Chapter 23 Blood and bone marrow
COMMON CLINICAL PROBLEMS FROM BLOOD AND BONE MARROW DISEASE
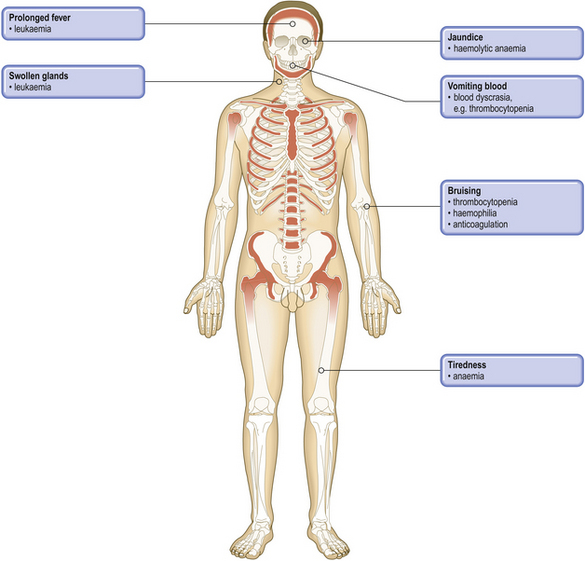
Pathological basis of haematological signs and symptoms
| Sign or symptom | Pathological basis |
|---|---|
| Tiredness, dyspnoea | Reduced oxygen-carrying capacity of blood due to anaemia |
| Mucosal pallor | Anaemia |
| Glossitis (sore mouth, smooth tongue) | Mucosal effects of haematinic deficiency |
| Spoon-shaped nails | Due to iron deficiency |
| Jaundice | Bilirubin accumulation from haemolysis |
| Abnormal tendencyto infections | Neutropenia, e.g. in leukaemia or hypoplastic anaemiaImmune deficiency, e.g. in myeloma, and due to chemotherapy in leukaemia and lymphoma |
| Splenomegaly | Due to expansion of haemopoiesis in myeloproliferative disorders, red cell pooling and destruction in haemolytic anaemias, infiltration in leukaemias and lymphomas |
| Also non-haematological causes, e.g. portal hypertension | |
| Lymphadenopathy | Non-neoplastic causes, e.g. infectious mononucleosisInfiltration with leukaemia or lymphoma |
| Bone pain and fractures | Osteoclast activation in myeloma |
| Purpura, bruising,mucosal or traumaticbleeding | Thrombocytopenia or platelet dysfunction |
| Bruising, muscle and joint bleeding, andtraumatic bleeding | Coagulation factor deficiency |
COMPOSITION, PRODUCTION AND FUNCTIONS OF BLOOD
Blood is a unique organ: it is fluid and comes into contact with almost all other tissues. The blood cells are non-cohesive and supported in the fluid medium of blood—the plasma. The blood cells comprise the non-nucleated erythrocytes and platelets, and the nucleated cells or leukocytes.
In addition to primary disease of the blood-forming organ—the bone marrow—many disease states produce secondary changes in the blood. For this reason, the counting and morphological examination of blood cells is routine in the clinical assessment of disease, frequently providing valuable diagnostic information.
CELLULAR COMPONENTS
The peripheral blood is investigated by microscopy of a droplet spread evenly over the surface of a glass slide—the blood film. Routinely, the blood film is treated with a combination of stains which allow identification of nuclear and cytoplasmic detail (Fig. 23.1).
Quantitation of blood cells is essential; in modern laboratories this is routinely performed by automated cell-counting equipment. The size and concentration of erythrocytes, and the leukocyte and platelet concentrations are measured. Haemoglobin is automatically measured. Also, the proportion of leukocytes of each category—the differential white cell count—is measured from cell size and granule content.
Erythrocytes
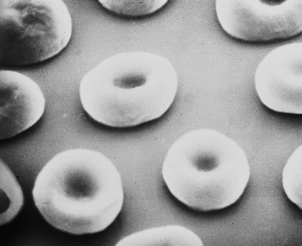
Erythrocytes (red blood cells) are deformable, non-nucleated and biconcave discs (Fig. 23.2). They are the most abundant blood cell. When blood is separated, by centrifugation, into cellular and plasma components, the red cell portion is approximately 45% of the total volume: this is the ‘packed cell volume’ or haematocrit.
The erythrocyte is a special oxygen-carrying cell because it is rich in haemoglobin. The cell membrane is composed of a phospholipid bilayer with integral proteins. The shape of the cell is maintained by structural proteins, such as spectrin, which form a cytoskeleton. Enzyme systems protect the haemoglobin from irreversible oxidation. The mature erythrocyte has no nuclear material, so new protein cannot be synthesised. The mature erythrocyte circulates for around 120 days before it is removed by the reticulo-endothelial system.
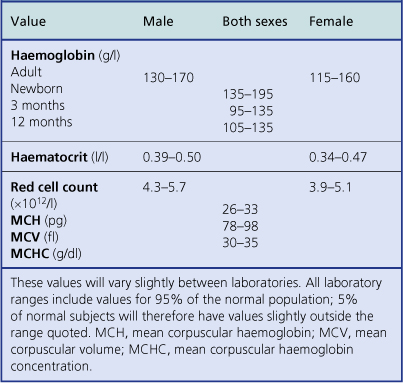
Absolute values
The absolute values are measures of red cell size and haemoglobin content which provide valuable information in the assessment of anaemia, as they provide diagnostic clues as to the likely cause. Absolute values are calculated from the red cell concentration, haemoglobin concentration and haematocrit as follows:

In the modern laboratory, automated cell counters provide these data on each blood sample analysed.
Morphology
The biconcave erythrocyte shape provides a large surface area for oxygen diffusion. By light microscopy erythrocytes appear as uniform round cells with central pallor. Up to 1% of cells stain with a purplish tinge and are of rather greater diameter. These are polychromatic cells; this purple staining is due to the residual ribonucleic acid (RNA) of the immature erythrocyte. These young cells become indistinguishable from the mature red cell population after 48 hours in the blood. When stained with a supravital stain (such as methylene blue) polychromatic cells are more easily identified by the presence of characteristic inclusions; they are then termed reticulocytes. The inclusions are remnants of RNA. When bone marrow production of erythrocytes is increased, the proportion of polychromatic cells, or reticulocytes, in the peripheral blood becomes greater than 1% or 100 × 109/l. This occurs most commonly in recovery from acute haemorrhage or when there is an increased rate of destruction of red cells, which is called haemolytic anaemia. Failure to produce a reticulocyte response to anaemia suggests that the patient has bone marrow failure or haematinic deficiency.
Changes in disease
Anaemia is present when the haemoglobin concentration is less than approximately 130g/l in a male or 115g/l in a female (Table 23.1); the haematocrit is also reduced. Conversely, polycythaemia describes an increased red cell concentration; it is usually accompanied by a raised haemoglobin concentration and haematocrit.
Anaemias may be simply classified according to red cell size (MCV) and haemoglobin content (MCH). This classification is of great diagnostic value in most common types of anaemia (Table 23.2). Further diagnostic information is obtained by the microscopic examination of the red cell morphology on a blood smear. Disease of the blood is frequently associated with increased variation in red cell size—anisocytosis—and the presence of erythrocytes of abnormal shape—poikilocytosis (Fig. 23.3). Increased erythrocyte anisocytosis and poikilocytosis are non-specific abnormalities present in many haematological and systemic disorders. An example is the marked aniso-poikilocytosis that occurs in the absence of a functioning spleen, due to surgical removal or secondary to disease. In this situation there are also inclusions in red cells. They are called Howell–Jolly bodies (Fig. 23.4) and are remnants of nuclear material that would normally be removed when newly formed erythrocytes released from bone marrow circulate for the first time through the spleen.

