[level-membership-for-pathology-category]
Chapter 25 Osteoarticular and connective tissues
Common clinical problems from osteoarticular and connective tissue disease
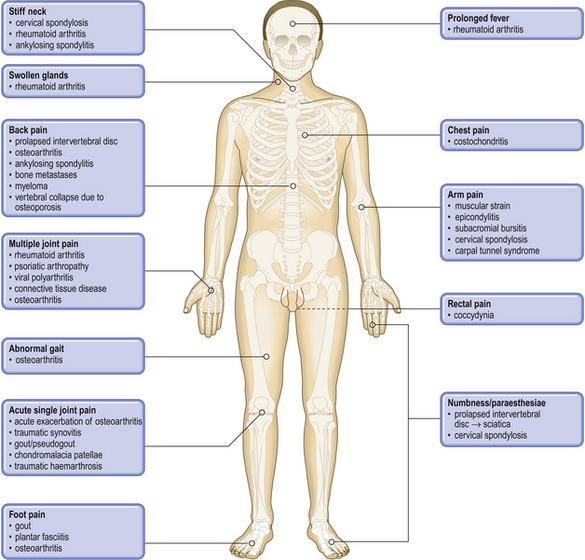
Pathological basis of clinical signs and symptoms of bone, joint and connective tissue diseases
| Sign or symptom | Pathological basis |
|---|---|
| Bone disease | |
| Pain | Stimulation of nerve endings in bone by: |
Fracture after trivial injuryBone weakening due to:
DeformityAbnormal bone growth/remodelling due to:
Hypercalcaemia
Joint disease PainStimulation of nerve endings in joint capsule and synovium by inflammation (arthritis) or abnormal load bearing/joint movementDeformityJoint swelling due to:
Restricted movement
Systemic features (e.g.subcutaneous nodules,lymphadenopathy)Arthritis mediated by immune mechanismsConnective tissue diseases Swelling
Joint painSynovial oedema and inflammation with stimulation of nerves in joint capsuleIschaemic lesionsVasculitisRestricted mobility of tissuesFibrosis or increased tissue tension due to inflammation
BONE
NORMAL STRUCTURE AND FUNCTION
Functions of bone
Bone has structural, protective and metabolic functions. The skeleton is divided into the axial (head, vertebral column, thoracic cage, shoulder and pelvic girdles) and the appendicular (limbs). The axial skeleton participates extensively in all three areas of function, whereas the appendicular skeleton has a primarily structural function. The structural functions of bone are to provide support and also insertion sites for muscles and ligaments. The skull and thoracic cage provide physical protection for the brain, thoracic and upper abdominal organs.
Bone has two major metabolic functions:
Structure of bone
Bone is characterised by its hard matrix. This matrix consists of two components, matrix proteins and mineral. The main structural protein in bone matrix is type I collagen. This protein provides the framework of the overall structure of bone. Within the collagen framework there is a mixture of many other proteins, some of which are thought to aid mineralisation; others mediate cell attachment. Another major group of proteins present in bone matrix are growth factors, such as the bone morphogenetic proteins and transforming growth factor beta (TGF-beta). These appear to be important in mediating the cellular events of bone remodelling. Proteoglycans are also present in bone matrix, but do not have the same major structural role in bone as they do in cartilage. The mineral component of bone matrix provides its structural resilience. Most of the mineral deposited in bone is in the form of a calcium phosphate complex known as hydroxyapatite. The precise mechanism by which bone mineral forms is unclear. The enzyme alkaline phosphatase, a major product of osteoblasts, and vitamin D metabolites are thought to be important in this process.
Despite its lifeless appearance, bone is a highly complex and dynamic cellular tissue. Bone contains two distinct types of cell, osteoclasts and the osteoblast family. Osteoclasts, the bone-resorbing cells, are mono- or multinucleated cells that are specialised members of the monocyte–macrophage lineage. These short-lived cells are recruited to the bone surface at sites of remodelling and destroy bone matrix by secreting hydrogen ions and proteolytic enzymes into a sealed space beneath the cell. The osteoblast family consists of: osteoblasts, which are bone forming cells; osteocytes, which form an interconnecting network throughout bone matrix; and lining cells, which cover metabolically inactive bone surfaces. Osteocytes are thought to be mechanosensory cells. The cells of the osteoblast family are unrelated to osteoclasts, being related more closely to fibroblasts, chondrocytes and adipocytes.
Osteoclasts and osteoblasts act together to control bone growth and metabolism through the bone remodelling cycle. This cycle, illustrated in Figure 25.1, forms the basis of bone metabolism.
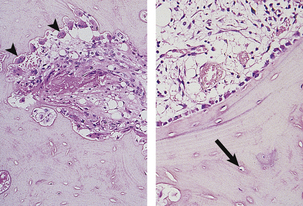
Fig. 25.1 The bone remodelling cycle. The bone remodelling cycle consists of resorption by osteoclasts (arrowheads in left-hand plate) which results in the removal of an area of bone matrix and is inevitably followed at the same site by resynthesis of bone matrix by osteoblasts (right-hand plate). Many osteoblasts become incorporated into the matrix they synthesise to become osteocytes (arrowed). Osteocytes are connected by a complex network of cytoplasmic processes not visible on these haematoxylin and eosin-stained sections.
Each remodelling cycle takes several weeks to complete. Approximately one million remodelling cycles are occurring within the adult human skeleton at any one time. These cycles are asynchronous, some being in resorption, some in formation. This continual process of remodelling renews the adult human skeleton approximately every 7 years. The functions of the remodelling cycle are to:
In the normal bone remodelling cycle, resorption and formation are coupled, with the same quantity of bone being formed as had been resorbed, except where extra bone matrix is called for by the requirements of growth. In pathological situations, the two processes can become uncoupled; for example, osteoporosis can result from uncoupled increases in bone resorption or decreases in bone formation resulting in a net loss of bone. All diseases of bone are associated with changes in bone remodelling of some type.
There are two types of mature bone, cortical and trabecular (sometimes known as cancellous bone). Cortical bone has a predominantly structural load-bearing function. It is the dense bone that forms the diaphyses (shafts) of long bones such as the femur and the outer surfaces of predominantly trabecular bones such as the vertebral bodies. Trabecular bone has some structural function, but contributes to the metabolic functions of bone far more than cortical bone. Because it is metabolically more active, it is far more prone to diseases involving or resulting from increased bone remodelling than cortical bone. For example, post-menopausal osteoporosis affects trabecular bone before it affects cortical bone, and deposits of metastatic carcinoma are far more common in sites occupied by trabecular bone.
Development and growth of bones
Most tissues and organs grow as a result of a general increase in the number of their constituent cells. Because the proteinaceous matrix is so heavily calcified, a similar process is not possible in bone. Thus the process of remodelling described above is essential for bone growth.
During development, bone is formed either directly in connective tissue, as in the skull (intramembranous ossification), or on pre-existing cartilage, as in the limb bones (endochondral ossification).
During intramembranous ossification the first bone that is laid down has a loose and rather haphazard arrangement. This ‘woven bone’ gradually matures into more organised and compact ‘lamellar’ bone.
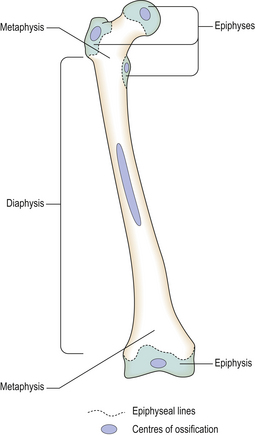
Endochondral ossification is a much more complicated process during which a cartilagenous template is converted into a bony structure with capacity for further growth. In each bone, ossification occurs at particular sites or centres of ossification situated in the shaft (diaphyseal centres) or towards the ends of the bone (epiphyseal centres) (Fig. 25.2). Ossification proceeds at different, but predictable, rates in each particular bone. In long bones, a plate of epiphyseal cartilage persists into adolescent or early adult life; this allows a continual increase in bone length. Skeletal growth through the growth plates is controlled by growth hormone and sex steroids with parathyroid hormone-related peptide (PTHrP) and insulin-like growth factor-1 (IGF-1) acting as paracrine (locally produced) growth factors. The overall shape and size of bone changes during growth and, to some extent, in adult life. This involves both osteoclastic bone resorption and enlargement of pre-existing, or the formation of new, bony trabeculae. Cortical bone grows in an analogous way through remodelling occurring at the periosteal and endosteal surfaces and within Haversian canals.
FRACTURES AND THEIR HEALING
 Types of fracture: simple (clean break); comminuted (multiple bone fragments); compound (breaking through overlying skin); complicated (involving adjacent structures—blood vessels, nerves, etc.); stress fractures (small linear fractures)
Types of fracture: simple (clean break); comminuted (multiple bone fragments); compound (breaking through overlying skin); complicated (involving adjacent structures—blood vessels, nerves, etc.); stress fractures (small linear fractures)


Causes of fractures
Fractures in normal bone are the result of substantial trauma, such as direct violence or a sudden unexpected fall. The precise site of fracture, the nature and direction of the fracture line, and the speed of the subsequent repair process depend very much on the age of the patient, the particular bone involved and the precise pattern of injury (Fig. 25.3).
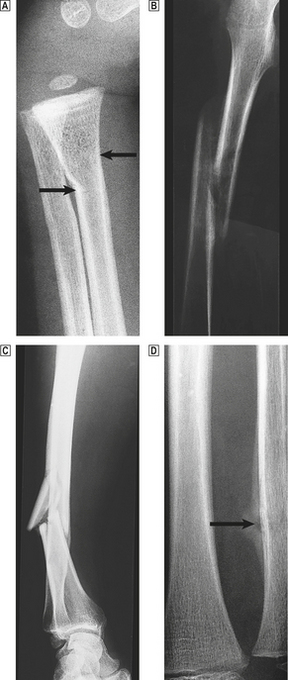
Fig. 25.3 Fracture types and fracture healing.  A greenstick fracture of the distal radius in a young child (arrowed).
A greenstick fracture of the distal radius in a young child (arrowed).  A displaced spiral fracture of the femur in a child.
A displaced spiral fracture of the femur in a child.  A comminuted fracture of the tibia. One fragment of bone has almost separated from the shaft.
A comminuted fracture of the tibia. One fragment of bone has almost separated from the shaft.  A healing fracture of the ulna. The site of the break is just visible and is surrounded by callus (arrowed).
A healing fracture of the ulna. The site of the break is just visible and is surrounded by callus (arrowed).
Repeated episodes of minor trauma, for example after marching, marathon running or training for sport, can produce small but often painful stress fractures. These usually occur in the long bones of the lower limbs but have also been described in the metatarsals, the upper limb, pelvis and spine. They usually heal satisfactorily after a short period of rest. Even professional athletes can develop these fractures.
Fractures occur more easily in bone that is structurally abnormal. They may occur after a trivial injury or minor fall or even spontaneously during normal activity. This is particularly common in patients with osteoporosis but also occurs in most forms of metabolic bone disease (e.g. in osteomalacia and rickets), in Paget’s disease and in bone infiltrated by malignant tumours. Fractures of this type are called pathological fractures.
Fracture healing
The first stage in fracture healing is the formation of a bony bridge between the separated fragments. When this is formed, and some rigidity has been regained, remodelling and restructuring gradually restore the normal contours of the fractured bone. This process and the factors that can interfere with it are described in Chapter 6.
The major causes of delayed fracture healing are:
All of these are well recognised by orthopaedic surgeons, who modify the treatment in individual cases in line with the particular pattern of injury, and the age and general health of the patient. For example, a fracture through the neck of the femur usually deprives the head of its normal blood supply and satisfactory fracture healing is unlikely to occur. Surgical treatment, such as excision of the head and replacement by a metallic prosthesis, is therefore essential. Fractures in which the overlying skin surface is broken (compound fractures) are liable to infection, whereas this is extremely uncommon in closed fractures. Healing will be substantially delayed if a wound infection develops.
In many fractures, healing can be accelerated by prompt and appropriate surgical treatment using internal fixation by nails, plates and screws or external fixator devices to hold the fractured fragments in an appropriate position; this often allows early mobilisation. Primary callus does form but is reduced in amount. Small gaps are filled by new woven bone. Dead bone is gradually revascularised and new Haversian bone grows in.
Surgical treatment is sometimes necessary for fractures in which the healing process has been delayed. The object is to ‘restart’ the primary callus response. This can sometimes be achieved by lifting flaps of periosteum close to the fracture site. In addition, local ‘grafting’ with the patient’s own bone or devitalised bone from another donor can promote bone healing, presumably through the action of bone morphogenetic proteins and other growth factors present in the graft matrix.
OSTEOPOROSIS AND METABOLIC BONE DISEASE
Normal calcium metabolism
The two major hormones that regulate calcium metabolism are vitamin D and parathyroid hormone (PTH). Vitamin D is not a vitamin in the strict sense of an essential dietary requirement, as it can also be synthesised photochemically in the skin. In reality, vitamin D is more like a steroid hormone precursor that can be derived from the diet. Its active metabolites function in a similar way to conventional hormones. Vitamin D must be metabolised by the liver to 25-hydroxyvitamin D3, and subsequently by the kidney to the active metabolite 1,25-dihydroxyvitamin D3. Receptors for vitamin D are present in a variety of cell types in the body; the physiological role of this vitamin may be much wider than is currently known. Expression of the receptors is subject to genetic variation within the population and may contribute to the individual differences in risk of developing metabolic bone disease. The combined effects of vitamin D and parathyroid hormone are:
These functions are complex and are demonstrated in Figure 25.4. This area of metabolism is still incompletely understood. There is evidence to suggest that there may be another pathway regulating phosphate transport involving fibroblast growth factor 23. PTH and 1,25-dihydroxyvitamin D3 also have important direct effects on bone: PTH stimulates both bone resorption and formation; 1,25-dihydroxyvitamin D3 promotes bone matrix mineralisation.
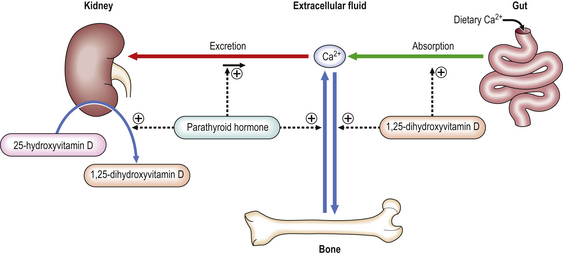
Fig. 25.4 Regulation of calcium metabolism. Calcium levels are maintained in the extracellular fluid within a narrow range of concentrations by absorption from the gut and excretion via the kidney with bone acting as a buffering reservoir. PTH raises calcium levels by increasing renal tubular calcium reabsorption, increasing release of calcium from bone matrix through osteoclastic resorption and indirectly increasing calcium absorption from the gut by increasing the metabolism of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D (the active form) in the kidney.
In contrast to PTH, calcitonin, a peptide hormone, appears to lower serum calcium, but usually only when it is pathologically elevated. The stimulus to its secretion is an increase in the serum calcium concentration; it is produced in specialised parafollicular cells (C-cells) of the thyroid. Its exact physiological action is uncertain but it has an inhibitory effect on osteoclasts.
Although vitamin D, PTH and, possibly, calcitonin are the most important factors controlling calcium and phosphate concentrations, and therefore normal bone integrity, several other factors are also involved. Glucocorticoids have a role in the regulation of skeletal growth but prolonged corticosteroid therapy often induces osteoporosis. Thyroid hormone deficiency, as in cretinism, is associated with several skeletal abnormalities. Sex steroids accelerate the closure of epiphyses, and growth hormone has an effect on the development and maturation of cartilage.
Osteoporosis






Osteoporosis is a disease in which there is a reduction in bone mass in the presence of normal mineralisation. It is diagnosed by radiological assessment of bone mineral density (generally measured by dual photon absorptiometry). The usual definition of osteoporosis is a bone mineral density measurement two standard deviations below the mean value for young adults of the same sex. Clinically, osteoporosis may present as a fragility fracture, loss of height, or stooping deformity (kyphosis or ‘dowager’s hump’) due to wedge fractures of the vertebral bodies. Sometimes osteoporosis is diagnosed, when clinically silent, by screening individuals thought to be at risk.
Clinically significant osteoporosis most often results from a combination of age-related bone loss and additional bone loss from another cause; by far the most common such cause is post-menopausal oestrogen withdrawal. Osteoporosis is a clinically silent disease until it is complicated by deformity or fractures.
Pathogenesis
Osteoporosis is caused by a loss of coupling in the bone remodelling process. This can be due to increased bone resorption, decreased bone formation, or both. The loss of coupling results in a net loss of bone volume. In contrast to osteomalacia (see below), mineralisation of bone is normal. Because of its greater metabolic activity, trabecular bone is usually affected more severely than cortical bone. This is particularly the case when increased bone resorption is the main pathogenetic mechanism.
The total bone mass of an individual is influenced by factors such as body build, race, gender, physical activity and general nutrition. Osteoporosis is more common in females than in males and is less common in blacks, who have a greater skeletal mass than whites or Asians. Osteoporosis can be assessed radiologically or by techniques based on the ability of bone to absorb photons released by a gamma-emitting isotope. These demonstrate a progressive loss of bone of 0.75–1% per annum in normal adults of both sexes from as early as 30 years of age. More importantly, there is an accelerated phase of bone loss of up to 1–3% per year in females in the 5–10 years following the menopause.
Localised osteoporosis is inevitable after immobilisation of any part of the skeleton. Even young, healthy males confined to bed after a limb fracture show substantial bone loss. Painful joints in patients with rheumatoid arthritis restrict movement, and osteoporosis often develops in adjacent bones, although this may also be the result of increased bone resorption due to inflammatory mediators produced in affected joints.
Complications
The major complications of osteoporosis are:
The commonest clinical feature of osteoporosis is the progressive loss of height that occurs with age. This is a direct result of compression of vertebrae. Sudden collapse or unequal compression of individual vertebral bodies can cause severe localised back pain and deformities such as kyphosis or scoliosis (Fig. 25.5).
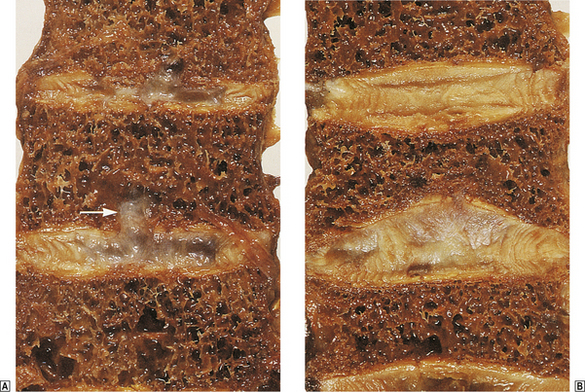
Fig. 25.5 Vertebral osteoporosis.  The lower thoracic vertebrae showing small protrusions of the intervertebral disc into the osteoporotic bone (arrowed).
The lower thoracic vertebrae showing small protrusions of the intervertebral disc into the osteoporotic bone (arrowed).  The lumbar spine. The vertebral body in the centre has collapsed and has a typical biconcave shape.
The lumbar spine. The vertebral body in the centre has collapsed and has a typical biconcave shape.
Wrist and hip fractures are common in elderly patients with osteoporosis. Although osteoporosis is the major underlying cause, other factors such as an increased tendency to fall and a loss of ‘protective neuromuscular reflexes’ (the ability to fall over safely) are also important. Hip fractures account for numerous hospital admissions and are a major source of disability and a frequent cause of death in the elderly.
Prevention and treatment
Osteoporosis is a major social and economic problem in the elderly, and preventive measures should begin in the middle-aged. Vertebral osteoporosis is reduced in women treated with oestrogens (hormone replacement therapy—HRT). Bisphosphonate drugs, which inhibit bone resorption, are more effective in preventing osteoporotic hip fractures. Regular exercise and an increased dietary intake of calcium also have beneficial effects.
Rickets and osteomalacia





