CHAPTER 96 Anatomy, Histology, Embryology, and Developmental Anomalies of the Small and Large Intestine
ANATOMY
MACROSCOPIC FEATURES
Colon and Rectum
The colon is a tubular structure approximately 30 to 40 cm in length at birth in the full-term infant. In the adult, the colon measures approximately 150 cm, about one quarter of the length of the small intestine. The diameter of the colon is greatest in the cecum (7.5 cm) and narrowest in the sigmoid (2.5 cm). The colon is continuous with the small intestine proximally at the ileocecal valve and ends distally at the anal verge (Fig. 96-1). The external appearance of the colon differs from that of the small intestine because the longitudinal muscle fibers of the colon coalesce into three discrete bands called taeniae, located at 120-degree intervals about the colonic circumference: taenia liberis, taenia omentalis, and taenia mesocolica. The taeniae start at the base of the appendix and extend continuously to the proximal rectum. Outpouchings of the colon, the haustra, are found between the taeniae. Semilunar folds characterize the mucosa between haustra. Small sacs of peritoneum filled with adipose tissue, the appendices epiploicae, are found on the external surface of the colon.
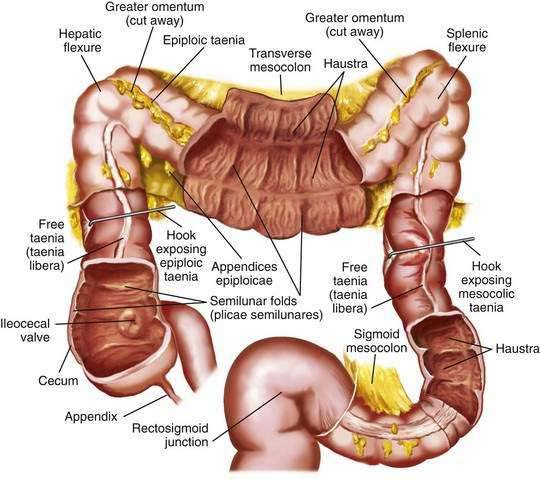
The most proximal portion of the colon, the cecum, lies in the right iliac fossa and projects downward as a blind pouch below the entrance of the ileum. The cecum is a sacculated structure 6 to 8 cm in length and breadth. Because of its large diameter, it is the part of the colon most apt to rupture with distal obstruction, and cecal tumors can grow to be quite large without producing symptoms of obstruction. The mobility of the cecum normally is fixed by a small mesocecum; an anomaly in fixation exists in 10% to 20% of people, especially women, predisposing them to cecal volvulus. The vermiform appendix is a blind outpouching of the ceum that begins inferior to the ileocecal valve. Appendiceal anatomy is discussed further in Chapter 116.
Anal Canal
The anal canal is approximately 5 cm in length in the adult and has discrete upper and lower demarcations. The anorectal ring is located proximally and is composed of the upper portion of the internal sphincter, the longitudinal muscle of the rectum, the deep portion of the external sphincter, and the puborectalis portion of the levator ani muscle; distally, the anal verge represents the transition of anoderm to true skin. The mucosa of the distal 3 cm of the rectum and the anal canal contains 6 to 12 redundant longitudinal folds called the columns of Morgagni, which terminate in the anal papillae. These columns are joined together by mucosal folds called the anal valves, which are situated at the dentate line. The muscularis mucosae disappears in the anorectal canal, and the inner circular coat of muscularis propria thickens to form the internal anal sphincter. The external anal sphincter surrounds the anal canal, and its fibers blend with those of the levator ani muscle to attach posteriorly to the coccyx and anteriorly to the perineal body. The anatomy and function of these muscles are described in more detail in Chapter 125.
Vasculature
The superior mesenteric artery delivers oxygenated blood to the distal duodenum, the jejunum and ileum, the ascending colon, and the proximal two thirds of the transverse colon. The remainder of the colon is supplied by branches of the inferior mesenteric artery. The arterial supply of the anal area is from the superior, middle, and inferior hemorrhoidal arteries, which are branches of the inferior mesenteric, hypogastric, and internal pudendal arteries respectively. Venous drainage of the anus is by both the systemic and portal systems. The internal hemorrhoidal plexus drains into the superior rectal veins and then into the inferior mesenteric vein, which, with the superior mesenteric vein, joins the splenic vein to form the portal vein. The distal anus drains by the external hemorrhoidal plexus through the middle rectal and pudendal veins into the internal iliac vein. (See Chapter 114 for additional discussion of the intestinal blood supply.)
Extrinsic Innervation
The autonomic nervous system—sympathetic, parasympathetic, and enteric—innervates the gastrointestinal tract. The sympathetic and parasympathetic nerves constitute the extrinsic nerve supply and connect with the intrinsic nerve supply, which is composed of ganglion cells and nerve fibers within the intestinal wall. Innervation of the small intestine and colon is discussed in detail in Chapters 97 and 98, respectively.
MICROSCOPIC FEATURES
General Considerations
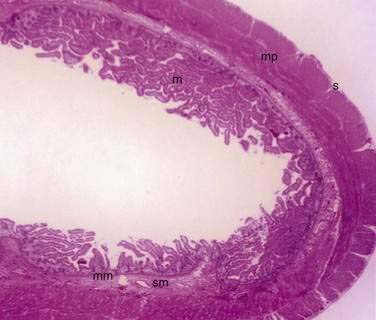
The small and large intestine share certain histologic characteristics. The wall of the small intestine and colon is composed of four layers: mucosa (or mucous membrane), submucosa, muscularis (or muscularis propria), and adventitia (or serosa) (Fig. 96-2).
Mucosa
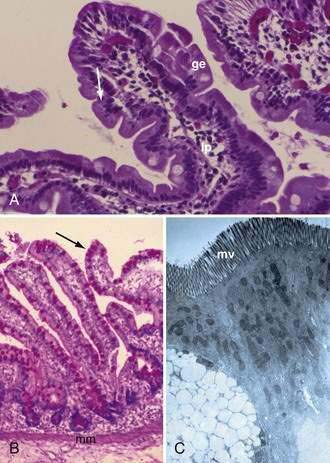
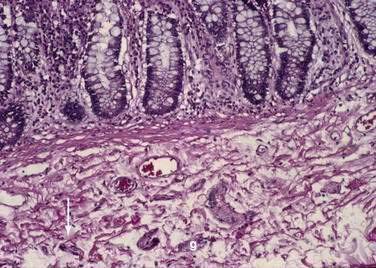
The mucosa is the innermost layer formed by glandular epithelium, lamina propria, and muscularis mucosae (Fig. 96-3A and B). The glandular epithelium forms cylindrical structures called crypts. The lamina propria, which supports the epithelium, is a layer of reticular connective tissue with elastin, reticulin, and collagen fibers, lymphocytes, plasma cells, and eosinophilic granulocytes, as well as lymphatics and capillaries. The muscularis mucosae consists of a thin layer of smooth muscle at the boundary of the mucosa and submucosa.
Signaling pathways such as Wnt, bone marrow protein (Bmp), PTEN/PI3K, Notch, hedgehog, platelet-derived growth factor, and SOX9 play important roles in the development of the intestinal epithelium.1–4
Wnt signaling plays a role in promoting cell proliferation; maintains stem cells in an undifferentiated state; defines compartmentalization into Paneth cells, proliferative, and differentiation zones along the crypt-villus axis; and directs early secretory lineage development as well as terminal differentiation of Paneth cells through the transcription factor SOX9.1
Bmps belong to the transforming growth factor-β family. Bmp signaling is important in intestinal development and homeostasis. It antagonizes crypt formation and stem cell self-renewal and has a role in directing maturation of all three secretory cell types (goblet, enteroendocrine, and Paneth). Bmp signaling in the mesenchyme plays a significant role in crypt morphogenesis; loss of Bmp leads to multiplication and elongation of crypts.2
PTEN/PI3K pathway plays a role in cell survival, proliferation, and growth.1
Notch proteins mediate cell fate decisions and pattern by regulating the helix-loop-helix factor that controls terminal differentiation. Notch directs development of absorptive cells and depletion of secretory lineage cells, and increases proliferation.1
The hedgehog (Hh) signaling pathway is important in crypt and villus morphogenesis and maintenance of stem cells.3 Both Sonic (Shh) and Indian (Ihh) play a role. Ihh is critical for the maintenance of intestinal stem cells, whereas Shh inhibits the growth of the villi. The contractile subepithelial pericryptal myofibroblasts represent a major target for Hh signaling. Hh signals sent to the epithelium-associated subepithelial myofibroblasts localize the precrypt structure and maintain the organization of the crypt-villus axis. Hh signaling also inhibits the proliferation or differentiation of smooth muscle and the proliferation compartment of the intestinal epithelium.3
Platelet-derived growth factor A stimulates mesenchymal condensation, proliferation, and evagination of overlying epithelium to form villi.3
Studies in animals also have contributed to the understanding of the molecular mechanism of the different pathways.1,2
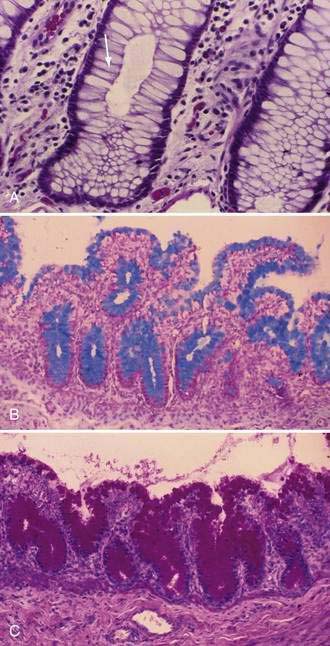
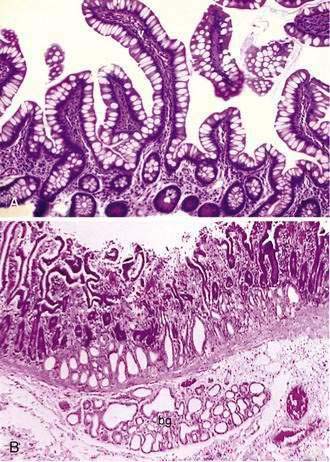
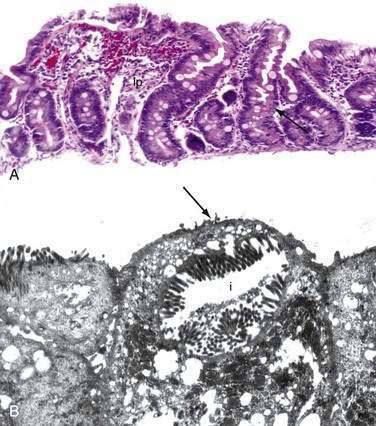
Stem cells are pluripotential cells located at the base of the intestinal crypts. Stem cells give rise to all types of mature intestinal epithelial cells and at the same time replenish themselves through self-renewal. Undifferentiated cells have fewer intracellular organelles and microvilli than do absorptive cells. The absorptive cells (see Fig. 96-3A) are high columnar cells with oval basal nuclei, eosinophilic cytoplasm, and a periodic acid–Schiff (PAS)–positive free surface, the brush border (see Fig. 96-3B). On electron microscopic examination, the brush border is seen to be composed of microvilli (see Fig. 96-3C), which are more numerous in the small intestinal than in the colonic epithelium. Small bowel enterocyte microvilli are estimated to increase the luminal surface area of the cell 14- to 40-fold. Goblet cells are oval or round, with flattened basal nuclei (Fig. 96-4A); their cytoplasm is basophilic, metachromatic (see Fig. 96-4B), and PAS positive (see Fig. 96-4C). Paneth cells are flask shaped and have an eosinophilic granular cytoplasm and a broad base positioned against the basement membrane (Fig. 96-5). Paneth cells contain zinc, antimicrobial peptides, and growth factors and secrete lysoenzymes. Enteric antimicrobial peptides produced by Paneth cells protect against intestinal infection and maintain enteric homeostasis.5 A cathelin-related antimicrobial peptide (CRAMP) identified in neonatal epithelium during the first weeks after birth, confers protection from Listeria monocytogenes.5
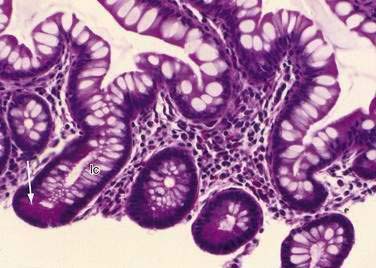
These neuroendocrine cells historically have been divided into argentaffin cells (granules able to reduce silver nitrate) and argyrophilic cells (granules that reduce silver nitrate only in the presence of a chemical reducer). Argentaffin cells stain positive with bichromate salts and also are called enterochromaffin cells. These cells are oval or triangular (also called “halo cells”) and have a basal position in relation to the remaining epithelial cells (Fig. 96-6A) and a pale cytoplasm filled with dark-stained granules. Variation in shapes and cell types has been detected with immunohistochemical staining. The unifying APUD concept—amine precursor, uptake, and decarboxylation—ascribes common characteristics to these neuroendocrine cells. APUD cells are a group of cells with a common embryonic neural crest origin and with similar cytochemical and electron microscopic features; however, embryologic and morphologic data support an endodermal origin of these cells.