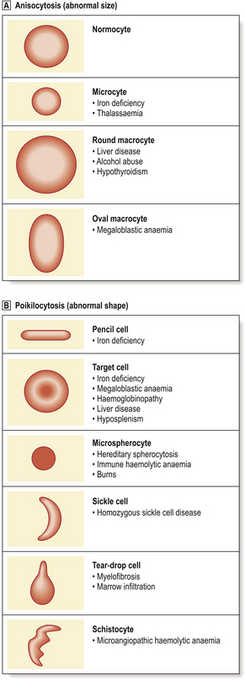
Fig. 23.3 Abnormalities of red cell size and shape.  Increased variation in size: anisocytosis.
Increased variation in size: anisocytosis.  Variation in shape: poikilocytosis.
Variation in shape: poikilocytosis.
In addition to Howell–Jolly bodies, red cells may contain other inclusions (Fig. 23.4) under certain circumstances. The basophilic stippling of the ‘stipple cell’ is due to the presence of residual RNA; stipple cells may be present in several anaemias, especially thalassaemia. Siderotic granules contain iron and may occur in states of iron overload, for example in chronically anaemic subjects who have received treatment by frequent transfusion of red cells. Occasionally, nucleated red cell precursors may escape into the peripheral blood; when these normoblasts are accompanied by immature neutrophil leukocytes the film is described as leukoerythroblastic. A leukoerythroblastic blood film results from gross marrow disturbance such as infiltration by malignancy, or fibrous tissue (myelofibrosis) or in severe anaemia due to deficiency of vitamin B12 or folate (megaloblastic anaemia).
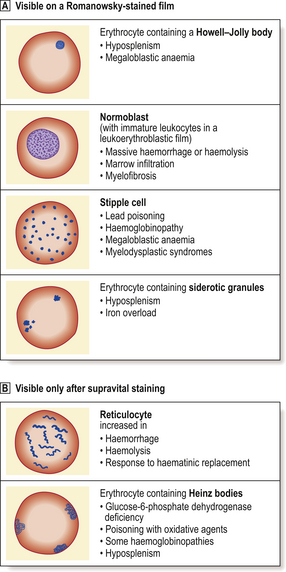
Supravital staining is used to detect the presence of reticulocytes, as described above. This technique also identifies another type of red cell inclusion—Heinz bodies. These inclusions represent denatured haemoglobin and are seen typically in certain haemolytic anaemias, especially those due to a deficiency in the protective enzyme systems such as glucose-6-phosphate dehydrogenase deficiency.
Leukocytes
The nucleated cells of the peripheral blood are termed white blood cells or leukocytes. Their primary role is protection against infection or infestation of the body. Morphologically, on a stained blood film, five varieties of leukocyte are identified.
The normal concentrations of these are:
These are typical values for healthy adults and older children. The normal counts differ in infants, who have a higher proportion of lymphocytes, for example.
Also, it is important to appreciate how such laboratory normal ranges are established in order to avoid misinterpretation. Cell counts are performed on a large number of healthy subjects and the range is determined from the population mean and two standard deviations above and below the mean. This dictates that, for a particular measurement, 2.5% of healthy subjects have a count just below the lower limit of ‘normal’ and a further 2.5% just above.
The granulocytes and monocytes are phagocytic leukocytes produced from precursor cells in the bone marrow. The lymphocytes are broadly composed of B-cells which mediate humoral immunity via the maturation to immunoglobulin-producing plasma cells; they are produced initially in the bone marrow and subsequently mature by antigen selection in the germinal centres of secondary lymphoid tissues; and T-cells which provide cell-mediated immunity such as killing virally infected cells. T-cells are produced and selected for antigen in the thymus gland. B- and T-cells circulate in the blood as small lymphocytes.
Neutrophil granulocytes
Neutrophils are the most numerous leukocytes in the blood of the healthy adult. The nucleus of the neutrophil granulocyte is characteristically segmented into up to five lobes and the nuclear chromatin stains densely (Fig. 23.1). The abundant cytoplasm stains pink and contains characteristic granules. Within the granules are enzymes, including myeloperoxidase, alkaline phosphatase and lysozyme. Neutrophils have a scavenging function and are of particular importance in defence against bacterial infection.
Neutrophil precursors and neutrophils spend 14 days in the bone marrow, whereas the half-life of neutrophils in the blood is only 6–9 hours. Peripheral blood counts therefore measure less than 10% of the total body neutrophils. Within the circulation the cells move between a circulating and a ‘marginating’ pool, margination being attachment to vascular endothelial cells. To perform their scavenging function, granulocytes irreversibly enter the tissues by penetrating endothelial cells modified by inflammatory mediators. Cytokine-stimulated endothelial cells present adhesion molecules which interact with neutrophils and facilitate their passage: one such is ICAM-1 (intercellular adhesion molecule 1).
Lymphocytes
The peripheral blood lymphocytes are small leukocytes with a round or only slightly indented nucleus and scanty sky-blue-staining cytoplasm which may contain an occasional pink- or red-staining granule. Circulating B- and T-cells are not distinguishable by morphology alone. Immunological staining shows that in health approximately 70% of circulating small lymphocytes are T-cells and 30% B-cells.
A small proportion of lymphocytes may be larger with abundant cytoplasm, sometimes referred to as ‘activated’ lymphocytes. These are believed to represent cells that have been stimulated, perhaps by foreign antigen. A more complete description of the classification and role of lymphocytes is to be found in Chapter 9.
Monocytes
Monocytes are the largest blood cells. The nucleus is oval or reniform but not lobed. The abundant cytoplasm stains pale blue and often contains pink granules; vacuoles are often present. The function of monocytes is similar to that of neutrophil granulocytes: they enter the tissues and, as tissue macrophages, are responsible for the phagocytosis and digestion of foreign material and dead tissue.
Eosinophil granulocytes
Eosinophil granulocytes have much larger red-staining granules. They contain enzymes, including a peroxidase. The nucleus is lobulated, but usually only two or three lobes are seen. The eosinophil is important in the mediation of the allergic response and in defence against parasitic infestation.
Basophil granulocytes
Basophil granulocytes are the least frequent leukocytes in normal blood. The granules are large, blue–black and obscure the bilobed nucleus; they contain heparin and histamine. Basophils are closely related to tissue mast cells but their function has not been determined precisely. They appear to be key mediators of immediate hypersensitivity reactions, involving release of histamine.
Changes in disease
Changes may be quantitative or qualitative; the former are more important and often of diagnostic value. Knowledge of the causes of increased numbers of the various leukocytes in the peripheral blood is useful clinically.
Quantitative changes
Leukocytosis means an increase in numbers of circulating white blood cells. Depending on the cause, there may be a polymorphonuclear leukocytosis (neutrophilia—increased neutrophil leukocytes), monocytosis, eosinophil leukocytosis (eosinophilia), basophil leukocytosis (basophilia) or lymphocytosis.
Causes of reactive neutrophil leukocytosis include:
Monocytosis may be reactive to:
Eosinophil leukocytosis may be reactive to:
Lymphocytosis is most commonly associated with an infection such as infectious mononucleosis, tuberculosis, etc.
In some disorders the leukocytosis may be extreme (for example 100 × 109/l), particularly in children. There may also be a tendency for immature leukocytes, particularly myelocytes and metamyelocytes, to appear in the peripheral blood. Severe bacterial infection may result in such an extreme reactive picture, which has in the past been referred to as a ‘leukaemoid reaction’ because of the similarity of the blood picture, with immature forms present, to that of chronic myeloid leukaemia. Occasionally, the lymphocyte series may be involved in such an extreme reactive process, especially during childhood viral infection.
A characteristic leukocytosis composed of ‘atypical’ lymphocytes is a feature of infectious mononucleosis (glandular fever). The infection is common in young adults and often manifests as a sore throat with enlarged lymph nodes and spleen and skin rash. It is due to infection with Epstein–Barr (EB) virus and is common between 15 and 25 years of age in developed countries, but occurs in young children in heavily populated developing countries. The major additional features are:
The atypical cells in peripheral blood are recognisable as lymphocytes but are much larger and have abundant cytoplasm and nuclear irregularities (Fig. 23.5). They are probably reactive T-lymphocytes responding to B-lymphocytes containing the virus, are detectable in blood about 7 days after the onset of illness and may persist for 6 weeks or more. Apparently fortuitously, but usefully, antibodies reactive against horse, sheep and ox red cells (heterophile antibodies) typically develop during the second week and may persist for a few months; they are detected in the Paul–Bunnell test or by more convenient commercial screening slide tests such as the ‘Monospot’ test, and are of diagnostic value. A very similar clinical and haematological (but not serological) picture can develop as a result of other infections, especially with human immunodeficiency virus (HIV), cytomegalovirus and toxoplasma.
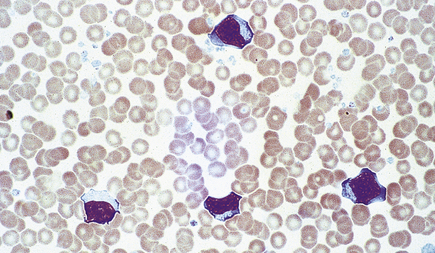
Fig. 23.5 Atypical mononuclear cells in infectious mononucleosis. These large T-lymphocytes have copious basophilic cytoplasm with irregular cell outline.
All of the above are examples of reactive leukocytosis. Increased white cell counts in peripheral blood, often with immature forms present, are also a typical feature of some primary disorders of the bone marrow, especially leukaemias and myeloproliferative disorders.
A reduction in circulating leukocytes is termed leukopenia. Most important is a deficiency of neutrophil granulocytes— neutropenia. Neutropenia is commonly seen in association with a reduction in other blood cells, that is, as part of a pancytopenia. Important causes of pancytopenia are:
Important causes of selective neutropenia are:
In cyclical forms the neutropenia is temporary and recurrent, often with a periodicity of 3–4 weeks. It is an uncommon condition.
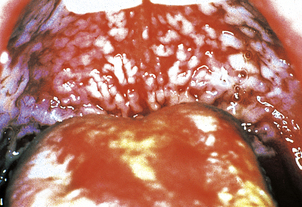
Neutropenia with counts of less than 0.5 × 109/l may result in severe sepsis, especially of the mouth, pharynx (Fig. 23.6) and peri-anal regions, and also in disseminated infection. This clinical picture is now most commonly seen in patients receiving drug or irradiation therapy for malignant disorders.
Qualitative changes
Qualitative leukocyte changes are less important than quantitative abnormalities. Defects of phagocytic cell function resulting in an increased tendency to bacterial infection are recognised, particularly as acquired defects after splenectomy, in leukaemic disorders and due to corticosteroid therapy. Congenital abnormalities of leukocyte function are uncommon. ‘Atypical’ lymphocytes in infectious mononucleosis have been described earlier. Other abnormalities of neutrophil morphology are also recognised (Fig. 23.7).
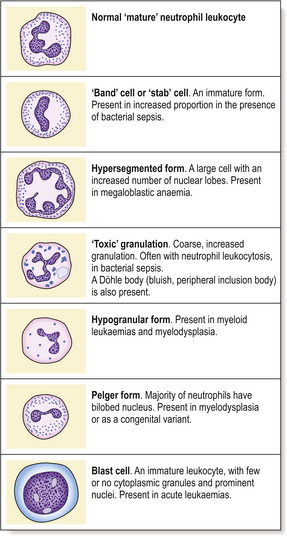
Deficiency of lymphocytes in blood is termed lymphopenia. It is often due to medication with immunosuppressive or cytotoxic drugs, for example. Lymphopenia is an important feature of infection with HIV.
Platelets
On a stained blood film platelets appear as non-nucleated fragments of granular cytoplasm, approximately one-fifth the diameter of erythrocytes and in a concentration of 150–400 × 109/l. Platelets are contractile and adhesive cells, the function of which is the maintenance of vascular integrity. Exposure of vascular subendothelial structures results in rapid adhesion of platelets to the exposed area and aggregation of platelets to each other in the formation of a primary haemostatic plug (Fig. 23.8). Platelets are rich in intracellular granules, which are released during stimulation. The most abundant granules, alpha granules, contain proteins and peptides, including von Willebrand factor, some coagulation factors and growth factors. Platelets deliver these to sites of vascular injury, where they contribute to clot formation and the repair process. Dense bodies are less abundant platelet granules and are rich in calcium, serotonin and adenine nucleotides.
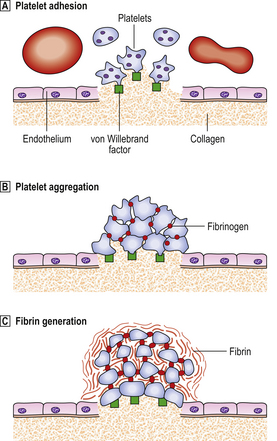
Fig. 23.8 The physiology of primary haemostasis.  Platelet adhesion. Exposure of subendothelial material causes activation of platelets, which change shape and produce pseudopodia, and rapidly adhere to the area via receptor sites which interact with von Willebrand factor (vWF). vWF is a multimeric protein synthesised by endothelial cells and megakaryocytes. It associates with a coagulation factor, factor VIII, in plasma.
Platelet adhesion. Exposure of subendothelial material causes activation of platelets, which change shape and produce pseudopodia, and rapidly adhere to the area via receptor sites which interact with von Willebrand factor (vWF). vWF is a multimeric protein synthesised by endothelial cells and megakaryocytes. It associates with a coagulation factor, factor VIII, in plasma.  Platelet aggregation. Platelets interact with each other via receptor sites which use fibrinogen as an intercellular bridge. Platelets contract and release granules which contain pro-aggregatory substances that promote the aggregation response. These include ADP, 5-hydroxytryptamine, fibrinogen and vWF. Metabolism of arachidonic acid, a fatty acid of the cell membrane, to the prostaglandin-like metabolite thromboxane A2 also promotes aggregation and, in addition, vasoconstriction.
Platelet aggregation. Platelets interact with each other via receptor sites which use fibrinogen as an intercellular bridge. Platelets contract and release granules which contain pro-aggregatory substances that promote the aggregation response. These include ADP, 5-hydroxytryptamine, fibrinogen and vWF. Metabolism of arachidonic acid, a fatty acid of the cell membrane, to the prostaglandin-like metabolite thromboxane A2 also promotes aggregation and, in addition, vasoconstriction.  Fibrin generation. Exposure of tissue factor activates the extrinsic coagulation system. Thrombin generation augments the platelet activation and activated platelets provide phospholipid, which is an essential co-factor at several points in the coagulation cascade.
Fibrin generation. Exposure of tissue factor activates the extrinsic coagulation system. Thrombin generation augments the platelet activation and activated platelets provide phospholipid, which is an essential co-factor at several points in the coagulation cascade.
A deficiency of blood platelets is termed thrombocytopenia, the causes and consequences of which are described on page 670. Thrombocytosis, or increased platelet numbers, may be due to a primary bone marrow problem or may be reactive. Examples of reactive thrombocytosis are seen in:•
Thrombocytosis may also occur in primary disorders of bone marrow—the myeloproliferative diseases and chronic myeloid leukaemia. Morphological platelet abnormalities are of minor importance, although ‘giant’ platelets, with a diameter exceeding that of an erythrocyte, are a feature of the myeloproliferative disorders rather than reactive thrombocytosis. Giant platelets are also a feature of some inherited syndromes, including those associated with mutations of the myosin heavy chain gene.
Blood count and morphology in disease
Changes in the blood are present in a wide range of diseases of other organs. These changes are most commonly reactive or secondary but may be useful in providing a clue to the presence and type of underlying disease, e.g. polymorphonuclear leukocytosis in bacterial sepsis; eosinophilia in some parasitic infections. In other cases the abnormalities of cell number and morphology are due to a primary haematological disorder.
BLOOD PLASMA
Plasma amounts to greater than 50% of blood volume. While changes in the innumerable constituents of plasma are outside the scope of this text, consideration of certain major plasma proteins is necessary for an understanding of the pathology of some blood and systemic disorders. The plasma proteins that are components of the blood coagulation and fibrinolytic systems are considered first.
Blood coagulation
For normal homeostasis, blood must be fluid; however, the capacity to minimise loss of blood through breaches of the vascular system is essential. The rapid plugging of defects in small vessels is the function of platelets (primary haemostasis, Fig. 23.8) but a more permanent and secure seal results from the generation of insoluble fibrillar fibrin from its soluble plasma protein precursor fibrinogen in the process of blood coagulation. Failure of primary haemostasis, due to platelet disorders, or of coagulation due to clotting factor deficiency or presence of a coagulation inhibitor, can each result in life-threatening haemorrhage. In contrast, inappropriate activation of platelets or blood coagulation may result in vascular occlusion, ischaemia and tissue death. A complex system of activators and inhibitors in plasma has therefore evolved in order to allow localised clot formation at sites of injury but to minimise the risk of inappropriate and undesirable clotting, i.e. thrombosis. These are the coagulation and fibrinolytic factors and their inhibitors (Fig. 23.9).
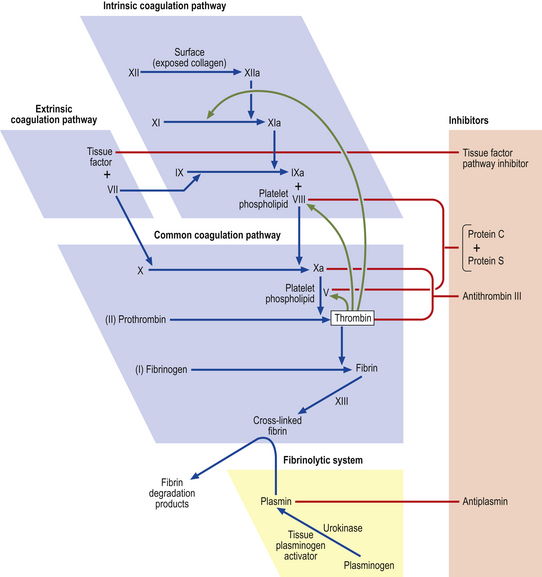
Fig. 23.9 The coagulation and fibrinolytic systems. Activators (blue boxes). These are enzyme precursors or co-factors. The nomenclature uses Roman numerals, the suffix ‘a’ designating the active enzyme. Ca2+ is required at several points in the cascade, as is phospholipid surface provided by activated platelets. Inhibitors. These have an anticoagulant action. Fibrinolytic system. This leads to plasmin generation. Plasmin has its own principal inhibitor, antiplasmin.
Important features of the haemostatic mechanism are as follows.
Coagulation inhibitors limit unwanted clotting and protect against vessel occlusion, especially in veins:
Although the scheme for the initial stages of coagulation activation can be conveniently divided into extrinsic and intrinsic pathways (Fig. 23.9), this is a simplification. Coagulation activation in vivo is initiated through tissue factor, an integral cell membrane protein which is not expressed by vascular endothelial cells in an unstimulated state but is expressed by subendothelial cells and smooth muscle as well as other cells. As soon as blood leaks from a vessel it is exposed to tissue factor. Tissue factor activates factor VII. The much slower pathway for fibrin generation through activation of factor XII on contact with subendothelial components is of minor importance. This partially explains the absence of any increased tendency to haemorrhage in subjects who are congenitally deficient in factor XII. It is the tissue factor–activated factor VII complex that rapidly activates factors X and IX, leading to thrombin generation. When the procoagulant stimulus is sufficiently strong, the degree of amplification through thrombin activation of factors V and VIII overcomes inhibition by activated protein C and fibrin generation proceeds.
The final step in clot formation is the stabilisation of fibrin by cross-linking through the activity of factor XIII.
In the laboratory, the function of the components of the coagulation system can be assessed by the time required for clotting of recalcified plasma prepared from a blood sample anticoagulated with sodium citrate. The citrate binds calcium ions, which are required at several points in the mechanism. Recalcification allows fibrin formation to take place. The two principal screening tests used in clinical practice are:
The pathology and consequences of deficiency of the components of the coagulation and fibrinolytic system are described on page 673.
Rheological considerations
Blood is a viscous fluid and changes in its physical properties accompany some diseases. The major determinant of blood viscosity is the haematocrit.
The plasma fibrinogen concentration is the major determinant of red cell aggregation and is second only to haematocrit as a factor in determination of blood viscosity. Other plasma protein molecules tend to be smaller and more symmetrical than fibrinogen and consequently have a much lesser effect on viscosity. However, when they are present in increased concentrations, blood viscosity may be affected. This may result in a hyperviscosity syndrome, in which there is stasis within the microcirculation and tissue anoxia. Cerebral dysfunction, with headache, visual disturbance and drowsiness progressing to coma may result. Very high plasma immunoglobulin concentration, which is a common feature of the malignant disorders multiple myeloma and macroglobulinaemia, is a common cause of the hyperviscosity syndrome. Numbers of leukocytes and platelets have little influence on blood flow in health. However, when leukocyte counts exceed 300 × 109/l, usually in leukaemia, flow may be adversely affected, resulting in clinical hyperviscosity.
The hyperviscosity syndrome represents an extreme abnormality of blood flow producing organ dysfunction. However, epidemiological studies suggest that even minor increases in blood viscosity, due to increased haematocrit or fibrinogen concentration, may result in a tendency to vascular occlusion, manifesting as an increased incidence of myocardial infarction and cerebral infarction. The concentration of plasma fibrinogen is a risk factor for atherosclerosis and arterial thrombosis that is at least as potent as the level of serum cholesterol. The interplay between rheological and haemostatic changes in thrombotic disease is not yet fully understood.
Erythrocyte sedimentation rate
The erythrocyte sedimentation rate (ESR) measures the rate at which red cells sediment by gravity in plasma in 1 hour and is a widely used laboratory test. Increased aggregation and sedimentation occur in the presence of high concentrations of immunoglobulin and fibrinogen. As the latter is an acute phase reactant, the ESR is increased in a wide variety of inflammatory and neoplastic conditions. It is an entirely non-specific test and a normal value for ESR can never be used to exclude the presence of significant disease. Direct measurement of plasma viscosity provides equally useful data and has replaced ESR measurement in some diagnostic laboratories.
HAEMOPOIESIS AND BLOOD CELL KINETICS
Haemopoiesis is the formation of blood cells.
Sites of haemopoiesis
In the adult, all blood cells are produced in the red marrow, which is restricted to the bones of the axial skeleton—vertebrae, ribs, sternum, skull, sacrum, pelvis and proximal femora. In these regions the bone marrow is composed of approximately 50% fat, within adipocytes, and 50% blood cells and their precursors (Fig. 23.10). The fatty marrow of other bones is capable of haemopoiesis when requirements for blood cells are increased in some diseases.
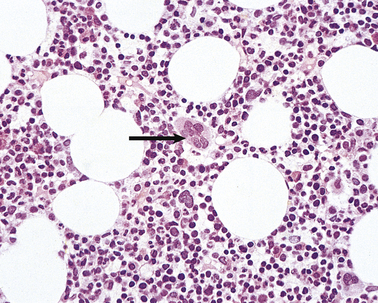
Fig. 23.10 Normal bone marrow. From a section of a bone marrow biopsy from the pelvis. Marrow cells are interspersed between fat spaces. The arrow points to a megakaryocyte.
In the infant and young child, practically all of the bones contain haemopoietically active marrow.
In fetal life, the liver and spleen are the major haemopoietic organs between about 6 weeks and 6–7 months’ gestation; the yolk sac is the main site before 6 weeks. In disease, the liver and spleen can again become haemopoietic organs, even in adult life; this development is referred to as extramedullary haemopoiesis and is particularly associated with the progressive fibrosis of bone marrow seen in the myeloproliferative disorders.
The bone marrow is examined histologically in two ways. Marrow can be aspirated through a needle inserted into a marrow cavity (usually sternum or pelvis), smeared on a slide and stained in a method similar to that for peripheral blood. Further information, particularly on the structure and cellularity of the marrow, can be obtained by preparation of sections of a marrow trephine biopsy: this is a core of tissue obtained using a wide-bore needle (Fig. 23.10).
Additional investigations carried out on bone marrow samples include staining of individual cells with monoclonal antibodies (immunophenotyping and immunohistochemistry) and genetic analysis including karyotyping, fluorescent in situ hybridisation (FISH) and molecular analysis of individual genes.
Haemopoietic stem cells
Studies of bone marrow in culture lead to the conclusion that erythrocytes, leukocytes (including lymphocytes) and platelets are derived from a common, self-replicating precursor cell or ‘pluripotential stem cell’. By a series of cell divisions, cells committed to each line are produced and further divisions result in mature cells—erythrocytes, granular leukocytes, megakaryocytes and T- and B-lymphocytes (Fig. 23.11). The pluripotential stem cells possess the ability to renew in addition to the capacity to differentiate. It is now clear that bone marrow also contains mesenchymal stem cells which can give rise to connective tissues such as fat cells, fibroblasts, bone and cartilage. The development and preferential survival of a malignant clone of haemopoietic cells, derived from mutated bone marrow stem cells, explains the pathological features of the leukaemias and myelodysplastic syndromes.
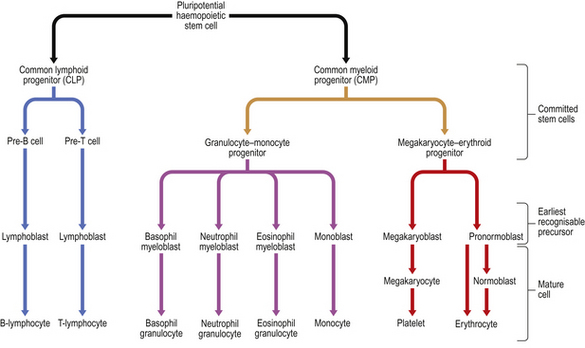
If human bone marrow is infused intravenously into a subject without functioning marrow, as during bone marrow transplantation treatment, normal blood cell production returns after a period of several weeks. This finding confirms the presence of pluripotential stem cells in bone marrow and also indicates that the microenvironment of the bone marrow is central to normal blood production; stem cells do not tend to thrive in other sites, and blood production resumes only in the marrow cavities after marrow infusion. Stem cells can be made to circulate in the peripheral blood. This is most conveniently achieved by the administration of one of the cytokines (most commonly G-CSF) responsible for stimulation of haemopoiesis—the colony stimulating factors. Using an extracorporeal centrifugation technique these cells can be harvested and used as an alternative to bone marrow cells in transplantation therapy—a ‘peripheral blood stem cell transplant’. Peripheral blood is used as a source of haemopoietic stem cells more commonly now than bone marrow because engraftment is faster and procurement of peripheral blood stem cells does not require the donor to have a general anaesthetic.
There is great interest in the possibility that marrow stem cells may be able to differentiate into diverse tissue types, including neuronal, muscle, liver and vascular cells. This is referred to as transdifferentiation or stem cell plasticity. Although definitive proof of this is still lacking, should it be confirmed it offers exciting new possibilities for treatment of common disorders through transfer of stem cells. Such studies are ongoing and to date show some benefit in treating damaged myocardium and improving blood flow to ischaemic limbs.
Erythropoiesis
The pronormoblast is the earliest red cell precursor that can be identified in the bone marrow. It is a large cell with prominent nucleoli within the nucleus. By a series of four cell divisions a fully haemoglobinised, non-nucleated erythrocyte is produced. During differentiation the nucleus becomes increasingly condensed and the cytoplasm contains increasing amounts of haemoglobin and less RNA; the early, intermediate and late normoblasts can be distinguished morphologically through the increasing haemoglobin content and progressive nuclear condensation (Fig. 23.12). The nucleus is eventually extruded, leaving a ‘polychromatic’ erythrocyte which remains in the marrow for a further 48 hours; it then circulates for approximately 48 hours before maturing in the spleen to an erythrocyte.
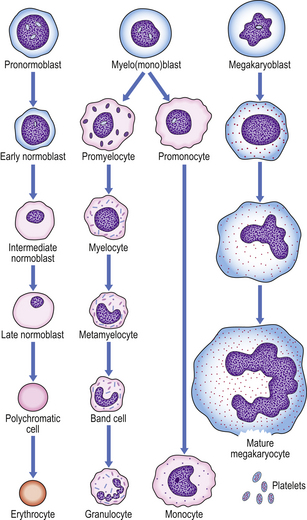
Fig. 23.12 Haemopoiesis. The later morphological stages of erythrocyte, megakaryocyte and myeloid cell development from the committed progenitor cell stage.
Only polychromatic erythrocytes and mature erythrocytes normally circulate. However, nucleated red cell precursors are present in the peripheral blood in some marrow disorders.
Leukopoiesis
The normal bone marrow contains many more myeloid than nucleated erythroid cells (around 5:1). In the granulocyte series these include the myeloblast, promyelocyte and myelocyte, which are capable of cell division, and the metamyelocyte and band cell, which are undergoing maturation without further division (Fig. 23.12). Maturation of granulocyte precursors involves a reduction in cell size, development of cytoplasmic granules, increased condensation of nuclear chromatin, and irregularity of nuclear outline.
Monoblasts cannot easily be distinguished from myeloblasts. They mature in the bone marrow to monocytes via a promonocyte stage. Peripheral blood contains mature granulocytes and monocytes only, in health. However, as previously mentioned, their precursors may enter the circulation in the presence of severe infection or bone marrow disease.
Megakaryocytopoiesis
Megakaryocyte maturation is unique. The earliest identifiable precursor, the megakaryoblast, is a large cell that undergoes nuclear replication without cell division, the cytoplasmic volume increasing as the nuclear material increases, in multiples of 2, up to 32N (Fig. 23.12). Cytoplasmic maturation occurs, often at the 8N stage, and platelets are released. Megakaryocytes are not seen in peripheral blood by routine methods.
Blood cell kinetics
Erythrocytes circulate for an average of 120 days and are then destroyed, predominantly in the bone marrow, but also in liver and spleen. There is no significant storage pool of erythrocytes in humans. In contrast, some 10 times more granulocytes are present in the bone marrow than in the peripheral blood, constituting a storage pool of leukocytes which can be mobilised rapidly in response to some stimuli, such as infection or tissue damage. Granulocytes spend only a few hours in the circulation before they enter the tissues, where they act as phagocytes, surviving for several days under normal circumstances. Monocytes also spend a limited time in the circulation, after which they enter the tissues and become tissue macrophages; they may survive for many months.
Platelets circulate for approximately 10 days. The spleen acts as a reservoir of platelets; some 30% are present in the spleen at any time.
Control of haemopoiesis
Peripheral blood cell counts are normally maintained within close limits. However, the ability of each cell line to respond appropriately to increased requirement is exemplified by the increased red cell production after haemorrhage, the granulocyte leukocytosis in response to sepsis and the enhanced platelet production that results from chronic bleeding.
Erythropoietin is a glycoprotein hormone, produced by peritubular fibroblasts in the kidney, that increases erythropoietic activity. The production of erythropoietin is increased in response to a reduced oxygen tension in the blood reaching the kidney. It results in an increase in the number of cells committed to the erythroid line, reduced maturation time and early release of erythrocytes from the bone marrow. Erythropoietin mediates the physiological response of the bone marrow to anaemia or hypoxia. In pathological states, failure of erythropoietin production is a major contributor to the anaemia of chronic renal failure and this can be corrected by erythropoietin administration; inappropriate erythropoietin production by some renal cysts and tumours results in secondary polycythaemia.
Numerous growth factors have been found to govern production of leukocytes in the bone marrow. They are synthesised mainly by T-lymphocytes, monocytes/macrophages, endothelial cells and fibroblasts of the bone marrow stroma. Examples are interleukins 1, 3 and 6 and the colony stimulating factors. GM-CSF increases stem cell commitment to granulocyte and monocyte production, G-CSF to granulocytes and M-CSF to monocytes. Recombinant forms of some of these cytokines are now in therapeutic use, particularly in cancer chemotherapy, where the duration of drug-induced neutropenia can be limited by cytokine administration.
Thrombopoietin, capable of the stimulation of platelet production, is synthesised principally in the liver. Analogues of thrombopoietin may be of use in stimulating platelet production in bone marrow failure states and as shown recently in immune thrombocytopenic purpura (ITP).
Haemoglobin
Structure, synthesis and metabolism
Some knowledge of haemoglobin structure and metabolism is necessary for an understanding of the pathology of the anaemias. Haemoglobin is the oxygen-carrying pigment. The haem group of haemoglobin is responsible for oxygen carriage and is composed of a protoporphyrin ring structure with an iron atom.
By 1 month of age red cell precursors synthesise predominantly haemoglobin A, composed of four haem groups and four polypeptide (globin) chains (Fig. 23.13), of which two molecular forms are present: alpha and beta chains. Haemoglobin A thus has the structure α2β2. Up to 2.5% of the haemoglobin in adults has delta chains (α2δ2)—haemoglobin A2; and up to 1% of the haemoglobin in adults has gamma chains (α2γ2)—haemoglobin F. Adult blood therefore has predominantly haemoglobin A with some A2 and F (Table 23.3).
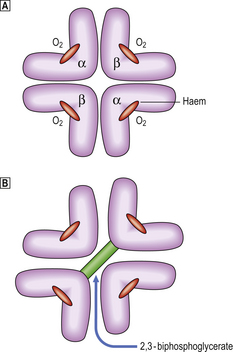
Fig. 23.13 Oxygenated and deoxygenated adult haemoglobin. Uptake and release of oxygen (O2) is associated with movement of the globin chains. On release of O2 the beta chains are moved apart, allowing entry of 2,3-BPG and a reduction in the affinity of the haemoglobin molecule for O2.

