CHAPTER 41 Anatomy, Histology, Embryology, and Developmental Anomalies of the Esophagus
ANATOMY AND HISTOLOGY
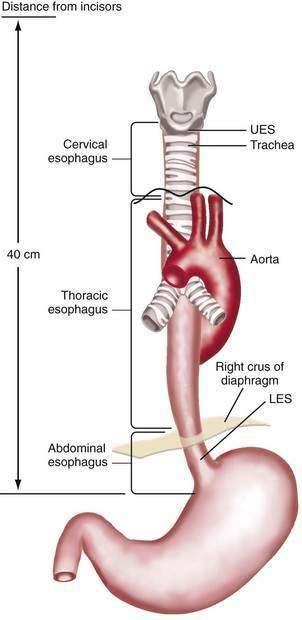
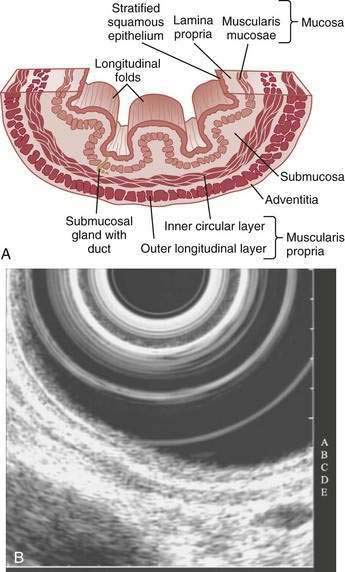
The esophagus acts as a conduit for the transport of food from the oral cavity to the stomach. To carry out this task safely and effectively, the esophagus is constructed as an 18- to 26-cm long hollow muscular tube with an inner “skin-like” lining of stratified squamous epithelium (Fig. 41-1). Between swallows the esophagus is collapsed, but the lumen distends up to 2 cm anteroposteriorly and 3 cm laterally to accommodate a swallowed bolus. Structurally, the esophageal wall is composed of four layers: innermost mucosa, submucosa, muscularis propria, and outermost adventitia; unlike the remainder of the gastrointestinal tract, the esophagus has no serosa.1,2 These layers are depicted anatomically and as viewed by endoscopic ultrasonography in Figure 41-2.
MUSCULATURE
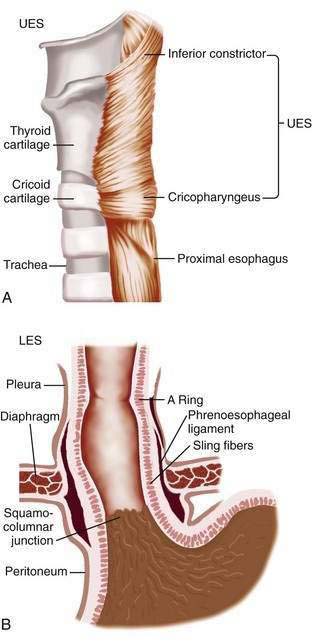
The muscularis propria is responsible for carrying out the organ’s motor function. The upper 5% to 33% is composed exclusively of skeletal muscle, and the distal 50% is composed of smooth muscle. In between is a mixture of both types.3 Proximally, the esophagus begins where the inferior pharyngeal constrictor merges with the cricopharyngeus, an area of skeletal muscle known functionally as the upper esophageal sphincter (UES) (Fig. 41-3A). The UES is contracted at rest and thereby creates a high pressure zone that prevents inspired air from entering the esophagus. Below the UES the esophageal wall comprises inner circular and outer longitudinal layers of muscle (see Fig. 41-2A). The esophageal body lies within the posterior mediastinum behind the trachea and left mainstem bronchus and swings leftward to pass behind the heart and in front of the aorta.1 At the T10 vertebral level the esophageal body leaves the thorax through a hiatus located within the right crus of the diaphragm (see Fig. 41-1). Within the diaphragmatic hiatus the esophageal body ends in a 2- to 4-cm length of asymmetrically thickened circular smooth muscle known as the lower esophageal sphincter (LES) (see Fig. 41-3B).4 The phrenoesophageal ligament, which originates from the diaphragm’s transversalis fascia and inserts on the lower esophagus, contributes to fixation of the LES within the diaphragmatic hiatus. This positioning is beneficial because it enables diaphragmatic contractions to assist the LES in maintenance of a high-pressure zone during exercise. The LES is contracted at rest, creating a high-pressure zone that prevents gastric contents from entering the esophagus. During swallowing, the LES relaxes to permit the swallowed bolus to be pushed by peristalsis from the esophagus into the stomach.
