CHAPTER 66 AMYOTROPHIC LATERAL SCLEROSIS
BACKGROUND
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder causing stereotypic motor impairment, commonly known in the United States as Lou Gehrig’s disease, after the famous baseball player stricken with ALS in the 1930s.1 Jean-Martin Charcot’s 1874 description of clinical and pathological features of ALS remains largely accepted today.2 Clinical and basic research since Charcot’s time have further refined the clinical features in ALS and provide evidence that ALS has multiple causes.3 Also apparent is that a number of neurological disorders share certain features with ALS, complicating the diagnostic evaluation of suspected ALS in some patients. Epidemiological aspects of ALS are summarized in Table 66-1.
| Worldwide prevalence | ≈4 per 100,000 |
| Annual incidence | 1-2 per 100,000 |
| Male-to-female ratio | ≈1.5-2.5:1.0 |
| Peak age at onset | 55-75 years |
Data from Kondo K: Epidemiology of motor neuron disease. In Leigh PN, Swash M, eds: Motor Neuron Disease: Biology and Management. London: Springer-Verlag, 1995, pp 19-33; and from Mitsumoto H, Chad D, Pioro E: Epidemiology. In Mitsumoto H, Chad DA, Pioro EP, eds: Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis, 1998, pp 18-33.
ALS is by definition a progressive motor disorder affecting upper motor neurons (UMNs) and lower motor neurons (LMNs), typically culminating in life-threatening complications of respiratory muscle weakness within 3 to 4 years after onset.4 However, the relative extent of UMN and LMN involvement differs among patients. Individual variation in rate of progression also is seen in ALS, from rapid decline over a period of months in a small proportion of patients to slow progression over 20 to 30 years in rare cases. Current pharmacotherapy at best yields only modest increase in survival.5 Management remains mainly supportive, but the effect of these interventions on quality of life can be significant.6
Nomenclature
Classic ALS is a mixed UMN and LMN disorder, but the term may also be applied to incomplete manifestations with only LMN or UMN signs or solely bulbar features. In “pure” form, however, these partial presentations also are recognized as disorders separate from ALS (Fig. 66-1).7 A proposed solution was to apply the general term motor neuron diseases to this range of presentations, ALS being one manifestation in the spectrum of adult motor neuron diseases (Table 66-2).8 Of the others, motor neuron disease with exclusively LMN features is classified as progressive muscular atrophy (PMA)9; generalized, purely UMN disease is classified as primary lateral sclerosis (PLS); and progressive bulbar palsy (PBP) is a UMN and/or LMN disorder restricted to the bulbar region. Most patients presenting with these syndromes eventually develop the full clinical picture of ALS, but approximately 10% of patients with adult motor neuron disease retain the diagnosis of PMA, PLS, or PBP.10 In life, these diagnoses are established on clinical grounds, because no confirmatory supportive test other than postmortem examination is available.11 The diagnostic challenge is exemplified by PMA, in which autopsy studies demonstrate UMN pathology, establishing the correct diagnosis of ALS. Diagnostic distinction is more than academic, inasmuch as prognosis differs for the various syndromes.12
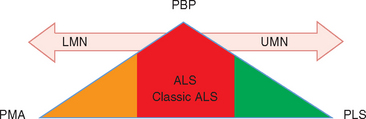
TABLE 66-2 Nomenclature of Idiopathic Adult Motor Neuron Disease
| Condition | Acronym | Features |
|---|---|---|
| Amyotrophic lateral sclerosis | ALS | Upper and lower motor neuron signs in limbs, trunk and bulbar regions |
| Progressive bulbar palsy | PBA | Upper and/or lower motor neuron signs in bulbar region only |
| Progressive muscular atrophy | PMA | Lower motor neuron signs of limb and trunk musculature; bulbar involvement late if at all; no upper motor neuron signs |
| Primary lateral sclerosis | PLS | Upper motor neuron signs in bulbar, limb and trunk regions; no lower motor neuron signs |
From Strong M, Rosenfeld J: Amyotrophic lateral sclerosis: a review of current concepts. Amyotroph Lateral Scler Other Motor Neuron Disord 2003; 4:136-143.
Diagnostic Criteria
The World Federation of Neurology Subcommittee on ALS in 1990 developed diagnostic criteria to standardize the assessment of patients with ALS for research trials. Referred to as the El Escorial criteria and first published in 1994, the guidelines divide the central nervous system into four regions—bulbar, cervical, thoracic, and lumbosacral—and rank the level of diagnostic certainty for ALS on the basis of signs found in each region (Table 66-3).13 Requirements for the diagnosis include the presence of UMN and LMN signs in multiple regions, evidence of progression, and absence of conditions that could otherwise account for the manifestation. Revised El Escorial criteria developed in 1998 and published in 2000 allow supportive evidence for the diagnosis of ALS to be obtained from electromyography (EMG) and created special guidelines for the diagnosis of familial ALS for cases in which confirmation by DNA testing is available (see Table 66-3).14
TABLE 66-3 El Escorial Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis (ALS): Original and Revised Criteria
| Level of Certainty | Original Criteria (1994) | Revised Criteria (2000) |
|---|---|---|
| Definite ALS | LMN and UMN signs in 3 regions | LMN and UMN signs in 3 regions |
| Definite familial ALS | Not used | LMN and UMN signs in ≥1 region plus laboratory identification of DNA mutation associated with ALS |
| Probable ALS | LMN and UMN signs in at least 2 regions; regions may be different, but some UMN signs must in part be rostral to LMN signs | LMN and UMN signs in at least 2 regions; regions may be different, but some UMN signs must in part be rostral to LMN signs |
| Probable ALS, laboratory supported | Not used | LMN and UMN signs in only 1 region or UMN signs alone are found, plus signs of active and chronic denervation on EMG in at least 2 limbs |
The guidelines divide the central nervous system into 4 regions: bulbar, cervical, thoracic, and lumbosacral.
EMG, electromyography; PBP, progressive bulbar palsy; PLS, primary lateral sclerosis; LMN, lower motor neuron; UMN, upper motor neuron.
