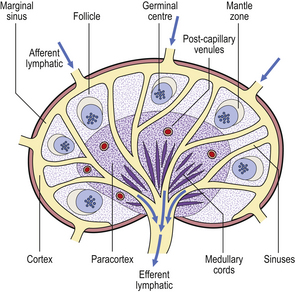
Fig. 9.8 Structure of a normal lymph node. The locations of T- and B-lymphocytes are shown in Figs 9.10–9.12.
The cortex contains primary follicles of B-lymphocytes, surrounded by T-cells in the ‘paracortex’ (Figs 9.9–9.11). There is a meshwork of dendritic cells that express MHC class II antigen throughout the lymph node, and these cells filter and present antigen to lymphoid cells. On antigen challenge, the ‘primary’ follicles of the lymph node develop into ‘secondary’ follicles which contain germinal centres. These comprise mainly B-cells with a few helper T-cells and a mantle zone of the original primary follicle B-cells. B-cells in a secondary follicle are antigen-activated and more mature; most have IgG on their surfaces, whereas those in the primary follicle and mantle zone bear both IgD and IgM. Activated B-cells migrate from the follicle to the medulla, where they develop into plasma cells (Fig. 9.12) in the medullary cords before releasing antibody into the efferent lymph.
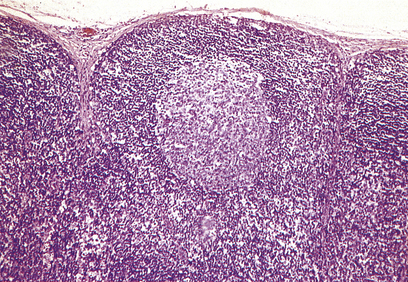

Fig. 9.10 A lymphoid follicle stained with a monoclonal antibody (anti-CD20) that reacts with B-cells. (Positive cells stain brown.)
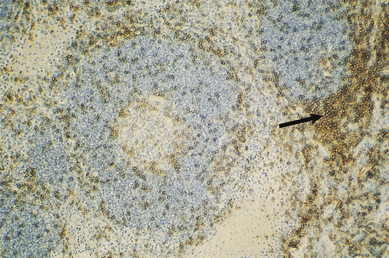
Fig. 9.11 Immunohistochemical identification of T-cells. This section, adjacent to that in Fig. 9.10, is stained with a monoclonal antibody that reacts with T-cells (anti-CD3). Cells of the paracortex are stained (arrow) and there are few T-cells in the follicle.
The majority of naive T-cells entering the lymph node will leave again immediately via efferent lymphatics. Naive T-cells that recognise specific antigen differentiate into effector T-cells before re-entering the circulation.
FUNCTIONAL ORGANISATION OF THE IMMUNE RESPONSE
 Processing and presentation of antigen to lymphocytes is performed by specialised antigen-presenting cells (APCs). The most efficient APCs are dendritic cells in lymph nodes
Processing and presentation of antigen to lymphocytes is performed by specialised antigen-presenting cells (APCs). The most efficient APCs are dendritic cells in lymph nodes Each B-lymphocyte is committed to the production of an antibody with a unique antigen-binding site—the idiotype
Each B-lymphocyte is committed to the production of an antibody with a unique antigen-binding site—the idiotype



Antigen presentation
The first stage of an immune response to any antigen is the processing and presentation of that antigen to lymphocytes by specialised antigen-presenting cells (APCs). T-cells cannot recognise antigen without it. The interaction between APCs and T-cells is strongly influenced by a group of cell-surface molecules that act as co-stimulators. Thus, CD80 and CD86 on the APC engage with their counter-receptors CD28 and CTLA-4 (cytotoxic T-lymphocyte antigen 4; CD152) on the T-cell surface (Fig. 9.6). Normal functioning of theco-stimulatory pathway is vital for T-cell activation. In the absence of a co-stimulatory signal, interaction between the APC and T-cell leads to T-cell unresponsiveness, or anergy. Antagonists to co-stimulatory molecules disrupt immune responses, an observation of potential therapeutic importance; for instance, antagonists to CTLA-4 are being used experimentally to treat severe autoimmune diseases and to prevent graft rejection.
Processed antigen is presented to T-cells alongside MHC class II antigens on the APC surface because T-cells do not recognise processed antigen alone. The most efficient APCs are the interdigitating dendritic cells found in the T-cell regions of a lymph node. Such cells have high concentrations of MHC class I and II molecules, co-stimulatory molecules (CD80, CD86) and adhesion molecules on their surfaces but limited enzymatic powers, so enabling effective processing and presentation of antigen without complete digestion.
Antibody production
Antibody production involves at least three types of cell: antigen-presenting cells, B-lymphocytes and helper T-cells.
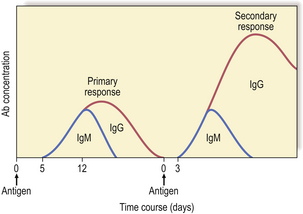
Antibodies are synthesised by B-cells and their mature progeny, called plasma cells (Fig. 9.12). B-cells are readily recognised because they express immunoglobulin on their surfaces. During development, B-cells first show intra-cellular mu chains and then surface IgM. These cells are able to switch from production of IgM to IgG, IgA or IgE as they mature, a process known as isotype switching (Fig. 9.13). This maturation sequence fits with the kinetics of an antibody response: the primary response is mainly IgM and the secondary response predominantly IgG (Fig. 9.14).
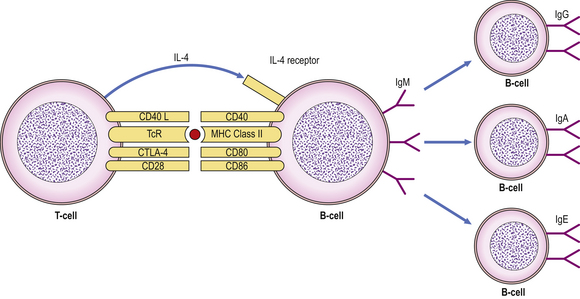
Fig. 9.13 Interaction between CD40L on T-cells and CD40 on B-cells leads to isotype switching under the influence of IL-4. (TcR, T-cell receptor.)
Isotype switching is mediated by the interaction of two important proteins: CD40 expressed on the B-cell surface engages with its ligand, CD40L (CD154), on activated T-cells (under the influence of IL-4) to induce B-cells to switch immunoglobulin production from IgM to IgG, IgA or IgE (Fig. 9.13). Deficiency of either CD40 or CD40L in humans leads to a severe immunodeficiency characterised by inability to switch from IgM to IgG antibody production.
Each B-cell is committed to the production of an antibody that has a unique Vh–Vl combination, the idiotype, and the surface immunoglobulin and secreted immunoglobulin are identical. Contact with antigen and factors released by helper T-cells (IL-4, -5, -6) stimulate the B-cell to divide and differentiate, generating more antibody-producing cells, all of which make the same antibody with the same idiotype. Simultaneously, a population of memory cells is produced which express the same surface immunoglobulin receptor. The result of these cell divisions is that a greater number of antigen-specific B-cells becomes available when the animal is exposed to the same antigen at a later date. This process, known as clonal expansion, helps to account for the amplified secondary response. As well as being quicker and more vigorous (Fig. 9.14), secondary responses are more efficient because the antibodies bind more effectively to the antigen, i.e. with higher affinity.
A minority of B-cells will respond directly to antigens called T-independent antigens, which have repeating, identical, antigenic determinants and provoke predominantly IgM antibody responses. B-cells, however, will not usually respond directly to antigen, even when presented by appropriate accessory cells. A second signal is needed to trigger the B-cell; this signal is normally provided by CD4+ helper T-cells.
T-cell help is antigen-specific. Only helper T-cells that have responded to antigen presented by macrophages can subsequently help B-cells already committed to that antigen. Helper T-cells recognise both antigen and MHC class II antigens as a complex on the presenting cells. They then recognise the same combination of antigen and class II molecule on the corresponding B-cell but co-stimulation is also required (Fig. 9.13). When helper T-cells meet an antigen for the first time, the limited number reacting with that antigen are activated to provide help for B-cells. They undergo blast transformation and proliferation, i.e. clonal expansion, so the immune response on second and subsequent exposures is quicker and more vigorous.
Other mechanisms help to improve this efficiency. Memory cells (which bear the surface marker CD45RO) have increased numbers of adhesion molecules (LFA-1, CD2, LFA-3, ICAM-1) plus a higher proportion of high-affinity receptors for the relevant antigen. Memory cells are therefore easily activated and produce high concentrations of IL-2 to recruit more helper T-cells. Thus T-cell memory is a combination of a quantitative increase of T-cells and a qualitative change in the efficiency of those T-cells.
Helper T-cells are further grouped into two distinct subsets depending on their cytokine profile. Th1 cells secrete TNF and interferon-gamma and mediate cellular immunity. In contrast, Th2 cells predominantly secrete IL-4, IL-5, IL-10 and IL-13 (Fig. 9.15) and are responsible for stimulating vigorous antibody production by B-cells. T-cells expressing cytokine profiles common to both Th1 and Th2 cells are designated Th0.
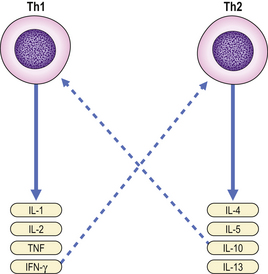
Fig. 9.15 Th1 and Th2 cells secrete different cytokines. Some cytokines provide inhibitory feedback (- – – -) on subsets of Th cells.
A Th1 cytokine profile provides protection against intracellular pathogens, while a Th2 profile is found in those diseases associated with overproduction of antibodies, especially IgE.
The effects of helper T-cells are balanced by those of functional suppressor T-cells that express the characteristic surface glycoprotein CD8.
Cell-mediated responses
Antigen-specific cell-mediated responses are carried out by T-lymphocytes. T-cells can lyse cells expressing specific antigens (cytotoxicity), release cytokines that trigger inflammation (delayed hypersensitivity), or regulate immune responses. These T-cell responses are mediated by distinct T-cell subpopulations: cytotoxicity is the role of cytotoxic T-cells and delayed hypersensitivity that of Th1 cells. These cells are responsible for fighting intracellular pathogens (all viruses, parasites and certain bacteria) which are inaccessible to antibodies.
Cytotoxic T-cells kill cells infected with virus (and possibly those tumour cells expressing recognisable tumour antigens). Such cytotoxicity is virus specific—only cells expressing the relevant viral proteins on their surfaces are killed. Since infected cells express surface viral proteins prior to the assembly of new virus particles and viral budding, cytotoxic T-cells are important in the recovery phase of an infection, destroying the infected cells before new virus particles are generated.
Cytotoxic T-cells recognise viral antigens together with MHC class I molecules. They show exquisite specificity for self-MHC antigens, in that they can only lyse cells expressing the same MHC class I molecules, i.e. express MHC restriction.
Regulatory T-cells are a subset of CD4+ T-cells with a distinct phenotype (CD4+, CD25+) under the control of a gene called FoxP3. These cells dampen down activation and expansion of self-reactive T-cells. Mutations in FoxP3 result in severe autoimmune disease and allergy.
NON-SPECIFIC EFFECTOR MECHANISMS

 Complement can be activated by antibody (the classical pathway), by bacterial cell walls (the alternative pathway) or by mannose-binding lectin (the lectin pathway)
Complement can be activated by antibody (the classical pathway), by bacterial cell walls (the alternative pathway) or by mannose-binding lectin (the lectin pathway)


Complement
Complement is a complex series of interacting plasma proteins which form a major effector system for antibody-mediated immune reactions. Many complement components exist as inactive precursors; once activated, the component may behave as an enzyme which cleaves several molecules of the next component in the sequence. Each precursor is cleaved into two or more fragments. The major fragment (usually designated ‘b’) has two biologically active sites: one for binding to cell membranes or the triggering complex, and the other for enzymatic cleavage of the next complement component. Minor cleavage fragments (designated ‘a’) have important biological properties in the fluid phase. Control of complement activation involves spontaneous decay of any exposed attachment sites and inactivation by specific inhibitors. The major purpose of the complement pathway is to remove or destroy antigen, either by direct lysis or by opsonisation.
Complement activation
Complement activation occurs in two sequential phases:
The critical step (Fig. 9.16) is cleavage of C3 by complement-derived enzymes called C3 convertases. The major fragment of activated C3—called C3b—mediates a number of vital biological activities, particularly opsonisation.
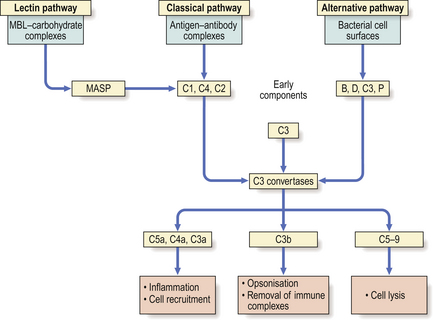
The cleavage of C3 is achieved via three main routes, the classical, alternative and lectin pathways, all of which generate C3 convertases but in response to different stimuli (Fig. 9.16).
Classical pathway activation
The classical pathway is activated when binding of IgM or IgG to antigen causes a conformational change in the Fc region of the antibody to reveal a binding site for the first component in the classical pathway, C1.
C1 is a macromolecular complex of three subcomponents—C1q, C1r and C1s. C1q is a collagen-like protein composed of six subunits. C1q reacts with Fc regions via its globular heads but attachment by two critically spaced binding sites is needed for activation. IgM is more efficient than IgG in activating C1q. IgA, IgD and IgE do not activate the classical pathway.
Once C1q is activated, C1r and C1s are sequentially bound to generate enzyme activity (C1 esterase) for C4 and C2, splitting both molecules into a and b fragments. The complex C4b2b is the classical pathway C3 convertase. C4b2b cleaves C3 into two fragments, one (C3a) possessing anaphylatoxic and chemotactic activity (see below) and one that binds to the initiating complex and promotes many of the biological properties of complement. The C4b2b3b complex so generated is an enzyme, C5 convertase, which initiates the final lytic pathway (the ‘attack’ sequence).
Alternative pathway activation
The central reaction in this pathway, as in the classical one, is the activation of C3. The alternative pathway, however, generates a C3 convertase without the need for antibody, C1, C4 or C2. Instead, the most important activators are bacterial cell walls and endotoxin (Fig. 9.16). Thus, the alternative pathway is responsible for innate defence against invading organisms, as it functions in the absence of preformed specific antibody.
The initial cleavage of C3 in the alternative pathway happens continuously and spontaneously (Fig. 9.16), generating a low level of C3b. C3b is then able to use factors D and B of the alternative pathway to produce the active enzyme ‘C3bBb’ which can break down more C3, providing still more C3b. In the absence of any regulation, this positive feedback loop would continue to cleave C3 until the supply was exhausted.
Regulation is provided by the control proteins, factors H and I. H competes with factor B for binding to C3b, and I then cleaves and inactivates the displaced C3b. Microbial agents that activate the alternative pathway circumvent the effects of factor H and I, and allow the pre-existing low-grade turnover to be amplified. This self-destructive property seems to depend on the carbohydrate composition of the bacterial cell wall.
Lectin pathway activation
The lectin pathway is initiated by mannose-binding lectin (MBL), a circulating protein that binds avidly to carbohydrate on the surface of certain micro-organisms. MBL is structurally related to C1q and activates complement through MASP (MBL-associated serine proteinase) which is similar to C1r and C1s of the classical pathway. The lectin pathway also contributes to innate immunity.