INNERVATION
The esophageal wall is innervated by parasympathetic and sympathetic nerves; the parasympathetics regulate peristalsis through the vagus nerve (Fig. 41-4). The cell bodies of the vagus nerve originate in the medulla. Those located within the nucleus ambiguus control skeletal muscle, and those located within the dorsal motor nucleus control smooth muscle. Medullary vagal postganglionic efferent nerves terminate directly on the motor endplate of skeletal muscle in the upper esophagus, whereas vagal preganglionic efferent nerves to smooth muscle in the distal esophagus terminate on neurons within Auerbach’s (myenteric) plexus, located between the circular and longitudinal muscle layers.3 A second neuronal sensory network, Meissner’s plexus, located within the submucosa, is the site of afferent impulses within the esophageal wall. These are transmitted to the central nervous system through vagal parasympathetic and thoracic sympathetic nerves. Sensory signals transmitted via vagal afferent pathways travel to the nucleus tractus solitarius within the central nervous system (see Fig. 41-4); from there nerves pass to the nucleus ambiguus and dorsal motor nucleus of the vagus nerve, where their signals may influence motor function.5
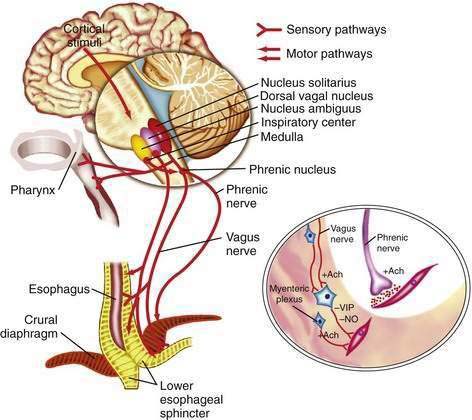
Pain sensation arising from the esophagus is typically triggered by stimulation of chemoreceptors in the esophageal mucosa or submucosa and/or mechanoreceptors in the esophageal musculature.6 Central perception then occurs when these impulses are transmitted to the brain by sympathetic and vagal afferents. Sympathetic afferents travel through the dorsal root ganglia to the dorsal horn of the spinal cord, and vagal afferents travel through the nodose ganglia to the nucleus tractus solitarius in the medulla. Information from sympathetic/spinal afferents then proceeds via the spinothalamic and spinoreticular pathways to the thalamus and reticular nuclei before transmission to the somatosensory cortex for pain perception and limbic system for pain modulation. Information from vagal afferents in the medulla also travels to the limbic system and frontal cortex for pain modulation. Furthermore, because the esophageal neuroanatomic pathways overlap with those of the heart and respiratory system, in clinical practice it may be difficult to discern the organ of origin for some chest pain syndromes.6
CIRCULATION
The arterial and venous blood supply to the esophagus is segmental. The upper esophagus is supplied by branches of the superior and inferior thyroid arteries, the midesophagus by branches of the bronchial and right intercostal arteries and descending aorta, and the distal esophagus by branches of the left gastric, left inferior phrenic, and splenic arteries.1–3 These vessels anastomose to create a dense network within the submucosa that probably accounts for the rarity of esophageal infarction. The venous drainage of the upper esophagus is through the superior vena cava, the midesophagus through the azygos veins, and the distal esophagus through the portal vein by means of the left and short gastric veins. The submucosal venous anastomotic network of the distal esophagus is important because it is where esophageal varices emerge in patients with portal hypertension.1–3
MUCOSA
On endoscopy the esophageal mucosa appears smooth and pink. Furthermore, the esophagogastric junction can be recognized by the presence of an irregular white Z-line (ora serrata) demarcating the interface between the lighter esophageal and the redder gastric mucosae. On biopsy, histology shows the esophageal mucosa to be lined by a nonkeratinized, stratified squamous epithelium (Fig. 41-5). This multilayered epithelium consists of three functionally distinct layers: stratum corneum, stratum spinosum, and stratum germinativum. The most lumen-oriented stratum corneum acts as a permeability barrier between luminal content and blood by having layers of pancake-shaped glycogen-rich cells connected laterally to each other by tight junctions and zonula adherens and having their intercellular spaces filled with a dense matrix of glycoconjugate material.7 The middle layer of stratum spinosum contains metabolically active cells with a spiny shape. The spiny shape is due to the numerous desmosomes connecting cells throughout the layer. Furthermore, this same desmosomal network maintains the structural integrity of the tissue. The basal layers of stratum germinativum contain cuboidal cells that occupy 10% to 15% of the epithelium’s thickness and are uniquely capable of replication.2 Consequently, basal cell hyperplasia, defined as basal cells occupying more than 15% of epithelial thickness, is common in gastroesophageal reflux disease, reflecting an increased rate of tissue repair2 (see Chapter 43). The esophageal epithelium contains a small number of other cell types including argyrophilic endocrine cells, melanocytes, lymphocytes, Langerhans cells (macrophages), and eosinophils. Neutrophils are not present in healthy epithelium.2
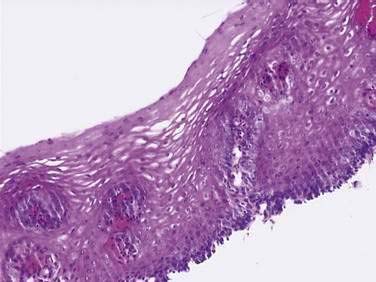
Figure 41-5. Esophageal epithelium. The human esophagus as shown on this biopsy specimen is lined by nonkeratinized stratified squamous epithelium. The cells of the surface (top) are long and flat and have a small nuclear-to-cytoplasmic ratio that contrasts with the cells of the basal layer (bottom), whose density, cuboidal shape, and large nuclear-to-cytoplasmic ratio account for their prominence. A subpopulation of these basal layer cells appears to have properties of esophageal stem cells.6a Rete pegs or dermal papillae containing elements of the lamina propria normally extend into the epithelium about one half the distance to the lumen.
(Courtesy of Pamela Jensen, MD, Dallas, Tex.)
Below the epithelium is the lamina propria, a loose network of connective tissue within which are blood vessels and scattered lymphocytes, macrophages, and plasma cells (see Fig. 41-5). The lamina propria protrudes at intervals into the epithelium to form rete pegs or dermal papillae. Normally these protrude to less than 50% of the epithelium’s thickness; when greater, it also is a recognized marker of gastroesophageal reflux disease.2 The muscularis mucosae is a thin layer of smooth muscle that separates the lamina propria above from the submucosa. Its functions are unclear.
SUBMUCOSA
The submucosa comprises a dense network of connective tissue, within which are blood vessels, lymphatic channels, neurons of Meissner’s plexus, and esophageal glands (see Fig. 41-2A). These glands, which vary as to number and distribution along the esophagus, consist of cuboidal cells organized as acini.8 They produce and secrete a lubricant, mucus, and factors such as bicarbonate and epidermal growth factor that are important for epithelial defense and repair. The secretions from these glands pass into tortuous collecting ducts that deliver them to the esophageal lumen.
EMBRYOLOGY
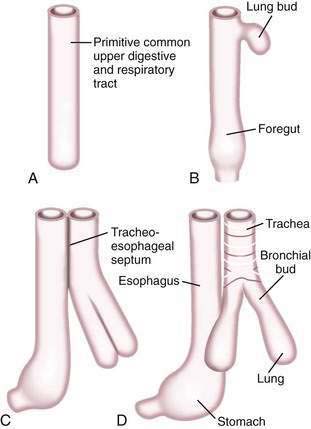
A brief review of the embryology of the upper digestive system is presented as a guide to understanding the origin of many of the developmental anomalies discussed in this chapter. In the developing fetus, the oropharynx and esophageal components of the gastrointestinal tract and the larynx, trachea, bronchi, and lungs of the respiratory tract develop from a common tube.3 By gestational week 4, this tube, composed of endoderm, develops a diverticulum on its ventral surface that is destined to become the epithelium and glands of the respiratory tract (Fig. 41-6A to D). This diverticulum subsequently elongates, becomes enveloped by splanchnic mesenchyme (future cartilage, connective tissue, and smooth muscle), and buds off to become the primitive respiratory tract. Concomitantly, the lumen of the dorsal tube, the primitive foregut, fills with proliferating (ciliated-columnar) epithelium. By week 10, vacuoles appear and subsequently coalesce within the primitive foregut to reestablish the lumen. By week 16, the columnar epithelium lining the primitive foregut and future esophagus is replaced by stratified squamous epithelium, a process that is complete by birth.