Data from Brooks BR: El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical Limits of Amyotrophic Lateral Sclerosis” workshop contributors. J Neurol Sci 1994; 124(Suppl):96-107; and from Brooks BR, Miller RG, Swash M, et al: El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1:293-299.
The El Escorial criteria in clinical practice can provide a framework for establishing the diagnosis of ALS, but they remain arbitrary guidelines. Patients’ conditions do not necessarily follow stepwise progression from the lower levels of diagnostic certainty to clinically definite ALS.15 Those without features required for a clinically definite diagnosis at presentation may die of the disease without developing signs that allow classification as clinically probable or definite ALS.
Internet Resources
The knowledge base for ALS is rapidly evolving. Sources of current information on the diagnosis and management of ALS and ongoing research trials include several internet sites, some of which offer downloadable patient information. A selection of these is listed in Table 66-4.
TABLE 66-4 Internet Resources for Information on Amyotrophic Lateral Sclerosis (ALS)
| Resource | Information Provided | World Wide Web Address |
|---|---|---|
| Amyotrophic Lateral Sclerosis Association (United States) | Information on management of ALS; lists of ongoing research trials | www.alsa.org |
| Amyotrophic Lateral Sclerosis/Motor Neurone Disease Association (United Kingdom) | Information on management of ALS; links to ongoing research trials | www.mndassociation.org |
| ALS Therapy Development Foundation (United States) | Extensive information on ongoing and completed medication studies in ALS | www.als-tdf.org |
| GeneTests (United States) | Publicly funded medical genetics information; access to information on genetic testing for ALS | www.geneclinics.org |
| Muscular Dystrophy Association (United States) | Information on management of ALS; lists of ongoing research trials | www.mdausa.org |
| National Institute of Neurological Disorders and Stroke (United States) | Information on ALS, including information in Spanish; information on ongoing research trials | www.ninds.nih.gov |
| World Federation of Neurology Research Group on Motor Neuron Diseases | Includes El Escorial ALS diagnostic criteria | www.wfnals.org |
CLINICAL FEATURES
Weakness with related symptoms and signs in ALS reflects loss of cortical, brainstem, and spinal motor neurons.16 Cortical, or UMN, pathology results in characteristic weakness and in UMN signs such as weakness, hyperreflexia, and pathological reflexes such as Hoffmann and Babinski signs. Anterior horn cell loss produces weakness, atrophy, and fasciculations.10
Initial motor deficits in ALS tend to arise focally, involving a single limb or orofacial muscles, and gradually extend to adjacent body regions.8 The ratio of patients with limb onset to those with bulbar onset is approximately 3:1.12,17,18 Diffuse onset of weakness is less common. With focal onset, deficits tend to progress to the corresponding opposite side of the body and ipsilaterally in a rostral or caudal direction.19 Rostral-caudal extension appears to occur more rapidly than caudal-rostral extension. Extension to a nonadjacent body region, such as from left lower limb to right upper limb, is atypical. Disease onset may be in the thoracic region, leading to stooped posture as a result of paraspinal muscle weakness and weakness of abdominal wall muscles.10,20
Fasciculations are characteristic of ALS but may be difficult to detect in some patients. Fasciculations appear to arise in the axon of diseased motor neurons in ALS but are not specific for the disorder; they are present in other chronic neurogenic disorders, endocrine/metabolic conditions, and in some normal persons.21–23 Those in normal persons, so-called benign fasciculations, tend to have restricted distribution, repetitively occurring in a single muscle rather than diffusely as in ALS. Exercise, stress, and fatigue may promote benign fasciculations in some healthy subjects. Normal neurological examination and electromyographic findings can provide assurance that fasciculations in this setting are benign.24 EMG may help identify fasciculation potentials that are not apparent on physical examination: for example, in obese patients.10
Dysphagia
Nutritional compromise and weight loss can occur in patients with ALS as a result of oropharyngeal weakness and dysphagia. UMN and/or LMN involvement may occur in the distributions of cranial nerve nuclei V, VII, IX, X, and XII. Dysphagia is an early or presenting symptom in 10% to 30% of patients with ALS.10 Nearly all such patients eventually experience dysphagia.25 Recognition of this complication is important in avoiding weight loss, possible malnutrition, and aspiration. Nutritional balance and composition with adequate fluid intake should match estimated dietary needs and swallowing ability.
Aspiration is a significant complication of oropharyngeal weakness, potentially leading to avoidance of certain foods or liquids and interfering with administration of oral medications.10 Symptoms of oropharyngeal weakness include fatigue with chewing, lodging of food in the gingival-buccal mucosa, sensation of food sticking in the throat when swallowing, coughing or choking during meals, sialorrhea, and leaking of liquids from the mouth during swallowing. Patients with severe oropharyngeal weakness may become at risk for aspiration of their own secretions.
Pseudobulbar affect refers to recurrent, involuntary outbursts of laughter or crying triggered by circumstances that normally would not provoke an overtly emotional response. This symptom can be distressing for patients and caregivers. The cause appears to be bilateral interruption of pathways between UMNs and bulbar nuclei, possibly including the cerebellum.26 The incidence of pseudobulbar affect increases gradually during the course of the disorder. In a study of 73 patients with a relatively long mean duration of disease (8.5 years), 49% exhibited pseudobulbar affect.27
Respiratory dysfunction in ALS tends to develop insidiously, although relatively rapid development of respiratory failure is reported in some cases. In rare cases, respiratory failure is the presenting sign of ALS.28–30 Respiratory impairment in ALS stems mainly from LMN weakness of the diaphragm and respiratory accessory muscles (Table 66-5). Exertional dyspnea and fatigue can be early symptoms, although patients may be asymptomatic despite respiratory muscle weakness demonstra ble on pulmonary function testing.31 Symptoms may be mitigated by reduced activity level.32 Dyspnea during conversation, frequent sighing, reduced speech volume, orthopnea, and weak cough can be indicators. Dyspnea with mild exertion or in association with meals is suggestive of significant respiratory muscle weakness. Postprandial dyspnea and orthopnea are attributable to pressure against the diaphragm from the abdominal contents.33

TABLE 66-5 Respiratory Muscles Affected by Amyotrophic Lateral Sclerosis
Rights were not granted to include this table in electronic media. Please refer to the printed book.