Osteomalacia is characterised by deficient mineralisation of the organic matrix of the skeleton. Rickets is the name given to osteomalacia affecting the growing skeleton of children; it results in characteristic deformities. Causes of osteomalacia, or rickets, include:
Aetiology
In the past, nutritional deficiency of vitamin D was a common cause of rickets in children and, occasionally, of osteomalacia in adults. In most communities, this has been eliminated by improvements in diet and by the addition of vitamin D to foodstuffs. The disease still occurs in some Asian communities in the UK; skin pigmentation impairs photochemical synthesis of vitamin D, and a constituent of chapatti flour interferes with calcium and phosphate absorption in the gut. As dietary rickets is becoming less common in Western countries, an increasing proportion of cases of rickets are due to congenital abnormalities in vitamin D metabolism. Disorders of this type are referred to as ‘vitamin D-resistant rickets’ because vitamin D supplements fail to generate active vitamin D metabolites.
Malabsorption of calcium and phosphate from the intestine is the commonest cause of osteomalacia in adults. The underlying cause is often coeliac disease, but occasional cases result from Crohn’s disease or extensive surgical resection of the small intestine. As the liver and kidney have important roles in the metabolism of vitamin D, renal and hepatic disorders may cause osteomalacia. This is uncommon in liver disease, but a complex pattern of bone disease that includes osteomalacia is seen in renal failure. Occasional patients treated with anticonvulsants, such as phenytoin, develop osteomalacia. These drugs induce liver enzymes that degrade vitamin D to inactive metabolites.
Diagnosis
The characteristic clinical deformities of rickets include:
Inadequate mineralisation of bone reduces its normal strength and allows deformities to develop, for example from pressure on the skull while lying in a cot, or on the limbs as they begin to bear weight. Calcification of epiphyseal cartilage is an essential step in the normal process of ossification in long bone. When the levels of vitamin D metabolites are low, calcification cannot occur and cartilaginous proliferation continues. This accounts for the enlargement of long bones and the ribs at growth plates.
The characteristic pathological feature in adults with osteomalacia is spontaneous incomplete fractures (‘Looser’s zones’), often in the long bones or pelvis. The main symptoms are bone pain and tenderness, and weakness of proximal limb muscles. Serum calcium levels may be reduced and serum alkaline phosphatase is increased (these biochemical abnormalities are usually absent in osteoporosis). A bone biopsy will demonstrate an increase in non-mineralised osteoid (Fig. 25.6).
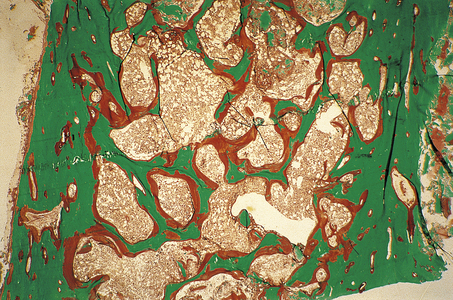
Fig. 25.6 Osteomalacia. This transiliac crest undecalcified bone biopsy, stained by the Goldner’s trichrome technique, was taken from a patient on renal dialysis suffering from osteomalacia due to a combination of low 1,25-dihydroxyvitamin D levels (a common consequence of renal failure) and aluminium toxicity (an iatrogenic complication of dialysis). Mineralised bone matrix is stained green and unmineralised matrix (osteoid) is stained red. The amount of osteoid present is approximately 20 times greater than normal.
Treatment and prevention
Uncomplicated rickets or osteomalacia will respond promptly to vitamin D treatment. Increased calcium intake may also be required to compensate for the flux of calcium into unmineralised bone matrix that occurs in response to vitamin D treatment. Intramuscular injection can overcome problems associated with malabsorption, and underlying disorders such as coeliac disease should be treated appropriately. A normal balanced diet will prevent rickets or osteomalacia, but many foodstuffs are now artificially supplemented with vitamin D.
Hyperparathyroidism and hypercalcaemia



Persistent elevation of fasting blood calcium, after correction has been made for the serum albumin concentration, is an important indication for further investigation. The major pathological causes are:
By far the commonest causes of hypercalcaemia are primary hyperparathyroidism and hypercalcaemia of malignancy.
In hyperparathyroidism (Ch. 17), increased secretion of parathyroid hormone stimulates calcium absorption in the intestine, reabsorption in the kidney and osteoclastic breakdown of bone.
In primary hyperparathyroidism the usual cause is a parathyroid adenoma or, occasionally, diffuse hyperplasia of the parathyroid glands. In contrast, in secondary hyperparathyroidism, prolonged hypocalcaemia stimulates parathyroid hyperplasia and eventually produces parathyroid enlargement. This is usually the result of renal failure or malabsorption secondary to coeliac disease. In occasional patients, secondary hyperparathyroidism is associated with hypercalcaemia. This has been called tertiary hyperparathyroidism and usually results from inappropriately high secretion of PTH by an adenoma arising in secondary hyperparathyroidism.
When obvious causes, such as malignant disease, sarcoidosis or drug therapy, have been excluded, it must be suspected that an otherwise fit patient with hypercalcaemia has primary hyperparathyroidism (Table 25.1).
Table 25.1 causes of hypercalcaemia
| Cause | Pathophysiology |
|---|---|
| Primary hyperparathyroidism | Abnormal PTH secretion from adenoma, hyperplasia or carcinoma of parathyroid glands |
| Malignant disease |
SarcoidosisProbable secretion of vitamin D metabolites from granulomas
Miscellaneous causes:
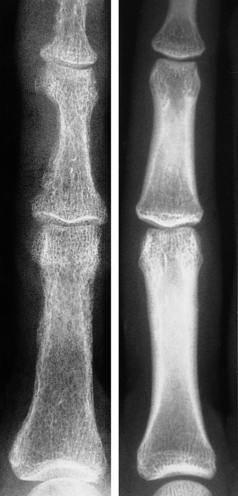
The advanced bone pathology associated with hyperparathyroidism is now rare. In the early stages there are subtle radiological changes such as subperiosteal resorption of phalangeal bone (Fig. 25.7) or characteristic changes around the teeth. As the disease progresses, cystic bone lesions may develop—osteitis fibrosa cystica (von Recklinghausen’s disease of bone). These are sometimes referred to as ‘brown tumours’, although they are not neoplasms. The brown appearance is the result of haemorrhage and there is often a marked associated osteoclastic reaction. Because PTH has anabolic as well as catabolic effects in bone, hyperparathyroidism does not usually cause generalised osteoporosis.
Bone disease in renal failure (renal osteodystrophy)
Most patients with chronic renal failure have clinical, radiological or pathological evidence of bone disease. There is no single bone disease that occurs in renal failure; in most patients it is a combination of osteomalacia with a variable degree of hyperparathyroidism. Other features include:
The most important pathophysiological changes in renal bone disease are summarised in Table 25.2. Several mechanisms have been suggested to account for the osteomalacia. In all forms of renal failure there is a decrease in the amount of functional renal tissue, and this may be directly responsible for the inadequate production of active vitamin D metabolites. An increased blood phosphate level (hyperphosphataemia) is frequent in renal failure, and this may directly inhibit enzymes responsible for vitamin D metabolism in the kidneys. In the past, haemodialysis fluids rich in aluminium were associated with aluminium deposition in organs such as brain and bone. In bone, aluminium inhibits the calcification of osteoid and contributes to osteomalacia in renal failure (Ch. 7).
Table 25.2 Mechanisms of renal bone disease (renal osteodystrophy)
| Feature of renal failure | Pathological effect in bone |
|---|---|
| Inadequate renal tissue | Impaired conversion of 25(OH)D3 to 1,25(OH)2D3 → Osteomalacia |
| High serum phosphate |
Prolonged haemodialysisInhibition of calcification of osteoid → OsteomalaciaSteroid therapy (e.g. for chronic glomerulonephritis)Osteoporosis Avascular necrosis of bone
Patients with chronic renal failure may have a low serum calcium. This is partly the result of impaired vitamin D metabolism, as vitamin D metabolites are essential for the proper absorption of calcium from the small intestine. The high serum phosphate also reduces the ionised fraction of plasma calcium. This acts as a stimulus to PTH production, and a degree of hyperparathyroidism is inevitable in severe renal failure. Patients with some forms of glomerulonephritis are treated with steroids and this may induce osteoporosis or, occasionally, areas of bone necrosis. Calcification of the soft tissues, or of blood vessel walls, is a further feature of chronic renal failure, particularly after prolonged haemodialysis. Longstanding disordered bone remodelling due to the combination of secondary hyperparathyroidism and osteomalacia can lead to alternating areas of thickened bone (osteosclerosis) and osteoporosis. This has a characteristic appearance (‘rugger jersey spine’).
OSTEOMYELITIS






Aetiology
Osteomyelitis is the result of a bacterial infection of bone. The typical patient is a young child who presents with pain in a long bone, sometimes with a misleading history of recent trauma. In the majority of cases the lesion develops in the metaphysis, the part of the shaft immediately adjacent to the epiphyseal plate. The rich capillary network and large venous channels in this area may favour the deposition of circulating micro-organisms and their subsequent growth. In children and adolescents, osteomyelitis is usually the result of Staphylococcus aureus bacteraemia, often secondary to a boil or other skin infections. Sometimes, the underlying cause of bacteraemia is not apparent. Osteomyelitis is also increasingly seen in elderly patients.
Before the introduction of antibiotics, tuberculous and even syphilitic osteomyelitis were common. Children with haemoglobinopathies, especially sickle cell disease, have an increased risk of osteomyelitis; unusual organisms, such as Salmonella, are sometimes responsible.
Osteomyelitis is a well-recognised complication of compound fractures, particularly if the wound in the overlying skin is extensive and there are necrotic bone splinters at the fracture ends. Osteomyelitis is not a complication of closed fractures.
Pathogenesis
The classical sequence of changes in osteomyelitis is as follows:
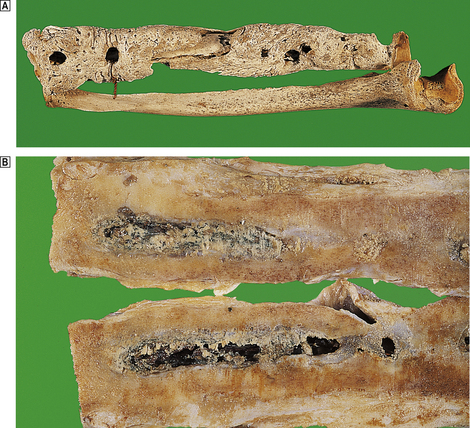
Fig. 25.8 Chronic osteomyelitis.  The radius and ulna of an 18th-century sailor. The ‘granular bone’ is the involucrum and the circular defects are ‘cloacae’ through which pus drained.
The radius and ulna of an 18th-century sailor. The ‘granular bone’ is the involucrum and the circular defects are ‘cloacae’ through which pus drained.  The cut surface of the femur of a 78-year-old male who received a shrapnel wound to his thigh in World War I. The pus drained through a sinus for the next 50 years! A thick bony involucrum surrounds a chronic inflammatory abscess in the marrow cavity.
The cut surface of the femur of a 78-year-old male who received a shrapnel wound to his thigh in World War I. The pus drained through a sinus for the next 50 years! A thick bony involucrum surrounds a chronic inflammatory abscess in the marrow cavity.
The development of a sequestrum is due to necrosis of bone caused by compression of blood vessels by the inflammatory process within the Haversian canals of the cortical bone. This event rarely occurs if antibiotic treatment is initiated early in the course of the disease. However, infections in bone can be difficult to eradicate, particularly if foreign material is present, for example following a penetrating injury. Commonly encountered ‘foreign materials’ in bone are joint prostheses, internal fracture fixation devices and other pieces of orthopaedic hardware. Bone infections associated with orthopaedic surgery are more common than primary osteomyelitis in many countries.
Brodie’s abscess is a distinctive clinical form of subacute pyogenic osteomyelitis. The lesion is solitary and, as in typical acute osteomyelitis, localised to the metaphysis.
Clinical features, laboratory investigations and treatment
Most patients with acute osteomyelitis present with localised bone pain and some tissue swelling. A dull continuous back pain, which increases on straining, is typical of vertebral osteomyelitis. The radiological changes are usually characteristic. Blood cultures are positive in some patients, but open biopsy of the lesion may be needed to ensure accurate bacteriological diagnosis. The commonest organisms are Staphylococcus aureus, Mycobacterium tuberculosis, Escherichia coli, pneumococcus or group A streptococcus. Wherever possible a precise bacteriological diagnosis must be made and treatment continued for several weeks.
PAGET’s DISEASE