DEVELOPMENTAL ANOMALIES
Congenital anomalies of the esophagus are relatively common (1 in 3000 to 4500 live births) and are due to either transmission of genetic defects or intrauterine stress that impedes fetal maturation. Esophageal anomalies are common in premature infants, and 60% have other anomalies, reflected by the term VACTERL (formerly VATER), a mnemonic for the association of anomalies of the vertebral, anal, cardiac, tracheal, esophageal, renal, and limb systems. Common specific defects include patent ductus arteriosus, cardiac septal defects, and imperforate anus.9
ESOPHAGEAL ATRESIA AND TRACHEOESOPHAGEAL FISTULA
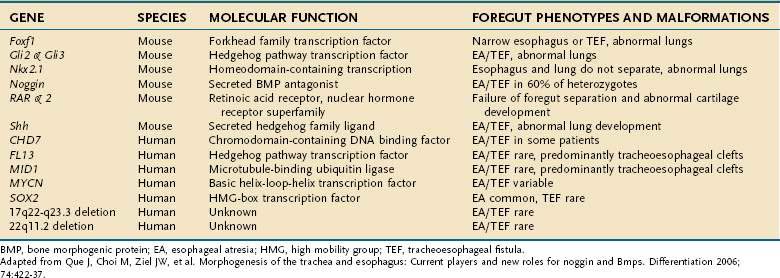
Esophageal atresia and tracheoesophageal fistulas are the most common developmental anomalies of the esophagus (Fig. 41-7). The former results from failure of the primitive foregut to recanalize and the latter from failure of the lung bud to separate completely from the foregut. Although the mechanisms are unclear, esophageal atresia and tracheoesophageal fistulas may result from genetic defects, such as those reported in mouse and humans (Table 41-1).10 In addition, experimental administration of the anticancer drug, adriamycin, into mouse or rat embryos commonly results in esophageal atresia and tracheoesophageal fistulas; and these defects may be accompanied by other anomalies that comprise the VACTERL group.11–12
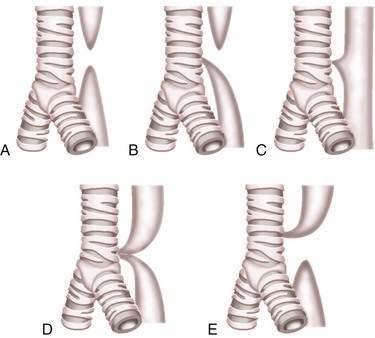
Esophageal atresia occurs as an isolated anomaly in only 7% of cases; the rest are accompanied by a form of tracheoesophageal fistula (distal-type fistula comprises 89% [see Fig. 41-7B] and the H-type fistula [see Fig. 41-7C] comprises 3% of cases).13 In isolated atresia, the upper esophagus ends in a blind pouch and the lower esophagus connects to the stomach (see Fig. 41-7A). The condition is suspected prenatally by the development of polyhydramnios (due to the inability of the fetus to swallow and so absorb amniotic fluid) or at birth by the regurgitation of saliva and a scaphoid (gasless) abdomen. Isolated atresia without a distal tracheoesophageal fistula results in a gasless abdomen because no pathway exists for inspired or swallowed air to enter the bowel. Furthermore, and as an indicator of high gastrointestinal obstruction, esophageal atresia results in the rapid onset of choking, coughing, and regurgitation on first feeding (Table 41-2). Once suspected, the diagnosis can be confirmed by failure to pass a nasogastric tube into the stomach and by a concurrent chest radiograph with air contrast in the upper esophageal segment (the air being introduced through a catheter positioned within the upper esophageal segment). In some instances, injection of 1 mL of barium into the obstructed segment helps with the diagnosis.
As mentioned, esophageal atresia usually is associated with a tracheoesophageal fistula, most often the distal type (see Fig. 40-7B).13 Thus, the atretic upper esophagus ends in a blind pouch and the trachea communicates with the distal esophageal segment. The clinical presentation with this configuration is usually similar to isolated esophageal atresia with the additional risk of aspiration pneumonia from refluxed gastric contents entering the trachea through the fistula (see Table 41-2). Nonetheless, distinction between an isolated atresia and one associated with a distal tracheoesophageal fistula is straightforward because the communication between the trachea and the esophagus results in a gas-filled abdomen, as shown on plain radiographs. In some instances, confirmation of the type of configuration is obtained by esophagography with or without bronchoscopy.
The three less common types of tracheoesophageal fistula are when the atretic upper esophagus communicates with the trachea, when both upper and lower segments of the atretic esophagus communicate with the trachea, and when an H-type fistula communicates with the trachea in a nonatretic esophagus (see Figs. 41-7E, D, and C, respectively). Because these types have in common the communication between upper esophagus and trachea, they all manifest clinically with signs and symptoms of recurrent (aspiration) pneumonia (see Table 41-2). Distinguishing among types, however, should not be difficult. Esophageal atresia accompanied by proximal tracheoesophageal fistula presents in infancy as recurrent pneumonia, and the presence or absence of bowel gas on a plain radiograph indicates whether an accompanying distal tracheoesophageal fistula exists. In contrast, in those with an H-type tracheoesophageal fistula without esophageal atresia, the diagnosis can be delayed until childhood or, at times, adulthood. Diagnosis of a suspected H-type fistula is usually made by esophagography, but this may be difficult owing to the small size of some communications.14 In such cases, detection may be improved by ingestion of methylene blue and searching by bronchoscopy for the blue-stained fistula site.