From Krvickas L: Pulmonary function and respiratory failure. In Mitsumoto H, Chad DA, Pioro EP, eds: Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis, 1998, pp 382-404.
Physical signs of respiratory compromise include tachypnea, use of accessory muscles, and dyssynchrony of chest/abdominal muscle movement, including paradoxical inward abdominal movement during chest expansion.34 Changes in laboratory tests such as increased hematocrit and respiratory acidosis with reduced serum chloride levels are relatively late indicators of severe respiratory compromise in ALS.10
Significant respiratory muscle weakness can be overlooked if not sought in the history and pulmonary function testing. Early identification of clinically significant respiratory muscle involvement in ALS is important in order to allow the patient adequate time to make decisions regarding potential use of mechanical ventilatory support.10
Sleep disturbance in ALS can be exacerbated by depression, limited mobility interfering with comfortable positioning, and respiratory muscle weakness. Mechanisms of weakness-related sleep disturbance include paresis of respiratory muscles, mechanical obstructive sleep apnea caused by pharyngeal muscle weakness and resulting hypopharyngeal collapse, and/or weakness of vocal cord abduction.10,35,36 Specific inquiry regarding changes in sleep habits, ability to sleep comfortably supine, and recurrent morning headache may be needed to identify these conditions.
DIFFERENTIAL DIAGNOSIS
A large number of disorders may enter into the differential diagnosis of suspected ALS.10 Differential diagnosis can be considered in terms of symptoms and by anatomical localization. An approach to a symptom-based differential diagnosis is shown in Table 66-6. Specific disorders in relation to anatomical localization are discussed as follows.
Several primary anterior horn cell diseases may resemble ALS with mainly LMN signs. All are less common than ALS. PMA, a sporadic LMN disorder, is one of these, occurring in about 10% of patients with motor neuron disease.37 ALS lacking UMN signs is clinically indistinguishable from PMA. Case series suggest that there are a higher male-to-female ratio in PMA and a better prognosis than in ALS, with longer survival and lower frequency of progression to significant bulbar dysfunction and respiratory compromise in PMA than in ALS. Ultimately, the determination that a patient with LMN disease has ALS rather than PMA can be made during the patient’s life only if UMN signs develop or at postmortem study through identification of pyramidal tract pathology.38–40
Adult-onset forms of the hereditary spinal muscular atrophies (SMA) may present diagnostic uncertainty of the type encountered with PMA.41 Several forms of adult-onset SMA are reported with autosomal recessive or autosomal dominant inheritance.42–44 Adult-onset SMA becomes apparent generally after age 20 years. Progression is slow, and the prognosis is better than that of ALS. Initial involvement typically is in the limbs. Generalized weakness may develop, but significant bulbar or respiratory weakness is rare.
Kennedy’s disease, or spinobulbar neuronopathy, is an X-linked inherited disorder in which LMN signs predominate.45,46 Progressive weakness affects bulbar and limb muscles with atrophy and fasciculations, the latter particularly in the orofacial muscles. Progression is slow and potentially compatible with a normal life span, but significant dysarthria, dysphagia, and respiratory impairment can develop. Gynecomastia and impotence may occur. Primary pathology involves LMNs, but sensory neurons also are involved. Sensory and other nonmotor abnormalities may be asymptomatic, however, and Kennedy’s disease is an important consideration in the differential of LMN presentations of ALS in men. Kennedy’s disease is caused by a trinucleotide (cytosine-adenine-guanine) repeat expansion in the X-linked androgen receptor gene.47 Needle electromyographic findings in Kennedy’s disease are similar to those of ALS, but in contrast to ALS, nerve conduction studies tend to show reduced sensory nerve action potential amplitudes.48 DNA testing for the trinucleotide mutation that causes the disorder can establish diagnosis of Kennedy’s disease.
Other uncommon LMN disorders that may resemble ALS include brachial amyotrophic diplegia, a progressive LMN condition of the upper limbs, and monomelic amyotrophy, a progressive LMN disorder affecting a single extremity, usually an upper limb.37,49–53 These conditions tend to progress over a few years and then stabilize, without spread to other body regions. Natural history data suggest that progression of disease outside of the initially involved limb or limbs is unlikely if spread is not evident within a few years after onset.
In rare cases, paraneoplastic motor neuron disease or motor neuronopathy occur with lymphoma.54,55 Weakness in most cases is exclusively LMN type, although some patients demonstrate probable or definite UMN signs that are compatible with ALS.56 Identification of lymphoma in this syndrome may be difficult. Paraproteinemia is a useful marker and, if present, should prompt cerebrospinal fluid examination. Increased cerebrospinal fluid protein and/or oligoclonal bands warrant consideration of bone marrow examination. The neurological disorder may stabilize with treatment of the lymphoma.56,57
Postpolio syndrome is the recurrence of weakness in patients with a history of poliomyelitis a decade or more after the initial infection.58,59 New or ongoing instability and degeneration of previously affected motor units is suspected, but the primary cause is not established.60 Involved muscle groups tend to be those originally affected, but previously unaffected muscles may be included. Progression is slow; periods of stabilization may be interspersed. Bulbar and respiratory function can be affected. Fatigue and musculoskeletal pain are significant in many patients. UMN signs generally are not found, although Babinski’s sign may be present. EMG shows chronic and active neurogenic changes. No specific diagnostic test is available for postpolio syndrome; distinction from LMN-type ALS is based on slow progression, absence of UMN signs, and history of poliomyelitis.