The membrane attack complex
There are two ways of producing the C5 splitting enzyme—the C5 convertase: in the classical pathway it is made up of C3b, C4b and C2b; in the alternative pathway it is composed of C3b, Bb and properdin (Fig. 9.16). Thereafter, the final lytic pathway of complement is the same, involving the sequential attachment of the components C5, C6, C7, C8 and C9, and resulting in lysis of the target cell. This target may be an invading organism or a virally infected cell. The lytic pathway complex binds to the cell membrane and a transmembrane channel is formed which leads to osmotic lysis of the cell.
Biological effects of complement
Complement-mediated lysis of antigen is dramatic but is not the most important role (Fig. 9.16). Instead, complement-dependent phagocytosis is crucial in defence. Micro-organisms coated (i.e. opsonised) with C3b can be bound by cells that possess receptors—called complement receptors type 1 (CR1)—for this ligand. CR1 receptors are present on phagocytic cells. Complement activation results in the release of the pro-inflammatory mediators C5a, C4a and C3a. These act as anaphylatoxins to increase vascular permeability, release vasoactive amines and induce smooth muscle spasm. C5a is a potent chemoattractant and stimulates neutrophils and macrophages to synthesise cytokines, undergo oxidative metabolism and release degradative enzymes.
Macrophages
Macrophages are the tissue equivalent of monocytes and together represent the mononuclear phagocytic system. Lymphocytes and macrophages are derived from closely related stem cells in the bone marrow but each cell lineage has different colony-stimulating factors. Monocytes circulate for only a few hours before entering the tissues where they may differentiate and live for weeks or months as mature macrophages. Tissue macrophages are heterogeneous in appearance, in metabolism and probably also in function; they include freely mobile alveolar and peritoneal macrophages, fixed Kupffer cells in the liver and those lining the sinusoids of the spleen. When found in other tissues, they are called histiocytes.
A major function of the mononuclear phagocyte system is the phagocytosis of invading organisms and other antigens. Macrophages have prominent lysosomal granules containing acid hydrolases and other degradative enzymes with which to destroy phagocytosed material. The material may be an engulfed viable organism, a dead cell, debris, an antigen or an immune complex. In order to carry out their functions effectively, macrophages must be ‘activated’; in this state they show increased phagocytic and killing activity. Stimuli include cytokines (see above), substances that bind to Fc receptors of IgG or soluble inflammatory mediators such as C5a. Toll-like receptors (TLRs) are pattern recognition receptors on macrophages and other cells that contribute to innate immunity. They recognise combinations of sugars, proteins and lipids on pathogens—such as Gram-negative bacterial lipopolysaccharide—and trigger inflammatory responses by up-regulating pro-inflammatory cytokines such as TNF or IL-1. Macrophages are also important for the presentation of antigen to other cells of the immune system, as described earlier.
Neutrophil polymorphonuclear leukocytes
Neutrophils play a major role in the body’s defence against acute infection. They synthesise and express adhesion receptors so that they can adhere to, and migrate out of, blood vessels into tissues. They do this in response to chemotactic agents produced at the site of inflammation; such substances include the chemokine CXCL8 (also called IL-8), complement-derived factors (such as C3a and C5a), lymphokines released by Th1 cells and chemokines produced by mast cells.
Neutrophils are phagocytic cells. Morphologically, the process of phagocytosis is similar in both neutrophils and mononuclear phagocytes. Neutrophils are also able to kill and degrade the substances they take in. This requires a considerable amount of energy and is associated with a ‘respiratory burst’ of oxygen consumption, increased hexose monophosphate shunt activity and superoxide production.
Antibody-dependent cell-mediated cytotoxicity (ADCC)
ADCC is a mechanism by which antibody-coated target cells are destroyed by cells bearing low-affinity receptors for the Fc fragment of IgG (FcγRIII)—natural killer cells, monocytes, neutrophils—with no involvement of the major histocompatibility complex. The mechanism of target cell destruction is not fully understood but includes the discharge of granules containing perforin and granzymes.
Natural killer cells
Natural killer (NK) cells look like large granular lymphocytes. They can kill target cells even in the absence of any antibody or antigenic stimulation. They do not need prior activation but already have the relevant recognition molecules on their surfaces. They are further activated non-specifically by mitogens, interferon and IL-12. NK cells are not immune cells in the strictest sense because, like macrophages, they are not clonally restricted; they show minimal specificity and have no memory. The range of their potential targets is broad. Animals and rare patients with deficient NK cell function have an increased incidence of certain tumours and viral infections. NK cells are therefore thought to be important in the early host response to viral infection and in ‘immune’ surveillance against tumours.
OUTCOMES OF IMMUNE RESPONSES





Once the immune response is initiated, the end result depends on the nature and localisation of the antigen, on whether the predominant response has been humoral or cell mediated, on the type of antibody provoked, and whether non-specific effector mechanisms have been involved.
Direct effects of antibody
Neutralisation is one direct effect of antibody, and IgM is particularly good at this. A number of antigens, including diphtheria toxin, tetanus toxin and many viruses, can be neutralised by antibody. Once neutralised, these substances are no longer able to bind to receptors in the tissues; the resulting antigen–antibody complexes are usually removed from the circulation and destroyed by macrophages.
Indirect effects of antibody
Opsonisation is the process by which an antigen becomes coated with substances (such as antibodies or complement) that make it more easily engulfed by phagocytic cells. The coating of soluble or particulate antigens with IgG antibodies renders them more susceptible to cells that have surface receptors for the Fc portions of IgG (FcγRIII). Neutrophils and macrophages have Fc receptors and can phagocytose IgG-coated antigens; however, this process is relatively inefficient if only Fc receptors are involved. The activation of complement by antibody (via the classical pathway) or by bacterial cell walls (via the alternative or lectin pathways) generates C3b on the surface of micro-organisms and makes them susceptible to binding by C3b receptors (CR1) on macrophages and neutrophils; C3 receptors are very efficient in triggering phagocytosis.
Killing of target cells
Target cells killed as a result of an immune response include organisms and cells bearing virally altered or tumour-specific antigens on their surfaces. They may be killed directly by antigen-specific mechanisms such as antibody and complement, antibody-dependent cell-mediated cytotoxicity or cytotoxic T-cells. Cytokine production results in activation of NK cells and macrophages.
Inflammation
Inflammation is defined as increased vascular permeability accompanied by infiltration of ‘inflammatory’ cells, initially neutrophil polymorphonuclear leukocytes and later macrophages, lymphocytes and plasma cells. Vascular permeability may be increased by complement fragments such as C3a or C5a. Some fragments (C3a, C5a and C567) also attract neutrophils and mobilise them from the bone marrow; cytokines generated by activated dendritic cells, T-cells and macrophages have similar properties. The triggering of mast cells via IgE also causes inflammation due to release of histamine and leukotrienes. Inflammation is covered in detail in Chapter 10.
IMMUNODEFICIENCY


 Patients with antibody deficiency present with recurrent bacterial infections of the respiratory tract
Patients with antibody deficiency present with recurrent bacterial infections of the respiratory tract Patients with defects in cellular immunity present with invasive and disseminated viral, fungal and opportunistic bacterial infections involving any organ
Patients with defects in cellular immunity present with invasive and disseminated viral, fungal and opportunistic bacterial infections involving any organ Infants with severe combined immunodeficiency (SCID) will die before the age of 2 years unless bone marrow transplantation is performed
Infants with severe combined immunodeficiency (SCID) will die before the age of 2 years unless bone marrow transplantation is performed Clinical presentations of defects in phagocytes and complement function show the dependence of humoral immunity on non-specific effector mechanisms
Clinical presentations of defects in phagocytes and complement function show the dependence of humoral immunity on non-specific effector mechanisms In some primary immunodeficiencies, the gene responsible has been identified and somatic gene therapy is possible
In some primary immunodeficiencies, the gene responsible has been identified and somatic gene therapy is possible Secondary immunodeficiency occurs when synthesis of key immune components is suppressed (e.g. bone marrow infiltration or infection with an immunosuppressive virus) or their loss is accelerated (e.g. nephrotic syndrome or protein-losing enteropathy)
Secondary immunodeficiency occurs when synthesis of key immune components is suppressed (e.g. bone marrow infiltration or infection with an immunosuppressive virus) or their loss is accelerated (e.g. nephrotic syndrome or protein-losing enteropathy)

Because the immune system evolved as a defence against infectious organisms, the most dramatic examples of its importance are provided by those disorders where one or more vital components of the immune system are missing or fail to function. Defects in immunity can be classified as primary, due to an intrinsic defect in the immune system, or secondary to an underlying condition (Fig. 9.17). These defects may involve specific or non-specific (innate) immune mechanisms. Underlying immunodeficiency should be suspected in every patient who has recurrent, persistent, severe or unusual infections, irrespective of age.
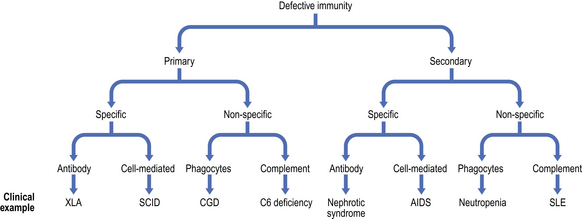
Fig. 9.17 Classification of immunodeficiency. (AIDS, acquired immune deficiency syndrome; CGD, chronic granulomatous disease; SCID, severe combined immunodeficiency; SLE, systemic lupus erythematosus; XLA, X-linked agammaglobulinaemia.)
Primary antibody deficiencies
Defects in antibody synthesis can involve all immunoglobulin classes (panhypogammaglobulinaemia) or only one class or subclass of immunoglobulin (selective deficiency). Antibody deficiency can occur in children or adults although the underlying physiological defects may differ.
In congenital forms of antibody deficiency, recurrent infections usually begin between 4 months and 2 years of age, because maternally transferred IgG affords passive protection for the first 3–4 months of life (Fig. 9.18). Some forms of primary antibody deficiency are inherited as X-linked or autosomal recessive traits; a history of affected relatives, especially boys, is therefore of diagnostic value. However, the average size of a family in developed countries is now so small that a negative family history does not exclude an inherited condition.
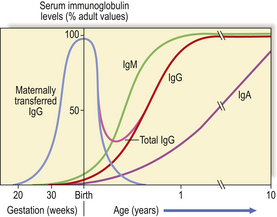
Fig. 9.18 Serum immunoglobulin levels in relation to age. Maternally transferred IgG has mostly been catabolised by 6 months. As the neonate synthesises IgG the level rises slowly but a physiological trough of IgG is seen between 3 and 6 months.
Recurrent infections of the upper and lower respiratory tracts occur in almost all antibody-deficient patients (Fig. 9.19). Many patients also present with skin sepsis (boils, abscesses or cellulitis), gut infection, meningitis, arthritis, splenomegaly or purpura. The commonest infecting organisms in antibody deficiency states are pyogenic bacteria such as staphylococci, Haemophilus influenzae and Streptococcus pneumoniae(Fig. 9.20). In general, these patients are not unduly susceptible to viral or fungal infections because cell-mediated immunity is preserved, but exceptions do occur.
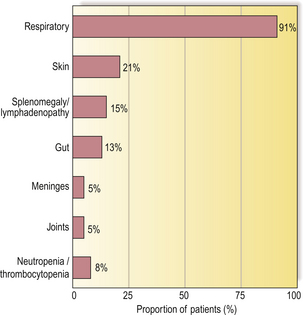
Fig. 9.19 Presenting infections/clinical signs in patients with primary antibody deficiency, irrespective of age.
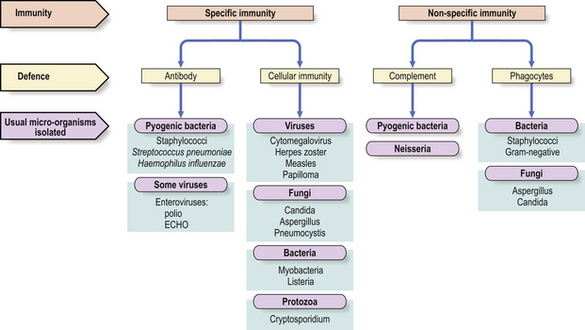
Fig. 9.20 Infections in immunodeficiency. Infections with certain micro-organisms are characteristic of various forms of immunodeficiency.
There are rarely any diagnostic physical signs of antibody deficiency, although examination often shows failure to thrive in children and the consequences of previous sepsis such as ruptured tympanic membranes or bronchiectasis. Measurement of serum immunoglobulin levels will reveal any gross quantitative abnormality but the ability of a patient to make antibody is a better guide to susceptibility to infection than total immunoglobulin levels. Some individuals fail to make specific antibody after test immunisation despite normal serum immunoglobulin levels. Measurements of IgG subclasses are of little clinical value unless backed up by test immunisation and detection of specific antibody responses.
Transient hypogammaglobulinaemia of infancy
Maternal IgG is actively transported across the placenta to the fetal circulation from the fourth month of gestational life, although maximum transfer takes place during the final 2 months. At birth, the infant has a serum IgG at least equal to that of the mother (Fig. 9.18); at first, catabolism of maternal IgG is only partly compensated by IgG synthesised by the newborn child. The period between 3 and 6 months of age represents a phase of ‘physiological hypogammaglobulinaemia’. The normal infant is not unduly susceptible to infection because functioning antibody is present despite the low IgG level, and T-cell function is intact. However, the trough in IgG is more severe and the risk of sepsis much greater if the gift of IgG acquired from the mother is severely reduced, as in extremely premature infants born around 28 weeks’ gestation or earlier.
X-linked agammaglobulinaemia (XLA)—Bruton’s disease
Boys with this condition usually present with recurrent pyogenic infections between the ages of 4 months and 2 years. The sites of infection and the organisms involved are similar to other types of antibody deficiency (Figs 9.19 and 9.20), although these patients are susceptible to enteroviruses. In almost all patients, circulating mature B-cells are absent but T-cells are normal and even increased. No plasma cells are found in the bone marrow, lymph nodes or gastrointestinal tract. The clinical diagnosis rests on the very low serum levels of all classes of immunoglobulin and the absence of circulating mature B-lymphocytes.
The gene responsible for XLA is found on the long arm of the X chromosome. Its product is a cytoplasmic enzyme known as Bruton’s tyrosine kinase, or Btk. In its absence pre-B-cells are unable to differentiate into B-cells. The identification of a mutation in the Btk gene confirms the diagnosis and enables asymptomatic female carriers to be identified and counselled. Management consists of replacement immunoglobulin for the affected boy.
Hyper-IgM syndrome
Some children with antibody deficiency have normal or high serum IgM levels. An X-linked form is due to a failure of CD40 ligand expression on CD4+ T-lymphocytes (Fig. 9.13). As a result, T-cells fail to trigger the switch from IgM to IgG or IgA production in antigen-stimulated B-cells with poor organisation of germinal centres in lymph nodes.
As well as bacterial infections, affected boys are susceptible to Pneumocystis jiroveci pneumonia because macrophage and dendritic cell function are also impaired. Replacement immunoglobulin therapy and co-trimoxazole prophylaxis (to prevent Pneumocystis infection) are required in the long term, and bone marrow transplantation is now considered to be the treatment of choice.