Treatment of esophageal atresia and tracheoesophageal fistulas is surgical, and the choice of procedure depends on the distance between the upper and lower esophageal segments. Short gaps permit end-to-end anastomosis, as do some long gaps after lengthening of the upper segment by either bougienage or intraoperative myotomy.13 If approximation of the two segments is not possible, the colon is interposed. The results of surgical correction of esophageal atresia are excellent when it exists as an isolated anomaly, with overall outcome determined by the gravity of accompanying genetic anomalies and by the birth weight of the infant.15
Long-term survival after successful repair of isolated esophageal atresia has steadily increased over the years and now approaches 90%. Notably, patients who survive until adulthood are at increased risk of developing gastroesophageal reflux disease (GERD),16–18 with the increased risk of GERD being due to abnormalities of esophageal motility and impaired acid clearance.19 In the largest follow-up to date, these patients, as adults, were found to have high rates of dysphagia (52%), reflux symptoms (63%), esophagitis (58%), Barrett’s metaplasia (11%), and strictures (42%)18; and anywhere from 6% to 45% require surgical fundoplication. Unfortunately, 15% to 30% of Nissen operations in these patients fail, usually resulting in reoperation.20 However, despite the increased incidence of Barrett’s metaplasia, a study of 272 such patients in Finland did not find an increased risk of esophageal or nonesophageal cancer.21
CONGENITAL ESOPHAGEAL STENOSIS
Esophageal stenosis is a rare anomaly, occurring in only 1 in every 25,000 live births.22 The stenotic segment varies from 2 to 20 cm in length and is usually located within the middle or lower third of the esophagus (Fig. 41-8A). The precise cause of the congenital stenosis is not entirely clear. Some patients (17% to 33%) have other associated anomalies, the most common being esophageal atresia (see Fig. 41-8B) and tracheoesophageal fistula.22 When resected, many stenotic walls contain tracheobronchial remnants (TBRs), which are sequestered respiratory tissue (hyaline cartilage, respiratory epithelium), suggesting its origin is incomplete separation of lung bud from primitive foregut.23 In other cases, stenosis results from fibromuscular hypertrophy associated with damage to the myenteric plexus with loss of the muscle-relaxing nitrinergic neural elements. A third subtype, a membranous diaphragm, is limited to the mucosa and does not involve the muscle layers.24
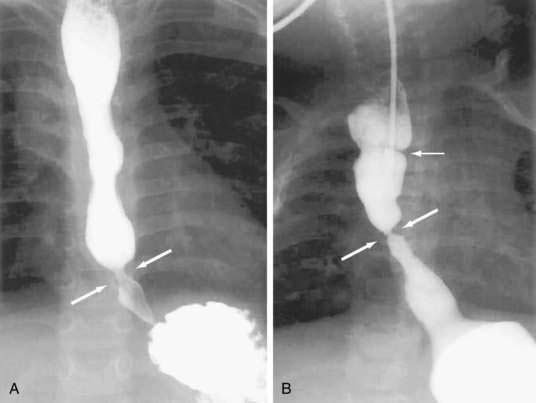
Although tight stenoses are symptomatic in infancy, most stenoses present with dysphagia and regurgitation in childhood when more solid food is ingested (see Table 41-2). The stenosis is best demonstrated by esophagography, which may reveal either an abrupt or tapered stricture. Dilatation of the esophagus proximal to the stenosis is commonly noted. Endoscopy may be of value by demonstrating normal mucosa in the stenotic region, helping to exclude an acquired cause for the stenosis. Endoscopic ultrasound (EUS) may show thickening of single or multiple layers of the esophageal wall and may demonstrate cartilaginous structures.25
Some patients improve after endoscopic-guided bougienage, although endoscopists should approach esophageal dilation carefully in these patients because chest pain and mucosal tears are commonly reported. Problematic stenoses require surgical resection of the involved segment. In general, congenital stenoses caused by TBRs rarely improve with bougienage, and therefore identification of this subtype by EUS may identify a group in need of surgical therapy.25 One novel surgical approach to this lesion is circular myectomy, a technique that involves stripping of the esophageal muscle layers containing the TBRs with preservation of the mucosal layer. This has the advantage of avoiding many of the potential complications associated with primary repair and end-to-end esophageal anastomosis.26
ESOPHAGEAL DUPLICATIONS
Congenital duplications of the esophagus occur in 1 in 8000 live births.2 They arise as epithelial-lined outpouchings off the primitive foregut and evolve to produce either cystic or tubular structures that do not communicate with the esophageal lumen. Cysts account for 80% of the duplications and are usually single fluid-filled structures.2 They may be found attached to the esophagus or to the tracheobronchial tree and are usually located within the right posterior inferior mediastinum. Some cysts are discovered while asymptomatic, manifesting as a mediastinal mass on a chest radiograph or a submucosal lesion on an esophagogram (Fig. 41-9A). Others manifest with symptoms from compression of structures adjacent to the tracheobronchial tree (coughing, stridor, tachypnea, cyanosis, wheezing, or chest pain) and of structures adjacent to the esophageal wall (dysphagia, chest pain, or regurgitation) (see Table 41-2).27
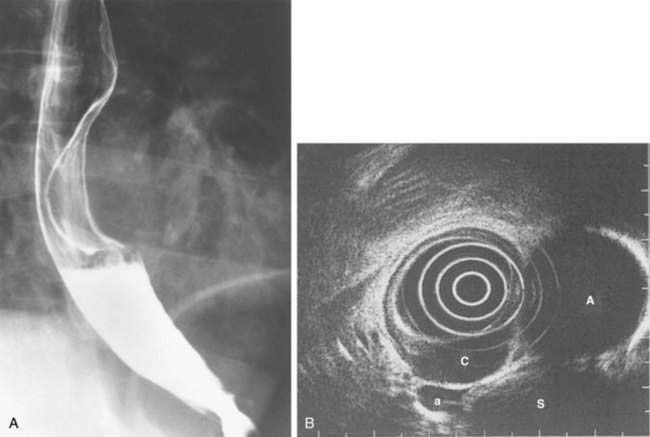
The diagnosis of an esophageal duplication cyst is supported by the demonstration of a cystic mass on computed tomography (CT), magnetic resonance imaging (MRI), or EUS (see Fig. 41-9B).28 Benign duplication cysts are anechoic by EUS, and for cysts in which the appearance is hypoechoic, fine-needle aspiration and cytologic evaluation of cyst contents may exclude malignancy. This approach may be particularly helpful in asymptomatic patients in whom the cysts are discovered incidentally on imaging or endoscopy.28 However, only surgical excision for pathologic assessment can exclude a cystic neoplasm. Surgical excision is also favored because it has low morbidity.29 Rarely, large duplication cysts can manifest with acute life-threatening respiratory symptoms. In this circumstance, emergent decompression can be achieved by radiologic or endoscopically guided needle aspiration.