Hexosaminidase A deficiency, a form of GM2 gangliosidosis, is an autosomal recessive condition with a range of neurological phenotypes, depending on the degree of residual enzyme activity.61 Complete absence or profound deficiency of hexosaminidase A results in the fatal infantile disorder Tay-Sachs disease. Less severe deficiency of hexosaminidase A can produce various childhood, juvenile, or adult-onset phenotypes, including one resembling a progressive LMN disease.62,63 Onset in the latter is typically in childhood, but clinical manifestation may be delayed until adulthood. Hexosaminidase A deficiency with prominent LMN features can suggest a sporadic LMN disorder. Clinical features such as childhood onset, slow progression, and associated signs such as tremor or developmental delay readily distinguish hexosaminidase A deficiency from ALS. EMG shows signs of chronic motor denervation, but nerve conduction studies usually show sensory nerve involvement, and brain computed tomography or magnetic resonance imaging (MRI) reveals cerebellar atrophy, at variance with ALS.
Multifocal motor neuropathy (MMN) with conduction block is a rare, immune-mediated disorder with a male-to-female ratio of 3:1 and mean age at onset of 40 years.64,65 The estimated incidence is 1 to 2 per 1,000,000, much lower than that of ALS. MMN can be an important consideration in the differential diagnosis ALS because of clinical overlap and the observation that up to 80% of affected patients respond to treatment. Weakness in MMN may be asymmetrical, beginning distally in the upper limbs. Tendon reflexes are usually reduced but may be retained and seem inappropriately brisk; pathological reflexes are not present. Nerve conduction studies show evidence of demyelination and conduction block in multiple motor nerves, generally not solely at common sites of entrapment. Needle electromyographic abnormalities may be similar to those of ALS but are found only in the territory of involved nerves, not diffusely.66 Approximately 50% of patients have serum immunoglobulin M antibodies that react with GM1 and other gangliosides.67,68 Elevated titers of ganglioside antibodies are found in some patients with ALS, but their presence nevertheless warrants thorough evaluation for possible motor conduction block.
Monoclonal gammopathy may be associated with peripheral neuropathy or polyradiculoneuropathy with mainly motor features, resembling the LMN presentation of ALS.69 Affected patients may have a lymphoproliferative disease potentially amenable to treatment.70 This syndrome can be difficult to distinguish from primary LMN disorders if sensory symptoms/signs are minimal or lacking. Muscle atrophy and fasciculations may be present, but UMN signs are not.71 Associated systemic features may be present and, when fully established, constitute the syndrome of polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes (POEMS). The monoclonal protein in these disorders usually is immunoglobulin G or Aκ, associated with one or more bony lesions (osteosclerotic or sclerotic/lytic). Nerve conduction studies compatible with demyelination and cerebrospinal fluid protein elevation potentially exceeding 100 mg/dL facilitate the distinction from ALS.70 Pathological significance of a monoclonal gammopathy in ALS can be difficult to establish, inasmuch as paraproteinemia is reported in nearly 10% of patients with motor neuron disorders in general, including clinically definite ALS.72
Other peripheral neuropathies that may resemble the LMN presentation of ALS include chronic inflammatory demyelinating polyneuropathy and unusual manifestations of Guillain-Barré syndrome with minimal paresthesia or sensory signs.73,74 Elevated cerebrospinal fluid protein, evidence of demyelination on nerve conduction studies, and nerve biopsy, if indicated, readily distinguish these conditions from ALS.75
Neuromuscular junction disorders that share certain features with ALS include the autoimmune diseases myasthenia gravis and Lambert-Eaton myasthenic syndrome (LEMS). Diffuse weakness may occur in both disorders, but myasthenia gravis has a predilection for ocular and bulbar muscles.76 Fasciculations are not a hallmark of either myasthenia gravis or LEMS. Motor nerve conduction studies of clinically weak muscles in myasthenia gravis tend to show a decrement on slow (2- to 3-Hz) repetitive stimulation. This may occur in ALS, but denervation on EMG distinguishes the latter. Unlike patients with ALS, approximately 85% of patients with myasthenia gravis have serum antibodies to the acetylcholine receptor, and approximately 40% of seronegative patients have antibodies that react with muscle-specific kinase.77,78
LEMS tends to produce limb girdle weakness with milder, if any, bulbar weakness. Increase in strength after repetitive use and autonomic dysfunction also are features of LEMS but not of ALS.79 Patients with LEMS tend to show an electromyographic decrement on slow repetitive stimulation. Motor amplitudes generally are low, and increase by more than 100% on stimulation immediately after brief (10- to 15-second) exercise. LEMS may occur as a primary autoimmune condition, but more often is associated with underlying cancer, particularly small cell lung carcinoma. A high proportion of patients with LEMS carry serum antibody against P/Q-type calcium channels.80
Inclusion body myositis (IBM), polymyositis, and dermatomyositis are inflammatory muscle diseases that in adults produce gradually progressive weakness without fasciculations or UMN signs.81,82 Clinical findings in IBM in particular may superficially resemble ALS with LMN features. IBM affects particularly knee extensors and flexor forearm muscles; neck muscle weakness and dysphagia also may occur. Polymyositis and dermatomyositis cause mainly proximal weakness. Characteristic skin rash is typical of dermatomyositis. Weakness may be asymmetrical, especially in IBM. EMG in these inflammatory myopathies typically reveals fibrillation potentials and myopathic motor unit potentials. In IBM, chronic neurogenic motor unit potential abnormalities are not uncommon. Muscle biopsy demonstrates inflammatory changes in all three, with rimmed vacuoles, amyloid deposition, and filamentous intranuclear inclusions in IBM. If inflammatory myopathy is a diagnostic consideration, muscle biopsy is indicated.
Other muscle disorders that may superficially resemble predominantly LMN presentations of ALS are listed in Table 66-7. Absence of fasciculations, myopathic features on EMG, and findings on muscle biopsy readily distinguish these from possible ALS.83
TABLE 66-7 Muscle Disorders in the Differential Diagnosis of Suspected Amyotrophic Lateral Sclerosis (ALS) with Lower Motor Neuron Features*
| Disease | Features Distinct from ALS†,‡ |
|---|---|
| Inclusion body myositis | — |
| Polymyositis | — |
| Dermatomyositis | Rash |
| Myotonic muscular dystrophy | Myotonia; characteristic facies |
| Oculopharyngeal dystrophy | Oculofacial signs generally established by the time limb weakness is symptomatic |
| Neck extensor myopathy (drop head syndrome) | Generally does not progress to generalized weakness |
| Nemaline myopathy | — |
| Acid maltase deficiency | — |
| McArdle’s disease | — |
| Phosphofructokinase deficiency | — |
| Debrancher deficiency | — |
| Carnitine deficiency | — |
| Mitochondrial myopathy | Ocular signs may distinguish this entity from ALS |
* Electromyography and muscle biopsy are generally useful in distinguishing these disorders from ALS.