Incidence and epidemiology
Paget’s disease is a disorder in which there is disorderly bone remodelling. There is considerable variation in its incidence both within and between different countries and racial groups. Despite intensive study, little is known of the cause of Paget’s disease and it is not regularly associated with any other common disorder. Electron microscopic studies have demonstrated probable viral inclusions in the nuclei of osteoclasts, possibly derived from measles or canine distemper virus, but no definite proof has been obtained. Paget’s disease has a distinctive epidemiology. It is most common in western Europe and those parts of the world to which western Europeans have emigrated. For unknown reasons, it is far more common in Lancashire (UK) than anywhere else in the world.
Clinicopathological features
The usual presenting complaints of patients with Paget’s disease are bone pain, deformities or fractures. Although the pelvis and spine are most frequently affected, deformities are most obvious in the long bones such as the tibia, which is characteristically bowed, and in the skull.
Serum calcium concentration is usually normal, but the alkaline phosphatase is markedly elevated, reflecting the osteoblastic activity. The histological changes of Paget’s disease consist of irregular trabecular bone, much of which is woven rather than lamellar, and areas of osteolysis with abnormally large osteoclasts. These changes reflect grossly disordered bone remodelling (Fig. 25.9).
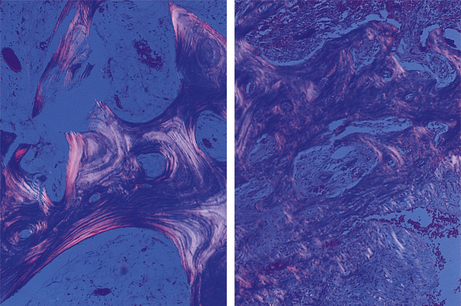
Fig. 25.9 Paget’s disease. In normal trabecular bone (left) viewed by polarised light, note the regular lamellar pattern. In bone affected by Paget’s disease (right) the lamellar pattern is largely replaced by irregular woven bone. Bone of this type is associated with deformity and poor mechanical function.
Complications
The complications of Paget’s disease are:
In many patients, Paget’s disease is completely asymptomatic and is unlikely to be diagnosed unless discovered as an incidental finding on X-ray. The commonest complications are deformities (Fig. 25.10) and bone pain. In some cases, the pain is the result of osteoarthritic degeneration of a related joint. The cause of the pain is uncertain, but interestingly responds well to treatments that inhibit bone resorption. Pagetoid bone is particularly susceptible to fracturing in the initial lytic phase. Enlargement in the sclerotic stage can lead to nerve or spinal cord compression. Deafness is the result of both VIIIth cranial nerve compression and distortion of the middle ear cavity. Occasional patients develop other cranial nerve palsies and, in advanced cases, paraplegia can result.
The most sinister complication of Paget’s disease is osteosarcoma; the majority of elderly patients with osteosarcoma do have Paget’s disease. As in younger patients, osteosarcoma develops in the long bones, particularly the humerus. The prognosis of osteosarcoma in Paget’s disease is especially poor. There is also an increased incidence of other forms of sarcoma.
Patients with Paget’s disease may also have heart failure. This is usually a simple coincidence of two common diseases of the elderly. However, the bone in patients with Paget’s disease is extremely vascular (Fig. 25.9), and blood flow in these areas is markedly increased. This may represent an example of ‘high output heart failure’ (Ch. 13). Paget’s disease is usually responsive to treatment with bisphosphonate drugs.
MISCELLANEOUS BONE DISORDERS
Achondroplasia
Achondroplasia is a single gene disorder transmitted as an autosomal dominant with almost complete penetrance and occurs in approximately 1 in 25000 births. The affected gene is for fibroblast growth factor receptor. The physical appearances of the patients are characteristic. The limbs are short, particularly the proximal portions of the arms, but the trunk is of normal length. There is a failure of proper ossification in bones that have developed from a cartilaginous template (endochondral ossification). In contrast, bones that develop from connective tissue (intramembranous ossification), such as the vault of the skull, are normal. Affected patients have normal intelligence and life-span. There is a wide variety of other congenital skeletal dysplasias, many of which are lethal in utero. Achondroplasia is the commonest non-lethal form.
Avascular necrosis of bone
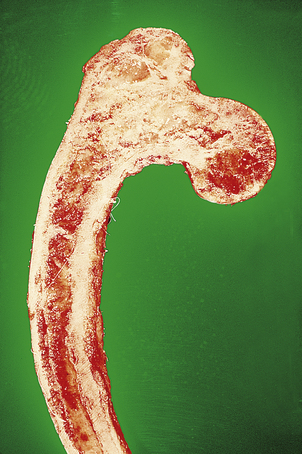
This usually presents with pain and limitation of joint movement. For anatomical reasons, fractures of bones such as the neck of the femur (Fig. 25.11) or the scaphoid deprive some areas of adjacent bone of their blood supply. Necrosis is then an inevitable consequence. Surgical treatment is therefore sometimes necessary to replace the fractured head of femur. The cause of other cases of avascular necrosis is less certain. Lesions occur in patients treated with corticosteroids, in sickle cell disease and other haemoglobinopathies. Similar lesions develop in divers and are probably the result of air embolism, associated with decompression.
 Compare the normal contours of the femoral neck on the right with the fracture on the left (arrowed).
Compare the normal contours of the femoral neck on the right with the fracture on the left (arrowed).  There is a displaced fracture of the left femoral neck (detected 8 weeks after a fall!) resulting in necrosis of the bone deprived of its blood supply.
There is a displaced fracture of the left femoral neck (detected 8 weeks after a fall!) resulting in necrosis of the bone deprived of its blood supply.Fibrous dysplasia
In this benign disorder of children and young adults, lesions composed of fibrous and bony tissue develop, usually in the ribs, femur, tibia or skull. It is sometimes asssociated with precocious puberty (McCune–Albright syndrome). Histologically, these lesions are composed of irregular masses of immature woven bone separated by a richly vascular fibrous stroma. Mature lamellar bone is not formed and this suggests that the lesion is a result of an arrest of bone maturation at the woven bone stage. Lesions do not usually enlarge after puberty, though some appear to be reactivated during pregnancy. Although the clinical and radiological findings are often diagnostic, lesions in long bones are often biopsied and the affected parts of ribs can be excised. In typical cases, the histological changes can be easily distinguished from true neoplastic disorders, such as osteosarcoma, fibrosarcoma or giant cell tumours.
Hypertrophic osteoarthropathy
This is an uncommon reactive condition in which there is clubbing, pain and swelling of the wrist and ankle joints, and subperiosteal new bone formation in the distal part of long bones. In the vast majority of affected patients there is an associated pulmonary carcinoma or a pleural mesothelioma. The underlying causes of both clubbing and hypertrophic osteoarthropathy are unknown, but in both cases there is a marked increase in blood flow in the distal portions of the limbs. Occasional cases regress after surgical treatment of the primary tumour.
Osteogenesis imperfecta
Osteogenesis imperfecta is a clinical syndrome characterised by fractures occurring as a result of mild or minimal trauma. In its most severe form it is fatal in utero or in the perinatal period. Mild forms compatible with normal development also occur, but many patients are severely disabled. Many cases are associated with mutations of the genes encoding the chains of type I collagen. Inheritance is usually autosomal dominant, but many cases result from sporadic new mutations. The uveal pigment of the eye is visible through the thinned sclera, which therefore appear blue in some forms of the disease.
BONE TUMOURS


Incidence and aetiology
Primary tumours of bone are uncommon and account for only 0.5% of all cancer deaths. Because of their rarity, these tumours are best managed in centres where there are orthopaedic surgeons, radiologists and histopathologists with sufficient experience of these lesions. Individual tumours tend to occur in particular age groups or in specific sites (Fig. 25.12).
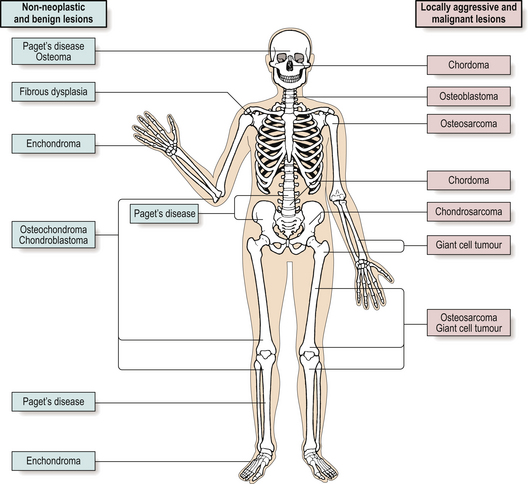
Classification
Benign tumours
The two commonest benign tumours of bone are the osteochondroma (exostosis) and the chondroma (enchondroma) which together make up over 50% of all benign bone tumours.
Osteochondroma
Patients with osteochondromas are usually under 20 years old and both sexes are affected. The lesions tend to develop near the epiphyses of limb bones, although they can form in any bone that develops from cartilage. Solitary lesions are benign, although they may recur if incompletely excised. Histologically, a thick cartilaginous cap is present, covering irregularly arranged bony trabeculae. There is an intermediate zone where the bone merges gradually into the overlying cartilage.
In diaphyseal aclasis there are multiple cartilage-capped exostoses and a substantial risk of associated chondrosarcoma. Malignancy should be suspected in any lesion that continues to grow after puberty. The incidence is approximately 1 per 2000 births and the lesion is transmitted as an autosomal dominant. Up to one-third of cases are the result of new mutations and the condition is probably a single gene disorder.
Chondroma (enchondroma)
Chondromas arise within the medullary cavity of the bones of the hands and feet and because of this are usually known as ‘enchondromas’. They are thought to develop from small nests of cartilage that are sometimes found close to the metaphysis. They usually present in patients aged 20–50 years and are more common in men. There may be slight swelling of the affected bone but the lesions are sometimes discovered incidentally on X-ray. Simple curettage of the lesion is usually curative, although some recur. Enchondromatosis (Ollier’s disease) is the counterpart of multiple exostoses. Numerous enchondromas develop and there is a considerable risk of chondrosarcoma. This disorder has no obvious genetic basis. Multiple enchondromas may also occur in Mafucci’s syndrome in association with multiple angiomas.
Other benign tumours
Other benign bone tumours are uncommon. An osteoma is a mass of abnormally dense bone, usually in the paranasal sinuses or the skull.
An osteoid osteoma is a solitary and characteristically painful lesion which usually affects the femur or tibia. The pain may be severe, worse at night, and characteristically relieved by aspirin. Histologically there is a central ‘nidus’ of vascular fibrous tissue containing bone trabeculae formed by benign osteoblasts.
Chondroblastoma and chondromyxoid fibroma are rare tumours of long bones with distinctive histological appearances. They are most common in the femur, tibia or humerus in patients between 10 and 30 years of age.
Locally aggressive or recurrent benign tumours
Some benign tumours of bone, such as giant cell tumour (‘osteoclastoma’) and osteoblastoma are locally aggressive or may recur after surgery.
Giant cell tumours make up as much as 5% of all bone neoplasms and most often occur at the end of the long bones. They probably originate from undifferentiated mesenchymal cells in the connective tissue framework of bone. Their histological appearance is distinctive. Giant cells are conspicuous and for this reason the tumour has also been known as osteoclastoma; however, these cells are not thought to be the neoplastic component but are non-neoplastic osteoclasts ‘recruited’ to the tumour by the neoplastic fibroblast-like component. Giant cell tumours can also be confused histologically with the bone lesions of primary hyperparathyroidism (‘von Recklinghausen’s disease of bone’), in which osteoclasts are plentiful.
Osteoblastomas are uncommon solitary tumours that involve vertebrae and, to a lesser extent, the long bones of the extremities, and are essentially large osteoid osteomas. The lesions are very vascular and show intense osteoblastic activity. Surgical treatment—usually by curettage—can be curative.
Chordomas arise from notochordal remnants, usually in the base of the skull or the sacral region. The constituent cells often have a characteristic ‘bubbly’ appearance due to cytoplasmic vacuolation. These tumours seldom metastasise but often recur locally.
Adamantinomas and ameloblastomas are histologically similar tumours. Ameloblastomas affect the jaw and have the capacity to produce tooth enamel but adamantinomas usually involve the tibia and do not produce enamel. The histological appearance is characteristic, with ribbons and cords of darkly staining cells arranged around a vascular fibrous stroma. Adequate surgical excision is often curative.
Malignant tumours
Although primary malignant tumours of bone are comparatively rare, they are always locally invasive and frequently metastasise. Patients are often young adults. The previously dismal prognosis of these tumours has been considerably improved in recent years with the advent of pre-operative radio- and chemotherapy, often in combination with limb-sparing surgery (Table 25.3).
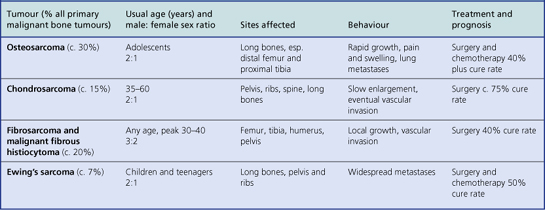
Osteosarcoma
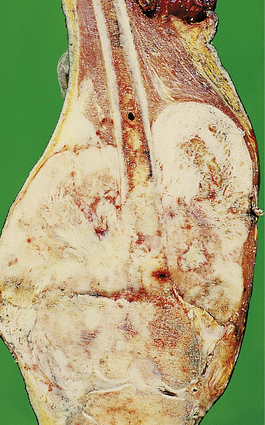
This aggressive malignant tumour usually affects adolescents and most often involves the distal femur, proximal tibia or humerus (Fig. 25.13). Occasional cases occur in the elderly, usually complicating Paget’s disease or previous radiation. Other cases are associated with familial cancer syndromes, notably the retinoblastoma syndrome and the Li–Fraumeni syndrome. Somatic mutations of the tumour suppressor genes that have germline mutations in these syndromes (RB1 and p53) are common in sporadic osteosarcomas. These tumours grow rapidly and often have a typical X-ray appearance. Osteosarcomas are characterised histologically by pleomorphic and mitotically active osteoblasts associated with osteoid; some variants are exceedingly vascular. Approximately 50% of patients can now be cured by a combination of surgery and intensive chemotherapy. Although amputation was previously necessary, local resection and the insertion of prosthetic joints is now usually possible.
Other malignant tumours
Chondrosarcomas, in contrast to osteosarcomas, grow slowly and arise not only in long bones but also in the pelvis, ribs and spine (Fig. 25.14). These tumours may be well differentiated and can resemble normal cartilage. Surgical excision is the treatment of choice, as radiotherapy and chemotherapy are usually ineffective.
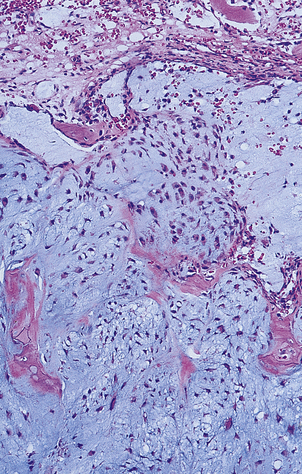
Fig. 25.14 Chondrosarcoma. The tumour has arisen in a pelvic bone of a 42-year-old female. Histologically the abnormal cartilage is much more cellular than normal.
Fibrosarcomas and malignant fibrous histiocytomas are spindle cell malignant tumours with characteristic histological appearances. These lesions probably arise from fibroblasts and collectively they make up the majority of soft tissue sarcomas. They also occur as primary bone lesions, with the long bones and pelvis most often affected. With adequate surgical treatment up to 40% of patients can be cured. Death is usually the result of blood-borne metastases.
Ewing’s sarcoma affects children and teenagers. The tumour is composed of small, darkly staining, undifferentiated cells whose exact origin (histogenesis) has puzzled pathologists for many years. It has a characteristic chromosomal translocation—t(11;22)(q24;q12)—that is also found in an equivalent soft tissue tumour (primitive neuro-ectodermal tumour or PNET). These are now recognised as being the same type of tumour. Males are affected more often than females, and the long bones, pelvis and ribs are the most frequent sites. Widespread metastases are frequent and the bone marrow is often involved.
Metastases and multiple myeloma
The commonest malignant tumours of bone are secondary metastatic deposits from carcinomas in other sites. The commonest primaries to cause skeletal metastases are breast, prostate, lung, kidney and thyroid. In the case of breast and prostate carcinomas, skeletal metastases may present early in the course of the disease and be associated with prolonged survival with appropriate treatment. Widespread and extensive bony lesions are also a feature of multiple myeloma. Most secondary deposits in bone cause bone breakdown (osteolysis) but some, particularly from carcinoma of the prostate, stimulate bone formation (osteosclerosis; Fig. 20.12, p. 541). Secondary deposits in bone are the commonest cause of hypercalcaemia in middle-aged and elderly patients and are a frequent cause of pathological fractures. Vertebral metastases can cause spinal cord compression.
JOINTS
NORMAL STRUCTURE AND FUNCTION
Joints permit mobility, but not all junctions between bones are designed to allow movement. At one extreme, the cranial sutures in adults are rigidly fixed while, at the other, the shoulder joint has an almost unlimited range of movement. Joints such as the symphysis pubis and the lower tibiofibular joint have limited movement but are firmly bound by fibrous and cartilaginous tissue. In contrast, the articulating surfaces of synovial joints are in contact but not in continuity.
The articular surfaces of synovial joints are covered by a thin layer of hyaline or, occasionally, fibrous cartilage, up to 3mm thick. In early life these surfaces are remarkably smooth, and slide and move against each other with very little friction. A viscous, clear synovial fluid lubricates the joint surfaces and supplies essential nutrients to the chondrocytes of the articular cartilage.
Synovial joints are enclosed by a tough fibrous capsule, which in turn is lined by a thin synovial membrane. Two types of cell—type A and type B—have been identified in the lining membrane. Type A synoviocytes are modified macrophages while type B are fibroblast-like cells and are responsible for synthesising and secreting the hyaluronic acid and other proteins of the synovial fluid. Ligaments are band-like thickenings of the joint capsule which not only provide stability, but, as with the cruciate ligaments of the knee joint, limit excessive mobility. Tendons, ligaments and joint capsules insert into bone, their collagen fibres becoming incorporated into the underlying bone. These insertions are called entheses and are prone to inflammation in the spondyloarthropathies (see below).
The bone immediately beneath the articular cartilage—the subchondral bone plate—provides the strength to withstand and cushion the repeated forces generated by joint movement. In weight-bearing joints, this plate is supported by an underlying ‘scaffold’ of bony trabeculae. If this supporting system is damaged, as in advanced osteoarthritis, the joint surfaces become deformed and movement is limited. The individual arteries and veins supplying joints and joint capsules have not been studied in detail and are seldom specifically named. Nevertheless, joints are richly vascular structures, particularly in acute inflammatory arthritis or during the active stages of rheumatoid disease. Joints such as the knee, the sternoclavicular and the temporomandibular have partial or complete discs of fibrocartilage called menisci, which either project into joint cavities or divide them into separate cavities. These may act as ‘cushions’ or ‘shock absorbers’. When these discs are damaged or torn, there is acute limitation of joint movement and surgical removal or repair is often necessary.
Joints have a rich innervation, usually derived from nerves supplying the adjacent muscular tissue. This arrangement allows a local reflex arc to be established between movement in an individual joint and the actions of surrounding muscles. There are many sensory nerve endings in the fibrous capsule of joints and in the bone underlying the articular surfaces. Any substantial pathological process involving a joint is likely to cause inflammatory cell infiltration and oedema of the adjacent joint capsule, if not of the articular surfaces themselves, and this leads to both pain and subsequent limitation of movement.
OSTEOARTHRITIS (OSTEOARTHROSIS)





