Anatomy and Development of The Thyroid
Development of the Thyroid Gland
Specification of the Thyroid Follicular Cells: The Thyroid Anlage
Early Stages of Thyroid Morphogenesis: Budding, Migration, and Lobulation of the Thyroid Primordium
Late Stages of Thyroid Morphogenesis: Functional Differentiation and Histogenesis
Anatomy of the Thyroid Gland
The thyroid gland was first described by Galen (130–210 ad) in his work “De Voce.” The gland was named thyroid by Thomas Whorton (1614–1673) because of its proximity to the thyroid cartilage.1 Despite its name (thyreòs in Greek means “shield”; also the German name Schilddrüse means “shield gland”), the characteristic shape of the thyroid, consisting of two lateral lobes connected by a narrow isthmus, is more reminiscent of a butterfly or a capital H than a shield (Fig. 1-1). The lateral lobes are 3 to 4 cm long and 15 to 20 mm wide and are located between the larynx and the trachea medially and the carotid sheath and the sternomastoid muscles laterally. The upper pole of the lobes reaches the level of the thyroid cartilage, while the lower pole reaches tracheal ring V-VI. The isthmus is 12 to 20 mm long, 20 mm wide, and crosses the trachea between ring I and II.
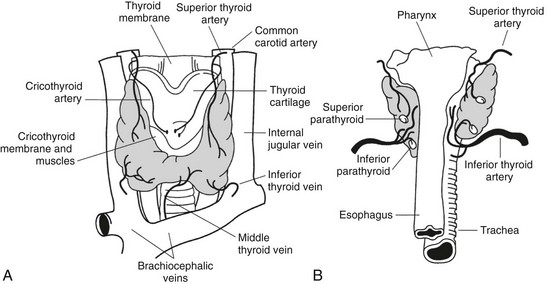
In a normal adult, the entire gland is approximately 6 to 7 cm wide, 3 to 4 cm long, and its weight ranges between 15 and 25 g. The thyroid is generally asymmetrical, the right lobe being even twice as large as the left and extending higher and lower in the neck than the left lobe. Interestingly, in patients with dextrocardia, the size of the lobes is reversed, suggesting that the asymmetry of the thyroid lobes could be connected to the position of the heart.2 A thin connective capsule encloses the thyroid. Fibrous septa are occasionally detached from this capsule and penetrate into the parenchyma to produce an incomplete lobulation. This inner capsule is connected to an outer capsule (also called the false capsule of the thyroid) that is continuous with the pretracheal fascia. Blood vessels, the parathyroids, and the recurrent laryngeal nerves are located in the space between the two capsules and are in close contact with the thyroid—the parathyroids on the posterior surface of the gland and the recurrent laryngeal nerves just medial to the lateral lobes.
Blood Supply
The thyroid is a highly vascularized organ with four arteries providing the gland with an abundant blood supply. Frequent anastomoses among these blood vessels and an arteriolar network are present on the surface of the gland; from this network, small arteries branch out and enter deeply into the tissue. The capillaries are localized in the interfollicular connective tissue, forming a basketlike network that surrounds each follicle. The capillary endothelial cells are fenestrated, like those of other endocrine glands. Each fenestration is about 50 nm in diameter. Stimulation with thyroid-stimulating hormone (TSH) increases the number and density of fenestrations.3 The veins emerge from the thyroid parenchyma and form a plexus of three groups of veins: the superior, middle, and inferior thyroid veins.
Lymphatics
A rich plexus of lymphatic capillaries surrounds the thyroid follicles and communicates with small lymphatic vessels found in the interlobular connective tissue. These deep blood vessels give rise to a surface network of lymphatics that drain into several groups of nodes. The uppermost group of nodes is situated just above the thyroid isthmus and is a systematic group consisting of one to five nodes called the Delphian nodes.2 These are readily felt when involved in cancer or Hashimoto’s thyroiditis. Other more variable groups of nodes in the thyroid area include the pretracheal nodes below the isthmus, those on the thyroid surface, and a last group found along the lateral vein, the recurrent laryngeal nerve, or along the carotid sheath.
Innervation
Thyroid innervation is very poor compared to that of other endocrine glands.3 The innervation of the thyroid is provided by sympathetic, parasympathetic, and peptidergic fibers, although few fibers enter the gland.4,5 Both sympathetic and parasympathetic fibers extend throughout the tissue among the follicles in close relation to the follicular cells or around blood vessels.
The regulator peptides detected in the thyroid are produced by both neural crest–derived parafollicular cells and intrathyroid peptidergic nerve fibers.6,7 Some neuropeptides, such as the vasoactive intestinal peptide (VIP), neuropeptide Y,8 substance P, or galanin, are exclusively produced in the nerve fibers distributed throughout the thyroid.9 Presumably, these neuropeptides regulate follicular cell functions via a paracrine pathway.
Anatomic Variants
The anatomic variants most frequently described in healthy individuals are caused by defects in the regression of the thyroglossal duct. This group of anomalies is generally characterized by the presence of an accessory lobe (pyramidal lobe) attached to the upper part of the isthmus of the thyroid (15% of the population).2 The pyramidal lobe is a rostral-directed stalk that results from the retention and growth of the caudal end of the thyroglossal duct. Other anomalies are associated with a defective atrophy of the duct. In some instances, the entire thyroglossal duct persists as an epithelial cord connecting the foramen cecum of the tongue to the larynx; in other cases, remainders of the duct form isolated or multiple cysts along the line of descent of the duct. Persistent portions of the thyroglossal duct may differentiate into thyroid tissue and form structures called accessory thyroids. The presence of accessory thyroids in addition to a normally located gland is characteristic of this anomaly.
A different developmental defect results in the formation of an ectopic thyroid gland (see “Ectopic Thyroid” later in this chapter). The other frequent anatomic variant (5% of the population) is a thyroid gland that has not developed as a unique mass2 but has the posterior part split into two distinct globes of thyroid tissue. Other anatomic anomalies are rare and found in less than 1% of healthy individuals. These variants include the absence of the isthmus (the thyroid consists of two independent lateral lobes, a physiologic condition in nonmammalian vertebrates10) or the absence of a significant portion of a lateral lobe, frequently the lower half of the left lobe.
The Thyroid Follicle
The thyroid gland displays a peculiar, highly-organized architecture characterized by the presence of spheroidal structures known as follicles. The follicles are supported by an interfollicular extracellular matrix and a capillary network. Thyroid follicles are composed of a single layer of epithelial cells (thyroid follicular cells) that surround a closed cavity (follicular lumen) filled with colloid, a concentrated solution of thyroglobulin.11 The follicle has been defined as the “morphofunctional unit” of the thyroid.12 As described later, thyroid follicular cells express a specific set of genes whose protein products perform functions which are essential for thyroid hormone biosynthesis. However, it is the follicular organization that, together with the polarity of the follicular cells, allows the biochemical steps required for thyroid hormone biosynthesis to occur as a functional chain of events (secretion of proteins in the follicular lumen as exocrine cells do; reabsorption and hydrolysis of proteins; release of hormones into blood by endocrine secretion13). The follicular architecture is absolutely required for thyroid function. In both mice14 and humans,15 T4 is detected only after the differentiation of the follicles.
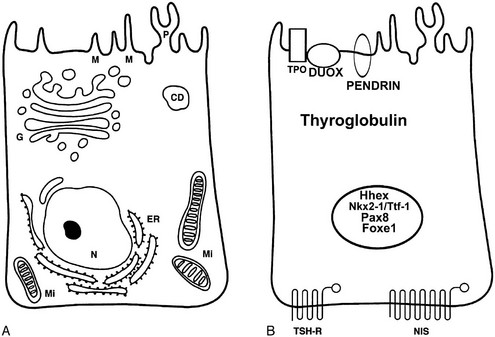
The follicular cell separates the follicular lumen (where hormone synthesis begins and pre-formed hormone is stored) from the bloodstream, from where iodide has to be uploaded and where hormones will be released at the end of the process. Follicular cells have a specific structural polarity established by molecular mechanisms that create specialized regions in the plasma membrane and cytoplasm.16,17 The surface of the cells is divided into two functionally distinct but physically contiguous regions: an apical and a basolateral domain. The apical domain faces the follicle lumen and displays a differentiated tissue-specific organization that is characterized by the presence of apical microvilli, pseudopods, and by the localization of thyroperoxidase (TPO),18 Na+ or Cl− channels,19,20 pendrin,21 and dual oxidase (Duox).22 The basal domain faces the extracellular matrix and is characterized by the expression of sodium/iodide symporter (NIS),23 Na+/K+ adenosine triphosphatase (ATPase),24 epidermal growth factor (EGF),25 and TSH receptors (TSHR).26 Junctional complexes between cells separate the apical and basal domains to prevent the mixing of the proteins that are sorted asymmetrically. Thyroid hormone synthesis requires basal to apical transport of iodide and thyroglobulin.27 Conversely, hormone secretion is based on apical to basal transport of thyroglobulin. In addition, follicular size is controlled by a bi-directional ion transport system. The basal cell plasma membrane is structurally and functionally connected by integrins to the basal lamina surrounding the follicle.28,29 The basal lamina consists of laminin, type IV collagen, and fibronectin30,31; a very thin connective space—less than 2 µm wide—separates the basal lamina from the endothelial capillary cells.
In the follicles, thyroid cells form a barrier between the extrafollicular space and the lumen. A tight barrier is critical because it promotes cell polarity, guarantees efficient transport, and prevents passive back-diffusion.32 The strong intercellular adhesion is mediated by a junctional complex that consists of tight junctions (zonula occludens), adherens junctions, and desmosomes. The three different types of cell junctions present an apical-to-basal distribution. In fact, the tight junctions are located close to the apical border of the cells, followed by the adherens junctions (located immediately below the tight junctions), and finally by the desmosomal junctions, which are positioned below the adherens junctions and occur on the plasma membrane. The anchoring junctions connect the cytoskeleton of a cell to the cytoskeleton of its neighbor or to the extracellular matrix. All these adhesion structures present the same overall organization: adhesive transmembrane proteins linked to the cytoskeleton by cytosolic adapter proteins.
Among the different types of cell junctions, the tight junctions are the structures that seal cells together. Tight junctions control the permeability of the paracellular space and define the boundary between the apical and basolateral domain of the follicular plasma membrane. They appear as a complex network of anastomosing fibrils consisting of junctional proteins responsible for cell-cell contact. This macromolecular complex interacts with different cellular structures. The major transmembrane proteins in a tight junction are occludin and claudins. The cytoplasmic portion of these proteins interacts with intracellular peripheral membrane proteins, such as ZO-1 and ZO-2,33 which link the junction to the microfilaments. The microtubules can also be functionally linked to the tight junctions via cytoskeletal-associated proteins34 such as cingulin, 7H6 antigen, and actin.
The epithelial adherens junctions, which as already said are located below the tight junctions, are formed by transmembrane adhesion proteins that belong to the cadherins family (cadherins and transmembrane Ca++-dependent adhesion molecules). In the thyroid tissue, as well as in other epithelial tissues,35 E-cadherin accumulates in adherens junctions and plays a crucial role in the induction of these stable adhesions, forming homophilic contacts between neighboring cells. E-cadherin is linked to an intracellular anchor protein, catenin, which in turns binds to the actin cytoskeleton. The catenins participate in intracellular signal transduction pathways,36 creating a functional bridge that couples physical adhesion to intracellular signaling events.
Desmosomal junctions, another type of anchoring junction, are the third type of junction present in epithelia. The transmembrane proteins found in desmosomal junctions are the desmogleins and desmocollins, members of the cadherins superfamily.37 The cytoplasmic tails of these adhesion proteins bind intracellular proteins (plakoglobin, desmosplakin), which in turn bind to the intermediate cytoskeleton filaments. These protein-protein interactions lead to the formation of a network through the tissue that connects different adjacent cells.38
Follicular cells in the epithelia communicate via gap junctions.39 Gap junctions are formed by channel-forming proteins (connexins). These channels allow inorganic ions and other small water-soluble molecules to pass directly from the cytoplasm of one cell to another, coupling the cells both electrically and metabolically.
Primary cultures of porcine thyroid follicular cells provided a useful in vitro model to understand the mechanisms leading to folliculogenesis. Follicular cells freshly isolated from pig thyroid glands and cultured in the absence of TSH aggregate only transiently.40 In the presence of TSH, these cells assemble epithelial junctions, polarize, and organize themselves into follicle-like structures in which apical poles of cells delineate a lumen cavity. The first and essential step for in vitro folliculogenesis is cell aggregation mediated by E-cadherin. During the first few hours in culture, E-cadherin expression increases in the lateral cell surface and accumulates in the subapical regions, where the adherens junctions will be assembled.41 ZO-1 and Na+/K+ ATPase are expressed. The earliest stage of cell-surface differentiation is marked by the redistribution of these two proteins: ZO-1 is recruited around the future pole of the cell, and ATPase is confined to the basal-lateral cell surface. At this stage, apical domain–associated proteins are detected in intracellular vacuoles that later fuse with the cell surface at the nascent apical pole. The follicular lumen is generated in two different steps, both of which are consequences of the polarized phenotype of thyroid cells. The first step is triggered by the lack of adhesive properties of the apical cell surface. The second step, which is required for the control of follicular size, is driven by a bi-directional ion transport system that secrets Cl− in a basal-to-apical direction and, conversely, absorbs Na+ in an apical-basal direction.12
The formation and maintenance of follicle-like structures in cultured porcine thyroid cells is dependent on TSH. In the absence of TSH or cyclic adenosine monophosphate (cAMP) stimulators, follicles undergo a morphologic conversion: thyroid cells spread and form epithelial monolayers.42 The activation of the ERK kinase signaling pathway is involved in this conversion. TSH, acting through its second messenger cAMP, inhibits ERK activation and represses the epithelial conversion.43 In addition, TSH modulates other steps required to maintain follicle organization.44 Stimulation of cAMP/PKA stabilizes E-cadherin-dependent cell-cell adhesions45 and inhibits the production of thrombospondin 1, a matricellular protein which acts as a negative modulator of cell-cell adhesion, thereby inhibiting the dissociation of tight and adherens junctions.46 Furthermore, TSH down-regulates the expression of TGF-β1,47 which induces the loss of epithelial polarization.48 TSH might also control follicular lumen generation because chloride channels, located at the apical pole, are regulated by cAMP.49
One limitation of the data on follicle formation from in vitro studies is that it cannot be directly extrapolated to comprehend tissue follicles in vivo. Follicles in the thyroid gland are surrounded by a basal lamina, which is absent in cultured follicles.41 Furthermore, cultured follicles invert their polarity and manifest a dramatic change in functional properties unless they are cultured with a gel consisting of extracellular matrix proteins.50 In addition, TSH is not required for the formation of follicles in vivo, since folliculogenesis is not affected in mutant mice with impaired TSH/TSHR signaling (see heading “TSHR”).
The Thyroid Cells
Thyroid Follicular Cells
When observed under the light microscope, thyroid follicular cells (TFCs) show a neutrophilic cytoplasm, a basal nucleus, and para-aminosalicylic acid (PAS)-positive vacuoles (phagosomes).51 Follicular cells appear as cuboidal epithelial cells whose height is approximately 15 µm. Cells become flatter (squamous) or higher (columnar) depending on whether or not they undergo TSH stimulation.