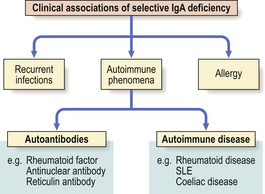
Selective IgA deficiency
This is the commonest primary defect of specific immunity with a prevalence of 1:700 in the UK. It is characterised by undetectable serum IgA levels with normal concentrations of IgG and IgA. Most IgA- deficient individuals are asymptomatic but selective IgA deficiency predisposes the individual to a variety of disorders (Fig. 9.21). About 20% of individuals make antibodies to IgA and some may develop adverse reactions following transfusions of blood or plasma.
Common variable immunodeficiency (CVID)
CVID embraces a group of disorders presenting as antibody deficiency in late childhood or adult life. Patients experience the same range of bacterial infections as other patients with antibody deficiency (Figs 9.19 and 9.20). CVID is much more common than XLA. About 20% of patients have affected relatives with CVID or selective IgA deficiency and several contributory genetic mutations have been identified. CVID patients also have a 40-fold increase in incidence of lymphoma.
Primary defects in cell-mediated immunity
Impairment of T-cell function is usually accompanied by variable degrees of B-cell dysfunction, reflecting the T–B-cell co-operation needed for efficient antibody production. Most defects are therefore combined immunodeficiencies and this is reflected in the wide range of infections experienced by such patients. Some examples are illustrated below.
Severe combined immunodeficiency
Infants in whom there is major failure of both T- and B-lymphocyte function have severe combined immunodeficiency (SCID). There are several genetic variants but all affected children present in the first few weeks or months of life with failure to thrive, chronic diarrhoea (‘gastroenteritis’) and respiratory infections. Usually, there is lymphopenia, which is often overlooked.
Immunisation with live vaccines and conventional blood transfusions must be avoided in patients with proven or suspected defects in cell-mediated immunity: live vaccines can lead to disseminated infection, and blood transfusion may result in graft-versus-host disease.
Infants with SCID die before they are 2 years old unless haemopoietic stem cell transplantation is undertaken. Some types of SCID caused by a specific genetic defect are prototypes for somatic gene therapy.
DiGeorge syndrome (‘Catch 22’ anomaly)
In this condition, severely affected neonates present with cardiovascular defects, hypocalcaemia, abnormal facies and thymic dysfunction. Most deaths are due to the heart lesions and not to the immunodeficiency, which is usually mild. The genetic defect (chromosome 22q11 deletion) results in impaired intrauterine development of the third and fourth pharyngeal pouches.
Primary defects in phagocyte function
Humoral immunity depends not only upon antibody synthesis but also upon effector mechanisms which eliminate antigen bound to antibody. Micro-organisms coated (i.e. opsonised) with IgG antibodies are readily bound and ingested by phagocytic cells. Thus, specific immunity requires non-specific effector mechanisms for its efficient operation; this partly explains similarities between the infectious complications experienced by patients with defects of antibody synthesis and those with neutrophil dysfunction (Fig. 9.20).
The major role of the neutrophil is to ingest, kill and digest invading micro-organisms, particularly bacteria and fungi. Failure to fulfil this role leads to infection. Defects in neutrophil function can be quantitative (neutropenia) or qualitative (neutrophil dysfunction). However, irrespective of the basic cause, the clinical features of infections are similar and certain generalisations are possible:
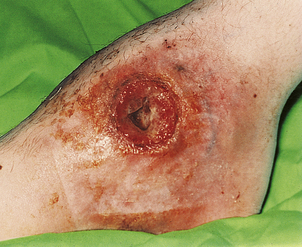
Chronic granulomatous disease
Chronic granulomatous disease (CGD) is a group of disorders resulting from a failure to produce high concentrations of toxic oxygen radicals during the ‘respiratory burst’ that accompanies activation of phagocytes. The classic type is inherited as an X-linked recessive disorder, and typically presents in the first 3 months of life as severe skin sepsis caused by Staphylococcus aureus or fungal infections with Candida albicans or Aspergillus fumigatus (Fig. 9.22). The resulting complications include regional lymphadenopathy, hepatosplenomegaly, hepatic abscesses and osteomyelitis. Affected organs show multiple abscesses and non-caseating giant-cell granulomas.
Primary complement deficiency
Inherited deficiencies of complement components are associated with characteristic clinical syndromes (Fig. 9.23). Many patients with C1, C4 or C2 deficiency present with a syndrome of malar flush, arthralgia, glomerulonephritis, fever or chronic vasculitis.
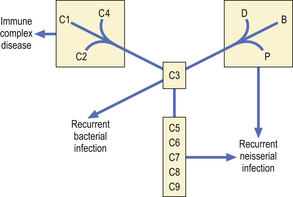
Patients with C3 deficiency occurring as a primary defect or secondary to deficiencies of factor H or factor I (see Fig. 9.16) have an increased susceptibility to life-threatening bacterial infections such as pneumonia, septicaemia and meningitis, illustrating the important role of C3 in defence against infection.
There is a striking association between deficiencies of C5, C6, C7, C8 or properdin and recurrent neisserial infection. Most patients present with recurrent meningococcal meningitis, less commonly with gonococcal septicaemia and arthritis. However, many patients experience only one episode of meningitis, or many years may elapse between attacks.
Hereditary angioedema is caused by deficiency of the inhibitor of the first component of complement (C1 inhibitor). Patients experience recurrent attacks of cutaneous, intestinal or laryngeal oedema which can be fatal if the airway is occluded.
Secondary immunodeficiency
Secondary causes of immunodeficiency are far more common than primary causes. Since levels of immune components represent the net balance of synthesis versus catabolism (or loss), low levels reflect either depressed production or accelerated catabolism or loss (Fig. 9.24). Protein loss severe enough to cause hypogammaglobulinaemia occurs mainly via the kidney (nephrotic syndrome) or through the gut (protein-losing enteropathy) in a variety of active inflammatory diseases such as Crohn’s disease or ulcerative colitis. In intestinal lymphangiectasia, the dilated lymphatics leak lymphocytes as well as protein.
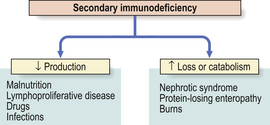
Impaired synthesis is exemplified by protein–energy malnutrition. Malnourished individuals show impaired specific antibody production following immunisation, and even more striking defects in cell-mediated immunity, phagocyte function and complement activity. Many of these defects reverse after adequate protein and calorie supplementation of the diet.
Patients with lymphoproliferative diseases are very prone to infection. The infection risk in patients with multiple myeloma is 5–10 times higher than in age-matched controls, while untreated chronic lymphocytic leukaemia is commonly associated with hypogammaglobulinaemia and recurrent chest infections, which tend to become more severe as the disease progresses. Non-Hodgkin’s lymphoma may be associated with defects of both humoral and cell-mediated immunity.
Immunosuppressive drugs affect many aspects of cell function. Lymphocyte and polymorph activity are often impaired, although severe hypogammaglobulinaemia is unusual. Patients taking drugs to prevent organ transplant rejection can develop unusual opportunistic infections.
Another iatrogenic form of secondary immune deficiency is that associated with splenectomy. Death occurs from sudden, overwhelming infection due to Streptococcus pneumoniae. The risk of death from infection following splenectomy is 1–2% over 15 years. All patients should receive immunisation with pneumococcal conjugate vaccine as well as penicillin prophylaxis.
In a number of infections, the micro-organism paradoxically suppresses rather than stimulates the immune system. Severe, though transient, impairment of cell-mediated immunity has been noted in many viral illnesses, particularly cytomegalovirus, measles, rubella, infectious mononucleosis and viral hepatitis; however, the most florid example is infection with the human immunodeficiency virus (HIV).
Acquired immune deficiency syndrome (AIDS)
AIDS is a worldwide (pandemic) form of immunodeficiency first recognised in 1981 and caused by the retroviruses human immunodeficiency virus (HIV) types 1 and 2 (Fig. 9.25). At the end of 2007, the number of people living with HIV worldwide was estimated to be 33 million, 2.5 million of whom had become newly infected during 2007, with 1.7 million of these living in Sub-Saharan Africa. About half of new HIV infections occur in women and 40% in young people aged from 15 to 24 years. Almost all new childhood infections are due to mother to child (vertical) transmission, before or during childbirth or through breastfeeding.
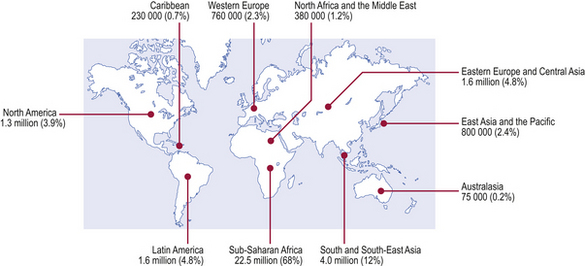
Fig. 9.25 People living with HIV/AIDS. Estimated number and distribution of people living with HIV/AIDS by December 2007 (estimated total over 33 million).
(Source: UNAIDS/WHO.)
Transmission of HIV
HIV is transmitted through sexual intercourse, both heterosexual and homosexual, and through the sharing of contaminated needles and syringes by intravenous drug abusers or via therapeutic procedures in areas of the world where reuse of contaminated equipment occurs. Other methods of transmission are through the receipt of infected blood or blood products, donated organs or semen. Cases of seroconversion and death among health care workers after needlestick injuries or blood splashes have also been reported (0.3% seroconvert).
Clinical spectrum of HIV infection
HIV produces a spectrum of disorders (Fig. 9.26) which are classified by the Centers for Disease Control in the USA. A transient, acute glandular fever-like illness (CDC I) may occur in 10–20% of patients within a few weeks of initial HIV infection and precedes seroconversion (i.e. production of antibodies to HIV). Peripheral blood shows many atypical lymphocytes and an increased number of CD8+ T-cells at this time. Most seropositive individuals then remain symptom-free (CDC II) for 2–10 years or more. Development of AIDS depends on the contribution of many co-factors, such as genetic background, repeated immune stimulation (multiple co-infections speed the rate of progression) and pregnancy. The most important prognostic factor for progression to AIDS is the concentration of HIV RNA in the blood—the viral load—at diagnosis.
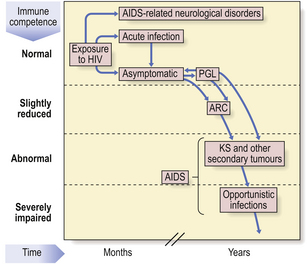
After this latent period, some individuals develop asymptomatic persistent generalised lymphadenopathy (PGL; CDC III), defined as enlarged nodes in two or more extra-inguinal sites persisting for at least 3 months in the absence of any illness associated with lymphadenopathy. Some seropositive people show constitutional and clinical features such as unexplained lymphadenopathy, diarrhoea, night sweats, oral candidiasis and weight loss—the AIDS-related complex (ARC; CDC IV A).
HIV is neurotropic: acute aseptic meningitis, encephalopathy, myelopathy and neuropathy have been reported around the time of seroconversion, whereas chronic meningitis, cerebral lymphoma, encephalopathy and dementia (CDC IV B) may occur later (Ch. 26). Up to 70% of AIDS patients suffer from HIV-related dementia which is probably a direct neurotropic effect of HIV.
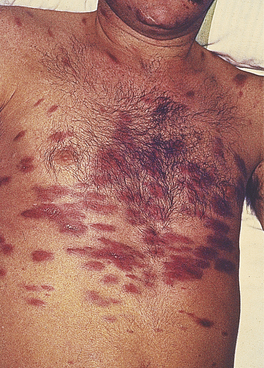
The dominant clinical manifestations of AIDS are opportunistic infections and tumours (CDC IV C and D). Typical infections include Pneumocystis jiroveci pneumonia (Fig. 9.27), cytomegalovirus or herpes simplex infections (Fig. 9.28), cerebral toxoplasmosis, atypical mycobacterial infections, systemic fungal infection and parasitic infestations of the gastrointestinal tract. The presentation in African patients may be characterised by a diarrhoea–wasting syndrome, called ‘slim’ disease, and opportunistic infection with tubercle bacillus, cryptococcus or cryptosporidium.
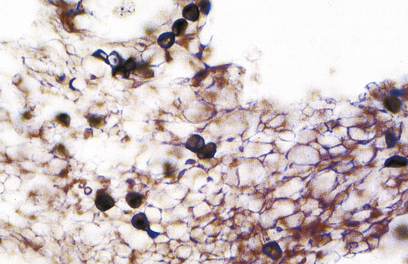
Fig. 9.27 Pneumocystis jiroveci pneumonia. A foamy exudate fills the alveoli. The methenamine silver stain shows the organism as black spheres.
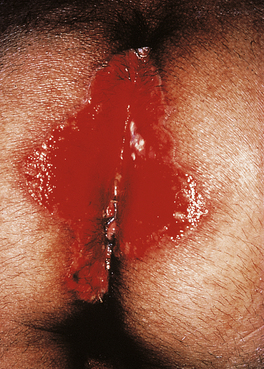
Common tumours are the consequences of the activities of oncogenic viruses operating in an immunocompromised host: Kaposi’s sarcoma (KS) (Fig. 9.29) is caused by human herpes virus type 8 and non-Hodgkin’s lymphoma by Epstein–Barr virus.
Immunopathogenesis of HIV infection
HIV enters susceptible cells through binding of viral envelope glycoprotein (gp120) to specific receptors on the cell surface, mainly the CD4 molecule itself, although other cell surface molecules—the chemokine receptors CXCR4 and CCR5—are also involved. Any cell bearing the CD4 antigen can be infected by HIV; typically these are helper T-cells but macrophages, glial cells of the central nervous system and epithelial cells of the gut can be infected via CCR5 or other HIV receptors.
The most striking effects of HIV are on T-lymphocyte-mediated responses. HIV replicates at a rate of 109–1010 new virions per day, resulting in up to 108 new mutants per day, so the immune system has an enormous task to limit HIV spread. The hallmark of disease progression is the inexorable fall in the absolute number of CD4+ T-cells, the result of the destructive, cytopathic effects of HIV. While HIV infection may be latent clinically for many years, the destruction of CD4+ cells takes place continuously within lymph nodes and other lymphoid organs, particularly gut-associated lymphoid tissue, until the virus can no longer be contained and reappears in the blood stream—HIV antigenaemia.
Therapeutic options
Knowledge of the way in which HIV gains access into CD4+ cells and its method of replication has led to exploration of potential therapies (Fig. 9.30).
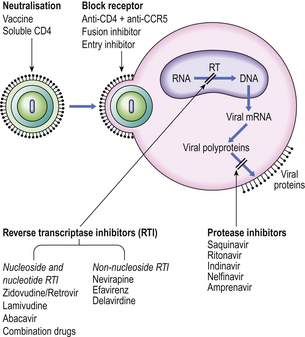
Fig. 9.30 Potential therapeutic options for preventing or eliminating HIV infection. (CCR5, chemokine receptor; RT, reverse transcriptase.)
Binding of virus to the CD4 antigen in the cell membrane might be blocked by antibody to the viral envelope or to CD4 or to the chemokine receptor CCR5. Traditional vaccines, using killed or attenuated organisms, are unlikely to be of value. The fragile nature of the HIV envelope makes it a poor immunogen, while its high mutation rate poses a problem in selecting a stable common epitope able to provoke a protective immune response. Safety is a major concern because a mutation of an attenuated HIV back to its virulent state would be catastrophic. The search for a candidate ‘AIDS vaccine’ continues.
However, an entry inhibitor drug blocks HIV from entering the host cell by binding CCR5 while fusion inhibitors block HIV from fusing with a cell’s membrane to enter and infect that cell.