The tubular esophageal duplication is far less common than its cystic counterpart. It is usually located within the esophageal wall, parallels the true esophageal lumen, and, in contrast to duplication cysts, communicates with the true lumen at either or both ends of the tube.27 Tubular duplications usually cause chest pain, dysphagia, or regurgitation in infancy, and the diagnosis is established by esophagography or endoscopy. Reconstructive surgery is indicated for patients who are symptomatic.27
VASCULAR ANOMALIES
Intrathoracic vascular anomalies are present in 2% to 3% of the population. Only rarely do they produce symptoms of esophageal obstruction despite evident vascular compression on an esophagogram. In infancy, most intrathoracic vascular anomalies manifest as respiratory symptoms from compression of the tracheobronchial tree. Later in childhood or adulthood, however, these same abnormalities can produce dysphagia and regurgitation, owing to esophageal compression (see Table 41-2).
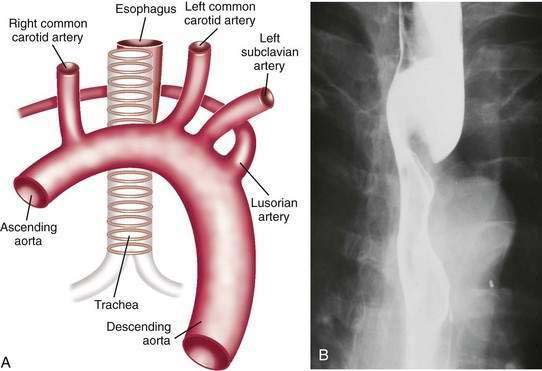
Dysphagia lusoria is the term given for symptoms arising from vascular compression of the esophagus by an aberrant right subclavian artery.30 The right subclavian artery in this circumstance arises from the left side of the aortic arch and courses from the lower left to the upper right side posterior to the esophagus (Fig. 41-10A). In 20% of cases the artery courses anterior to the esophagus.31 It is estimated that arteria lusoria is present in 0.7% of the general population on the basis of autopsy studies. Typically the diagnosis is established by barium esophagogram, which shows the characteristic pencil-like indentation at the level of the third and fourth thoracic vertebrae (see Fig. 41-10B).30 Confirmation is by CT, MRI, arteriography, or EUS.31 Given the frequency with which such lesions are asymptomatic, endoscopy or esophageal manometry may be desirable to exclude other causes of dysphagia. During endoscopy the right radial pulse may diminish or disappear from instrumental compression of the right subclavian artery. Esophageal manometry has demonstrated a high-pressure zone at the location of the aberrant artery.32 Symptoms usually respond to simple modification of the diet to meals of soft consistency and small size. When necessary, surgery relieves the obstruction by reanastomosing the aberrant artery to the ascending aorta.32
ESOPHAGEAL RINGS
The distal esophagus may contain two “rings,” the A and B (Schatzki’s) ring, that demarcate anatomically the proximal and distal borders of the esophageal vestibule (Fig. 41-11A). The A (muscular) ring is located at the proximal border. It is a broad (4 to 5 mm) symmetrical band of hypertrophied muscle that constricts the tubular esophageal lumen at its junction with the vestibule. In this location the A ring, which is covered by squamous epithelium, corresponds to the upper end of the lower esophageal sphincter.33 The A ring is rare, and because it varies in caliber on esophagography depending on the degree of esophageal distention, it is generally asymptomatic. Occasionally an A ring is found in association with dysphagia for solids and liquids (see Table 41-2).33 Symptomatic A rings can be treated by passage of a 50-French mercury-weighted esophageal dilator or by injection of botulinum toxin.34
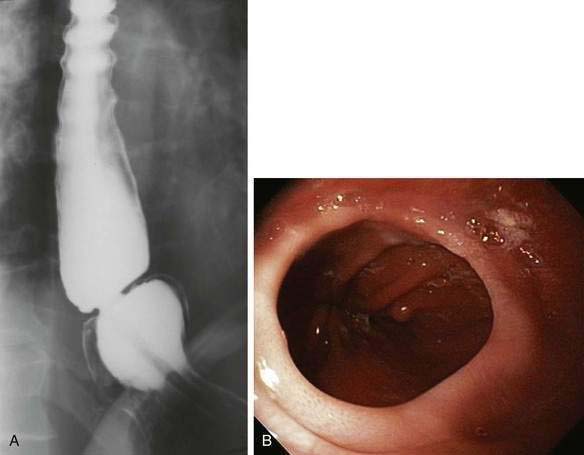
The B ring, otherwise known as the mucosal or Schatzki’s ring, is very common, and found in 6% to 14% of subjects having a routine upper gastrointestinal series.35 A recent review of more than 10,000 upper endoscopies found a Schatzki ring in 4% of cases.36 On barium study it is always found in association with a hiatal hernia and is recognized as a thin (2-mm) membrane that constricts the esophageal lumen at the junction of the vestibule and gastric cardia (see Fig. 41-11A). The Schatzki ring has squamous epithelium on its upper surface and columnar epithelium on its lower surface and so demarcates the squamocolumnar junction. The ring itself is composed of only mucosa and submucosa; there is no muscularis propria. Schatzki’s rings can be congenital or acquired, and a relationship to gastroesophageal reflux disease is likely (see Chapter 43).35
Most B rings are asymptomatic, yet when the diameter of the esophageal lumen is narrowed to less than or equal to 13 mm, rings commonly are the cause of intermittent dysphagia for solids or unheralded acute solid-food impactions (see Table 41-2).37 Identification of symptomatic rings on esophagography or endoscopy is generally not difficult (see Fig. 41-11B), although attention should be paid to adequately distend the distal esophagus.35 In some instance, the obstructing ring is best demonstrated radiographically by its ability to trap a swallowed marshmallow or a barium tablet.