† Fasciculations and upper motor neuron signs are not found in these disorders.
‡ As in ALS, mild creatine phosphokinase elevation may be present.
Modified from Mitsumoto H, Chad D, Pioro E: The differential diagnosis of ALS. In Mitsumoto H, Chad DA, Pioro EP, eds: Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis, 1998, pp 87-121.
Neurological disease manifesting solely with UMN signs and no associated evidence of LMN pathology may indicate the initial stages of ALS. However, the differential diagnosis of this syndrome includes other forms of motor neuron disease, such as PLS, and conditions dominated by UMN weakness and spasticity, such as hereditary spastic paraparesis.
PLS is a rare sporadic disorder affecting corticobulbar and corticospinal neurons, found in 2% to 4% of patients in larger motor neuron disease series.84,85 The age at onset is similar to that of ALS.86 Spasticity and weakness may begin in the bulbar region or lower limbs, usually the latter. Physical findings may initially be asymmetrical (i.e., hemiparesis).86 Gradual progression to initially uninvolved regions is typical. The concept of PLS as a specific disease entity is supported by autopsy data demonstrating solely UMN pathology in patients given this diagnosis while alive.85,87 However, natural history data include reports of patients with an initial diagnosis of PLS who after years of follow-up developed LMN signs compatible with ALS.9 Furthermore, mild creatine kinase elevation is reported in some patients who otherwise meet criteria for diagnosis of PLS; needle EMG may reveal signs of active and chronic motor denervation; and muscle biopsy may demonstrate neurogenic changes.84,88 Also, patients given a diagnosis of PLS while alive may at postmortem demonstrate anterior horn cell disease.89 Nosology aside, it is clear that the diagnosis of PLS, even if LMN signs eventually develop, tends to progress more slowly than in patients meeting diagnostic criteria for ALS.84,85 Like that of ALS, diagnosis of PLS rests on history, clinical findings, and periodic reassessment.
Hereditary spastic paraparesis causes a progressive spastic weakness of the lower limbs, rarely extending to the upper limbs and bulbar region.90–92 Inheritance may be autosomal dominant, autosomal recessive, or X-linked; more than 20 genetic loci are identified.93 Onset usually occurs in childhood or early adulthood, earlier than expected for ALS. Bulbar or significant upper limb involvement generally is not a feature of hereditary spastic paraparesis. Urinary symptoms and large fiber sensory involvement may occur, readily distinguishing hereditary spastic paraparesis from ALS.
Mixed UMN and LMN or mainly UMN disorders resembling ALS are uncommon but are important to consider in the workup of patients with possible ALS. Spondylotic cervical spinal stenosis may produce myelopathy and associated LMN signs in the upper limbs.94,95 Thyrotoxicosis can produce a syndrome with mixed UMN and LMN signs attributed to combined myopathy, myelopathy, and motor neuropathy.94 Vitamin B12 deficiency, multiple sclerosis, syringomyelia, adrenal leukodystrophy, human T cell lymphotropic virus I myelopathy, and human immunodeficiency virus myeloneuropathy may superficially suggest ALS with UMN signs.96–102 Identification of these conditions is aided by signs and/or symptoms of neurological involvement beyond solely UMN and anterior horn cell pathology.
Neurological disorders with motor features confined to the bulbar region may resemble a bulbar presentation of ALS. PBP, by definition a UMN and/or LMN disorder limited to bulbar muscles, is included in this differential diagnosis.103 Distinction of PBP from ALS with bulbar onset is established by the presence of LMN and/or UMN signs outside the bulbar region. Physical findings establishing the diagnosis of ALS usually develop in PBP.
Disorders suggestive of bulbar-onset ALS that potentially cause mixed UMN/LMN signs include structural lesions such as tumors in the brainstem or foramen magnum.104 Cerebrovascular disease and demyelinating disease such as multiple sclerosis may produce similar signs and symptoms. Associated symptoms may help rule out ALS, but brain imaging, particularly MRI, as a rule is indicated.83 Cerebrovascular events can generally be ruled out on the basis of their acute onset and nonprogressive course, but some ALS patients report acute symptom onset, and early on it may be unclear whether the disorder is progressive.
Exclusively bulbar weakness may occur in myopathic conditions, neuromuscular transmission disorders, cranial neuropathies, and brainstem structural or demyelinating diseases. Ptosis is characteristic of myopathies such as oculopharyngeal dystrophy and of neuromuscular transmission disorders such as myasthenia gravis.76,105 Ophthalmoparesis, similarly, is not present in ALS except in advanced disease, generally in ventilator-dependent patients approaching a locked-in state.106,107
A variety of conditions have been reported in rare instances to produce an ALS-like disorder. These include lead exposure or other heavy metal exposure, connective tissue disease such as Sjögren’s disease, Lyme disease, and hyperparathyroidism.108 Data supporting these associations are limited, and investigation of these diagnoses seems warranted only in the event of associated clinical or laboratory indications or, in the case of heavy metal toxicity, a history of exposure.
AMYOTROPHIC LATERAL SCLEROSIS WITH ATYPICAL FEATURES
Patients with ALS occasionally have neurological findings in addition to UMN and LMN involvement, also referred to as ALS-plus.14 These findings include dementia, extrapyramidal signs, autonomic features, and sensory signs and/or symptoms. Diagnosis of these syndromes as variants of ALS requires exclusion of potential causes for the variant features. Two of these, dementia and extrapyramidal features, are mentioned briefly here. Clinical management of motor aspects of the syndromes is similar to that of classic ALS.