Inhibition of viral replication can be achieved by inhibiting activity of reverse transcriptase (RT) as this is a unique retroviral enzyme with no mammalian equivalent. Such inhibitors fall into two groups: nucleoside and nucleotide RT inhibitors and non-nucleoside RT inhibitors. Protease inhibitors also prevent the assembly of new infectious virions. Current management uses combinations of drugs from different therapeutic groups—called highly active antiretroviral therapy, or HAART—to control viral replication and limit progression of immune deficiency.
HYPERSENSITIVITY REACTIONS

 Immediate hypersensitivity (type I) reactions result from the binding of antigen by IgE on mast cells or basophils
Immediate hypersensitivity (type I) reactions result from the binding of antigen by IgE on mast cells or basophils Antibody to cell-bound antigen (type II) causes cell destruction by activating complement or promoting phagocytosis
Antibody to cell-bound antigen (type II) causes cell destruction by activating complement or promoting phagocytosis Type III reactions result from deposition or formation of immune complexes in tissues, particularly the skin, joints and kidneys. Chronic immune complex nephritis accounts for most cases of glomerulonephritis in humans
Type III reactions result from deposition or formation of immune complexes in tissues, particularly the skin, joints and kidneys. Chronic immune complex nephritis accounts for most cases of glomerulonephritis in humans Type I, II and III reactions are caused by antibodies: type IV reactions are caused by T-lymphocytes
Type I, II and III reactions are caused by antibodies: type IV reactions are caused by T-lymphocytes

Unfortunately, the recognition of foreign antigen by the immune system can cause incidental tissue damage as well as the intended destruction of the antigen. Such reactions are called ‘hypersensitivity’ reactions; Gell and Coombs defined four main types:
| Type I: | immediate hypersensitivity, or ‘allergy,’ due to activation of IgE antibody on mast cells or basophils |
| Type II: | antibody to cell-bound antigen |
| Type III: | immune complex reactions |
| Type IV: | delayed hypersensitivity mediated by T-cells. |
Immediate hypersensitivity (type I)
Immediate hypersensitivity (type I) reactions are those in which antigen interacts with IgE bound to tissue mast cells or basophils (Fig. 9.31).
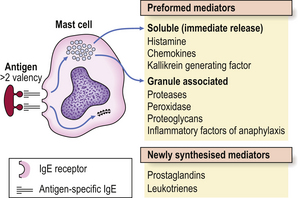
IgE is embedded in the membranes of mast cells, exposing the antigen-binding sites of the molecule to the microenvironment of the cell. Exposure to specific antigen bridges two adjacent IgE molecules and this bridging effect triggers the mast cell to release its mediators. There are two groups of mediators: those that are preformed and those that are newly synthesised (Fig. 9.31).
The preformed mediators include histamine, lysosomal enzymes, chemokines and heparin. Because they are preformed, immediate (type I) hypersensitivity reactions are rapid: clinically the effects begin within 5–10 minutes and peak around 30 minutes. This is well illustrated by skin prick tests: if the antigen is pricked or scratched into the skin of an allergic individual a ‘wheal and flare’ reaction rapidly appears (Fig. 9.32). IgE responses are usually directed against antigens that enter at epithelial surfaces, i.e. inhaled or ingested antigens.
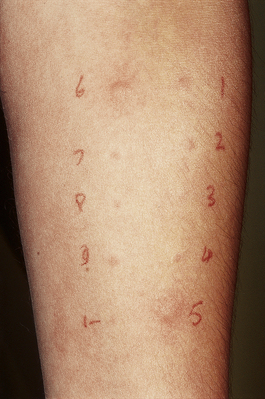
Fig. 9.32 Positive skin prick tests showing ‘wheal and flare’ responses to house dust mite (number 5) and dog dander (number 6) in a patient with allergic rhinitis.
Allergic diseases are common: about 15–20% of the population has some form of allergy. Such patients are frequently atopic: atopy defines an inherited tendency for overproduction of IgE antibodies to common environmental antigens. Several genes predispose to this familial tendency but environmental factors must be involved because there is only 50% concordance in monozygotic twins. Typical atopic disorders include seasonal allergic rhinitis (‘hay fever’), asthma and atopic eczema. However, life-threatening reactions can occur if the antigen enters the systemic circulation or if the patient has very high levels of circulating IgE antibodies. Generalised degranulation of IgE-sensitised mast cells and basophils leads to sudden hypotension, severe bronchoconstriction and collapse, a condition called anaphylaxis. Common allergens are bee and wasp venom, antibiotics (e.g. penicillin), peanuts and latex. Similar reactions that are not mediated by IgE antibodies are called anaphylactoid: the same mast cell mediators are responsible but the stimulus for their release differs. Substances inducing anaphylactoid reactions act directly on mast cells; they include anaesthetic induction agents and radiological contrast media.
Complex ‘allergic’ conditions such as asthma or eczema cannot be explained solely on the basis of IgE-mediated release of mediators. T-lymphocytes play a major role in the activation and/or recruitment of IgE antibody-producing B-cells, mast cells and eosinophils, the cellular triad involved in allergic inflammation. Two major subsets of helper T-cells (Th) have been identified by their profile of cytokine secretion. Sensitised T-cells found in bronchial biopsies and bronchoalveolar lavage fluid from allergic individuals are of the Th2 subset, and cytokines IL-4 and IL-13 produced by these cells stimulate IgE production (Fig. 9.33). When an atopic subject is exposed to the relevant antigen, immunological recognition can occur both via the T-cell receptor and via IgE bound to mast cells.
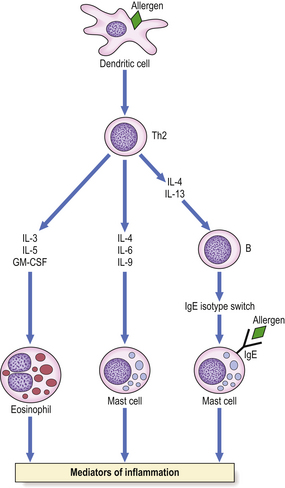
Fig. 9.33 Cytokines produced by Th2 cells. Immunological recognition can occur via the T-cell receptor or via IgE on mast cells.
Once the lining of the airways becomes inflamed it is susceptible to any irritant, such as airways cooling, tobacco smoke, diesel particles or sulphur dioxide. Thus, bronchial hyper-responsiveness is the hallmark of asthma. Much of the inflammatory damage is induced by eosinophils which contain major basic protein (MBP) capable of damaging epithelial cells of the airways. Damage to the epithelium by MBP, cytokines and mediators also exposes sensory nerve endings in the basement membrane and further increases irritability through neural triggering.
Antibody to cell-bound antigen (type II)
Type II hypersensitivity reactions are triggered by antibodies reacting with antigenic determinants which form part of the cell membrane. The consequences of the reaction depend on whether or not complement or accessory cells are involved and whether the metabolism of the cell is affected. IgM or IgG antibodies are typically implicated (Fig. 9.34).
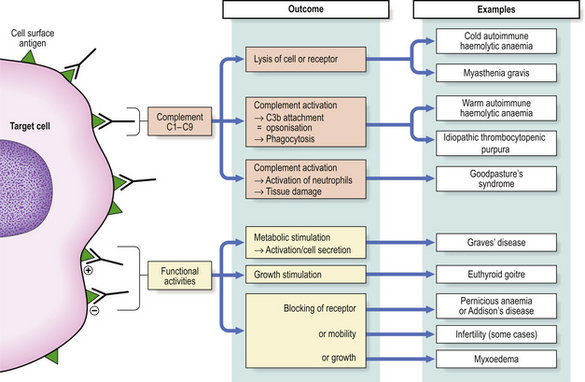
Fig. 9.34 Type II hypersensitivity. Antibodies to cell-bound antigen can result in cell death or interference with cell functions.
Many examples of type II hypersensitivity involve drugs or their metabolites which have bound to the surface of red blood cells or platelets to form highly immunogenic epitopes. Antibodies formed against the drug or its metabolite inadvertently destroy the cell as well—‘bystander lysis’—resulting in haemolytic anaemia or thrombocytopenic purpura. The same mechanism is responsible for certain autoimmune disorders where the target antigen is intrinsic (i.e. self) antigen rather than extrinsic (Fig. 9.34). Under these circumstances, auto-antibodies can also cause disease by binding to the functional sites of self-antigens, such as receptors for hormones or neurotransmitters, so mimicking or blocking the action of the hormone without causing inflammation or tissue damage. Some textbooks regard stimulation of cell function by antibody as a separate category of hypersensitivity—type V: an example is Graves’ disease where antibodies against the thyroid stimulating hormone receptor drive overproduction of thyroid hormones by the cell.
Immune complex hypersensitivity (type III)
Type III reactions result from the deposition or formation of immune complexes in the tissues. Localisation of immune complexes depends on their size, their electrostatic charge, and the nature of the antigen. If they accumulate in the tissues in large quantities, they may activate complement and accessory cells and produce extensive tissue damage (Fig. 9.35).
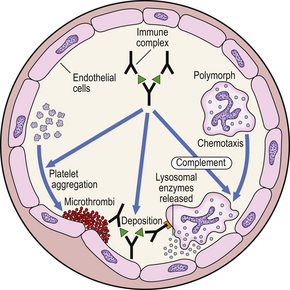
A classic example is the Arthus reaction, an experimental model where an antigen is injected into the skin of an animal that has been previously sensitised (Fig. 9.36). The reaction of preformed antibody with this antigen results in high concentrations of local immune complexes; these cause complement activation and neutrophil attraction and result in local inflammation 6–24 hours after the injection.
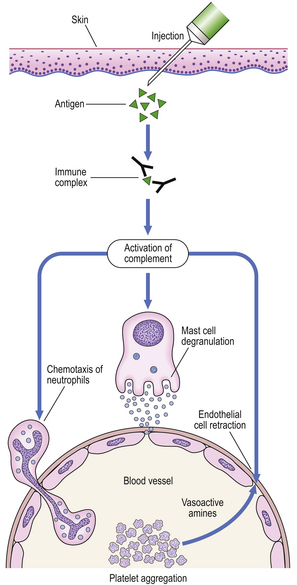
Fig. 9.36 The Arthus reaction. Intradermal injection of antigen results in local immune complex formation with complement activation.
Acute ‘one-shot’ serum sickness is another example; in this condition, urticaria, arthralgia and glomerulonephritis occur about 10 days after initial exposure to the antigen. This is the time when IgG antibody, produced in response to antigen stimulation, reacts with remaining antigen to form circulating, soluble immune complexes (Fig. 9.37). As these damaging complexes are formed, the antigen concentration is rapidly lowered; the process continues only as long as circulating antigen persists and is usually self-limiting.
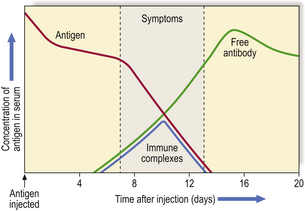
Such reactions were common when antisera raised in animals were injected repeatedly into humans to neutralise bacterial toxins, e.g. tetanus antitoxin. The animal serum immunoglobulins were highly immunogenic and resulted in an IgG antibody response to the foreign serum, hence ‘serum sickness’. This reaction is now rare because animal serum is no longer used in this way.
Acute post-streptococcal glomerulonephritis (Ch. 21) is caused by a similar mechanism. It occurs 10–12 days after a streptococcal infection of the throat or skin and results in deposition of immune complexes of IgG and C3 in the glomerular basement membrane. Streptococcal antigens are rarely found in the complexes but antigenic fragments from certain ‘nephritogenic’ strains of streptococci bind to the glomerular basement membrane, so localising antibody to this site.
Chronic immune complex nephritis accounts for most cases of chronic glomerulonephritis in humans. When compared with the ‘one-shot’ model, chronic immune complex formation and deposition will occur if:
Persistent antigen exposure is most likely to occur if the antigen is a micro-organism capable of replication despite a host response, a medically prescribed drug, or an auto-antigen.
Delayed-type hypersensitivity (type IV)
Type IV reactions are mediated by T-lymphocytes which react with antigen and release interleukin-2, interferon-gamma and other Th1 cytokines. Once T-cells have been sensitised by primary exposure, secondary challenge is followed by a delayed-type hypersensitivity reaction (DTH), a local inflammatory response which takes 2–3 days to develop clinically. Histologically, these reactions consist of infiltrating T-lymphocytes, macrophages and occasional eosinophils. Experimentally, DTH can be transferred by T-lymphocytes but not by serum, i.e. antibodies are not involved.
A classic example of DTH is the tuberculin reaction. If a small amount of purified protein derivative (PPD) of tubercle bacilli is injected intradermally (Mantoux or Heaf test) into non-immune individuals, there is no effect. However, in individuals with cell-mediated immunity to tubercle bacilli, as a result of previous tuberculous infection or immunisation with BCG (bacille Calmette–Guérin, a live but non-virulent strain of Mycobacterium bovis), an area of reddening and induration develops after 24–48 hours (Fig. 9.38). The dermis of the reaction site becomes infiltrated by lymphocytes and macrophages around small blood vessels, with oedema and vascular dilatation.
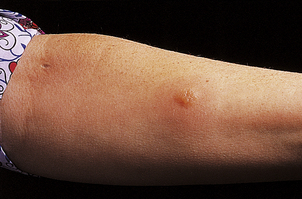
DTH may result from the normal cell-mediated immune response to infection with viruses, fungi and certain bacteria, notably Mycobacterium tuberculosis and Mycobacterium leprae. If macrophages are unable to destroy ingested organisms, they may undergo differentiation into epithelioid cells or multinucleate giant cells. A collection of these cells forms a granuloma (Fig. 9.39). Local tissue damage is an unwanted side-effect of this otherwise protective immune response. If the DTH response is absent or impaired, however, T-lymphocytes are unable to localise the invading micro-organism and patients develop invasive, aggressive disseminated disease, such as acute miliary tuberculosis (Fig. 9.40) or lepromatous leprosy.
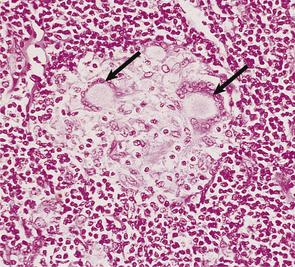

Contact dermatitis (Ch. 24) to occupational and other antigens is also a type IV reaction. Agents that do this are of relatively low molecular weight (<1 kD) and not immunogenic in their own right; instead, they are highly reactive molecules that bind covalently to skin or tissue proteins. The sensitising chemical is known as a hapten and the host protein as the carrier. The range of potential sensitising antigens is wide. Two phases of pathogenesis are recognised: the induction phase and the elicitation phase. In the induction phase, antigen-presenting cells in the skin—Langerhans’ cells—bind the hapten–carrier protein complex and present it to T-lymphocytes in association with MHC class II antigen. Induction of T-cells usually occurs after months of exposure to small amounts of antigen. Re-exposure to the relevant antigen triggers the elicitation phase where effector T-cells migrate to the skin to meet the protein complex presented by Langerhans’ cells in the epidermis with consequent cytokine release and skin inflammation.
The diagnosis of the offending agent is made by patch testing. A suspected contact sensitiser is applied to normal skin on the patient’s back and covered for 48 hours. The reaction site is inspected after 2 and 4 days. In a positive response, there is inflammation and induration at the test site.
Delayed-type hypersensitivity is also a key mechanism underpinning the rejection of transplanted tissues and organs (p. 193).