Electron microscopy (Fig. 1-2) reveals the characteristic features of cells actively engaged in protein synthesis.52 The rough endoplasmic reticulum and the Golgi apparatus are the dominant organelles in the cell. The apical surface of the cell is covered with thin microvilli or pseudopods that protrude into the follicular lumen. A distinctive feature of follicular cells engaged in protein synthesis is the presence of several vesicles localized in the apical or subapical cytoplasm. The smaller (150 to 200 nm) vesicles are exocytotic vesicles containing newly synthesized thyroglobulin. The fusion of these vesicles with the apical plasma membrane leads to the delivery of thyroglobulin into the follicle lumen. TPO and hydrogen peroxide–producing enzymes are localized to the luminal side of the follicular cell membrane,18 thus allowing the iodination process to occur. The larger vesicles (500 to 4000 nm) are filled with dense material called colloid droplets that are the result of the uptake of the iodinated thyroglobulin stored in the follicular lumen. TSH induces the uptake of thyroglobulin from the follicle lumen, increasing the number of colloid droplets.53 The reabsorption of the colloid involves a macropinocytosis mechanism whose first step is the formation of pseudopods at the apical pole. The pseudopods close, and a portion of the colloid is internalized into the cell.54
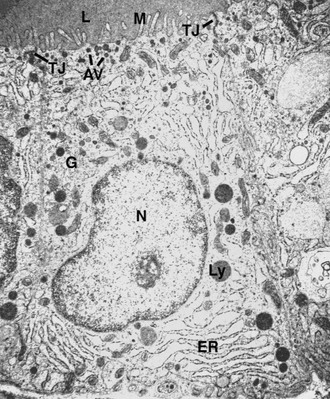
FIGURE 1-2 Electron micrograph of a rat follicular cell. AV, Apical vesicles; ER, endoplasmic reticulum; G, Golgi apparatus; L, lumen; Ly, lysosome; M, microvilli; N, nucleus; Tg, thyroglobulin; TJ, tight junction. (Courtesy Professor L. Nitsch.)
At the molecular level (Fig. 1-3), a follicular cell can be identified by the presence of a specific set of proteins—and corresponding mRNAs—indispensable for its specialized functions.55 Among such proteins, thyroglobulin (Tg) and TPO are remarkably specific, being detectable exclusively in thyroid follicular cells. Other proteins, such as TSHR, NIS, pendrin, and Duox, though expressed in thyroid follicular cells, are also present in other tissues. The exclusive or prevalent expression of genes necessary for thyroid hormone biosynthesis in thyroid follicular cells appears to be due to a combination of transcription factors unique to this cell type.55 These transcription factors (Nkx2-1/Ttf-1, Pax8, Foxe1) have subsequently been found to be important in controlling not only the differentiation but also the morphogenesis of the gland. The molecular features and the functions of these factors will be described further on (see “Molecular Genetics of Thyroid Gland Development”).
C Cells
C cells represent the other endocrine cellular type present in the thyroid gland of mammals. They synthesize and secrete calcitonin,56 a polypeptide hormone involved in calcium metabolism. In lower vertebrates, C cells form an organ called the ultimobranchial gland that is separate from the thyroid gland. Quail chick chimera experiments have demonstrated that C cells of avian species derive from neural crest cells,57 which during embryonic development colonize the ultimobranchial body, a transient organ in mammals, to finally disperse into the thyroid gland.58
C cells are known as parafollicular cells because of their distribution among follicular cells. However, in spite of their name, not all C cells are located between follicular cells and the basement membrane (a real parafollicular position); they are also found among the follicles (interfollicular) or in an intrafollicular position. In fact, C cells are found dispersed as individual cells in small groups closely associated to follicular cells, and in more complex structures consisting of both follicular and C cells. The number of parafollicular cells in the thyroid differs among species.59 In humans, the number of C cells decreases with age: the neonatal thyroid has 10 times more C cells than the adult thyroid, where they are fewer than 1% of the follicular cells. These cells are usually distributed in the upper two thirds of the lateral lobe in intrafollicular and parafollicular positions.60
C cells are characterized by clear cytoplasm and small, compact nuclei. Electron microscopy reveals that these cells contain cytoplasmic secretory granules 100 to 200 nm in diameter.61 At the molecular level, C cells are identified by the presence of calcitonin. The calcitonin/calcitonin gene-related peptide (CGRP) gene encodes four proteins: calcitonin, CGRP, katacalcin I and katacalcin II. The splicing of the first three exons to the fourth gives rise to an mRNA that encodes for a protein precursor which is subsequently processed to give calcitonin and a peptide, katacalcin I. CGRP and katacalcin II are the products of alternative splicing and are by far less abundant than calcitonin in C cells. CGRP is a 37-amino-acid vasoactive peptide with unknown effect on calcium metabolism.62 Interestingly, C cells, although different from follicular cells in function and embryologic derivation, express Nkx2-1/Ttf-1,63 which is a distinctive marker of thyroxine-producing cells. Nkx2-1/Ttf-1 is also present in immature C cells, in the migrating ultimobranchial body, and in the cells of the fourth pharyngeal pouch.14,64,65
Parafollicular cells share several biological features with other neuroendocrine cells that originate from the neural crest. Indeed, parafollicular cells express neuroendocrine markers such as neurospecific enolase and chromogranin A and a large number of regulatory peptides and their receptors,66 including somatostatin, serotonin, cholecystokinin2-receptor (CCK2R), gastrin releasing peptide, thyrotropin-releasing hormone (TRH), and helodermin. Whether different subpopulations of parafollicular cells synthesize different sets of regulatory factors has not yet been demonstrated. However, there is some evidence of functional heterogeneity within mammalian parafollicular cells.7 In neonatal rats, 90% of calcitonin-producing cells coexpress somatostatin, while in adults this factor is detected in only 1% of parafollicular cells.66 Also, in humans calcitonin and somatostatin are co-localized in few parafollicular cells.67 However, somatostatin is detected in almost all C-cell carcinomas in rats.66 Similarly, in many human medullary thyroid carcinomas, C cells can express somatostatin.68 The functional relevance of the production of these regulatory peptides by C cells is still unclear. These biologically active peptides might regulate thyroid function in a paracrine pathway, because parafollicular cells are distributed among follicular cells and often tightly adhered to them. Somatostatin, calcitonin, CGRP, and katacalcin inhibit thyroid hormone secretion, while gastrin-releasing peptide and helodermin stimulate this process.7 The expression of CCK2R, which binds cholecystokinin and gastrin,69 and the observation that gastrin induces calcitonin secretion suggest an interrelationship between calcium homeostasis and gastrointestinal hormones. In addition, the presence of CCK2 in thyroid tissues allows one to hypothesize that CCK2 and its receptor are both involved in an autocrine loop required for C-cell function.69
Ultimobranchial Body–Derived Epithelial Cells
In addition to the common thyroid follicles, other epithelial structures are evident in the mammalian thyroid. These structures, described as a “second kind of thyroid follicles,”70 rarely display a clear follicular organization. The finding that the second kind of thyroid follicles are absent in the avian thyroid (where ultimobranchial bodies never merge with the thyroid) has suggested that these structures represent remnants of the ultimobranchial body of endodermal origin.71
In humans, ultimobranchial-body remnants known as solid cell nests (SCN)72 are frequently present in the thyroid gland and are preferentially located in the middle and upper third of the lobes.72 SCN appear as para- or intrafollicular clusters and cords of epithelial cells clearly separated from the follicles by a basal lamina.
SNC are composed of two cell types: C cells and “main” cells, the most important cell population of these structures. The presence of C cells is consistent with the common ultimobranchial derivation of both SNC and C cells. In most cases, the SNC are found mixed with another structure known as a mixed follicle, in which follicular cells and main cells underline a lumen filled with colloid-like material.72,73 Main cells, polygonal in shape, show squamoid features, with oval nuclei and eosinophilic cytoplasm lacking intercellular bridges. The molecular phenotype of main cells is peculiar. These cells express p63,73 Bcl2 and telomerase.74 They do not express markers of differentiation such as Nkx2-1/Ttf-1, Tg, calcitonin, and CGRP. It is worth noting that recent studies in mouse embryos have revealed that Nkx2-1/Ttf-1–negative, p63-positive cells are present both in the epithelium of the fourth pharyngeal pouch and in the ultimobranchial body, confirming the SNC are ultimobranchial-body remnants.64 The presence in the main cells of both telomerase and p63, a transcription factor present in basal/stem cells of several multilayered epithelia and absent in differentiated cells, has suggested the hypothesis that these cells could be a source of multipotent cells able to differentiate towards either Tg- or calcitonin-producing cells. In addition, it has been suggested that main cells could be the cells of origin of a subset of papillary thyroid carcinoma.75
Development of the Thyroid Gland
The adult thyroid gland in mammals is assembled from two different embryologic structures. This composite origin reflects the dual endocrine function of the gland. The thyroglobulin-producing follicular cells derive from a small group of endodermal cells of the primitive pharynx (the thyroid anlage). The calcitonin-producing parafollicular cells are neural crest–derived cells from the ultimobranchial bodies. The ultimobranchial bodies are transient embryonic structures originated from the fourth pharyngeal pouch. The thyroid anlage and the ultimobranchial bodies migrate from their original sites, reach their final position anterior to the trachea, and fuse to form the definitive thyroid gland. The thyroid follicles derive from the thyroid anlage cells, while the C cells are scattered within the interfollicular space. After this early ontogenetic phase, the thyroid begins to function at a basal level; subsequent differentiation of the hypothalamic nuclei and the organization of the pituitary-portal vascular system guarantee the maturation of the thyroid-system function.76
Here we will describe in detail the morphologic and molecular aspects of thyroid development in mice (summarized in Fig. 1-4). These data can reasonably be extended to humans; the known relevant differences will be highlighted.
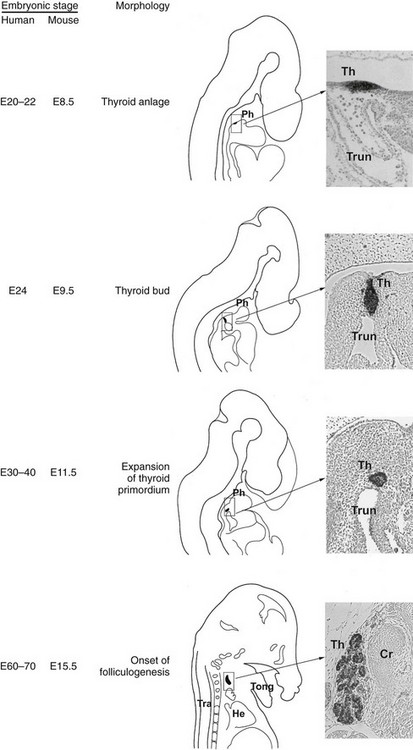
FIGURE 1-4 Thyroid development. In the middle is a schematic representation of the different steps of thyroid organogenesis in mouse embryos; on the left, the correspondent embryonic stages (E, embryonic day) in mice and humans are indicated; on the right relevant sagittal sections of mouse embryos stained with an anti-Nkx2-1/Ttf-1 are shown. Cr, Cricoid cartilage; He, heart; Ph, pharynx; Th, thyroid; Tong, tongue; Tra, trachea; Trun, truncus arteriosus.
Specification of The Thyroid Follicular Cells: The Thyroid Anlage
While the inductive mechanisms by which endodermal cells produce liver, pancreas, and lung cell lineage begin to be detailed, the events that commit a group of multipotent endodermal cells to a thyroid fate are almost unknown. The first morphologic consequence of thyroid specification is the appearance of the thyroid anlage (also called thyroid placode). In the mouse (19.5-day-long gestation) it is visible at embryonic day (E)8 to 8.5 and in humans at E22.77 In both the species, the thyroid anlage appears as a midline endodermal thickening in the ventral wall of the primitive pharynx caudal to the region of the first branchial arch that forms the tuberculum impar78 (see Fig. 1-4). The cells of the thyroid anlage simultaneously express four transcription factors: Hhex,Nkx2-1/Ttf-1, Pax8 and Foxe1.79 This unique molecular signature hallmarks these cells, which can be defined as the precursors of the TFCs.
The genetic program directing the specification of thyroid precursor cells is rather obscure. Any genes relevant in the patterning process of the foregut, such as Nodal, members of the Gata family, or Sox genes could play a role in thyroid specification. As yet, there have been no reports of either cell-fate mapping studies or genetically modified mice clarifying the early steps of the thyroid specification. Actually, mouse embryos carrying targeted inactivation of genes involved in foregut patterning usually show developmental arrest at stages that preclude the assessment of the thyroid anlage. In zebrafish, Bon and Gata5, two transcription factors that are downstream effectors of Nodal activity, seem to be specifically involved in the commitment of thyroid fate. Indeed, in the absence of these factors, endodermal cells form a reduced gut tissue but do not contribute to the thyroid.80
The study of the developing thyroid in mice has shown that at E8.5, the earliest identifiable thyroid anlage appears in close apposition to the aortic sac, the cardiac region that gives rise to the embryonic heart outflow and pharyngeal arteries. This observation suggests that short-range inductive signals from cardiac mesoderm or from the endothelial lining of the aortic sac could have an inductive role to specify undifferentiated endodermal cells towards a thyroid fate. In mice, alterations in the foregut have been observed as a consequence of an impaired heart development81; furthermore, it has been reported that signals from cardiac tissue adjacent to the endodermal layer are crucial in the early stages of liver, pancreas, and lung development.82–84
Data obtained from zebrafish are proving invaluable for the study of thyroid specification. The close association between thyroid anlage and aortic sac is maintained also in zebrafish.85 In this species it has been demonstrated that in the absence of the basic Helix-loop-helix (bHLH) transcription factor Hand2, expressed in the cardiac mesoderm surrounding the site of initiation of thyroid development, the endoderm appears to be normal, but thyroid precursor cells are not present. Experiments strongly suggest both that Hand2 has a non–cell-autonomous role in thyroid specification and that FGF proteins participate in this pathway.85 Interestingly, in mice it has been proved that FGF signals are required for correct thyroid development, but the role of these factors in thyroid specification has never been demonstrated.86
Consistent with the hypothesis of a role of cardiac tissue in thyroid development are the findings that cardiac malformations represent the most frequent birth defects associated with thyroid dysgenesis in humans,87 and DiGeorge syndrome is characterized by both congenital heart defects and an increased risk of congenital hypothyroidism.88
Early Stages of Thyroid Morphogenesis: Budding, Migration, and Lobulation of The Thyroid Primordium
After the specification stage, the developing thyroid undergoes very rapid changes in its appearance. In mice, by E8.5 the thyroid anlage first appears as a multilayered epithelium. Shortly after, it forms a bud that, evaginating from the floor of the pharynx, invades the surrounding mesenchyma as an endodermal extroflexion close to the aortic sac. In these early stages of morphogenesis, the expansion of the thyroid primordium does not seem to be due to the proliferation of cells of the primitive thyroid anlage; indeed, these cells show a very low cell proliferation rate as compared to that of the cells of the surrounding endoderm. Other cells from the pharyngeal endoderm could be recruited into the developing thyroid, thus contributing to the increase of the number of thyroid progenitor cells.89 At E10.5, the thyroid bud has a flask-shaped structure still connected to the floor of the pharynx by a thin cord, the thyroglossal duct, a transient narrow channel. One day later, the thyroid bud is visible as a caplike shape that has migrated caudally into the mesenchyme, losing all connections with the floor of the pharynx (see Fig. 1-4). At this stage, the developing thyroid begins to expand laterally, the first step of the process that eventually leads to the formation of the two lobes. At E12.5, the thyroid primordium appears as an elongated structure, extended laterally, in contact with the third pharyngeal arch arteries that will participate in the formation of the definitive carotid vessels.
By E13, the thyroid bud continues its downward relocation into the mesenchyme and approaches the ultimobranchial bodies that are accomplishing their ventro-caudal migration from the fourth pharyngeal pouch. By E13.5, the developing thyroid, already formed by rudimental lobes connected by a thin central portion, reaches its definitive pretracheal position where it merges with the ultimobranchial bodies containing the precursors of C cells derived from the neural crest.78 Once the gland reaches its final location, the two rudimentary paratracheal lobes expand, and by E15 and 16, the thyroid gland assumes its definitive shape (see Fig. 1-4). In the last stages of thyroid organogenesis, the gland increases further in size, probably due to the high proliferation of TFCs.