Dementia, characteristically a frontotemporal type, is reported in about 5% of patients with ALS, although data suggest that the incidence may be higher.18,109,110 Clinically similar familial and sporadic forms occur; one phenotype is linked to chromosome 9.111 Associated cognitive dysfunction includes impaired executive function, emotional lability, decreased speech output, perseveration, and disinhibition. These may develop before or concurrently with motor abnormalities in ALS or manifest after motor deficits are established.
Extrapyramidal signs, including bradykinesia, rigidity, tremor, and postural instability may be present with features of ALS.112,113 One such ALS variant is the ALS-parkinsonismdementia complex of Guam, largely confined to western Pacific islands (Guam, New Guinea, and the Kii Peninsula of Japan).114,115 Patients with this form of ALS often have parkinsonismdementia. Its occurrence outside of this geographic distribution has been reported,116–118 and its incidence on the western Pacific islands may be declining.119 The pathogenesis is not established; dietary exposure to a cycad-derived toxin may contribute. Other forms of ALS with extrapyramidal involvement include rare familial extrapyramidal syndromes.120,121
PATHOGENESIS
The cause of sporadic ALS is unknown, although data are suggestive of an interaction of genetic and acquired mechanisms leading to neuronal death.1,122 Causative genes are identified for some inherited forms of ALS, but the molecular pathogenesis is not understood. ALS appears to be a multisystem disorder with selective vulnerability of UMNs and LMNs.
Pathology of ALS is characterized by loss of LMNs in bulbar motor nuclei and the spinal anterior horn.16 UMN changes include myelin pallor in the corticospinal tract and variable pathological changes in motor cortex, such as loss of pyramidal neurons, including Betz cells, and motor cortex gliosis. LMN nuclei characteristically spared are those innervating external ocular muscles (cranial nerves III, IV, and VI) and those innervating pelvic floor/sphincter muscles (Onuf’s nucleus).
Molecular pathogenesis of ALS appears to involve more than one distinct mechanism.1,3 Basic research data suggest that oxidative damage, neurofilament disorganization, glutamate excitotoxicity, formation of intracellular aggregates, and the failure of protein degradation are important components. It is unknown whether more than one independent mechanism is necessary and sufficient to cause sporadic ALS. Potential pathogenic mechanisms are referred to in relation to therapy in the following discussion.
Genetic factors in ALS include disease-causing mutations in familial ALS and genes that appear to modify disease risk in sporadic ALS. About 10% of cases of ALS are familial, mostly autosomal dominant, although autosomal recessive and X-linked familial ALS also occur.120,123 Genetic linkage is established for a growing number of familial ALS types (Table 66-8). Reports of families with ALS not linked to known loci indicate further heterogeneity.
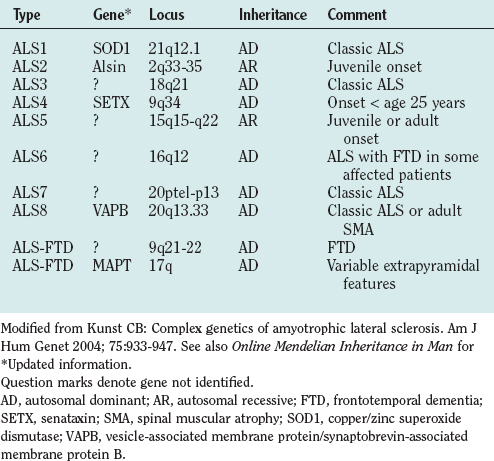
Approximately 20% of patients with familial ALS have ALS1, a mutation in the copper/zinc superoxide dismutase gene (SOD1) linked to chromosome 21q12.1.124 More than 100 pathogenic SOD1 mutations are known.125 Inheritance with all but one of these is autosomal dominant. The phenotype is compatible with classic ALS, but age at onset and severity may vary within families, and penetrance may be less than 100%.120
Other genes account for the 80% of cases of familial ALS not linked to SOD1 (see Table 66-8).126–132 Better understanding of the molecular basis of familial ALS is expected to lead to improved understanding of the pathogenesis of sporadic ALS.
The development of ALS or course of the disease in sporadic and familial ALS may be modified by genes that are not by themselves pathogenic. These include vascular endothelial growth factor120 and the survival motor neuron genes 1 and 2 on chromosome 5.133–136
Environmental factors have been considered as potential risk factors for ALS, but a pathogenic basis is not yet established for any of these.5,115 Putative dietary risks include high dietary fat, low fiber intake, and high dietary glutamate. Current smoking is associated with the threefold risk increase; past cigarette smoking confers a twofold risk. Athletic conditioning and reduced body mass index (BMI) may confer increased risk, although physical activity per se apparently does not.137 Risk is higher for urban dwellers. Isolated examples of geographic clustering of sporadic ALS raise questions of environmental or environmental-genetic factors, but no proved factors are identified. A slightly increased but unexplained risk of ALS is seen in military personnel.138 Necessary interaction of environmental factors in genetically susceptible individuals may underlie the failure to more strongly link specific environmental factors with sporadic ALS.