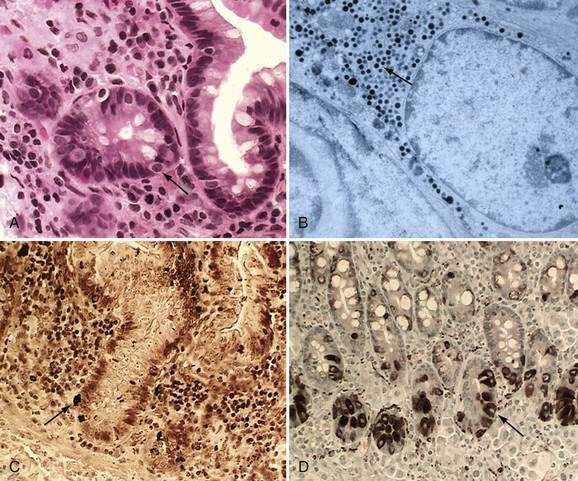
Ultrastructurally, enteroendocrine cells contain membrane-bound granules with variably sized electrodense cores (see Fig. 96-6B), averaging 100 to 250 nm in diameter, and consisting of large dense-core vesicles and smaller, synaptic-type microvesicles. Neurosecretory granules can be demonstrated with the Grimelius stain by light microscopy as dark granules (see Fig. 96-6C), or, more specifically, by immunofluorescence, and with immunohistochemical stains such as neuron-specific enolase, chromogranin, and synaptophysin. Chromogranin enables visualization of the large dense-core vesicles, and synaptophysin targets the small synaptic-like microvesicles (see Fig. 96-6D).6 Vesicular monoamine transporter 1 (VMAT1) and 2 (VMAT2) are two isoforms of the adenosine triphosphate (ATP)–dependent vesicular monoamine transporters. These antigens, derived from both the large and small dense-core vesicles, are expressed differentially in small dense-core vesicles. Both are expressed in neuroendocrine cells, but VMAT1 is restricted to serotonin-producing enterochromaffin cells, and VMAT2 is expressed in histamine-producing cells, enterochromaffin-like cells, and pancreatic islet cells.7 Specific immunohistochemical stains allow for identification of individual protein products of the neuroendocrine cells.
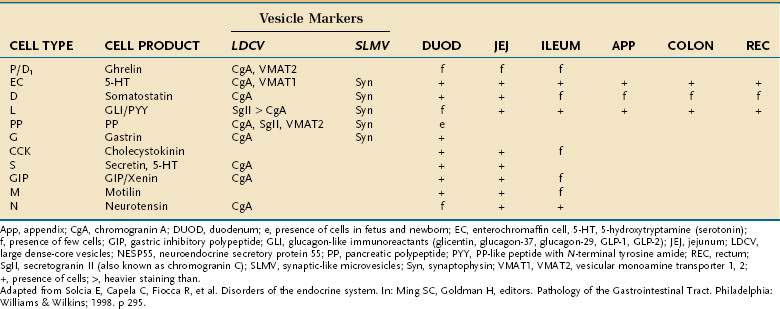
Besides releasing hormones in the blood, neuroendocrine cells also regulate secretion, absorption, motility, mucosal cell proliferation, and possibly immunobarrier control.6 Electron microscopy and immunohistochemistry have led to the identification of a variety of cell types (Table 96-1). Designation according to the nature of the stored peptide is preferable to characterization of neuroendocrine cells by letters. Serotonin-producing enterochromaffin cells, vasoactive intestinal polypeptide (VIP), and somatostatin D cells are distributed throughout the small and large intestine. Gastrin-, ghrelin-, gastric inhibitory peptide (GIP)-, secretin-, and cholecystokinin-producing cells are found predominantly in the stomach and proximal small intestine; peptide YY-, glucagon-like peptide (GLP)-1-, GLP-2-, and neurotensin-secreting cells are found in the ileum.8
Table 96-1 Enteroendocrine Cells of the Intestinal Tract: Cell Types and Products, Vesicle Markers, and Distribution
Neuroendocrine cells originate from a common precursor cell in the intestinal crypt. The earliest cell fate is regulated by the Notch signaling pathway (see earlier). Math1 is the first factor involved in endocrine specification, followed by neurogenin3.8 Pax4 and Pax6, paired ox homeodomain transcription factors, and Nkx2.2 also are required for neuroendocrine differentiation.8,9 As mature neuroendocrine cells migrate to the tip of the villi, they undergo apoptosis and are extruded into the lumen.
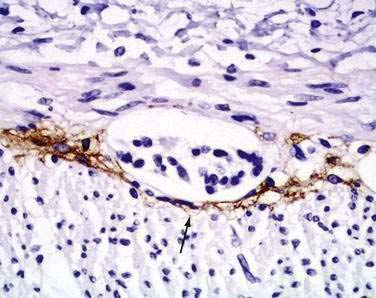
Interstitial cells of Cajal (ICC) are present in both the small intestine and the colon and are mesenchymal cells, located in the myenteric plexus, the muscularis propria, and the submucosa (Fig. 96-7). The distribution of the ICC is similar in children and in adults although a difference in their distribution is seen in fetuses of different gestational ages.10 Recognized as the pacemaker cells of the intestine, the ICC regulate intestinal motility by generating slow waves and determining frequency of smooth muscle contraction; they also amplify the neuronal signals, mediate neurotransmission from enteric motor neurons to smooth muscle cells, and set the smooth muscle membrane potential gradient. The ICC are spindle shaped or stellate, with long ramified processes, and have large, oval light-staining nuclei with sparse perinuclear cytoplasm. The ICC express the receptor for tyrosine kinase (c-Kit) or CD117 which is necessary for their maintenance. Serotonin regulates the number of the ICC by increasing their proliferation.11 Immunohistochemical stains that use antibodies against c-Kit allow the ICC to be labeled. The distribution and onset of appearance of these cells in the gastrointestinal tract have been described.10
Muscularis or Muscularis Propria
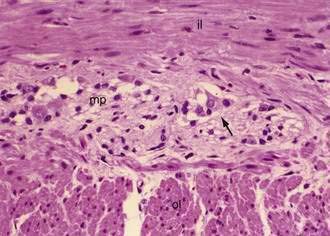
The muscularis propria, mainly responsible for contractility, consists of two layers of smooth muscle: an inner circular coat and an outer longitudinal coat arranged in a helicoidal pattern. A prominent nerve fiber plexus called the myenteric plexus, or Auerbach’s plexus, is found between these two muscle layers (Fig. 96-8). Parasympathetic and postganglionic sympathetic fibers terminate in parasympathetic ganglion cells, and postganglionic parasympathetic fibers terminate in smooth muscle.
Small Intestine
The mucosa of the small intestine is characterized by mucosal folds (plicae circulares, or valves of Kerckring) and villi. The mucosal folds are composed of mucosa and submucosa. Villi are mucosal folds that decrease in size from the proximal to distal small intestine and are of different shapes in the various segments of the small intestine: they may be broad, short, or leaf-like in the duodenum; tongue-like in the jejunum; and finger-like more distally (Fig. 96-9A). The villous pattern also may vary in different ethnic groups. Thus, for example, biopsy specimens from Africans, Indians, South Vietnamese, and Haitians have shorter and thicker villi, an increased number of leaf-shaped villi, and more mononuclear cells in comparison with specimens from North Americans.
Intestinal villus morphogenesis begins when mesenchymal aggregates impinge on the basal aspect of the epithelium to produce primitive folds. By nine to 10 weeks of gestation, the pseudostratified squamous epithelium converts to a single layer of columnar cells that lines mesenchymal stalks or the lamina propria.12 During mid- to late gestation, the basic tissue architecture of the intestine is established through epithelial-mesenchymal interaction. Induced by signals from mesoderm-derived mesenchyme, the endoderm-derived epithelium evaginates to form villi and intervillus regions. The intervillus region eventually invaginates into the mucosa to form crypts.1 Contractile subepithelial pericryptal myofibroblasts contribute mechanically to crypt formation and are the major source of instructive signals to the epithelium.3
Two types of glands are present in the small intestine: Brunner’s glands and crypts of Lieberkühn (intestinal crypts). Brunner’s glands are submucosal glands (see Fig. 96-9B) found primarily in the first portion of the duodenum and in decreased numbers in the distal duodenum; their function is to secrete a bicarbonate-rich alkaline secretion that helps neutralize gastric chyme. In children these glands also may be present in the proximal jejunum. Brunner’s glands open into the intestinal crypts and morphologically resemble pyloric glands.
Crypts of Lieberkühn are tubular glands that extend to the muscularis mucosae (see Fig. 96-5). The crypts are occupied mainly by undifferentiated cells and Paneth cells. Cells are generated at the crypt base and proceed to migrate toward the villus. During this migration, these cells mature and differentiate into a secretory lineage (goblet cells, enteroendocrine cells, Paneth cells) and enterocytes. The commitment of the stem cells to differentiate is acquired in the upper third of the crypt, where cells lose their ability to divide. The constant renewal of enterocytes is regulated by human acyl-coenzyme A synthetase.13
Most types of enteroendocrine cells are present in the duodenum. Cells that produce ghrelin, gastrin, cholecystokinin, motilin, neurotensin, GIP, and secretin are restricted to the small intestine.6
The proportions of these cells differ in the villi and crypts, as well as in different segments of the intestine. Ninety percent of the villus epithelial cells are absorptive cells intermingled with goblet and enteroendocrine cells. The proportion of goblet to absorptive cells is increased in the ileum. The ICC are more abundant in the myenteric plexus of the small bowel than in the colon.10
Colon
Colonic epithelial cells are generated from stem cells at the base of the crypts and migrate toward the intestinal lumen after three to five days, on initiation of apoptosis. Most epithelial cells undergo apoptosis when they lose contact with the extracellular matrix and are shed into the lumen through caspase activation. Caspase activation is responsible for the cleavage of essential intracellular proteins leading to apoptosis and therefore loss of anchorage.14
Anal Canal
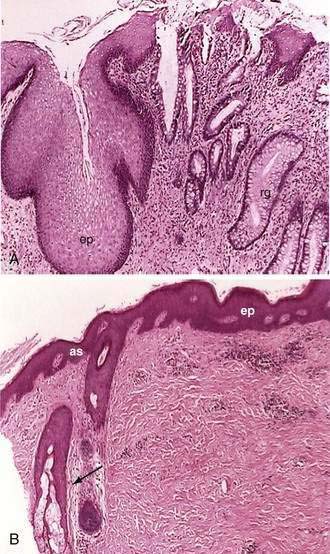
Microscopically the anal canal is divided into three zones: proximal, intermediate or pectinate, and distal or anal skin. The proximal zone is lined by stratified cuboidal epithelium, and the transition with the rectal mucosa, which is lined by high columnar mucus-producing cells, is called the anorectal histologic junction (Fig. 96-10A). The intermediate or pectinate zone is lined by stratified squamous epithelium but without adnexae (e.g., hair, sebaceous glands) and also is referred to as anoderm. Its proximal margin, in contact with the proximal zone, is called the dentate line; its distal margin, in contact with the anal skin, constitutes the pectinate line, also referred to as the mucocutaneous junction (see Fig. 96-10B). The anal skin is lined by squamous stratified epithelium and contains hair and sebaceous glands.