In later fetal and early neonatal life haemoglobin F predominates. In early fetal life three other haemoglobins are present: Gower 1, Gower 2 and Portland (Table 23.3).
The whole haemoglobin molecule is thus composed of a tetramer of globin chains, each with a haem group. The complex structure of the molecule is responsible for its oxygen (O2) binding characteristics, the globin chains moving against each other during transfer of O2. The affinity of the haemoglobin molecule for O2 is also controlled by its ability to bind the metabolite 2,3-biphosphoglycerate (2,3-BPG) produced during anaerobic respiration. When 2,3-BPG enters the haemoglobin molecule as the beta chains pull apart during release of O2, the affinity for O2 of the haemoglobin–2,3-BPG complex is reduced, allowing O2 to be given up more readily. Haemoglobin F cannot bind 2,3-BPG and thus has a relatively high O2 affinity, facilitating O2 transfer from maternal blood across the placenta.
At the end of the erythrocyte life-span haemoglobin is metabolised, with conservation of iron and amino acids. Iron is carried by plasma transferrin to the bone marrow and utilised in the synthesis of haem. Globin is degraded to its constituent amino acids, which enter the general pool. Liver, gut and kidneys are all involved in excretion of products of haem breakdown as derivatives of bilirubin.
In the congenital disorders collectively known as haemoglobinopathies, the rate of synthesis of one globin chain type is defective (the thalassaemias) or an abnormal chain is synthesised (the sickle haemoglobinopathies and other haemoglobin variants).
FUNCTIONS OF THE BLOOD
From a consideration of the preceding sections the functions of blood and the major pathological consequences of blood and marrow disease will be apparent.
Oxygen transport is the primary function of the red blood cells. Failure of red cell production, or loss or dysfunction thereof, results in tissue hypoxia affecting the metabolism of all organs.
The cells responsible for host defence against infection are carried, in the blood, from the bone marrow to sites of infection. Infections with bacteria, viruses and fungi are the predictable results of a failure to produce normal leukocytes in adequate numbers.
The primary haemostatic and coagulation mechanisms allow the transport functions of blood to operate without risk of exsanguination from breaches of the vascular compartment. Failure of these leads to spontaneous haemorrhage, whereas a defect in the control mechanisms can result in thrombosis and vascular occlusion.
The diseases that interfere with the function of the blood and their pathological consequences are now described.
ANAEMIAS




Anaemia is present when the haemoglobin level falls below around 130g/l in a male or 115g/l in a female. The different lower limits of normal haemoglobin concentration for neonates, infants and children should be noted (Table 23.1).
The consequences of anaemia are dependent upon the speed of onset. Thus the rapid loss of 10% or more of the circulating blood volume through haemorrhage will result in shock, i.e. the failure of adequate perfusion of all tissues and organs, with consequent hypoxia. In this situation the subject may not initially be anaemic, as both red cells and plasma are lost through haemorrhage. The plasma component is more rapidly replaced, however, and anaemia will be present after several hours have elapsed.
Anaemia that develops more gradually is better tolerated. A haemoglobin concentration as low as 20g/l may be consistent with survival if it develops over a protracted period. The inevitable result of anaemia, however, is a reduction in the oxygen-carrying capacity of the blood and thus chronic tissue hypoxia.
The general consequences of anaemia are due to the tissue hypoxia, which can result in fatty change, especially in the myocardium and liver, and even infarction. Lethargy, increased breathlessness on exertion, and new or worsened ischaemic phenomena are typical clinical features. Breathlessness at rest implies the development of heart failure, a result of severe anaemia. Expansion of the red marrow is present in those anaemias where a marrow response is possible—generally the haemolytic anaemias. Other features are specific to anaemias resulting from a particular mechanism, such as the jaundice of haemolytic anaemias, or are specific to anaemia of a particular type, such as the nail changes of iron deficiency anaemia. Such pathological features are described in the relevant sections.
A low haemoglobin concentration usually reflects a reduction in the body red cell mass. An important exception is pregnancy, when both red cell mass and plasma volume increase, but the latter to a greater degree. This process results in a haemoglobin concentration in blood that is lower than in the non-pregnant state in the presence of a relatively increased red cell mass and overall oxygen-carrying capacity; this condition is often referred to as the physiological ‘anaemia’ of pregnancy. The increased red cell mass during pregnancy is necessary to support the increased metabolic requirement of the mother and fetus. The reason for the expansion of the plasma compartment is obscure, but it may be explained in part by a need for increased skin perfusion for heat loss due to the increased metabolic rate.
Expansion of the plasma volume, resulting in dilutional anaemia, may also occur when the spleen is pathologically enlarged. (The spleen appears to exert a controlling influence on plasma volume.) Other mechanisms also operate in this situation, however, as described under hypersplenism (p. 654).
Classification
Table 23.4 outlines a classification of anaemias. Anaemias are divided into two categories: those where anaemia is due to failure to produce erythrocytes, and those in which erythrocyte loss is increased but production is normal (or usually increased, in response to the anaemia). While useful, this categorisation is an oversimplification, as both mechanisms are present in some anaemias. Thus, in the megaloblastic states, cell production is defective due to lack of vitamin B12 or folic acid for nucleic acid synthesis but, in addition, the erythrocytes that are produced are abnormal and of diminished survival. In thalassaemia, cell production is not optimal due to abnormal haemoglobin synthesis, and there is also increased erythrocyte destruction.
Table 23.4 A classification of anaemias
| Type | Cause |
|---|---|
| Production failure anaemia | |
| Haematinic deficiency | Insufficiency of iron, vitamin B12 or folic acid |
| Anaemia of chronic disorders | Infection, inflammation and neoplasia |
| Dyserythropoiesis | |
Hypoplasia Marrow infiltration
Increased red cell loss, lysis or pooling
Haemolysis due toabnormality outsidethe red cell
Hypersplenism
The myeloid and megakaryocytic lines are also involved in some anaemias due to failure of haemopoiesis (megaloblastic anaemia, hypoplastic anaemia) but not in others (iron deficiency anaemia).
Despite these qualifications, the classification described is useful as an aid to determining the cause of the anaemia.
PRODUCTION FAILURE ANAEMIAS
The most commonly encountered anaemias are in the production failure group.
Haematinic deficiency
Haematinics are dietary factors essential for either haemoglobin synthesis or erythrocyte production.
Iron deficiency





Iron deficiency is the commonest cause of anaemia worldwide. It is also the commonest cause of a microcytic hypochromic blood picture, the others being thalassaemias and (rarely) sideroblastic anaemias.
Iron metabolism
Iron is an essential requirement. It is also one of the commonest elements present in the Earth’s crust. Excessive iron deposited in the tissues is, however, toxic, causing damage to the myocardium, pancreas and liver in particular (Ch. 16). As the body has no active method for iron excretion, iron status is controlled largely by its absorption; the capacity to absorb iron is, however, limited and any tendency to increased loss of iron, due to haemorrhage, is highly likely to result in a negative iron balance and iron deficiency. These factors explain the high prevalence of iron deficiency.
Normally, at least 60% of the body iron is in the haemoglobin of erythroid cells. Approximately 30% is stored within the reticulo-endothelial system (also known as the mononuclear phagocyte system), especially in the bone marrow, as ferritin and haemosiderin. A small proportion of total body iron is present in other tissues, especially muscle, and iron-containing enzymes. This tissue iron is relatively conserved during states of iron deficiency. Only a small fraction of the total body iron is in transport, attached to the carrier protein transferrin.
Ferritin is a protein–iron complex. The protein, apoferritin, is a shell made up of 22 subunits. The core is composed of ferric oxyhydride. Haemosiderin consists of partially degraded ferritin aggregates. Ferritin is present in all tissues, but especially in the macrophages of the bone marrow and spleen and in hepatocytes. A small amount is detectable in plasma and, as it is derived from the storage pool of body iron, its concentration is thus an accurate indicator of body iron stores. Low serum ferritin concentration is a useful confirmatory test for iron deficiency. However, because ferritin is an acute phase response protein, the concentration in plasma is not a reliable guide to body iron stores in the presence of infection, inflammation and neoplasia. In those situations serum ferritin may be normal or high despite tissue iron depletion.
Ferritin is water soluble and not visible by light microscopy; haemosiderin is insoluble and forms yellow granules. When exposed to potassium ferrocyanide (Perls’ stain) the granules are blue–black. Examination of aspirated bone marrow stained with Perls’ stain can therefore be used to assess body iron stores reliably. When iron stores are normal, haemosiderin is visible, mainly in the reticulo-endothelial cells of the bone marrow. In iron overload, most of the iron is in the form of haemosiderin and can be easily identified.
Transferrin is an iron-binding beta-globulin responsible for iron transport and delivery to receptors on immature erythroid cells. Each molecule of transferrin can bind two atoms of iron, but normally the transferrin is only one-third saturated (thus the serum iron concentration is normally one-third of the total serum iron-binding capacity). Transferrin is reutilised after delivering its iron. A low transferrin saturation is therefore diagnostic of iron deficiency while high levels are a feature of iron overload with deposition of iron in tissues.
In order to maintain iron balance, sufficient iron must be absorbed to replace that lost from the urinary and gastrointestinal tracts as shed cells and in sweat, together with any extra requirements.
Thus, requirements vary with circumstances, extra iron being required for growth during childhood, for the fetus and placenta and expansion of maternal red cell mass during pregnancy, and to compensate for menstrual loss of women of child-bearing age.
As a Western diet contains only 10–20 mg of iron per day and only a maximum of one-third of this can be absorbed, excess losses of iron of just a few milligrams will inevitably result in negative iron balance and eventual depletion of iron stores. One millilitre of blood contains 0.5 mg iron. Thus, loss of 10 ml of blood daily will inevitably exceed the capacity to absorb sufficient iron, even from a good diet. This explains the finding of some degree of iron depletion in 25% or more of menstruating women.
Iron absorption takes place in the duodenum and upper jejunum. Haem iron is present in meat and readily absorbed, with little effect from other dietary components. Inorganic iron in vegetables and cereals is mostly trivalent and may be complexed to amino acids and organic acids, from which it must be released and reduced to the divalent state for absorption. HCl produced by the stomach and ascorbic acid in food favour its absorption. In contrast, phosphates and phytates in some foods form precipitates and prevent absorption.
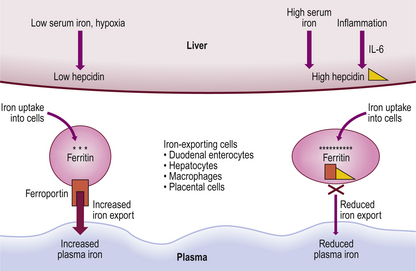
Mechanisms controlling the rate of iron absorption are becoming better understood. Major influences are the total body iron stores and rate of erythropoiesis. Thus, if iron stores are replete a smaller proportion of available iron is absorbed; when erythropoiesis is more active, due to premature red cell destruction for example, extra iron is absorbed even though total stores may be high. This is a feature in thalassaemia, and iron overload may ensue. A major regulator of iron balance is the liver protein hepcidin. Iron loading leads to rapid production of hepcidin by the liver which, in turn, inhibits intestinal iron absorption and movement of iron from stores. This is achieved by hepcidin downregulating the plasma membrane transfer protein ferroportin. Iron is consequently trapped in iron-exporting cells, including duodenal enterocytes. Plasma iron levels subsequently fall. Conversely, low iron levels lead to downregulation of liver hepcidin and increased iron transfer from the gut and iron-exporting cells so that plasma iron levels rise (Fig. 23.14).
Mechanisms of iron deficiency
In developed countries, iron deficiency in the non-menstruating and non-pregnant adult most frequently results from chronic blood loss, often from the gastrointestinal tract. As it is possible to lose several millilitres of blood daily into the gut lumen without marked change in appearance of the stool, such blood loss is frequently occult. Iron deficiency anaemia is thus commonly a presenting feature of lesions within the gastrointestinal tract (Fig. 23.15). In clinical practice, when iron deficiency anaemia occurs in the face of a reasonable diet and no excessive menstrual loss it is mandatory to perform a careful assessment of the gastrointestinal tract.
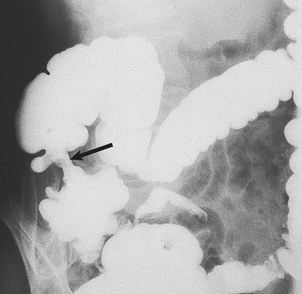
Fig. 23.15 Carcinoma of caecum causing iron deficiency anaemia. An annular carcinoma can be seen as a lesion that causes narrowing of the barium-filled bowel (arrowed) and from which blood loss has occurred.
Causes of iron deficiency are:
More than one factor may operate concurrently. Thus a poor-quality vegetarian diet is highly likely to result in iron deficiency in a menstruating female. In a male or post-menopausal female, failure to ingest or absorb any iron would result in complete depletion of iron stores only after 3 or more years (1 mg/day). Malnutrition or malabsorption is thus rarely the sole cause of iron deficiency, although it may be an important contributory factor.
The microcytic hypochromic anaemia is a late stage in iron deficiency; it does not occur until iron stores are severely depleted. The microcyte results from an extra cell division, in addition to the normal four, during red cell production. Increasing cytoplasmic haemoglobin concentration normally acts as an inhibitor of normoblast division. The failure of haemoglobin synthesis that results from iron deficiency therefore allows extra mitoses to occur, with the production of small erythrocytes. The same mechanism is responsible for the microcytes in thalassaemia, another disorder of haemoglobin synthesis.
Blood and bone marrow changes
The typical blood picture is one of microcytic, hypochromic red cells, with increased anisocytosis and poikilocytosis; elongated ‘pencil’ or ‘cigar’ cells are typically present (Figs 23.3 and 23.16). The proportion of polychromatic cells (or reticulocytes) is low for the degree of anaemia, indicating an inability of the bone marrow to respond due to lack of iron for haemoglobin synthesis. The platelet count is often raised, especially if chronic bleeding is present. The leukocytes are typically normal.
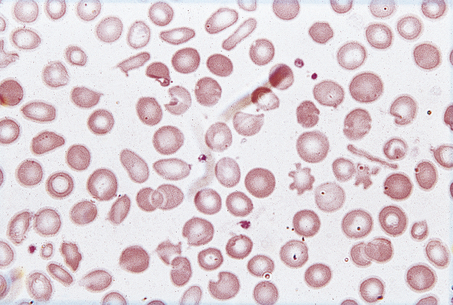
Fig. 23.16 The blood in iron deficiency: microcytic hypochromic anaemia. There is central pallor of the erythrocytes and poikilocytosis with elongated (pencil or cigar) cells.
Occasionally, a mixture of microcytic, hypochromic erythrocytes and macrocytic cells is seen. This is termed a dimorphic picture and occurs in mixed deficiency of iron and folic acid or vitamin B12. The MCV and MCH may be misleadingly normal.
A mixture of microcytic, hypochromic cells and normocytes is present in iron deficiency responding to iron replacement or after transfusion of a subject with iron deficiency anaemia (Fig. 23.17). In the former circumstance, mildly increased polychromasia (and reticulocytosis) may be present.
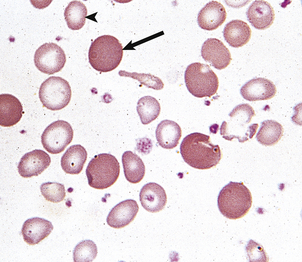
Fig. 23.17 A dimorphic blood film in iron deficiency anaemia responding to oral iron therapy. Hypochromic microcytes (arrowhead) and normocytes (arrow) are present.
Abnormalities are also present in the bone marrow. The nucleated red cell precursors are small in diameter and the cytoplasm is frequently ragged—micronormoblastic erythropoiesis. Staining for haemosiderin (Perls’ stain) reveals its absence from macrophages and normoblasts.
Important biochemical changes in the blood are a fall in serum iron and increase in total iron-binding capacity (representing a compensatory increased transferrin concentration). Saturation of iron-binding capacity is thus reduced to 10% or less, from the normal 33%. The serum ferritin is generally markedly reduced, corresponding to severely depleted body iron content. This situation contrasts with the anaemia associated with chronic inflammatory disease or neoplasia (‘anaemia of chronic disorders’), where red cells are often normocytic but may be mildly microcytic. The serum iron may also be low but this is misleading as iron stores are normal. In contrast to iron deficiency, total iron-binding capacity is usually reduced in anaemia of chronic disorders, and serum ferritin is often raised due to the presence of inflammation or malignancy.
Changes in other organs and tissues
In addition to the manifestations of chronic anaemia, a variety of epithelial changes may be present in chronic iron deficiency:
The cause of these phenomena is unknown. Angular cheilitis (Fig. 23.18), painful fissuring of the mouth corners, is common but not specific: it occurs in dental malocclusion, most often due to poorly fitting dentures. Smooth tongue is also common (Fig. 23.18). Gastric achlorhydria appears to be an occasional result, as well as a contributory cause, of iron deficiency.
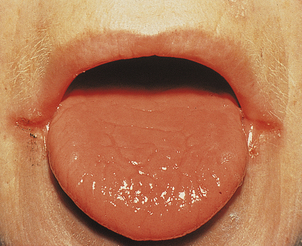
Dysphagia (difficulty in swallowing) due to the presence of a web or fold of mucosa in the post-cricoid region is an uncommon association of iron deficiency. The combination has been termed Paterson–Kelly or Plummer–Vinson syndrome and is important mainly because the mucosal abnormality is premalignant, carcinoma occasionally developing at the site.
Koilonychia (spoon-shaped nails) of chronic tissue iron depletion is typical but only rarely seen.
The pathological changes of iron deficiency are reversed by adequate replacement therapy by the oral route.
Vitamin B12 and folate deficiency