Osteoarthritis is a remarkably common, disabling, degenerative disease which usually affects large weight-bearing joints. The inevitable pain, and limitation of movement, associated with this disease is a major cause of morbidity in almost all societies.
Epidemiology
About 20% of elderly men and women have significant osteoarthritic joint disease. Pain and limitation of movement are the most important symptoms, particularly in the hip, the knee and the joints of the cervical spine (cervical spondylosis).
Certain occupations are associated with a high incidence of osteoarthritis in particular joints. Coal miners develop osteoarthritis in elbow joints, golfers in the first metatarsophalangeal joint of the foot, and footballers in the knees. Some patients with premature osteoarthritis of the hip have previous congenital dislocation in this joint. Similarly, any obvious deformity or previous fracture is an important predisposing cause. Patients with pre-existing bone disease, such as Paget’s disease, gout or acromegaly, or other forms of arthritis such as rheumatoid disease, are at risk for secondary osteoarthritis. Nevertheless, in the majority of patients there are no obvious predisposing factors. This is particularly true of vertebral osteoarthritis, which, although often asymptomatic, can produce severe pain and disability.
Pathogenesis
The term ‘osteoarthritis’ is a misnomer; the role of inflammation in the pathogenesis of this disease appears to be minimal. It is primarily a degenerative disease for which the term ‘osteoarthrosis’ would be more appropriate. The earliest changes are fragmentation and fibrillation of the normally smooth surface of the articular cartilage (Fig. 25.15). Change of this degree is very common with ageing and may not necessarily progress to symptomatic osteoarthritis. Only in a proportion of joints does the process of degeneration progress to ‘joint failure’. Progressive osteoarthritis may occur because of the predisposing factors listed above. However, in the majority of cases, the reason for progression is not clear. There is increasing evidence that genetic factors are important. As the disease progresses, there is increasing loss of articular cartilage accompanied by abortive attempts at regeneration. Because the overall structural integrity of the cartilage has been lost (Fig. 25.16), this new cartilage formation has no structural benefit and eventually the full thickness of the cartilage may be lost, resulting in bone articulating against bone (eburnation). The mechanisms underlying the cartilage degradation are still poorly understood.
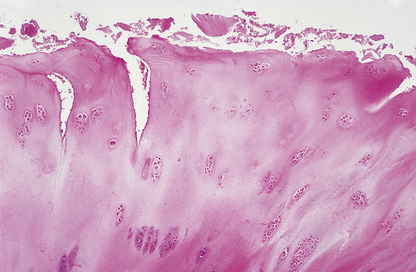
Fig. 25.15 Early osteoarthritis. Histology of a section through the articular surface showing ‘fibrillation’ and fissuring.
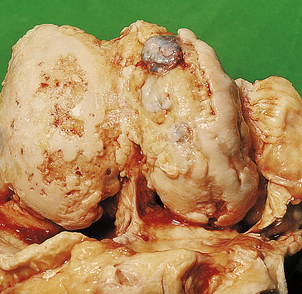
Fig. 25.16 Osteoarthritis. This view of the articular surfaces of the femoral condyles shows advanced osteoarthritis. There are areas of complete cartilage loss with eburnated subchondral bone.
The second series of changes in the development of osteoarthritis involves the subchondral bone plate. This plate not only defines the contours of the articular surface but also contributes to the strength and resilience of the joint. Loss of articular cartilage may be a stimulus to reactive proliferation of this plate. In turn, this leads to ever increasing damage to the residual articular cartilage.
Eventually, the articular surface becomes deformed and in many joints this is the major cause of limitation of movement. There is progressively more severe disorganised bone remodelling, leading to loss of the normal cortical and trabecular patterns of subchondral bone and the development of cystic lesions often visible on X-rays. Bony outgrowths (osteophytes) develop at the margins of the articular surface. These are a characteristic feature of osteoarthritis but serve no useful function. In joints such as the hip they limit the range of movement, and in the distal interphalangeal joints they produce characteristic nodular swellings called Heberden’s nodes. In cervical spondylosis, a variant of osteoarthritis affecting the cervical spine, symptoms result from osteophytic outgrowths impinging on nerve roots as they leave the intervertebral foramina.
Clinicopathological features
The major symptoms of osteoarthritis are pain on joint movement, stiffness during inactivity and audible creaking of joints, often accompanied by a palpable crepitus. Although there is no primary synovial pathology in osteoarthritis, small joint effusions are common and histologically the synovium shows slight hyperplasia and focal areas of chronic inflammation, often as a reaction to calcified debris shed from the articular surfaces.
The diagnosis of osteoarthritis is made on the basis of clinical examination and characteristic radiological appearances. Almost all of the pathological features of osteoarthritis can be identified on a plain X-ray. One of the earliest changes is loss of joint space: the articular surfaces of the bone appear close together when articular cartilage has been lost. Reactive proliferation of the subchondral bone plate occurs and there are deformities of the articular surface. All routine laboratory investigations are normal.
Treatment of osteoarthritis causing the failure of major joints is by replacement of the joint by a prosthesis. Prostheses themselves can fail by loosening of their attachment to the bone into which they are inserted. This can occur as a result of infection, often by low virulence organisms such as Staphylococcus albus, or by aseptic loosening. Aseptic loosening is due to localised bone resorption resulting from cytokine production by macrophages activated by prosthesis debris.
RHEUMATOID DISEASE




Rheumatoid arthritis is a common, systemic, progressive and often disabling chronic inflammatory disorder. Unlike osteoarthritis, the pathological changes are not restricted to joints. Inflammatory lesions can develop in many tissues, including the heart and pericardium, lungs, blood vessels, skin and subcutaneous tissues, eye, and salivary and lacrimal glands. For this reason, the disorder is more correctly termed rheumatoid disease, but the arthritis is the first and generally most disabling feature.
Aetiology and pathogenesis
The main epidemiological and pathogenetic features of rheumatoid disease are:
Immunological abnormalities
Rheumatoid disease is associated with a variety of immunological abnormalities. These involve both the cellular and the humoral arms of the immune system.
Humoral
Most patients have considerably raised levels of serum immunoglobulins and have a characteristic auto-antibody directed against autologous immunoglobulin, called rheumatoid factor. The exact role of this antibody in the pathogenesis of rheumatoid arthritis is uncertain (it is possible that rheumatoid factors are simply markers of a loss of immunological self-tolerance) but at the least it is associated with the progression of the disease. Generally, patients with severe arthritis and evidence of multisystem disease have high titres of this antibody. In contrast, seronegative patients often have a milder form of disease. There is no definite evidence that aggregates of these immunoglobulins can act as immune complexes and trigger acute and chronic inflammatory reactions in joints and other tissues. The nature of the underlying stimulus to rheumatoid factor production is completely unknown, but many patients with severe rheumatoid disease have generalised hyperplasia of lymphoid tissue with prominent lymphadenopathy and splenomegaly.
Cellular
There is increasing evidence to suggest that rheumatoid arthritis is a disease driven by cellular, rather than humoral, immune mechanisms. The T-helper lymphocyte populations present in rheumatoid joints are oligoclonal, and may be autoreactive. These cells probably contribute to the process that causes macrophages, macrophage-like type A synoviocytes and other inflammatory cells to secrete cytokines such as tumour necrosis factor alpha and interleukin-1. Thus type IV rather than type II or type III hypersensitivity may be the predominant mechanism. Tumour necrosis factor alpha and interleukin-1 are potent stimulators of synoviocyte proliferation and cartilage resorption and are known to be present in joint fluid and pannus tissue (see below and Fig. 25.19) from rheumatoid joints. Pro-inflammatory cytokines can induce the production of other cytokines, possibly contributing to the progressive nature of rheumatoid disease. Of particular significance, these cytokines are potent inducers of proteolytic enzyme production by synoviocytes. These cytokines are of central importance in the pathogenesis of rheumatoid arthritis, and specific cytokine antagonist drugs are coming into clinical use.
Infectious agents
Although the nature of the immunological abnormalities in rheumatoid disease is becoming clearer, very little progress is being made in identifying the factors that initiate these changes. Micro-organisms have been implicated for many years and it is now beyond doubt that Lyme arthritis (see below) is infectious and that erythrovirus B19 (formerly parvovirus B19) can induce arthritis. Bacteria, viruses and mycoplasmas have been suggested as causes of rheumatoid disease but there is no conclusive proof that rheumatoid disease is caused by infection. Joints affected by rheumatoid are, however, vulnerable to secondary bacterial infection, a potentially serious complication.
Clinicopathological features
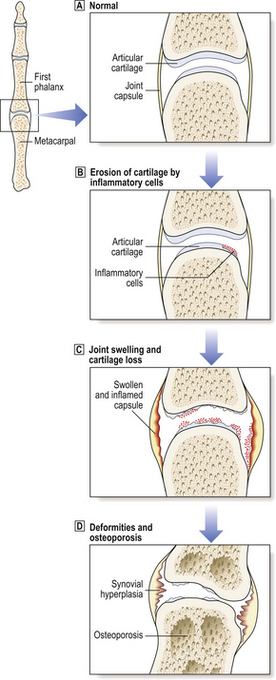
The two major changes in rheumatoid arthritis are a chronic inflammatory synovitis and progressive erosion of the articular cartilage (Figs 25.17 and 25.18). In the early stages of the disease there is pain and swelling, chiefly of the hands and the distal metatarsal joints of the foot. As the disease becomes established, the knees, ankles, hips, cervical spine and temporomandibular joints are affected. Joints are tender, and often swollen.
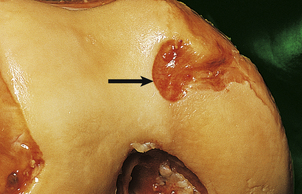
Fig. 25.17 Early rheumatoid disease. A layer of chronic inflammatory tissue (‘pannus’, arrowed) has eroded the articular surface of the femoral condyle.
The synovial infiltrates include lymphocytes, plasma cells, macrophages and occasional polymorphs (Fig. 25.19). In the earlier stages there is only a mild increase in inflammatory cells, but in the established disease large nodular masses of lymphocytes and macrophages are characteristic. True granulomas are not usually identified in the synovium but may occasionally be seen in the adjacent joint capsule. A layer of chronically inflamed fibrous tissue (pannus) slowly extends from the synovial margin, eroding the articular cartilage as it spreads (Fig. 25.17).
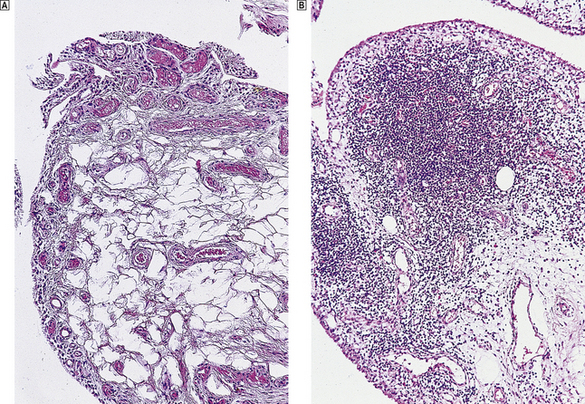
Fig. 25.19 Rheumatoid arthritis.  Normal synovium.
Normal synovium.  Note the dense lymphocytic infiltrates in the synovial biopsy from a patient with active rheumatoid arthritis.
Note the dense lymphocytic infiltrates in the synovial biopsy from a patient with active rheumatoid arthritis.
Osteoporosis often develops in the bones immediately adjacent to affected joints, particularly in the fingers. This is often the first radiological change of rheumatoid disease, but small pocket-like erosions of the peri-articular surface are also seen (Fig. 25.20). In chronically diseased joints the articular cartilage is extensively eroded and fragmented and the resultant debris is a further source of synovial inflammation. The loss of articular cartilage, along with damage to the joint capsule and peri-articular structures, causes deformities (Fig. 25.20) and secondary osteoarthritis can develop, particularly in the knee.
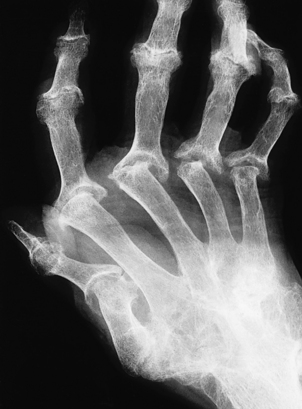
Fig. 25.20 Radiological features of rheumatoid arthritis. There is prominent ulnar deviation in longstanding rheumatoid disease. There are erosions of the distal metacarpal bones and of most of the carpal bones.
Extra-articular features
The extra-articular features of rheumatoid disease are:
Subcutaneous rheumatoid nodules develop in up to one-third of patients with rheumatoid disease, but most of these are severe and progressive cases. Typically, they involve the extensor surfaces of the forearm, less commonly the dorsum of the foot. These locations suggest that everyday incidental trauma contributes to their development. The nodules vary in size but are usually 20–40mm in maximum dimension. Their histological structure is characteristic: central areas of necrotic collagen are surrounded by palisades of fibroblasts and macrophages.
Many patients with rheumatoid disease have clinically obvious lymphadenopathy. In some, the spleen can be felt—rheumatoid disease is one of the commoner causes of splenomegaly. A chronic inflammatory fibrinous pericarditis is a frequent finding in rheumatoid disease and occasionally chronic inflammatory granulomas can develop within the myocardium.
The wide range of pulmonary pathology includes rheumatoid nodule formation within the parenchyma of the lung and chronic interstitial fibrosis. Chronic inflammatory infiltrates can develop in both the lacrimal and salivary glands, impairing tear and saliva production. The resulting dryness in both the eyes and mouth (Sjögren’s syndrome) is often persistent and irritating. Patients complain of a gritty feeling in the eyes and of photophobia. Both uveitis and scleritis are important ocular manifestations of rheumatoid disease (Fig. 25.21). The uveal tract, like the skin, the glomerulus of the kidney and the joints, has a very high blood flow per unit mass of tissue and this may contribute to the deposition of immune complexes and the subsequent inflammatory reaction. Paradoxically, uveitis can occur many years after the onset of rheumatoid arthritis, sometimes when the disease is in a quiescent phase. Vasculitis is a sinister feature and implies a poor prognosis. It is clinically most obvious in the fingers, particularly the nail beds.
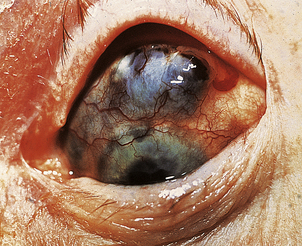
Fig. 25.21 Scleritis in rheumatoid arthritis. The inflammation has caused thinning of the scleral connective tissue, revealing the underlying pigmentation of the choroid.
Increasingly, treatment of rheumatoid arthritis is aimed at limiting joint damage, with immunosuppressive drugs such as methotrexate becoming the treatment of choice. Corticosteroids are also effective, but their use is limited by their many side-effects. Newly developed specific inhibitors of cytokines, such as tumour necrosis factor alpha, show considerable promise in the treatment of severe rheumatoid disease.
Laboratory investigations
In contrast to the situation in osteoarthritis, there are a wide range of haematological and immunological abnormalities in rheumatoid disease:
Patients with severe symptoms in the early stages of the disease, or a poor response to anti-inflammatory drug therapy, tend to progress more rapidly. Careful evaluation of radiographs of the hand is also important, as the early development of bony erosions is another poor prognostic feature. If a remission of disease can be obtained by drug treatment, it can sometimes be sustained for months or even years.
Prognosis and complications
At the onset of the disease it is very difficult to determine the prognosis for any individual patient. Approximately 10% of patients who develop rheumatoid arthritis will become severely disabled, dependent on others for some or all of their normal daily activities. In contrast, at least 20% of patients have only slight symptoms and relatively mild disability. They may have mild intermittent swelling of joints and difficulty in movements such as unscrewing lids. The remaining 70% of patients have varying degrees of disability and most will require some form of drug therapy, at least during exacerbations of disease.
Although rheumatoid disease can be relentless and progressive, it is seldom directly responsible for the death of an individual. Infections are more common in patients with rheumatoid disease than in the general population; these include septic arthritis, pneumonia, suppurative pericarditis and septicaemia. Blood culture is essential in a seriously ill patient with rheumatoid disease. Rheumatoid disease can be complicated by reactive systemic (AA) amyloidosis (Ch. 7).
Rheumatoid disease in children
At the turn of the 20th century, chronic inflammatory arthritis with systemic manifestations was described by Sir George Still (1868–1941), a paediatrician at Great Ormond Street Hospital. He emphasised that the disease occurred ‘between the first and second dentition’ and that there were similarities to rheumatoid disease in adults. It is now appreciated that there are many different forms of juvenile chronic arthritis and that only some of these are strictly similar to rheumatoid disease in adults.
Clinicopathological features
In juvenile chronic arthritis there are prominent systemic manifestations, often early in the course of the disease. These include pyrexia, skin rashes, lymphadenopathy, splenomegaly, pericarditis and pleurisy, and some can occur even before the onset of the arthritis. As in the classical rheumatoid arthritis of adults, several joints are affected. The joint symptoms and synovial pathology are very similar to those of adult rheumatoid arthritis. A chronic inflammatory pannus may form and erode articular cartilage. Fortunately, articular cartilage is somewhat thicker in children and this tends to protect the underlying bone.
Laboratory investigations and prognosis
As in adults, the erythrocyte sedimentation rate (ESR) is raised and a normochromic normocytic anaemia may be present. Although there may be hyperglobulinaemia, specific tests for rheumatoid factor are usually negative; this is the most important laboratory investigation distinguishing juvenile chronic arthritis from typical rheumatoid disease. A subgroup of adolescent children with chronic arthritis do have rheumatoid factor, and these children often have a progressive form of disease and require early treatment. In at least three-quarters of cases, the disease remits, either spontaneously or after drug treatment, and there is often no residual joint damage. In contrast, some patients may have a progressive arthritis and there can be substantial and permanent joint deformity.
Very little is known of the cause of juvenile chronic arthritis. The principles of treatment are much the same as in adult rheumatoid disease.
SPONDYLOARTHROPATHIES
This is a puzzling group of related disorders of unknown aetiology, including ankylosing spondylitis and Reiter’s disease. These diseases are characterised by enthesitis and a strong association with the HLA-B27 tissue type. In classical cases of ankylosing spondylitis and Reiter’s disease, there are well-defined clinical signs and symptoms and, even if the pathogenesis is obscure, pathological changes have been identified in a number of different organs. In some systemic disorders, including psoriasis, ulcerative colitis, Crohn’s disease, infectious dysentery and chlamydial urethritis, there may be a chronic and disabling arthropathy resembling true ankylosing spondylitis or Reiter’s disease.
Ankylosing spondylitis