Nerves
The intrinsic nervous system (enteric nervous system) consists of subserosal, muscular, and submucosal plexuses. The subserosal plexus contains a network of thin nerve fibers, without ganglia, that connects the extrinsic nerves with the intrinsic plexus. The myenteric plexus, or Auerbach’s plexus, is situated between the outer and inner layers of the muscularis propria (see Fig. 96-8); it consists of ganglia and bundles of unmyelinated axons that connect with the ganglia forming a meshwork. These axons originate from processes of the ganglion cells and extrinsic vagus and sympathetic ganglia. The deep muscular plexus is situated on the mucosal aspect of the circular muscular layer of the muscularis propria. It does not contain ganglia; it innervates the muscularis propria and connects with the myenteric plexus. The submucosal plexus, or Meissner’s plexus, consists of ganglia and nerve bundles. The nerve fibers of this plexus innervate the muscularis mucosae and smooth muscle in the core of the villi. Fibers from this plexus also form a mucosal plexus that is situated in the lamina propria and provides branches to the intestinal crypts and villi. The ganglion cells of the submucosal plexus are distributed in two layers: one is adjacent to the circular muscular layer of the muscularis propria; the other is contiguous to the muscularis mucosae. Ganglion cells are large cells, isolated or grouped in small clusters called ganglia (Fig. 96-11). Ganglion cells have an abundant basophilic cytoplasm, a large vesicular round nucleus, and a prominent nucleolus. Ganglion cells are scarce in the physiologically hypoganglionic segment 1 cm above the anal verge.
EMBRYOLOGY
MOLECULAR REGULATION OF INTESTINAL MORPHOGENESIS
The induction of endoderm appears to be governed by nodal or transforming growth factor-β signaling.15 Specification is initiated by transcription factors expressed in the different regions of the intestinal tube. Thus, PDX1 specifies the duodenum, CDXC the small intestine, and CDXA the large intestine and rectum.16 Differentiation of the gastrointestinal tract depends on the interaction between the endoderm and mesoderm through the Hox code. Signaling from the mesoderm to endoderm is regulated by the Hox genes that encode homeodomain-containing transcription factors. Induction of the Hox code in the mesoderm results from expression of Shh through the endoderm of the midgut and hindgut. Shh is a signaling molecule that acts as a morphogen or form-producing substance in a variety of organ systems. When prompted by this code, the mesoderm instructs the endoderm to form the various components of the midgut and hindgut regions, for example, the small bowel, cecum, colon, and cloaca.16 As indicated by animal studies, Hox genes contribute to the subdivision of the intestine, and formation of the ileocecal valve that separates the small and the large intestine. Shh also plays a crucial role in the development of the hindgut.17
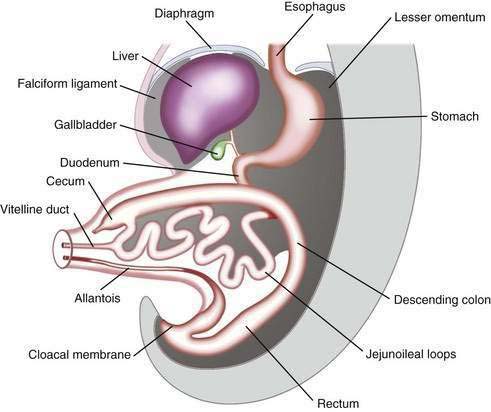
The primitive gut results from incorporation of the endoderm-lined yolk sac cavity into the embryo, following embryonal cephalocaudal and lateral folding. The primitive gut is composed of a blind-ended tube in the cephalic and caudal portions of the embryo, which is the progenitor of the foregut and hindgut; the midgut (Fig. 96-12) is connected to the yolk sac by the vitelline duct. The endoderm gives rise to the epithelial lining of the gastrointestinal tract; muscle, connective tissue, and peritoneum originate from the splanchnic mesoderm. During the ninth week of development, the epithelium begins to differentiate from the endoderm with villus formation and differentiation of epithelial cell types. Organogenesis is complete by 12 weeks of gestation.
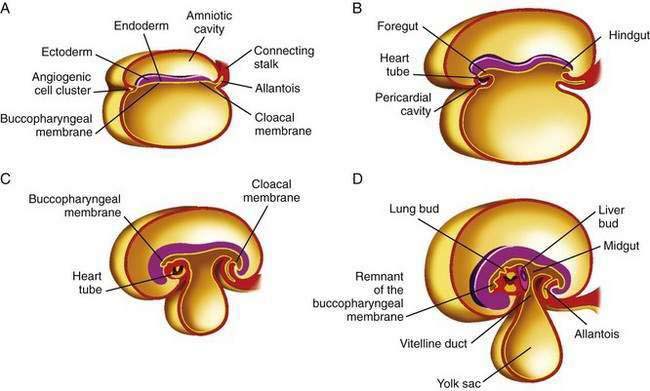
Figure 96-12. Formation of the foregut, midgut, and hindgut (see text for details).
(From Sun B, editor. Langman’s Medical Embryology. 9th ed. Philadelphia: Lippincott Williams & Wilkins; 2004.)
Initially the foregut, midgut, and hindgut are in broad contact with the mesenchyma of the posterior abdominal wall. The intraembryonic cavity is in open communication with the extraembryonic cavity. Subsequently the intraembryonic cavity loses its wide connection with the extraembryonic cavity. By week five of embryonic development, splanchnic mesoderm layers are fused in the midline and form a double-layered membrane, the dorsal mesentery, between the right and left halves of the body cavity. The mesoderm surrounds the intestinal tube and suspends it from the posterior body wall, allowing it to hang into the body cavity. The caudal portions of the foregut, the midgut, and most of the hindgut thus are suspended from the abdominal wall by the dorsal mesentery extending from the duodenum to the cloaca. The dorsal mesentery forms the mesoduodenum in the duodenum, the dorsal mesocolon in the region of the colon, and the mesentery proper in the region of the jejunum and ileum.16
SPECIFIC STRUCTURES AND SYSTEMS
Duodenum
The duodenum originates from the terminal portion of the foregut and cephalic part of the midgut. With rotation of the stomach, the duodenum becomes C-shaped and rotates to the right; the fourth portion becomes fixed in the left upper abdominal cavity. The mesoduodenum fuses with the adjacent peritoneum; both layers disappear, and the duodenum becomes fixed in its retroperitoneal location. The lumen of the duodenum is obliterated during the second month of development by proliferation of its cells; this phenomenon is shortly followed by recanalization. Because the foregut is supplied by the celiac artery and the midgut by the superior mesenteric artery, the duodenum is supplied by both arteries and therefore is relatively protected from ischemic injury.16
Midgut
In a 5-week embryo, the midgut is suspended from the dorsal abdominal wall by a short mesentery and communicates with the yolk sac by way of the vitelline duct. The midgut gives rise to the duodenum distal to the ampulla, to the entire small bowel, and to the cecum, appendix, ascending colon, and the proximal two thirds of the transverse colon. The midgut rapidly elongates with formation of the primary intestinal loop. The cephalic portion of this loop, which communicates with the yolk sac by the narrow vitelline duct, gives rise to the distal portion of the duodenum, the jejunum, and a portion of the ileum; the distal ileum, cecum, appendix, ascending colon, and proximal two thirds of the transverse colon originate from the caudal limb. During week 6 of embryonic development, the primary intestinal loop enters the umbilical cord (physiologic umbilical herniation) (Fig. 96-13), and by week 10 it re-enters the abdominal cavity. The proximal portion of the jejunum is the first portion of the intestine to re-enter the abdominal cavity and becomes located on the left side; the subsequent loop that re-enters the abdominal cavity locates to the right. The cecal bud is the last segment to re-enter the abdominal cavity. The cecum originates as a small dilatation of the caudal limb of the primary intestinal loop by approximately 6 weeks of development. Initially it lies in the right upper quadrant; then it descends to the right iliac fossa, placing the ascending colon and hepatic flexure in the right side of the abdominal cavity. The appendix originates from the distal end of the cecal bud. Because the appendix develops during descent of the colon, its final position frequently is retrocecal or retrocolonic.
The primary intestinal loop rotates counterclockwise for approximately 270 degrees around an axis formed by the superior mesenteric artery. This rotation occurs in three stages (Fig. 96-14): the first stage occurs between six and eight weeks (90 degrees), the second stage is at nine weeks (180 degrees), and the third stage is at 12 weeks of gestation (270 degrees). Elongation of the bowel continues, and the jejunum and ileum form a number of coiled loops within the peritoneal cavity.16
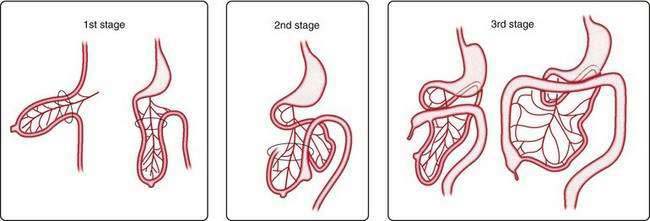
Figure 96-14. The three stages of normal intestinal rotation (see text for details).
(From Gosche JR, Touloukian RJ. Congenital anomalies of the midgut. In: Wyllie R, Hyams JS, editors. Pediatric Gastrointestinal Disease. Pathophysiology, Diagnosis, Management. 2nd ed. Philadelphia: WB Saunders; 1999.)
Mesentery
When the caudal limb of the primitive intestine moves to the right side of the abdominal cavity, the dorsal mesentery twists around the origin of the superior mesenteric artery. After the ascending and the descending portions of the colon reach their final destinations, their mesenteries fuse with the peritoneum of the posterior abdominal wall, and they become retroperitoneal organs. The appendix, cecum, and descending colon retain their free mesentery. The transverse mesocolon fuses with the posterior wall of the greater omentum. The mesentery of the jejunum and ileum at first is in continuity with the ascending mesocolon; after the ascending colon becomes retroperitoneal, the mesentery only extends from the duodenum to the ileocecal junction.16
Arterial System
Vascular endothelial growth factor (VEGF)-A and its receptors, VEGFR-1 and VEGFR-2, are important for endothelial cell proliferation, migration, and sprouting. Angiopoietins and their receptors, Tie1 and Tie2, play a role in remodeling and maturation of the developing vasculature. Mutation in Tie2 has been reported in vascular dysmorphogenesis. Vascular malformation is briefly discussed in Chapter 36.
Lymphatic System
Lymphatics originate from endothelial budding of veins, after which the peripheral lymphatic system spreads by endothelial sprouting into the surrounding tissues and organs. Flt4 (also known as VEGFR-3), a receptor for VEGF, plays a role in development of the vascular as well as the lymphatic systems. Overexpression of VEGF-C, a ligand of Flt4, results in hyperplasia of lymphatic vessels in transgenic mice. The homeobox gene Prox1 is essential for normal development of the lymphatic system based on animal studies. Homeobox genes contain a conserved sequence of 183 nucleotides. The proteins encoded by homeobox-containing genes act as regulatory molecules that control the expression of other genes. Several families of homeobox-containing genes are known, including the murine Hox family, which has been implicated in pattern formation during embryogenesis. Disruption of this gene in mice causes chyle-filled intestine. Abnormalities in the development of the lymphatic system can result in lymphangiectasia (see Chapter 28).
Enteric Nervous System
The enteric nervous system originates from vagal, truncal, and sacral neural crest cells. Most of the enteric nervous system cells derive from the vagal and truncal neural crest, enter the foregut mesenchyma, and colonize the developing intestine in a cephalocaudal direction. The truncal neural crest gives rise to ganglia of the proximal stomach, whereas the vagal neural crest supplies ganglia to the entire intestine including the rectum; this colonization is complete by 13 weeks of embryonic development. A small component of the enteric nervous system originates from sacral neural crest cells. These cells form extraintestinal pelvic ganglia that colonize the hindgut mesenchyma before the arrival of the vagal-derived neural crest cells.18 The normal development of the enteric nervous system depends on the survival of cells developed from the neural crest, and their proliferation, movement, and differentiation into neurons and glial cells. Microenvironmental, genetic, or molecular mechanisms may intervene in these processes (see “Disturbance in the Enteric Nervous System”).