Vitamin B12 and folic acid are essential co-factors for blood cell production. Deficiency of either results in macrocytic anaemia with characteristic pathological appearances in the bone marrow described as megaloblastic haemopoiesis. Megaloblastic anaemias are common, being second in incidence only to iron deficiency and the so-called anaemia of chronic disorders among production failure anaemias. Some other disorders may be associated with macrocytosis (Fig. 23.3) but megaloblastic haemopoiesis is most commonly due to deficiency of vitamin B12 or folate.
Vitamin B12 deficiency
Vitamin B12 metabolism
Vitamin B12 is necessary for DNA synthesis. Deoxyadenosyl-cobalamin is the main form of vitamin B12 in tissues and methylcobalamin is the main form in plasma. These forms differ only in the type of chemical group (deoxyadenosyl or methyl) attached to the cobalt atom which is located at the centre of a corrin ring, to which a nucleotide portion is attached. (The corrin ring is similar to the porphyrin ring of haem.) The vitamin is known to be a coenzyme in the methylation of homocysteine to methionine and also in conversion of methylmalonyl CoA to succinyl CoA. During the former reaction, methylcobalamin loses its methyl group and this is replaced from methyltetrahydrofolic acid, the principal form of folic acid in plasma. The tetrahydrofolic acid is essential for the generation of deoxythymidine monophosphate, a precursor of DNA. Metabolism of vitamin B12 and of folate are thus closely related and essential for nucleic acid production (Fig. 23.19).
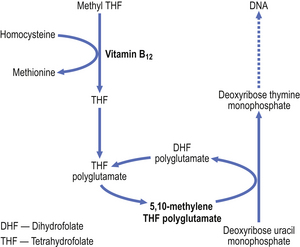
Vitamin B12 is present in foods of animal origin. It cannot be synthesised by higher animals but is produced by micro-organisms. Animals obtain the vitamin from bacterially contaminated foods. Cereals, fruit and vegetable foods contain no vitamin B12 unless they have undergone bacterial contamination. Milk and eggs contain sufficient vitamin B12 for human needs (1–2 mg daily) and thus dietary deficiency can occur only if a strictly vegetarian (vegan) diet is consumed. Nutritional vitamin B12 deficiency (in contrast to dietary folate deficiency) is thus rarely encountered.
Vitamin B12 released from food in the stomach becomes bound to a glycoprotein produced by gastric parietal cells —intrinsic factor. The complex of cobalamin and intrinsic factor binds to receptors on the mucosal cells of the terminal ileum, where vitamin B12 is absorbed and intrinsic factor remains in the lumen of the bowel. In the absence of intrinsic factor, cobalamin cannot be absorbed.
Vitamin B12 is transported to the tissues attached to a plasma-binding protein—transcobalamin II. Another transcobalamin (transcobalamin I), synthesised by neutrophil granulocytes, binds the greater proportion of plasma vitamin B12 but does not liberate it efficiently. The function of transcobalamin I-bound vitamin B12 is unknown.
Body stores of vitamin B12 amount only to some 2–3mg. However, only 1μg daily is required for normal DNA synthesis. Several years must therefore have elapsed before a deficiency state develops, even in the absence of absorption of the vitamin. Twenty micrograms or more per day is available in a mixed diet.
Mechanisms of vitamin B12 deficiency
Causes of vitamin B12 deficiency are:
Addisonian pernicious anaemia accounts for by far the majority of cases of megaloblastic anaemia due to deficiency of vitamin B12. Other causes are uncommon. Some cases occur after gastric resection, usually total gastrectomy. Blind loops of bowel have previously been the result of gastric surgery and this is now a rare cause of vitamin B12 deficiency, with improvements in the medical and surgical management of gastric and duodenal disease.
Addisonian pernicious anaemia is a common disorder in which chronic atrophic gastritis and failure of intrinsic factor synthesis lead to malabsorption of vitamin B12 and, after several years, the development of megaloblastic anaemia. Untreated, this was severe and eventually fatal; the condition was indeed ‘pernicious’ but is now easily corrected by injections of vitamin B12.
Pernicious anaemia is due to an autoimmune process, resulting in atrophy of the chief and parietal glands of the stomach, with consequent failure of acid and intrinsic factor production. An auto-antibody to parietal cells is present in the serum in the majority of cases, but is not specific to this disorder. Antibodies to intrinsic factor are present in 50–70% of cases and much more specific to pernicious anaemia. Absence of anti-intrinsic factor antibodies does not exclude the diagnosis of pernicious anaemia; however, in cases where it is not identified, malabsorption of vitamin B12 can be detected by the Schilling test which measures absorption of an oral dose both in isolation and combined with intrinsic factor. Malabsorption that is corrected by the addition of intrinsic factor is due to pernicious anaemia; malabsorption that is not corrected is due to another cause.
The disease is rather more common in females and rarely presents before 30 years of age, although an uncommon childhood form is occasionally seen. The patient may have another autoimmune disorder such as thyroid disease or vitiligo. There is an association with blue eyes and premature greying of hair.
Blood and bone marrow changes
In contrast to iron deficiency, the defect in DNA synthesis affects all cell lines, and pancytopenia is frequently present. The MCV is high, and oval macrocytes are visible on the blood film (Figs 23.3 and 23.20). In megaloblastic anaemia, a reduction in the number of mitoses during red cell development, due to impaired DNA synthesis with normal RNA and protein synthesis, results in the production of macrocytes. The degree of polychromasia on the blood film is not appropriate to the severity of anaemia, because the marrow is unable to respond.
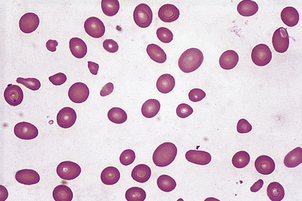
Fig. 23.20 A macrocytic blood film in megaloblastic anaemia. Oval macrocytes and neutrophil nuclear hypersegmentation (Fig. 23.7) are typical.
A proportion of neutrophil leukocytes have exaggerated lobulation of the nucleus and are often large (neutrophil hypersegmentation). Rarely, the blood picture is leukoerythroblastic.
The bone marrow is hypercellular and the stained smears reveal the characteristic megaloblastic change of the developing red cells (Fig. 23.21): the red cells are larger than normal at each stage of development; nuclear chromatin has a very open appearance, with little condensation, and nuclear development lags behind that of the cytoplasm; thus well-haemoglobinised cells with an immature nucleus are a feature. Multilobed polymorphonuclear leukocytes may be seen, as well as particularly large metamyelocytes and band cells. Megakaryocytes may also appear abnormal.
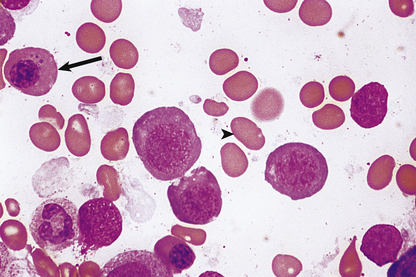
Fig. 23.21 Bone marrow appearances in megaloblastic anaemia from a vitamin B12-deficient patient. The megaloblasts are extremely large red cell precursors and the nucleus has a very open, speckled pattern. Although some of the megaloblasts are well haemoglobinised, the nucleus is still present, suggesting nuclear/cytoplasmic developmental asynchrony (arrow). The more mature non-nucleated red cells are also large and oval in shape—oval macrocytes (arrowhead).
Biochemical abnormalities detectable in the serum include unconjugated hyperbilirubinaemia and increased concentration of lactic dehydrogenase. These changes are due to increased cell breakdown within the marrow, called ineffective erythropoiesis, and the premature removal of macrocytes, in the reticulo-endothelial system. The serum concentration of vitamin B12 is reduced.
Changes in other organs and tissues
Lesions of the nervous system are a frequent feature of vitamin B12 deficiency from any cause. Myelin degeneration of the posterior and lateral columns of the spinal cord is typical and often associated with a peripheral neuropathy affecting sensory neurones. This subacute combined degeneration of the cord causes spasticity, reduced coordination and impaired sensation in the lower limbs and may be present despite normal haemoglobin levels, although the megaloblastic erythropoiesis is always detectable. Conversely, extreme megaloblastic change and profound pancytopenia may be present without evidence of damage to the nervous system from vitamin B12 deficiency. Optic atrophy and cerebral changes resulting in psychiatric disease are less common accompaniments of deficiency of vitamin B12. The cause may be failure of synthesis of S-adenosyl methionine necessary for myelin formation. Deficiency of folate is not generally associated with the neurological features seen in cobalamin deficiency, although psychiatric abnormalities may occur.
Mucosal abnormalities may be present. Atrophic glossitis is a common feature. In pernicious anaemia there is atrophy of the glands of the gastric body affecting chief cells and parietal cells; there is replacement by mucus-secreting goblet cells. The intestinal epithelial cells are often larger than normal, reflecting megaloblastic change akin to that in the bone marrow.
In addition to the above, changes may be present in the heart and elsewhere due to the chronic hypoxia of severe anaemia. Cardiomyopathy is a particularly important feature; transfusion is tolerated badly due to volume overload and may result in fatal cardiac failure.
The clinical features of B12 deficiency are explained by the pathology, although it is unusual for all features to be present together:
Treatment is by parenteral (intramuscular) administration of vitamin B12. Oral replacement is ineffective in pernicious anaemia due to the deficiency of intrinsic factor.
The haematological abnormalities are completely reversed by vitamin B12 replacement; however, the neuropathology and associated clinical features may only be partly corrected. The gastric atrophy and achlorhydria are primary features in pernicious anaemia, not secondary to the deficiency state, and as such do not reverse on treatment of the deficiency. There is a life-long slightly increased risk of carcinoma of the stomach.
The haematological response is manifested by a marked increase in the reticulocyte count 2–3 days after administration of vitamin B12 and maximal at 7 days; the rise is proportional to the severity of the anaemia. White cell and platelet counts recover within several days and haemoglobin increases at about 10 g/l each week, with an accompanying fall in the MCV to normal values. Erythropoiesis is already normoblastic within 48 hours of starting replacement therapy.
Folic acid deficiency
Deficiency of folic (pteroylglutamic) acid, the parent compound of folates, causes a macrocytic anaemia with megaloblastic haemopoiesis identical to that resulting from deficiency of cobalamin.
Folate metabolism
Folates are required for DNA synthesis. Folate poly-glutamates (pteroylglutamic acid with extra glutamic acid residues) are the main intracellular forms. However, all dietary folates are metabolised to the monoglutamate methyltetrahydrofolate during absorption from the gut and are transported in this form. Folates are necessary for single carbon unit transfer reactions in amino acid interconversions, in purine synthesis and, crucially, in the thymidylate synthetase reaction.
Humans cannot synthesise folates de novo. Vegetables and fruits are especially rich in folates as polyglutamate conjugates, but most foods contain some folate. Absorption occurs in the proximal jejunum. Dietary polyglutamates are, however, very sensitive to heat, and cooking can markedly deplete foods of their available folate.
Body stores of folate, mainly in the liver, are modest, amounting to some 10mg. As up to 200μg is required daily, a deficiency state can develop within weeks, in contrast to deficiency of vitamin B12. Furthermore, folate requirements are markedly increased in pregnancy and in some diseases associated with increased cell turnover, such as chronic haemolysis.
Mechanisms of folic acid deficiency
Causes of folate deficiency are:
Whereas malnutrition is an unusual cause of deficiency of vitamin B12, it is the most common mechanism of folate deficiency. It is most prevalent in the elderly. Overcooking of food and lack of fresh foods contribute.
During pregnancy, folate and iron deficiency may occur if no supplements are given. In contrast, vitamin B12 deficiency is almost unknown, as fertility is impaired in vitamin B12 deficiency and the commonest cause, pernicious anaemia, is a disease of late middle age and after.
In some disorders the folate deficiency is likely to be multifactorial, as in malignant disease, where lack of appetite with resultant malnutrition may aggravate folate deficiency secondary to increased utilisation of folate by the malignant tissues.
Phenytoin and phenobarbital used long term as anticonvulsants probably impair folate absorption and may interfere with folate metabolism.
Some anti-cancer drugs act as folic acid antagonists. Methotrexate inhibits the enzyme dihydrofolate reductase, thus depleting tetrahydrofolate. The antimalarial pyrimethamine acts similarly. Trimethoprim acts as a folate inhibitor in bacteria but is ineffective as an inhibitor in humans.
Blood and bone marrow
In folic acid deficiency, blood and bone marrow changes are indistinguishable from those in vitamin B12 deficiency. The concentration of folic acid in serum and erythrocytes (red cell folate) is reduced.
Oral folic acid supplements result in a complete reversal of the pathological features. Even in malabsorption states, sufficient folate can be absorbed from pharmacological doses. The time course of the response is identical to that in vitamin B12 deficiency.
Contrasting features of vitamin B12 and folate deficiency are listed in Table 23.5.
Table 23.5 Comparison of features of vitamin B12 and folic acid deficiency states
| Feature | Cobalamin (vitamin B12) deficiency | Folate deficiency |
|---|---|---|
| Nutritional deficiency | Uncommon | Common |
| Onset | Slow (years) | More rapid (weeks) |
| Revealed by increased demands | Never | Frequently |
| Absorption | In terminal ileum as a complex with intrinsic factor. Gastric and terminal ileal disease (e.g. autoimmune gastritis, Crohn’s disease) may cause deficiency | In jejunum. Jejunal disease (e.g. coeliac disease) may cause deficiency |
| Drug-related | Never | May be due to anticonvulsant therapy. Anti-metabolites induce a similar deficiency |
| Spinal cord and peripheral nerve degeneration | Frequent | None |
Megaloblastic anaemia is the result of deficiency of vitamin B12 or folate in the vast majority of instances. However, other causes include drugs and congenital defects. Prolonged anaesthesia with N2O causes inactivation of vitamin B12 by oxidising the cobalt moiety and has resulted in pancytopenia with megaloblastic erythropoiesis. Cases of congenital deficiency of enzymes involved in cobalamin or folate metabolism or purine or pyrimidine synthesis are extremely rare. However, several anti-neoplastic drugs act by inhibition of synthesis of purine or pyrimidine (hydroxyurea, cytosine arabinoside) and can cause reversible marrow pathology similar to megaloblastic haemopoiesis.
Anaemia of chronic disorders
Anaemia of chronic disorders is one of the most common anaemias. It is found in association with a range of chronic inflammatory diseases, especially connective tissue disorders, chronic infections such as osteomyelitis or tuberculosis, and malignancies such as carcinoma and lymphoma. Anaemia is not severe; the haemoglobin concentration is 80 g/l or greater and the red cells are normocytic and normochromic. A degree of microcytosis and hypochromia may be present, but never to the degree seen in iron deficiency. Bone marrow iron is plentiful. If the underlying chronic disorder remits, the anaemia resolves. It may also respond to pharmacological doses of erythropoietin.
The disorder may represent a cytokine-induced failure of transfer of iron from reticulo-endothelial cells to normoblasts. This is supported by recent evidence showing that the inflammatory cytokine IL-6 stimulates the production of hepcidin from the liver which then inhibits release of iron from iron-exporting cells (Fig. 23.14). It has also been shown that some cytokines, such as tumour necrosis factor (TNF), can induce apoptosis of erythroblasts. Indeed, treatment of rheumatoid disease with anti-TNF leads to improvement in anaemia. This type of anaemia occurs in disorders frequently associated with other types of anaemia, resulting in a complicated picture. For example, in rheumatoid arthritis iron deficiency often accompanies anaemia of chronic disorders due to the gastric irritation and bleeding caused by anti-inflammatory medications. There may be folate deficiency due to poor diet, and hypersplenism and immune haemolytic anaemia may also be present.
Dyserythropoietic anaemias
The term dyserythropoietic anaemias is used to describe disorders where anaemia is at least in part due to production failure, but haematinic deficiency is not present and marrow cellularity is normal or increased. There are prominent morphological abnormalities of bone marrow cells, often affecting erythroid, myeloid and megakaryocytic cell lines. Most of these conditions are classified as myelodysplastic syndromes (MDS) and described in the section on MDS below.
Sideroblastic anaemias
The term sideroblastic anaemias describes a rather diverse and uncommon group of anaemias in which a defect of haem synthesis is present and a characteristic cell is seen in the bone marrow—the ring sideroblast. This cell is a nucleated red cell precursor that has granules of haemosiderin within mitochondria surrounding the nucleus, visible on staining with Perls’ reagent. Causes include:
Although deficiency of vitamin B6 (pyridoxine) causes a similar anaemia in animals, it is never the cause in humans. However, vitamin B6 antagonism can result from antituberculous therapy, and other sideroblastic anaemias occasionally respond partially to pharmacological doses of vitamin B6.
RARS/RCMD-RS are primary acquired sideroblastic anaemias which are classified among the myelodysplastic syndromes. These are neoplastic disorders of bone marrow in which dyserythropoiesis is prominent. They are described on page 663.
Hypoplastic anaemia
Hypoplastic (aplastic) anaemia is pancytopenia (anaemia, neutropenia and thrombocytopenia) resulting from bone marrow hypoplasia of variable severity. Hypoplastic anaemia probably results from failure or suppression of pluripotent stem cells. Very occasionally, the defect appears to affect cells committed to the erythroid series only, when ‘pure red cell aplasia’ results.
The cause is often unknown; however, it is occasionally congenital or due to poisoning, an associated viral infection or to iatrogenic causes. Thus, hypoplastic anaemia may be:
Most cases caused by anti-neoplastic drugs are reversible. Aplasia is also often a feature of the rare disorder paroxysmal nocturnal haemoglobinuria, a clonal disorder of bone marrow.
In idiopathic forms of hypoplastic anaemia there is strong evidence that T-lymphocytes are involved in the suppression of stem cells. As such idiopathic aplastic anaemia is recognised as a form of autoimmune disease. Some 15% of patients have an associated hepatitis as demonstrated by rises in liver transaminases, also thought to be autoimmune. Immunosuppressive therapy with anti-thymocyte globulin (ATG) is successful in idiopathic aplastic anaemia and is the mainstay of treatment for most patients.
The reason for an idiosyncratic response to some drugs is unknown. Chloramphenicol, an antibiotic, and gold, used in treatment of rheumatoid arthritis, are especially likely to produce marrow aplasia, often irreversible. Infection with parvovirus causes a transient suppression of erythropoiesis; this suppression is brief and clinically insignificant in otherwise healthy subjects. However, where red cell survival is markedly shortened, as in sickle cell disease, such infection may cause a catastrophic fall in haemoglobin. Aplastic anaemia is a rare late complication of viral hepatitis.
There is anaemia (normocytic or slightly macrocytic), leukopenia (including lymphopenia in severe cases) and thrombocytopenia. There is reduced polychromasia, especially in relation to the degree of anaemia, and the reticulocyte count is very low. Morphologically abnormal cells are not a feature.
Marrow aspiration often fails. Trephine biopsy reveals increased fat spaces and little residual marrow activity, although a few small clusters of haemopoietic cells occasionally remain.
Clinically, anaemia, infections and bleeding due to thrombocytopenia occur. Splenomegaly and lymphadenopathy are absent. Without successful treatment of the aplasia, severe forms are fatal within months. Spontaneous remission occasionally occurs. Immunosuppression is the mainstay of treatment for idiopathic aplastic anaemia. Bone marrow transplantation can be curative but is generally reserved for children and young adults with severe aplasia.
Anaemia due to bone marrow infiltration
Not infrequently, carcinoma and lymphoma involve the bones and bone marrow (Fig. 23.22). A leukoerythroblastic blood picture may result. In carcinomatosis, numerous other factors are likely to be contributory to the anaemia, such as bleeding from carcinoma of the gastrointestinal tract, folate deficiency and chemotherapy.
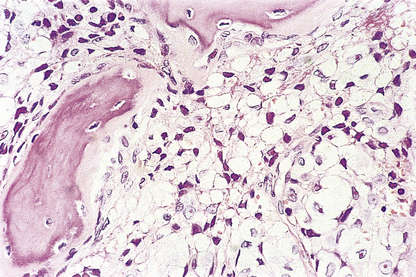
Fig. 23.22 Carcinoma cells infiltrating bone marrow. Sheets of non-haemopoietic carcinoma cells replace normal bone marrow.
In myelofibrosis the marrow is replaced by reticulin and collagen. Fibrosis of the marrow is also a feature of other myeloproliferative disorders and some other malignant marrow infiltrates.
Other causes of marrow infiltration are very uncommon, e.g. Gaucher’s disease, a metabolic defect where glucocerebroside accumulates in the reticulo-endothelial cells of many organs (Ch. 7).
ANAEMIAS DUE TO INCREASED CELL LOSS, LYSIS OR POOLING
The haemolytic states are the main members of the group of anaemias due to increased cell loss, lysis or pooling. However, anaemia due to acute blood loss and the pancytopenia of hypersplenism are also conveniently included.
A fall in haemoglobin of much greater than 10g/l per week must indicate the presence of haemorrhage or haemolysis, as complete cessation of erythropoiesis would result in a rate of fall of no more than 10g/l per week. An exception is the rapid fall in haematocrit due to infusion of cell-free fluids in a dehydrated subject.
Acute blood loss anaemia
Chronic haemorrhage, usually gastrointestinal, causes anaemia by depletion of iron stores. Acute blood loss may result initially in a state of cardiovascular collapse, as described in an earlier section. Following adjustment to the plasma volume over a period up to 48 hours, anaemia will be apparent. The blood picture is normocytic and normochromic, and an increased number of polychromatic erythrocytes and reticulocytes in the days following a brisk haemorrhage reflects increased haemopoiesis. Transient leukocytosis and thrombocytosis commonly occur.
Haemolytic anaemias

 Classified into hereditary red cell disorders and acquired haemolytic states due to a defect outwith the red cell
Classified into hereditary red cell disorders and acquired haemolytic states due to a defect outwith the red cell Important hereditary haemolytic disorders include sickle cell disease, thalassaemias and spherocytosis
Important hereditary haemolytic disorders include sickle cell disease, thalassaemias and spherocytosis