The term ‘spondylitis’ implies an inflammatory disorder of the spine, whereas ‘spondylosis’ is used to describe the commoner degenerative osteoarthritic change.
Epidemiology
Fully developed ankylosing spondylitis is rare and occurs in only 1 in 1000 middle-aged male adults, and rather fewer females. About 90% of patients with unequivocal ankylosing spondylitis have the human leukocyte antigen B27—one of the strongest disease associations with a particular HLA haplotype. Once this association was established, surveys were undertaken to determine what proportion of the 5–10% of the Caucasian population who are B27 positive had signs of spondylitis. Depending on the stringency of the criteria used for diagnosis, up to 15% of these patients have some evidence of mild spondylitis but only 1–2% of severe spondylitis. The first symptoms usually occur before the age of 30 years.
Clinicopathological features
Information on the underlying pathological changes can be obtained only by sequential radiological studies or postmortem examination of the occasional patients who die in the early stages of the disease. The inflammatory process begins at the entheses where ligaments are attached to vertebral bone. As these lesions heal, there is reactive new bone formation in the adjacent ligaments and sclerosis of the underlying bone. The earliest changes are often present in the sacro-iliac joints and may be detected by careful radiological examination or computed tomography. Pain may be produced if the lower portion of the sacrum is depressed forward with the patient lying face down. Fusion of the vertebral bodies inhibits both flexion and rotation, and this is particularly disabling when the cervical segment is affected. Some patients develop fixed spinal deformities.
The symptoms and lesions strongly associated with ankylosing spondylitis are:
At least 30% of patients with typical ankylosing spondylitis have a peripheral arthropathy. There are no specific histological features that distinguish this from other low-grade forms of arthropathy, but the clinical distribution is distinctive. Lower limb joints and the shoulders are often involved, but the lower arm, particularly the hands, is usually spared. The arthritis may begin before, together with, or some time after the first back symptoms.
Ankylosing spondylitis is one of the diseases that is associated with uveitis. In most cases, the anterior part of the eye—the iris and ciliary body—is affected, and choroidal changes are less common. The cause of the majority of cases of uveitis is unknown but between 10% and 20% will have some evidence of spondylitis. Uveitis is also associated with inflammatory bowel disease, and there is no doubt that both Crohn’s disease and ulcerative colitis are more common in those with ankylosing spondylitis than in the general population. The best-recognised cardiovascular complication of spondylitis is aortic incompetence, but this is present in only 1–2% of longstanding cases. Pathologically, there is a chronic inflammatory aortitis, usually restricted to the valve ring and ascending aorta.
Reiter’s disease
During World War I, a German physician, Hans Reiter, described the combination of arthritis, conjunctivitis and urethritis developing in a soldier shortly after an attack of dysentery. Most of the patients are males and the onset of the illness is usually abrupt. The arthritis usually affects lower limb joints and classically there is swelling and inflammation at the insertion of the Achilles tendon. The urethritis is usually due to chlamydial infection, and the conjunctivitis is only a minor feature of the illness.
The underlying pathological mechanisms in Reiter’s disease are unknown. Chlamydial infections usually respond to tetracycline, but the arthritis can persist for months and occur at irregular intervals. Up to one-half of severely affected patients subsequently develop signs of spondylitis and, as most of these are HLA-B27 positive, it is very likely that the underlying disease process is the same as in true ankylosing spondylitis.
Psoriasis and arthritis
Psoriasis is a common and chronic skin disorder affecting up to 1% of Caucasians (Ch. 24). About 5% of patients with psoriasis have arthropathy, typically involving the distal interphalangeal joints. Again, if there is evidence of spondylitis, patients with arthropathy and psoriasis are usually HLA-B27 positive.
Arthritis and bowel disease
A low-grade peripheral arthropathy is a well-recognised, though comparatively rare, complication of Salmonella, Shigella and Yersinia gastroenteritis. HLA-B27-positive patients have a substantially increased risk of developing this complication. The underlying pathological mechanisms are unknown. Arthritis occurs in up to 20% of patients with ulcerative colitis and Crohn’s disease which may manifest itself as sacro-ileitis or fully developed ankylosing spondylitis.
DEGENERATIVE DISEASE OF INTERVERTEBRAL DISCS
Degenerative softening of the fibrocartilaginous intervertebral discs is a very common cause of back pain. It usually affects adults and clinical symptoms are exacerbated by heavy straining when lifting or by poor posture. Discs in the lumbar spine are affected most commonly.
The softened nucleus pulposus can herniate vertically into an adjacent vertebral body, forming a Schmorl’s node (Fig. 25.5) which may be radiologically evident. More seriously, the disc material may herniate posterolaterally through the surrounding annulus fibrosus, forming a protrusion that impinges upon the nerve emanating from the intervertebral foramina. This is the prolapsed intervertebral disc, or so-called ‘slipped disc’, that causes severe pain radiating into the territory supplied by the compressed nerve (e.g. ‘sciatica’ when the pain radiates across the buttock and down the leg); motor nerve conduction may also be impaired. In many cases the symptoms resolve with analgesia and/or physiotherapy or spinal manipulation. Recurrences are common. Surgical removal of the degenerate disc material may be necessary in some patients. The natural history of the disease is for the nucleus pulposus to degenerate spontaneously over time, thus relieving the symptoms. With increasing age the naturally restricted movement of the spine reduces the probability of disc herniation.
INFECTIVE ARTHRITIS
Most cases of septic arthritis are the result of bacterial infection. In some viral diseases there is an associated arthritis, or at least arthralgia (joint pain) is a prominent symptom. In contrast, fungal infections of joints are extremely rare.
Organisms responsible include:
Septic arthritis is the result of blood-borne spread from a focus of infection elsewhere. The epiphyseal plate forms a very effective barrier and it is unusual for an area of osteomyelitis in the metaphysis to spread and involve adjacent joints. This occasionally occurs in the hip, where the metaphysis may lie within the joint capsule.
As with osteomyelitis, children are affected more commonly than adults but the reasons for this are uncertain. Most cases are the result of staphylococcal or streptococcal infection. Both pneumococci and gonococci have a tendency t o involve joints, and a septic arthritis can follow a bacteraemia or septicaemia associated with these organisms. Haemophilus influenzae arthritis is restricted to young children. Gram-negative septicaemia may cause inflammatory arthritis, particularly in drug addicts.
Diabetes mellitus, rheumatoid arthritis and immunosuppressive treatment are all risk factors for septic arthritis. Similarly, intra-articular injections of corticosteroids can be followed by inflammatory arthritis, and rigorous asepsis is essential during these procedures, particularly in patients with rheumatoid disease.
Clinicopathological features
The symptoms of infective arthritis are pain, tenderness, swelling and erythema. Although most cases of bacterial arthritis involve a single joint (monoarthritis), a small proportion of staphylococcal infections and the majority of cases of gonococcal arthritis involve two or more joints. As there is usually an associated septicaemia, patients are obviously ill, are usually pyrexial and may have rigors.
There are no diagnostic X-ray features, although radiographs are essential to exclude fractures or other bony injury. The diagnosis can be made only by aspirating and culturing the joint fluid. Special culture techniques are required for gonococci and mycobacteria, and full clinical details must therefore be given to the laboratory. Gram-stained preparations of synovial fluid may give a pointer to the causative organism and suggest appropriate antibiotic treatment before culture results are available. Initially, parenteral antibiotics should be given and oral antibiotics continued for 4–6 weeks. With adequate and prompt treatment, surgical drainage is not usually necessary, but should be considered if symptoms persist. The heavy polymorph infiltrates inevitably cause some superficial destruction of articular cartilage, and joint-space narrowing may become visible in radiographs. There may also be evidence of osteoporosis in the bones immediately adjacent to the joint.
Uncommon forms of infective arthritis
Gonococcal arthritis
This is usually a disorder of females or homosexual males, in whom the primary genital infection has been overlooked. Characteristically, it is a polyarthritis, frequently involving the hands, wrists and knees. Gonococcal arthritis is probably the commonest cause of infective arthritis in teenagers and young adults. Only in a minority of cases can gonococci be isolated from the synovial fluid.
Tuberculous arthritis
Approximately 1% of patients with tuberculosis have bony involvement as a result of spread from an established focus in the lungs or elsewhere. Involvement of the synovial membrane and peri-articular tissues produces a persistent arthritis with or without typical caseating granulomatous lesions. An inflammatory pannus may form and erode the articular cartilage. Tuberculous arthritis affects the hip and knee in children, but the vertebral column is most commonly involved in adults. In the vertebral column the associated osteomyelitis causes extensive bony destruction, and wedging or complete collapse of vertebrae may result (Pott’s disease of the spine). The lower thoracic and lumbar spine are most commonly affected. Infection may spread along fascial planes, particularly around the psoas muscle, producing a full-blown psoas abscess. Synovial fluid culture may produce a diagnosis.
Arthritis due to spirochaetes
Lyme disease was first described in the eastern United States in 1977. Epidemics of the disease have now been reported in many different areas of the world. The presenting symptom is a migratory erythematous rash—erythema chronicum migrans. An arthritis follows weeks, or months, after the initial infection. Epidemiological studies have now shown that ticks of the genus Ixodes are responsible for transmitting the causative agent, a spirochaete named Borrelia burgdorferi. Immune complexes containing antibody directed against this organism have been detected in joint fluid and may play a role in the development of the arthritis. Antibiotic therapy given early in the course of the disease can prevent the joint disease.
Virus-associated arthritis
Many different virus infections are associated with a transient arthritis, or at least distinct pain within joints. In rubella infections, arthritis of the hands and wrists is common and the virus has been isolated from affected joints. A mild arthralgia may persist for several weeks. Occasionally, transient arthritis follows rubella vaccination. An arthritis may be a feature early in the course of viral hepatitis and is more severe with hepatitis B infection. Arthralgia can also be a feature of infectious mononucleosis.
Infective discitis
Occasionally, intervertebral discs can be the site of bacterial infections. This is usually due to Staphylococcus aureus, Mycobacterium tuberculosis or Brucella abortus. Because of the avascular nature of disc tissue, antibiotic penetration is poor and surgical removal of the disc may be necessary.
RHEUMATIC FEVER




Rheumatic fever is a disease of disordered immunity characterised by inflammatory changes in the heart (Ch. 13) and joints, and in some cases associated with neurological symptoms (chorea). The disease is common in India, the Middle East and Central Africa. Although it is now rare in Europe and North America, occasional clusters of cases do occur and several recent outbreaks in the United States have emphasised that the disease must be considered in any child or adolescent with joint pain, skin rashes or unexplained fever. Polyarthritis is the presenting feature in over 75% of cases and usually involves the large joints of the wrists, elbows, knees and ankles. The arthritis characteristically ‘flits’ from joint to joint, involving each for 2–4 days, and may cause severe pain. In the acute phase the inflammation involves the endocardium, the myocardium and the pericardium (‘pancarditis’). Heart murmurs are common and children can die from cardiac failure.
Most patients have had a recent sore throat, typically a group A beta-haemolytic streptococcal infection. Disappointingly there have been no recent advances in the understanding of the underlying immunopathology. For many years it has been known that there is a strong antibody reaction to the streptococcus and it is thought that this may cross-react with as yet unknown antigens in connective tissues, especially in the heart and joints.
GOUT





Gout is one of the most clearly documented conditions in medical history. Evidence of gouty arthritis has been detected in Egyptian mummies, and the condition is clearly described in the writings of Hippocrates and other Greek and Roman physicians. It appears to have been common (or perhaps a fashionable diagnosis) in the 17th and 18th centuries and in the popular imagination is associated with corpulence and alcoholism.
Pathogenesis
The underlying biochemical mechanisms in gout are well understood (Ch. 7), although the exact reasons why the majority of patients develop a raised uric acid level are uncertain. The mechanisms and causes are:
At least 5% of middle-aged males have a serum uric acid greater than 0.5mmol/l, but less than 5% of these will ever develop clinical signs of gout. In over 75% of patients who present with gout, there is a decrease in uric acid clearance by the kidney but the underlying cause of this is not known. In a few patients, there appears to be an idiopathic increase in the rate of purine synthesis, leading in turn to increased uric acid production. The increased cellular turnover associated with a wide variety of different malignant disorders and other diseases is a common cause of secondary gout. Most patients receiving chemotherapy are now treated prophylactically with xanthine oxidase inhibitors in order to minimise the hyperuricaemia associated with the cellular necrosis and metabolic breakdown of nucleic acids induced by cytotoxic drugs.
The stimulus to the acute inflammatory reaction in acute gout is the deposition of monosodium urate crystals in the synovium and adjacent connective tissues of the joints. The exact mechanisms leading to this are poorly understood. Gout is most common in the metatarsophalangeal joint of the great toe, and gravitational factors could play a part in promoting crystal deposition. Acute gout also occurs in the ankle but is comparatively uncommon in the knee and hips. In acute gout, the joint fluid contains numerous polymorphs, and crystals can frequently be detected by polarised light microscopy. Microcrystals of monosodium urate can absorb a variety of immunoglobulins, complement components, fibrinogen and fibronectin, and these may encourage their phagocytosis by polymorphs. Regulatory cytokines, such as interleukin-1, almost certainly have some role in promoting inflammation. There is a rapid turnover of neutrophils within acutely inflamed gouty joints, largely because phagocytosed microcrystals have a toxic effect on cellular membranes. This is in itself a potent acute inflammatory stimulus, and in acute gout there is often an associated cellulitis.
Clinicopathological features
The clinicopathological features of gout are as follows:
Gout presents as an acute inflammatory monoarthritis. In over two-thirds of patients the metatarsophalangeal joint is affected. The onset can be surprisingly abrupt. Affected joints are warm and tender and exquisitely painful (Fig. 25.22). There may be associated pyrexia, and the white cell count and ESR are generally raised. The clinical diagnosis is usually obvious, and prompt treatment with non-steroidal anti-inflammatory drugs relieves symptoms within hours. Occasionally, several joints can be involved simultaneously and this can be diagnostically misleading.
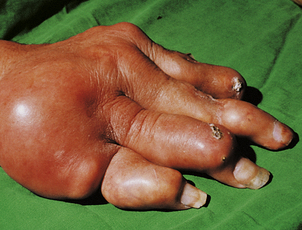
Fig. 25.22 Acute gout. Note the marked oedema and inflammation. There are areas of early ulceration at the tips of the index and ring fingers.
There may be long intervals between acute attacks in individual patients. Most patients are treated with allopurinol, which suppresses uric acid synthesis by inhibiting xanthine oxidase.
Renal disease is the most serious complication of gout. For poorly understood reasons, the incidence of renal calculi in gout varies from country to country. In western Europe it is of the order of 10% and gout should be considered in any patient who presents with renal colic. Mild proteinuria is found in a proportion of patients but very few progress to chronic urate nephropathy and renal failure. Those that do so have usually received inadequate treatment or have a strong familial history of severe gout. Urate crystal deposition in renal tubular epithelium induces cellular necrosis, chronic interstitial nephritis and fibrosis.
In chronic tophaceous gout, large deposits of uric acid occur within joints or in the soft tissues, particularly around the pinna of the ear. In these patients, there are substantial X-ray changes, with soft tissue swelling, calcification of urate deposits and even erosions of phalangeal bone.
Gout is associated with obesity, alcoholism, hypertension, ischaemic heart disease, various forms of hyperlipoproteinaemia and impaired glucose tolerance. However, the majority of patients who have a raised blood uric acid level will never develop gout, or any of its complications.
PYROPHOSPHATE ARTHROPATHY
Pyrophosphate arthropathy, also known as ‘pseudogout’ or ‘chondrocalcinosis’, results from the deposition of calcium pyrophosphate crystals in joint cartilage. Occasionally, an acute arthritis results and this can mimic true gout.
The cause of the pyrophosphate deposition is unknown. It is very much an age-related phenomenon and is more common in the elderly. There is an association with hyperparathyroidism and haemochromatosis, and occasional familial cases are described. Pyrophosphate arthropathy can also complicate true gout.
Clinicopathological features
In most cases, pyrophosphate deposition produces no clinical symptoms. Minor degrees of cartilaginous calcification, particularly in the knee, are a common finding in X-rays taken in the elderly. In advanced cases, there is a characteristic linear area of calcification—evidence that the mid-zone of the articular cartilage is particularly susceptible to pyrophosphate deposition. If crystals are shed into the joint cavity in sufficient numbers, an acute inflammatory arthritis results. This can usually be distinguished from true gout: if joint fluid is examined the crystals can be distinguished from urate by their weak positive, rather than strong negative, birefringence.
There is a strong association between chondrocalcinosis and osteoarthritic joint disease. One explanation of this is that crystal deposition predisposes the articular cartilage to degenerative change and subsequent florid osteoarthritis. Alternatively, pyrophosphate crystals may be preferentially deposited in cartilage previously injured by early osteoarthritis. There is also a form of osteoarthritis associated with deposition of calcium hydroxyapatite crystals in and around knee and shoulder joints (‘pseudopseudogout’). This usually affects middle-aged or elderly women.
As most cases of chondrocalcinosis are asymptomatic, no particular treatment is indicated. Acute pseudogout is best treated by intra-articular corticosteroids. Anti-inflammatory drugs and colchicine are less effective than in true gout. No prophylactic measures are available to prevent recurrent attacks but these are uncommon.
JOINT INVOLVEMENT IN SYSTEMIC DISEASE
An arthritis, or at least some degree of joint involvement, is a common feature of many systemic disorders (Table 25.4). In some of these, the arthritis is the direct result of the primary pathological process involving the synovium.
Table 25.4 Systemic diseases and joint changes
| Underlying disease | Clinical features | Pathology |
|---|---|---|
| Acromegaly | Episodic painful swelling of small joints, e.g. in hands | Periosteal new bone formation |
| Amyloidosis | Arthropathy of shoulders, knees and wrists, usually secondary to myeloma | Amyloid deposition in synovium |
| Behçet’s disease |
ClubbingCharacteristic swelling of nail beds
Hypertrophic pulmonary osteoarthropathy (HPOA)Arthropathy of wrists, ankles, feet
SarcoidosisSmall joint polyarthropathy, especially fingersSarcoidal granulomas in synovium
CONNECTIVE TISSUES
TRAUMATIC AND DEGENERATIVE CONDITIONS OF CONNECTIVE TISSUES
In contrast to the remarkable healing capacity of bone, most other connective tissues are rather limited in their ability to repair. Injuries to connective tissue structures can occur as a result of single episodes of trauma, such as a ligament rupture, or chronic damage due to overuse or repetitive stress.
Single episode trauma to connective tissue structures results either in healing by scarring, such as a skeletal muscle tear, or in a chronic injury, such as a meniscal tear or cruciate ligament rupture. Chronic trauma to tendons, ligaments or joint capsules results in neovascularisation and increased proteoglycan content in the tissue. These processes cause chronic pain and can result in impaired movement. There are several distinct clinical syndromes that result from these pathological changes such as ‘tennis elbow’, ‘housemaid’s knee’ and the carpal tunnel syndrome. In some of these conditions, inflammation of bursae or tendon sheaths is the main pathological process. Where repetitive injury, usually to a tendon sheath, causes local proteoglycan accumulation, this can lead to the formation of cysts referred to as ganglia/ganglion cysts.
CONNECTIVE TISSUE DISEASES
There is no agreed definition of a ‘connective tissue disease’. It is a convenient general term which covers a wide variety of disorders:
These disorders have the following common features:
These disorders were originally termed ‘collagen diseases’, and this emphasises that cutaneous and subcutaneous changes are often prominent clinical features. The clinical and pathological features of some of the more important connective tissue diseases are summarised in Table 25.5.
Systemic lupus erythematosus