CLINICAL IMPLICATIONS
Table 96-2 summarizes the different congenital clinical entities that result from disturbances in embryologic development. Gastrointestinal malformations can be associated with extraintestinal defects when genes such as those that determine left-right asymmetry are involved. The CFC1 gene plays a role in establishing left-right axis. Mutations of this gene have been reported in extrahepatic biliary atresia, in the polysplenia syndrome (inferior vena cava abnormalities, preduodenal portal vein, intestinal malrotation, and situs inversus), and in right-sided stomach and congenital heart disease.19,20
Table 96-2 Causes of Abnormalities in Normal Embryologic Development
| Body wall | |
| Omphalocele | Failure of the intestine to return to the abdominal cavity after its physiologic herniation |
| Gastroschisis | Weakening of the abdominal wall |
| Mesentery | |
| Mobile cecum | Persistence of mesocolon |
| Volvulus | Failure of fusion of mesocolon with posterior abdominal wall |
| Vitelline duct | |
| Meckel’s diverticulum | Persistence of the vitelline duct (see Fig. 96-17) |
| Omphalomesenteric cyst | Focal failure of vitelline duct obliteration |
| Patent omphalomesenteric duct | Total failure of vitelline duct obliteration |
| Rotation | |
| Malrotation | Failure of rotation of the proximal midgut; distal midgut rotates 90 degrees clockwise |
| Nonrotation | Failure of stage 2 rotation (see Fig. 96-18) |
| Reverse rotation | Rotation of 90 degrees instead of 270 degrees |
| Proliferation | |
| Duplication | Abnormal proliferation of the intestinal parenchyma |
| Intestinal atresia and stenosis | |
| “Apple-peel” atresia | Coiling of proximal jejunum distal to the atresia around the mesenteric remnant |
| Duodenum | Lack of recanalization |
| Small and large intestine | Vascular “accident” |
| Anorectum | Disturbance in hindgut development |
| Enteric nervous system | |
| Hirschsprung’s disease | Failure of migration of ganglion cells; microenvironment changes |
| Intestinal neuronal dysplasia | Controversial |
| Pseudo-obstruction | Multifactorial (see Chapter 120) |
| Miscellaneous | |
| Intestinal epithelial dysplasia | Abnormalities of the basement membrane |
| Microvillus inclusion disease | Defective protein trafficking and abnormal cytoskeletal and microfilament function |
| Other genetic defects | |
| Congenital chloride diarrhea | Abnormal Cl−-HCO3− exchange in the ileum and colon |
| Congenital glucose or galactose malabsorption | Absence of Na+-glucose cotransporter for glucose and galactose |
| Congenital lactase deficiency | Decrease in lactase-phlorizin hydrolase |
| Congenital sodium diarrhea | Defective sodium-proton exchange |
| Congenital sucrase/isomaltase deficiency | Abnormal intracellular transport, aberrant processing, and defective function of sucrase or isomaltase |
| Cystic fibrosis | Defective cystic fibrosis transmembrane conductance regulator |
ABNORMALITIES IN NORMAL EMBRYOLOGIC DEVELOPMENT
ABDOMINAL WALL
Omphalocele
Omphalocele is a congenital hernia involving the umbilicus. It is covered by an avascular sac composed of fused layers of amnion and peritoneum (Fig. 96-15). The umbilical cord usually is inserted into the apex of the sac, and the blood vessels radiate within the sac wall. Although a central defect is present in the skin and the linea alba, the remainder of the abdominal wall is intact, including the surrounding musculature. Because a small occult omphalocele of the umbilical cord may not be observed at birth, it is recommended that the umbilical cord be tied at least 5 cm from the abdominal wall at the time of delivery. Close inspection of the umbilical cord before clamping will avoid clamping an occult omphalocele.
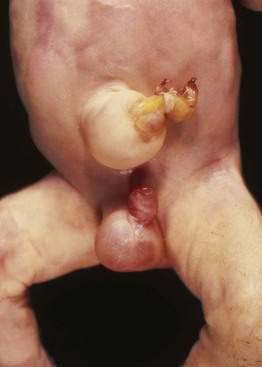
Figure 96-15. A newborn with an omphalocele. Note the translucent sac-like structure with its attached umbilical cord.
With a large omphalocele, the liver and spleen frequently are outside the abdominal cavity. Associated anomalies occur in about 75% of children with omphalocele and include chromosomal abnormalities such as trisomy 13 or 18, nonchromosomal syndromes such as Beckwith-Wiedemann syndrome (mental retardation, hepatomegaly, large body stature, hypoglycemia), fetal valproate syndrome, exstrophy of the bladder or cloaca, and OEIS (omphalocele, exstrophy of the bladder, imperforate anus, spinal defect). Malformations of the musculoskeletal, cardiovascular, and central nervous systems, also can occur.21,22
Gastroschisis
Gastroschisis is an abdominal wall defect most commonly located to the right of an intact umbilical cord (Fig. 96-16). The incidence of gastroschisis is approximately 1 in 10,000 births overall, but approaches 7 in 10,000 among mothers younger than 20 years of age. Gastroschisis occurs more frequently in whites and in Hispanic infants than in other races or ethnicities. In gastroschisis, a sac is absent, and the extruded bowel is “padded” and thickened along its length from its extended exposure to the amniotic fluid. Histologically, the bowel usually is normal. Atresia occurs in 10% to 15% of children with gastroschisis. Almost all infants with gastroschisis also exhibit malrotation. Whereas prematurity is more common in children born with gastroschisis than it is in children with omphalocele, extraintestinal anomalies are much more common with omphalocele than they are with gastroschisis. The morbidity and mortality in patients with gastroschisis are largely related to intestinal atresia; other congenital anomalies also have been reported in a small number of patients.21,22 Gastroschisis may be complicated by necrotizing enterocolitis, with all its attendant short-term and long-term complications.
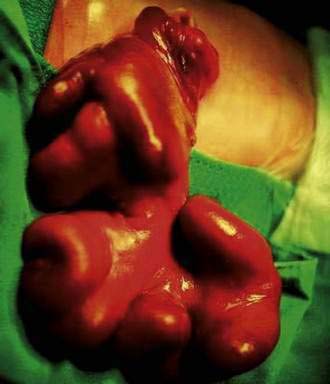
Most children with gastroschisis can undergo primary closure safely; however, for the child with significant intestinal atresia as a complication of gastroschisis, bowel exteriorization and secondary closure often are preferred treatment. It is crucial to conserve intestinal length in these children. Adhesive small bowel obstruction is a frequent and a serious complication, especially in the first year of life.23
MECKEL’S DIVERTICULUM AND OTHER VITELLINE DUCT ABNORMALITIES
Persistence of the ductal communication between the intestine and the yolk sac beyond the embryonic stage may result in several anomalies of the omphalomesenteric (vitelline) duct (Fig. 96-17) including (1) a blind omphalomesenteric duct, or Meckel’s diverticulum; (2) a central cystic dilatation in which the duct is closed at both ends but patent in its center, an omphalomesenteric or vitelline cyst; (3) an umbilical-intestinal fistula (see Fig. 96-17A), resulting from the duct remaining patent throughout its length; and (4) complete obliteration of the duct, resulting in a fibrous cord or ligament extending from the ileum to the umbilicus, as an omphalomesenteric band.24 In approximately 1% to 4% of all infants, a remnant of the embryonic yolk sac is retained, making the omphalomesenteric or vitelline duct the most common site of congenital gastrointestinal anomaly. Between the fifth and seventh weeks of gestation, the omphalomesenteric duct, which has connected the embryo to the yolk sac, attenuates, involutes, and separates from the intestine. Before this separation, the epithelium of the yolk sac develops an appearance similar to that of the gastric mucosa. Under normal circumstances the omphalomesenteric duct becomes a thin fibrous band that fragments and is absorbed spontaneously during the fifth to tenth week of gestation. Partial or complete failure of involution of the duct results in the variety of retained structures described above.
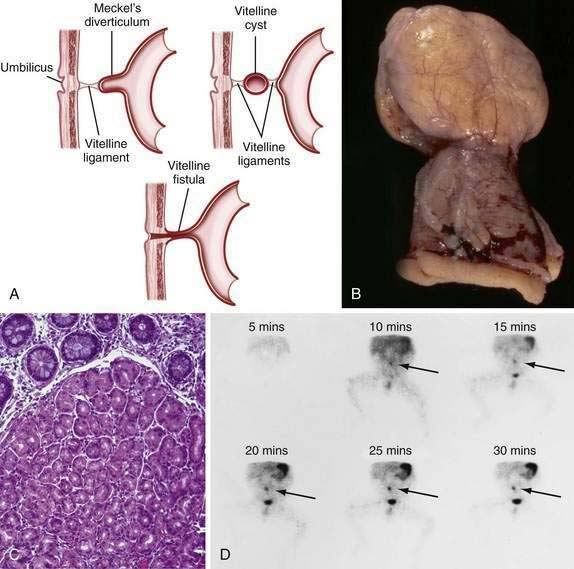
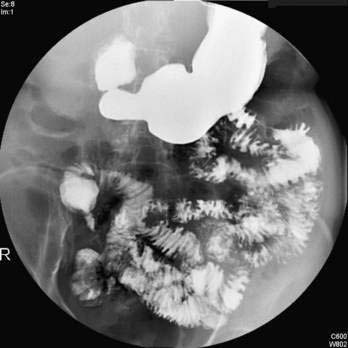
A Meckel’s diverticulum is an antimesenteric outpouching of the ileum that usually is found approximately 2 feet from the ileocecal junction (see Fig. 96-17B). It occurs in 1.2% to 2% of the population and has a male-to-female ratio of 3 : 1.25 Meckel’s diverticula account for 67% of all omphalomesenteric duct remnants.24 Length of the diverticulum varies, ranging from 1 to 10 cm. Ectopic gastrointestinal mucosa—duodenal, gastric, biliary or colonic, or aberrant pancreatic tissue—is present in about 50% of Meckel’s diverticula; most common is ectopic gastric mucosa, accounting for 80% to 85% of all Meckel’s diverticula–associated ectopic tissue (see Fig. 96-17C).
Diverticulitis of a Meckel’s diverticulum occurs as a result of acute inflammation. Most commonly, affected patients are diagnosed as having acute appendicitis, and the diagnosis of Meckel’s diverticulitis is made at exploratory laparotomy. Perforation occurs in approximately one third of patients with Meckel’s diverticulitis and may result from peptic ulceration.26 A chronic form of Meckel’s diverticulitis (Meckel’s ileitis) may mimic the presentation of Crohn’s disease of the ileum.
Meckel’s diverticulum may be an incidental finding.25 The presence of a Meckel’s diverticulum always should be considered in an infant or child with significant painless rectal bleeding. Standard abdominal plain films, barium contrast studies, and ultrasonographic imaging rarely are helpful in making the diagnosis. Because bleeding almost always is from ectopic gastric mucosa within the diverticulum, the Meckel’s scan, which allows imaging of the gastric mucosa, should be the initial diagnostic study (see Fig. 96-17D). Uptake of the 99mTc-pertechnetate is by the mucus-secreting cells of the gastric mucosa, not the parietal cells. Unfortunately, this study has only 85% sensitivity and 95% specificity.
Omphalomesenteric (or Vitelline) Cyst
Omphalomesenteric (or vitelline) cyst is more common in men and is characterized by a mucosa-lined intestinal cystic mass within the center of a fibrous cord.24 Infection of the cyst or intestinal obstruction may result.
Patent Omphalomesenteric (Vitelline) Duct
Patent omphalomesenteric (vitelline) duct represents a persistent connection between the distal ileum and the umbilicus. This fistula has a male-to-female ratio of 5 : 1, and accounts for 6% to 15% of omphalomesenteric duct remnants. The diagnosis usually is made in the first few weeks of life after separation of the umbilical cord from the newborn umbilicus. Foul-smelling discharge from the umbilicus occurs.27 Examination of the umbilicus reveals either an opening or a polypoid mass resulting from limited prolapse of the patent omphalomesenteric duct. Definitive diagnosis can be made by fistulography. Complications of this type of fistula include prolapse of the patent duct, or of the duct and the attached ileum, through the umbilicus, which may lead to partial intestinal obstruction. Prolapse should not be mistaken for an umbilical polyp, because excision of involved tissue might result in perforation. Resection is warranted.27
MALROTATIONS
Because anomalies of intestinal rotation may remain asymptomatic throughout life, their true incidence is unknown; a prevalence of 1 in 500 live births has been reported.28 Symptoms usually manifest within the first month of life, with bilious emesis and abdominal distention, but presentation may be delayed in mild cases to the fourth decade of life. Patients may have cramping abdominal pain, vomiting, diarrhea, abdominal tenderness, and blood or even mucosal tissue in the stool from ischemia. If ischemia is allowed to progress, peritonitis and hypovolemic shock may develop, potentially culminating in death. Delay in surgery in patients with ischemic injury may result in a short bowel, necessitating chronic total parenteral nutrition therapy and eventually small bowel transplantation, with or without liver transplantation. Most adult patients with anomalies of intestinal rotation have chronic symptoms for several months or years before diagnosis.