DIAGNOSTIC STUDIES
Neurophysiological Evaluation
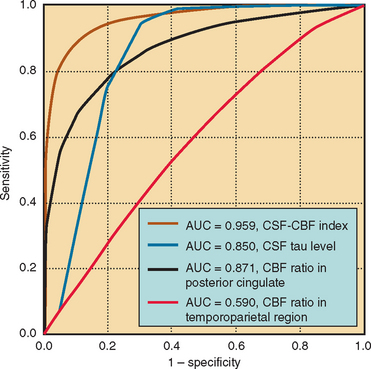
Nerve conduction studies and EMG are central to establishing the diagnosis of ALS and investigating other possible diagnoses.139,140 In the revised El Escorial criteria, nerve conduction studies are required for the diagnosis of ALS “principally to define and exclude other disorders of peripheral nerve, neuromuscular junction and muscle that may mimic or confound the diagnosis of ALS. These studies should generally be normal or near normal.”14 In accordance with this guideline, nerve conduction data in ALS typically show normal sensory amplitudes, normal or reduced motor amplitudes, and normal conduction velocities and distal latencies.140 Abnormalities outside this range do not preclude ALS if accounted for by additional conditions, such as superimposed focal entrapment neuropathy. Evidence of motor conduction block should be sought, especially in patients with predominantly LMN features, because this is a key marker for multifocal motor neuropathy with conduction block (Fig. 66-2).141,142 Sensory responses in the latter disorder are expected to be normal. Marked motor denervation associated with reduced motor amplitude may be accompanied by mild motor conduction velocity slowing as a result of loss of fast conducting axons. Sensory amplitudes and distal latencies may be mildly abnormal in patients with ALS but should be interpreted cautiously, and alternative causes should be investigated. General guidelines for interpretation of nerve conduction abnormalities in ALS have been published.143
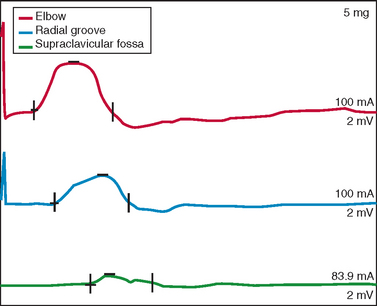
Repetitive motor nerve stimulation at low rates (i.e., 2 to 3 Hz) is indicated if myasthenia gravis or LEMS is a consideration. Decremental response is expected with stimulation of clinically weak muscles in these disorders but is reported in ALS, particularly if motor amplitudes are low.144 In ALS, this defect apparently arises from the nerve terminal–neuromuscular junction. Associated signs of motor denervation on EMG distinguish this finding in ALS from neuromuscular junction disorders. Single fiber EMG may be useful if neuromuscular junction disease is suspected and standard EMG is not suggestive of motor denervation. Increase in amplitude of the motor response by more than 200%, immediately after isometric exercise (or 20- to 50-Hz stimulation), termed postexercise facilitation, is observed with LEMS.79
EMG aids in demonstrating the presence of LMN involvement in ALS and is central to published diagnostic guidelines used in establishing the diagnosis (Table 66-9; see Table 66-3).13,14,140 The revised El Escorial criteria advise electrophysiological studies to confirm signs of LMN dysfunction, identify electrophysiological evidence of LMN dysfunction in clinically uninvolved regions, and exclude other pathophysiological processes. Evidence of LMN dysfunction supports the diagnosis of ALS but is not itself diagnostic of the disorder, inasmuch as similar abnormalities can be found in a wide range of conditions that involve the LMN, including peripheral neuropathic, radicular, and anterior horn cell disorders.139
TABLE 66-9 El Escorial Guidelines for Electrophysiological Testing in the Diagnosis of Amyotrophic Lateral Sclerosis (ALS)
| Nerve Conduction Studies | Expected in ALS | Features Suggestive of Other Diagnoses |
|---|---|---|
| Motor | CV is normal unless CMAP is small | Evidence of motor conduction block |
| CV < 70% of LLN | ||
| DL > 30% above ULN | ||
| F-wave or H-wave latency > 30% above ULN | ||
| Decrement > 20% on repetitive stimulation | ||
| Sensory | Normal, but may be abnormal with peripheral nerve disease/entrapment coexisting with ALS; lower limb sensory responses may be difficult to elicit in the elderly | Abnormal sensory nerve conduction studies (i.e., as a result of peripheral neuropathy, entrapment |
| Needle Electromyography (EMG) | ||
| Signs of active and chronic denervation* on needle EMG examination in at least 2 of the 4 CNS regions | Brainstem | ≥1 Muscle; i.e., tongue, face, jaw |
| Cervical | ≥2 Limb muscles innervated by different roots/peripheral nerves | |
| Thoracic | Paraspinal region below T6 or abdominal muscles | |
| Lumbosacral | ≥2 Limb muscles innervated by different roots/peripheral nerves | |
| Other Electrophysiological Testing | ||
| Modality | Expected in ALS | Features Suggestive of Other Diagnoses |
|---|---|---|
| Somatosensory evoked potentials | Normal | Evoked response latencies > 20% above ULN |
| Autonomic testing | Normal | Significant abnormality |
| Electronystagmography | Normal | Significant abnormality |
CMAP, compound muscle action potential; CNS, central nervous system; CV, conduction velocity; DL, distal latency; EMG, electromyographic; LLN, lower limit of normal; ULN, upper limit of normal.
* Signs of active denervation: fibrillation potentials; positive sharp waves; signs of chronic denervation: large motor unit potentials; reduced recruitment (firing rates of individual potentials > 10Hz), unstable motor unit potentials (see Fig. 66-3C); fasciculation potentials not required, but absence raises doubts regarding diagnosis.
Modified from Mitsumoto H, Chad D, Pioro E: Diagnostic evaluation of ALS. In Mitsumoto H, Chad DA, Pioro EP, eds: Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis, 1998, pp 125-128.