Asymptomatic B rings require no treatment, and those producing dysphagia are effectively treated by passage of either a single, large (≥50-French), mercury-weighted dilator or a series of such dilators of progressively larger diameter.38 Early studies reported that 32% of patients required repeat dilation after 1 year.35 More recent studies report much lower rates (13%), perhaps due to the more routine use of both larger dilators and a short course of postdilation antireflux therapy.39 In a randomized placebo-controlled study of 44 patients with symptomatic Schatzki’s rings, maintenance therapy with omeprazole resulted in a 40% reduction in the need for redilation after a mean follow-up of 35 months.40 Symptomatic rings that are refractory to dilation have been successfully treated by endoscopic means using electrocautery incision.41 A randomized, controlled trial of standard bougie dilation versus electrocautery incision for symptomatic Schatzki rings has demonstrated that the two therapies have comparable initial success rates but that endoscopic incision had a longer duration of symptom resolution.42
A syndrome in which multiple esophageal rings are found has been described.43 The term “corrugated ringed esophagus” generally implies a condition in which the muscular rings are concentric and evenly spaced over a long segment of the esophagus, usually starting proximally and extending, in some instances, the entire length of the organ. These rings may not be apparent on barium studies but on endoscopy persist despite maximal air insufflation. Another defining characteristic of the corrugated ringed esophagus is that dilation is difficult with mucosal tears and perforations being common. For this reason, it is recommended that dilation in patients with solid-food dysphagia be limited to a maximum diameter of a 40-French bougie. Support for the corrugated ringed esophagus being a congenital anomaly is based on the large male predominance (75% of reported cases) and history of solid-food dysphagia, often with impactions, beginning in early childhood.43 Several studies have proposed that many of these cases are a manifestation of eosinophilic esophagitis (see Chapter 27), and a few studies suggest an association with GERD. The former is readily diagnosed by esophageal biopsy showing greater than 15 to 20 eosinophils per high-power field and the latter by response to acid suppression therapy.
ESOPHAGEAL WEBS
Esophageal webs are developmental anomalies characterized by one or more thin horizontal membranes of stratified squamous epithelium within the upper (cervical) esophagus and midesophagus. Unlike rings these anomalies rarely encircle the lumen but instead protrude from the anterior wall, extending laterally but not to the posterior wall (Fig. 41-12A and B). Webs are common in the cervical esophagus and are best demonstrated on an esophagogram with the lateral view. In up to 5% of cases they are identified in an asymptomatic state, but when they are symptomatic they cause dysphagia for solids (see Table 41-2).44 Webs are fragile membranes and so respond well to esophageal bougienage with mercury-weighted dilators.
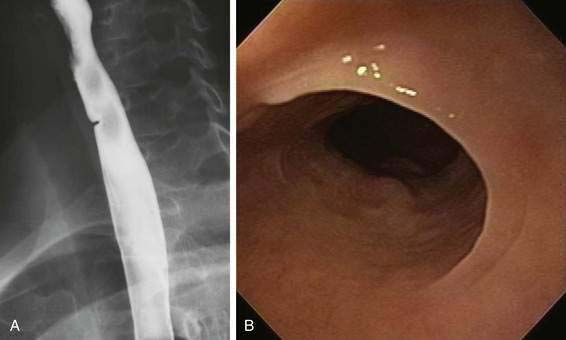
An association among cervical esophageal webs, dysphagia, and iron deficiency anemia in adults has been described as the Plummer-Vinson or Paterson-Kelly syndrome.44 The syndrome, although uncommon, occurs primarily in women. Recent reports have shown an association between Plummer-Vinson syndrome and celiac disease.45 It is an important syndrome because it identifies a group of patients at increased risk for squamous carcinoma of the pharynx and esophagus.44 Correction of iron deficiency in Plummer-Vinson syndrome may result in resolution of the associated dysphagia as well as disappearance of the web.44
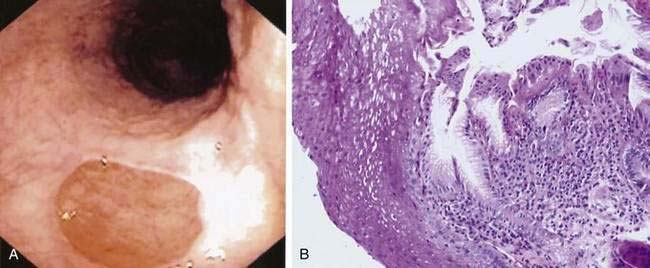
HETEROTOPIC GASTRIC MUCOSA (INLET PATCH)
The inlet patch refers to the appearance on endoscopy of a small (0.5 to 2 cm) distinctive, velvety red island of heterotopic gastric mucosa amid a lighter pink squamous mucosa, generally localized immediately below the upper esophageal sphincter (Fig. 41-13A). When sought, an inlet patch is found in up to 10% of endoscopies, and biopsy specimens reveal gastric fundic- or antral-type mucosa (see Fig. 41-13B).46 The fundic-type mucosa contains chief and parietal cells and thus in some specimens retains the capacity for acid secretion.47 Similar to gastric mucosa in the stomach, the inlet patch may be infected with Helicobacter pylori.48 However, inlet patches are usually asymptomatic and unassociated with disease and thus require no treatment. A possible association with globus pharyngeus was suggested in a recent pilot study in which this symptom was improved after ablation of inlet patches using argon plasma coagulation.49 In rare instances, an inlet patch is found in association with an esophageal web or stricture50 or ulcer, the latter resulting in bleeding or perforation.46 In addition, although adenocarcinoma arising in an inlet patch is a rare complication, a total of 24 such cases have been reported in the literature.46
Achildi O, Grewal H. Congenital anomalies of the esophagus. Otolaryngol Clin North Am. 2007;40:219-44. (Ref 3.)
Atmatzidis K, Papaziogas B, Pavlidis T, et al. Plummer-Vinson syndrome. Dis Esophagus. 2003;16:154-7. (Ref 44.)
Cioffi U, Bonavina L, De Simone M, et al. Presentation and surgical management of bronchogenic and esophageal duplication cysts in adults. Chest. 1998;113:1492-6. (Ref 29.)