Classification
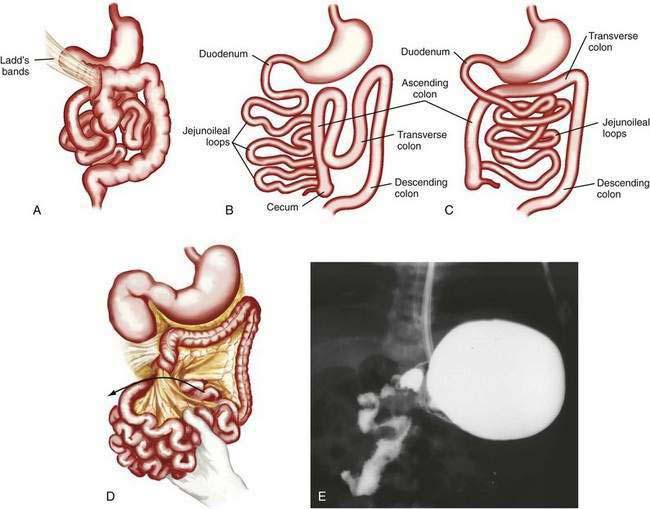
Anomalies of rotation usually are characterized by the stage in the rotational process at which normal embryonic development of the midgut has been interrupted. Most anomalies of midgut rotation occur during the second stage of rotation and have been characterized as nonrotation, reverse rotation, and malrotation (Fig. 96-18). Of these, nonrotation is most common and reflects a complete failure of the second stage of rotation. With this anomaly the intestinal tract occupies the same position in the abdomen as it does in an eight-week-old embryo; the small intestine is located to the right of the midline and the colon is positioned to the left.
Associated Abnormalities
Associated anomalies are seen in 30% to 60% of patients with defects in intestinal rotation. Nonrotation of the midgut is a significant finding in patients with omphalocele, gastroschisis, and diaphragmatic hernia. Rotation defects are seen in approximately 30% to 50% of infants with duodenal or jejunal atresia and in 10% to 15% of children with intestinal pseudo-obstruction; they also are associated with a variety of other conditions including Hirschsprung’s disease, esophageal atresia, biliary atresia, annular pancreas, meconium ileus, intestinal duplications, mesenteric cysts, Meckel’s diverticulum, urologic anomalies, and imperforate anus.29
Anomalies of rotation can cause acute or chronic intermittent obstruction from volvulus (see Fig. 96-18D,E). Venous and lymphatic obstruction, also from volvulus, can lead to malabsorption and abnormalities in intestinal motility. Patients may fail to thrive and present with chylous ascites and other symptoms and signs of lymphangiectasia resulting from chronic lymphatic obstruction.
Diagnosis and Management
If time allows, diagnosis can be made by upper gastrointestinal contrast examination and delineation of the site of the duodenojejunal junction. Findings on ultrasonography may suggest malrotation if the superior mesenteric vein is seen located to the left of the superior mesenteric artery, in contradistinction to the normal anatomy. In the child with acute onset of bilious vomiting and peritoneal signs, no diagnostic studies should be performed if they delay surgical intervention. In the full-term infant with bilious emesis, anomalies of rotation should be considered first and foremost, to avoid the morbidity and mortality associated with these lesions. Ladd’s procedure, including dividing Ladd’s bands, if present, widening of the mesentery, appendectomy, and fixation of the small intestine on the right and the colon on the left side of the abdomen, is the operation of choice.30
PROLIFERATION
Enteric Duplication
Enteric duplications are rare with an incidence of 1 in 4500 births. Enteric duplications are either tubular or spherical; the tubular type communicates with the normal intestinal tract, whereas the spherical type does not. Tubular duplications may join the intestine at one or at both of its ends. Except for duodenal duplications, duplications occur on the mesenteric side of the bowel, and a common blood supply and muscular coat are shared by the duplicated segment and the adjacent bowel. Duplication cysts may be completely isolated and have their own exclusive blood supply. Small bowel duplications often contain pancreatic tissue or gastric mucosa; the latter can be diagnosed by 99mTc radioisotopic imaging.31
The etiology of duplications is unclear, but may involve a defect in intestinal recanalization. Enteric duplications occur throughout the gastrointestinal tract but are most common in the ileum.31 Gastric duplications occur least commonly. Depending on the site of the duplication, and whether ectopic gastric mucosa is present (seen in approximately 50% of the cases), complications include intestinal hemorrhage, ulceration, perforation, intestinal obstruction, volvulus, intussusception, infection, pancreatitis, jaundice, hematobilia, and cutaneous enteric fistulas. Duplication of the rectum is the most common of the large bowel duplications and may be associated with constipation or obstipation. Colonic duplications frequently involve the entire colon. Occasionally, large bowel duplications affect several segments of the colon, leaving “skip areas” of normal colon. A high percentage of children with duplications have associated malformations. Adenocarcinoma, neuroendocrine carcinoma, and squamous carcinoma have been documented with gastric, small bowel, and colonic duplications,31,32 and carcinoid has been described in duplications of the rectum.
An intra-abdominal mass may be appreciated in a child with intestinal duplication, either by abdominal palpation or on rectal examination. Stool may contain occult blood from ulcerated ectopic gastric mucosa or ischemic damage. Other symptoms and signs include abdominal distention, constipation, vomiting, and respiratory distress.33 Generalized peritonitis can be the first manifestation of a perforated duplication cyst. In adults, acute abdomen, intra-abdominal mass, symptoms of colonic diverticulitis and chronic abdominal pain have been observed.34
INTESTINAL ATRESIA AND STENOSIS
Of all of the congenital anomalies of the midgut, atresias and stenoses occur most frequently. Intestinal atresia refers to a congenital complete obstruction of the intestinal lumen, whereas stenosis indicates a partial or incomplete obstruction. Atresias occur more commonly than do stenoses, and small bowel atresias have a reported incidence rate of 1 in 1500 live births.35 Small bowel atresias are more common in black infants, low birth weight infants, and twins. Jejunoileal atresias are distributed equally throughout the jejunum and ileum, and multiple atresias are found in up to 20% of children. Colonic atresia occurs infrequently and accounts for less than 10% of all atresias.
In the duodenum, atresia results from failure of recanalization of the solid stage of duodenal development, whereas in the remaining small intestine and colon, atresia is the result of intestinal ischemia. Evidence of a vascular “accident” is noted in 30% to 40% of infants with atresia; proposed mechanisms include volvulus, constriction of the mesentery in a tight abdominal wall defect such as gastroschisis, internal hernia, intussusception, and obstruction with perforation. Jejunoileal atresia may follow maternal use of ergotamine (in Cafergot for headaches) or cocaine taken during pregnancy and also is associated with congenital rubella. Atresias also may result from low-flow states and placental insufficiency35; in such cases, evidence of a vascular accident will be absent. Absence of fibroblastic growth factor 10 may result in intestinal atresia.36,37 In familial cases of jejunoileal atresia there is probably a disruption of a normal embryonic pathway, making this type of atresia a true embryologic malformation rather than an acquired lesion.38
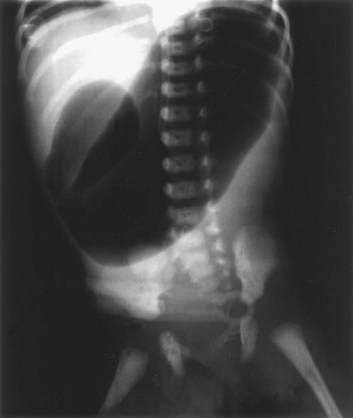
Clinically, the presentation is that of a proximal intestinal obstruction with bilious vomiting on the first day of life, usually without abdominal distention. With gastric dilatation, the epigastrium may appear to be full by inspection and palpation. Excessive retention of gastric bile–stained fluid is typical. Duodenal obstruction is diagnosed easily by abdominal films revealing a typical “double bubble” sign with a paucity of small intestinal air (Fig. 96-19). Mothers of infants with duodenal obstruction often have polyhydramnios, and uterine ultrasonography may even demonstrate a double bubble in the unborn fetus. Vomiting, abdominal distention, delayed meconium passage, and jaundice are more frequent with jejunoileal than duodenal atresia.39
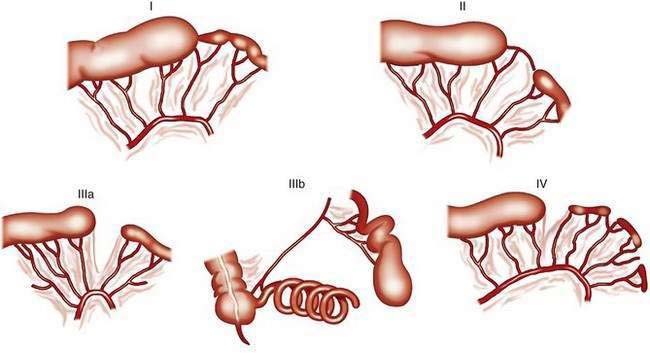
The classification system of Grosfeld and colleagues comprises five different types of jejunoileal and colonic atresias (Fig. 96-20).39a In the “apple-peel” atresia or “Christmas tree” deformity (type IIIb), proximal atresia with wide separation of the bowel loops is associated with absence of the distal superior mesenteric artery. The distal ileum receives its blood supply by retrograde perfusion through the ileocolic artery. Type IIIb atresias account for less than 5% of all atresias. Atresias are far more common than stenoses, with a frequency ratio of 15 : 1. With the exception of multiple atresias and perhaps the apple-peel atresia, heredity appears to be of little significance in most cases.
Approximately 50% of children with duodenal atresia have associated malformations. Of this group, 30% have Down syndrome.39 Major anomalies occur less frequently with jejunoileal atresias and colonic atresias than with duodenal atresia. The most common anomalies are malrotation, volvulus, and gastroschisis, all of which can cause intestinal ischemia in utero.40 Extragastrointestinal anomalies associated with atresias include cardiovascular, pulmonary, and renal malformations, and skeletal deformities. Prematurity is common, ranging in incidence from 25% in ileal atresias to 40% in jejunal lesions; 50% percent of babies with multiple atresias are born prematurely. If the obstruction occurs beyond the ampulla of Vater, bilious or feculent vomiting with abdominal distention is seen. The presence of meconium in the colon is uncommon at surgery, but variable amounts may be noted. With distal obstruction, abdominal films may demonstrate multiple dilated air-filled bowel loops. If perforation has occurred in utero, extraluminal air and intraperitoneal calcifications or calcifications within the scrotal sac may be present, suggesting meconium peritonitis. A “soap bubble” appearance of the ileum may suggest meconium ileus (cystic fibrosis). Air-fluid levels rarely are seen in meconium ileus. Prenatal ultrasonographic findings in jejunoileal atresia include dilated bowel and polyhydramnios.41
ANORECTUM
Anorectal malformations comprise a wide spectrum of diseases that can involve the male and female anus and rectum as well as the urinary and genital tracts.42 Anorectal malformations occur in 1 in 4000 to 5000 newborns and are more common among boys and in children with Down syndrome.43
During normal development, after appearance of the urorectal septum, migration of the primitive anus down the posterior wall of the cloaca may occur. Some experts postulate that a craniocaudal fusion of the lateral urorectal ridges occurs from the walls of the cloaca. Migration of the anus is completed when the urorectal septum reaches the perineum. Anorectal malformations during the fourth to twelfth weeks of gestation are believed to result from failure of migration of the anus and excessive fusion. Vascular accidents, maternal diabetes, and maternal ingestion of thalidomide, phenytoin, and trimethadione all have been proposed causes. Defective development of the dorsal cloaca also has been implicated44 and distal 6q deletions have been reported in sacral or anorectal malformations.45 Alteration in Shh signaling also may play a role in producing abnormal notochord development and sacral or anorectal malformations.46,47 Anorectal malformations may occur with higher frequency in infants born after in vitro fertilization.48
Different types of anorectal malformations are illustrated in Figure 96-21. Anorectal malformations are divided into low (infra- or translevator), high (supralevator), and intermediate categories. A functional and practical classification of these malformations, the Wingspread classification, is summarized in Table 96-3A. The classification in Table 96-3B is designed, according to Pena,49 to increase the physician’s awareness of the possibility of the presence of these lesions, as well as to establish therapeutic priorities.