In humans, as described in mice, at an early stage of embryonic life, the thyroid anlage appears as a bud invading the mesenchyma at E26; in a few days it appears as a migrating primordium, connected to the pharynx by the thyroglossal duct that disappears around E37. At this stage, the thyroid bud acquires a bilobed shape. The developing thyroid merges with the ultimobranchial bodies by the sixth week and reaches its final position in front of the trachea around the seventh week.90
The translocation of TFC precursors to reach the sublaryngeal position is a process that lasts for almost 4 days in the mouse embryo and almost 4 weeks in the human embryo. Budding and translocation from the gut tube is a developmental process shared by many endoderm-derived organs.91 In the case of the thyroid, since the definitive location is rather distant from the site of primitive specification, this process mostly involves active migration of the precursors, even if other morphogenetic events occurring in the neck region and in the mouth92 could contribute to the definitive location of the thyroid. The molecular mechanisms involved in the movement of the thyroid primordium are still a matter of debate. In many processes of embryogenesis, such as gastrulation, neural-crest migration, and heart formation, cell migration is involved. However, in these processes, migrating cells lose the epithelial phenotype and acquire mesenchymal features.93 This phenomenon, called epithelial-mesenchymal transition, is hallmarked by an increased expression of N-cadherin and the down-regulation of E-cadherin. In contrast, TFC precursors seem to use a different and yet unidentified pathway to move, because they maintain their epithelial phenotype throughout the entire translocation process and never acquire a mesenchymal identity.94 The expression of transcription factors such as either Hhex or Pax8 or Nkx2-1/Ttf-1 is not sufficient for thyroid migration, while Foxe1 plays a crucial role because the presence of this factor in the thyroid bud is required to allow the cells to move.79,95
The genetic basis of the formation of two symmetrical lateral lobes (lobulation) is beginning to be understood. When the lobulation process begins at E11.5, the developing thyroid is in contact with the third pharyngeal arch arteries that will participate in the formation of the definitive carotid vessels located most closely to the mature thyroid lobes. Signals originating from adjacent vessels or factors regulating vasculogenesis could instruct the process of lobulation by non–cell-autonomous mechanisms. The study of animal models seems to confirm this hypothesis. Mice deficient in either Shh (a key regulator of embryogenesis)96 or TBX (a factor regulated by Shh itself)88 display a disturbed morphogenetic patterning of vessels adjacent to the developing thyroid. In these mutated embryos, as a consequence of the absence of caudal pharyngeal arch arteries, the thyroid bud is never in close contact with vessels, and the lobulation process is impaired. The thyroid fails to separate into two distinct lobes, maintaining the shape of a single tissue mass throughout development. In line with these findings is the observation that thyroid dysgenesis is not uncommon in patients affected by DiGeorge syndrome,97 characterized by congenital anomalies of the heart and great vessels. However, the inductive signals from adjacent tissues must interact with events restricted to the thyroid cells to accomplish the lobulation process. Indeed, thyroid hemiagenesis is very frequent in mice double heterozygous for the null allele of Nkx2-1/Ttf-1 and Pax8, genes expressed in thyroid precursor cells and absent in other structures close to the developing thyroid.98
Late Stages of Thyroid Morphogenesis: Functional Differentiation and Histogenesis
When the thyroid primordium reaches the sublaryngeal position, TFC precursors accomplish their functional differentiation. Notably, the normal final location of thyroid follicular cells in front of the trachea is not an essential requirement for functional differentiation, since an ectopic, sublingual thyroid expresses thyroglobulin in both human patients99 and mutated mice.95
The functional differentiation of TFCs is hallmarked by the expression of a series of proteins essential for thyroid hormone biosynthesis, such as Tg, TPO, TSHR, NIS, Duox, and pendrin. This program requires almost 3 days, between E14 and E16.5, and results in the differentiation of the thyroid primordium in a functional thyroid gland that is able to produce and release hormones. Tg and Tshr genes are expressed by E14100; TPO and NIS, the two key enzymes involved in the process of Tg iodination appear between E15 and E15.5,101 probably because their expression is absolutely dependent on the pathway activated by the binding of TSH to its receptor, TSHR.101 Duox appears at E15.5102 and finally, thyroxine by E16.5.14
Alongside functional differentiation, the thyroid gland accomplishes its peculiar histologic organization. An inductive role of the stromal component surrounding follicular cells can be hypothesized in histogenesis. Accordingly, follicular cells, when explanted from a developing chick thyroid, can organize a correct histologic pattern in vitro only if co-cultured in the presence of fibroblasts obtained from the capsule of a thyroid gland.103 By E15.5, TFCs start to form small rudimentary follicles, as revealed by the expression of ZO-1, a tight-junction marker. At E16.5, the gland displays an evident follicular organization. The histogenesis is complete in late fetal life between E17 and E18: the thyroid parenchyma is organized into small follicles surrounded by a capillary network, enclosing thyroglobulin in their lumen.89 At birth, the thyroid gland is able to produce and release thyroid hormones, though the regulation of its growth and function by the hypothalamic-pituitary axis is fully active only after birth.104
The molecular mechanisms involved in the differentiation of the human thyroid are not much different from those found in mice. Functional differentiation of TFCs requires almost 3 weeks. It begins after the developing thyroid is located in front of the trachea, at E48, when TFCs express Tg and TSHR; T4 synthesis is detected at the 10th week.90 In humans, the establishment of the characteristic histologic organization lasts several weeks and can be divided into three phases: the precolloid, the beginning colloid, and the follicular growth, which occur at 7 to 10, 10 to 11, and after 11 weeks of gestation, respectively.76 In the precolloid phase, small intracellular canaliculi develop as an accumulation of colloid material. These small canaliculi enlarge, and the colloid organizes itself into extracellular spaces. In the last phase, primary follicles are clearly visible, and the fetal thyroid is able to concentrate iodide and synthesize thyroid hormones. The human thyroid continues to expand until term, and contrary to mice, the hypothalamic-pituitary-thyroid axis starts functioning at mid-gestation.
It is widely accepted that thyroglobulin-producing cells are derived from the endodermal cells of the thyroid anlage. However, the thyroid is assembled from both thyroid anlage and ultimobranchial bodies. Because of this composite origin, the question arises whether the neural crest–derived cells of ultimobranchial bodies (fated to become calcitonin-producing parafollicular cells) could also differentiate towards thyroglobulin-producing cells. In fish, amphibians, and birds, Tg- and calcitonin-producing cells are found in separate gland organs. In addition, lineage studies in zebrafish suggest that ultimobranchial bodies do not contribute to the development of the thyroid, which derives completely from the endodermal cells of the thyroid anlage.105
In mammals, where endodermal cells from thyroid anlage and ultimobranchial bodies merge in the definitive gland, the contribution of the different cell lineages to the TFC population is still controversial. In the past, embryologists considered the ultimobranchial bodies as the lateral anlage of the thyroid, whose cells were fated to differentiate towards the typical follicular cells and become a definitive component of the mature gland.106 This hypothesis is consistent with the report of patients displaying thyroid tissue in the submandibular region and no detectable thyroid tissue in the normal median position.107 Furthermore, structures appearing as colloid-containing follicles have been observed in ultimobranchial bodies which fail to fuse with the thyroid bud (persistent ultimobranchial body).108 Data suggesting that thyroid follicular cells could originate from ultimobranchial bodies are supported by the study of some murine models displaying persistent ultimobranchial bodies.109–112 In mice, the size of the follicular thyroid appears smaller than would be expected if only cells contributed by ultimobranchial bodies were missing. However, it is worth noting that the expression of follicular cell-specific genes (such as Tg or TPO) in the ultimobranchial bodies has not been described. Thus, there is no conclusive evidence that these cells can differentiate in cells producing thyroid hormones.
Ultimobranchial Bodies Development
By E10 in mice, the fourth pharyngeal pouch is evident for the first time. It appears as a lateral extroflexion of the primitive foregut expressing both the transcription factor Islet1 (Isl)113 and protein gene product (PGP)9.5.114,115 Shortly after, the caudal portion of the pouch grows, and at E11.5 the fourth pharynx-branchial duct is pinched off, forming an ultimobranchial body primordium visible as an ovoid vesicle with a lumen lined by a columnar epithelium identified by the simultaneous presence of Isl1, PGP9.5, and Nkx2-1/Ttf-1.64,114,115 By E11.5 ultimobranchial body primordia migrate, and at E13 they appear as solid clusters of cells in contact with the midline primordium of the thyroid. By E14.5, ultimobranchial body cells begin to disperse within the thyroid parenchyma, and 1 day later only remnants of ultimobranchial bodies can be distinguished in the thyroid gland. C cells complete their differentiation program through the expression of a series of proteins according to a precise temporal pattern: the basic helix-loop-helix transcription factor Mash1 is expressed by E12.5; the neuronal markers TuJ1, CGRP, and somatostatin by E14.5. One day later, the expression of Mash1 disappears, and calcitonin-producing cells can be detected between follicular cells. During the late stages of thyroid morphogenesis, the expression of Isl1 decreases,113 while calcitonin-producing cells gradually increase in number.114,115
In humans, the development of ultimobranchial bodies is similar to that of mice. At E24 the ultimobranchial body primordia appear as an outpouching of the ventral component of the fourth pharyngeal pouch. At this stage, the primordium of parathyroid IV is visible as a dorsal evagination of the same pouch. Some authors describe a transient fifth pouch as the endodermal origin of the ultimobranchial body.116 Probably the shape of the ultimobranchial body anlage itself, which appears as an incomplete pouch, has generated these different interpretations. By E35 the ventral extroflexion is a long-necked flask still attached to the pharynx; a few days later, the ultimobranchial body primordium loses its connection with the pharyngeal cavity, starts its migration, and at E40 reaches the posterior surface of the median thyroid. A connective layer separates these two buds, which display a different histologic organization: the lateral bud is composed of a compact mass of cells, while the median bud is composed of interconnecting sheets of epithelium. Finally, at E55 the ultimobranchial bodies are incorporated with the lateral lobes of the thyroid, and the cells from both structures mix with each other.
The genetic mechanisms that allow the developing thyroid and ultimobranchial body to recognize each other and fuse are beginning to be understood. Ultimobranchial bodies are absent in Splotch mutant mice,117 a strain characterized by an impaired migration of neural crest cells due to a loss-of-function mutation in Pax3, a gene expressed in the migrating neural crest cells.118 The absence of ultimobranchial bodies is also reported in Pax9 null mice.119 Pax9 is expressed in the entire pharyngeal endoderm and has been reported to be involved in the regulation of epithelial-mesenchymal interactions that are crucial for the correct morphogenesis of both teeth119 and thymus.120 Ultimobranchial body defects have been reported in mice carrying mutations in Hox3 paralog genes. In Hoxa-3 null mice,110 the migration of neural crest cells is not impaired and C cells differentiate correctly, but their number is significantly reduced. In many cases, ultimobranchial bodies fail to fuse with the thyroid bud and remain as bilateral vesicles composed exclusively of calcitonin-producing cells (persistent ultimobranchial bodies). The phenotype appears more severe in mice carrying various mutant combinations in Hoxa3 and its paralogs Hoxb3 and Hoxd3.111 These data indicate that Hox3 paralogs do not play a direct role in the migration and differentiation of C cells but control the correct development of the ultimobranchial body and its fusion to the ventral thyroid primordium. Another gene, Eya1, expressed in the pharyngeal arches mesenchyme and in the endoderm pouches, could control the interactions between ultimobranchial bodies and the thyroid primordium. Indeed, mice in which the Eya1 gene has been inactivated show fewer calcitonin-producing cells and persistent ultimobranchial bodies.112 Thus both Hoxa3 and Eya1 seem to control the merging of ultimobranchial bodies and thyroid primordium, a step in thyroid gland organogenesis that only occurs in mammals. Recently it has been demonstrated that Nkx2-1/Ttf-1is required for the survival of ultimobranchial body cells during migration, but it is not necessary for ultimobranchial body formation.64 Nkx2-1/Ttf-1 functions are in part dosage sensitive. Indeed, Nkx2-1/Ttf-1+/− mice display an abnormal fusion of the ultimobranchial bodies with the thyroid diverticulum. Ultimobranchial body cells are incompletely incorporated into the thyroid parenchyma and remain at the dorsal part of the thyroid lobe.64 A phenotype similar to that has been reported in mice defective for Hox3 genes.
Differentiation of c Cells
It is generally accepted that C-cell precursors do not originate from the endodermal epithelium of the pouches but derive from the cells of the neural crest that, during early development, colonize the ventral part of the fourth pharyngeal pouches. The ontogenesis and differentiation of C cells were initially studied in birds, using quail chick chimeras as models.57 The analysis of this model demonstrated that avian C-cell precursors, derived from the neural crest, colonize the ultimobranchial bodies. Neural crest cells originate early in embryonic life at the boundary between neural and non-neural ectoderm. The cells undergo an epithelial-mesenchymal transition, delaminate from the neural tube, migrate, and reach different areas of the embryo, where they differentiate into a variety of cell types. In birds, C-cell precursors probably originate from the vagal region of the neural crest that also gives rise to serotonergic enteric neurons. Indeed, mature C cells and serotonergic enteric neurons share some biochemical and morphologic features.121
In birds, the thyroid diverticulum does not fuse with the ultimobranchial bodies, which remain as distinct glands,122 but in mammals, the thyroid primordium and ultimobranchial bodies merge in the definitive thyroid gland. Experimental transplantation and ablation studies in mice123 suggest that precursors of C cells colonize the mesenchyme of the fourth pharyngeal pouch around E9 to E9.5 and a day later are localized in the endodermal layer of the pouch. However, no experimental evidence of the neural crest origin of these cells is available. Recently, fate-mapping techniques using neural crest–specific transgenes have challenged the ectodermal origin of C cells in mice,114 and it has been proposed that these cells can be derived from the endodermal epithelium of the fourth pharyngeal pouch. Consistent with this hypotheses are the findings that precursors of C cells express Isl1, a gene expressed along the endoderm and absent in neural crest cells.113
The genetic pathway controlling the differentiation of C cells is unknown. It is worth noting that expression of neuronal genes (such as TuJ1, CGRP, and somatostatin) in precursor C cells follows the expression of Mash1.114,115 This gene is involved in the differentiation of autonomic neurons.124 The relevance of Mash1 for C-cell differentiation is confirmed by the finding that Mash1 null mutant mice lack thyroid C cells.115 These mutant mice show a normal formation and migration of ultimobranchial bodies, but precursor C cells degenerate before they become differentiated C cells. It has been suggested that the transmembrane receptor tyrosine kinase Ret could also be involved in C-cell differentiation. Indeed, Ret is expressed in precursors of C cells at early stages of development, and the expression of this receptor continues in adult C cells. The role of Ret in the development of C cells has not yet been elucidated. At E18, calcitonin-producing cells are present in the thyroid of Ret null mouse embryos, even though the number is reduced compared to a wild type thyroid. These data suggest that only a subgroup of C cells (or their progenitor) require Ret/GDNF signaling to differentiate or to survive.125
Molecular Genetics of Thyroid Gland Development
The discovery of transcription factors, which regulate the expression of follicular cell-specific genes in the mature thyroid and are also expressed in the thyroid primordium, offered a useful tool to explore the genetic basis of the developmental process of the thyroid gland. At E8.5 epithelial cells fated to become thyroid follicular cells are unequivocally individualized in the endodermal layer of the primitive pharynx. These cells are characterized by the expression of Nkx2-1/Ttf-1,100 Foxe1,126 Pax8,127 and Hhex.128 Although these transcription factors are also expressed in other embryonic tissues, the coexpression of all four is only seen in the presumptive thyroid anlage when the thickening of proliferating cells appears in the midline of the floor of the primitive pharynx. When the thyroid diverticulum forms and begins its migration, only the thyroid primordium still expresses Nkx2-1/Ttf-1, Foxe1, Pax8 and Hhex, while the thyroglossal duct does not.100 These four factors remain expressed and are a hallmark of differentiated thyroid follicular cells (Table 1-1). Their expression is down-regulated only after transformation of the cells.129 The hypothesis that the expression of these four factors is required at early stages of thyroid morphogenesis has been confirmed by studies on both animal models and patients affected by thyroid dysgenesis.86,130 However, the presence of these genes is not sufficient to guarantee a correct organogenesis of the gland. Mutations in other genes, both thyroid-enriched and ubiquitous, have been demonstrated to impair the development of the thyroid. Here we summarize what we know at the moment about the molecular genetics of thyroid development, mainly as deduced by the phenotype of knockout animals.