Systemic lupus erythematosus (SLE) is, more than any other connective tissue disease, a multisystem disorder. Females are affected more often than males, in some age groups by a ratio of up to 10:1. The incidence is higher in blacks and Asians than in Caucasians. The peak age of onset is usually between 20 and 30 years.
Aetiology and immunopathology
The cause of SLE is unknown. The first immunological abnormality was detected in the 1940s. It was found that leukocytes from patients with SLE phagocytosed nuclear debris produced by agitating a sample of freshly drawn blood. It was subsequently shown that most patients with SLE have circulating auto-antibodies directed against nuclear antigens. When serum containing these antibodies is added to tissue sections, such as liver or kidney, they bind to nuclear components and this can be visualised by immunofluorescence. Many other auto-antibodies have now been detected, reacting with both nuclear and cytoplasmic antigens. As these antibodies are characterised, it is becoming clear that they are directed against a relatively small range of epitopes, components of both nucleic acids and cytoplasmic phospholipids:
The immune system is capable of developing immunogenicity or tolerogenicity towards any given antigen. Which of these events occur depends upon the balance of immunogenic and tolerogenic cytokine pathways, which are poorly understood. As in rheumatoid disease, the auto-antibodies of SLE may be manifestations of a generalised loss of self-tolerance. None of these antibodies is SLE-specific and most have been detected in other forms of autoimmune disease. However, there is more evidence to suggest that immune complexes are important in the pathogenesis of SLE than there is in rheumatoid disease.
The precise trigger for loss of self-tolerance in SLE is mysterious. The antihypertensive drug hydralazine can cause an SLE-like illness.
No other environmental or occupational factors have been implicated in SLE. It is not obviously associated with any particular human leukocyte antigen but there is a strong familial tendency. Relatives of SLE patients have a substantially increased incidence of the disorder and there is a greater than 50% concordance in monozygotic twins.
Clinicopathological features
Spontaneous remissions and exacerbations are common in SLE and some patients can be managed for long periods without specific treatment. Signs and symptoms can occur in almost every system:
Arthralgia is often the presenting symptom and in the early stages may resemble rheumatoid arthritis. The hands, knees and ankles are most affected, and a series of joints may be involved in succession (flitting arthralgia). Synovial biopsy shows a low-grade inflammatory synovitis but, unlike the situation in rheumatoid disease, pannus does not form and cartilaginous erosions are rare.
The most characteristic cutaneous change in SLE is a symmetrical erythematous facial (‘butterfly’) rash (Fig. 25.23) which may be precipitated by exposure to sunlight (photosensitivity). Raynaud’s phenomenon, urticaria, hyperpigmentation and cutaneous ulcers are other less common features.
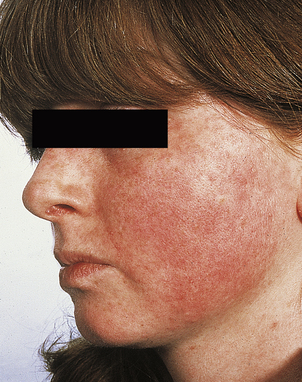
Non-specific early signs in SLE include mild pyrexia, normochromic normocytic anaemia, leukopenia, muscle pain, lymphadenopathy and splenomegaly. Although pleurisy and pericarditis are common postmortem findings, they rarely produce overt clinical symptoms. Non-bacterial thrombotic vegetations (Libman–Sacks endocarditis) may form on mitral or aortic valves and can be detected by echocardiography. Murmurs may result, but the vegetations seldom erode the valve leaflets or cause significant embolic disease.
Renal involvement is one of the most important features and patients should be regularly screened for proteinuria. The underlying lesion is glomerulonephritis, which may be of minimal change, focal, membranous or diffuse proliferative type (Ch. 21). Although some patients do progress to renal failure, not all forms are necessarily associated with a poor prognosis.
Neurological changes are more common in SLE than in any other connective tissue disease. Psychiatric symptoms are the commonest abnormality but some of these may be the result of treatment with steroids. Severe headaches and convulsions occur in some patients. The small sub-group who present with recurrent transient ischaemic attacks or definite episodes of cerebral infarction generally have circulating antibodies to cardiolipin—the so-called ‘antiphospholipid antibody syndrome’. Anti-cardiolipin antibodies are also associated with recurrent spontaneous abortions. The neuropathological changes in SLE are rather non-specific but occasional cases have evidence of cerebral arteritis or thrombosis.
Polyarteritis nodosa


Polyarteritis nodosa (PAN) is one of the most florid of the inflammatory disorders of arteries, collectively known as arteritis (Ch. 13). The main features are:
Almost any artery can be affected but, because arthralgia and abdominal pain are frequent early symptoms, these patients are often referred to rheumatologists or gastroenterologists.
Aetiology and immunopathology
The cause of PAN is not fully understood. There is increasing evidence to suggest hepatitis B virus is important in the development of this disease, although this may not apply to all forms of PAN-like vasculitis. The incidence seems to be increasing but this may be because milder forms of vasculitis are now recognised. The florid acute inflammatory changes with associated fibrinoid necrosis resemble experimental models of immune complex vasculitis. Circulating immune complexes may be elevated in patients with vasculitis. Complement levels can be reduced and abnormal deposits of immunoglobulins are sometimes present in affected vessels in biopsy specimens. Many patients with systemic vasculitis have circulating anti-neutrophil cytoplasmic antibodies (ANCA). They are not specific for any particular disorder but are most strongly associated with Wegener’s granulomatosis (a severe destructive vasculitis affecting the nasal cavity, lungs and kidneys) and PAN, and may not be present in patients in clinical remission. It is not known whether these antibodies are involved directly in the disease process or are a secondary result of the associated vascular damage.
Clinicopathological features
Most patients present with non-specific signs of a generalised illness, such as pyrexia of unknown origin (PUO), malaise and myalgia. Arthralgia, abdominal pain, vomiting and diarrhoea are common in the early stages of the disease, and other non-specific signs include hypertension and pericarditis.
The underlying vasculitic process (Ch. 13) can involve almost any medium-sized artery. The disease was originally named because of the tender nodules produced by involvement of subcutaneous arteries, but these are quite different from rheumatoid nodules and occur in only a small proportion of cases. Peripheral gangrene can result from arteritis of digital vessels. Polyarteritis nodosa is a cause of mononeuritis multiplex—a pattern of peripheral neuropathy where individual nerves are affected because of disease of the nutrient arteries (Ch. 26). Symptoms can be motor, sensory or mixed.
Renal disease is one of the leading causes of death in PAN (Ch. 21). There is a well-established association between PAN, pulmonary disease and eosinophilia (Churg–Strauss syndrome). Classically, there are prominent asthmatic symptoms, with cough and dyspnoea. In severe cases, pulmonary arteritis leads to areas of infarction and there may be haemoptysis.
In contrast to the case in SLE, central nervous system involvement is comparatively rare in PAN. Only occasional patients develop cerebral arteritis.
Laboratory investigations
Typical haematological findings in PAN include a mild normochromic normocytic anaemia, a moderate leukocytosis and an absolute eosinophilia. ESR and other acute phase reactants are raised. Although gamma globulin levels may be increased, auto-antibodies such as rheumatoid factor and antinuclear antibodies are usually negative.
A firm diagnosis can be made histologically or by arteriography. Biopsy of a skin lesion or a nerve associated with an area of cutaneous anaesthesia or a muscle biopsy may show the histological appearances of necrotising arteritis (Ch. 13). Selective angiography of the major abdominal branches, such as the coeliac axis or mesenteric artery, may reveal typical aneurysms.
The outlook in PAN is worst in patients with definite evidence of multisystem involvement. Patients with limited forms of the disease often do well and may need little or no treatment. It is quite possible that several disease processes are presently included under the label of ‘polyarteritis nodosa’.
Polymyalgia rheumatica
Polymyalgia rheumatica (PMR) is an important disorder of the elderly. It is more common in females than in males, and patients usually present with persistent muscular pain in the shoulders and hips, lethargy and tiredness. These rather non-specific symptoms occur in most rheumatic disorders, and PMR may therefore be confused with rheumatoid arthritis, cranial arteritis (see below), polyarteritis nodosa or various forms of polymyositis. In most patients these disorders can be excluded, and PMR is considered to be a disease in its own right.
The underlying pathological changes are incompletely understood. Myositis does not occur, but there is evidence from magnetic resonance imaging (MRI) that bursitis occurs in active disease. In typical cases, the ESR is greater than 100mm/h, but there are no other consistent laboratory abnormalities. There may be a mild normochromic or hypochromic anaemia and non-specific elevations of immunoglobulins. Circulating auto-antibodies, such as rheumatoid factor and antinuclear antibody, are usually absent and creatine phosphokinase levels are normal.
Although PMR is a somewhat non-specific clinical and pathological entity, it is a common and important condition which can affect up to 1% of elderly patients. Clinical benefit can be obtained with steroid treatment, but there are inevitable side-effects if this is continued for long periods. At least 15% of patients with polymyalgia rheumatica have cranial arteritis and in these patients a positive temporal artery biopsy is of great value in justifying long-term treatment. In other patients steroids are usually given for at least a year and then discontinued gradually.
Cranial arteritis
Cranial arteritis (also known as temporal or ‘giant cell’ arteritis) is an important, and not uncommon, disease of the elderly. It affects arteries of the head and neck region and, unlike almost all other arterial diseases, responds rapidly and predictably to treatment with anti-inflammatory drugs such as corticosteroids.
Aetiology and pathogenesis
Epidemiological surveys in different communities have demonstrated that the incidence of the disease varies between 50 and 150 cases per 100000 population. The disease is commonest in Caucasians but occurs in all races. There is no satisfactory explanation for the preferential involvement of the arteries of the head and neck. Involvement of the superficial temporal artery is responsible for scalp tenderness, but it is disease of the ciliary and ophthalmic arteries that may produce blindness. The carotid arteries and, occasionally, the aorta may be affected but this is usually clinically silent. The disease appears to be a distinct entity, and the histological appearances and clinical course of the disorder are very different from those of other inflammatory conditions of arteries, such as polyarteritis nodosa, although there may be some relationship with Takayasu’s arteritis. This is a form of arteritis, usually affecting the thoracic aorta and its branches, that is best known in Japan. Cranial arteritis has no obvious genetic basis and is not associated with auto-antibody production.
Clinicopathological features
Because the superficial temporal artery is accessible for biopsy, the histological features are well described (Ch. 13).
Most patients present with headache or scalp tenderness, and in any elderly patient these symptoms should suggest a diagnosis of cranial arteritis. Jaw claudication is another important symptom. There is a well-recognised overlap between the symptoms of cranial arteritis and polymyalgia rheumatica. Careful palpation of the temporal arteries is essential in any patient with polymyalgia and, conversely, musculoskeletal symptoms are not uncommon in cases of cranial arteritis. The precise relationship between these disorders is uncertain, particularly as the pathological changes in the two disorders are rather dissimilar. Fortunately, steroid therapy is the treatment of choice in each disorder.
Laboratory investigations
Temporal artery biopsy should be performed urgently in any patient with suspected cranial arteritis. Ideally, 20–30mm of superficial temporal artery should be removed, preferably before steroid therapy has started. However, because of the risk of blindness due to involvement of the ophthalmic arteries, steroid therapy should not be delayed while awaiting the result of the biopsy, because of the risk of rapid progression to blindness. A high ESR is characteristic but in a few patients this may be only slightly elevated. There are no other characteristic laboratory findings, although a mild normochromic normocytic anaemia is sometimes present. Up to 40% of patients with good clinical evidence of cranial arteritis have a negative temporal artery biopsy.
Polymyositis and dermatomyositis
Polymyositis is a chronic inflammatory disorder of skeletal muscle of unknown cause. The typical patient is a female in late middle age with a history of progressive muscular weakness, often commencing in the shoulder or neck muscles. There is associated pain and tenderness but muscle wasting is not a feature in the early stages.
In active polymyositis there is a florid chronic inflammatory infiltrate in affected muscles (Fig. 25.24) with extensive associated degeneration of both type 1 and type 2 fibres (Ch. 26). Many of the clinical features suggest a viral infection but no particular virus has been consistently isolated. Auto-antibodies to a variety of nuclear antigens have been detected in up to 25% of patients. Rheumatoid factor and antinuclear antibody are usually absent. The diagnosis is best made on a combination of clinical findings, raised levels of muscle enzymes such as creatine phosphokinase (CPK), electromyography and muscle biopsy. Serial estimation of CPK may give a clue to the prognosis in individual patients. The majority make an uneventful recovery, albeit over a period of months or years. Oesophageal disease produces a troublesome dysphagia, and cardiac involvement is not uncommon. The mortality rate is between 5% and 15% and is usually the result of respiratory failure, aspiration pneumonia or, occasionally, heart failure.
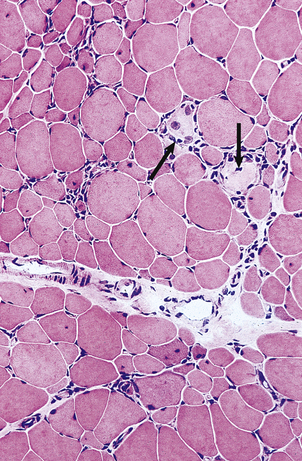
Fig. 25.24 Polymyositis. A low-power view of a muscle biopsy showing irregular infiltrates of inflammatory cells. Some muscle fibres are undergoing necrosis (arrows).
In dermatomyositis a variety of skin changes occur in association with an otherwise typical polymyositis. There is a diffuse erythema involving the upper part of the body, prominent swelling of the eyelids and a variable purple ‘heliotrope’ discoloration of the eyelids (Ch. 24). The muscular symptoms are more prominent in patients with dermatological changes.
At least 10% of middle-aged or elderly patients with polymyositis have evidence of malignant disease, most commonly carcinoma of the bronchus, gastrointestinal tract or breast. The polymyositis may occur months or years before the first symptoms of the underlying carcinoma, and for this reason all patients with genuine polymyositis should be screened for the common malignancies. Although polymyositis and dermatomyositis are more common in females, this ratio is reversed when there is an associated malignancy.
Scleroderma