The haemolytic anaemias are those in which a major feature is a reduction in red cell life-span. In severe haemolysis red cell survival may be reduced from the normal 120 days to less than 1 day. Although erythropoiesis will increase, anaemia is inevitable under such circumstances. Even in the presence of normal marrow function and adequate supplies of haematinics, the maximum potential increase in red cell production is some six times the normal rate. In the presence of a defect of red cell production, as in folate deficiency or thalassaemia major, the severity of the anaemia is increased in relation to the degree of shortening of red cell survival.
Classification and incidence
Haemolytic anaemias can be divided usefully into those due to a defect of the red cell itself and those due to an abnormality outside the red cell (Table 23.4). Almost all of the former are hereditary; an exception is the uncommon acquired disease paroxysmal nocturnal haemoglobinuria (PNH). Those due to mechanisms ‘outside’ the red cell are acquired disorders.
The relative incidence of haemolytic anaemias is highly variable geographically. In the UK the acquired haemolytic states, especially autoimmune haemolytic anaemias, are relatively common disorders. Worldwide, however, thalassaemia, sickle cell disease and malaria are of major importance.
Consequences of haemolysis
In addition to the particular pathological and clinical features of the various haemolytic diseases, certain consequences of the haemolytic process and the response to it are common to all types of haemolytic disorder. These consequences are:
Red cell destruction occurs predominantly in the reticulo-endothelial tissues of the spleen and liver. Splenomegaly is therefore common in chronic haemolytic anaemia, and hepatomegaly may also be present. Within the spleen there is congestion within the cords and deposition of haemosiderin.
Less commonly, the red cells are destroyed within the circulation. Examples are haemolytic red cell antibody in major blood group mismatch, the presence of a foreign surface such as a (malfunctioning) artificial heart valve, malaria and glucose-6-phosphate dehydrogenase deficiency. Particular features of intravascular haemolysis are the presence of free haemoglobin in plasma and urine (haemoglobinaemia, haemoglobinuria), of methaemalbumin in plasma (oxidised haem bound to albumin) and of haemosiderin in urine (in shed renal tubular cells that have reabsorbed haemoglobin from the tubular contents; the haem is incorporated into haemosiderin).
Haemolytic anaemia due to red cell defects
The major components of the erythrocyte are haemoglobin, enzymes involved in protection of haemoglobin from oxidant stress, and the plasma membrane. Abnormalities of each of these components can be a cause of chronic haemolytic anaemia.
Defects of the red cell membrane
Hereditary spherocytosis and hereditary elliptocytosis include several disorders in which diminished red cell survival is due to a defect in one of the structural proteins of the erythrocyte membrane such as spectrin. Inheritance is dominant. Spherocytosis is the most common cause of hereditary haemolytic anaemia among Caucasians in the UK. Spherocytes are not confined to hereditary spherocytosis however; they are also present in the blood film in immune haemolytic anaemia (p. 639).
In hereditary spherocytosis, biconcave erythrocytes are released from the marrow but they rapidly lose membrane and therefore assume a spherical shape. Spherocytes are of reduced deformability, which impedes their traverse through the splenic microcirculation. The cells are retained for long periods in the splenic cords. They become metabolically stressed by glucose lack and acidosis, and are eventually prematurely phagocytosed. The abnormal red cells in these disorders are more sensitive than normal to lysis under osmotic stress. This increased osmotic fragility is of diagnostic value.
Anaemia is usual but varies in severity between affected kindreds. The blood film has many spherocytes (Fig. 23.23); they appear smaller than normocytes and denser, with loss of the central pallor. Polychromatic cells are increased. General features of chronic haemolysis are also present. Haemolysis tends to be less severe in elliptocytosis.
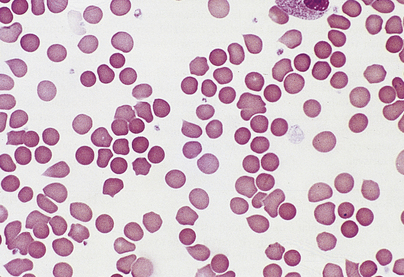
The clinical features are variable and are those of chronic extravascular haemolysis. Pigment gallstones commonly develop. The disorder can be subclinical. Occasionally, transient red cell aplasia secondary to parvovirus infection can develop, when several family members may be affected by aplasia simultaneously.
Removal of the spleen results in resolution of the anaemia, confirming the role of the spleen in the haemolytic process. Splenectomy is reserved for cases in which symptoms of anaemia are intolerable.
Defects of red cell enzymes
Defects of red cell enzymes render the erythrocyte susceptible to damage by oxidant compounds. The generation of reduced glutathione by the metabolic activity of the red cell normally inactivates oxidants. Reduced glutathione is generated by the hexose monophosphate shunt of the Embden–Meyerhof glycolytic pathway, which is the source of: energy, as ATP, necessary for maintenance of red cell shape, volume and flexibility; NADH for reduction of oxidised haemoglobin; and 2,3-biphosphoglycerate (2,3-BPG) for the regulation of the oxygen affinity of haemoglobin.
Deficiency of several of the enzymes involved in these reactions has been identified. Only two are of pathological and major clinical significance: glucose-6-phosphate dehydrogenase deficiency and pyruvate kinase deficiency.
Glucose-6-phosphate dehydrogenase deficiency
Deficiency or defect of glucose-6-phosphate dehydrogenase (G6PD) results in impaired reduction of glutathione. Reduced glutathione protects haemoglobin and red cell membrane from oxidative damage. Inherited G6PD deficiency is an uncommon cause of anaemia in the UK but is among the most common genetic disorders worldwide. It is a sex-linked disorder: female heterozygotes are usually asymptomatic and may have some protection from falciparum malaria; this probably explains the high prevalence of the disorder in many parts of the world.
The common isoenzymes are traditionally designated ‘type B’, the most common, ‘type A’ and ‘type A-minus’, found among American blacks (30% and 11% respectively). Type A differs from type B by a single amino acid substitution and is functionally normal. Type A-minus has an additional amino acid substitution resulting in decreased red cell enzyme activity and disease. Typically there is a tendency to the development of an acute haemolytic episode associated with the ingestion of an oxidant drug (for example some antimalarials and antibiotics) and with other stresses such as surgery or infection. Clinically, a self-limiting episode of anaemia and jaundice develops. Treatment centres on avoidance of exposure to known oxidant drugs.
A further variant is found in Mediterranean populations and is associated with the acute haemolytic tendency known as favism, where ingestion of the fava (broad) bean results in acute haemolysis. The responsible oxidant compound has not yet been identified. Again, oxidant drugs, surgical stress and infections may also lead to haemolysis.
Many other less common genetic variants have been recognised. Some result in a more chronic haemolytic state or neonatal jaundice due to haemolysis.
The blood picture during haemolytic crisis includes increased poikilocytosis with contracted red cells, ‘bite’ cells and ‘blister’ cells (poikilocytes with bite-shaped defects or surface blebs). Oxidised, denatured haemoglobin is seen as red cell inclusions (Heinz bodies) attached to the cell membrane, when blood is stained supravitally as in the reticulocyte preparation. Haemolysis is generally self-limiting because of the rapid outpouring of new red cells, with higher G6PD content, from the marrow in response to the falling haemoglobin. The blood picture is normal between haemolytic episodes.
Treatment consists essentially of avoidance of known precipitating factors for haemolysis. Health is generally good between haemolytic episodes.
Pyruvate kinase deficiency
Pyruvate kinase (PK) deficiency is an autosomal recessive disorder which results in congenital chronic haemolytic anaemia. The blood film has increased poikilocytosis. The chronic anaemia is associated with increased erythrocyte 2,3-BPG because of the site of the metabolic block. This situation results in reduced oxygen affinity of haemoglobin and increased oxygen delivery to the tissues; the anaemia is thus less symptomatic than would be expected from its severity. No specific treatment is available.
Haemoglobinopathies (abnormal haemoglobins)
Abnormal haemoglobins are caused by a single point mutation in the genetic code resulting in an amino acid substitution in the alpha or beta globin chain of haemoglobin
A. Variant haemoglobins can be readily identified by their electrophoretic mobility (Fig. 23.24). Several hundred variant haemoglobins have been identified but few are clinically significant and almost all of those involve beta chain substitutions. Depending on the site of the substitution, four main types of functional defect result:
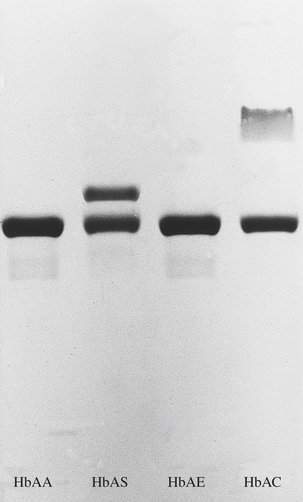
Fig. 23.24 An example of haemoglobin electrophoresis: acid haemoglobin electrophoresis (pH 6.0). This procedure clearly separates haemoglobins S and C from A. The carrier for haemoglobin E cannot be distinguished from AA but would be by performing the procedure at alkaline pH.
The first defect is the most common. HbS is very common worldwide, as are three related haemoglobins: C, D and E.
Sickle cell disease



Substitution of valine for glutamic acid in position 6 in the beta chain of globin results in a haemoglobin (HbS) that undergoes aggregation and polymerisation at low oxygen tensions. In the homozygote for sickle cell disease, where the majority of the haemoglobin content of the erythrocytes is HbS, this results in distortion of the red cells, which acquire a sickle shape. The consequence of this distortion and the predominant features of sickle cell disease are a chronic haemolytic anaemia and microvascular occlusion, causing ischaemic tissue damage. The results of the latter dominate the clinical picture.
The gene for HbS is common in the West and Central African populations, the Mediterranean, Middle East and some parts of the Indian subcontinent. Carriage of the gene may confer some protection against falciparum malaria.
The gene is carried by 8% of black Americans and 30% of black Africans. The heterozygous state, or sickle cell trait, results in less than 40% HbS, the remainder being mostly normal HbA. Two major bands are therefore present on electrophoresis of haemoglobin: one corresponding to HbS and one to HbA. The carrier is clinically and haematologically essentially normal, sickling occurring only very uncommonly and only under conditions of severe hypoxia. Haematuria is an occasional feature, due to renal papillary necrosis from focal sickling in the renal medulla. Hypoxic sickling in heterozygotes is an avoidable risk of general anaesthesia.
In the homozygote the haemoglobin concentration is low (70–90g/l). Sickle cells and target cells are present on the blood film, as are features of hyposplenism in the adult (Fig. 23.25). (Splenomegaly due to chronic haemolysis is present during childhood but the spleen shrinks progressively due to microvascular occlusion and infarction.) Even in infancy children with sickle cell disease are functionally hyposplenic with an increased risk of capsulate organism infection. The bone marrow is hyperplastic with erythroid hyperplasia. Extramedullary erythropoiesis in the liver and, occasionally, other sites is a minor feature. Pathological changes in other organs result from the effects of local ischaemia (Fig. 23.26). Haemoglobin electrophoresis reveals a characteristic single band of HbS.
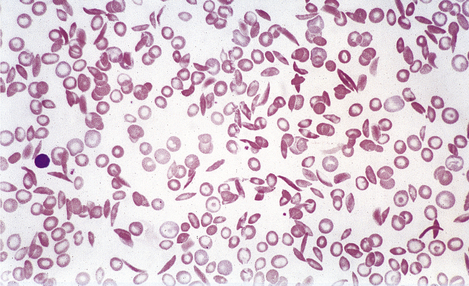
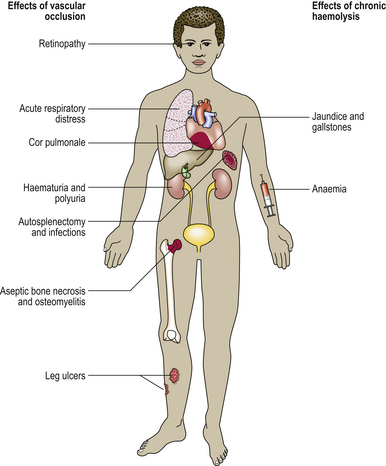
Clinical features. These are predictable from the above. There is anaemia and jaundice from infancy. Sickle ‘crises’ of various clinical types occur from an early age. Vascular occlusion with resultant ischaemia causes severe pain, often in the long bones, abdomen or chest. Ischaemic stroke is common. There is convincing evidence that transcranial arterial Doppler ultrasonography can identify children at increased risk of stroke manifested by increased Doppler flow rates. Introduction of an exchange red cell transfusion programme can reduce this risk. Acute sequestration of sickle cells in the liver or (in children) spleen may cause pain and acute exacerbation of anaemia. Between episodes of crisis, health may be good. Cholecystitis, due to the presence of pigment stones, is a frequent occurrence. As in pyruvate kinase deficiency, oxygen affinity of the haemoglobin is low and symptoms of anaemia mild, due to the relatively enhanced O2 delivery to tissues. Premature death, often from respiratory complications, may occur in early middle age, but longer survival is a feature in some populations.
Treatment. This is essentially conservative, with avoidance of factors known to precipitate crises, especially hypoxia, and provision of warmth and rehydration during crises. Pregnancy may be complicated by an increased tendency to acute sickle crises. Use of vasoconstrictor drugs is contraindicated—this is an issue when local anaesthetic containing adrenaline (epinephrine) is being considered. Exchange transfusion of red cells to reduce the proportion of cells capable of sickling is a useful treatment in severe exacerbations such as acute chest syndrome where lung tissue is often sequestered following the development of infection. In sickle cell disease, a relatively high proportion of HbF in erythrocytes is associated with a less severe clinical course. Hydroxyurea is a drug used in chemotherapy of some malignancies that increases HbF. It reduces the tendency to acute sickle crises in some patients with sickle cell disease.
Haemoglobin C, D and E. These are also the result of point mutations in the beta chain gene. In the homozygous state they produce mild chronic haemolysis with splenomegaly but without the occlusive manifestations of sickle cell disease. They are commonly found in West Africa, India and South-East Asia respectively. Due to their geographical distribution, the gene for HbC is often inherited with that for HbS. HbS-C disease behaves as a mild sickle disease with a particular tendency to venous thrombosis.
Thalassaemias





In contrast to the abnormal haemoglobin states described above, where a structurally abnormal globin is synthesised but at a normal rate, in thalassaemia the globin chains are of normal composition, but the rate at which one of the globin chains (alpha or beta) is synthesised is reduced. In alpha-thalassaemia the alpha globin chain synthesis is so affected; in beta-thalassaemia the beta chain is affected. Accumulation of an excess of the unaffected globin chains results in damage to the developing and mature erythrocytes.
Again, in contrast to ‘variant haemoglobin’ conditions (such as sickle cell disease) where point mutations affecting coding regions underlie the disorders, in thalassaemias the genetic lesions are of a regulatory nature, affecting the normal expression of the globin structural genes.
Each chromosome 16 has a pair of alpha globin genes, thus each cell has four genes coding for alpha globin, all of them functional. The genes for beta globin, as well as those for gamma and delta, are located in close linkage on chromosome 11.
In the alpha-thalassaemia syndromes there is deletion of all four genes or of three of the four. In alpha-thalassaemia trait, there is deletion of two or only one gene (Table 23.6).
Table 23.6 The alpha-thalassaemia disorders
| Number of globin genes deleted | Syndrome | Clinicopathological features |
|---|---|---|
| 4 | Hydrops fetalis | Death in utero. Congestive cardiac failure secondary to an extreme degree of anaemia. Free gamma chains form tetramers (γ4, Hb Bart’s) |
| 3 | Haemoglobin H disease | Free beta chains form tetramers (HbH, β4). Moderate microcytic, hypochromic anaemia. HbH inclusions visible in erythrocytes on supravital staining. Splenomegaly |
| 2 | Alpha-thalassaemia trait | Normal haemoglobin concentration. Low MCV and MCH. Occasional HbH inclusion visible. A subclinical disorder, resembling beta-thalassaemia minor |
| 1 | Alpha-thalassaemia trait | Normal haematology or slightly reduced MCV |
More than 200 genetic defects responsible for beta-thalassaemia have now been described, predominantly point mutations, in contrast to the deletions that cause much alpha-thalassaemia. The type of defect tends to vary between racial groups. Some defects result in an absence of chain synthesis (β0); in others, chain synthesis is severely restricted but present (β+).
Alpha-thalassaemia
This is an uncommon cause of anaemia in the UK. Red cells are microcytic. Haemoglobin H disease (Table 23.6) is seen mainly in Asian populations. HbH is identifiable on electrophoresis in the 3-gene deletion disorder and HbH inclusion bodies are visible in red cells stained supravitally (as in the reticulocyte preparation) in the 3- and 2-gene deletion forms. Electrophoresis is normal in alpha-thalassaemia trait and the conditions can be confirmed only by direct measurement of rate of synthesis of alpha and beta chains.
Beta-thalassaemia major (Mediterranean or Cooley’s anaemia)
This is a severe disorder due to the inheritance of two genes for beta-thalassaemia—β+/β+, β0/β0 or occasionally β+/β0. The beta-thalassaemia genes are most frequent in Mediterranean countries, the Middle East and parts of Africa and South-East Asia.
The blood picture is that of a severe microcytic, hypochromic anaemia (haemoglobin concentration 30–60g/l) developing from 3 to 6 months of age, when beta-chain production would have normally replaced the great majority of gamma chain production, leading to the dominance of Hb A(α2β2) and only 1% residual fetal haemoglobin (α2γ2).
In response to the defective haemoglobin synthesis and haemolysis the red bone marrow is dramatically expanded with gross erythroid hyperplasia. As a result, cortical bone is thinned and new bone deposits on the outer aspect, especially in the skull vault, maxilla and frontal facial bones (Fig. 23.27). Cortical thinning and fractures may develop in the long bones, vertebrae and ribs. The spleen is grossly enlarged, with expansion of the reticulo-endothelial elements and extramedullary erythropoiesis. The liver is similarly affected. Iron overload is apparent and often gross.
Fig. 23.27 X-ray appearances of the skull in beta-thalassaemia major.  Characteristic ‘hair-on-end’ appearances of the skull vault due to marrow expansion.
Characteristic ‘hair-on-end’ appearances of the skull vault due to marrow expansion.  Normal subject.
Normal subject.
Haemoglobin electrophoresis reveals absent or markedly reduced haemoglobin A. Small (normal) amounts of haemoglobin A2 are present and the remainder of the haemoglobin is F.
Predictably, the clinical features are those of severe anaemia, including growth retardation, and iron overload secondary to red cell transfusion and a tendency to absorb excess iron as a result of the dyserythropoietic state. Secondary iron overload may result in failure of sexual development due to iron deposition in the pituitary and endocrine organs, as well as diabetes mellitus, liver and heart failure. Facial deformities result from the bone changes. Iron chelation therapy to reduce tissue iron is a crucial part of management and has been shown to prolong life when used in trials in thalassaemia major patients. Commonly, it is introduced once serum ferritin has reached 1000mg (approximately 20–25 units of red cells transfused). Until recently iron chelation could only be efficiently delivered by near continuous subcutaneous infusion of the iron-chelating drug desferrioxamine. However, there are now two orally administered iron chelators available:deferiprone and deferasirox. Bone marrow transplantation has been curative.
Beta-thalassaemia minor
This is mild and most commonly subclinical. The characteristic pathology is gross microcytic and hypochromic change on the blood film with normal or slightly raised red cell count and normal haemoglobin concentration. Mild anaemia may be present during pregnancy, when the condition is often first diagnosed, as it is often the first occasion when the healthy carrier undergoes a routine blood count. The blood picture is very similar to that of iron deficiency but the MCV and MCH are disproportionately low for the level of haemoglobin. Iron stores are normal or high. Bone changes and hepatosplenomegaly are absent. Haemoglobin electrophoresis reveals a raised haemoglobin A2 concentration (>2.5%). Diagnosis is important in order that genetic counselling can be offered.
Thalassaemia intermedia
The term thalassaemia intermedia describes disease of intermediate severity, often not requiring transfusion and compatible with prolonged survival. Hepatosplenomegaly and iron overload are present. It is genetically heterogeneous, some cases being severely affected heterozygotes, others homozygotes with an unusually mild beta-chain deficiency.
Occasional patients are doubly heterozygous for beta-thalassaemia and HbS. These patients have a variant of sickle cell disease, often with prominent and persisting splenomegaly.
Paroxysmal nocturnal haemoglobinuria
Paroxysmal nocturnal haemoglobinuria is an acquired disorder in which chronic haemolysis is due to a clonal abnormality of erythrocytes that renders them abnormally sensitive to complement lysis. Due to an acquired mutation of the PIG-A gene within a stem cell clone, blood cells lack an enzyme required for the synthesis of a phosphatidyl inositol which anchors several proteins to the red cell membrane, including some responsible for complement degradation. Leukocytes and platelets are also affected. The condition is rare and often chronic. Aplastic anaemia, chronic haemolytic anaemia and venous thrombosis in the portal, hepatic or cerebral veins are major features. Haemoglobinuria occurring at night or in early morning is not a common feature, despite the name (nocturnal) of the disorder. It seems to result from a decrease in plasma pH during the night which promotes complement activation.
The presence of haemosiderinuria and tendency of erythrocytes to lyse at low pH, which activates complement (acid lysis or Ham’s test), are useful diagnostically. More commonly nowadays flow cytometry is used to demonstrate the missing phosphatidyl inositol-anchored proteins on cell surfaces, e.g. absence of CD59 or CD55 on red cell surfaces or absence of CD14 on monocyte surfaces. Until recently treatment has been supportive. However, there has been success using an anti-complement (C5) humanised monoclonal antibody (eculizumab). Recent trials of this antibody have shown that it decreases the rate of haemolysis, decreases the red cell transfusion requirement and improves patient quality of life. Historically, death is often ultimately due to sepsis or thrombosis.
Haemolytic anaemia due to a defect outside the red cell
Haemolytic anaemias due to a defect outside the red cell are all acquired disorders.
Immune haemolytic anaemias