Table 1-1
Expression of Relevant Genes and Capacity to Produce Thyroid Hormones at Different Stages of Thyroid Development in Mice
Nkx2-1/Ttf-1
Nkx2-1/Ttf-1 (formerly TTF-1, for thyroid transcription factor 1, or T/EBP) is a transcription factor that recognizes and binds to specific DNA sequences via a 61-amino-acid long DNA binding domain called a homeodomain. The homeodomain sequence is conserved from the fruit fly to humans. Nkx2-1/Ttf-1 was initially identified in a rat thyroid cell line131 as a nuclear protein able to bind to specific sequences in the Tg promoter. The corresponding cDNA was subsequently cloned, and comparative sequence analyses demonstrated that Nkx2-1/Ttf-1 has a considerable degree of homology to the Drosophila NK-2 class of homeodomain proteins.
Nkx2-1/Ttf-1 is a member of the Nkx2 class of transcription factors and is encoded by a single gene whose official name is Nkx2-1 in mice and NKX2-1 in humans, located on chromosome 12 and on chromosome 14q13,132 respectively. The gene is formed by at least three exons that express multiple transcripts.133,134 The most abundant is the shorter isoform, a 2.3-kilobase (kb) mRNA which encodes a 42-kD protein 371 amino acids long in humans.135 In the lung, a longer protein has also been detected; however, the biological relevance of the various proteins is unclear. Functional studies have determined that the homeodomain is responsible only for the binding to DNA,136 while the transactivating activity resides in two domains, N and C, localized at the NH2 and COOH ends of the protein, respectively.137 These two domains appear redundant in in vitro assay but could have different functions in vivo. In fact, a number of interactors of Nkx2-1/Ttf-1 have been identified that bind specifically to either the N or C domain. Nkx2-1/Ttf-1 is phosphorylated in several serine residues.138 This posttranslation modification could regulate Nkx2-1/Ttf-1 activity. Indeed, a mouse model harboring an unphosphorylated Nkx2-1/Ttf-1 allele shows impaired differentiation of both thyroid and lung.139
The expression pattern of Nkx2-1/Ttf-1 has been exhaustively studied in rodents. Nkx2-1/Ttf-1 is expressed in the thyroid, lung, and brain. In mouse embryos, Nkx2-1/Ttf-1 is detected in the developing thyroid as soon as the thyroid anlage is visible (E8.5).100 Interestingly, Nkx2-1/Ttf-1 is present also in the epithelial cells of the four pharyngeal pouches forming the ultimobranchial body.64 In the adult thyroid, Nkx2-1/Ttf-1 maintains its expression in both follicular and parafollicular C cells.63 During embryonic life, Nkx2-1/Ttf-1 is detected in the epithelial cells of the developing trachea and lungs; in adults, it is present in bronchiolar epithelial Clara cells and in type II alveolar cells. As for the brain, Nkx2-1/Ttf-1 is expressed in some areas of the developing diencephalon, such as the hypothalamic areas and the infundibulum from which the neurohypophysis develops.100 Nkx2-1/Ttf-1 expression in adult hypothalamus is faint but is up-regulated before the onset of puberty.140 In human embryos, the expression pattern of Nkx2-1/Ttf-1 is not different from that of mice except that Nkx2-1/Ttf-1 is not detected in the fourth pharyngeal pouch.77
Gene targeting experiments have allowed in vivo study of the role of this transcription factor. In the absence of Nkx2-1/Ttf-1, newborn mice immediately die at birth and are characterized by impaired morphogenesis of both lung and brain and lack of thyroid and the entire pituitary141 (Fig. 1-5). In Nkx2-1/Ttf-1 null newborns, the lungs appear as dilated saclike structures without normal pulmonary parenchyma141; furthermore, the trachea has a reduced number of cartilage rings and is not separated from the esophagus.142 In the case of the brain, alterations in the ventral region of the forebrain are evident.143 In addition, both neurohypophysis and anterior hypophysis are absent. As a consequence of the lack of signals from the pituitary, the morphogenesis of adrenal glands is impaired too.141
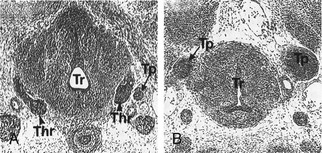
FIGURE 1-5 Thyroid gland phenotype in Nkx2-1/Ttf-1 mouse embryos. Transversal sections of E13 wt (A) and Nkx2-1/Ttf-1−/− (B) mouse embryos stained with hematoxylin/eosin. In Nkx2-1/Ttf-1 null embryos, the thyroid primordium is absent. Thr, Thyroid; Tr, trachea; Tp, Thymic primordium. (From Kimura S, Hara Y, Pineau T et al: The T/ebp null mouse: Thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev 10:60–69, 1996.)
In Nkx2-1/Ttf-1 null embryos, the thyroid anlage forms in its correct position, but at an early stage, the morphogenesis of the gland is impaired. The thyroid primordium by E10 is hypoplastic; subsequently it undergoes degeneration probably due to apoptosis.144 The ultimobranchial bodies undergo the same process; they form but degenerate by E12.64 Hence, Nkx2-1/Ttf-1 is dispensable for the initial commitment of both follicular and parafollicular thyroid cells. However, both cell types that will form the thyroid gland require Nkx2-1/Ttf-1 for their survival. In the absence of this transcription factor, no thyroid rudiment is detectable. The genetic pathways defective in Nkx2-1/Ttf-1 null embryos have been only partially elucidated. In the case of the developing thyroid, we know that Nkx2-1/Ttf-1 is part of a network that includes Pax8, Hhex, and Foxe1. Actually, the expression of these factors is slightly (Pax8) or strongly (Hhex and Foxe1) reduced in the absence of Nkx2-1/Ttf-1.79
We do not know which genes are controlled by Nkx2-1/Ttf-1 factor in the thyroid primordium. A detailed analysis of the phenotype of other tissues, such as lung and brain, reveals that when Nkx2-1/Ttf-1 is absent, the expression of Bmp4, a TGF-β-related peptide growth factor that is required for the proximal-distal patterning of the lung bud, is abolished.142 In Nkx2-1/Ttf-1 null embryos, Fgf8 is down-regulated in the infundibulum of the pituitary, probably causing the regression of the pituitary primordium in these embryos.145 These data indicate that both in the lung and in the pituitary, signaling molecules that are important for a further specification of preformed structures are controlled by Nkx2-1/Ttf-1. A similar role could be hypothesized also for the developing thyroid. The finding that Fgfr2 is expressed in the thyroid bud suggests that Nkx2-1/Ttf-1 could regulate the survival of the precursor thyroid cells through an Fgf-dependent mechanism.86
In Nkx2-1/Ttf-1 null mice, the thyroid primordium disappears before TFC precursors accomplish their differentiation. For this reason, information on the function of this factor in differentiated TFC is scarce. A mutated mouse in which Nkx2-1/Ttf-1 has been disrupted only in the thyroid gland in the middle of organogenesis presents a variable phenotype: either the thyroid follicles appear atrophic, or the thyroid shows a reduced number of dilated follicles.146 This indicates the requirement of Nkx2-1/Ttf-1 in adult life to maintain the follicular structure of the gland.
Other studies on culture cell lines can offer data on the role of Nkx2-1/Ttf-1 in terminal differentiated cells. In the promoters of thyroid-specific or enriched genes, such as Tg, TPO, Tshr and NIS, consensus sequences recognized by Nkx2-1/Ttf-1 have been identified. Nkx2-1/Ttf-1 has also been shown to be able to bind in vitro to its own 5′-flanking region.147 In both Tg and TPO proximal promoters, multiple Nkx2-1/Ttf-1 binding sites are present, and transfection assays have demonstrated that binding of Nkx2-1/Ttf-1 to these sites leads to the activation of Tg or TPO promoters.
Pax8
Pax8 (paired box gene 8) is a member of a family of transcription factors characterized by the presence of a 128-amino-acid domain that recognizes and binds to specific DNA sequences. This DNA binding domain is called a paired domain because it was first identified in the Drosophila segmentation gene as paired. Pax8 was identified in the mouse as a protein expressed in the developing thyroid gland.127 Further studies demonstrated that the Pax8 paired domain recognizes and binds to a single site present in the Tg and in TPO promoters.148
The gene encoding Pax8 is located on chromosome 2 in mice,89,127 and the human orthologue, PAX8, is located on chromosome 2q12-q14.149 It consists of 12 exons150 that encode for different alternative spliced transcripts,151 at least six different transcripts in the mouse and five in humans. All the isoforms generated share the paired domain located near the amino terminus and differ in their carboxy-terminal regions. Pax8a, the most abundant protein isoform, is 457 amino acids long in mice127 and 450 amino acids long in humans.152
Pax8 is expressed in kidney, in the nervous system, and in the thyroid.127 At an early stage in the developing kidney, Pax8 is expressed in the nephrogenic cord and in mesonephric tubules; then it is present in the cortex of the metanephros. In the adult kidney, Pax8 is clearly detected in the medullar zone. In embryos, Pax8 is transiently expressed in the myelencephalon, through the entire length of the neural tube, otic vesicle, and at the midbrain-hindbrain boundary127 but is no longer detected by E12.5. As for the thyroid, Pax8 is expressed in the thyroid follicular cells and in their precursors from the early stages of gland morphogenesis.
Recently it has been reported that Pax8 is expressed in the uterine epithelium, the luminal epithelium of the oviduct, and vagina in 3-week-old as well as sexually mature female mice.153,154 In males, a strong Pax8 mRNA expression was observed in the epithelium of the epididymis.155 In humans, PAX8 is detected in the developing thyroid, kidneys, otic vesicle, and central nervous system at E3277 and in both luminal and glandular epithelium of the endometrium.154
Studies in cultured thyroid cells156,157 have suggested that Pax8 synthesis is regulated by TSH by a cAMP-mediated mechanism. However, mutated mice in which the TSH/TSHR signaling is abolished do not show a reduced expression of Pax8.101
Analysis of Pax8−/− mice65 (Fig. 1-6) offers the unique possibility of studying the in vivo role of this transcription factor. Pax8 null pups are born without any apparent brain or kidney defects. On the contrary, the thyroid appears as a rudimental structure lacking thyroid follicular cells, composed almost completely of calcitonin-producing C cells. The animals are affected by a severe hypothyroidism, show growth retardation, and die within 2 to 3 weeks after birth. The administration of thyroxine to these mice leads to their survival. However, in T4-treated female Pax8−/− animals, the development of the reproductive system is severely affected: the uterus appears as remnants of myometrium lacking endometrial structures, and the vaginal opening does not occur at all.154
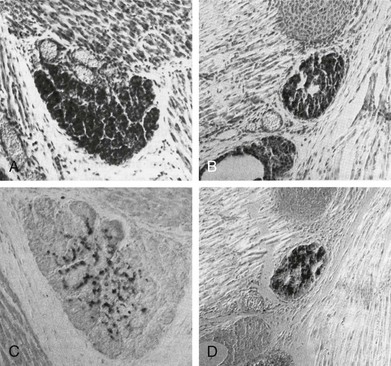
FIGURE 1-6 Thyroid gland phenotype in Pax8−/− mouse embryos. Sagittal sections of E18 wt (A, C) and Pax8−/− (B, D) mouse embryos stained with hematoxylin/eosin (A, B) and anticalcitonin-specific antibody (C, D). The thyroid gland of Pax8−/− embryos is smaller than that of wt and composed almost wholly of C cells. (From Mansouri A, Chowdhury K, Gruss P: Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet 19:87–90, 1998.)
In Pax8 null embryos, the thyroid anlage is correctly formed, evaginates from the endoderm, and begins to migrate into the mesenchyme. However, by E11 the thyroid bud is much smaller compared to that of a wild-type. In addition, in the absence of Pax8, other transcription factors such as Foxe1 and Hhex are down-regulated in the precursors of thyroid cells.79 At E12.5, thyroid follicular cells are not detectable.65 During morphogenesis, Pax8 holds a specific upstream role in the genetic regulatory cascade which controls thyroid development; it is required for the survival of the thyroid precursor cells and to maintain the tissue-specific gene expression program. In particular, Foxe1 seems to be tightly regulated by Pax8, which is necessary for the onset of its expression.79
In Pax8 null mice, mature TFCs are absent, making it difficult to reveal the role of this factor in the control of adult thyroid function. All the available data on Pax8 functions in differentiated cells come from studies on cell lines in culture. Functional assays have demonstrated that Pax8 is required to activate the TPO promoter and to a lesser extent the Tg promoter148 and NIS enhancer.158 In addition, Pax8 is able to bind in vitro to the 5′-flanking region of Foxe1 and Duox genes.147 Pax8 activates the expression of the endogenous genes encoding Tg, TPO, and NIS at their chromosomal locus.159 These data suggest that Pax8 has an important role in the maintenance of functional differentiation in thyroid cells.159
Pax8 and Nkx2-1/Ttf-1 are coexpressed only in thyroid cells. This finding has suggested some interaction between these two factors. In fact, coimmunoprecipitation experiments in thyroid cells have shown that Nkx2-1/Ttf-1 and Pax8 form a protein complex in vivo. Furthermore, Nkx2-1/Ttf-1 and Pax8 also have functional interactions, since the two factors cooperate in a synergistic manner in activating Tg promoter.160
Foxe1
Foxe1 (formerly called TTF-2 for thyroid transcription factor 2) was originally identified as a thyroid-specific nuclear protein that can bind to a sequence present on both Tg and TPO promoters under insulin, insulin-like growth factor 1 (IGF-1), or TSH stimulation.161 Subsequently, rat Foxe1 cDNA was cloned, allowing characterization of the salient features of this protein.126 Foxe1 belongs to the winged helix/forkhead family of transcription factors characterized by a 100 amino acid–long DNA binding domain162 homologous to that of the Drosophila forkhead gene.163 The official name for the genetic locus encoding this transcription factor is Foxe1 in mice (located on chromosome 4126) and FOXE1 in humans (located on chromosome 9q2295,164). Foxe1 is an intronless gene coding for a 42 kD protein that is phosphorylated.165 The protein contains an alanine stretch of variable length. In humans, the most frequent FOXE1 allele contains 14 alanine residues and is 371 amino acids long.166
Like Nkx2-1/Ttf-1 and Pax8, Foxe1 is detected in the thyroid primordium, and its expression is maintained in TFCs during all stages of development and in adulthood. However, during embryonic life, Foxe1 has a wide domain of expression. Indeed, at early stages of development, Foxe1 is detected in the endodermal epithelium lining the primitive pharynx, the arches and the foregut and, transiently, in Rathke’s pouch. Subsequently, Foxe1 is expressed in tissues which are developed from the pharynx and pharyngeal arches: thyroid, tongue, epiglottis, palate, choanae and esophagus. In addition, Foxe1 is also detected in the whiskers and hair follicles which derive from the ectoderm.165 Studies in mutant mice have shown that Foxe1 is tightly regulated in the thyroid bud by Pax8 and in the pharyngeal cells by Shh.79 In humans, FOXE1 is expressed in the thyroid, foregut, embryonic thymus,77 in the outer follicular hair sheath, and the seminiferous tubules of prepubertal testis.167
Analysis of Foxe1 null mice (Fig. 1-7) revealed the role of this transcription factor during thyroid development. Targeted inactivation of Foxe1 showed that homozygous Foxe1−/− mice are born at the expected Mendelian ratio but die within 48 hours of birth. These mice display a severe cleft palate, probably responsible for the perinatal death, an absent thyroid or ectopic thyroid, lack of thyroid hormones and elevated TSH levels in the bloodstream.95 The early stages of thyroid morphogenesis, when the thyroid anlage is the formed, are not affected in Foxe1−/− embryos. However, at E10, thyroid precursor cells are still on the floor of the pharynx in Foxe1 null embryos, whereas in wild-type embryos the thyroid primordium begins to descend towards its final location. At later stages of development, in the absence of Foxe1, thyroid follicular cells either disappear or form a small thyroid remnant still attached to the pharyngeal floor. In this case, the cells are able to complete their differentiation program, as tested by their ability to synthesize thyroglobulin. These data indicate that during embryonic life, Foxe1 has a specific role in controlling the migration of thyroid follicular cell precursors, most likely by controlling the expression of target genes required for the migration process, but this is not relevant for the specification and differentiation of the thyroid anlage. In addition, Foxe1 could be involved in the survival of TFCs, since in many Foxe1 null embryos, the thyroid primordium disappears.79,95
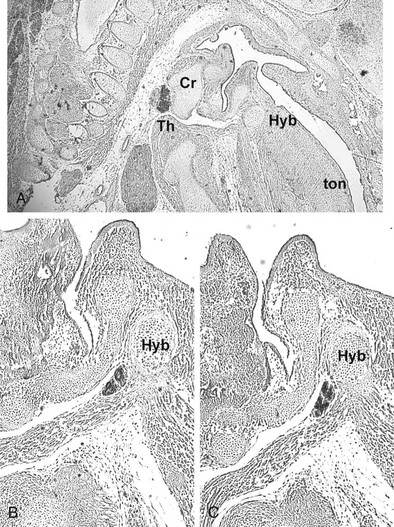
FIGURE 1-7 Thyroid gland phenotype in Foxe1−/− mouse embryos. Sagittal sections of E15.5 Foxe1+/− (A) and Foxe1−/− (B, C) embryos stained with an anti-Nkx2-1/Ttf-1 (A, B) or antithyroglobulin (C) antibody. The thyroid gland of Foxe1−/− embryo is in a sublingual position. Cr, Cricoid cartilage; Hyb, hyoid bone; Th, thyroid; ton, tongue. (From De Felice M, Ovitt C, Biffali E et al: A mouse model for hereditary thyroid dysgenesis and cleft palate. Nat Genet 19:395–398, 1998.)