AUTOIMMUNITY AND AUTOIMMUNE DISEASE





Autoimmunity is an immune response against a self (auto) antigen or antigens. Autoimmune disease is tissue damage or disturbed physiological function resulting from an auto-immune response. This distinction is important as autoimmune responses can occur without resulting disease. Proof that autoimmunity causes a particular disease requires a number of criteria to be met, similar to Koch’s postulates for micro-organisms in infectious diseases:
Patterns of autoimmune disease
Autoimmune diseases can affect any organ in the body although certain systems, such as endocrine glands, seem particularly susceptible. They are conventionally classified into organ-specific and non-organ-specific disorders.
Organ-specific autoimmune diseases
These affect a single organ; one or another endocrine gland is commonly involved. The antigen targets may be molecules expressed on the surface of living cells (particularly hormone receptors) or intracellular molecules, particularly intracellular enzymes (Table 9.4).
Table 9.4 Some examples of self-antigens and associated autoimmune diseases
| Self-antigen | Disease |
|---|---|
| Hormone receptors | |
Neurotransmitter receptor Acetylcholine receptorMyasthenia gravisCell adhesion molecules Epidermal cell adhesion moleculesBlistering skin diseasesPlasma proteins
Other cell surface antigens
Intracellular enzymes
Intracellular molecules involved in transcription and translation
Auto-antibodies to some self-antigens are involved in disease pathogenesis (e.g. acetylcholine receptor antibodies); others are useful diagnostic markers but not pathogenic (e.g. anti-mitochondrial enzyme antibodies).
Non-organ-specific autoimmune diseases
Non-organ-specific disorders affect multiple organs and are usually associated with autoimmune responses against self-molecules which are widely distributed through the body, particularly intracellular molecules involved in transcription and translation of the genetic code.
Epidemiology of autoimmune disease
Around 3% of the population has an autoimmune disease. Many chronic disabling diseases are considered to have an autoimmune basis, including multiple sclerosis, rheumatoid arthritis and insulin-dependent diabetes mellitus. Autoimmune diseases show clustering within families but are rare in childhood; the peak years of onset are from 15 to 65 years, the major exception being the childhood-onset form of diabetes mellitus.
There are striking sex differences in the risk of developing an autoimmune disease. Almost all are more common in women, and for some autoimmune diseases the risk may be eight times greater. A notable exception is ankylosing spondylitis, which is much commoner in young men.
Immunological tolerance
Autoimmune responses are similar to immune responses to non-self-antigens. Both are driven by antigen, involve the same immune cell types and produce tissue damage by the same effector mechanisms. The key point is to understand what regulatory mechanisms prevent autoimmune responses occurring in everyone.
The immune system can generate a vast diversity of different T-cell antigen receptors and immunoglobulin molecules by differential genetic recombination. This process produces many antigen-specific receptors capable of binding to self-molecules. To avoid autoimmune disease, the T- and B-cells bearing these self-reactive molecules must be either eliminated or down-regulated so that the immune system is made specifically unreactive—tolerant—to self-antigens. Because T-cells (in particular CD4+ T-cells) have a central role in controlling nearly all immune responses, the process of T-cell tolerance is of greater importance in avoidance of autoimmunity than B-cell tolerance, since most self-reacting B-cells will not be able to produce auto-antibodies unless they receive appropriate T-cell help.
Those processes that induce specific tolerance arise inside the thymus (thymic tolerance) or outside (peripheral tolerance).
Thymic tolerance
T-cell development in the thymus plays a major role in eliminating T-cells capable of recognising peptides from self-proteins. The principles of positive and negative selection are explained above (p. 167).
This process of thymic education is only partially successful. Thymic tolerance can fail if self-peptides are not expressed at a sufficient level in the thymus to induce negative selection. Most peptides found bound to MHC molecules in the thymus are from either ubiquitous intracellular or membrane-bound proteins present in the extracellular fluid. Thymic tolerance is induced to some but not all tissue-specific proteins (such as might be found in the brain, muscle, joints, islets of Langerhans, etc.) and it is not surprising that autoreactive T-cells can be detected in healthy people.
Peripheral tolerance
A second level of control over potentially autoreactive cells is termed peripheral tolerance. Several mechanisms are involved.
Immunological ignorance
Some self-antigens are effectively invisible to the immune system—immunological ignorance. This occurs because the antigen is sequestered in an avascular organ such as the vitreous humour of the eye. Immunological ignorance also occurs because CD4+ T-cells will only recognise antigens presented in association with MHC class II molecules. The very limited distribution of these molecules, confined to professional antigen-presenting cells, means that most organ-specific molecules will not be presented at levels high enough to induce T-cell activation. To prevent large amounts of self-antigen from gaining access to antigen-presenting cells, debris from self-tissue breakdown must be cleared rapidly and destroyed. This is achieved by cell death through apoptosis, so preventing widespread spilling of cell contents, together with a variety of scavenger mechanisms that mop up cell debris.
Self-antigens and lymphocytes are also kept separate by the restricted routes of lymphocyte circulation which limit naive lymphocytes to secondary lymphoid tissue and the blood.
Anergy
Naive CD4+ T-cells need two signals to become activated and initiate an immune response: an antigen-specific signal through the T-cell antigen receptor and a second, non-specific co-stimulatory signal, usually signalled by CD28 (on the T-cell) binding to CD80 or CD86 on the stimulator (see Fig. 9.6). If the T-cell receives both signals, then it will become activated and proliferate and produce cytokines. If no co-stimulatory molecules are engaged, then stimulation through the T-cell receptor alone leads to apoptosis or a state of longstanding unresponsiveness called anergy. Expression of these co-stimulatory molecules is tightly controlled and confined to specialised antigen-presenting cells such as dendritic cells. Given their distributions, interaction between CD4+ cells and dendritic cells is only likely to occur in secondary lymphoid tissues such as lymph nodes. The restricted expression of co-stimulatory molecules means that even if a T-cell recognises a tissue-specific peptide– MHC molecule complex (e.g. an antigen derived from a pancreatic islet cell), then anergy rather than activation is likely to follow, as no antigen-presenting cell will be available in healthy tissue to provide the co-stimulatory signal. T-cells also express cell surface molecules similar in structure to co-stimulatory molecules but which exert a negative effect on T-cell activation (see Fig. 9.6). CTLA-4 (CD152), which has a similar structure to CD28 and binds to the same ligands (CD80 or CD86), induces anergy or apoptosis and may be important in terminating an immune response to self-antigens.
Regulation and suppression
Self-reactive T-cells may be actively suppressed by inhibitory populations of T-cells that recognise the same antigen—so-called regulatory or suppressor T-cells with a distinctive CD4+ CD25+ phenotype. The best defined mechanism involves cytokines produced by antigen stimulation which either inhibit or alter the activation of nearby T-cells. For example, a Th2 response may specifically inhibit a Th1 response through IL-10 production (see Fig. 9.15).
B-cell tolerance
B-cell tolerance operates at a peripheral rather than a central level and is less complete than T-cell tolerance. The production of self-reactive antibodies is limited mainly by the lack of T-cell help for self-antigens, despite the fact that new B-cells are being produced continuously from bone marrow precursors and many of these are autoreactive.
Breakdown of tolerance
For autoimmune responses to occur, the key mechanisms of immunological tolerance outlined above must be broken down.
Overcoming peripheral tolerance
This can result from inappropriate access of self-antigens to antigen-presenting cells, inappropriate or increased local expression of co-stimulatory molecules, or alterations in the ways in which self-molecules are presented to the immune system (Fig. 9.41). All of these are more likely to happen when inflammation or tissue damage is present. The increased activity of proteolytic enzymes in inflammatory sites can cause both intra- and extracellular proteins to be broken down, leading to high concentrations of peptides being presented to responsive T-cells. These novel peptides are known as cryptic epitopes. The structures of self-peptides may also be altered by viruses, free radicals or ionising radiation, thus bypassing previously established tolerance. For antigens that are sequestered from the immune system (e.g. in the eye), sufficient antigen may be released by tissue damage to initiate an immune response.
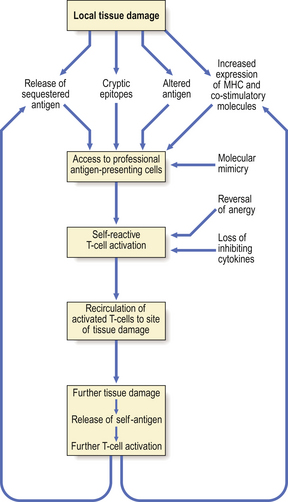
Molecular mimicry
Structural similarity between self-proteins and microbial antigens may trigger an autoimmune response. In systemic infection, this cross-reactivity will cause expansion of the responsive T-cell population recognising the self-peptide if local conditions allow. The process is known as molecular mimicry (Table 9.5).
| Microbial antigen | Self-antigen with similar structure | Disease in which consequent molecular mimicry may play a role |
|---|---|---|
| Group A streptococcal M protein | Antigen found in cardiac muscle | Rheumatic fever |
| Bacterial heat shock proteins | Self heat shock proteins | Links suggested with several autoimmune diseases but none proven |
| Coxsackie B4 nuclear protein | Pancreatic islet cell glutamate decarboxylase | Insulin-dependent diabetes mellitus |
| Campylobacter jejuni glycoproteins | Myelin-associated gangliosides and glycolipids | Guillain–Barré syndrome |
Once tolerance has broken down, the resulting process of inflammation may allow presentation of further peptides. The immune response broadens and local tissue damage accelerates. This domino-like process is known as epitope spreading. This is best demonstrated in experimental models, where immunisation with a single peptide from a protein found in myelinated nerve sheaths (known as myelin basic protein or MBP) can lead to widespread inflammation in the central nervous system with an immune response against many peptides found in both MBP and other CNS proteins. This implies that, once the barrier of tolerance is broken down, autoimmune responses may be easier to sustain.
Aetiology of autoimmune disease
In autoimmune diseases, interactions between genetic and environmental factors are critically important (Fig. 9.42).
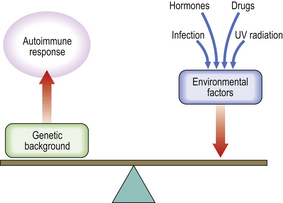
Fig. 9.42 Interactions between genetic and environmental factors are important in the aetiology of autoimmune diseases.
Genetic factors
Twin and family studies have confirmed a genetic contribution in all autoimmune diseases studied. Multiple autoimmune diseases may cluster within the same family, and subclinical autoimmunity is common among family members. The genetic contribution to autoimmune disease usually involves multiple genes but some single-gene defects involve defects in apoptosis.
The strongest and best characterised associations involve alleles of the major histocompatibility complex (MHC), as might be expected from the central role of the products of many of these genes in T-cell function, and the involvement of other MHC genes in control of immunity and inflammation (Table 9.6).
Table 9.6 Common autoimmune diseases and their major HLA associations
| HLA association | Disease | Relative risk |
|---|---|---|
| B27 |
DR2Goodpasture’s syndrome16DR3
DR4Insulin-dependent diabetes mellitus6
Environmental factors
Environmental triggers in autoimmunity include:
Hormones
Females are far more likely than males to develop most autoimmune diseases and hormonal factors must play a major role in this gender difference. Most autoimmune diseases have their peak age of onset within the reproductive years and evidence implicates oestrogens as triggering factors. Removal of the ovaries inhibits the onset of spontaneous autoimmunity in animal models of systemic lupus erythematosus (SLE), while administration of oestrogen accelerates the onset of disease.
Infection
The relationship between infection and autoimmunity is clearest in the situation of molecular mimicry but infection of an organ may also cause upregulation of co-stimulatory molecules and altered patterns of antigen breakdown and presentation. Autoimmune diseases tend to be less common in parts of the world that carry a high burden of parasitic diseases and other infections. In some animal models of autoimmunity, the development of disease can be dramatically inhibited by keeping the animals in a laboratory environment with a high prevalence of infection. Keeping the same animals in germ-free conditions promotes the development of autoimmunity for reasons that are not clear. Attempts to identify hidden infections in autoimmune diseases such as rheumatoid arthritis and multiple sclerosis have been unsuccessful.
Drugs
Drug-induced autoimmunity may involve mechanisms comparable to molecular mimicry, whereby the drug or a drug–self-molecule complex has a structural similarity to self and hence allows bypass of peripheral tolerance. Some drugs (e.g. penicillamine) have the ability to bind directly to the peptide-containing groove in MHC molecules and a direct capacity to induce abnormal T-cell responses.
Drug-mediated autoimmunity affects only a small proportion of those treated and is probably genetically determined. For example, HLA-DR2 is associated with penicillamine-induced myasthenia gravis, whereas DR3 is associated with nephritis. Genetic variation in drug metabolism is important. The best example is the relationship between drug-induced systemic lupus erythematosus and the rate of acetylation of the triggering drug: slow acetylators are prone to SLE. It seems likely that this partial defect in metabolism may allow the formation of immunogenic conjugates between drug and self-molecules. Drugs may also have intrinsic adjuvant or immunomodulatory effects that disturb normal tolerance mechanisms; for example, thyroid autoimmunity can occur after treatment with interferon-alpha.
Ultraviolet radiation
Exposure to ultraviolet (UV) radiation (usually in the form of sunlight) is a well-defined trigger for skin inflammation and sometimes systemic involvement in SLE (Fig. 9.43). UV radiation can modify self-antigens, so enhancing their immunogenicity, or lead to apoptotic death of cells within skin. Apoptosis is associated with cell surface expression of auto-antigens usually found only within cells that are then able to bind related auto-antibodies and trigger tissue damage.
Mechanisms of tissue damage
Tissue damage in autoimmune disease is mediated by antibody or immune complexes or CD4+ T-cell-mediated activation of macrophages or cytotoxic T-cells, or a combination of these mechanisms.
In addition to their destructive effects, auto-antibodies can also cause disease by binding to the functional sites of self-antigens such as hormone receptors, neurotransmitter receptors and plasma proteins. These auto-antibodies either mimic or block the action of the endogenous ligand for the self-protein, and thus cause abnormalities in function without necessarily causing inflammation or tissue damage. This phenomenon is best characterised in endocrine autoimmunity (Table 9.4; Fig. 9.34).
Treatment of autoimmune diseases
The treatment of autoimmune disease is based either on suppression of the damaging immune response or on replacement of the function of the damaged organ (Fig. 9.44).
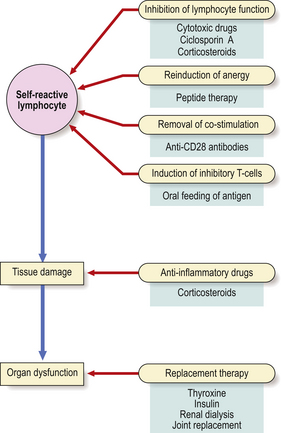
Replacement of function
This is the usual treatment in those autoimmune diseases with failure of the affected endocrine organ, such as hypothyroidism or insulin-dependent diabetes mellitus.
Suppression of the autoimmune response
Immunosuppression before irreversible tissue damage occurs is vital, but the early detection of preclinical autoimmunity presents a major challenge. In many autoimmune diseases, such as SLE, rheumatoid arthritis and autoimmune kidney disease, immunosuppression is the only means of preventing severe disability or death. However, all forms of immunosuppression are limited by their lack of specificity and toxic side-effects. Some newer strategies (Fig. 9.44) have been developed in animal models for selective suppression of autoimmune responses and are being evaluated in clinical trials.