Deurloo JA, Ekkelkamp S, Schoorl M, et al. Esophageal atresia: Historical evolution of management and results in 371 patients. Ann Thorac Surg. 2002;73:267-72. (Ref 13.)
Hirano I, Gilliam J, Goyal RK. Clinical and manometric features of the lower esophageal muscular ring. Am J Gastroenterol. 2000;95:43-9. (Ref 33.)
Jalil S, Castell DO. Schatzki’s ring. A benign cause of dysphagia in adults. J Clin Gastroenterol. 2002;35:295-8. (Ref 35.)
Janssen M, Baggen MGA, Veen HF, et al. Dysphagia lusoria: Clinical aspects, manometric findings, diagnosis, and therapy. Am J Gastroenterol. 2000;95:411-16. (Ref 30.)
Mittal RK, Balaban DH. The esophagogastric junction. N Engl J Med. 1997;336:924-32. (Ref 4.)
Que J, Choi M, Ziel JW, et al. Morphogenesis of the trachea and esophagus: Current players and new roles for noggin and Bmps. Differentiation. 2006;74:422-37. (Ref 10.)
Sgouros SN, Vlachogiannakos J, Karamanolis G, et al. Long-term acid suppressive therapy may prevent the relapse of lower esophageal (Schatzki’s) rings: A prospective, randomized, placebo-controlled study. Am J Gastroenterol. 2005;100:1929-34. (Ref 40.)
Skandalakis JE, Ellis H. Embryologic and anatomic basis of esophageal surgery. Surg Clin North Am. 2000;80:85-155. (Ref 1.)
Takamizawa S, Tsugawa C, Mouri N, et al. Congenital esophageal stenosis: Therapeutic strategy based on etiology. J Pediatr Surg. 2002;37:197-201. (Ref 25.)
Taylor ACF, Breen KJ, Auldist A, et al. Gastroesophageal reflux and related pathology in adults who were born with esophageal atresia: A long-term follow-up study. Clin Gastroenterol Hepatol. 2007;5:702-6. (Ref 18.)
Von Rahden BHA, Stein HJ, Becker K, et al. Heterotopic gastric mucosa of the esophagus: Literature-review and proposal of a clinicopathologic classification. Am J Gastroenterol. 2004;99:543-51. (Ref 46.)
Wills JC, Hilden K, DiSario JA, Fang JC. A randomized, prospective trial of electrosurgical incision followed by rabeprazole versus bougie dilation followed by rabeprazole of symptomatic esophageal (Schatzki’s) rings. Gastrointest Endosc. 2008;67:808-13. (Ref 42.)
1. Skandalakis JE, Ellis H. Embryologic and anatomic basis of esophageal surgery. Surg Clin North Am. 2000;80:85-155.
2. The normal anatomy of the esophagus. In: Fenoglio-Preiser CM, editor. Gastrointestinal pathology. An atlas and text. 2nd ed. Philadelphia: Lippincott-Raven; 1999:15-29.
3. Achildi O, Grewal H. Congenital anomalies of the esophagus. Otolaryngol Clin North Am. 2007;40:219-44.
4. Mittal RK, Balaban DH. The esophagogastric junction. N Engl J Med. 1997;336:924-32.
5. Hornby PJ, Abrahams TP. Central control of lower esophageal sphincter relaxation. Am J Med. 2000;108:90S.
6. Orlando RC. Esophageal perception and noncardiac chest pain. Gastroenterol Clin North Am. 2004;33:25-33.
6a. Kalabis J, Oyama K, Okawa T, et al. A subpopulation of mouse esophageal basal cells has properties of stem cells with the capacity for self-renewal and lineage specification. J Clin Invest. 2008;118:3860-9.
7. Orlando RC. Pathophysiology of gastroesophageal reflux disease: Esophageal epithelial resistance. In: Castell DO, Richter JE, editors. The esophagus. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 1999:409-19.
8. Long JD, Orlando RC. Esophageal submucosal glands: Structure and function. Am J Gastroenterol. 1999;94:818-24.
9. Keckler SJ, St. Peter SD, Valusek PA, et al. VACTERL anomalies in patients with esophageal atresia: An updated delineation of the spectrum and review of the literature. Pediatr Surg Int. 2007;23:309-13.
10. Que J, Choi M, Ziel JW, et al. Morphogenesis of the trachea and esophagus: Current players and new roles for noggin and Bmps. Differentiation. 2006;74:422-37.
11. Ioannides AS, Chaudhry B, Henderson DJ, et al. Dorsoventral patterning in oesophageal atresia with tracheo-oesophageal fistula: Evidence from a new mouse model. J Pediatr Surg. 2002;37:185-91.
12. Gillick J, Mooney E, Giles S, et al. Notochord anomalies in the adriamycin rat model: A morphologic and molecular basis for the VACTERL association. J Pediatr Surg. 2003;38:469-73.
13. Deurloo JA, Ekkelkamp S, Schoorl M, et al. Esophageal atresia: Historical evolution of management and results in 371 patients. Ann Thorac Surg. 2002;73:267-72.
14. Brookes JT, Smith MC, Smith RJ, et al. H-type congenital tracheoesophageal fistula: University of Iowa experience 1985 to 2005. Ann Otol Rhinol Laryngol. 2007;116:363-8.
15. Orford J, Cass DT, Glasson MJ. Advances in the treatment of oesophageal atresia over three decades: The 1970s and the 1990s. Pediatr Surg Int. 2004;20:402-7.
16. Krug E, Bergmeijer JHLJ, Dees J, et al. Gastroesophageal reflux and Barrett’s esophagus in adults born with esophageal atresia. Am J Gastroenterol. 1999;94:2825-8.
17. Deurloo JA, Ekkelkamp S, Bartelsman JFWM, et al. Gastroesophageal reflux. Prevalence in adults older than 28 years after correction of esophageal atresia. Ann Surg. 2003;238:686-9.
18. Taylor ACF, Breen KJ, Auldist A, et al. Gastroesophageal reflux and related pathology in adults who were born with esophageal atresia: A long-term follow-up study. Clin Gastroenterol Hepatol. 2007;5:702-6.