 Direct antiglobulin (Coombs’) test is positive, indicating erythrocyte sensitisation with immunoglobulin or complement
Direct antiglobulin (Coombs’) test is positive, indicating erythrocyte sensitisation with immunoglobulin or complement

Immune haemolytic anaemias are due to red cell damage by an antibody. The phenomenon may be autoimmune, as in idiopathic and drug-induced autoimmune haemolytic anaemias and cold antibody disorders, or alloimmune (where the antibody forms to an antigen foreign to that individual), as in haemolysis due to mismatched blood transfusion and that in haemolytic disease of the newborn (Table 23.7). In all cases the presence of antibody or complement on the red cell surface is confirmed by the direct antiglobulin (or Coombs’) test which uses antibodies to human immunoglobulin or complement raised in an animal to cause in vitro agglutination of red cells sensitised with antibody or complement in vivo.
Table 23.7 The immune haemolytic anaemias
| Autoimmune | ||
|---|---|---|
| ‘Warm antibody’ | ‘Cold antibody’ | Alloimmune |
Mismatched blood transfusion
Haemolytic disease of the newbornDrug-relatede.g. methyl dopa
In some (the more common) instances of autoimmune haemolysis the auto-antibody is IgG and most reactive at 37°C—‘warm antibody’ autoimmune disorders. In ‘cold antibody’ autoimmune disorders an IgM antibody is active at 4°C, becoming less active at higher temperatures, but is still able to bind complement and agglutinate red cells at the temperature (c. 30°C) of the peripheral tissues (hands, feet, nose, ears).
Antibody-coated cells bind to macrophages of the reticulo-endothelial system via Fc receptors. Partial phagocytosis results and the erythrocyte loses some membrane. In order to maintain cellular integrity after this reduction of surface area, a sphere is formed. Such spherical red cells are less deformable than normal; they eventually become trapped in the spleen and are removed by phagocytosis.
‘Warm antibody’ immune haemolytic anaemia
In ‘warm antibody’ immune haemolytic anaemia, the autoantibody is usually IgG and may or may not bind complement. Red cell destruction occurs in the cells of the reticulo-endothelial system, especially the spleen. Most cases are idiopathic, occurring at any age. There may be a family history of autoimmune disease. In about one-third of instances the process is initiated by a drug or it occurs in association with some other disease, particularly a lymphoproliferative disorder, or collagen vascular disease such as systemic lupus erythematosus or rheumatoid arthritis (Ch. 25).
Drugs can cause the disorder by one of three mechanisms (Fig. 23.28). Withdrawal of the drug results in resolution of the disorder.
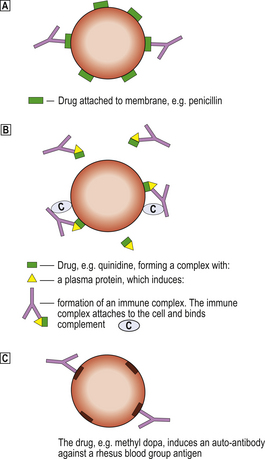
Fig. 23.28 Three mechanisms of drug-induced immune haemolysis.  The drug acts as a hapten.
The drug acts as a hapten.  The drug forms an immune complex which attaches non-specifically to red cells.
The drug forms an immune complex which attaches non-specifically to red cells.  The drug induces an auto-antibody.
The drug induces an auto-antibody.
The blood picture in warm antibody haemolysis is that of a chronic anaemia with microspherocytes and increased polychromasia (and reticulocytosis). The degree of anaemia is very variable within and between cases but may be extremely severe. Erythroid hyperplasia is marked in the bone marrow; megaloblastic erythropoiesis may supervene, as in all haemolytic anaemias, due to increased folate requirements. The spleen is moderately enlarged and congested. Features of an underlying disorder, such as lymphoma, may also be present.
Clinical features and treatment.The clinical features are those of haemolytic anaemia—pallor, jaundice and splenomegaly. In those instances where a drug cannot be implicated and withdrawn, treatment by immunosuppression with corticosteroids and other immunomodulatory drugs is employed. In some cases splenectomy is successful in reducing the rate of haemolysis.
‘Cold antibody’ immune haemolytic anaemias
In ‘cold antibody’ immune haemolytic anaemias, the IgM antibody attaches to red cells in the peripheral circulation and complement is bound. On re-entering the central circulation the IgM antibody may become detached, but complement activation leads to red cell destruction in the reticulo-endothelial system. The main consequences of this sequence of events are agglutination of erythrocytes in cooler areas which causes sluggish flow and reduced oxygen saturation, and chronic haemolysis. Severity relates particularly to the thermal amplitude of the antibody, that is its activity at temperatures up to 30°C.
The pathological features are those of chronic haemolysis with a tendency to marked agglutination of red cells on the blood film. If the film is prepared at 37°C the agglutination is no longer present. The reticulocyte count is increased.
Clinical features and treatment.The clinical features are of anaemia and of discoloration (blueness) and coldness of the fingers, toes, nose and ears, occasionally progressing to ischaemia and ulceration. Many cases occur spontaneously in older adults. The disorder is chronic and often mild. It occurs as an unusual complication of lymphoma, and also, rarely and transiently, in infectious mononucleosis (glandular fever) and mycoplasma pneumonia.
The degree of haemolysis can be reduced by maintenance of a warm environment. Steroids and splenectomy are rarely successful, probably because complement-sensitised cells tend to be destroyed at other sites, especially the liver.
Haemolytic disease of the newborn
Haemolytic disease of the newborn, a previously common disorder, is due to passage across the placenta of maternal IgG antibodies which are reactive against, and cause destruction of, the fetal red cells. This disorder requires the inheritance by the fetus of a red cell antigen from the father which is not present on the maternal red cells, thus provoking antibody development in the mother. Antibodies against the D antigen of the rhesus blood group system are most commonly implicated, but with improvements in management classical rhesus haemolytic disease is now much less common and an increased proportion of cases are due to antibodies to other antigens in the rhesus system, to the A antigen of the ABO system or occasionally to other antibodies.
The prevalence of negativity for the rhesus D antigen varies according to race: approximately 15% for Caucasians, 8% for African–Americans, 4% for Africans, 1% for Native Americans and <1% for Asians. Such individuals who are negative for the rhesus D antigen can become sensitised to produce anti-D. Passage of fetal red cells into the maternal circulation occurs normally at delivery or as a result of miscarriage or operative intervention during pregnancy and these D-positive cells sensitise a D-negative mother. Further stimulation of antibody production occurs in subsequent pregnancies with a D-positive fetus. IgG antibody then crosses the placenta from mother to fetus and causes immune destruction of fetal red cells. Thus, the disorder does not manifest in the first pregnancy. The pathogenesis is similar for other antibodies; however, the fetus may be affected in the first pregnancy in ABO haemolytic disease of the newborn, where IgG antibody to A or B on fetal red cells develops in a group O mother.
Clinicopathological features.The pathological features are those of a haemolytic anaemia of variable severity occurring in utero. In the most severe cases, associated with a high titre of anti-D, the result is death in utero from ‘hydrops fetalis’; the fetus is extremely pale and oedematous and has gross hepatosplenomegaly, the result of severe anaemia with cardiac and hepatic failure and increased extramedullary erythropoiesis. In less severe examples the neonate is pale and jaundiced at birth, with hepatosplenomegaly.
The blood picture is that of anaemia, polychromasia with increased reticulocytes and often nucleated red cells in the peripheral blood. The direct antiglobulin test on the neonatal red cells is positive, indicating that they are coated with antibody. When unconjugated bilirubin levels are very high, bile pigment becomes deposited in the central nervous system, especially the basal ganglia, causing severe damage, known as kernicterus. The bilirubin levels rise rapidly after birth due to immaturity of the liver, with further central nervous system damage. Spasticity and mental retardation may be the clinical consequences of this damage.
In some cases of haemolytic disease of the newborn due to anti-D, and most due to anti-A, the disease is mild, with neonatal anaemia and mild jaundice.
Management.The incidence of the disorder has been reduced by the prophylactic removal of fetal cells entering the maternal circulation before sensitisation can occur. This is achieved by injection of anti-D into the D-negative mother.
Management of the affected fetus centres on provision of unsensitised red cells by intra-uterine transfusion and removal of bilirubin by exchange blood transfusion postnatally. Mildly affected neonates are treated by phototherapy, in which exposure to light of an appropriate wavelength degrades bilirubin.
Haemolysis due to mismatched blood transfusion
Haemolytic transfusion reaction constitutes a second type of alloimmune haemolysis. Severe reactions result from transfusion of red cells possessing an antigen (e.g. ABO group antigens) to which the recipient possesses complement binding antibody of IgG or IgM class.
Microangiopathic haemolytic anaemia
The term microangiopathic haemolytic anaemia describes the dramatic haematological picture that occurs when haemolysis is caused by physical trauma to erythrocytes as they are forced through narrow or damaged areas in the microvasculature. Characteristic cells are present on the blood film, especially schistocytes (Fig. 23.3). This type of process is commonly present in disseminated intravascular coagulation (p. 663); the erythrocytes are damaged on fibrin strands deposited in small blood vessels. It is also a feature of the haemolytic uraemic syndrome, thrombotic thrombocytopenic purpura (p. 671), malignant hypertension and of the extensive vasculitis in systemic lupus erythematosus. In many of these conditions, thrombocytopenia is also present, due to platelet consumption in microthrombi formed on damaged endotheium.
Similar erythrocyte damage without microvascular lesions occurs in march haemoglobinuria, originally described in soldiers after prolonged marching; red cell damage presu-mably occurs in the feet. An analogous situation has been described in marathon runners, bongo drummers and exponents of karate! In most of these situations the haemolysis is not chronic, and splenomegaly and other features of chronic red cell destruction are absent. The direct antiglobulin test is negative, as antibody is not involved in the pathogenesis.
Schistocytes and haemolysis, sometimes catastrophic, are occasionally the result of red cell injury from a malfunctioning mechanical heart valve or other vascular prostheses.
Other causes of haemolytic anaemia
Extensive burns are associated with haemolysis, in part due to direct heat damage of erythrocytes in blood vessels of the burned areas, and in part due to a microangiopathic mechanism. Snake bites, spider bites and chemicals are occasional causes.
Infection with clostridia is a rare cause of haemolysis. Malarial infection is common and results in haemolytic anaemia (Fig. 23.29). Schizonts escape by rupturing the erythrocytes in which they have matured. In chronic malarial infection, extreme splenomegaly is often present. Histologically, there is marked congestion and expansion of reticulo-endothelial cells; macrophages contain parasites and red cells, and are laden with malarial pigment.
Hypersplenism
Hypersplenism is defined as anaemia (often accompanied by leukopenia and thrombocytopenia) secondary to splenic enlargement (Ch. 22). This anaemia is in part due to a haemolytic component, presumed to be due to increased red cell sequestration in the enlarged spleen, with enhanced phagocytosis by macrophages. However, other mechanisms contribute: the plasma volume increases in proportion to the degree of splenic enlargement, for reasons that are not understood. This results in a dilutional anaemia. Pooling of blood cells also occurs within the spleen.
Hypersplenism is associated with splenomegaly from any cause, such as portal hypertension and collagen vascular disease. Hypersplenism in rheumatoid arthritis has the eponym Felty’s syndrome. The blood picture in hypersplenism is that of a pancytopenia with no specific features. The haemoglobin concentration would rarely be less than 80g/l and the platelet count less than 60 × 109/l due to hypersplenism alone.
NEOPLASTIC DISORDERS OF THE BONE MARROW
Classification of bone marrow malignancies
Bone marrow malignancies are classified according to
their presentation (acute or chronic), their tissue distribution (e.g. leukaemia in blood and marrow or lymphoma in lymph nodes and other tissues) and their histogenesis (e.g. myeloid, lymphoid). The World Health Organization has refined the classification of tumours of the haemopoietic and lymphoid tissues. The objectives of the WHO classification are to offer pathologists, haematologists, oncologists and geneticists worldwide a system of classification of human haemopoietic neoplasms that is based on their histopathological and genetic features. This classification takes into account tissue and cell morphology, immunological characteristics of the malignant cells and, where known, specific acquired genetic aberrations associated with the malignancies. The advantages of this approach to classification are improved reliability in diagnosis, better prognostic information with the possibility of tailored therapy for a given prognostic group and, finally, more reliable characterisation of patients entered into trials of therapy.
Summary of the major WHO subtypes of tumours of haematopoietic and lymphoid tissues