PRINCIPLES OF ORGAN TRANSPLANTATION







Transplantation of living cells, tissues or organs is well established as a routine practice. Cells (e.g. red blood cells in transfusion), tissues (e.g. skin grafting in extensively burned patients), or whole organs (such as kidney, heart, pancreas or liver) may be successfully transferred between genetically dissimilar individuals (allogeneic grafting). The outcome depends on the degree of ‘matching’ of the relevant transplantation antigens between the two individuals and the success of therapeutic immunosuppressive measures to prevent rejection.
In contrast, grafting of a person’s tissue from one part of their body to another (autologous grafting) is always successful, provided there are no surgical setbacks.
Transplantation across a species barrier, for instance from pigs to humans (xenogeneic grafting), is a focus of interest because of the shortage of human organs.
Histocompatibility genetics
The surfaces of all human cells express a series of molecules that are recognised by other individuals as foreign antigens. The most important antigens in organ transplantation are called ‘histocompatibility antigens’, or human leukocyte antigens (HLAs), and have a crucial role in determining survival of transplanted organs. In humans, they are encoded by a segment of chromosome 6 known as the major histocompatibility complex (MHC) (Fig. 9.4). Additional antigenic systems (minor histocompatibility antigens) play a more minor role in transplantation and are largely ignored. MHC antigens are described in detail above (p. 165).
The HLA system is extremely polymorphic; that is, it has multiple alternative forms (alleles) of the gene at each known locus. The close proximity of the HLA loci means that their antigenic products tend to be inherited together as an HLA haplotype. Because one haplotype is inherited from each parent, there is a one-in-four chance that siblings will possess identical pairs of haplotypes (Fig. 9.45).
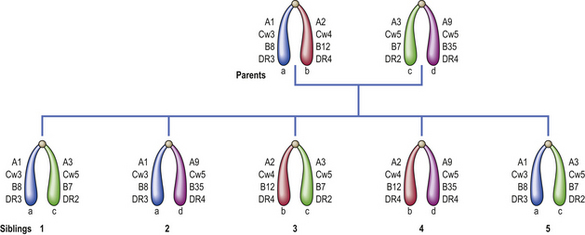
Kidney transplantation
Kidney transplantation is the treatment of choice for most patients with end-stage renal failure and illustrates the principles underpinning solid organ allografts.
Selection of recipient and donor
In kidney transplantation, organs can come from cadavers or living related donors. Because humans have two kidneys (unlike most other solid organs), a relative may choose to donate one to the recipient. Relatives must be screened clinically and psychologically, and ABO and HLA typed so that the most suitable donor can be chosen.
The selection of a donor kidney is rigorous (Table 9.7). Knowing the ABO blood group and HLA type of a cadaver kidney,national and international registers of potential recipients can be searched by computer to find an ABO-compatible patient who matches the donor at as many loci as possible. Once the recipient has been selected, the recipient’s serum is then cross-matched against the donor’s lymphocytes. If the patient has cytotoxic antibodies to donor MHC class I antigens (positive T-cell cross-match), then the kidney is unsuitable for that recipient.
Table 9.7 Selection of donor and recipient in kidney transplantation
| Recipient selection |
Kidney selectionCadaver donor
Living donor
Once the donor kidney has been removed, it is perfused with cold physiological fluids. Provided cooling begins within 30 minutes of cutting the renal blood supply (warm ischaemia time), the kidney has an excellent chance of functioning in the recipient. The duration of the perfusion (cold ischaemia time) should be less than 48hours.
The post-transplantation period
The transplanted kidney is usually sited in the iliac fossa. Great care is taken with the vascular anastomosis and implantation of the ureter. Once the vascular anastomoses are complete, the graft often starts to function immediately.
Renal function may deteriorate immediately after surgery for several reasons. Acute tubular necrosis can occur due to low blood pressure in either the recipient or the donor. If this happens, the recipient can be dialysed until renal function recovers. Alternatively, poor renal function may indicate hyperacute rejection (see below) or urinary obstruction, which must be relieved surgically.
It is crucial to distinguish rejection from infection, as the treatment differs. Rejection can be detected by percutaneous fine-needle aspiration of the transplant. Immunosuppressive therapy is vital to prevent graft rejection.
Clinical rejection
Rejection of the organ graft may be:
Hyperacute rejection occurs minutes to hours following revascularisation of the graft. It is due to preformed circulating cytotoxic antibody which reacts with MHC class I antigens in the donor kidney. Activation of complement results in an influx of polymorphonuclear leukocytes, platelet aggregation, obstruction of the blood vessels, and ischaemia. Histologically, the microvasculature becomes plugged with leukocytes and platelets, resulting in infarction. The kidney swells dramatically and is tender. Renal function declines; oliguria or anuria follows. There is no successful therapy and the kidney must be removed.
Acute rejection occurs a few weeks or months following transplantation. Early diagnosis is important because prompt treatment with intravenous methylprednisolone and/or anti-CD3 antibody reverses renal damage. Histologically, there is a mononuclear infiltrate in the renal cortex, and necrosis of arterial walls; after successful treatment, the inflammatory infiltrate clears. Acute rejection is associated with increased expression of MHC class I and class II antigens in inflamed grafts, and with early infiltration of CD8+ T-lymphocytes.
Chronic rejection is seen after months or years of good renal function. There is slowly progressive renal failure and hypertension. Dominant histological findings are thickening of the glomerular basement membrane, hyalinisation of the glomeruli, interstitial fibrosis, and proliferation of endothelial cells. This picture of chronic allograft nephropathy must be distinguished from recurrence of the original glomerular disease.
Immunopathology of graft rejection
CD4+ T-lymphocytes play a central role in rejection.
The rejection process has two parts (Fig. 9.46):
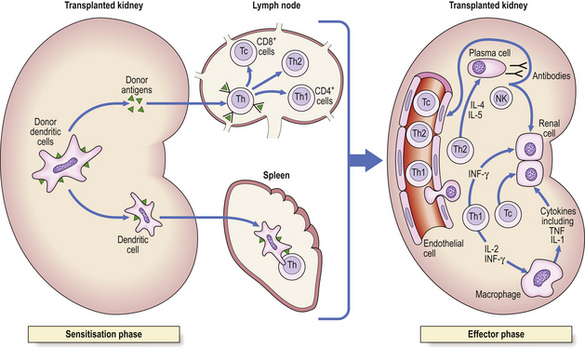
In the afferent phase, donor MHC molecules found on ‘passenger leukocytes’ (dendritic cells) within the graft are recognised by the recipient’s CD4+ T-cells, a process called allorecognition, which takes place either in the graft itself or in the lymphoid tissue of the recipient.
The effector phase of rejection is orchestrated by CD4+ T-cells which enter the graft parenchyma and recruit effector cells responsible for the tissue damage of rejection, namely macrophages, CD8+ T-cells, natural killer cells and B-lymphocytes. The most important cytokines in graft rejection are interleukin-2 and interferon-gamma.
Not all parts of the graft need to be attacked for rejection to occur. The critical targets are the endothelium of the microvasculature and the specialised parenchymal cells of the organ, such as renal tubules, pancreatic islets of Langerhans or cardiac myocytes.
Immunosuppression
Immunosuppressive drugs are used to prevent graft rejection. Azathioprine is inactive until metabolised by the liver but then affects all dividing cells, including lymphocytes, by inhibiting DNA synthesis. Corticosteroids have their main immunosuppressive effects on macrophage activity but are ineffective alone in preventing the early phase of rejection.
Graft rejection has been reduced dramatically by the discovery of ciclosporin A, tacrolimus and comparable agents. Ciclosporin is a powerful drug on its own or in combination with azathioprine and prednisolone.
Monoclonal antibodies that destroy T-lymphocytes in the graft recipient can also be used as immunosuppressive agents.
Graft survival
Long-term graft survival is closely correlated with the degree of HLA matching, particularly at the class II locus. About 50% of fully matched cadaveric grafts survive 17 years but mismatched grafts survive for only 8 years.
Complications
Patients have an increased susceptibility to infection after transplant. Major causes of infection in the first month after transplantation are those related to surgical wounds, indwelling cannulae, or post-operative lung infections. The effects of such infections, however, are often considerable in the immunocompromised host. After 1–4 months of immunosuppression, cytomegalovirus dominates a picture that includes various fungal, viral and protozoal infections. Infections occurring beyond 4 months fall into three main groups:
A late complication of organ transplantation is recurrence of the original disease. This should always be considered in patients in whom there is functional deterioration following long periods of stable graft function. The incidence of malignancy, particularly lymphoma, in transplant recipients is 40 times greater than in the general population. Both lymphoma and Kaposi’s sarcoma are more common when profound immune suppression is used, and are related to persistent viral stimulation with Epstein–Barr virus or human herpes virus 8 respectively.
Transplanted patients also have an increased risk of acute myocardial infarction. This may be linked to hypertension, hypertriglyceridaemia or insulin-resistant diabetes, as these conditions are often present before transplantation and are aggravated by steroids.
Transplantation of other organs
Liver transplantation
The results of human liver transplantation have improved dramatically in the last 15 years. Hepatic surgery poses unique problems; these include the bleeding tendency of a recipient with liver failure and the technically difficult surgery required to revascularise a grafted liver. However, compared with transplants of other organs, rejection episodes are milder and require less immunosuppression. Hepatitis C is the commonest indication for transplantation; other indications include biliary atresia, hepatocellular carcinoma, primary biliary cirrhosis and alcoholic cirrhosis (Ch. 16).
Heart transplantation
Unlike renal transplantation, there is no satisfactory long-term support available if the donated heart is rejected. Consequently, early diagnosis of rejection is crucial. Electrocardiographic changes are closely monitored and serial endomyocardial biopsies show lymphocytic infiltration with increased MHC class I expression by myocardial cells.
A major post-operative problem is accelerated atherosclerosis in the graft coronary arteries. This is the major cause of death in patients who survive more than 1 year.
Pancreatic transplantation
Improvements in surgical technique and better immunosuppression have resulted in 90% survival at 1 year of transplanted vascularised pancreatic grafts but longer-term results are disappointing.
Skin grafting
Allogeneic skin grafting in humans is useful in providing skin cover in severely burned patients. HLA typing is not required because the endogenous immunosuppressive effect of severe burns allows prolonged survival of unmatched skin. Although the graft is finally rejected, the short-term protective barrier afforded by covering burns during this time is vital to the patient in resisting infection.
Corneal grafting
Corneas are obtained from cadaveric donors. There is no need to HLA type or systemically immunosuppress the recipient because corneal rejection does not occur unless the graft becomes vascularised. In grafts that do become vascularised following chemical burns or chronic viral infection, HLA matching significantly improves survival.
Bone marrow transplantation
The transplantation of haemopoietic stem cells (HSCT) from bone marrow offers the only chance of cure for many patients with a wide range of disorders:
Graft rejection is common, but HSCT has the unique and often fatal complication of graft-versus-host disease (GVHD), in which the grafted immunocompetent cells recognise the host as foreign and mount an immunological attack.
Selection and preparation of patients
Theoretically, any abnormality of bone marrow stem cells is correctable by transplantation.
Preparation for transplantation begins 10 days before grafting. Measures to reduce infection risk include strict reverse-barrier nursing, decontamination of the skin and gut, the use of appropriate antibiotics and antimycotics, and immunoglobulin replacement if necessary.
The grafting procedure is straightforward; small amounts of marrow are taken from multiple sites under general anaesthetic. Bone spicules are removed by filtration through graded sieves.
Commonly confused conditions and entities relating to immunology and immunopathology
| Commonly confused | Distinction and explanation |
|---|---|
| Interleukins, cytokines, lymphokines and chemokines |
Atopy, allergy and anaphylaxisAll three terms apply to type I (immediate) hypersensitivity or allergy. Atopy is an inherited tendency to produce IgE antibodies to environmental allergens. Exposure of the individual to an allergen may result in allergy as a result of mast cell degranulation. A severe, systemic, life-threatening allergic reaction is termed anaphylaxis.Monocytes, macrophages and histiocytes
Graft rejection and graft-versus-host diseaseGraft rejection is an immunologically mediated reaction against a transplanted tissue or organ. Graft-versus-host disease occurs when immunologically active cells in the donor tissue (e.g. bone marrow transplant) damage the recipient’s tissues.Cells can then be given either without fractionation or after removal of immunocompetent T-lymphocytes responsible for graft-versus-host disease (see below). Cells are then transplanted by intravenous infusion.
A successful graft is indicated by a rise in the peripheral white cell count and the appearance of haemopoietic precursors in the marrow 10–20 days after transplantation.
Complications
Three major problems dominate the post-transplant period:
Failure of engraftment can be due to using insufficient haemopoietic stem cells or rejection of the grafted cells by the host. Patients with some residual immunity (e.g. as in leukaemia in remission or partial immune deficiencies) require immune suppression— or conditioning—prior to grafting to ensure that rejection does not occur. Patients with no immune function (e.g. severe combined immune deficiency) do not, in theory, require conditioning as they are unable to reject the graft.
Serious bacterial, fungal and viral infections occur despite the elaborate measures aimed at reducing their incidence and severity. Infection with cytomegalovirus (CMV) is a common cause of death; evidence of CMV reactivation is seen in 75% of patients who are CMV positive before transplant.
Graft-versus-host disease (GVHD) occurs in most patients who receive allogeneic bone marrow transplants. About 7–14 days later, a skin rash, fever, hepatosplenomegaly, bloody diarrhoea and breathlessness develop. Skin biopsy shows lymphocytic infiltration with vascular cuffing and basal cell degeneration. The mortality of GVHD is considerable; over 70% of those with severe GVHD and about one-third with mild GVHD will die. Treatment requires an increase in immunosuppression but, once established, GVHD is very difficult to eradicate.
Prevention of GVHD involves ways to eliminate or reduce the numbers of immunocompetent T-cells in the engrafted bone marrow. T-cell-specific monoclonal antibodies can help deplete the T-cell load.
Peripheral blood stem cell transplantation
Because pluripotent stem cells can be mobilised from peripheral blood using colony-stimulating factors, stem cell transplants can theoretically be used as an alternative to bone marrow. There is interest in using umbilical cord blood as a source of stem cells in view of its ready availability. Allogeneic or autologous stem cell transplantation is under investigation for the management of autoimmune diseases such as rheumatoid arthritis and multiple sclerosis, for solid tumours such as breast cancer and neuroblastoma, and for haematological malignancies.
ACKNOWLEDGEMENT
The following figures are reproduced and modified, with permission, from Essentials of Clinical Immunology (5th edn), Blackwell Science, Oxford: 9.1, 9.2, 9.3, 9.4, 9.5, 9.6, 9.7, 9.13, 9.14, 9.15, 9.18, 9.20, 9.23, 9.24, 9.31, 9.33, 9.34, 9.37, 9.41, 9.44 and 9.46.
Albiger B., Dahlberg S., Henriques-Normark B., Normark S.. Role of the innate immune system in host defence against bacterial infections: focus on the Toll-like receptors. Journal of Internal Medicine. 2007;261:511-528.
Andrews P.A.. Renal transplantation. British Medical Journal. 2002;324:530-534.
Belkaid Y.. Regulatory T cells and infection: a dangerous necessity. Nature Reviews. Immunology. 2007;7:875-888.
Carter R.H.. B cells in health and disease. Mayo Clinic Proceedings. 2006;81:377-384.
Changanti R K 2006 Complement deficiencies. http://www.emedicine.com/med/topic419.htm
Chapel H.M., Haeney M.R., Misbah S.A., Snowden H.N.. Essentials of clinical immunology, 5th edn. Oxford: Blackwell Science, 2006. http://www.immunologyclinic.com
Copelan E.A.. Medical progress: haematopoietic stem-cell transplantation. New England Journal of Medicine. 2006;354:1813-1826.