In differentiated thyroid cell lines, the transcription of the Foxe1 gene is under the tight control of TSH and cAMP as well as that of insulin and IGF-1.126,168 These data suggest that Foxe1 plays an important role in controlling the interactions between hormone signaling pathway and thyroid specific gene expression. In the developing thyroid these controls do not seem to be effective, since in mutated mice which lack TSH, growth hormone (GH), and IGF-1, the expression of Foxe1 is not affected.101
Hhex
Hhex (formerly known as Hex for hematopoietically expressed homeobox or Prh for proline-rich homeobox) is a homeodomain-containing transcription factor that was first identified in multipotent hematopoietic cells.169 It was successively demonstrated that Hhex is expressed in other tissues, including the thyroid.128
Hhex is encoded by a gene called Hhex in mice and HHEX in humans, located on chromosome 19 and chromosome 10q23.32, respectively. The gene has 4 exons and codes for a protein 270 amino acids long. Hhex is an orphan homeobox-containing gene because the sequence of its homeodomain responsible for binding to DNA shows some differences with respect to other homeodomains.169 Outside the homeodomain, Hhex contains an N-terminal proline-rich region and a C-terminal acid region. These two regions are probably involved in repressing the transcription of the target genes.
In mouse embryos, Hhex is expressed very early in the primitive and then in the definitive endoderm. At E7.0, Hhex is detected in the developing blood islands, in the ventral foregut endoderm at E8.5, and in the endothelium of the developing vasculature and heart at E9.0. Hhex is an early marker of thyroid cells, since it is already expressed in the thyroid anlage at E8.5128 at the same stage in which Nkx2-1/Ttf-1, Foxe1, and Pax8 are detected. In the adult, in addition to the thyroid, Hhex expression is maintained in liver and lungs.
The analysis of mutated mice has revealed that this factor is absolutely necessary in many developmental processes. Hhex−/− embryos die at mid-gestation (between E13.5 and E15.5) and show severe defects in liver, forebrain, heart, and thyroid morphogenesis.170,171 In Hhex null embryos, thyroid precursor cells are present and express Nkx2-1/Ttf-1, Pax8, and Foxe1 until E9. At E10, the thyroid primordium is composed of a few cells which express neither Nkx2-1/Ttf-1 nor Foxe1 nor Pax8.170 Hence, Hhex guarantees the survival of TFC precursors and maintains the expression of Foxe1, Nkx2-1/Ttf-1, or Pax8. However, in the absence of either Nkx2-1/Ttf-1 or Pax8, the expression of Hhex is down-regulated.
As for Nkx2-1/Ttf-1, Pax8, and Foxe1, a conditional knockout mouse will be a useful tool to elucidate the role of Hhex in the adult thyroid gland. Studies in differentiated thyroid cells suggest that the network ruling Hhex and the other thyroid-specific transcription factors is complex. Hhex seems to be regulated by Nkx2-1/Ttf-1,172 and the overexpression of Hhex partly inhibits Tg promoter activity. These data are consistent with the hypothesis that Hhex is a transcriptional repressor in thyroid cells as reported in other systems.173
Fgfr2
Fgfr2-IIIb (fibroblast growth factor receptor 2, isoform IIIb) is a tyrosine kinase receptor present in many types of epithelial cells. This receptor is recognized and activated by specific peptide growth factors (Fgf1, Fgf3, Fgf7 and Fgf10) expressed in the mesenchyme.174 Interactions of Fgfr2 with its cognate ligands mediate epithelium-mesenchyme cross-talk and are involved in important processes of organogenesis.174 The relevance of Fgfr2/Fgf interaction in thyroid morphogenesis is demonstrated by the absence of the thyroid gland in mutant mice expressing a soluble dominant negative form of Fgfr2-IIIb175 and in mice deficient for this isoform.176 The stage at which thyroid morphogenesis is first impaired has not been elucidated yet. Since Fgfr2 is expressed in the thyroid primordium starting at E11.5, it is possible that Fgfr2/Fgf signaling is required after budding for the progression of the differentiation programs.
Nkx2-5, Nkx2-6, and Nkx2-3
Nkx2-5 is present in the ventral region of the pharynx and in the thyroid anlage by E8.5177; however, after E11.5, the expression of Nkx2-5 is no longer detected in the thyroid bud.178 In Nkx2-5−/− embryos, thyroid morphogenesis occurs, but the thyroid bud appears to be smaller than that of wild-type embryos.178 It is not possible to study the role of this factor in differentiated thyroid morphogenesis, because Nkx2-5−/− embryos die at an early stage of development.
Other genes of the Nkx2 family are present in the primitive pharynx and the thyroid anlage. Nkx2-6 is transiently expressed in the endodermal layer of the midline region of the pharynx at E8.5.177 Nkx2-3 is strongly expressed in the developing thyroid and disappears at birth.177 However, neither Nkx2-6179 nor Nkx2-3 null mice show any apparent thyroid phenotype.89,177
TSHR
TSHR (thyroid-stimulating hormone receptor), a member of the family of G protein–coupled receptors,180 is encoded by the gene Tshr in mice and TSHR in humans, located on chromosome 12 and on chromosome 14q31, respectively. The gene, formed by 10 exons, is translated into a protein 765 amino acids long, expressed on the basolateral membrane of TFCs and in a few other tissues. The binding of TSH to TSHR triggers a signaling pathway that regulates many functions of the thyroid gland in postnatal life. Here we describe the role of the TSH/Tshr pathway during thyroid organogenesis.
In the mouse embryo, TSHR is detected in TFC precursors between E14 and 14.5,100,181 when the developing thyroid has reached its final position and Tg is expressed. At later stages of development, Tshr expression increases and remains expressed in adult life.
The availability of mice that carry mutations in the Tshr gene allowed elucidation of the role of TSH/TSHR signaling during embryonic life.101 Both Tshrhyt/hyt (carrying a loss-of-function mutation in the Tshr gene182) and Tshr−/− adult mice183 display a severe hypothyroidism with an hypoplastic thyroid. During embryonic life, in the absence of a functional TSHR, the developing thyroid does not show any alterations in either size or histologic structure. However, the expression of both TPO mRNA and NIS is strongly down-regulated.101 Thus, TSH/TSHR signaling is required to complete the differentiation program of the thyroid follicular cell. However, the TSH-induced cAMP pathway is not involved in controlling the growth of the embryonic gland, while this pathway is the main regulator of the growth of the adult thyroid. Other growth factors expressed during embryogenesis, such as IGF-1 or EGF, able to promote the growth of thyroid cells in culture, could also be involved in controlling the proliferation of immature thyroid cells. It is worth noting that the requirements for the growth of the embryonic thyroid seem to be different between mouse and human; in the latter, TSH/TSHR signaling during fetal life is necessary for the development of thyroid.104
Hoxa3, Eya1, and Tbx
Hoxa3 belongs to the Hox family of transcription factors characterized by the presence of a homeodomain as the DNA binding domain. These factors regulate the regionalization of the embryo along its major axes and are also involved in the morphogenesis of several structures. Hoxa3 is expressed in the pharynx, in the developing thyroid, and in the mesenchymal, endodermal, and neural crest–derived cells of the fourth pharyngeal pouch.110
Mice in which Hoxa3 has been disrupted lack the thymus and parathyroid and show persistent ultimobranchial bodies and variable thyroid anomalies,109,110 including hypoplasia, hemiagenesis, reduction of the number of follicular cells, and absence of isthmus. The thyroid phenotype is exacerbated in mice carrying various mutant combinations in Hoxa3 and its paralogs Hoxb3 and Hoxd3.111 It is thought that the impaired organogenesis of the thyroid is secondary to defects in the ultimobranchial bodies. This hypothesis is confirmed by other findings. Mice deprived of Eya1—a transcription factor expressed in the ultimobranchial bodies but not in the developing thyroid—display a thyroid phenotype that is almost identical to that displayed in Hoxa3 mutants.112 Furthermore, mice carrying mutations in Pax3117 or Endothelin-1,184 both molecules implicated in the development of neural crest–derived structures, show a defective thyroid organogenesis. Finally, in Tbx null embryos, the ultimobranchial bodies fail to develop, and the thyroid gland appears as a hypoplastic single-tissue mass.88 Tbx is a transcription factor expressed in the non–neural crest mesoderm and endoderm of the pharyngeal arches but not in thyroid precursor cells. It is worth noting that hypothyroidism is detected in a number of patients affected by velocardiofacial syndrome/DiGeorge syndrome, which is most likely associated with haploinsufficiency of the Tbx1 gene.185 Taken together, these observations suggest a relevant role for non–cell-autonomous factors in thyroid development.
Insights from Human Diseases
In 85% of congenital hypothyroidism (CH) cases, the most frequent endocrine disorder in newborns, the condition is due to thyroid dysgenesis (TD). The term thyroid dysgenesis indicates an ectopic or hypoplastic thyroid as well as the absence of thyroid (thyroid agenesis or athyreosis).186 In addition, the term also includes thyroid malformations, such as hemiagenesis, which are not associated with apparent thyroid dysfunction.187 Thyroid dysgenesis is due to anomalies during gland organogenesis: defects in growth and/or differentiation of the thyroid primordium can result in an absent or hypoplastic thyroid; an impaired migration of thyroid precursor cells causes an ectopic gland.
The study on the molecular genetics of thyroid dysgenesis seems to be a useful tool in the elucidation of the mechanisms underlying thyroid development, since in many cases it has confirmed data obtained in animal models and has provided new insights into thyroid morphogenesis and differentiation.86,90,188 Here we will focus on genes known (or candidate) to be responsible for this phenotype (Table 1-2). The mutations thus far identified in patients with congenital hypothyroidism associated with TD account only for a very small number of cases. However, these cases could be much more frequent than hitherto identified because mutations which might arise in specific regulatory elements were not searched for in the published studies.
Table 1-2
Currently Available Mouse Models of Thyroid Dysgenesis and Summary of Known and Potential Genes Involved in the Pathogenesis of the Disease
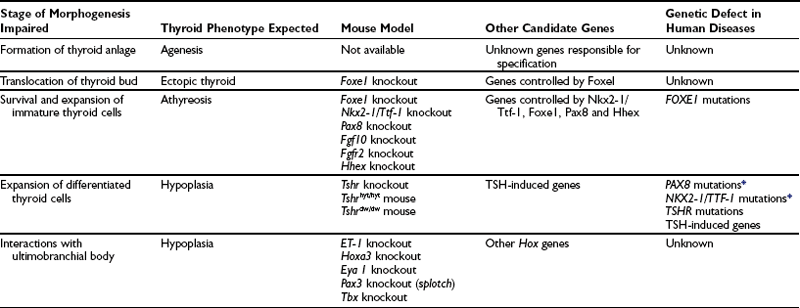
*In humans, unlike mice, the abnormal phenotype is detected in heterozygotes.
Agenesis and Athyreosis
Both athyreosis and agenesis indicate the absence of thyroid tissue. However, agenesis should be used to define the absence of the gland due to a defective initiation of thyroid morphogenesis.86 The term athyreosis indicates a dysgenesis characterized by the disappearance of the thyroid due to alterations in any step following the specification of the thyroid anlage. In several patients with athyreosis, the presence of cystic structures (probably resulting from the degeneration of TFC clusters189) suggests that thyroid specification was not impaired in these subjects.
FOXE1 and Thyroid Dysgenesis
Homozygous loss-of-function mutation in the FOXE1 gene was first reported190 in two siblings affected by syndromic congenital hypothyroidism characterized by athyreosis, cleft palate, bilateral choanal atresia, and spiky hair (Bamforth-Lazarus syndrome191). After this report, a different mutation in FOXE1 has been recorded in two siblings with athyreosis and a less severe extrathyroidal phenotype.192 A third loss-of-function mutation within the FOXE1 forkhead domain has been described193 in a child displaying CH with eutopic thyroid tissue and extrathyroidal defects. The variable thyroid phenotype displayed by patients carrying FOXE1 mutations could be due to different effects of the various mutations. The phenotype is consistent with the expression domain of FOXE1 and partially overlaps that displayed by Foxe1 null mice. However, in mice, the absence of this factor causes either athyreosis or defects in thyroid migration, whereas in humans, FOXE1 mutations associated with an ectopic thyroid have not been described.
Ectopic Thyroid
The ectopic thyroid is a consequence of an impaired thyroid bud migration. More than 50% of patients affected by CH show an ectopic thyroid, but to date no known genes have been associated with this dysgenesis.194 Familial cases of TD have been described, in which affected members show either ectopic thyroid or athyreosis.195 This observation raises the possibility that athyreosis and thyroid ectopy have a common underlying mechanism. This hypothesis is consistent with the data from mice lacking the Foxe1 protein, which show either ectopy with a very small thyroid or no thyroid at all.95
Recently, four subjects affected by CH with TD-carrying heterozygous mutations in the NKX2-5 gene have been described. In three out of the four patients, the presence of an ectopic thyroid suggests that NKX2-5 mutations might have a specific pathogenic role in this thyroid malformation.178
PAX8 and Thyroid Dysgenesis
Mutations in the DNA binding domain of the PAX8 gene have been found in sporadic and familial cases of CH with TD.196–200 Patients show a variable phenotype ranging from mild to severe hypoplasia of the thyroid in the presence of elevated levels of TSH in the bloodstream. Since the phenotype shows discrepancy even in familial cases in which related individuals carry the same mutation, effects of other modifier genes could be hypothesized. In two patients, renal hemiagenesis has been reported.199
Affected individuals are heterozygous for the mutations, and in the familial cases, the mode of inheritance is dominant. Thus in humans, both PAX8 alleles are necessary for correct thyroid morphogenesis, and a reduced dosage of the gene product (haploinsufficiency) causes dysgenesis; in contrast, Pax8+/− mice display a normal phenotype.65 The reason for this discrepancy is unknown.
NKX2-1/TTF-1 and Thyroid Dysgenesis
Heterozygous point mutations or chromosomal deletions in the NKX2-1/TTF-1 locus have been found in patients affected by a complex syndrome characterized by thyroid alterations, choreoathetosis, and lung abnormalities.201–206
All affected individuals are heterozygous for the mutations. The dominant effect of NKX2-1/TTF-1 mutations in humans could be due to a haploinsufficiency. Accordingly, in mice Nkx2-1/Ttf-1 functions are partially dosage-sensitive. Functional studies have revealed that Nkx2-1/Ttf-1+/− mice present decreased coordination and mild hyperthyrotropinemia.204,206 NKX2-1/TTF-1+/− subjects display a highly variable thyroid phenotype. In many patients, thyroid alterations have not been reported; in other cases, patients show elevated TSH levels with mild or severe hypoplasia of the gland. There is no clear correlation between the phenotype severity and the type of mutations. In addition, a variable phenotype has also been reported in the same familial cluster. As in the case of PAX8 disease, other modifier genes could be responsible for the incomplete penetrance and variability of the phenotype.