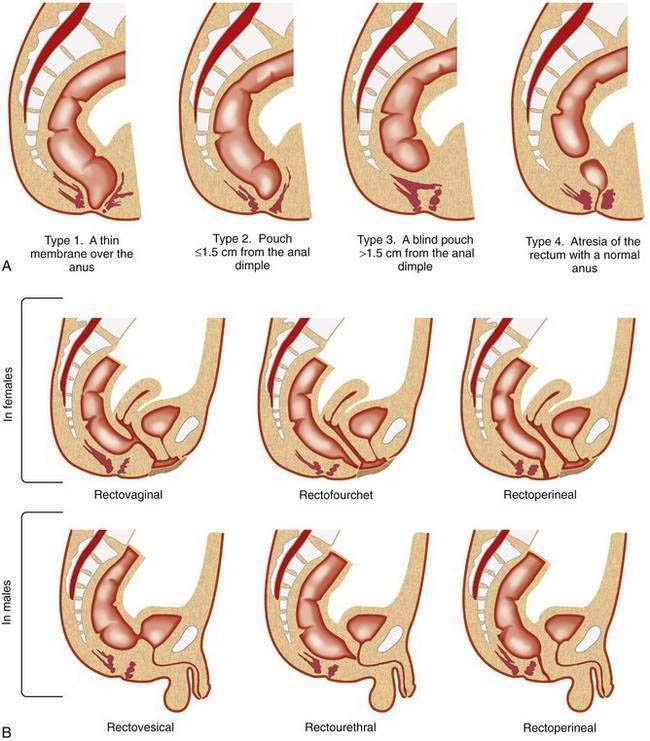
| Wingspread Classification | |
|---|---|
| MALE | FEMALE |
| Low* | |
| Anocutaneous fistula | Anovestibular fistula |
| Anal stenosis | Anal stenosis |
| Anocutaneous fistula | |
| Intermediate† | |
| Anal agenesis without fistula | Anal agenesis without fistula |
| Rectobulbar urethral fistula | Rectovaginal fistula |
| Rectovestibular fistula | |
| High‡ | |
| Anorectal agenesis | Anorectal agenesis |
| With rectoprostatic urethral fistula | With rectovaginal fistula |
| Without fistula | Without fistula |
| Rectal agenesis | Cloaca |
| Classification Based on the Need for Colostomy49 | |
|---|---|
| MALE | FEMALE |
| Colostomy Not Required | Colostomy Not Required |
| Perineal (cutaneous) fistula | Perineal (cutaneous) fistula |
| Colostomy Required | Colostomy Required |
| Rectourethral fistula | Vestibular fistula |
| Bulbar | |
| Prostatic | |
| Rectovesical fistula | Persistent cloaca |
| Imperforate anus without fistula | Imperforate anus without fistula |
| Rectal atresia | Rectal atresia |
* Low: infra-, or translevator.
† Intermediate: between high and low.
Rectourethral Fistula
In rectourethral fistula, by far the most frequent anorectal malformation in male children, the rectum descends through a portion of the pelvic floor musculature but focally deviates anteriorly and communicates with the posterior urethra. This fistula may end in the lower posterior (bulbar) or in the upper posterior (prostatic) urethra.49 Prenatal echogenic calcifications within the bowel, due to a mixture of meconium and urine, should suggest an anorectal malformation with rectourinary fistula and bladder outlet obstruction.50 Children with prostatic urethral fistulas more commonly have sacral and urologic defects (60%) than do children with bulbar prostatic fistula (30%). Eighty-five percent of children with rectourethral bulbar fistula achieve fecal continence after repair, compared with 60% of children with rectoprostatic fistula.
Persistent Cloaca
In the complex defect of persistent cloaca, the rectum, vagina, and urethra are fused into a single common channel that opens into one perineal orifice situated at the site of what should be the opening of the normal urethra. Prognosis depends on the intactness of the sacrum and the length of the common channel. Prognosis is better in those children with a shorter common channel (less than 3 cm) than in those with a common channel longer than 3 cm; the latter have a higher incidence of urologic anomalies.51 Associated urologic problems are an important consideration with persistent cloaca. For example, urologic emergencies from obstructive uropathy are common, and hydrocolpos may compress the opening of the ureters, resulting in bilateral megaureters and massive vesicoureteral reflux.
Associated Abnormalities
Other associated abnormalities have been reported in 70% of children with anorectal malformation (Table 96-4).42,43 Anorectal malformations occur in malformation syndromes and with chromosomal anomalies.43,52
Table 96-4 Common Abnormalities Associated with Anorectal Malformations
Data adapted from Cho S, Moore SP, Fangman T. One hundred three consecutive patients with anorectal malformations and their associated anomalies. Arch Pediatr Adolesc Med 2001; 155:587-91.
The higher and more complex the anorectal defect, the greater the chance of severe urologic anomalies (72%); sacral abnormalities also are frequent. Children with a persistent cloaca or rectovesical fistula have a 99% chance of having an associated genitourinary anomaly, whereas less than 10% of children with low fistula have such abnormalities. Overall, patients with additional anomalies are more likely to have high lesions than are patients with isolated anorectal malformations.43 Boys with low and high anorectal malformation have a high incidence of genital and gastrointestinal anomalies, whereas urologic anomalies are more frequent in girls with high anorectal malformations.53 Long-term bowel dysfunction occurs in one third of boys with perineal fistula.
In the first 24 hours of life, a decision should be made whether a child needs a colostomy or simple anoplasty. The presence of an associated defect, either urologic or cardiac, that might be life threatening requires immediate evaluation. A cloaca with a common channel shorter than 3 cm can be repaired by posterior sagittal intervention, whereas a common channel longer than 3 cm requires a laparotomy.51
ENTERIC NERVOUS SYSTEM
Hirschsprung’s Disease
Hirschsprung’s disease (HD) is due to a congenital absence of ganglion cells in both the submucosal (Meissner’s) and myenteric (Auerbach’s) plexuses. Aganglionosis extends continuously for a variable distance proximal to the internal sphincter. Short-segment HD is most common with a transition zone from aganglionic colon to ganglionic colon at the level of the sigmoid. In long-segment HD the entire colon and even the small intestine may lack ganglia. With an incidence of 1 in 5000 live births, approximately 700 new cases of HD occur each year in the United States. The incidence is lowest in Hispanic and highest in Asian individuals. Approximately 10% of babies with Down syndrome have HD. Deletion of 17q21 and other chromosomal anomalies also have been reported.54 Familial occurrence has been reported in about 7% of cases. Familial cases have a male predominance with an increased incidence of long-segment aganglionosis. Affected families carry a high risk of familial recurrence of long-segment HD.55 HD is seen most commonly in full-term infants but on occasion does occur in premature births. In the short-segment type, a 4 : 1 male preponderance is observed, and in the long-segment type, the ratio is reduced to about 2 : 1. Short-segment HD accounts for nearly 90% of cases in childhood, and long-segment HD accounts for the remainder. It is rare that ultrashort-segment HD manifests in the pediatric population, but it does explain certain cases of chronic constipation that come to attention in adulthood.
Pathogenesis
Colonic Microenvironment Changes
The genetics of HD have now been characterized.18 Inheritance of the disease can be autosomal dominant, autosomal recessive, or polygenic. Penetration of mutations generally is low and depends on the extent of aganglionosis in affected family members. RET (rearranged during transfection) mutation penetrance is incomplete and sex dependent. It appears that the mutation, although increasing a child’s odds of having HD, is not predictive of the specific abnormality. Alterations of several genes have been implicated (Table 96-5).56–59
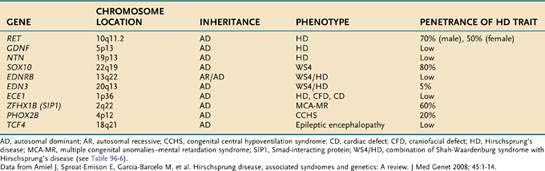
RET, a proto-oncogene that codes for a receptor tyrosine kinase protein (c-Kit), is the major susceptibility gene in HD, and maps to chromosome 10q11.2. More than 100 mutations of this gene have been identified and reduced c-Kit levels in the colon of patients with HD have been observed.54 Identified gene mutations currently account for only approximately half of all cases of HD, but it is recommended that RET exon 10 mutation analysis be done in all children with HD18; germline RET mutations also can cause multiple endocrine neoplasia type IIA (MEN-IIA). Although the test results will be negative in the vast majority of cases, the significance of identifying MEN-IIA mutation carrier status for that individual and family appear to justify such testing.54 Mutation of the RET has been noted in familial and sporadic HD.
Congenital birth defects are found 5% to 33% of patients with HD.54 Although HD usually occurs as an isolated event, in 30% of the patients it may be part of a syndrome (Table 96-6).
Table 96-6 Some Congenital Anomalies and Syndromes Associated with Hirschsprung’s Disease
MEN, multiple endocrine neoplasia.
Data from Amiel J, Sproat-Emison E, Garcia-Barcelo M, et al. Hirschsprung disease, associated syndromes and genetics: A review. J Med Genet 2008; 45:1-14.
Diagnosis
A contrast enema performed on an unprepared colon will show the distal narrowed hypertonic segment of bowel (usually seen best in a lateral projection). The transition zone between the narrowed distal and dilated proximal intestine will be seen in the most common form of HD—the rectosigmoid form (Fig. 96-22A)—but may not be seen with long- or ultrashort-segment intestinal involvement. In ultrashort-segment HD, a radiologic picture indistinguishable from that in functional constipation with dilated bowel extending to the anus usually is seen. The transition zone may not be evident in rectosigmoid HD if the patient has undergone cleansing enemas or colonic irrigation before the study. Although it has been suggested that the transition zone may not be evident in the first six weeks of life, it almost always is noted in the neonate with partial bowel obstruction.
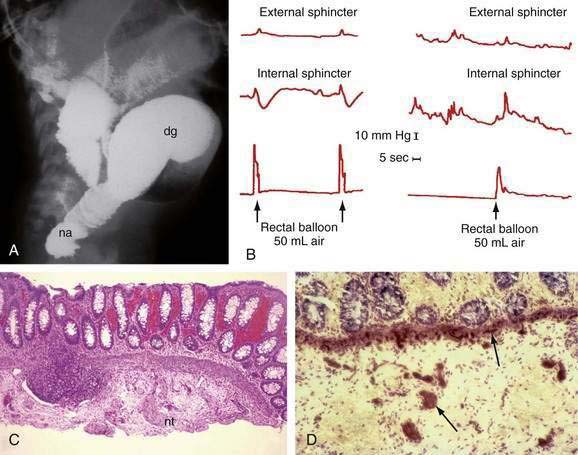
Anal manometry is the most reliable method by which the gastroenterologist can make the diagnosis of ultrashort-segment HD. A normal physiologic response to distention of the rectum is relaxation of (smooth muscle) internal sphincter pressure. In HD, not only does rectal distention fail to induce internal sphincter relaxation, but a paradoxical rise in external sphincter pressure often is seen (see Fig. 96-22B). Sufficient volumes of air must be used to stimulate rectal distention for a reliable study. A false-positive result most commonly is caused by a capacious rectum in a constipated child or with megacolon, in which case balloon distention may not stimulate the reflex. Up to 20% of normal children have a falsely absent reflex, especially if they are premature or of low birth weight. Nonetheless, a positive response such as internal sphincter relaxation is strong evidence against HD.
Suction biopsy of the rectal mucosa is the most reliable method of diagnosis, except in patients with ultrashort-segment HD. The biopsy capsule should be placed at least 2 cm above the mucocutaneous junction in infants and 3 cm above the junction in older children to avoid the physiologic hypoganglionic zone. To be certain of the absence of ganglion cells in the submucosal plexus, an experienced pathologist may need to review many serial sections. Hyperplastic sympathetic nerve fibers and proliferating Schwann cells are associated findings (see Fig. 96-22C), but can be absent in total aganglionosis.
Controversy exists regarding the type of stains necessary to make a diagnosis of HD. Because acetylcholinesterase is increased in the muscularis mucosae and lamina propria in the aganglionic segment (see Fig. 96-22D), staining for this enzyme has been used for many years. This technique requires fresh, non–formalin-fixed tissue and technical expertise; at best, this stain is confirmatory. False-positive and false-negative reports have been documented in total colonic aganglionosis.60 A variety of histochemical staining methods have been proposed for the identification of ganglion cells, but all are expensive, time-consuming, and unnecessary.
In the neonate, considerations in the differential diagnosis of HD include other causes of intestinal obstruction, such as meconium ileus, ileal atresia, meconium plug syndrome, and the microcolon seen in infants of diabetic mothers. When symptoms and signs of enterocolitis are present, diagnostic possibilities in the neonate also include primary necrotizing enterocolitis, HD-associated enterocolitis, milk protein–induced colitis (see Chapter 9), and sepsis with possible disseminated intravascular coagulation.