Davidson A., Diamond B.. Autoimmune diseases. New England Journal of Medicine. 2001;345:340-350.
Delves P.J., Roitt I.M.. The immune system. New England Journal of Medicine. 2000;343(37–49):108-117.
Jiang H., Chess L.. Mechanisms of disease: regulation of immune responses by T cells. New England Journal of Medicine. 2006;354:1166-1176.
Kay A.B.. Allergy and allergic diseases. New England Journal of Medicine. 2001;344(30–37):109-113.
Klein J., Sato A.. The HLA system. New England Journal of Medicine. 2000;343(702–709):782-786.
Letvin N.L.. Progress and obstacles in the development of an AIDS vaccine. Nature Reviews. Immunology. 2006;6:930-939.
Ley K., Laudanna L., Cybulsky M.I., Nourshaugh S.. Getting to the site of inflammation: the leukocute adhesion cascade update. Nature Reviews. Immunology. 2007;7:678-689.
Marodi L., Notarangelo L.D.. Immunological and genetic bases of new primary immunodeficiencies. Nature Reviews. Immunology. 2007;7:851-861.
Medzhitov R.. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819-826.
Miller S.D., Turley D.M., Podojil J.R.. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nature Reviews. Immunology. 2007;7:665-677.
National Cancer Institute 2006 Understanding Cancer Series: The Immune System. www.cancer.gov/cancertopics/understandingcancer/immunesystem
Newburger P.E. Disorders of neutrophil number and function. Hematology. American Society of Hematology. Education Program 2006;104-110
Uniting the world against AIDS (UNAIDS):http://www.unaids.org
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Chapter 9 Immunology and immunopathology
DEFENCE AGAINST INFECTION
The immune system evolved as a defence against infectious diseases. Individuals with deficient immune responses, if untreated, succumb to infections in early life. There is, therefore, a selective evolutionary pressure for an efficient immune system. Specific immunity is called into play only when micro-organisms bypass non-specific or innate mechanisms.
Non-specific defences
Many non-specific mechanisms prevent invasion of the body by micro-organisms:
Innate immunity
The innate immune system is activated by pattern recognition receptors on dendritic cells recognising conserved polysaccharide molecular patterns on microbes. Key components include:
Specific immunity
The immune system has four essential features:
A specific or adaptive immune response consists of two parts: a specific response to the particular antigen and a non-specific augmentation of the effect of that response. For the specific response there is a quicker and larger response the second time that a particular antigen is encountered; memory of the initial specific immune response provides the efficiency.
The immune system has to recognise all pathogens, past and future, and must have considerable diversity of response. This diversity is partly genetic (germline encoded) and partly generated by somatic mutation during maturation of the immune system.
Immune responses, both innate and adaptive, have two phases: first the recognition phase, involving antigen-presenting cells and T-lymphocytes (see Key molecules), in which the antigen is recognised as foreign; and second the effector phase, in which antibodies and effector T-lymphocytes eliminate the antigen, often by recruiting innate mechanisms such as complement or macrophage activation.
KEY MOLECULES
Antigens are substances able to provoke an immune response and react with the products of that response Antibodies are immunoglobulin molecules produced by plasma cells. Antigen-binding properties reside in the Fab fragments, while effector functions lie in the Fc fragment Major histocompatibility complex (MHC) antigens are of two main types—class I and class II. They play a fundamental role in the normal immune response by presenting antigenic peptides to T-cells Helper T-cells recognise antigen in association with MHC class II molecules, while cytotoxic T-cells recognise antigen associated with MHC class I T-cell receptors will recognise antigen only as part of a complex of antigenic peptide and the MHC molecule—a process termed MHC restrictionAntigens
Antigens are substances able to provoke an immune response and react with the immune products. They react both with the T-cell recognition receptor and with antibody. An antigenic molecule may have several antigenic determinants (epitopes); each epitope can bind with an individual antibody, and a single antigenic molecule can therefore provoke many antibody molecules with different binding sites. Some low molecular weight molecules, called haptens, are unable to provoke an immune response themselves, although they can react with existing antibodies. Such substances need to be coupled to a carrier molecule in order to have sufficient epitopes to be antigenic. For some chemicals, such as drugs, the carrier may be a host protein—called an auto-antigen. The tertiary structure, as well as the amino acid sequence, is important in determining antigenicity.
Antigens are conventionally divided into thymus-dependent and thymus-independent antigens. Thymus-dependent antigens require T-cell participation to provoke the production of antibodies; most proteins are examples. Thymus-independent antigens require no T-cell co-operation for antibody production; they directly stimulate specific B-lymphocytes by cross-linking antigen receptors on the B-cell surface but provoke poor immunological memory. Such antigens include bacterial cell wall polysaccharides.
Factors other than the intrinsic properties of the antigen also influence the quality of the immune response. These include:
Substances that improve a host’s immune response to a separate antigen are called adjuvants; these are routinely used in immunisation programmes in childhood.
Superantigen is the term given to foreign proteins that simultaneously activate large numbers of T-lymphocytes carrying a particular T-cell receptor V-beta gene (see T-cell receptors). Widespread T-cell activation results in florid cytokine release, as exemplified by toxic shock syndrome induced by certain streptococcal toxins.
Antibody
Humoral immunity is dependent on the production of antibodies and their actions. All antibodies belong to the immunoglobulin class of proteins and are produced by plasma cells, themselves derived from B-lymphocytes. The basic structure of an immunoglobulin molecule is shown in Figure 9.2. It has a four-chain structure: two identical heavy (H) chains (molecular weight 50 kD) and two identical light (L) chains (mol wt 25 kD). There are two alternative types of light chain, known as kappa and lambda; an antibody molecule has either two kappa or two lambda light chains, never one of each. In contrast, there are five types of heavy chain, each with important functional differences (Table 9.1). The heavy chains determine the class (isotype) of the antibody and the physiological function of the antibody molecule. Once the antigen-binding site has reacted with its antigen, the molecule undergoes a change in the conformation of its heavy chains in order to take part in effector functions (Table 9.1).
Fig. 9.2 Basic structure of an immunoglobulin molecule. The two identical light chains and two identical heavy chains are held together by disulphide bonds. (Fab, fragment antigen binding; Fc, fragment crystallisable; VL, variable domain of a light chain; VH, variable domain of a heavy chain.)
The amino (N) terminal regions of the heavy and light chains include the antigen-binding sites. The amino acid sequences of these N- terminal domains vary between different antibody molecules of the same isotype and are known as variable (V) regions. Most of these differences reside in three hypervariable areas of the molecule, each only 6–10 amino acid residues long. In the folded molecules, these hypervariable regions in each chain come together, with their counterparts on the other pair of heavy and light chains, to form the antigen-binding site. The structure of this part of the antibody molecule is unique to that molecule and is known as the idiotypic determinant. In any individual, about 106–107 different antibody molecules could be made up by 103 different heavy chain variable regions associating with 103 different light chain variable regions. Somatic mutation during multiple divisions of B-lymphocytes generates further diversity of around 1014 antibody specificities.
IgM is the oldest class of immunoglobulin in evolutionary terms. It is a large molecule consisting of five basic units held together by a joining (J) chain; it penetrates poorly into tissues on account of its large size (Table 9.1). The major physiological role of IgM is intravascular neutralisation of organisms (especially viruses) aided by its 10 antigen-binding sites. IgM also has multiple complement-binding sites; this results in excellent complement activation and lysis of the organism or removal of the antigen–antibody–complement complexes by complement receptors on phagocytic cells. It is the first class of antibody to be formed in response to an initial encounter with an antigen (primary immune response).
IgG is a smaller immunoglobulin which penetrates tissues easily. It is the most abundant immunoglobulin in the plasma and extracellular fluid. It is the only immunoglobulin that crosses the placenta to provide immune protection to the neonate; this is an active process involving specific placental receptors for the Fc portion of the IgG molecule. Polymorphs and macrophages also have surface receptors for the Fc fragment of IgG; thus binding of IgG to particulate antigen promotes adhesion of these cells and subsequent phagocytosis of the antigen.
There are four subclasses of IgG: IgG1 and IgG3 activate complement efficiently and are responsible for clearing most protein antigens; IgG2 and IgG4 react predominantly with carbohydrate antigens (in adults).
IgA is sometimes referred to as ‘mucosal antiseptic paint’. It is secreted locally by plasma cells in the intestinal and respiratory mucosa and is an important constituent of breast milk. It consists of two basic units (a dimer) linked by a ‘joining’ or J chain. The addition of a ‘secretory component’ prevents digestion of the immunoglobulin molecule by enzymes present in intestinal or bronchial secretions. Secretory component is a fragment of the polymeric immunoglobulin receptor synthesised by epithelial cells and transports secretory IgA from the mucosa into the lumen of the gut or bronchi.
There is little free IgD or IgE in serum or normal body fluids. These two classes mainly act as cell receptors. IgD is synthesised by antigen-sensitive B-lymphocytes and acts as a cell surface receptor for antigen. IgE is produced by plasma cells but taken up by specific IgE receptors on mast cells and basophils. IgE probably evolved as a way of expelling intestinal parasites via mast cell degranulation.
T-cell receptors
Like B-cells, each T-cell is committed to a given antigen, which it recognises by one of two types of T-cell receptor (TCR). T-cells have either alpha/beta TCR (a heterodimer of alpha and beta chains) or gamma/delta TCR (a heterodimer of gamma and delta chains). Alpha/beta TCRs predominate in adults, although 10% of T-cells in epithelial structures are of the gamma/delta TCR type. Each type of TCR is associated with several transmembrane proteins which make up the cluster differentiation 3 (CD3) molecule (Fig. 9.3) to form the CD3–TCR complex responsible for taking the antigen recognition signal inside the cell (transduction). The CD3 antigen is widely used as a marker of mature T-cells in diagnostic and investigative pathology.
Fig. 9.3 The structure of the T-cell receptor (TCR). The variable regions of the alpha (α) and beta (β) chains make up the T idiotype. The TCR is closely associated on the cell surface with the CD3 molecule.
The TCR complex recognises small processed antigen peptides in the context of major histocompatibility complex (MHC) class I and II antigens (see below), depending on the type of T-cell. Helper T-cells recognise MHC class II molecules in association with foreign antigen and use the CD4 molecule to enhance binding and intracellular signalling. Cytotoxic T-cells recognise antigen associated with MHC class I molecules and use CD8 molecules for increased binding and signalling. However, recognition of processed antigen alone is not enough to activate T-cells. Additional signals through soluble interleukins are needed; some of these are generated during ‘antigen processing’.
Major histocompatibility complex antigens
Histocompatibility antigens were so named because of the vigorous reactions they provoked during mismatched organ transplantation. However, these antigens play a fundamental role in the normal immune response by presenting antigenic peptides to T-cells. Human major histocompatibility complex (MHC) antigens are also known as human leukocyte antigens(HLAs). MHC antigens are cell surface glycoproteins of two basic types: class I and class II (Fig. 9.4). They exhibit extensive genetic polymorphism with multiple alleles at each locus. As a result, genetic variability between individuals is very great and most unrelated individuals possess different HLA molecules. This means that it is very difficult to obtain perfect HLA matches between unrelated persons for transplantation.
The antigen-specific receptor of an individual T-cell (TCR) will only recognise antigen as part of a complex of antigenic peptide and that individual’s MHC. This process of dual recognition of peptide and MHC molecule is known as MHC restriction because the MHC molecule restricts the ability of the T-cell to recognise antigen. T-cells from one person will not co-operate with antigen-presenting cells from a person of different HLA type.
MHC class I antigens are subdivided into three groups: A, B and C. Each group is controlled by a different gene locus within the major histocompatibility complex on chromosome 6 (Fig. 9.4). The products of the genes at all three loci are chemically similar. MHC class I antigens (Fig. 9.5) are made up of a heavy chain (alpha) controlled by a gene in the relevant MHC locus, associated with a smaller chain called beta-2-microglobulin, controlled by a gene on chromosome 15. The differences between individual MHC class I antigens are due to variations in the alpha chains; the beta-2-microglobulin component is constant. The detailed structure of class I antigens was determined by X-ray crystallography. This shows that small antigenic peptides are tightly bound to a groove in the surface alpha chains.
MHC class II antigens have a folded structure similar to class I antigens with the peptide-binding groove found between the alpha and beta chains (Fig. 9.5). Whereas class I molecules are expressed by most nucleated cells, expression of class II molecules is restricted to dendritic cells, B-lymphocytes, activated T-cells, macrophages, inflamed vascular endothelium and some epithelial cells. However, other cells (e.g. thyroid, pancreas, gut epithelium) can be induced to express class II molecules under the influence of interferon-gamma released during inflammation. In humans, there are three groups of class II antigen: the loci are known as HLA-DP, HLA-DQ and HLA-DR.
MHC class III antigens (Fig. 9.4) constitute early complement proteins C4 and C2. Other inflammatory proteins, e.g. tumour necrosis factor (TNF), are encoded in adjacent areas.
Accessory and co-stimulatory molecules
T-cell activation needs more than just binding between the T-cell receptor and the MHC class II molecule and processed antigen complex on the antigen-presenting cell.
Accessory and co-stimulatory molecules are needed for efficient binding and signalling (Fig. 9.6). Each accessory molecule has a corresponding protein, or ligand, to which it binds. The interaction between antigen-presenting cells and T-cells is strongly influenced by accessory molecules which function as co-stimulators; for example, CD80 and CD86 on the activated dendritic cell engage with their counter-receptors CD28 and CTLA-4 (CD152) on the T-cell surface (Fig. 9.6). A functional co-stimulatory pathway is essential for T-cell activation. In the absence of a co-stimulatory signal, interaction between the dendritic cell and T-cell leads to T-cell unresponsiveness (a state called anergy).
Fig. 9.6 Accessory and co-stimulatory molecules on T-lymphocytes and their ligands on antigen-presenting cells (APCs).
Adhesion molecules mediate cell-to-cell adhesion as well as adhesion between leukocytes and endothelial cells, and are grouped into two main families: integrins and selectins.
The migration of leukocytes to sites of inflammation depends on three key sequential steps mediated by adhesion molecules:
Integrins are subdivided into five families (beta-1 to beta-5 integrins) which mediate binding of lymphocytes and monocytes to the endothelial adhesion receptor called vascular cell adhesion molecule (VCAM-1). Defective expression of certain integrins is associated with a severe immunodeficiency characterised by marked neutrophil leukocytosis because neutrophils are unable to migrate from blood vessels into sites of infection.
The selectin family comprises three glycoproteins designated by the prefixes E- (endothelial), L- (leukocyte) or P- (platelet) to denote the cells on which they were first described. Selectins bind strongly to carbohydrate molecules on leukocytes and endothelial cells, and regulate the homing of these cells to sites of inflammation.
Cytokines
Cytokines are soluble mediators secreted by lymphocytes (lymphokines) or by macrophages/monocytes (monokines). They act as stimulatory or inhibitory signals between cells.
Cytokines that act between cells of the immune system are called interleukins; those that induce chemotaxis of leukocytes are called chemokines. All cytokines share common features:
Among the array of cytokines produced by macrophages and T-cells, interleukin-1 (IL-1) and interleukin-2 (IL-2) have a pivotal role in amplifying immune responses. IL-1 acts on a wide range of targets, including T- and B-cells (Table 9.2). In contrast, the effects of IL-2 are restricted largely to lymphocytes: it has a trophic effect on T-cells, IL-2 receptor-bearing B-cells and natural killer (NK) cells. The considerable overlap between individual cytokines and interleukins is summarised in Table 9.3.