TSHR and Thyroid Dysgenesis
Almost 40 years ago, Stanbury207 hypothesized that TSH unresponsiveness could cause CH without goiter. However, mutations in the TSHR gene were identified 15 years ago208 and shown to be responsible for TD-associated congenital hypothyroidism. Among the molecular defects causing TD, mutations in the TSHR gene represent the most frequent find. Patients affected with CH show increased TSH secretion and mild or severe thyroid hypoplasia.209 In many cases, the thyroid size appears normal, and only an asymptomatic hyperthyrotropinemia characterizes these individuals. Athyreosis or ectopic thyroid have never been found in patients carrying TSHR mutations. These observations confirm that the TSHR-induced pathway is not involved in the early stages of thyroid development. All the affected individuals are homozygous or compound heterozygous for loss-of-function mutations. Consistently in the familial forms, the disease is inherited as an autosomal recessive trait.
Phylogenesis of the Thyroid Gland
The morphologic bases of thyroid function are the same in all vertebrates, as reflected by the conserved follicular structure throughout vertebrate evolution. On the contrary, the gross anatomy of the thyroid gland appears different among vertebrate classes.10 In placental mammals and in some reptiles, the thyroid is composed of two lobes connected by an isthmus which crosses the trachea. In nonplacental mammals, birds, and amphibians, the thyroid consists of two isolated lobes. In cartilaginous fish and in some teleosts (mostly marine ones), the thyroid is massed into a compact organ. In the remaining marine and almost all freshwater teleosts, the thyroid consists of nonencapsulated follicles scattered in the subpharyngeal connective tissue.122 Often heterotopic follicles are found in nonpharyngeal areas such as kidneys, heart, esophagus, or spleen in these animals. Interestingly, the ability of thyroid cells to proliferate in these ectopic areas has been shown to be genetically controlled in platyfish, and low iodine intake represents a strong inductive stimulus towards this condition.210 Even though iodine-concentrating activity and iodoamino acid production are ancient phenomena that have also been described in nonvertebrate animals, only cyclostomes (such as the lamprey) have a bona fide thyroid characterized by follicles scattered in the subpharyngeal connective tissue.
The ontogeny of the thyroid follows the same pattern in all vertebrates, including teleosts211: the thyroid anlage arises from an outpouching of the primitive pharynx, migrates caudally, and reaches its definitive position. The development of the thyroid in cyclostomes follows a different pattern from that used by the higher vertebrates. Thyroid follicles of the adult lamprey derive from the endostyle, an organ present only during larval life. In the larval lamprey, at the early stages of development, the endostyle forms as a ciliated groove in the ventral part of the pharynx. During development the groove becomes a cylinder and then a complex structure connected to the pharyngeal cavity by the ductus hypobranchialis.212 The epithelium of the endostyle differentiates into different types of cells, a group of which displays the ability to trap iodine, peroxidase activity, and expression of thyroxine.213 These cells are fated to become “typical” follicular cells only after metamorphosis.214
A structure similar to the lamprey endostyle is present in the ventral pharynx of protochordates such as cephalochordates (amphioxus) and urochordates (such as ascidians). This organ, which is also called the endostyle, is composed of different cell types, the major part of which are involved in the food-capturing process.212 More than a century ago, the homology between the cyclostome endostyle and the thyroid suggested the hypothesis that the endostyle of protochordates was a primitive antecedent of the vertebrate thyroid gland.215,216 This hypothesis, based on morphologic observation, has gained support from the presence of iodine trapping,217 thyroperoxidase activity,218 and Ci-Duox (the Ciona homolog vertebrate Duox) in a group of cells of the endostyle of protochordates. Thus, these cells can be considered “protothyroid” cells.212 The molecular mechanisms involved in the differentiation of thyroid cells of mammals are conserved throughout the evolution of protochordates to vertebrate chordates. In urochordates (Ciona intestinalis), the group of cells that express the TPO homologs219 also express Ci-FoxE (Foxe1) homolog220 but not Ci-Nkx2-1/Ttf-1.221,222 On the contrary, in amphioxus, both Nkx2-1/Ttf-1 and TPO homologous genes are coexpressed in the same cells of the endostyle,218 while the Foxe1 homolog Amphi-FoxE4 is not detected in the endostyle but in another pharyngeal-derived structure.223 These data suggest that during the evolution of protochordates, a group of genes whose function was to contribute to the specification of the anterior part of the gut were selected as part of the genetic program that leads to thyroid specification.
References
1. Werner, S. Historical resumè. In: Ingbar SH, Braverman LE, eds. The Thyroid. Philadelphia: Lippincott; 1986:3–6.
2. Netter FH: Anatomy of the thyroid and parathyroid glands. In The CIBA Collection of Medical Illustrations, vol 4, Summit, NJ, 1965, CIBA Pharmaceutical Products, pp 41–70.
3. Fujita, H. Functional morphology of the thyroid. Int Rev Cytol. 1988;113:145–185.
4. Romeo, HE, Gonzalez Solveyra, C, Vacas, MI, et al. Origins of the sympathetic projections to rat thyroid and parathyroid glands. J Auton Nerv Syst. 1986;17:63–70.
5. Cauna, N, Naik, N. The distribution of cholinesterases in the sensory ganglia of man and some mammals. J Histochem Cytochem. 1963;11:129–138.
6. Sundler, F, Grunditz, T, Hakanson, R, et al. Innervation of the thyroid. A study of the rat using retrograde tracing and immunocytochemistry. Acta Histochem Suppl. 1989;37:191–198.
7. Sawicki, B. Evaluation of the role of mammalian thyroid parafollicular cells. Acta Histochem. 1995;97:389–399.
8. Grunditz, T, Hakanson, R, Rerup, C, et al. Neuropeptide Y in the thyroid gland: neuronal localization and enhancement of stimulated thyroid hormone secretion. Endocrinology. 1984;115:1537–1542.
9. Ahrèn, B. Regulatory peptides in the thyroid gland—a review on their localization and function. Acta Endocrinol. 1991;124:225–232.
10. Gorbman, A, Bern, HA. Thyroid gland. In: Gorbman A, Bern HA, eds. A Textbook of Comparative Endocrinology. New York: Willey; 1962:99–173.
11. Mauchamp, J, Mirrione, A, Alquier, C, et al. Follicle-like structure and polarized monolayer: role of the extracellular matrix on thyroid cell organization in primary culture. Biol Cell. 1998;90:369–380.
12. Yap, AS, Stevenson, BR, Armstrong, JW, et al. Thyroid epithelial morphogenesis in vitro: a role for butamide-sensitive Cl− secretion during follicular lumen development. Exp Cell Res. 1994;213:319–326.
13. Romagnoli, P, Herzog, V. Transcytosis in thyroid follicle cells: regulation and implications for thyroglobulin transport. Exp Cell Res. 1991;194:202–209.
14. Meunier, D, Aubin, J, Jeannotte, L. Perturbed thyroid morphology and transient hypothyroidism symptoms in Hoxa5 mutant mice. Dev Dyn. 2003;227:367–378.
15. Shepard, T. Onset of function in the human fetal thyroid: biochemical and radioautographic studies from organ culture. J Clin Endocrinol Metab. 1987;27:945–958.
16. Drubin, DG, Nelson, J. Origins of cell polarity. Cell. 1996;84:335–344.
17. Matter, K, Mellman, I. Mechanism of cell polarity: sorting and transport in epithelial cells. Curr Opin Cell Biol. 1994;6:545–554.
18. Ekholm, R. Biosynthesis of thyroid hormones. Int Rev Cyt. 1990;120:243–288.
19. Bourke, JR, Sand, O, Abel, KC, et al. Chloride channels in the apical membrane of thyroid epithelial cells are regulated by cyclic AMP. J Endocrinol. 1995;147:441–448.
20. Bourke, JR, Abel, KC, Huxham, GJ, et al. Sodium channel heterogeneity in the apical membrane of porcine thyroid epithelial cells. J Endocrinol. 1996;149:101–108.
21. Royaux, IE, Suzuki, K, Mori, A, et al. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology. 2000;141:839–845.
22. De Deken, X, Wang, D, Many, MC, et al. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem. 2000;275:23227–23233.
23. Paire, A, Bernier-Valentin, F, Selmi-Ruby, S, et al. Characterization of the rat thyroid iodide transporter using anti-peptide antibodies. J Biol Chem. 1997;272:18245–18249.
24. Gerard, C, Gabrion, J, Verrier, B, et al. Localization of the Na+/K+-ATPase and of an amiloride sensitive Na+ uptake on thyroid epithelial cells. Eur J Cell Biol. 1985;38:134–141.
25. Westermack, K, Westermack, B, Karslsson, A, et al. Localization of epidermal growth factor receptors on porcine thyroid follicle cells and receptor regulation by thyrotropin. Endocrinology. 1986;118:1040–1046.
26. Costagliola, S, Rodien, P, Many, MC, et al. Genetic immunization against the human thyrotropin receptor causes thyroiditis and allows production of monoclonal antibodies recognizing the native receptor. J Immunol. 1998;160:1458–1465.
27. Ericson, L. Exocytosis and endocytosis in the thyroid follicle cell. Mol Cell Endocrinol. 1981;22:1–24.
28. Lohi, J, Leivo, I, Franssila, K, et al. Changes in the distribution of integrins and their basement membrane ligands during development of human thyroid follicular epithelium. Histochem J. 1997;29:337–345.
29. Vitale, M, Casamassima, A, Illario, M, et al. Cell-to-cell contact modulates the expression of the beta1 integrins in primary cultures of thyroid cells. Exp Cell Res. 1995;220:124–129.
30. Burgi-Saville, ME, Gerber, H, Peter, HJ, et al. Expression patterns of extracellular matrix components in native and cultured normal human thyroid tissue and in human toxic adenoma tissue. Thyroid. 1997;7:347–356.
31. Andre, F, Filippi, P, Feracci, H. Merosin is synthesized by thyroid cells in primary culture irrespective of cellular organization. J Cell Sci. 1994;107:183–193.
32. Gumbiner, B. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357.
33. Denker, BM, Nigam, S. Molecular structure and assembly of the tight junction. Am J Physiol. 1998;274:1–9.
34. Yap, AS, Stevenson, BR, Abel, KC, et al. Microtubule integrity is necessary for the epithelial barrier function of cultured thyroid cell monolayers. Exp Cell Res. 1995;218:540–545.
35. Boller, K, Vestweber, D, Kemler, R. Cell-adhesion molecule uvomorulin is localized in the intermediate junctions of adult intestinal epithelial cells. Cell Biol. 1985;100:327–332.
36. Kemler, K. From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 1993;9:317–321.
37. Buxton, RS, Magee, A. Structure and interactions of desmosomal and other cadherins. Semin Cell Biol. 1992;3:157–167.
38. Kowalczyk, AP, Bornslaeger, EA, Norvell, SM, et al. Desmosomes: intercellular adhesive junctions specialized for attachment of intermediate filaments. Int Rev Cytol. 1999;185:237–302.
39. Munari-Silem, Y, Rousset, B. Gap junction-mediated cell-to-cell communications in endocrine glands-molecular and functional aspects: a review. Eur J Endocrinol. 1996;135:251–264.
40. Yap, AS, Manley, S. Contact inhibition of cell spreading: a mechanism for the maintenance of thyroid cell aggregation in vitro. Exp Cell Res. 1993;208:121–127.
41. Yap, AS, Stevenson, BR, Keast, JR, et al. Cadherin-mediated adhesion and apical membrane assembly define distinct steps during thyroid epithelial polarization and lumen formation. Endocrinology. 1995;136:4672–4680.
42. Yap, AS, Manley, SW. Thyrotropin inhibits the intrinsic locomotility of thyroid cells organized as follicles in primary culture. Exp Cell Res. 1994;214:408–417.
43. Yap, AS. Initiation of cell locomotility is a morphogenetic checkpoint in thyroid epithelial cells regulated by ERK and PI3-kinase signals. Cell Motil Cytoskeleton. 2001;49:93–103.
44. Yap, AS, Abel, KC, Bourke, JR, et al. Different regulation of thyroid cell-cell and cell-substrate adhesion by thyrotropin. Exp Cell Res. 1992;202:366–369.
45. Nilsson, M, Fagman, H, Ericson, L. Ca2+-dependent and Ca2+-independent regulation of the thyroid epithelial junction complex by protein kinases. Exp Cell Res. 1996;225:1–11.
46. Pellerin, S, Croizet, K, Rabilloud, R, et al. Regulation of the three-dimensional organization of thyroid epithelial cells into follicle structures by the matricellular protein, thrombospondin-1. Endocrinology. 1999;140:1094–1103.
47. Gartner, R, Schopohl, D, Schaefer, S, et al. Regulation of transforming growth factor beta 1 messenger ribonucleic acid expression in porcine thyroid follicles in vitro by growth factors, iodine, or delta iodolactone. Thyroid. 1997;7:633–640.
48. Toda, S, Matsumura, S, Fujitani, N, et al. Transforming growth factor beta 1 induces a mesenchyma-like cell shape without epithelial polarization in thyrocytes and inhibits thyroid folliculogenesis in collagen gel culture. Endocrinology. 1997;138:5561–5575.
49. Paire, A, Bernier-Valentin, F, Rabilloud, R, et al. Expression of alpha- and beta subunits and activity of Na+K+-ATPase in pig thyroid cells in primary culture: modulation by thyrotropin and thyroid hormones. Mol Cell Endocrinol. 1998;146:93–101.
50. Tacchetti, C, Zurzolo, C, Monticelli, C, et al. Functional properties of normal and inverted rat thyroid follicles in suspension culture. J Cell Physiol. 1986;126:93–98.
51. Halmi, N. Anatomy and histochemistry. In: Ingbar SH, Braverman LE, eds. The Thyroid. Philadelphia: Lippincott; 1986:24–36.
52. Fujita, H. Fine structure of the thyroid gland. Int Rev Cytol. 1975;40:197–280.
53. Engstrom, G, Ericson, L. Effect of graded dose of thyrotropin on exocytosis and early phase of endocytosis in the rat thyroid. Endocrinology. 1981;108:399–405.
54. Bernier-Valentine, F, Kostrouch, Z, Rabilloud, R, et al. Analysis of the thyroglobulin internalization process using in vitro reconstituted follicles: evidence for a coated vesicle-dependent endocytic pathway. Endocrinology. 1991;129:2194–2201.
55. Damante, G, Tell, G, Di Lauro, R. A unique combination of transcription factors controls differentiation of thyroid cells. Prog Nucleic Acid Res Mol Biol. 2001;66:307–356.
56. Pearse, A. The cytochemistry of the thyroid C cells and their relationship to calcitonin. Proc Roy Soc Biol. 1966;164:478–487.
57. Le Douarin, N, Fontaine, J, LeLievre, C. New studies on the neural crest origin of the avian ultimobranchial glandular cells. Interspecific combinations and cytochemical characterization of C cells based on the uptake of biogenic amine precursors. Histochemie. 1974;38:297–305.
58. Pearse, AG, Caralheira, A. Cytochemical evidence for an ultimobranchial origin of rodent thyroid C cells. Nature. 1967;214:929–930.
59. Martin-Lacave, M, Conde, E, Montenero, C, et al. Quantitative changes in the frequency and distribution of the C-cell population in the rat thyroid gland with age. Cell Tissue Res. 1992;270:73–77.
60. Wolfe, HJ, DeLellis, RA, Voelkel, EF, et al. Distribution of calcitonin-containing cells in the normal neonatal human thyroid gland: a correlation of morphology with peptide content. J Clin Endocrinol Metab. 1975;41:1076–1081.
61. Neve, P, Wollman, S. Fine structure of ultimobranchial follicles in the thyroid gland of the rat. Anat Rec. 1971;171:259–272.