Intestinal Neuronal Dysplasia
Intestinal neuronal dysplasia (IND) is a motility disorder that manifests with intestinal obstruction or severe chronic constipation; characteristic biopsy findings include an increased number of enlarged ganglia and neural hypertrophy (Fig. 96-23A).62 In addition, acetylcholinesterase activity is increased in the lamina propria and muscularis mucosae. A full-thickness surgical biopsy specimen is often necessary to diagnose IND. IND has been reported as an isolated lesion affecting especially premature infants, or infants with a history of formula protein intolerance, ileal stenosis, or small left colon–meconium plug syndrome.
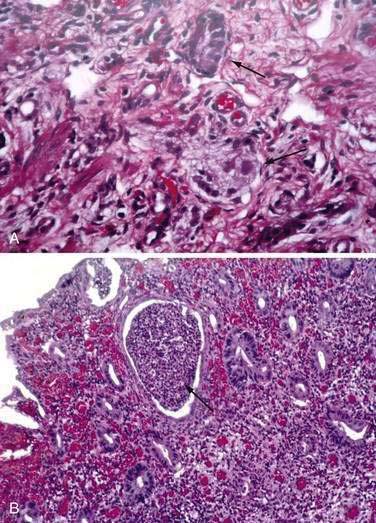
Three types of IND have been defined. IND type A usually manifests acutely in the neonatal period as severe constipation and enterocolitis. Biopsy features include mucosal inflammation (see Fig. 96-23B), ulceration with hyperplastic neural changes limited to the myenteric plexus, and increased acetylcholinesterase activity in the lamina propria and muscularis mucosae. The submucosal plexus in this type of intestinal neuronal dysplasia is histologically normal.
The pathogenesis of IND is controversial. In some patients it is congenital malformation, whereas in others it is an acquired phenomenon. IND also can be seen in association with other syndromes such as neurofibromatosis or MEN-IIB, in proximal-segment HD, and with congenital anomalies predominantly of the gastrointestinal tract.63 Other associated conditions include cystic fibrosis, microvillus inclusion disease, congenital anomalies, lipoblastomatosis, and inflammatory bowel disease. Therefore, IND may not represent a well-defined entity but rather may constitute a secondary phenomenon related either to age or to obstruction or inflammation.64 IND can resolve with age.
Chronic Intestinal Pseudo-obstruction
Congenital forms of neuropathic and myopathic pseudo-obstruction are rare and sporadic, perhaps representing new mutations (see Chapter 120). In these situations, a family history of pseudo-obstruction is lacking, as are any associated syndromes and evidence of other predisposing factors such as toxins, infections, ischemia, or autoimmune disease. Children with chromosomal abnormalities such as Down syndrome, as well as those with MEN-III or with Duchenne’s muscular dystrophy, may suffer from pseudo-obstruction.
MISCELLANEOUS AND GENETIC DEFECTS
Microvillus Inclusion Disease
Congenital microvillus atrophy, also known as microvillus inclusion disease, is an autosomal recessive disorder that may manifest with severe diarrhea shortly after birth and is characterized by atrophy of the intestinal villi and characteristic electron microscopic findings.65 Although the prevalence of microvillus inclusion disease is not known, it is reported to be the most common cause of familial intractable diarrhea.66 A female gender predominance has been observed, and consanguinity is reported in 20% of cases. The incidence of microvillus inclusion disease may be higher among Navajo Indians and persons from the Middle East. Defective protein trafficking and abnormal cytoskeletal and microfilament function have been proposed as possible etiologies.67 A blockage in the transport pathway from the Golgi apparatus leads to fusion of the small vesicles into microvillus inclusions.68 Secretory diarrhea is severe, with intolerance to oral feeding and unresponsiveness to most therapeutic modalities.
The wall of the small intestine is paper-thin in microvillus inclusion disease. The mucosa of the duodenum and small bowel is characterized by villus atrophy, hypoplastic or normal crypts, and normal or decreased cellularity of the lamina propria (Fig. 96-24A). The absence of the brush border membrane is demonstrated by lack of linear staining with PAS, carcinoembryonic antigen (CEA), and CD10.69 These stains also visualize the microvillus inclusions on light microscopy.
Evaluation by electron microscopy reveals ultrastructural abnormalities of the microvillus membrane, including disruption or absence of the brush border membrane, shortening and absence of the microvilli, and microvillus inclusions (see Fig. 96-24B). Although these lesions are most commonly noted in biopsies from the small intestine, microvillus inclusions also may be seen in specimens from the rectum and colon.
Total parenteral nutrition must be used to prolong survival. The secretory diarrhea persists but becomes less voluminous. Small bowel transplantation should be considered because without it, microvillus inclusion disease is fatal.70
Intestinal Epithelial Dysplasia
Intestinal epithelial dysplasia (IED), also known as tufting enteropathy, is a congenital enteropathy with early onset, severe intractable diarrhea, and characteristic microscopic findings.71 In IED, there is a variable degree of villus atrophy. Surface epithelial cells are arranged in tufts with a round apex. Tufts can also been seen in the colonic mucosa. These epithelial cells have an abnormal expression of E-cadherin and do not contain inclusions on electron microscopic examination. In the basement membrane, heparin sulfate proteoglycan is increased, and laminin is faint and irregular.71
The diarrhea is secretory, malabsorption intractable, and growth is impaired. Several cases of IED have been associated with congenital anomalies.71 Nonspecific punctate keratitis is observed in more than 60% of patients with IED.
IED is characterized by a basement membrane with abnormal distribution of 2 β1 integrin adhesion molecules along the crypt-villus axis.71 Tufts result from nonapoptotic epithelial cells that are no longer in contact with the basement membrane.
Congenital Glucose and Galactose Malabsorption
Familial glucose and galactose malabsorption, transmitted as an autosomal recessive trait, due to mutation in the SGlLT1 gene, is characterized by an absence of the active transport carrier protein (Na+-glucose cotransporter) for glucose and galactose.72 Ingestion of any formula containing glucose or galactose results in severe life-threatening watery diarrhea in the newborn period. Stools are strongly positive for reducing substances. Neither blood nor white blood cells are present in the stool. Findings on biopsy of the small bowel and colon are normal. Discontinuation of formula containing glucose, galactose, or lactose (lactose is metabolized to glucose and galactose) and institution of a fructose-containing formula with resultant therapeutic benefit usually are sufficient to make a clinical diagnosis of glucose or galactose malabsorption. Diarrhea abruptly ceases and the newborn begins to thrive when fructose-containing formula feedings are instituted. Some reports indicate that the severity of the diarrhea from glucose or galactose malabsorption diminishes with age because of the increased capacity of the intestinal flora to metabolize glucose.
Congenital Chloride Diarrhea
Congenital chloride diarrhea is an autosomal recessive disorder of intestinal Cl-HCO3 exchange caused by mutations of the SLC26A3 gene.73 The chloride-bicarbonate exchange mechanism in the ileum and colon is reversed, and chloride is actively secreted, resulting in a chloride-rich diarrhea. The baby with congenital chloride diarrhea often is premature and may present with an ileus or absence of passage of meconium. Watery diarrhea with a high stool chloride content and low stool pH is lifelong. Increased absorption of bicarbonate may result in dehydration, a hypochloremic metabolic alkalemia, hyponatremia, and marked hypokalemia. The stool contains no blood, no white blood cells, and no reducing substances. Urinary chloride is low. Biopsy specimens of the small intestine and colon are normal. Treatment is fluid and electrolyte replacement. Acid reduction with proton pump inhibitors has been tried with variable results.
Congenital Sodium Diarrhea
Congenital sodium diarrhea is caused by defective sodium or proton exchange.74 Patients have acidemia and hyponatremia. The stool concentration of HCO3 and sodium are increased.
Abdullah F, Arnold MA, Nabaweesi R, et al. Gastroschisis in the United States 1988-2003: Analysis and risk categorization of 4344 patients. J Perinatol. 2007;27:50-5. (Ref 22.)
Amiel J, Sproat-Emison E, Garcia-Barcelo M, et al. Hirschsprung disease, associated syndromes and genetics: A review. J Med Genet. 2008;45:1-14. (Ref 54.)
Auclair BA, Benoit YD, Rivard N, et al. Bone morphogenetic protein signaling is essential for terminal differentiation of the intestinal secretory lineage. Gastroenterology. 2007;133:887-96. (Ref 2.)
Cho S, Moore SP, Fangman T. One hundred three consecutive patients with anorectal malformations and their associated anomalies. Arch Pediatr Adolesc Med. 2001;155:587-91. (Ref 43.)
Fairbank TJ, Sala FG, Kanard R, et al. The fibroblast growth factor pathway serves a regulatory role in proliferation and apoptosis in the pathogenesis of intestinal atresia. J Pediatr Surg. 2006;41:132-6. (Ref 36.)
Goulet O, Salomon J, Ruemmele F, et al. Intestinal epithelial dysplasia (tufting enteropathy). Orphanet J Rare Dis. 2007;2:20-29. (Ref 71.)
Karnak I, Ocal T, Senocak ME, et al. Alimentary tract duplication in children: Report of 26 years experience. Turk J Pediatr. 2000;42:118-25. (Ref 33.)
Madison BB, Braunstein K, Kuizon E, et al. Epithelial hedgehog signals pattern the intestinal crypt-villus axis. Development. 2005;132:279-89. (Ref 3.)
Mastroiacovo P, Lisi A, Castilla EE, et al. Gastroschisis and associated defects: An international study. Am J Genet A. 2007;143:660-71. (Ref 21.)
Meier-Ruge WA, Ammann K, Bruder E, et al. Updated results on intestinal neuronal dysplasia (INDB). Eur J Pediatr Surg. 2004;14:384-91. (Ref 62.)
Pena A. Imperforate anus. In: Wyllie R, Hyams JS, editors. Pediatric Gastrointestinal Disease. Pathophysiology, Diagnosis, Management. 2nd ed. Philadelphia: WB Saunders; 1999:499. (Ref 49.)
Penco JM, Murillo JC, Hernandez A, et al. Anomalies of intestinal rotation and fixation: Consequences of late diagnosis beyond two years of age. Pediatr Surg Int. 2007;23:723-30. (Ref 29.)
Schonhoff SE, Giel-Moloney M, Leiter AB. Minireview: Development and differentiation of gut endocrine cells. Endocrinology. 2004;145:2639-44. (Ref 8.)
Scoville DH, Sato T, He XC, Li L. Current view: Intestinal stem cells and signaling. Gastroenterology. 2008;134:849-64. (Ref 1.)
1. Scoville DH, Sato T, He XC, Li L. Current view: Intestinal stem cells and signaling. Gastroenterology. 2008;134:849-64.
2. Auclair BA, Benoit YD, Rivard N, et al. Bone morphogenetic protein signaling is essential for terminal differentiation of the intestinal secretory lineage. Gastroenterology. 2007;133:887-96.
3. Madison BB, Braunstein K, Kuizon E, et al. Epithelial hedgehog signals pattern the intestinal crypt-villus axis. Development. 2005;132:279-89.
4. Mori-Akiyama Y, van den Born M, van Es JH, et al. SOX9 is required for the differentiation of Paneth cells in the intestinal epithelium. Gastroenterology. 2007;133:539-46.
5. Menard S, Foerster V, Lotz M, et al. Developmental switch of intestinal antimicrobial peptide expression. J Exp Med. 2008;205:183-96.
6. Rindi G, Leiter AB, Kopin AS, et al. The “normal” endocrine cell of the gut. Changing concepts and new evidence. Ann N Y Acad Sci. 2004;1014:1-14.
7. Jakobsen AM, Andersson P, Saglik G, et al. Differential expression of vesicular monoamine transporter (VMAT) 1 and 2 in gastrointestinal endocrine tumors. J Pathol. 2001;195:463-72.
8. Schonhoff SE, Giel-Moloney M, Leiter AB. Minireview: Development and differentiation of gut endocrine cells. Endocrinology. 2004;145:2639-44.
9. Desai S, Loomis Z, Pugh-Bernard A, et al. Nkx2.2 regulates cell fate choice in the enteroendocrine cell lineage of the intestine. Dev Biol. 2008;313:58-66.
10. Ahmed A, Yee H, Grecco MA, Kahn E. Distribution of c-Kit positive interstitial cells of Cajal in the gastrointestinal tract of fetuses and children [abstract]. Modern Pathol. 2001;14:3P.