Scleroderma describes a spectrum of diseases of uncertain cause. The disorder is most common in young and early middle-aged females who usually present with Raynaud’s phenomenon, polyarthritis and thickening and tightness of the skin (Table 25.6). This affects the hands and fingertips first, and movements of the finger joints are impaired. Facial changes are also common; the mouth appears small and the peri-oral skin is taut and creased (Fig. 25.25). Careful observation of the progress of the disease gives a good indication of the prognosis. If the disease spreads to the upper arms, legs or the flank there is a strong chance that a generalised form of disease, systemic sclerosis, may develop. In contrast, in other patients the disease is entirely limited to the skin and is characterised by well-defined plaques or bands of subcutaneous fibrosis (‘morphoea’). The most complete manifestation of the disease is the CREST syndrome (cutaneous calcinosis, Raynaud’s phenomenon, oesophgeal dysfunction, sclerodactyly and telangiectasia).
Table 25.6 Clinical and pathological features of scleroderma and systemic sclerosis
| Organ/system | Clinical features | Pathology |
|---|---|---|
| Skin |
Joints
Low-grade inflammatory synovitisGastrointestinal tract
Submucosal fibrosis and muscular atrophy in lower oesophagusCardiovascular systemRaynaud’s phenomenonPericarditis
Kidneys
LungsDyspnoeaInterstitial inflammation and fibrosis
The underlying pathological changes include vascular injury, perivascular accumulation of mononuclear cells and deposition of connective tissue. In systemic sclerosis the process may extend to the gastrointestinal tract. A frequent symptom is dysphagia and there may be submucosal fibrosis and muscular atrophy in the lower oesophagus.
Little is known of the cause of scleroderma. Some circulating auto-antibodies have now been detected but they have not been as well characterised as in diseases such as systemic lupus erythematosus or rheumatoid disease. The pathological changes in small arteries and arterioles resemble those of systemic hypertension, but only 20% of patients with scleroderma have raised blood pressure.
Mixed connective tissue disease (MCTD)
Connective tissue diseases share many common clinical and pathological features. In clinical practice, it is usually possible to attach a specific diagnostic label to the majority of patients. The term ‘overlap syndrome’ or mixed connective tissue disease (MCTD) is used when there are distinct overlapping features of systemic lupus erythematosus (SLE), systemic sclerosis, rheumatoid arthritis, polymyositis or even polyarteritis nodosa. The usual features are:
All patients with MCTD have a strongly positive fluorescent test for antinuclear antibodies. The exact nature of the nuclear antigen is unknown, but there is evidence that several different nuclear matrix proteins may be involved.
Because true MCTD is uncommon there is comparatively little information on clinical and pathological progression. In some patients a full clinical remission occurs, but others develop into typical scleroderma or SLE. In these patients the outlook is much the same as if an MCTD phase had not occurred.
CONNECTIVE TISSUE TUMOURS
Connective tissue tumours are, with a few exceptions, relatively uncommon. However, those classified as malignant (i.e. sarcomas) are important because they are often deeply located, for example in the retroperitoneum, resulting in late clinical presentation and difficulties in ensuring complete surgical removal. As a general rule, the more deeply a connective tissue tumour is situated, the less likely it is to be benign.
Benign connective tissue tumours
Benign connective tissue tumours include:
Lipomas are the commonest connective tissue tumours, usually subcutaneous and comprising morphologically mature adipocytes. They are usually solitary, but may be multiple, well circumscribed and located in subcutaneous fat. Less common variants include angiolipoma characterised by numerous blood vessels, spindle cell lipoma in which there is an admixture of fibroblasts, and myelolipoma containing haemopoietic cells.
Although angiomas are benign in the sense that they do not actively invade surrounding tissues, they can be troublesome clinically and prone to recurrences requiring further surgery. These problems arise because the margins of the lesion are often blurred, merging with the surrounding normal tissue.
Leiomyomas in connective tissues (they are commoner in the uterus and gut) arise from smooth muscle cells in the walls of blood vessels and, in the skin, from arrector pili muscles.
Fibrous histiocytomas are neoplasms of fibroblast-like cells that also contain varying numbers of macrophages. Benign fibrous histiocytomas occur most commonly in the skin (dermatofibroma; Ch. 24).
Granular cell tumours are of uncertain histogenesis. Although benign, they are usually poorly circumscribed. They are generally located superficially and, curiously, when they occur beneath squamous epithelium (in skin, tongue, etc.) they often induce florid epithelial hyperplasia morphologically mimicking carcinoma.
Malignant connective tissue tumours (sarcomas)
Malignant connective tissue tumours include:
Liposarcomas are the commonest. The incidence of tumours designated ‘malignant fibrous histiocytoma’ varies according to the belief of local histopathologists in this controversial entity; in places where its existence is accepted, the incidence is often as high as that of liposarcoma. However, most of these tumours are either poorly differentiated examples of other types of sarcoma or essentially undifferentiated sarcomas.
The behaviour of sarcomas is strongly influenced by their histological grade. Low-grade sarcomas, such as well-differentiated liposarcomas, rarely metastasise. High-grade sarccomas frequently metastasise to the lungs. Overall, the long-term survival rate for soft tissue sarcomas is approximately 50%. Surgical excision, usually followed by radiotherapy, is the treatment of choice for most types of sarcoma. Many of them grow as ‘spindle cell tumours’ or ‘small blue round cell tumours’, the latter particularly in children (the ‘blue’ refers to their appearance with haematoxylin and eosin staining), with few identifying characteristics to betray their true
Commonly confused conditions and entities relating to osteoarticular and connective tissue pathology
| Commonly confused | Distinction and explanation |
|---|---|
| Rheumatic and rheumatoid | Both terms denote disorders affecting joints, but of different aetiologies. Rheumatic fever is an immunologically mediated post-streptococcal illness affecting the heart and joints. Rheumatoid disease is an autoimmune disorder causing arthritis, completely unrelated to rheumatic fever. |
| Osteoporosis and osteomalacia | Both conditions cause bone weakening. In osteoporosis there is a reduction in the bone protein matrix; however, that which is formed is adequately mineralised. In osteomalacia, the bone matrix is normal but there is insufficient mineralisation. |
| Osteomalacia and rickets | Both conditions are due to defective mineralisation of the skeleton. Osteomalacia is the name given to the appearances in the adult skeleton. Rickets occurs in children; the defective mineralisation leads to deformities of the growing skeleton. |
| Gout and pseudogout | Both are crystal arthropathies. In gout the crystals are urate. In pseudogout, the crystals are calcium pyrophosphate. |
| Systemic sclerosis and multiple sclerosis | Systemic sclerosis (sclerosis = hardening) is a disorder of connective tissue. Multiple sclerosis is a demyelinating disorder of the central nervous system. |
histogenesis. Immunohistochemistry may reveal the histogenesis of the tumour. However, because several of these tumours (e.g. synovial sarcoma, myxoid liposarcoma) have characteristic chromosomal translocations, cytogenetics is increasingly becoming the most important diagnostic investigation.
Some of these tumours merit specific comments. Synovial sarcomas usually occur, as the name suggests, close to joints and have a biphasic growth pattern comprising spindle cells and epithelial cells. They are, however, soft tissue carcinosarcomas and are not derived from synovium. Epithelioid sarcomas are rare tumours occurring on the limbs and have an unusual tendency to arise from and grow along fascial sheaths. Peripheral neuroectodermal tumours occur most commonly in children and are cytogenetically identical to Ewing’s sarcoma of bone.
Tumour-like lesions of connective tissues
Some nodular connective tissue lesions are characterised by cellular proliferation but they do not fulfil all the criteria of neoplasia. These include:
Fibromatoses are tumour-like proliferations of myofibroblasts. These cells have contractile properties; this explains the puckering and tethering associated with some variants of fibromatoses. For example, palmar fibromatosis (Dupuytren’s contracture) leads to permanent flexion of the fingers. Other variants of fibromatosis include musculoaponeurotic fibromatosis and desmoid tumours. Many examples of fibromatosis have abnormalities of the Wnt signalling pathway that can be demonstrated histologically through nuclear localisation of the signalling molecule beta-catenin. Desmoid and intra-abdominal fibromatoses can be associated with Gardner’s syndrome and familial adenomatous polyposis.
Nodular fasciitis occurs superficially as a rapidly growing nodule with alarming histological appearances mimicking sarcoma. Myositis ossificans, as its name suggests, is characterised by inflammation in muscle and ossification. The clinical significance of these benign lesions is that they can be mistaken, both clinically and histologically, for malignant lesions, resulting in clinically disastrous overtreatment.
Cope A .P., Schulze-Koops H., Aringer M.. The central role of T cells in rheumatoid arthritis. Clinical and Experimental Rheumatology. 2007;25:S4-11.
Ebeling P .R.. Osteoporosis in men. New England Journal of Medicine. 2008;358:1474-1482.
Goldring M.B., Goldring S.R.. Osteoarthritis. Journal of Cellular Physiology. 2007;213:626-634.
Hakim A., Clunie G.J.A., Haq I.. Oxford Handbook of Rheumatology, 2nd edn. Oxford: Oxford University Press; 2006.
Lane N.E.. Osteoarthritis of the hip. New England Journal of Medicine. 2007;357:1413-1421.
Tung S., Iqbal J.. Evolution, aging, and osteoporosis. Annals of the New York Academy of Sciences. 2007;1116:499-506.
Weiss S.W., Goldblum J.R.. Enzinger and Weiss’s soft tissue tumors, 5th edn. St Louis: Mosby; 2007.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Chapter 25 Osteoarticular and connective tissues
Common clinical problems from osteoarticular and connective tissue disease
Pathological basis of clinical signs and symptoms of bone, joint and connective tissue diseases
| Sign or symptom | Pathological basis |
|---|---|
| Bone disease | |
| Pain | Stimulation of nerve endings in bone by: |
Fracture after trivial injuryBone weakening due to:
DeformityAbnormal bone growth/remodelling due to:
Hypercalcaemia
Joint disease PainStimulation of nerve endings in joint capsule and synovium by inflammation (arthritis) or abnormal load bearing/joint movementDeformityJoint swelling due to:
Restricted movement
Systemic features (e.g.subcutaneous nodules,lymphadenopathy)Arthritis mediated by immune mechanismsConnective tissue diseases Swelling
Joint painSynovial oedema and inflammation with stimulation of nerves in joint capsuleIschaemic lesionsVasculitisRestricted mobility of tissuesFibrosis or increased tissue tension due to inflammation
BONE
NORMAL STRUCTURE AND FUNCTION
Functions of bone
Bone has structural, protective and metabolic functions. The skeleton is divided into the axial (head, vertebral column, thoracic cage, shoulder and pelvic girdles) and the appendicular (limbs). The axial skeleton participates extensively in all three areas of function, whereas the appendicular skeleton has a primarily structural function. The structural functions of bone are to provide support and also insertion sites for muscles and ligaments. The skull and thoracic cage provide physical protection for the brain, thoracic and upper abdominal organs.
Bone has two major metabolic functions:
Structure of bone
Bone is characterised by its hard matrix. This matrix consists of two components, matrix proteins and mineral. The main structural protein in bone matrix is type I collagen. This protein provides the framework of the overall structure of bone. Within the collagen framework there is a mixture of many other proteins, some of which are thought to aid mineralisation; others mediate cell attachment. Another major group of proteins present in bone matrix are growth factors, such as the bone morphogenetic proteins and transforming growth factor beta (TGF-beta). These appear to be important in mediating the cellular events of bone remodelling. Proteoglycans are also present in bone matrix, but do not have the same major structural role in bone as they do in cartilage. The mineral component of bone matrix provides its structural resilience. Most of the mineral deposited in bone is in the form of a calcium phosphate complex known as hydroxyapatite. The precise mechanism by which bone mineral forms is unclear. The enzyme alkaline phosphatase, a major product of osteoblasts, and vitamin D metabolites are thought to be important in this process.
Despite its lifeless appearance, bone is a highly complex and dynamic cellular tissue. Bone contains two distinct types of cell, osteoclasts and the osteoblast family. Osteoclasts, the bone-resorbing cells, are mono- or multinucleated cells that are specialised members of the monocyte–macrophage lineage. These short-lived cells are recruited to the bone surface at sites of remodelling and destroy bone matrix by secreting hydrogen ions and proteolytic enzymes into a sealed space beneath the cell. The osteoblast family consists of: osteoblasts, which are bone forming cells; osteocytes, which form an interconnecting network throughout bone matrix; and lining cells, which cover metabolically inactive bone surfaces. Osteocytes are thought to be mechanosensory cells. The cells of the osteoblast family are unrelated to osteoclasts, being related more closely to fibroblasts, chondrocytes and adipocytes.
Osteoclasts and osteoblasts act together to control bone growth and metabolism through the bone remodelling cycle. This cycle, illustrated in Figure 25.1, forms the basis of bone metabolism.
Fig. 25.1 The bone remodelling cycle. The bone remodelling cycle consists of resorption by osteoclasts (arrowheads in left-hand plate) which results in the removal of an area of bone matrix and is inevitably followed at the same site by resynthesis of bone matrix by osteoblasts (right-hand plate). Many osteoblasts become incorporated into the matrix they synthesise to become osteocytes (arrowed). Osteocytes are connected by a complex network of cytoplasmic processes not visible on these haematoxylin and eosin-stained sections.
Each remodelling cycle takes several weeks to complete. Approximately one million remodelling cycles are occurring within the adult human skeleton at any one time. These cycles are asynchronous, some being in resorption, some in formation. This continual process of remodelling renews the adult human skeleton approximately every 7 years. The functions of the remodelling cycle are to:
In the normal bone remodelling cycle, resorption and formation are coupled, with the same quantity of bone being formed as had been resorbed, except where extra bone matrix is called for by the requirements of growth. In pathological situations, the two processes can become uncoupled; for example, osteoporosis can result from uncoupled increases in bone resorption or decreases in bone formation resulting in a net loss of bone. All diseases of bone are associated with changes in bone remodelling of some type.
There are two types of mature bone, cortical and trabecular (sometimes known as cancellous bone). Cortical bone has a predominantly structural load-bearing function. It is the dense bone that forms the diaphyses (shafts) of long bones such as the femur and the outer surfaces of predominantly trabecular bones such as the vertebral bodies. Trabecular bone has some structural function, but contributes to the metabolic functions of bone far more than cortical bone. Because it is metabolically more active, it is far more prone to diseases involving or resulting from increased bone remodelling than cortical bone. For example, post-menopausal osteoporosis affects trabecular bone before it affects cortical bone, and deposits of metastatic carcinoma are far more common in sites occupied by trabecular bone.
Development and growth of bones
Most tissues and organs grow as a result of a general increase in the number of their constituent cells. Because the proteinaceous matrix is so heavily calcified, a similar process is not possible in bone. Thus the process of remodelling described above is essential for bone growth.
During development, bone is formed either directly in connective tissue, as in the skull (intramembranous ossification), or on pre-existing cartilage, as in the limb bones (endochondral ossification).
During intramembranous ossification the first bone that is laid down has a loose and rather haphazard arrangement. This ‘woven bone’ gradually matures into more organised and compact ‘lamellar’ bone.
Endochondral ossification is a much more complicated process during which a cartilagenous template is converted into a bony structure with capacity for further growth. In each bone, ossification occurs at particular sites or centres of ossification situated in the shaft (diaphyseal centres) or towards the ends of the bone (epiphyseal centres) (Fig. 25.2). Ossification proceeds at different, but predictable, rates in each particular bone. In long bones, a plate of epiphyseal cartilage persists into adolescent or early adult life; this allows a continual increase in bone length. Skeletal growth through the growth plates is controlled by growth hormone and sex steroids with parathyroid hormone-related peptide (PTHrP) and insulin-like growth factor-1 (IGF-1) acting as paracrine (locally produced) growth factors. The overall shape and size of bone changes during growth and, to some extent, in adult life. This involves both osteoclastic bone resorption and enlargement of pre-existing, or the formation of new, bony trabeculae. Cortical bone grows in an analogous way through remodelling occurring at the periosteal and endosteal surfaces and within Haversian canals.
FRACTURES AND THEIR HEALING
Types of fracture: simple (clean break); comminuted (multiple bone fragments); compound (breaking through overlying skin); complicated (involving adjacent structures—blood vessels, nerves, etc.); stress fractures (small linear fractures)Causes of fractures
Fractures in normal bone are the result of substantial trauma, such as direct violence or a sudden unexpected fall. The precise site of fracture, the nature and direction of the fracture line, and the speed of the subsequent repair process depend very much on the age of the patient, the particular bone involved and the precise pattern of injury (Fig. 25.3).
Fig. 25.3 Fracture types and fracture healing. A greenstick fracture of the distal radius in a young child (arrowed). A displaced spiral fracture of the femur in a child. A comminuted fracture of the tibia. One fragment of bone has almost separated from the shaft. A healing fracture of the ulna. The site of the break is just visible and is surrounded by callus (arrowed).
Repeated episodes of minor trauma, for example after marching, marathon running or training for sport, can produce small but often painful stress fractures. These usually occur in the long bones of the lower limbs but have also been described in the metatarsals, the upper limb, pelvis and spine. They usually heal satisfactorily after a short period of rest. Even professional athletes can develop these fractures.
Fractures occur more easily in bone that is structurally abnormal. They may occur after a trivial injury or minor fall or even spontaneously during normal activity. This is particularly common in patients with osteoporosis but also occurs in most forms of metabolic bone disease (e.g. in osteomalacia and rickets), in Paget’s disease and in bone infiltrated by malignant tumours. Fractures of this type are called pathological fractures.
Fracture healing
The first stage in fracture healing is the formation of a bony bridge between the separated fragments. When this is formed, and some rigidity has been regained, remodelling and restructuring gradually restore the normal contours of the fractured bone. This process and the factors that can interfere with it are described in Chapter 6.
The major causes of delayed fracture healing are:
All of these are well recognised by orthopaedic surgeons, who modify the treatment in individual cases in line with the particular pattern of injury, and the age and general health of the patient. For example, a fracture through the neck of the femur usually deprives the head of its normal blood supply and satisfactory fracture healing is unlikely to occur. Surgical treatment, such as excision of the head and replacement by a metallic prosthesis, is therefore essential. Fractures in which the overlying skin surface is broken (compound fractures) are liable to infection, whereas this is extremely uncommon in closed fractures. Healing will be substantially delayed if a wound infection develops.
In many fractures, healing can be accelerated by prompt and appropriate surgical treatment using internal fixation by nails, plates and screws or external fixator devices to hold the fractured fragments in an appropriate position; this often allows early mobilisation. Primary callus does form but is reduced in amount. Small gaps are filled by new woven bone. Dead bone is gradually revascularised and new Haversian bone grows in.