62. Minvielle, S, Giscard-Dartevelle, S, Cohen, R, et al. A novel calcitonin carboxyl-terminal peptide produced in medullary thyroid carcinoma by alternative RNA processing of the calcitonin/calcitonin gene-related peptide gene. J Biol Chem. 1991;266:24627–24631.
63. Suzuki, K, Kobayashi, Y, Katoh, R, et al. Identification of thyroid transcription factor-1 in C cells and parathyroid cells. Endocrinology. 1998;139:3014–3017.
64. Kusakabe, T, Hoshi, N, Kimura, S. Origin of the ultimobranchial body cyst: T/ebp/Nkx2.1 expression is required for development and fusion of the ultimobranchial body to the thyroid. Dev Dyn. 2006;235:1300–1309.
65. Mansouri, A, Chowdhury, K, Gruss, P. Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet. 1998;19:87–90.
66. Martin-Lacave, I, Rojas, F, Bernabe, R, et al. Comparative immunohistochemical study of normal, hyperplastic and neoplastic C cells of the rat thyroid gland. Cell Tissue Res. 2002;309:361–368.
67. Kameda, Y, Oyama, H, Endoh, M, et al. Somatostatin immunoreactive C cells in thyroid glands from various mammalian species. Anat Rec. 1982;204:161–170.
68. Neonakis, E, Thomas, GA, Davies, HG, et al. Expression of calcitonin and somatostatin peptide and mRNA in medullary thyroid carcinoma. World J Surg. 1994;18:588–593.
69. Blaker, M, de Weerth, A, Schulz, M, et al. Expression of the cholecystokinin 2-receptor in normal human thyroid gland and medullary thyroid carcinoma. Eur J Endocrinol. 2002;146:89–96.
70. Wollman, SH, Neve, P. Postnatal development and properties of ultimobranchial follicles in the rat thyroid. Anat Rec. 1971;171:247–258.
71. Calvert, R. Structure of rat ultimobranchial bodies after birth. Anat Rec. 1974;181:561–580.
72. Harach, H. Solid-cell nests of the thyroid. J Pathol. 1988;155:191–200.
73. Reis-Filho, JS, Preto, A, Soares, P, et al. p63 expression in solid-cell nests of the thyroid: further evidence for a stem cell origin. Mod Pathol. 2003;16:43–48.
74. Preto, A, Cameselle-Teijeiro, J, Moldes-Boullosa, J, et al. Telomerase expression and proliferative activity suggest a stem cell role for thyroid solid cell nests. Mod Pathol. 2004;17:819–826.
75. Burstein, DE, Nagi, C, Wang, BY, et al. Immunohistochemical detection of p53 homolog p63 in solid cell nests, papillary thyroid carcinoma, and Hashimoto’s thyroiditis: a stem cell hypothesis of papillary carcinoma oncogenesis. Hum Pathol. 2004;35:465–473.
76. Fisher, DA, Dussault, JH, Sach, J, et al. Ontogenesis of hypothalamic-pituitary-thyroid function and metabolism in man, sheep and rat. Recent Prog Hormone Res. 1976;33:59–116.
77. Trueba, SS, Auge, J, Mattei, G, et al. PAX8, TITF1 and FOXE1 gene expression patterns during human development: new insights into human thyroid development and thyroid dysgenesis associated malformations. J Clin Endocrinol Metab. 2004;90:455–462.
78. Kaufman, MH, Bard, J. The thyroid. In: Kaufman MH, Bard J, eds. The Anatomic Basis of Mouse Development. San Diego: Academic Press; 1999:165–166.
79. Parlato, R, Rosica, A, Rodriguez-Mallon, A, et al. An integrated regulatory network controlling survival and migration in thyroid organogenesis. Dev Biol. 2004;276:464–475.
80. Elsalini, OA, von Gartzen, J, Cramer, M, et al. Zebrafish Hhex, Nk2.1a, and Pax2.1 regulate thyroid growth and differentiation downstream of nodal-dependent transcription factors. Dev Biol. 2003;263:67–80.
81. Cai, CL, Liang, X, Shi, Y, et al. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–889.
82. Kumar, M, Melton, D. Pancreas specification: a budding question. Curr Opin Genet Dev. 2003;13:401–407.
83. Lammert, E, Cleaver, O, Melton, D. Role of endothelial cells in early pancreas and liver development. Mech Dev. 2003;120:35–43.
84. Serls, A, Doherty, S, Parvatiyar, P, et al. Different thresholds of fibroblast growth factors pattern the ventral foregut into liver and lung. Development. 2005;132:35–47.
85. Wendl, T, Adzic, D, Schoenebeck, JJ, et al. Early developmental specification of the thyroid gland depends on han-expressing surrounding tissue and on FGF signals. Development. 2007;134:2871–2879.
86. De Felice, M, Di Lauro, R. Thyroid development and its disorders: genetics and molecular mechanisms. Endocr Rev. 2004;25:722–746.
87. Olivieri, A, Stazi, MA, Mastroiacovo, P, et al. A population-based study on the frequency of additional congenital malformations in infants with congenital hypothyroidism: data from the Italian Registry for Congenital Hypothyroidism (1991–1998). J Clin Endocrinol Metab. 2002;87:557–562.
88. Fagman, H, Liao, J, Westerlund, J, et al. The 22q11 deletion syndrome candidate gene Tbx1 determines thyroid size and positioning. Hum Mol Genet. 2007;16:276–285.
89. Fagman, H, Andersson, L, Nilsson, M. The developing mouse thyroid: embryonic vessel contacts and parenchymal growth pattern during specification, budding, migration, and lobulation. Dev Dyn. 2006:235.
90. Polak, M, Sura-Trueba, S, Chauty, A, et al. Molecular mechanisms of thyroid dysgenesis. Horm Res. 2004;62:14–21.
91. Hogan, B, Zaret, K. Development of the endoderm and its tissue derivatives. In: Rossant J, ed. Mouse Development: Patterning, Morphogenesis, and Organogenesis. New York: Academic Press; 2002:301–310.
92. Hilfer, SR, Brown, JW. The development of pharyngeal endocrine organs in mouse and chick embryos. Scan Electron Microsc. 1984;4:2009–2022.
93. Thiery, JP, Sleeman, J. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142.
94. Fagman, H, Grande, M, Edsbagge, J, et al. Expression of classical cadherins in thyroid development: maintenance of an epithelial phenotype throughout organogenesis. Endocrinology. 2003;144:3618–3624.
95. De Felice, M, Ovitt, C, Biffali, E, et al. A mouse model for hereditary thyroid dysgenesis and cleft palate. Nat Genet. 1998;19:395–398.
96. Fagman, H, Grande, M, Gritli-Linde, A, et al. Genetic deletion of sonic hedgehog causes hemiagenesis and ectopic development of the thyroid in mouse. Am J Pathol. 2004;164:1865–1872.
97. Burke, BA, Johnson, D, Gilbert, EF, et al. Thyrocalcitonin-containing cells in the DiGeorge anomaly. Hum Pathol. 1987;4:355–360.
98. Amendola, E, De Luca, P, Macchia, PE, et al. A mouse model demonstrates a multigenic origin of congenital hypothyroidism. Endocrinology. 2005;146:5038–5047.
99. Hartzband, PI, Diehl, DL, Lewin, KJ, et al. Histological characterization of a lingual mass using thyroglobulin immunoperoxidase staining. J Endocrinol Invest. 1984;7:221–223.
100. Lazzaro, D, Price, M, De Felice, M, et al. The transcription factor TTF-1 is expressed at the onset of thyroid and lung morphogenesis and in restricted regions of the foetal brain. Development. 1991;113:1093–1104.
101. Postiglione, MP, Parlato, R, Rodriguez-Mallon, A, et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proc Natl Acad Sci U S A. 2002;99:15462–15467.
102. Milenkovic, M, De Deken, X, Jin, L, et al. Duox expression and related H2O2 measurement in mouse thyroid: onset in embryonic development and regulation by TSH in adult. J Endocrinol. 2007;192:615–626.
103. Hilfer, SR, Stern, M. Instability of the epithelial-mesenchymal interaction in the eight-day embryonic chick thyroid. J Exp Zool. 1971;178:293–305.
104. De Felice, M, Postiglione, MP, Di Lauro, R. Mini review: thyrotropin receptor signaling in development and differentiation of the thyroid gland: insights from mouse models and human diseases. Endocrinology. 2004;145:4062–4067.
105. Alt, B, Reibe, S, Feitosa, NM, et al. Analysis of origin and growth of the thyroid gland in zebrafish. Dev Dyn. 2006;235:1872–1883.
106. Weller, G. Development of the thyroid, parathyroid and thymus glands in man. Contrib Embryol. 1933;24:93–140.
107. Kumar, R, Gupta, R, Bal, CS, et al. Thyrotoxicosis in a patient with submandibular thyroid. Thyroid. 2000:363–365.
108. Harach, H. Thyroglobulin in human thyroid follicles with acid mucins. J Pathol. 1991;164:261–263.
109. Chisaka, O, Musci, TS, Capecchi, MR. Developmental defects of the ear, cranial nerves and hindbrain resulting from targeted disruption of the mouse homeobox gene Hox-1.6. Nature. 1992;355:516–520.
110. Manley, NR, Capecchi, M. The role of Hoxa-3 in mouse thymus and thyroid development. Development. 1995;121:1989–2003.
111. Manley, NR, Capecchi, M. Hox group 3 paralogs regulate the development and migration of the thymus, thyroid, and parathyroid glands. Dev Biol. 1998;195:1–15.
112. Xu, PX, Zheng, W, Laclef, C, et al. Eya1 is required for the morphogenesis of mammalian thymus, parathyroid and thyroid. Development. 2002;129:1033–1044.
113. Westerlund, J, Andersson, L, Carlsson, T, et al. Expression of Islet1 in thyroid development related to budding, migration, and fusion of primordia. Dev Dyn. 2008;237:3820–3829.
114. Kameda, Y, Nishimaki, T, Chisaka, O, et al. Expression of the epithelial marker E-cadherin by thyroid C cells and their precursors during murine development. Histochem Cytochem. 2007;55:1075–1088.
115. Kameda, Y, Nishimaki, T, Miura, M, et al. Mash1 regulates the development of C cells in mouse thyroid glands. Dev Dyn. 2007;236:262–270.
116. Merida-Velasco, JA, Garcia-Garcia, JD, Espin-Ferra, J, et al. Origin of the ultimobranchial body and its colonizing cells in human embryos. Acta Anat. 1989;73:325–330.
117. Franz, T. Persistent truncus arteriosus in the Splotch mutant mouse. Anat Embryol. 1989;180:457–464.
118. Epstein, JA, Li, J, Lang, D, et al. Migration of cardiac neural crest cells in Splotch embryos. Development. 2000;127:1869–1878.
119. Neubüser, A, Peters, H, Balling, R, et al. Antagonistic interactions between FGF and BMP signaling pathways: a mechanism for positioning the sites of tooth formation. Cell. 1997;90:247–255.
120. Hetzer-Egger, C, Schorpp, M, Haas-Assenbaum, A, et al. Thymopoiesis requires Pax9 function in thymic epithelial cells. Eur J Immunol. 2002;32:1175–1181.
121. Clark, MS, Lanigan, TM, Page, NM, et al. Induction of a serotonergic and neuronal phenotype in thyroid C-cells. J Neurosci. 1995;15:6167–6178.
122. Gorbman, A. Comparative anatomy and physiology. In: Ingbar SI, Braverman LE, eds. The Thyroid. Philadelphia: Lippincott; 1986:43–52.
123. Fontaine, J. Multistep migration of calcitonin cell precursors during ontogeny of the mouse pharynx. Gen Comp Endocrinol. 1979;37:81–92.
124. Guillemot, F. Analysis of the role of basic-helix-loop-helix transcription factors in the development of neural lineages in the mouse. Biol Cell. 1995;84:3–6.
125. Lindahl, M, Timmusk, T, Rossi, J, et al. Expression and alternative splicing of mouse Gfra4 suggest roles in endocrine cell development. Mol Cell Neurosci. 2000:522–533.
126. Zannini, M, Avantaggiato, V, Biffali, E, et al. TTF-2, a new forkhead protein, shows a temporal expression in the developing thyroid which is consistent with a role in controlling the onset of differentiation. EMBO J. 1997;16:3185–3197.
127. Plachov, D, Chowdhury, K, Walther, C, et al. Pax8, a murine paired box gene expressed in the developing excretory system and thyroid gland. Development. 1990;110:643–651.
128. Thomas, PQ, Brown, A, Beddington, R. Hex: a homeobox gene revealing peri-implantation asymmetry in the mouse embryo and an early transient marker of endothelial cell precursors. Development. 1998;125:85–95.
129. Francis-Lang, H, Zannini, MS, De Felice, M, et al. Multiple mechanisms of interference between transformation and differentiation in thyroid cells. Mol Cell Biol. 1992;12:5793–5800.
130. De Felice, M, Di Lauro, R. Murine models for the study of thyroid gland development. Endocr Dev. 2007;10:1–14.
131. Civitareale, D, Lonigro, R, Sinclair, AJ, et al. A thyroid-specific nuclear protein essential for tissue-specific expression of the thyroglobulin promoter. EMBO J. 1989;8:2537–2542.
132. Guazzi, S, Price, M, De Felice, M, et al. Thyroid nuclear factor 1 (TTF-1) contains a homeodomain and displays a novel DNA binding specificity. EMBO J. 1990;9:3631–3639.
133. Lonigro, R, De Felice, M, Biffali, E, et al. Expression of thyroid transcription factor 1 gene can be regulated at the transcriptional and posttranscriptional levels. Cell Growth Differ. 1996;7:251–261.
134. Hamdan, H, Liu, H, Li, C, et al. Structure of the human Nkx2.1 gene. Biochim Biophys Acta. 1998;1396:336–348.
135. Ikeda, K, Clark, JC, Shaw-White, JR, et al. Gene structure and expression of human thyroid transcription factor-1 in respiratory. J Biol Chem. 1995;270:8108–8114.
136. Damante, G, Di Lauro, R. Several regions of Antennapedia and thyroid transcription factor 1 homeodomains contribute to DNA binding specificity. Proc Natl Acad Sci U S A. 1991;88:5388–5392.
137. De Felice, M, Damante, G, Zannini, MS, et al. Redundant domains contribute to the transcriptional activity of thyroid transcription factor 1 (TTF-1). J Biol Chem. 1995;270:26649–26656.
138. Zannini, MS, Acebron, A, Felice, MD, et al. Mapping and functional role of phosphorylation sites in the thyroid transcription factor 1 (TTF-1). J Biol Chem. 1996;271:2249–2254.
139. De Felice, M, Silberschmidt, D, DiLauro, R, et al. TTF-1 phosphorylation is required for peripheral lung morphogenesis, perinatal survival, and tissue-specific gene expression. J Biol Chem. 2003;278:35574–35583.
140. Lee, BJ, Cho, GJ, Norgren, RB, Jr., et al. TTF-1, a homeodomain gene required for diencephalic morphogenesis, is postnatally expressed in the neuroendocrine brain in a developmentally regulated and cell-specific fashion. Mol Cell Neurosci. 2001;17:107–126.
141. Kimura, S, Hara, Y, Pineau, T, et al. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 1996;10:60–69.
142. Minoo, P, Su, G, Drum, H, et al. Defects in tracheoesophageal and lung morphogenesis in Nkx2.1(−/−) mouse embryos. Devel Biol. 1999;209:60–71.
143. Sussel, L, Marin, O, Kimura, S, et al. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370.
144. Kimura, S, Ward, JD, Minoo, P. Thyroid-specific enhancer-binding protein/transcription factor 1 is not required for the initial specification of the thyroid and lung primordia. Biochimie. 1999;81:321–328.
145. Takuma, N, Sheng, HZ, Furuta, Y, et al. Formation of Rathke’s pouch requires dual induction from the diencephalon. Development. 1998;125:4835–4840.
146. Kusakabe, T, Kawaguchi, A, Hoshi, N, et al. Thyroid-specific enhancer-binding protein/NKX2.1 is required for the maintenance of ordered architecture and function of the differentiated thyroid. Mol Endocrinol. 2006;20:1796–1809.