19. Sistonen SJ, Koivusalo A, Lindahl H, et al. Cancer after repair of esophageal atresia: Population-based long-term follow-up. J Pediatr Surg. 2008;43:602-5.
20. Tomaselli V, Volpi ML, Dell’Agnola CA, et al. Long-term evaluation of esophageal function in patients treated at birth for esophageal atresia. Pediatr Surg Int. 2003;19:40-3.
21. Bergmeijer JHLJ, Tibboel D, Hazebroek FWJ. Nissen fundoplication in the management of gastroesophageal reflux occurring after repair of esophageal atresia. J Pediatr Surg. 2000;35:573-6.
22. Amae S, Nio M, Kamiyama T, et al. Clinical characteristics and management of congenital esophageal stenosis: A report of 14 cases. J Pediatr Surg. 2003;38:565-70.
23. Zhao LL, Hsieh WS, Hsu WM. Congenital esophageal stenosis owing to ectopic tracheobronchial remnants. J Pediatr Surg. 2004;39:1183-7.
24. Ramesh JC, Ramanujam TM, Jayaram G. Congenital esophageal stenosis: Report of three cases, literature review, and a proposed classification. Pediatr Surg Int. 2001;17:188-92.
25. Takamizawa S, Tsugawa C, Mouri N, et al. Congenital esophageal stenosis: Therapeutic strategy based on etiology. J Pediatr Surg. 2002;37:197-201.
26. Saito T, Ise K, Kawahara Y, et al. Congenital esophageal stenosis because of tracheobronchial remnant and treated by circular myectomy: A case report. J Pediatr Surg. 2008;43:583-5.
27. Berrocal T, Torres I, Gutierrez J, et al. Congenital anomalies of the upper gastrointestinal tract. Radiographics. 1999;19:855-72.
28. Fazel A, Moezardalan K, Varadarajulu S, et al. The utility and the safety of EUS-guided FNA in the evaluation of duplication cysts. Gastrointest Endosc. 2005;62:575-80.
29. Cioffi U, Bonavina L, De Simone M, et al. Presentation and surgical management of bronchogenic and esophageal duplication cysts in adults. Chest. 1998;113:1492-6.
30. Janssen M, Baggen MGA, Veen HF, et al. Dysphagia lusoria: Clinical aspects, manometric findings, diagnosis, and therapy. Am J Gastroenterol. 2000;95:1411-16.
31. De Luca L, Bergman JGHM, Tytgat GNJ, et al. EUS imaging of the arteria lusoria: Case series and review. Gastrointest Endosc. 2000;52:670-3.
32. Levitt B, Richter JE. Dysphagia lusoria: A comprehensive review. Dis Esophagus. 2007;20:455-60.
33. Hirano I, Gilliam J, Goyal RK. Clinical and manometric features of the lower esophageal muscular ring. Am J Gastroenterol. 2000;95:43-9.
34. Varadarajulu S, Noone T. Symptomatic lower esophageal muscular ring: Response to Botox. Dig Dis Sci. 2003;48:2132-4.
35. Jalil S, Castell DO. Schatzki’s ring. A benign cause of dysphagia in adults. J Clin Gastroenterol. 2002;35:295-8.
36. Mitre MC, Katzka DA, Brensinger CM, et al. Schatzki ring and Barrett’s esophagus: Do they occur together? Dig Dis Sci. 2003;49:770-3.
37. Byrne KR, Panagiotakis PH, Hilden K, et al. Retrospective analysis of esophageal food impaction: Differences in etiology by age and gender. Dig Dis Sci. 2006;52:717-21.
38. Mann NS. Single dilation of symptomatic Schatzki ring with a large dilator is safe and effective. Am J Gastroenterol. 2001;96:3448-9.
39. Scolapio JS, Pasha TM, Gostout CJ, et al. A randomized prospective study comparing rigid to balloon dilators for benign esophageal strictures and rings. Gastrointest Endosc. 1999;50:13-17.
40. Sgouros SN, Vlachogiannakos J, Karamanolis G, et al. Long-term acid suppressive therapy may prevent the relapse of lower esophageal (Schatzki’s) rings: A prospective, randomized, placebo-controlled study. Am J Gastroenterol. 2005;100:1929-34.
41. DiSario JA, Pedersen PJ, Bichis-Canoutas C, et al. Incision of recurrent distal esophageal (Schatzki) ring after dilation. Gastrointest Endosc. 2002;56:244-8.
42. Wills JC, Hilden K, DiSario JA, Fang JC. A randomized, prospective trial of electrosurgical incision followed by rabeprazole versus bougie dilation followed by rabeprazole of symptomatic esophageal (Schatzki’s) rings. Gastrointest Endosc. 2008;67:808-13.
43. Lee GSC, Craig PI, Freiman JS, et al. Intermittent dysphagia to solids associated with a multiringed esophagus: Clinical features and response to dilatation. Dysphagia. 2007;22:55-62.
44. Atmatzidis K, Papaziogas B, Pavlidis T, et al. Plummer-Vinson syndrome. Dis Esophagus. 2003;16:154-7.
45. Jessner W, Vogelsang H, Puspok A, et al. Plummer-Vinson syndrome associated with celiac disease and complicated by postcricoid carcinoma and carcinoma of the tongue. Am J Gastroenterol. 2003;98:1208-9.
46. Von Rahden BHA, Stein HJ, Becker K, et al. Heterotopic gastric mucosa of the esophagus: Literature-review and proposal of a clinicopathologic classification. Am J Gastroenterol. 2004;99:543-51.
47. Galan AR, Katzka DA, Castell DO. Acid secretion from an esophageal inlet patch demonstrated by ambulatory pH monitoring. Gastroenterology. 1998;115:1574-6.
48. Gutierrez O, Akamatsu T, Cardona H, et al. Helicobacter pylori and heterotopic gastric mucosa in the upper esophagus (the inlet patch). Am J Gastroenterol. 2003;98:1266-70.
49. Meining A, Bajbouj M, Preeg M, et al. Argon plasma ablation of gastric inlet patches in the cervical esophagus may alleviate globus sensation: A pilot trial. Endoscopy. 2006;38:566-70.
50. Ward EM, Achem SR. Gastric heterotopia in the proximal esophagus complicated by stricture. Gastrointest Endosc. 2003;57:131-3.