Surgical treatment is sometimes necessary for fractures in which the healing process has been delayed. The object is to ‘restart’ the primary callus response. This can sometimes be achieved by lifting flaps of periosteum close to the fracture site. In addition, local ‘grafting’ with the patient’s own bone or devitalised bone from another donor can promote bone healing, presumably through the action of bone morphogenetic proteins and other growth factors present in the graft matrix.
OSTEOPOROSIS AND METABOLIC BONE DISEASE
Normal calcium metabolism
The two major hormones that regulate calcium metabolism are vitamin D and parathyroid hormone (PTH). Vitamin D is not a vitamin in the strict sense of an essential dietary requirement, as it can also be synthesised photochemically in the skin. In reality, vitamin D is more like a steroid hormone precursor that can be derived from the diet. Its active metabolites function in a similar way to conventional hormones. Vitamin D must be metabolised by the liver to 25-hydroxyvitamin D3, and subsequently by the kidney to the active metabolite 1,25-dihydroxyvitamin D3. Receptors for vitamin D are present in a variety of cell types in the body; the physiological role of this vitamin may be much wider than is currently known. Expression of the receptors is subject to genetic variation within the population and may contribute to the individual differences in risk of developing metabolic bone disease. The combined effects of vitamin D and parathyroid hormone are:
These functions are complex and are demonstrated in Figure 25.4. This area of metabolism is still incompletely understood. There is evidence to suggest that there may be another pathway regulating phosphate transport involving fibroblast growth factor 23. PTH and 1,25-dihydroxyvitamin D3 also have important direct effects on bone: PTH stimulates both bone resorption and formation; 1,25-dihydroxyvitamin D3 promotes bone matrix mineralisation.
Fig. 25.4 Regulation of calcium metabolism. Calcium levels are maintained in the extracellular fluid within a narrow range of concentrations by absorption from the gut and excretion via the kidney with bone acting as a buffering reservoir. PTH raises calcium levels by increasing renal tubular calcium reabsorption, increasing release of calcium from bone matrix through osteoclastic resorption and indirectly increasing calcium absorption from the gut by increasing the metabolism of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D (the active form) in the kidney.
In contrast to PTH, calcitonin, a peptide hormone, appears to lower serum calcium, but usually only when it is pathologically elevated. The stimulus to its secretion is an increase in the serum calcium concentration; it is produced in specialised parafollicular cells (C-cells) of the thyroid. Its exact physiological action is uncertain but it has an inhibitory effect on osteoclasts.
Although vitamin D, PTH and, possibly, calcitonin are the most important factors controlling calcium and phosphate concentrations, and therefore normal bone integrity, several other factors are also involved. Glucocorticoids have a role in the regulation of skeletal growth but prolonged corticosteroid therapy often induces osteoporosis. Thyroid hormone deficiency, as in cretinism, is associated with several skeletal abnormalities. Sex steroids accelerate the closure of epiphyses, and growth hormone has an effect on the development and maturation of cartilage.
Osteoporosis
Osteoporosis is a disease in which there is a reduction in bone mass in the presence of normal mineralisation. It is diagnosed by radiological assessment of bone mineral density (generally measured by dual photon absorptiometry). The usual definition of osteoporosis is a bone mineral density measurement two standard deviations below the mean value for young adults of the same sex. Clinically, osteoporosis may present as a fragility fracture, loss of height, or stooping deformity (kyphosis or ‘dowager’s hump’) due to wedge fractures of the vertebral bodies. Sometimes osteoporosis is diagnosed, when clinically silent, by screening individuals thought to be at risk.
Clinically significant osteoporosis most often results from a combination of age-related bone loss and additional bone loss from another cause; by far the most common such cause is post-menopausal oestrogen withdrawal. Osteoporosis is a clinically silent disease until it is complicated by deformity or fractures.
Pathogenesis
Osteoporosis is caused by a loss of coupling in the bone remodelling process. This can be due to increased bone resorption, decreased bone formation, or both. The loss of coupling results in a net loss of bone volume. In contrast to osteomalacia (see below), mineralisation of bone is normal. Because of its greater metabolic activity, trabecular bone is usually affected more severely than cortical bone. This is particularly the case when increased bone resorption is the main pathogenetic mechanism.
The total bone mass of an individual is influenced by factors such as body build, race, gender, physical activity and general nutrition. Osteoporosis is more common in females than in males and is less common in blacks, who have a greater skeletal mass than whites or Asians. Osteoporosis can be assessed radiologically or by techniques based on the ability of bone to absorb photons released by a gamma-emitting isotope. These demonstrate a progressive loss of bone of 0.75–1% per annum in normal adults of both sexes from as early as 30 years of age. More importantly, there is an accelerated phase of bone loss of up to 1–3% per year in females in the 5–10 years following the menopause.
Localised osteoporosis is inevitable after immobilisation of any part of the skeleton. Even young, healthy males confined to bed after a limb fracture show substantial bone loss. Painful joints in patients with rheumatoid arthritis restrict movement, and osteoporosis often develops in adjacent bones, although this may also be the result of increased bone resorption due to inflammatory mediators produced in affected joints.
Complications
The major complications of osteoporosis are:
The commonest clinical feature of osteoporosis is the progressive loss of height that occurs with age. This is a direct result of compression of vertebrae. Sudden collapse or unequal compression of individual vertebral bodies can cause severe localised back pain and deformities such as kyphosis or scoliosis (Fig. 25.5).
Fig. 25.5 Vertebral osteoporosis. The lower thoracic vertebrae showing small protrusions of the intervertebral disc into the osteoporotic bone (arrowed). The lumbar spine. The vertebral body in the centre has collapsed and has a typical biconcave shape.
Wrist and hip fractures are common in elderly patients with osteoporosis. Although osteoporosis is the major underlying cause, other factors such as an increased tendency to fall and a loss of ‘protective neuromuscular reflexes’ (the ability to fall over safely) are also important. Hip fractures account for numerous hospital admissions and are a major source of disability and a frequent cause of death in the elderly.
Prevention and treatment
Osteoporosis is a major social and economic problem in the elderly, and preventive measures should begin in the middle-aged. Vertebral osteoporosis is reduced in women treated with oestrogens (hormone replacement therapy—HRT). Bisphosphonate drugs, which inhibit bone resorption, are more effective in preventing osteoporotic hip fractures. Regular exercise and an increased dietary intake of calcium also have beneficial effects.
Rickets and osteomalacia
Osteomalacia is characterised by deficient mineralisation of the organic matrix of the skeleton. Rickets is the name given to osteomalacia affecting the growing skeleton of children; it results in characteristic deformities. Causes of osteomalacia, or rickets, include:
Aetiology
In the past, nutritional deficiency of vitamin D was a common cause of rickets in children and, occasionally, of osteomalacia in adults. In most communities, this has been eliminated by improvements in diet and by the addition of vitamin D to foodstuffs. The disease still occurs in some Asian communities in the UK; skin pigmentation impairs photochemical synthesis of vitamin D, and a constituent of chapatti flour interferes with calcium and phosphate absorption in the gut. As dietary rickets is becoming less common in Western countries, an increasing proportion of cases of rickets are due to congenital abnormalities in vitamin D metabolism. Disorders of this type are referred to as ‘vitamin D-resistant rickets’ because vitamin D supplements fail to generate active vitamin D metabolites.
Malabsorption of calcium and phosphate from the intestine is the commonest cause of osteomalacia in adults. The underlying cause is often coeliac disease, but occasional cases result from Crohn’s disease or extensive surgical resection of the small intestine. As the liver and kidney have important roles in the metabolism of vitamin D, renal and hepatic disorders may cause osteomalacia. This is uncommon in liver disease, but a complex pattern of bone disease that includes osteomalacia is seen in renal failure. Occasional patients treated with anticonvulsants, such as phenytoin, develop osteomalacia. These drugs induce liver enzymes that degrade vitamin D to inactive metabolites.
Diagnosis
The characteristic clinical deformities of rickets include:
Inadequate mineralisation of bone reduces its normal strength and allows deformities to develop, for example from pressure on the skull while lying in a cot, or on the limbs as they begin to bear weight. Calcification of epiphyseal cartilage is an essential step in the normal process of ossification in long bone. When the levels of vitamin D metabolites are low, calcification cannot occur and cartilaginous proliferation continues. This accounts for the enlargement of long bones and the ribs at growth plates.
The characteristic pathological feature in adults with osteomalacia is spontaneous incomplete fractures (‘Looser’s zones’), often in the long bones or pelvis. The main symptoms are bone pain and tenderness, and weakness of proximal limb muscles. Serum calcium levels may be reduced and serum alkaline phosphatase is increased (these biochemical abnormalities are usually absent in osteoporosis). A bone biopsy will demonstrate an increase in non-mineralised osteoid (Fig. 25.6).
Fig. 25.6 Osteomalacia. This transiliac crest undecalcified bone biopsy, stained by the Goldner’s trichrome technique, was taken from a patient on renal dialysis suffering from osteomalacia due to a combination of low 1,25-dihydroxyvitamin D levels (a common consequence of renal failure) and aluminium toxicity (an iatrogenic complication of dialysis). Mineralised bone matrix is stained green and unmineralised matrix (osteoid) is stained red. The amount of osteoid present is approximately 20 times greater than normal.
Treatment and prevention
Uncomplicated rickets or osteomalacia will respond promptly to vitamin D treatment. Increased calcium intake may also be required to compensate for the flux of calcium into unmineralised bone matrix that occurs in response to vitamin D treatment. Intramuscular injection can overcome problems associated with malabsorption, and underlying disorders such as coeliac disease should be treated appropriately. A normal balanced diet will prevent rickets or osteomalacia, but many foodstuffs are now artificially supplemented with vitamin D.
Hyperparathyroidism and hypercalcaemia
Persistent elevation of fasting blood calcium, after correction has been made for the serum albumin concentration, is an important indication for further investigation. The major pathological causes are:
By far the commonest causes of hypercalcaemia are primary hyperparathyroidism and hypercalcaemia of malignancy.
In hyperparathyroidism (Ch. 17), increased secretion of parathyroid hormone stimulates calcium absorption in the intestine, reabsorption in the kidney and osteoclastic breakdown of bone.
In primary hyperparathyroidism the usual cause is a parathyroid adenoma or, occasionally, diffuse hyperplasia of the parathyroid glands. In contrast, in secondary hyperparathyroidism, prolonged hypocalcaemia stimulates parathyroid hyperplasia and eventually produces parathyroid enlargement. This is usually the result of renal failure or malabsorption secondary to coeliac disease. In occasional patients, secondary hyperparathyroidism is associated with hypercalcaemia. This has been called tertiary hyperparathyroidism and usually results from inappropriately high secretion of PTH by an adenoma arising in secondary hyperparathyroidism.
When obvious causes, such as malignant disease, sarcoidosis or drug therapy, have been excluded, it must be suspected that an otherwise fit patient with hypercalcaemia has primary hyperparathyroidism (Table 25.1).
Table 25.1 causes of hypercalcaemia
| Cause | Pathophysiology |
|---|---|
| Primary hyperparathyroidism | Abnormal PTH secretion from adenoma, hyperplasia or carcinoma of parathyroid glands |
| Malignant disease |
SarcoidosisProbable secretion of vitamin D metabolites from granulomas
Miscellaneous causes:
The advanced bone pathology associated with hyperparathyroidism is now rare. In the early stages there are subtle radiological changes such as subperiosteal resorption of phalangeal bone (Fig. 25.7) or characteristic changes around the teeth. As the disease progresses, cystic bone lesions may develop—osteitis fibrosa cystica (von Recklinghausen’s disease of bone). These are sometimes referred to as ‘brown tumours’, although they are not neoplasms. The brown appearance is the result of haemorrhage and there is often a marked associated osteoclastic reaction. Because PTH has anabolic as well as catabolic effects in bone, hyperparathyroidism does not usually cause generalised osteoporosis.
Bone disease in renal failure (renal osteodystrophy)
Most patients with chronic renal failure have clinical, radiological or pathological evidence of bone disease. There is no single bone disease that occurs in renal failure; in most patients it is a combination of osteomalacia with a variable degree of hyperparathyroidism. Other features include:
The most important pathophysiological changes in renal bone disease are summarised in Table 25.2. Several mechanisms have been suggested to account for the osteomalacia. In all forms of renal failure there is a decrease in the amount of functional renal tissue, and this may be directly responsible for the inadequate production of active vitamin D metabolites. An increased blood phosphate level (hyperphosphataemia) is frequent in renal failure, and this may directly inhibit enzymes responsible for vitamin D metabolism in the kidneys. In the past, haemodialysis fluids rich in aluminium were associated with aluminium deposition in organs such as brain and bone. In bone, aluminium inhibits the calcification of osteoid and contributes to osteomalacia in renal failure (Ch. 7).
Table 25.2 Mechanisms of renal bone disease (renal osteodystrophy)
| Feature of renal failure | Pathological effect in bone |
|---|---|
| Inadequate renal tissue | Impaired conversion of 25(OH)D3 to 1,25(OH)2D3 → Osteomalacia |
| High serum phosphate |
Prolonged haemodialysisInhibition of calcification of osteoid → OsteomalaciaSteroid therapy (e.g. for chronic glomerulonephritis)Osteoporosis Avascular necrosis of bone
Patients with chronic renal failure may have a low serum calcium. This is partly the result of impaired vitamin D metabolism, as vitamin D metabolites are essential for the proper absorption of calcium from the small intestine. The high serum phosphate also reduces the ionised fraction of plasma calcium. This acts as a stimulus to PTH production, and a degree of hyperparathyroidism is inevitable in severe renal failure. Patients with some forms of glomerulonephritis are treated with steroids and this may induce osteoporosis or, occasionally, areas of bone necrosis. Calcification of the soft tissues, or of blood vessel walls, is a further feature of chronic renal failure, particularly after prolonged haemodialysis. Longstanding disordered bone remodelling due to the combination of secondary hyperparathyroidism and osteomalacia can lead to alternating areas of thickened bone (osteosclerosis) and osteoporosis. This has a characteristic appearance (‘rugger jersey spine’).
OSTEOMYELITIS
Aetiology
Osteomyelitis is the result of a bacterial infection of bone. The typical patient is a young child who presents with pain in a long bone, sometimes with a misleading history of recent trauma. In the majority of cases the lesion develops in the metaphysis, the part of the shaft immediately adjacent to the epiphyseal plate. The rich capillary network and large venous channels in this area may favour the deposition of circulating micro-organisms and their subsequent growth. In children and adolescents, osteomyelitis is usually the result of Staphylococcus aureus bacteraemia, often secondary to a boil or other skin infections. Sometimes, the underlying cause of bacteraemia is not apparent. Osteomyelitis is also increasingly seen in elderly patients.
Before the introduction of antibiotics, tuberculous and even syphilitic osteomyelitis were common. Children with haemoglobinopathies, especially sickle cell disease, have an increased risk of osteomyelitis; unusual organisms, such as Salmonella, are sometimes responsible.
Osteomyelitis is a well-recognised complication of compound fractures, particularly if the wound in the overlying skin is extensive and there are necrotic bone splinters at the fracture ends. Osteomyelitis is not a complication of closed fractures.
Pathogenesis
The classical sequence of changes in osteomyelitis is as follows:
Fig. 25.8 Chronic osteomyelitis. The radius and ulna of an 18th-century sailor. The ‘granular bone’ is the involucrum and the circular defects are ‘cloacae’ through which pus drained. The cut surface of the femur of a 78-year-old male who received a shrapnel wound to his thigh in World War I. The pus drained through a sinus for the next 50 years! A thick bony involucrum surrounds a chronic inflammatory abscess in the marrow cavity.
The development of a sequestrum is due to necrosis of bone caused by compression of blood vessels by the inflammatory process within the Haversian canals of the cortical bone. This event rarely occurs if antibiotic treatment is initiated early in the course of the disease. However, infections in bone can be difficult to eradicate, particularly if foreign material is present, for example following a penetrating injury. Commonly encountered ‘foreign materials’ in bone are joint prostheses, internal fracture fixation devices and other pieces of orthopaedic hardware. Bone infections associated with orthopaedic surgery are more common than primary osteomyelitis in many countries.
Brodie’s abscess is a distinctive clinical form of subacute pyogenic osteomyelitis. The lesion is solitary and, as in typical acute osteomyelitis, localised to the metaphysis.
Clinical features, laboratory investigations and treatment
Most patients with acute osteomyelitis present with localised bone pain and some tissue swelling. A dull continuous back pain, which increases on straining, is typical of vertebral osteomyelitis. The radiological changes are usually characteristic. Blood cultures are positive in some patients, but open biopsy of the lesion may be needed to ensure accurate bacteriological diagnosis. The commonest organisms are Staphylococcus aureus, Mycobacterium tuberculosis, Escherichia coli, pneumococcus or group A streptococcus. Wherever possible a precise bacteriological diagnosis must be made and treatment continued for several weeks.
PAGET’s DISEASE
Incidence and epidemiology
Paget’s disease is a disorder in which there is disorderly bone remodelling. There is considerable variation in its incidence both within and between different countries and racial groups. Despite intensive study, little is known of the cause of Paget’s disease and it is not regularly associated with any other common disorder. Electron microscopic studies have demonstrated probable viral inclusions in the nuclei of osteoclasts, possibly derived from measles or canine distemper virus, but no definite proof has been obtained. Paget’s disease has a distinctive epidemiology. It is most common in western Europe and those parts of the world to which western Europeans have emigrated. For unknown reasons, it is far more common in Lancashire (UK) than anywhere else in the world.
Clinicopathological features
The usual presenting complaints of patients with Paget’s disease are bone pain, deformities or fractures. Although the pelvis and spine are most frequently affected, deformities are most obvious in the long bones such as the tibia, which is characteristically bowed, and in the skull.
Serum calcium concentration is usually normal, but the alkaline phosphatase is markedly elevated, reflecting the osteoblastic activity. The histological changes of Paget’s disease consist of irregular trabecular bone, much of which is woven rather than lamellar, and areas of osteolysis with abnormally large osteoclasts. These changes reflect grossly disordered bone remodelling (Fig. 25.9).
Fig. 25.9 Paget’s disease. In normal trabecular bone (left) viewed by polarised light, note the regular lamellar pattern. In bone affected by Paget’s disease (right) the lamellar pattern is largely replaced by irregular woven bone. Bone of this type is associated with deformity and poor mechanical function.
Complications
The complications of Paget’s disease are:
In many patients, Paget’s disease is completely asymptomatic and is unlikely to be diagnosed unless discovered as an incidental finding on X-ray. The commonest complications are deformities (Fig. 25.10) and bone pain. In some cases, the pain is the result of osteoarthritic degeneration of a related joint. The cause of the pain is uncertain, but interestingly responds well to treatments that inhibit bone resorption. Pagetoid bone is particularly susceptible to fracturing in the initial lytic phase. Enlargement in the sclerotic stage can lead to nerve or spinal cord compression. Deafness is the result of both VIIIth cranial nerve compression and distortion of the middle ear cavity. Occasional patients develop other cranial nerve palsies and, in advanced cases, paraplegia can result.
[/not-level-membership-for-pathology-category]