147. D’Andrea, B, Iacone, R, Di Palma, T, et al. Functional inactivation of the transcription factor Pax8 through oligomerization chain reaction. Mol Endocrinol. 2006;20:1810–1824.
148. Zannini, MS, Francis-Lang, H, Plachov, D, et al. Pax-8, a paired domain-containing protein, binds to a sequence overlapping the recognition site of a homeodomain and activates transcription from two thyroid-specific promoters. Mol Cell Biol. 1992;12:4230–4241.
149. Stapleton, P, Weith, A, Urbanek, P, et al. Chromosomal localization of 7 Pax genes and cloning of a novel family member, Pax-9. Nature Genet. 1993;3:292–298.
150. Okladnova, O, Poleev, A, Fantes, J, et al. The genomic organization of the murine Pax 8 gene and characterization of its basal promoter. Genomics. 1997;15:452–461.
151. Poleev, A, Wendler, F, Fickenscher, H, et al. Distinct functional properties of three human paired-box-protein, PAX8, isoforms generated by alternative splicing in thyroid, kidney and Wilms’ tumors. Eur J Biochem. 1995;228:899–911.
152. Poleev, A, Fickenscher, H, Mundlos, S, et al. PAX8, a human paired box gene: isolation and expression in developing thyroid, kidney and Wilms’ tumors. Development. 1992;116:611–623.
153. Ferretti, E, Arturi, F, Mattei, T, et al. Expression, regulation, and function of paired-box gene 8 in the human placenta and placental cancer cell lines. Endocrinology. 2005;146:4009–4015.
154. Mittag, J, Winterhager, E, Bauer, K, et al. Congenital hypothyroid female Pax8-deficient mice are infertile despite thyroid hormone replacement therapy. Endocrinology. 2007;148:719–725.
155. Wistuba, J, Mittag, J, Luetjens, CM, et al. Male congenital hypothyroid Pax8−/− mice are infertile despite adequate treatment with thyroid hormone. J Endocrinol. 2007;192:99–109.
156. Fabbro, D, Pellizzari, L, Mercuri, F, et al. Pax-8 protein levels regulate thyroglobulin gene expression. J Mol Endocrinol. 1998;21:347–354.
157. Van Renterghem, P, Vassart, G, Christophe, D. Pax 8 expression in primary cultured dog thyrocyte is increased by cyclic AMP. Biochim Biophys Acta. 1996;1307:97–103.
158. Ohno, M, Zannini, MS, Levy, O, et al. The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat Sodium/Iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol Cell Biol. 1999;19:2051–2060.
159. Pasca di Magliano, M, Di Lauro, R, Zannini, MS. Pax8 has a key role in thyroid cell differentiation. Proc Natl Acad Sci U S A. 2000;97:13144–13149.
160. Di Palma, T, Nitsch, R, Mascia, A, et al. The paired domain-containing factor Pax8 and the homeodomain-containing factor TTF-1 directly interact and synergistically activate transcription. J Biol Chem. 2003;278:3395–3402.
161. Santisteban, P, Acebron, A, Polycarpou-Schwarz, M, et al. Insulin and insulin-like growth factor 1 regulate a thyroid-specific nuclear protein that binds to the thyroglobulin promoter. Mol Endocrinol. 1992;6:1310–1317.
162. Kaestner, KH, Lee, KH, Schlondorff, J, et al. Six members of the mouse forkhead gene family are developmentally regulated. Proc Natl Acad Sci U S A. 1993;90:7628–7631.
163. Lai, E, Prezioso, VR, Tao, WF, et al. Hepatocyte nuclear factor 3 alpha belongs to a gene family in mammals that is homologous to the Drosophila homeotic gene forkhead. Genes Dev. 1991;5:416–427.
164. Chadwick, BP, Obermayr, F, Frischau, A. FKHL15, a new human member of the forkhead gene family located on chromosome 9q22. Genomics. 1997;41:390–396.
165. Dathan, N, Parlato, R, Rosica, A, et al. Distribution of the Titf2/Foxe1 gene product is consistent with an important role in the development of foregut endoderm, palate, and hair. Dev Dyn. 2002;224:450–456.
166. Macchia, PE, Mattei, MG, Lapi, P, et al. Cloning, chromosomal localization and identification of polymorphisms in the human thyroid transcription factor 2 gene (TITF2). Biochimie. 1999;81:433–440.
167. Sequeira, M, Al-Khafaji, F, Park, S, et al. Production and application of polyclonal antibody to human thyroid transcription factor 2 reveals thyroid transcription factor 2 protein expression in adult thyroid and hair follicles and prepubertal testis. Thyroid. 2003;13:927–932.
168. Ortiz, L, Zannini, MS, Di Lauro, R, et al. Transcriptional control of the forkhead thyroid transcription factor TTF-2 by thyrotropin, insulin and insulin-like growth factor 1. J Biol Chem. 1997;272:23334–23339.
169. Crompton, MR, Bartlett, TJ, MacGregor, AD, et al. Identification of a novel vertebrate homeobox gene expressed in haematopoietic cells. Nucleic Acids Res. 1992;20:5661–5667.
170. Martinez Barbera, JP, Clements, M, Thomas, P, et al. The homeobox gene Hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development. 2000;127:2433–2445.
171. Hallaq, H, Pinter, E, Enciso, J, et al. A null mutation of Hhex results in abnormal cardiac development, defective vasculogenesis and elevated Vegfa levels. Development. 2004;131:5197–5209.
172. Puppin, C, D’Elia, AV, Pellizzari, L, et al. Thyroid-specific transcription factors control Hex promoter activity. Nucleic Acids Res. 2003;31:1845–1852.
173. Tanaka, T, Inazu, T, Yamada, K, et al. DNA cloning and expression of rat homeobox gene, Hex, and functional characterization of the protein. Biochem J. 1999;339:111–117.
174. De Moerlooze, L, Spencer-Dene, B, Revest, J, et al. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127:483–492.
175. Celli, G, LaRochelle, WJ, Mackem, S, et al. Soluble dominant-negative receptor uncovers essential roles for fibroblast growth factors in multi-organ induction and patterning. EMBO J. 1998;17:1642–1645.
176. Revest, JM, Spencer-Dene, B, Kerr, K, et al. Fibroblast growth factor receptor 2-IIIb acts upstream of Shh and Fgf4 and is required for limb bud maintenance but not for the induction of Fgf8, Fgf10, Msx1, or Bmp4. Dev Biol. 2001;231:47–62.
177. Biben, C, Wang, CC, Harvey, RP. NK-2 class homeobox genes and pharyngeal/oral patterning: Nkx2-3 is required for salivary gland and tooth morphogenesis. Int J Dev Biol. 2002;46:415–422.
178. Dentice, M, Cordeddu, V, Rosica, A, et al. Missense mutation in the transcription factor NKX2-5: a novel molecular event in the pathogenesis of thyroid dysgenesis. J Clin Endocrinol Metab. 2006;91:1428–1433.
179. Tanaka, M, Yamasaki, N, Izumo, S. Phenotypic characterization of the murine Nkx2.6 homeobox gene by gene targeting. Mol Cell Biol. 2000;20:2874–2879.
180. Parmentier, M, Libert, F, Maenhaut, C, et al. Molecular cloning of the thyrotropin receptor. Science. 1989;246:1620–1622.
181. Brown, RS, Shalhoub, V, Coulter, S, et al. Developmental regulation of thyrotropin receptor gene expression in the fetal and neonatal rat thyroid: relation to thyroid morphology and to thyroid-specific gene expression. Endocrinology. 2000;141:340–345.
182. Stuart, A, Oates, E, Hall, C, et al. Identification of a point mutation in the thyrotropin receptor of the hyt/hyt hypothyroid mouse. Mol Endocrinol. 1994;8:129–138.
183. Marians, RC, Ng, L, Blair, HC, et al. Defining thyrotropin-dependent and independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc Natl Acad Sci U S A. 2002;99:15776–15781.
184. Kurihara, Y, Kurihara, H, Maemura, K, et al. Impaired development of the thyroid and thymus in endothelin-1 knockout mice. J Cardiovasc Pharmacol. 1995;26:13–16.
185. Liao, J, Kochilas, L, Nowotschin, S, et al. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum Mol Genet. 2004;13:1577–1585.
186. Fisher, DA, Klein, A. Thyroid development and disorders of thyroid function in the newborn. New Engl J Med. 1981;304:702–712.
187. Maiorana, R, Carta, A, Floriddia, G, et al. Thyroid hemiagenesis: prevalence in normal children and effect on thyroid function. J Clin Endocrinol Metab. 2003;88:1534–1536.
188. Van Vliet, G. Development of the thyroid gland: lessons from congenitally hypothyroid mice and men. Clin Genet. 2003;63:445–455.
189. Marinovic, D, Garel, C, Czernichow, P, et al. Additional phenotypic abnormalities with presence of cysts within the empty thyroid area in patients with congenital hypothyroidism with thyroid dysgenesis. J Clin Endocrinol Metab. 2003;88:1212–1216.
190. Clifton-Bligh, RJ, Wentworth, JM, Heinz, P, et al. Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat Genet. 1998;19:399–401.
191. Bamforth, JS, Hughes, IA, Lazarus, JH, et al. Congenital hypothyroidism, spiky hair, and cleft palate. J Med Genet. 1989;26:49–60.
192. Castanet, M, Park, SM, Smith, A, et al. A novel loss-of-function mutation in TTF-2 is associated with congenital hypothyroidism, thyroid agenesis and cleft palate. Hum Mol Genet. 2002;11:2051–2059.
193. Baris, I, Arisoy, AE, Smith, A, et al. A novel missense mutation in human TTF-2 (FKHL15) gene associated with congenital hypothyroidism but not athyreosis. J Clin Endocrinol Metab. 2006;91:4183–4187.
194. Grueters, A, Krude, H, Biebermann, H. Molecular genetic defects in congenital hypothyroidism. Eur J Endocrinol. 2004;151:U39–U44.
195. Gagne, N, Parma, J, Deal, C, et al. Apparent congenital athyreosis contrasting with normal plasma thyroglobulin levels and associated with inactivating mutations in the thyrotropin receptor gene: are athyreosis and ectopic thyroid distinct entities? J Clin Endocrinol Metab. 1998;83:1771–1775.
196. Congdon, T, Nguyen, LQ, Nogueira, CR, et al. A novel mutation (Q40P) in PAX8 associated with congenital hypothyroidism and thyroid hypoplasia: evidence for phenotypic variability in mother and child. J Clin Endocrinol Metab. 2001;86:3962–3967.
197. Komatsu, M, Takahashi, T, Takahashi, I, et al. Thyroid dysgenesis caused by PAX8 mutation: the hypermutability with CpG dinucleotides at codon 31. J Pediatr. 2001;139:597–599.
198. Macchia, PE, Lapi, P, Krude, H, et al. PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet. 1998;19:83–86.
199. Meeus, L, Gilbert, B, Rydlewski, C, et al. Characterization of a novel loss of function mutation of PAX8 in a familial case of congenital hypothyroidism with in-place, normal-sized thyroid. J Clin Endocrinol Metab. 2004;89:4285–4291.
200. Vilain, C, Rydlewski, C, Duprez, L, et al. Autosomal dominant transmission of congenital thyroid hypoplasia due to loss-of-function mutation of PAX8. J Clin Endocrinol Metab. 2001;86:234–238.
201. Doyle, DA, Gonzalez, I, Thomas, B, et al. Autosomal dominant transmission of congenital hypothyroidism, neonatal respiratory distress, and ataxia caused by a mutation of NKX2-1. J Pediatr. 2004:145.
202. Iwatani, N, Mabe, H, Devriendt, K, et al. Deletion of NKX2.1 gene encoding thyroid transcription factor 1 in two siblings with hypothyroidism and respiratory failure. J Pediatr. 2000;137:272–276.
203. Krude, H, Schutz, B, Biebermann, H, et al. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2-1 haploinsufficiency. J Clin Invest. 2002;109:475–480.
204. Moeller, LC, Kimura, S, Kusakabe, T, et al. Hypothyroidism in thyroid transcription factor 1 haploinsufficiency is caused by reduced expression of the thyroid-stimulating hormone receptor. Mol Endocrinol. 2003;17:2295–2302.
205. Moya, CM, Perez de Nanclares, G, Castano, L, et al. Functional study of a novel single deletion in the TITF1/NKX2.1 homeobox gene that produces congenital hypothyroidism and benign chorea but not pulmonary distress. Clin Endocrinol Metab. 2006;91:1832–1841.
206. Pohlenz, J, Dumitrescu, A, Zundel, D, et al. Partial deficiency of thyroid transcription factor 1 produces predominantly neurological defects in humans and mice. J Clin Invest. 2002;109:469–473.
207. Stanbury, JB, Rocmans, P, Buhler, UK, et al. Congenital hypothyroidism with impaired thyroid response to thyrotropin. N Engl J Med. 1968;279:1132–1136.
208. Sunthornthepvarakui, T, Gottschalk, ME, Hayashi, Y, et al. Brief report: resistance to thyrotropin caused by mutations in the thyrotropin-receptor gene. N Engl J Med. 1995;332:155–160.
209. Park, SM, Chatterjee, VK. Genetics of congenital hypothyroidism. J Med Genet. 2005;42:379–389.
210. Eales, J. Thyroid functions in cyclostomes and fishes. In: Barrington E, ed. Hormones and Evolution. New York: Academic Press; 1979:341–436.
211. Wendl, T, Lun, K, Mione, M, et al. Pax2.1 is required for the development of thyroid follicles in zebrafish. Development. 2002;129:3751–3760.
212. Ericson, LE, Fredriksson, G. Phylogeny and ontogeny of the thyroid gland. In: Greer M, ed. The Thyroid Gland. New York: Raven; 1990:1–35.
213. Kluge, B, Renault, N, Rohr, KB. Anatomical and molecular reinvestigation of lamprey endostyle development provides new insight into thyroid gland evolution. Dev Genes Evol. 2005;215:32–40.
214. Wright, GM, Youson, JH. Transformation of the endostyle of the anadromous sea lamprey, Petromyzon marinus L., during metamorphosis. I. Light microscopy and autoradiography with 125I1. Gen Comp Endocrinol. 1976;30:243–257.
215. Müller, W. Ueber die Hypobranchialrinne der Tunicaten und deren Vorhandensein bei Amphioxus und den Cyklostomen. Jena Z Med Naturw. 1873;7:327–332.
216. Dohrn, A. Thyroidea bei Petromyzon, Amphioxus und den Tunicaten. Mitt Zool Stat Neapel. 1885;6:49–92.
217. Dunn, A. Properties of an iodinating enzyme in ascidian endostyle. Gen Comp Endocrinol. 1980;40:484–493.
218. Ogasawara, M. Overlapping expression of amphioxus homologs of the thyroid transcription factor 1 gene and thyroid peroxidase gene in the endostyle: insight into evolution of the thyroid gland. Dev Genes Evol. 2000;210:231–242.
219. Ogasawara, M, Di Lauro, R, Satoh, N. Ascidian homologs of mammalian thyroid peroxidase genes are expressed in the thyroid-equivalent region of the endostyle. J Exp Zool. 1999;285:58–69.
220. Ogasawara, M, Satou, Y. Expression of FoxE and FoxQ genes in the endostyle of Ciona intestinalis. Dev Genes Evol. 2003;213:416–419.
221. Ristoratore, F, Spagnuolo, A, Aniello, F, et al. Expression and functional analysis of Cittf1, an ascidian NK-2 class gene, suggest its role in endoderm development. Development. 1999;126:149–159.
222. Ogasawara, M, Shigetani, Y, Suzuki, S, et al. Expression of thyroid transcription factor-1 (TTF-1) gene in the ventral forebrain and endostyle of the agnathan vertebrate, Lampetra japonica. Genesis. 2001;30:51–58.
223. Yu, JK, Holland, LZ, Jamrich, M, et al. AmphiFoxE4, an amphioxus winged helix/forkhead gene encoding a protein closely related to vertebrate thyroid transcription factor-2: expression during pharyngeal development. Evol Dev. 2002;4:9–15.