11. Wouters MM, Gibbons SJ, Roeder JL, et al. Exogenous serotonin regulates proliferation of interstitial cells of Cajal in mouse jejunum through 5-Ht2B receptors. Gastroenterology. 2007;133:897-906.
12. Kim BM, Mao J, Taketo MM, Shivdasani RA. Phases of canonical Wnt signaling during the development of mouse intestinal epithelium. Gastroenterology. 2007;133:529-38.
13. Gassler N, Roth W, Funke B, et al. Regulation of enterocyte apoptosis by acyl-coA synthetase 5 splicing. Gastroenterology. 2007;133:587-98.
14. Hofmann C, Obermeier F, Artinger M, et al. Cell-cell contacts prevent anoikis in primary human colonic epithelial cells. Gastroenterology. 2007;132:587-600.
15. Cano DA, Hebrok M, Zenker M. Pancreatic development and disease. Gastroenterology. 2007;132:745-62.
16. Sadler TW. Digestive System. Langman’s Medical Embryology, 10th ed. Philadelphia: Lippincott Williams & Wilkins; 2006.
17. Mandhan P, Quan QB, Beasley S, Sullivan M. Sonic hedgehog, BMP4, and Hox genes in the development of anorectal malformations in ethylenethiourea-exposed fetal rats. J Pediatr Surg. 2006;41:2041-5.
18. Gariepy E. Developmental disorders of the enteric nervous system: Genetic and molecular bases. J Pediatr Gastroenterol Nutr. 2004;39:5-11.
19. Jacquemin E, Cresteil D, Raynaud N, Hadchouel M. CFC1 gene mutation and biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr. 2002;34:326-7.
20. Bamford RN, Roessler E, Burdine RD, et al. Loss-of-function mutations in the EGF-CF gene CFC1 are associated with human left-right laterality defects. Nat Genet. 2000;26:365-9.
21. Mastroiacovo P, Lisi A, Castilla EE, et al. Gastroschisis and associated defects: An international study. Am J Genet A. 2007;143:660-71.
22. Abdullah F, Arnold MA, Nabaweesi R, et al. Gastroschisis in the United States 1988-2003: Analysis and risk categorization of 4344 patients. J Perinatol. 2007;27:50-5.
23. Heinrich K, Huemmer HP, Reingruber B, Weber PG. Gastroschisis and omphalocele: Treatments and long-term outcomes. J. Pediatr Surg Int. 2008;24:167-73.
24. Sawada F, Yoshimura R, Ito K, et al. Adult case of an omphalomesenteric cyst resected by laparoscopic-assisted surgery. World J Gastroenterol. 2006;12:825-7.
25. Zani A, Eaton S, Rees CM, Pierro A. Incidentally detected Meckel diverticulum: To resect or not to resect? Ann Surg. 2008;247:276-81.
26. McKay R. High incidence of symptomatic Meckel’s diverticulum in patients less than fifty years of age: An indication for resection. Am Surg. 2007;73:271-5.
27. Nursal TZ, Yildirim S, Tarim A, Noyan T. Laparoscopic resection of patent omphalomesenteric duct in an adult. Surg Endosc. 2002;16:1638.
28. Palanivelu C, Rangarajan M, Shetty AR, Jani K. Intestinal malrotation with midgut volvulus presenting as acute abdomen in children: Value of diagnostic and therapeutic laparoscopy. J Laparoendosc Adv Surg Tech A. 2007;17:490-2.
29. Penco JM, Murillo JC, Hernandez A, et al. Anomalies of intestinal rotation and fixation: Consequences of late diagnosis beyond two years of age. Pediatr Surg Int. 2007;23:723-30.
30. Malek MM, Burd RS. Surgical treatment of malrotation after infancy: A population-based study. J Pediatr Surg. 2005;40:285-9.
31. Kuraoka K, Nakayama H, Kagawa T, et al. Adenocarcinoma arising from a gastric duplication cyst with invasion to the stomach: A case report and with literature review. J Clin Pathol. 2004;57:428-31.
32. Horne G, Ming-Lum C, Kirkpatrick AW, Parker RL. High-grade neuroendocrine carcinoma arising in a gastric duplication cyst: A case report with literature review. Int J Surg Pathol. 2007;15:187-91.
33. Karnak I, Ocal T, Senocak ME, et al. Alimentary tract duplication in children: Report of 26 years experience. Turk J Pediatr. 2000;42:118-25.
34. Simsek A, Zeybek N, Yagci G, et al. Enteric and rectal duplication cysts in the adult. ANZ J Surg. 2005;75:174-6.
35. Komuro H, Amagai T, Hori T, et al. Placental vascular compromise in jejunoileal atresia. J Pediatr Surg. 2004;39:1701-5.
36. Fairbank TJ, Sala FG, Kanard R, et al. The fibroblast growth factor pathway serves a regulatory role in proliferation and apoptosis in the pathogenesis of intestinal atresia. J Pediatr Surg. 2006;41:132-6.
37. Fairbank TJ, Kanard R, Del Moral PM, et al. Colonic atresia without mesenteric vascular occlusion. The role of the fibroblast growth factor 10 signaling pathway. J Pediatr Surg. 2005;40:390-6.
38. Shorter NA, Georges A, Perenyi A, Garrow E. A proposed classification system for familial intestinal atresia and its relevance to the understanding of the etiology of jejunoileal atresia. J Pediatr Surg. 2006;41:1822-5.
39. Ozturk H, Ozturk H, Gedik S, et al. A comprehensive analysis of 51 neonates with congenital intestinal atresia. Saudi Med J. 2007;28:1050-4.
39a. Grosfeld JL, Ballantine TVN, Shoemaker R. Operative management of intestinal atresia and stenosis based on pathologic findings. J Pediatr Surg. 1979;14:368.
40. Komuro H, Hori T, Amagai T, et al. The etiologic role of intrauterine volvulus and intussusception in jejunoileal atresia. J Pediatr Surg. 2004;39:1812-14.
41. Wax JR, Hamilton T, Cartin A, et al. Congenital jejunal and ileal atresia: natural prenatal sonographic history and association with neonatal outcome. J Ultrasound Med. 2006;25:337-42.
42. Levitt MA, Pena A. Anorectal malformations. Orphanet J Rare Dis. 2007;2:33-49.
43. Cho S, Moore SP, Fangman T. One hundred three consecutive patients with anorectal malformations and their associated anomalies. Arch Pediatr Adolesc Med. 2001;155:587-91.
44. van der Putte SC. Anal and ano-urogenital malformations: A histopathological study of “imperforate anus” with a reconstruction of the pathogenesis. Pediatr Dev Pathol. 2006;9:280-96.
45. Titomanlio L, Giurgea I, Baumann C, et al. A locus for sacral/anorectal malformations maps to 6q25.3 in a 0.3 Mb interval region. Eur J Hum Genet. 2006;14:971-4.
46. Mandhan P, Sullivan M, Quan QB, Beasley S. The contribution of the sonic hedgehog cascade in the development of the enteric nervous system in fetal rats with anorectal malformations. J Pediatr Surg. 2007;42:2080-5.
47. Mo R, Kim JH, Zhang J, et al. Anorectal malformations caused by defects in sonic hedgehog signaling. Am J Pathol. 2001;159:765-74.
48. Midrio P, Nogare CD, Di Gianantonio E, Clementi M. Are congenital anorectal malformations more frequent in newborns conceived with assisted reproductive techniques? Reprod Toxicol. 2006;22:576-7.
49. Pena A. Imperforate anus. In: Wyllie R, Hyams JS, editors. Pediatric Gastrointestinal Disease. Pathophysiology, Diagnosis, Management. 2nd ed. Philadelphia: WB Saunders; 1999:499.
50. Rolle U, Faber R, Robel-Tillig E, et al. Bladder outlet obstruction causes fetal enterolithiasis in anorectal malformation with rectourinary fistula. J Pediatr Surg. 2008;43:11-13.
51. Pena A, Levitt MA, Hong A, Midulla P. Surgical management of cloacal malformations: A review of 339 patients. J Pediatr Surg. 2004;39:470-9.
52. Stoll C, Alembik Y, Dott B, Roth MP. Associated malformations in patients with anorectal anomalies. Eur J Med Genet. 2007;50:281-90.
53. Ratan SK, Rattan KN, Pandey RM, et al. Associated congenital anomalies in patients with anorectal malformations—A need for developing a uniform practical approach. J Pediatr Surg. 2004;39:1706-11.
54. Amiel J, Sproat-Emison E, Garcia-Barcelo M, et al. Hirschsprung disease, associated syndromes and genetics: A review. J Med Genet. 2008;45:1-14.
55. Moore SW, Zaahl MG. A review of genetic mutation in familial Hirschsprung’s disease in South Africa towards genetic counseling. J Pediatr Surg. 2008;43:325-9.
56. Crone SA, Negro A, Trumpp A, et al. Colonic epithelial expression of ErbB2 is required for postnatal maintenance of the enteric nervous system. Neuron. 2003;37:29-40.
57. de Pontual L, Pelet A, Trochet D, et al. Mutations of the RET gene in isolated and syndromic Hirschsprung’s disease in human disclose major and modifier alleles at a single locus. J Med Genet. 2006;43:419-23.
58. van de Putte T, Maruhashi M, Francis A, et al. Mice lacking Zfhx1b, the gene that codes for the Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of Hirschsprung disease–mental retardation syndrome. Am J Hum Genet. 2003;72:465-70.
59. Dauger S, Pattyn A, Lofaso F, et al. Phox2b controls the development of peripheral chemoreceptors and afferent visceral pathways. Development. 2003;130:6635-42.
60. Nakao M, Suita S, Taguchi T, et al. Fourteen-year experience of acetylcholinesterase staining for rectal mucosa biopsy in neonatal Hirschsprung’s disease. J Pediatr Surg. 2001;36:1357-63.
61. Anitha M, Joseph I, Ding X, et al. Characterization of fetal and postnatal enteric neuronal cell lines with improvement in intestinal function. Gastroenterology. 2008;134:1424-35.
62. Meier-Ruge WA, Ammann K, Bruder E, et al. Updated results on intestinal neuronal dysplasia (INDB). Eur J Pediatr Surg. 2004;14:384-91.
63. Martucciello G, Torre M, Pini Prato A, et al. Associated anomalies in intestinal neuronal dysplasia. J Pediatr Surg. 2002;37:219-23.
64. Skaba R, Frantiova M, Horak J. Intestinal neuronal dysplasia. Eur J Gastroenterol Hepatol. 2006;18:699-701.
65. Sherman P, Mitchel D, Cutz E. Neonatal enteropathy: Defining the causes of protracted diarrhea of infancy. J Pediatr Gastroenterol Nutr. 2004;38:16-26.
66. Amosu MO, Mousa HM, Luquette M. Clinical challenges and images in GI. Microvillous inclusion disease. Gastroenterology. 2007;132:1225. 1639
67. Rund C, Carmichael B, Harris AA, Schaffner V. Small bowel biopsy in a 6-week-old infant. Microvillous inclusion disease in the small bowel. Arch Pathol Lab Med. 2006;130:e19-e21.
68. Mierau G, Wills EJ, Wyatt-Ashmead, et al. Microvillous inclusion disease: Report of a case with atypical features. Ultrastruct Pathol. 2001;25:517-21.
69. Groisman GM, Amar M, Livne E. CD10: A valuable tool for the light microscopic diagnosis of microvillous inclusion disease (familial microvillous atrophy). Am J Surg Pathol. 2002;26:902-7.
70. Ruemmele FM, Jan D, Lacaille F, et al. New perspectives for children with microvillous inclusion disease: Early small bowel transplantation. Transplantation. 2004;77:1024-8.
71. Goulet O, Salomon J, Ruemmele F, et al. Intestinal epithelial dysplasia (tufting enteropathy). Orphanet J Rare Dis. 2007;2:20-9.
72. Wright EM, Turk E, Martin MG. Molecular basis for glucose-galactose malabsorption. Cell Biochem Biophys. 2002;36:115-21.
73. Hihnala S, Hoeglund P, Lammi J, et al. Long-term clinical outcome in patients with congenital chloride diarrhea. J Pediatr Gastroenterol Nutr. 2006;42:369-75.
74. Mueller T, Wijmenga C, Phillips AD, et al. Congenital sodium diarrhea is an autosomal recessive disorder of sodium/proton exchange but unrelated to known candidate genes. Gastroenterology. 2000;119:1506-13.