The lymphomas are described in Chapter 22. The myelodysplastic syndromes are described on pages 663–664.
LEUKAEMIAS
Leukaemias are neoplastic proliferations of white blood cell precursors. This proliferation results in the common features of leukaemia:
Bone marrow failure with anaemia, neutropenia and thrombocytopenia is the most important consequence, particularly in the acute leukaemias.
Aetiology
In the majority of cases the cause is unknown. Leukaemias represent neoplastic monoclonal proliferations of cells within the bone marrow and blood. Whether or not the cell of origin is a pluripotent stem cell or a more committed cell in each type remains contentious. However, current evidence suggests that in most cases of acute myeloblastic leukaemia (AML) a pluripotential stem cell is mutated. The exception to this is acute promyelocytic leukaemia (APL), which seems to arise from a more committed myeloid progenitor cell. In acute lymphoblastic leukaemia (ALL) the transforming events occur in a very primitive B-cell that has not yet developed the capacity to produce immunoglobulin; most cases of chronic myeloid (or granulocytic) leukaemia (CML), where megakaryocytes and erythroid cells are involved as well as leukocytes, derive from a pluripotent stem cell. Most cases of chronic lymphocytic leukaemia (CLL) are of B-cell origin, but these cells are more differentiated than those in ALL.
In acute leukaemia the typical cells—‘blast’ cells—accumulate as a result of a combination of proliferation but failure of maturation (Fig. 23.30). In CML the abnormal myeloid stem cells also accumulate, but maturation still occurs, with increased numbers of mature myeloid cells in blood and bone marrow, as well as blast cells.
Fig. 23.30 Blast cells in acute lymphoblastic leukaemia. Blast cells are relatively large leukocytes with fine chromatin, nucleoli and basophilic cytoplasm.
It seems likely that several predisposing factors acting together trigger the onset of the disease in most cases. These triggers act by inducing a series of mutations in certain key genes involved in regulating cell proliferation and differentiation. Such genes are known as oncogenes if they promote tumour development and as tumour suppressor genes if their normal, un-mutated form protects against tumour development. In some leukaemias genetic material is exchanged between two genes (translocation), leading to the development of a novel fusion gene which acts as an oncogene. The best understood examples of these types of mutation in leukaemogenesis include the t(9;22) translocation in CML, in which the fusion gene is a tyrosine kinase, BCR-ABL (Fig. 23.31), and the t(15;17) translocation in acute promyelocytic leukaemia. Such genetic mutations are often first recognised by the identification of gross cytogenetic abnormalities at the chromosome level, for example the Philadelphia chromosome is the small chromosome 22 produced by the exchange of genetic material with chromosome 9. With more sophisticated molecular techniques such as fluorescent in situ hybridisation (FISH) and gene array technology it is becoming clear that all leukaemias have altered genes. Already such abnormalities are being used to produce targeted therapies, for example imatinib mesilate (Glivec) in CML (p. 649), and to identify patients with a good prognosis who do not necessarily need a stem cell transplant, for example AML patients with t(15;17) or t(8;21).
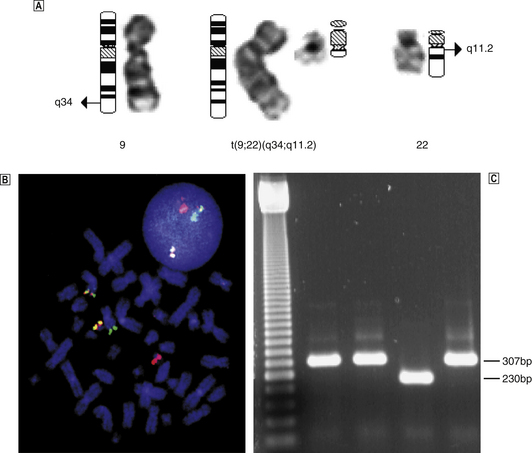
Fig. 23.31  Philadelphia chromosome. The Philadelphia chromosome produced by t(9;22) (the small derivative chromosome centre right).
Philadelphia chromosome. The Philadelphia chromosome produced by t(9;22) (the small derivative chromosome centre right).  BCR-ABL rearrangement identified by FISH (yellow) because of co-localised BCR (red) and ABL (green), and
BCR-ABL rearrangement identified by FISH (yellow) because of co-localised BCR (red) and ABL (green), and  as a single band of 230 kb by PCR. (FISH, fluorescent in situ hybridisation.)
as a single band of 230 kb by PCR. (FISH, fluorescent in situ hybridisation.)
(Courtesy of David Stevenson.)
Certain factors known to initiate leukaemic transformation are:
Acute leukaemias
Acute leukaemias arise from mutations in haemopoietic stem cells. The leukaemic clone of cells proliferates but loses the ability to differentiate into mature blood cells. This imbalance between proliferation and differentiation in acute leukaemia leads to the accumulation of blast cells in the bone marrow and the hallmark clinical features of bone marrow failure. Two broad types of acute leukaemia are recognised: acute lymphoblastic leukaemia (ALL), most common in childhood, and acute myeloblastic leukaemia (AML), most common in adults. The incidence of ALL and AML with age is shown in Figure 23.32.
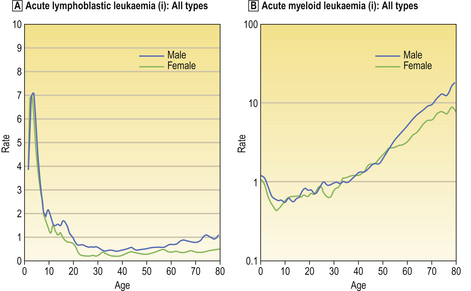
Fig. 23.32 The age-specific incidence rates per 100000 population for  ALL and
ALL and  AML in England and Wales 1984–1993. (Leukaemia Research Fund, with permission.)
AML in England and Wales 1984–1993. (Leukaemia Research Fund, with permission.)
Acute lymphoblastic leukaemia
ALL is most common between 2 and 4 years of age. It is the commonest cause of cancer death in childhood. Recent data suggest that one of the predisposing genetic mutations for childhood ALL actually occurs in utero and that further genetic mutations in the first years of life lead to the disease.
ALL blasts were described morphologically by the French– American–British (FAB) classification according to their size, nuclear:cytoplasmic ratio and whether or not there is marked cytoplasmic vacuolation, as L1, L2 or L3 cells. Routine use of panels of monoclonal antibodies has shown that the majority of cases of ALL derive from B-cell precursors, and the use of these antibodies can classify ALL according to the degree of maturation along the B-cell pathway, i.e. common ALL and pre-B ALL, which can be of L1 or L2 morphology, or B-ALL which is of L3 morphology and now known as Burkitt’s leukaemia because of its biological similarity to Burkitt’s lymphoma. The WHO classification distinguishes cases as precursor B-lymphoblastic leukaemia/lymphoma and precursor T-lymphoblastic leukaemia/lymphoma. A number of acquired genetic abnormalities are recognised in ALL. These include the occurrence of a Philadelphia chromosome caused by the t(9;22) translocation in 2% of children but 20–30% of adults with ALL, hypodiploid or hyperdiploid (>50) numbers of chromosomes, t(1;19), t(12;21) and rearrangements of the myeloid/lymphoid or mixed lineage leukaemia (MLL) gene on chromosome 11q.
Acute myeloblastic leukaemia
The incidence of AML increases steadily with age. In older people AML is more likely to develop from an existing bone marrow disorder such as myelodysplastic syndrome (MDS), whereas the majority of younger patients present with de novo AML.
The WHO classification of AML distinguishes subtypes based on biological homogeneity and clinical relevance. As such there are four major categories:1.AML with recurrent genetic abnormalities (including t(8;21), t(15;17), deletion 11q and inversion 16) 2.AML with multilineage dysplasia 3.AML, therapy related (including following exposure to alkylating agents or topoisomerase II inhibitors) 4.AML not otherwise categorised (including cases with evidence of some myeloid, monocytic, erythroid or megakaryocytic differentiation).In all cases there must be at least 20% blast cells in the bone marrow for AML to be diagnosed.
Prognosis in AML is related to acquired cytogenetic abnormalities. As acknowledged in the WHO classification, some of these abnormalities occur in specific subtypes of AML. Patients with balanced translocations t(15;17) in acute promyelocytic leukaemia (APL), t(8;21) and inversion of chromosome 16 respond very well to treatment and have a long-term cure rate of 70–80%. Much is now known about the genes altered by these chromosome rearrangements. For example, in APL with t(15;17) the genes rearranged are PML/RARA. The retinoic acid receptor alpha gene (RARA) in its fused form with PML leads both to the development of this leukaemia and its response to pharmacological doses of all-trans-retinoic acid (ATRA), which is now part of standard therapy for this subtype of AML. At the other end of the spectrum are cytogenetic abnormalities that carry a very poor prognosis with cure rates of less than 20%. These include loss of a whole chromosome 5 or 7 or complex multiple cytogenetic abnormalities. These cases occur more commonly in elderly people, following on from MDS or in so-called treatment-related AML after exposure to chemotherapy for previous cancers. A significant majority of patients have no detectable chromosomal abnormality. However, genetic abnormalities are increasingly recognised in this group of patients using molecular techniques. Examples include internal tandem duplication of the FLT3 gene in 30% of cases which carries a poor prognosis, and a mutation in the nucleophosmin gene which leads to the aberrant location of the protein in the cytoplasm rather than the nucleolus and which carries a relatively favourable prognosis.
Blood and bone marrow changes in acute leukaemia
In peripheral blood the white cell count is usually increased but can be decreased or normal despite massive marrow infiltration with blast cells. Counts greater than 100 × 109/l can occur. Irrespective of the total white cell count, a majority of nucleated cells in the blood are leukaemic blasts. In AML, cells containing diagnostic rod-like granular structures (Auer rods) may be present, as may hypogranular polymorphonuclear variants and pseudo-Pelger cells (Fig. 23.7). Anaemia is present, usually normocytic and normochromic. Thrombocytopenia is marked, particularly in AML.
Bone marrow cellularity is markedly increased. Blast cells constitute at least 20% of nucleated cells present, and often greater than 80%. Extension into areas of previously fatty marrow may occur. Gross bone erosion with fractures is not generally a feature of acute leukaemia. Karyotype analysis reveals abnormalities in the leukaemic blasts, with gains and losses of whole chromosomes as well as translocations.
Changes in other organs
Lymph nodes, liver and spleen may be infiltrated with leukaemic blast cells in all types of acute leukaemia. Lymph node enlargement is generally mild and nodes remain discrete, although in some cases of ALL massive involvement of mediastinal lymph nodes is a feature. Splenic enlargement, where present, is also minor in contrast to that in chronic leukaemias. Histologically, there is effacement of normal node architecture by sheets of leukaemic blasts and focal or diffuse infiltration of the spleen.
A diffuse infiltrate of leukaemic cells may also be present in most other organs. Evidence of bacterial, fungal or viral infection may be apparent, as may haemorrhage secondary to thrombocytopenia.
Meningeal infiltration in ALL is an important feature. Leukaemic blasts within the central nervous system are protected from chemotherapeutic agents by the blood–brain barrier. Perivascular aggregates of blast cells later form diffuse lesions and plaques which may result in compression of adjacent nerve tissue.
Infiltration of the gums (Fig. 23.33) and skin is a peculiar feature of the monocytic types of AML.
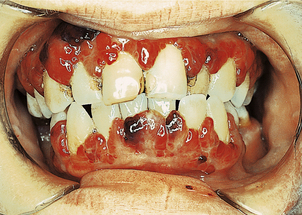
Severe, life-threatening coagulation failure occurs in APL, probably due to coagulation activation and consumption of clotting factors by activators released from the granules of the leukaemic promyelocytes and excess fibrinolysis.
Clinical course
The onset is often very rapid and progression to death from anaemia, haemorrhage or infection occurs within weeks if no treatment is given. The features are those of marrow failure, with anaemia, infection due to neutropenia and mucocutaneous bleeding due to severe thrombocytopenia (Fig. 23.34).
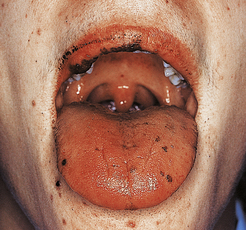
Infections are typically with bacteria and fungi. Septicaemias, pneumonia and skin sepsis are common (Fig. 23.35). Fungal infections can be local, such as in the oral cavity (Fig. 23.6), but fungal septicaemia and organ invasion occur. Systemic fungal infection is often fatal (Fig. 23.36). The situation may be exacerbated, especially in AML, by transient but prolonged aplasia induced by highly myelotoxic chemotherapeutic agents. The clinical course is, however, often less catastrophic in childhood ALL.
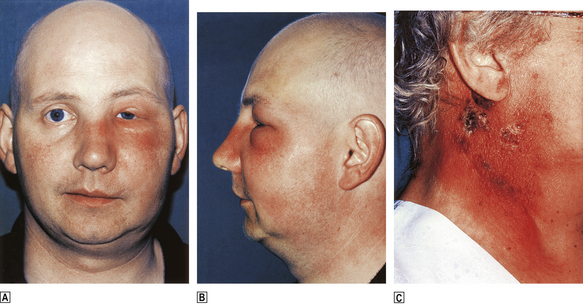
Fig. 23.35 Bacterial infection in AML.  and
and  Peri-orbital cellulitis caused by Pseudomonas aeruginosa (with permission) and streptococcal cellulitis.
Peri-orbital cellulitis caused by Pseudomonas aeruginosa (with permission) and streptococcal cellulitis.
Fig. 23.36 Fatal disseminated fungal infection in AML.  Fungal pneumonia.
Fungal pneumonia.  and
and  Multiple lung and brain fungal abscesses.
Multiple lung and brain fungal abscesses.  Aspergillus fumigatus grown from lung abscess.
Aspergillus fumigatus grown from lung abscess.
Treatment
Treatment of acute leukaemia is directed by individual prognostic assessment. The aim is to offer curative treatment where possible and minimise the long-term complications in groups of patients who have a high cure rate, while intensifying the treatment or using new modes of treatment in those groups of patients who presently do badly. Therefore, as an example of this principle, children and young adults with good prognosis ALL or AML will not be routinely offered bone marrow transplantation, while those with poor prognosis disease may be offered bone marrow transplantation as part of initial treatment. Treatment is by chemotherapeutic agents in combination to clear the blood, bone marrow and other sites of leukaemic blasts as far as is possible. The first one or two courses of treatment are aimed at producing a state of remission, in which the blood counts are normal and there are less than 5% blast cells in the bone marrow as identified by light microscopy. However, light microscopy is not very sensitive and in remission there can still be in the order of 109 leukaemic cells in the marrow. Further courses of chemotherapy are given to consolidate the remission and reduce this leukaemic burden further. In poor prognostic disease it seems chemotherapy alone cannot overcome the leukaemic burden and the additional allogeneic immune attack provided by a stem cell transplant is required to eradicate the disease. This attack is called graft-versus-leukaemia and is mediated by the donor’s engrafted T-lymphocytes. New approaches to leukaemia management include more accurate monitoring of minimal residual disease (disease that cannot be detected by conventional microscopy) by PCR or flow cytometry and acting on the results of such tests. This has been particularly successful in childhood ALL and in APL. New modes of therapy, such as the addition of antibodies targeted against the leukaemia cells, e.g. calicheamicin bound to anti-CD33 (gemtuzumab ozogamicin) in AML, and techniques to harness the graft-versus-leukaemia effect whilst reducing the toxicity of the transplant procedure by using intense T-cell immunosuppression and less myelosuppression (so-called reduced intensity conditioned allografts) are showing promise.
Intensive support by transfusion of blood products and use of antibacterial and antifungal agents is necessary to support the patient during the treatment while bone marrow function is suppressed. Survival is months or a few years in adults, with an increasing proportion of long-term survivors with advances in therapy. The outlook is much better in childhood ALL and in adults with good prognosis AML, where significant cure rates are now achieved.
Chronic leukaemias
The important differences between chronic and acute leukaemia are shown in Table 23.8.
Table 23.8 Acute versus chronic leukaemia
| Acute | Chronic |
|---|---|
| Leukaemic cells do not differentiate | Leukaemic cells retain ability to differentiate |
| Bone marrow failure | Proliferation without bone marrow failure |
| Rapidly fatal if untreated | Survival for a few years |
| Potentially curable | Not presently curable without bone marrow transplant |
Chronic myeloid (granulocytic) leukaemia
Although a ‘chronic’ leukaemia, the natural history of CML is that of a fatal disorder with a median survival in the pre-imatinib era of about 5 years in patients not eligible for allogeneic bone marrow transplantation. It occurs in all age groups. Normal bone marrow is replaced by an abnormal clone derived from a pluripotential stem cell which, in the majority of cases, is characterised by the presence of a karyotypic abnormality, the Philadelphia chromosome (reciprocal translocation of part of the long arm of chromosome 22 to another chromosome, usually 9) (Fig. 23.31). Erythroid, megakaryocytic and B-lymphocyte cell lines all carry the defect, as well as the granulocytic series. In most cases the disease eventually enters a more aggressive phase due to the emergence and dominance of a clone of myeloid cells that have now lost the ability to differentiate. The disease, called blast crisis, then bears a close resemblance to AML (or less commonly ALL) and is fatal.
Blood and bone marrow changes
Leukocytosis is a uniform feature, with occasional cell counts in excess of 300 × 109/l. The cell picture in the blood can superficially resemble that in a bone marrow aspirate, with myelocytes, promyelocytes, myeloblasts and normoblasts present as well as large numbers of band cells and mature polymorphonuclear granulocytes (Fig. 23.37). Basophilia is common. Platelets are increased (sometimes over 1000 ×109/l), normal or reduced. A normochromic anaemia is often present.
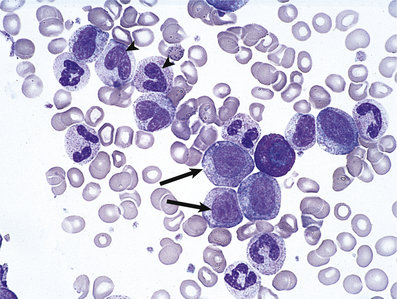
Fig. 23.37 The blood in chronic myeloid leukaemia. Myelocytes (arrows) and metamyelocytes (arrowheads) enter the circulation.
The leukocytes are abnormal, as exemplified by an absence or severe reduction of their content of alkaline phosphatase, a feature unique to CML and of diagnostic value. Serum vitamin B12 is elevated due to production of binding protein by the granulocyte series.
The bone marrow is hypercellular with marked reduction of fat spaces; granulocytopoiesis predominates. In the acute, terminal phase increased numbers of blast cells become evident in blood and bone marrow, and anaemia and thrombocytopenia are more marked.
Changes in other organs
The spleen is enlarged, often massively, due to infiltration by CML cells (Fig. 23.38); it may fill the abdominal cavity and extend into the pelvis. Areas of infarction are present due to the rapid enlargement outstripping the available blood supply. Hepatomegaly is also frequently present. Infiltration in other organs is an occasional feature. Infection and bleeding are not common in the chronic phase.
Clinical course
Symptoms may be mild in the chronic phase and are essentially those of anaemia and massive splenomegaly (abdominal fullness and pain from splenic infarction). Rarely, a hyperviscosity state may develop when the white count is greater than 300 × 109/l. In the acute phase the clinical features are those of acute leukaemia.
Treatment
The treatment of BCR-ABL-positive CML has seen a remarkable change in recent times. With detailed understanding of the molecular structure of the causative oncogene BCR-ABL, a drug called imatinib mesilate—trade name Glivec—has been developed. The drug is a small molecule that binds to the ATP-binding site of BCR-ABL and inhibits the function of the protein. In a large randomised clinical trial (IRIS), this oral drug has had very dramatic positive results with >95% of patients achieving complete haematological remission within 12 months and 87% achieving a complete cytogenetic remission by 60 months. After 5 years of follow-up, the estimated overall survival is 89% of patients treated with imatinib. A trial of imatinib with monitoring of response milestones is now regarded as the standard therapy for all patients newly diagnosed with BCR-ABL-positive chronic phase CML. Some patients will fail to meet the response milestones whilst on imatinib and some who do respond subsequently develop resistance to the drug because of mutations in the ATP-binding site or multiple copies of the BCR-ABL gene. Such patients who are young enough, fit enough and have a suitable donor can be cured by an allogeneic stem cell transplant. Newer tyrosine kinase inhibitors with a broader range of cellular targets (dasatinib) or more potent anti-BCR-ABL activity (nilotinib) are being introduced for patients who fail imatinib therapy and for whom stem cell transplantation is not available. Imatinib and the newer tyrosine kinase inhibitors are also more effective in treating patients in blast crises than conventional chemotherapy. It is too early to know if imatinib or its successors will cure patients with CML; however, the introduction of imatinib is a landmark event and marks the proof of principle that a clear understanding of the pathogenesis of a disease at a molecular level can lead to the design of effective targeted treatment. If tyrosine kinase inhibitor therapy is not available, the proliferative features of CML can be controlled with oral hydroxycarbamide and the chronic phase prolonged with the use of alpha-interferon with or without cytosine arabinoside.
Chronic lymphocytic leukaemia
Aetiology
CLL is a chronic lymphoproliferative disorder with features similar to those of a low-grade lymphoma but with predominant blood and marrow involvement. During the last few years there has been significant new understanding of the pathology of this disease. The disease process is a relentless accumulation of B-lymphocytes that appear resistant to apoptosis. In the majority of cases it is a considerably less aggressive disorder than are the other leukaemias. This common form of the disease is a disease of the elderly. It is slowly progressive, usually following a predictable clinical course (Fig. 23.39) with a median survival of 25 years and often does not require therapy. In this form of the disease the malignant B-cell has undergone rearrangement of its immunoglobulin genes and the cell has also passed through the germinal centre of the lymph node and been selected for antigen by hypermutation of its rearranged immunoglobulin genes. Such cells therefore are the leukaemic equivalent of memory B-cells. In a proportion of patients the disease is much more aggressive in its behaviour with resistance to chemotherapy and a much shortened median survival of 8–9 years. In these cases the leukaemic B-cell has not been selected for antigen by hypermutation of the immunoglobulin genes and is the leukaemic equivalent of a naive B-cell. In both forms of the disease lymphocytes accumulate in blood, marrow, liver and spleen until the total lymphoid mass is expanded up to 100-fold.
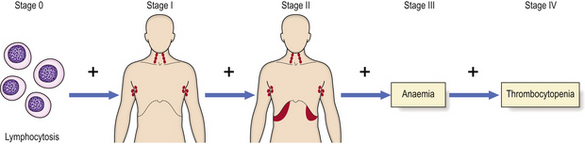
Blood and bone marrow changes
Leukocytosis is present; up to 99% of nucleated cells are small lymphocytes (Fig. 23.40) of B-cell origin in most instances. The lymphocyte count is between 5 × 109/l and more than 300 × 109/l. The CLL cells tend to fragment during preparation of the blood film, producing many ‘smear cells’ (Fig. 23.40). Anaemia (normocytic) and thrombocytopenia are late developments (Fig. 23.39). However, in up to 10% of cases a secondary autoimmune haemolytic anaemia develops, with reticulocytosis and microspherocytes and a positive direct antiglobulin test result. Serum immunoglobulins are low in the later stages of the disease.
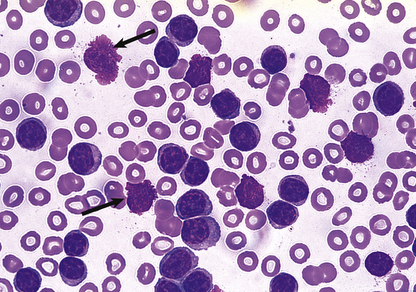
Fig. 23.40 The blood in chronic lymphocytic leukaemia. Numerous small lymphocytes and ‘smear’ cells (arrows) are characteristic.
The bone marrow is hypercellular, with progressive replacement of normal tissue by small lymphocytes, resulting eventually in anaemia and thrombocytopenia.
Advanced stage non-Hodgkin’s lymphoma (NHL) may result in blood and marrow involvement superficially resembling CLL. However, extensive involvement usually occurs late in the course of the disease and the lymphoma cells are morphologically distinct from the lymphocytes of CLL. Immunophenotyping of B-CLL cells shows a distinct pattern of antigen staining, with the leukaemic cells staining positively for the B-cell antigen CD19 and also for CD5 and CD23. This pattern of staining is useful for helping to distinguish CLL from other cases of B-NHL appearing in the blood which lack staining for CD5 and/or CD23. Cases of CLL, as with other leukaemias, have acquired cytogenetic abnormalities within the leukaemic cells. The common ones are deletions of 13q, 11q and 17p (loss of p53) and trisomy 12. As with acute leukaemia, these abnormalities have prognostic significance, with deletion of 11q and of p53 carrying a poorer prognosis.
Changes in other organs
The lymph nodes, liver and spleen are characteristically involved. In nodes and spleen the normal architecture becomes completely effaced by the infiltrate of monomorphic small lymphocytes, and similar cells are present in the portal tracts of the liver. Occasionally, the predominant presentation is with lymph node involvement with little or no evidence of disease in the blood and marrow, when it is termed small cell lymphocytic lymphoma (WHO-CLL/SCLL).
Clinical course and treatment
For the good prognostic form of the disease the clinical course is protracted; it is summarised in Figure 23.39. The protracted course means that many cases are diagnosed as a result of routine blood tests or clinical examination for some other reason. Elderly patients with this form of the disease often die from an unrelated cause. Many of these patients do not require treatment at all or for many years. Treatment is indicated for the development of significant cytopenias, bulky lymphadenopathy or hepatosplenomegaly, or systemic symptoms such as loss of weight or night sweats. Successful first-line treatment options include single agent chlorambucil or fludarabine alone or in combination with cyclophosphamide.
For the aggressive form of the disease occurring in younger patients and in patients with relapsed disease the outlook is less favourable. Treatment should still be initiated only when there are clinical indications, and treatment regimens involving intensive combinations of chemotherapy, such as fludarabine and cyclophosphamide with the anti-CD20 antibody rituximab, CHOP combination chemotherapy, stem cell transplantation, and the anti-CD52 monoclonal antibody alemtuzumab are therapeutic options to be considered.
Patients with autoimmune manifestations including autoimmune haemolysis and immune thrombocytopenic purpura are treated with corticosteroids.
Other leukaemias
The prolymphocytic variant of CLL is more aggressive and responds poorly to therapy. Splenomegaly is massive. The leukaemic cells in blood and bone marrow are larger and of more primitive appearance than is the case in CLL. T-PLL (T-cell prolymphocytic leukaemia) occurs in younger subjects than does the more common B-cell CLL described above. Skin involvement is common. T-PLL has been shown to respond well to alemtuzumab.
Hairy-cell leukaemia is a rare B-cell leukaemia of the middle-aged and elderly. The characteristic cells in the blood have cytoplasmic projections or ‘hairs’. Pancytopenia is typical, as is splenomegaly, which may be gross. A characteristic feature of the peripheral blood count is a marked monocytopenia. The marrow is diffusely infiltrated by the malignant cells and marrow fibrosis is present. The disorder may run a chronic course and is of particular interest because there are a number of very effective treatments, including interferon, cladribine and deoxycoformycin. Splenectomy is also useful in management and can relieve cytopenias, but is used less frequently now with these newer effective therapies.
MYELODYSPLASTIC SYNDROMES
This is a group of neoplastic conditions of bone marrow in which there is dysplastic haemopoiesis, resulting in marked morphological abnormalities in blood cells, and a tendency to progress to AML. As such they are pre-leukaemic disorders. Their hallmark is the presence of a cellular bone marrow with cytopenias in the peripheral blood. The pathogenesis appears to be an abnormal clone arising from a mutated stem cell. The combination of the high marrow cellularity and blood cytopenias may be explained by development of a mutated clone with predisposition to apoptosis. In some cases the myelodysplastic syndrome is secondary to stem cell damage from prior treatment of unrelated malignancies with chemotherapy or radiotherapy and is therefore iatrogenic.
Blood and bone marrow changes
There is anaemia, usually macrocytic, with leukopenia and thrombocytopenia. Abnormal cells such as poikilocytes and neutrophils with poorly developed or absent cytoplasmic granules are commonly present (Fig. 23.7). Bone marrow appearances are dysplastic with changes resembling those seen in megaloblastic anaemia—megaloblastoid change. Typical appearances include binucleate normoblasts, ring sideroblasts, megakaryocytes with a single round nucleus and neutrophils with absent granules and poorly developed nuclear segmentation including bilobed forms (pseudo-Pelger–Huet cells). Leukaemic blast cells may be present but constitute less than 20% of the marrow cells, unless there has been progression to acute leukaemia. Dysplasia is considered to be significant in a given cell line if 10% of the marrow nucleated cells are dysplastic. Abnormalities of marrow chromosomes are commonly present, consistent with the malignant and clonal nature of the disorders.
Subclassification is possible depending on the number of cell lineages involved, the presence of ring sideroblasts at greater than 15% of nucleated erythroblasts and whether or not there are increased leukaemic blasts. The WHO classification, therefore, recognises subtypes involving the red cell lineage without increases in blast cells; refractory anaemia (RA) or refractory anaemia with ring sideroblasts (RARS) as distinct from those involving two or more cell lineages without increases in blast cells; refractory cytopenias and multilineage dysplasia (RCMD) and refractory cytopenias with multilineage dysplasias and ring sideroblasts (RCMD-RS). If blasts are increased the term refractory anaemia with increased blasts is used: RAEB-1 (blasts 5–9%), RAEB-2 (blasts 10–19%). In another variant of myelodysplasia there is a marked excess of monocytes in the blood—chronic myelomonocytic leukaemia (CMML). Because of the combination of features of myeloproliferative disease and myelodysplasia the WHO uses the term ‘MDS/myeloproliferative crossover syndrome’ for CMML and some other variants of myelodysplasia.
Clinical features
Myelodysplasia occurs in the elderly most commonly—median age over 65 years. Anaemia is usually the most troublesome problem and occurs in some 80% of patients. Infections and bleeding also occur. Splenomegaly is rare, except in CMML. Progression to acute leukaemia occurs in around 30%. The mainstay of treatment is largely supportive with red cell transfusions and treatment of infections. In some cases the anaemia responds to treatment with recombinant erythropoietin with or without granulocyte colony-stimulating factor (G-CSF).
AML-type chemotherapy is used for some patients with excess blasts and can achieve transient remissions. Increasingly, allogeneic stem cell transplantation offers a hope of cure, with reduced intensity conditioning regimens allowing older patients to benefit from this approach. Important new developments include the recognition that patients with the cytogenetic abnormality of del 5q rerspond very well to the drug lenalidomide with relief of anaemia and transfusion dependence and the achievement of cytogenetic remissions. In addition, the demethylating agents azacitidine and decitabine improve cytopenias and survival, perhaps by switching on differentiating genes inhibited by DNA methylation. Overall, survival varies with the subtype, ranging from a few years for RA and RARS to less than 1 year in those with excess blasts.
NON-LEUKAEMIC MYELOPROLIFERATIVE DISORDERS