Table 9.2 Actions of interleukin-1
| Target cell | Effect |
|---|---|
| T-lymphocytes |
B-lymphocytes
Neutrophils
Proliferation/activationOsteoclastsReabsorption of boneHepatocytesAcute-phase protein synthesisHypothalamusProstaglandin-induced feverMuscleProstaglandin-induced proteolysis
Table 9.3 Cytokines and their actions
| Cytokine | Source | Action |
|---|---|---|
| IL-2 | T-cells | Proliferation and maturation of T-cells, induction of IL-2 receptors and activation of NK cells |
|
Induction of MHC class II, FcR and IL-2R on B- and T-cells; induction of isotype switching in B-cells; facilitate IgE production (mainly IL-4) | |
| IL-6 | Macrophages, fibroblasts, T-cells, mast cells | Growth and differentiation of T-, B- and haemopoietic cells; acute phase response |
| IL-8 | Monocytes/macrophages, T-cells, fibroblasts, keratinocytes, endothelial cells, NK cells | Chemotaxis of neutrophils |
| IL-10 | T-cells, monocytes, keratinocytes | Inhibitory cytokine production |
| IL-12 | B-cells, monocytes/macrophages | Synergism with IL-2; regulates IFN-gamma production |
| IL-13 | T-cells | Actions overlap with IL-4 |
| GM-CSF | T-cells, macrophages, endothelial cells | Stimulates growth of polymorphs and mononuclear progenitors |
| IFN-gamma | T-cells, NK cells | Activation of macrophages, endothelial cells and NK cells; increases expression of MHC class I and II; inhibits IgE production |
| TNF | Macrophages, T-cells, B-cells | Promotion of inflammation; interferes with catabolism in muscle and fat |
STRUCTURAL ORGANISATION OF THE IMMUNE SYSTEM
T- and B-lymphocyte development
All lymphoid cells originate in the bone marrow although the nature of the uncommitted lymphoid stem cell remains unclear (Fig. 9.7). An understanding of the developmental pathway is important, not only to clarify the physiology of the normal immune response but also because some leukaemias and immunodeficiency states reflect maturation arrest of cells in their early stages of development. Lymphoid progenitors destined to become T-lymphocytes migrate from the bone marrow into the cortex of the thymus where further differentiation into mature T-cells occurs. Passage of T-cells from the thymic cortex to the medulla is associated with the acquisition of characteristic surface glycoprotein molecules so that medullary thymocytes resemble mature peripheral blood T-cells. T-cell development in the thymus is characterised by a process of positive selection whereby T-cells that recognise and bind with low affinity to fragments of self-antigen in association with self-MHC molecules proceed to full maturation. In contrast, T-cells that do not recognise self-MHC or that recognise and bind with high affinity to self-antigen are selected out—negative selection—and do not develop further. Negatively selected T-cells kill themselves by apoptosis, i.e. programmed cell death. This process is an important mechanism in preventing autoimmune disease. In summary, the thymus selects out the useful, neglects the useless and destroys the harmful, i.e. autoreactive T-cells.
In contrast, B-cell development occurs in the bone marrow and depends on the secretion of cytokines by stromal cells.
Primary and secondary lymphoid organs
The thymus and the bone marrow are primary lymphoid organs. They contain cells undergoing a process of maturation from stem cells to antigen-sensitive but antigen-restricted cells. This process of maturation is independent of antigenic stimulation. In contrast, secondary lymphoid organs are those that contain antigen-reactive cells in the process of recirculating through the body. They include the lymph nodes, spleen and mucosa-associated lymphoid tissues. Antigenic stimulation changes the relative proportions of mature cell types in secondary tissues.
Peripheral T- and B-cells circulate in a characteristic pattern through the secondary lymphoid organs. Most of the recirculating cells are T-cells and the complete cycle takes about 24 hours; some B-cells, including long-lived memory B-cells, also recirculate. Lymphocyte circulation is strongly influenced by chemokine receptors on lymphocyte surfaces which act as homing agents. Adhesion molecules direct cells to their respective ligands on high endothelial venules of lymph nodes.
Lymph node architecture is well adapted to its function (Fig. 9.8). Lymphatic vessels draining the tissues penetrate the lymph node capsule and drain into the marginal sinus from which a branching network of sinuses passes through the cortex to the medulla and into the efferent lymphatic. This network provides a filtration system for antigens entering the node from peripheral tissue.
Fig. 9.8 Structure of a normal lymph node. The locations of T- and B-lymphocytes are shown in Figs 9.10–9.12.
The cortex contains primary follicles of B-lymphocytes, surrounded by T-cells in the ‘paracortex’ (Figs 9.9–9.11). There is a meshwork of dendritic cells that express MHC class II antigen throughout the lymph node, and these cells filter and present antigen to lymphoid cells. On antigen challenge, the ‘primary’ follicles of the lymph node develop into ‘secondary’ follicles which contain germinal centres. These comprise mainly B-cells with a few helper T-cells and a mantle zone of the original primary follicle B-cells. B-cells in a secondary follicle are antigen-activated and more mature; most have IgG on their surfaces, whereas those in the primary follicle and mantle zone bear both IgD and IgM. Activated B-cells migrate from the follicle to the medulla, where they develop into plasma cells (Fig. 9.12) in the medullary cords before releasing antibody into the efferent lymph.
Fig. 9.10 A lymphoid follicle stained with a monoclonal antibody (anti-CD20) that reacts with B-cells. (Positive cells stain brown.)
Fig. 9.11 Immunohistochemical identification of T-cells. This section, adjacent to that in Fig. 9.10, is stained with a monoclonal antibody that reacts with T-cells (anti-CD3). Cells of the paracortex are stained (arrow) and there are few T-cells in the follicle.
The majority of naive T-cells entering the lymph node will leave again immediately via efferent lymphatics. Naive T-cells that recognise specific antigen differentiate into effector T-cells before re-entering the circulation.
FUNCTIONAL ORGANISATION OF THE IMMUNE RESPONSE
Processing and presentation of antigen to lymphocytes is performed by specialised antigen-presenting cells (APCs). The most efficient APCs are dendritic cells in lymph nodes Each B-lymphocyte is committed to the production of an antibody with a unique antigen-binding site—the idiotypeAntigen presentation
The first stage of an immune response to any antigen is the processing and presentation of that antigen to lymphocytes by specialised antigen-presenting cells (APCs). T-cells cannot recognise antigen without it. The interaction between APCs and T-cells is strongly influenced by a group of cell-surface molecules that act as co-stimulators. Thus, CD80 and CD86 on the APC engage with their counter-receptors CD28 and CTLA-4 (cytotoxic T-lymphocyte antigen 4; CD152) on the T-cell surface (Fig. 9.6). Normal functioning of theco-stimulatory pathway is vital for T-cell activation. In the absence of a co-stimulatory signal, interaction between the APC and T-cell leads to T-cell unresponsiveness, or anergy. Antagonists to co-stimulatory molecules disrupt immune responses, an observation of potential therapeutic importance; for instance, antagonists to CTLA-4 are being used experimentally to treat severe autoimmune diseases and to prevent graft rejection.
Processed antigen is presented to T-cells alongside MHC class II antigens on the APC surface because T-cells do not recognise processed antigen alone. The most efficient APCs are the interdigitating dendritic cells found in the T-cell regions of a lymph node. Such cells have high concentrations of MHC class I and II molecules, co-stimulatory molecules (CD80, CD86) and adhesion molecules on their surfaces but limited enzymatic powers, so enabling effective processing and presentation of antigen without complete digestion.
Antibody production
Antibody production involves at least three types of cell: antigen-presenting cells, B-lymphocytes and helper T-cells.
Antibodies are synthesised by B-cells and their mature progeny, called plasma cells (Fig. 9.12). B-cells are readily recognised because they express immunoglobulin on their surfaces. During development, B-cells first show intra-cellular mu chains and then surface IgM. These cells are able to switch from production of IgM to IgG, IgA or IgE as they mature, a process known as isotype switching (Fig. 9.13). This maturation sequence fits with the kinetics of an antibody response: the primary response is mainly IgM and the secondary response predominantly IgG (Fig. 9.14).
Fig. 9.13 Interaction between CD40L on T-cells and CD40 on B-cells leads to isotype switching under the influence of IL-4. (TcR, T-cell receptor.)
Isotype switching is mediated by the interaction of two important proteins: CD40 expressed on the B-cell surface engages with its ligand, CD40L (CD154), on activated T-cells (under the influence of IL-4) to induce B-cells to switch immunoglobulin production from IgM to IgG, IgA or IgE (Fig. 9.13). Deficiency of either CD40 or CD40L in humans leads to a severe immunodeficiency characterised by inability to switch from IgM to IgG antibody production.
Each B-cell is committed to the production of an antibody that has a unique Vh–Vl combination, the idiotype, and the surface immunoglobulin and secreted immunoglobulin are identical. Contact with antigen and factors released by helper T-cells (IL-4, -5, -6) stimulate the B-cell to divide and differentiate, generating more antibody-producing cells, all of which make the same antibody with the same idiotype. Simultaneously, a population of memory cells is produced which express the same surface immunoglobulin receptor. The result of these cell divisions is that a greater number of antigen-specific B-cells becomes available when the animal is exposed to the same antigen at a later date. This process, known as clonal expansion, helps to account for the amplified secondary response. As well as being quicker and more vigorous (Fig. 9.14), secondary responses are more efficient because the antibodies bind more effectively to the antigen, i.e. with higher affinity.
A minority of B-cells will respond directly to antigens called T-independent antigens, which have repeating, identical, antigenic determinants and provoke predominantly IgM antibody responses. B-cells, however, will not usually respond directly to antigen, even when presented by appropriate accessory cells. A second signal is needed to trigger the B-cell; this signal is normally provided by CD4+ helper T-cells.
T-cell help is antigen-specific. Only helper T-cells that have responded to antigen presented by macrophages can subsequently help B-cells already committed to that antigen. Helper T-cells recognise both antigen and MHC class II antigens as a complex on the presenting cells. They then recognise the same combination of antigen and class II molecule on the corresponding B-cell but co-stimulation is also required (Fig. 9.13). When helper T-cells meet an antigen for the first time, the limited number reacting with that antigen are activated to provide help for B-cells. They undergo blast transformation and proliferation, i.e. clonal expansion, so the immune response on second and subsequent exposures is quicker and more vigorous.
Other mechanisms help to improve this efficiency. Memory cells (which bear the surface marker CD45RO) have increased numbers of adhesion molecules (LFA-1, CD2, LFA-3, ICAM-1) plus a higher proportion of high-affinity receptors for the relevant antigen. Memory cells are therefore easily activated and produce high concentrations of IL-2 to recruit more helper T-cells. Thus T-cell memory is a combination of a quantitative increase of T-cells and a qualitative change in the efficiency of those T-cells.
Helper T-cells are further grouped into two distinct subsets depending on their cytokine profile. Th1 cells secrete TNF and interferon-gamma and mediate cellular immunity. In contrast, Th2 cells predominantly secrete IL-4, IL-5, IL-10 and IL-13 (Fig. 9.15) and are responsible for stimulating vigorous antibody production by B-cells. T-cells expressing cytokine profiles common to both Th1 and Th2 cells are designated Th0.
Fig. 9.15 Th1 and Th2 cells secrete different cytokines. Some cytokines provide inhibitory feedback (- – – -) on subsets of Th cells.
A Th1 cytokine profile provides protection against intracellular pathogens, while a Th2 profile is found in those diseases associated with overproduction of antibodies, especially IgE.
The effects of helper T-cells are balanced by those of functional suppressor T-cells that express the characteristic surface glycoprotein CD8.
Cell-mediated responses
Antigen-specific cell-mediated responses are carried out by T-lymphocytes. T-cells can lyse cells expressing specific antigens (cytotoxicity), release cytokines that trigger inflammation (delayed hypersensitivity), or regulate immune responses. These T-cell responses are mediated by distinct T-cell subpopulations: cytotoxicity is the role of cytotoxic T-cells and delayed hypersensitivity that of Th1 cells. These cells are responsible for fighting intracellular pathogens (all viruses, parasites and certain bacteria) which are inaccessible to antibodies.
Cytotoxic T-cells kill cells infected with virus (and possibly those tumour cells expressing recognisable tumour antigens). Such cytotoxicity is virus specific—only cells expressing the relevant viral proteins on their surfaces are killed. Since infected cells express surface viral proteins prior to the assembly of new virus particles and viral budding, cytotoxic T-cells are important in the recovery phase of an infection, destroying the infected cells before new virus particles are generated.
Cytotoxic T-cells recognise viral antigens together with MHC class I molecules. They show exquisite specificity for self-MHC antigens, in that they can only lyse cells expressing the same MHC class I molecules, i.e. express MHC restriction.
Regulatory T-cells are a subset of CD4+ T-cells with a distinct phenotype (CD4+, CD25+) under the control of a gene called FoxP3. These cells dampen down activation and expansion of self-reactive T-cells. Mutations in FoxP3 result in severe autoimmune disease and allergy.
NON-SPECIFIC EFFECTOR MECHANISMS
Complement can be activated by antibody (the classical pathway), by bacterial cell walls (the alternative pathway) or by mannose-binding lectin (the lectin pathway)Complement
Complement is a complex series of interacting plasma proteins which form a major effector system for antibody-mediated immune reactions. Many complement components exist as inactive precursors; once activated, the component may behave as an enzyme which cleaves several molecules of the next component in the sequence. Each precursor is cleaved into two or more fragments. The major fragment (usually designated ‘b’) has two biologically active sites: one for binding to cell membranes or the triggering complex, and the other for enzymatic cleavage of the next complement component. Minor cleavage fragments (designated ‘a’) have important biological properties in the fluid phase. Control of complement activation involves spontaneous decay of any exposed attachment sites and inactivation by specific inhibitors. The major purpose of the complement pathway is to remove or destroy antigen, either by direct lysis or by opsonisation.
Complement activation
Complement activation occurs in two sequential phases:
The critical step (Fig. 9.16) is cleavage of C3 by complement-derived enzymes called C3 convertases. The major fragment of activated C3—called C3b—mediates a number of vital biological activities, particularly opsonisation.
The cleavage of C3 is achieved via three main routes, the classical, alternative and lectin pathways, all of which generate C3 convertases but in response to different stimuli (Fig. 9.16).
Classical pathway activation
The classical pathway is activated when binding of IgM or IgG to antigen causes a conformational change in the Fc region of the antibody to reveal a binding site for the first component in the classical pathway, C1.
C1 is a macromolecular complex of three subcomponents—C1q, C1r and C1s. C1q is a collagen-like protein composed of six subunits. C1q reacts with Fc regions via its globular heads but attachment by two critically spaced binding sites is needed for activation. IgM is more efficient than IgG in activating C1q. IgA, IgD and IgE do not activate the classical pathway.
Once C1q is activated, C1r and C1s are sequentially bound to generate enzyme activity (C1 esterase) for C4 and C2, splitting both molecules into a and b fragments. The complex C4b2b is the classical pathway C3 convertase. C4b2b cleaves C3 into two fragments, one (C3a) possessing anaphylatoxic and chemotactic activity (see below) and one that binds to the initiating complex and promotes many of the biological properties of complement. The C4b2b3b complex so generated is an enzyme, C5 convertase, which initiates the final lytic pathway (the ‘attack’ sequence).
Alternative pathway activation
[/not-level-membership-for-pathology-category]