The non-leukaemic myeloproliferative disorders are listed in Table 23.9. This list is something of an oversimplification as intermediate forms exist. More importantly, progression in an individual from one such disorder to another within the group is well recognised.
Table 23.9 The non-leukaemic myeloproliferative diseases
| Disorder | Pathology of bone marrow | Clinical features |
|---|---|---|
| Myelofibrosis |
Polycythaemia rubra vera
Essential thrombocythaemia
Myeloproliferative disorders most often represent a neoplastic proliferation of a marrow myeloid stem cell with differentiation to the mature form(s) (in contrast to the acute myeloid leukaemias, where maturation is very limited). The normal control mechanisms governing the cell line(s) involved are no longer active, allowing accumulation of erythrocytes, platelets or leukocytes. Proliferation of megakaryocytes produces growth factors causing a secondary overgrowth of fibroblasts (myelofibrosis).
The molecular basis of the myeloproliferative diseases has recently been significantly clarified by the recognition of the common occurrence of point mutations in the cell signalling gene JAK2 kinase. This leads to stem cells proliferating independently of the normal regulatory signals (e.g. erythropoietin). Some 95% of cases of polycythaemia rubra vera carry the mutation and this is rapidly becoming a crucial diagnostic tool. The other myeloproliferative diseases carry the mutation less frequently: essential thrombocythaemia ~40% and myelofibrosis ~40%. Interestingly, cases of ET that are JAK2 mutation-positive tend towards having a higher red cell count and haematocrit and this supports the notion that the mutations favour a polycythaemic phenotype.
Polycythaemia rubra vera
Polycythaemia is an increase in the concentration of red cells above normal, usually with a corresponding increase in haemoglobin concentration and haematocrit. In polycythaemia rubra vera (PRV) it is an idiopathic, primary condition.
The body red cell mass and plasma volume can be accurately assessed by isotopic labelling techniques. Normal ranges are 25–35ml/kg for red cell mass (22–32ml/kg in females) and 35–45ml/kg for plasma volume.
Blood and bone marrow changes
The haemoglobin concentration is raised, often to 200g/l or more, with haematocrit values of up to 75%. Red cell mass may be as high as 80ml/kg. However, iron deficiency is not uncommon, partly due to increased requirements and partly to a bleeding tendency with chronic gastrointestinal blood loss due to production of functionally abnormal platelets. In such circumstances of iron-deficient polycythaemia, haemoglobin and haematocrit may be normal or even low, but the red cell count is still high.
Thrombocytosis and neutrophil leukocytosis are present in up to 50% of cases. Serum vitamin B12 and uric acid are increased, the former due to production of binding protein by myeloid cells, the latter due to increased cell turnover. The bone marrow is hypercellular. Erythroid hyperplasia is present. Megakaryocytes may be prominent and increased reticulin deposition is common. Molecular analysis reveals the V617F mutation in the JAK2 kinase gene in virtually all cases with the full-blown PRV phenotype. Some cases in which only pure erythrocytosis exists harbour an alternative mutation in the JAK2 kinase gene.
Changes in other organs
The spleen is enlarged in 75% of cases, usually to a moderate extent. Splenic sinuses are engorged. Extramedullary haemopoiesis may be present: normoblasts and cells of the developing myeloid series are present in the spleen and often the liver. Infarction of heart, brain and spleen is common due to the high blood viscosity and poor flow.
Haemorrhagic lesions may be a feature, especially in the gastrointestinal tract. Peptic ulceration is common in PRV, for unknown reasons.
Clinical features
Clinical features correspond to the pathological changes described above. The skin is plethoric and cyanosis is common. Itching is typical and usually exacerbated by changes in temperature, as after bathing. The conjunctival vessels appear congested, as are retinal vessels. Hyperviscosity results in headache and lethargy. The spleen is palpable. Acute gout may be a presenting feature. Evidence of mucosal bleeding or of thrombosis (particularly arterial) may be present. Myeloproliferative PRV must be distinguished from other causes of polycythaemia, in which splenomegaly and pancytosis are not features (see below).
Treatment is by venesection and/or myelosuppression with hydroxycarbamide or, less commonly nowadays, the alkylating agent busulfan. Aspirin is of proven benefit in reducing all-cause mortality. In the elderly radiophosphorus (32p) is still occasionally used. Survival is for many years. Progression to a myelofibrotic state is common and transformation to acute myeloid leukaemia may occur, especially following 32p treatment.
Secondary and low plasma volume polycythaemias
Polycythaemia is considered significant when the haematocrit is above 0.52 for a man or above 0.47 for a woman. Most cases of polycythaemia are due not to PRV, but to secondary causes that result in appropriate increases in erythropoietin production:
Any disorder resulting in chronic hypoxia results in stimulation of erythropoietin production and secondary polycythaemia, as in severe chronic bronchitis, emphysema or alveolar hypoventilation for any reason. Congenital heart disease in which a right-to-left shunt is present is a potent cause; haemoglobin concentrations of 200g/l are not uncommon. Cigarette smokers have a higher haematocrit than non-smokers, due in part to the carbon monoxide in tobacco smoke. In these situations the polycythaemia is frequently not symptomatic and the blood and bone marrow are otherwise normal. Treatment is rarely necessary.
Very rarely the following renal disorders and tumours are associated with inappropriate erythropoietin production and secondary polycythaemia:
Polycythaemia may also result from a reduction in the plasma volume with normal red cell mass (low plasma volume polycythaemia). This situation occurs chronically in so-called stress polycythaemia, also known as Gaisbock’s syndrome, where the plasma volume may be 30ml/kg or less. It is a common disorder, especially in middle-aged, overweight male heavy smokers. It is associated with an increased risk of arterial occlusion causing myocardial infarction and stroke. The pathogenesis is obscure.
Myelofibrosis
Also known as myelosclerosis, myelofibrosis(MF) is characterised by the predominant features of gross marrow fibrosis with massive extramedullary haemopoiesis in liver and spleen. The fibrosis is reactive (a polyclonal proliferation of fibroblasts is present). Factors released from pathological megakaryocytes which proliferate in the bone marrow are thought to be the stimulus to the fibroblastic response. It is a chronic disorder of late middle age and beyond.
Blood and bone marrow changes
Anaemia is usually present; platelets and leukocytes are often increased, but become subnormal eventually. The blood film is typically leukoerythroblastic. Characteristic poikilocytes with a tear-drop shape are a consistent finding (Fig. 23.3).
Bone marrow cannot be aspirated. Trephine biopsy reveals variable cellularity with increased reticulin, progressing to massive deposition of collagen. Megakaryocytes are often increased. Bony trabeculae may be expanded.
Changes in other organs
The spleen is invariably enlarged, often to a massive degree. Lymphoid follicles are preserved but the red pulp is expanded with diffuse areas of extramedullary haemopoiesis. The liver is often enlarged, with obvious foci of haemopoiesis present. Occasionally, lymph nodes are also involved. The liver involvement may result in portal hypertension, causing oesophageal varices and ascites.
Clinical features
Symptoms are caused by the anaemia and massive splenomegaly. Symptoms of hypermetabolism may also be present, especially weight loss and night sweats. Sclerosis of bones may be apparent on X-ray examination. Many patients have a history of polycythaemia rubra vera or essential thrombocythaemia; in others, the onset is insidious. With supportive therapy (blood transfusion), survival is often a few years. If the enlarged spleen is troublesome, splenectomy can be safely performed, surprisingly without exacerbation of the anaemia, though sometimes leading to significant hepatomegaly. There are reports of improved blood counts and reduction in splenic size with thalidomide. Some patients have achieved a remission with resolution of marrow fibrosis following allogeneic stem cell transplantation, suggesting a graft-versus-disease effect.
Essential thrombocythaemia
Essential thrombocythaemia, a myeloproliferative disorder, is an important cause of thrombocytosis. The diagnosis is being made more frequently as an incidental finding now that automated cell counters are routinely used.
Blood and bone marrow changes
The platelet count is raised, often to 1000 × 109/l and even to 3000 × 109/l. Neutrophil leukocytosis may also be a feature. ‘Giant’ platelets and megakaryocyte fragments may be present. Anaemia, when present, is due to iron deficiency from chronic blood loss. Howell–Jolly bodies and other features of hyposplenism may be apparent due to splenic infarction. Bone marrow cellularity is normal or increased, megakaryocytes predominate and some increase in marrow reticulin is common. Mutations in the JAK2 kinase gene are found in about 40% of cases.
Changes in other organs
The spleen may be enlarged but is usually normal or reduced in size due to infarction. Ischaemic changes in the area supplied by digital arteries may be present, as may evidence of infarction in other organs. Paradoxically, haemorrhagic lesions also occur, often in the gastrointestinal tract.
Clinical features
The disorder may be asymptomatic for many years. Painful ischaemic lesions of the digits are an occasional feature. Paradoxical haemorrhage, which may be serious, occurs particularly in association with platelet counts over 1000 × 109/l. Treatment with aspirin and hydroxyurea or the megakaryocyte-suppressing drug anagrelide is effective and survival prolonged. Progression to myelofibrosis may occur.
PLASMA CELL NEOPLASMS
Plasma cells are the immunoglobulin-producing cells resulting from terminal differentiation of B-cells and are normally identifiable in the bone marrow. Diffuse neoplastic, monoclonal proliferation of plasma cells throughout the red marrow is characteristic of the disorder multiple myeloma. When the proliferation is more localised an apparently discrete plasma cell tumour develops, usually in bone, but also in soft tissue of the airways and head and neck—solitary plasmacytoma. Monoclonal proliferation of IgM-producing plasma cells and lymphoplasmacytoid cells in the reticulo-endothelial organs, bone marrow, liver and spleen is present in a subgroup of non-Hodgkin’s lymphoma (NHL) called lymphoplasmacytic lymphoma—the combination of NHL (usually lymphoplasmacytic type) with an IgM paraprotein, with or without signs of hyperviscosity, is known as Waldenström’s macroglobulinaemia.
Multiple myeloma





Multiple myeloma is a common neoplastic disease affecting especially the elderly; almost all cases occur after the age of 40 years. Multifocal plasma cell tumours erode the bones of the axial skeleton; the plasma cells synthesise a monoclonal immunoglobulin or light chain, referred to as the M-component or paraprotein in plasma. The M-component is present in over 99% of cases of multiple myeloma; it is most commonly IgG (60% of cases) but may be IgA or immunoglobulin light chains only (kappa or lambda). IgD and IgE M-components are unusual and IgM types are much more commonly a feature of Waldenström’s macroglobulinaemia. In two-thirds of cases of IgG and IgA myeloma, a large excess of free light chains is produced in addition to the complete immunoglobulin molecule, presumably due to a functional defect in the malignant plasma cells. While immunoglobulins cannot pass the glomerular filter, free light chains are small enough to enter the urine, where they are called Bence Jones protein.Free light chains can now be measured accurately in serum and urine by nephelometric methods. The result expressed as a serum free light chain ratio (kappa/lambda) is a useful tumour marker in assessing response to treatment. The plasma concentration of the unaffected immunoglobulins is often markedly suppressed (‘immune paresis’).
Paraprotein formation is not unique to multiple myeloma; a monoclonal immunoglobulin is present occasionally in CLL and lymphomas and, rarely, in carcinomatous disease. Furthermore, a proportion of elderly subjects are found to have a stable paraprotein without immune paresis and without the other features of multiple myeloma or lymphoproliferative disease—so-called ‘monoclonal gammopathy of uncertain significance’ (MGUS). Only 1% per year of patients with MGUS progress to multiple myeloma.
The pathology of the bone disease in myeloma is becoming better understood and leading to improved treatments for this catastrophic manifestation of the disease (Fig. 23.41). The osteolytic destruction of the axial skeleton (sites of haemopoiesis in adults) results from malignant plasma cells stimulating osteoclasts to erode bone. This leads to lytic lesions, pathological fractures, generalised osteoporosis and hypercalcaemia. For some time it has been recognised that chemical messengers (cytokines) produced from the interaction of malignant plasma cells with their microenvironment stimulate the osteoclast activity. Such cytokines were known as osteoclast activating factors (OAFs) and are now known to include interleukin-1 and -6.
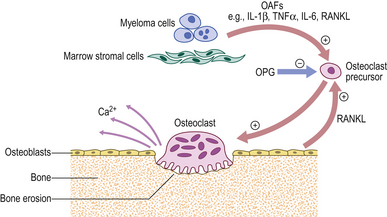
Fig. 23.41 The pathogenesis of bone disease in multiple myeloma. (OAF, osteoclast activating factors; OPG, osteoprotegerin.)
Recently another system of messengers has been shown to be important in the development of the bone disease in myeloma. A protein called RANKL is expressed by osteoblasts and plasma cells; it binds to its ligand RANK on the osteoclast surface and stimulates the osteoclast to erode bone. A second protein called osteoprotegerin (OPG) normally blocks this interaction. However, in myeloma, it is suggested that the levels of OPG are reduced and this RANKL stimulation of bone erosion goes unchecked. This understanding has produced successful treatments to limit the bone disease. A group of drugs called bisphosphonates (e.g. clodronate and pamidronate) directly inhibit osteoclasts and are used routinely in myeloma. Recombinant OPG is being used in trials.
Blood and bone marrow changes
Anaemia is common. The blood film often has rouleaux formation: a tendency for the erythrocytes to adhere to each other and form columns one cell across in the blood film, due to the presence of a high concentration of immunoglobulin. The anaemia is normocytic, but automated cell counters may suggest a high MCV, probably due to rouleaux formation. In advanced disease, pancytopenia is present. Abnormal plasma cells are only occasionally seen in the peripheral blood.
The marrow is hypercellular; 10–90% of the cells are morphologically abnormal plasma cells, including multinucleate forms (Fig. 23.42). Increased numbers of osteoclasts actively resorbing bone may be seen on trephine biopsy.
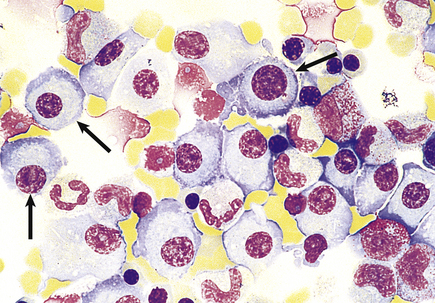
Fig. 23.42 The bone marrow in multiple myeloma. An infiltrate of atypical plasma cells (arrows) is a major diagnostic criterion.
The plasma cell infiltrate and discrete tumours are present in those bones normally containing red marrow, especially the skull, ribs, vertebrae and pelvis (Fig. 23.43). The distal long bones and those of the extremities are rarely involved. Generalised osteoporosis is common.
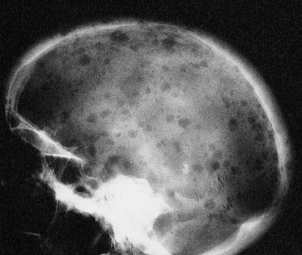
Changes in other organs
Renal involvement is present in over half of the cases. The most common abnormality is the presence of protein casts in the distal convoluted and collecting tubules with surrounding giant cells and atrophy of tubular cells—‘Bence Jones or myeloma kidney’. Metastatic calcification, changes of pyelonephritis and primary amyloid may also be present in the kidneys. Systemic amyloidosis (Ch. 7) is present in 10% of cases, particularly in the tongue, heart and peripheral nerves, as well as the kidneys.
Clinical features
The clinical features in multiple myeloma are outlined in Figure 23.44. Not all are present in every case. Bone pain is present in the majority and is often severe. Renal failure is common and prognostically sinister. Hyperviscosity is especially associated with IgA paraproteins because of the physical characteristics of IgA.
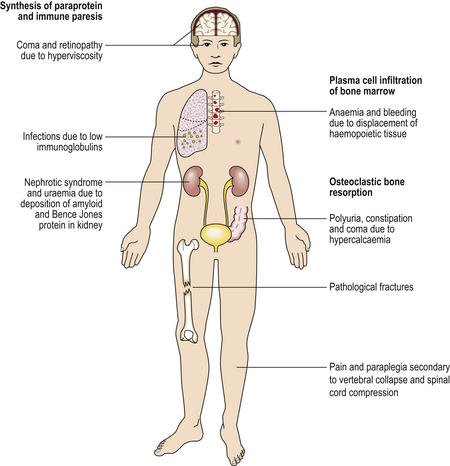
Treatment involves management of acute problems, including hypercalcaemia, cord compression and renal failure, and the routine use of bisphosphonates to limit bone disease. The disease is not curable but significant improvements in therapy have occurred in recent times. A number of initial approaches to therapy are successful and include single agent chemotherapy (melphalan) or combination chemotherapy such as vincristine, doxorubicin (Adriamycin), dexamethasone (VAD) or cyclophosphamide, thalidomide, dexamethasone (CTD). These are presently being compared in the UK Myeloma IX trial. Radiotherapy provides effective control of localised bone pain.
Randomised data have shown a survival advantage for high-dose melphalan therapy and autologous stem cell transplantation which is now offered routinely to younger patients. Relapsed disease has been successfully treated with thalidomide and new agents including the proteasome inhibitor bortezomib and lenalidomide. Survival is improving with these innovations in treatment to a median of 4–5 years.
Solitary plasmacytoma
Solitary tumours composed of malignant plasma cells identical in morphology to those in multiple myeloma occasionally arise in bone or extra-osseous sites. A paraprotein may be synthesised by the cells of the tumour. Solitary plasmacytoma of the bone may progress to multiple myeloma in some 60% of cases.
Waldenström’s macroglobulinaemia
In Waldenström’s macroglobulinaemia the marrow, lymph nodes, liver and spleen are infiltrated by cells with morphology between lymphocytes and plasma cells, which synthesise a monoclonal IgM. The disorder is uncommon and tends to occur in the later years. It is recognised as a low-grade NHL (lymphoplasmacytic) with moderate lymph node enlargement and hepatosplenomegaly, and does not produce the characteristic osteolytic bone lesions and hypercalcaemia of multiple myeloma. Hyperviscosity is common and occurs at lower paraprotein concentrations than is the case in myeloma, due to the physical characteristics of the IgM molecule. Visual deterioration, lethargy, bleeding tendency and disturbance of consciousness result. Hyperviscosity is treated with plasma exchange.
Survival for many years follows treatment with alkylating agents. Newer treatments include cladribine and rituximab.