EMG in ALS generally demonstrates fasciculation potentials, fibrillation potentials, and long-duration, high-amplitude, and complex voluntary motor unit potentials with reduced recruitment (Fig. 66-3).139,140 Fibrillation and fasciculation potentials in particular are expected in ALS. Large and/or complex motor unit potentials reflect chronic motor denervation and reinnervation in affected motor units. Recruitment changes signal loss of motor units. In the investigation of possible ALS, examination of muscles that do not appear to be involved may reveal subclinical LMN involvement. Examination of thoracic paraspinal muscles and rectus abdominis is useful in identifying thoracic region motor denervation.20,145
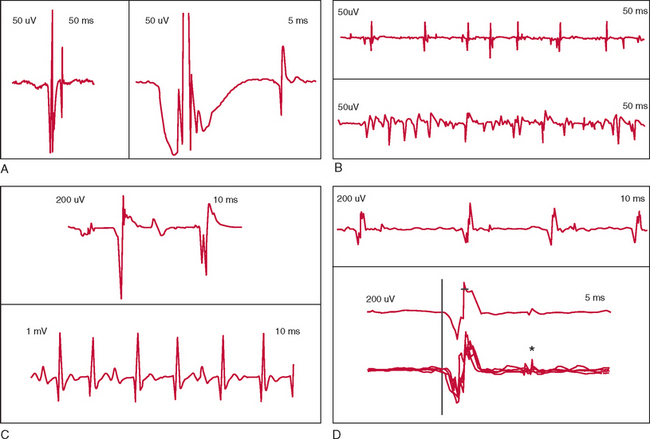
Motor unit estimates refers to various neurophysiological techniques developed for quantification of the number of motor units in skeletal muscles.109 Motor unit estimates have been studied in the evaluation and follow-up of ALS, but their use remains primarily investigative.146 Motor evoked potentials have been studied as a means of identifying UMN pathology in ALS, although their role for this indication is not established.147,148 Somatosensory evoked potentials may be abnormal in ALS, but the sensitivity and utility of these techniques are significantly less than those of EMG.149,150
Several MRI techniques have been investigated for their potential to demonstrate ALS-specific abnormalities.151–153 Brain MRI in ALS may reveal increased signal in the corticospinal tract using certain sequences, such as T2-weighted imaging and fluid-attenuated inversion recovery imaging sequences. Magnetic resonance spectroscopy allows localized, quantitative assessment of brain biochemistry and is reported to show signs of UMN involvement in proportion to clinical UMN signs. Data are suggestive of the utility of magnetic resonance diffusion tensor imaging or T1-weighted spinecho magnetization transfer contrast-enhanced imaging for detecting corticobulbar and corticospinal abnormalities.154 However, the sensitivity and specificity of these imaging techniques are not yet established for routine use in the diagnostic evaluation of ALS.
Clinical Laboratory Testing
Clinical laboratory testing is indicated in the evaluation of ALS in order to identify conditions other than ALS that may manifest similarly. Normal results aid in supporting the diagnosis of ALS. Standard tests also help to identify medically significant comorbid conditions. Specialized tests for neoplastic conditions or infectious diseases may be indicated, depending on history, clinical findings, and results of initial routine testing (Table 66-10).155
TABLE 66-10 Clinical Laboratory Evaluation of Amyotrophic Lateral Sclerosis
| Special Studies | Qualifiers |
|---|---|
| Ganglioside antibodies (GM1; asialo-GM1) | Mainly LMN signs |
| Kennedy’s disease: androgen receptor CAG mutation DNA testing | Mainly LMN signs, sensory symptoms/signs, gynecomastia |
| Leukocyte hexosaminidase A assay | Mental changes, sensory symptoms/signs |
| Anti-HIV antibody | HIV risk factors or workup suggests infectious disease |
| Anti-HTLV I/II antibody | Ascending myeloneuropathy; possible infectious disease |
| Urine metals screen | Heavy metals exposure |
| Adrenal myeloneuropathy: very-long-chain fatty acid levels | Onset < age 40 years, adrenal insufficiency, sensory symptoms/signs |
| Serum parathyroid hormone level | Serum calcium level elevated |
ANA, antinuclear antibody; CAG, cytosine-adenine-guanine; ENA, antibody to extractable nuclear antigen; HIV, human immunodeficiency virus; HTLV, human T cell lymphotropic virus; LMN, lower motor neuron; RF, rheumatoid factor; TSH, thyroid-stimulating hormone.
Muscle and Nerve Biopsy
Neither muscle nor peripheral nerve biopsy is essential for the diagnosis of ALS. Muscle biopsy may be warranted to confirm the presence of LMN involvement or if workup results raise the possibility of primary muscle disease.14,156 Nerve biopsy may be indicated if peripheral nerve disease is suspected.14
MANAGEMENT
Pharmacological Treatment
Of the variety of therapeutic agents tested in clinical trials, none to date has yielded improvement in motor function or significantly slowed disease progression in ALS (Table 66-11),5 although the antiglutamate agent riluzole modestly extends survival.157 Drug development in ALS research has followed advances in basic research, especially since the 1990s. Availability of transgenic mouse and rat models for ALS that overexpress mutant forms of human copper/zinc superoxide dismutase (SOD1) and greater understanding of mechanisms contributing to neuronal death in ALS allow more systematic evaluation of potential therapies. A number of therapeutics ineffective in human studies were originally beneficial in mouse models of ALS. Differences in the pathogenesis of human ALS and animal models of the disease may be responsible for this inconsistency, although interspecies differences in response to the medication, and in some trials, the method of drug delivery or dosage may be relevant. Human studies of potential therapeutic agents extend from current concepts of disease pathogenesis in ALS, including excitotoxicity with excessive glutamate activity in the brain and spinal cord, neuroinflammation with microglial activation, and oxidative stress.
TABLE 66-11 Therapeutic Agents Studied in Amyotrophic Lateral Sclerosis (ALS)
| Agent | Results in ALS Clinical Trials |
|---|---|
| Antioxidant | All negative |
| Betacarotene | |
| Coenzyme Q10 | |
| N-acetylcysteine | |
| Selegiline | |
| Vitamin C | |
| Vitamin E | |
| Calcium Channel Blocker | All negative |
| Nimodipine | |
| Verapamil | |
| Anti-inflammatory | |
| Celecoxib | Negative |
| Cyclophosphamide | Negative |
| Interferon | Negative |
| Intravenous immune globulin | Negative |
| Minocycline | Trial in progress |
| Levamisole | Negative |
| Plasmapheresis | Negative |
| Total lymphoid irradiation | Negative |
| Antiexcitotoxic | |
| Riluzole | Modest extension of survival (≈10%) |
| Branched-chain amino acids | Negative |
| Dextromethorphan | Negative |
| Gabapentin | Negative |
| Lamotrigine | Negative |
| Topiramate | Negative |
| Trophic Factor | |
| BDNF | Negative |
| CNTF | Negative |
| GDNF | Withdrawn during phase 1 testing |
| IGF-1 | Efficacious in 1 of 2 trials; third trial under way |
| Other | |
| Creatine | Negative |
| Ceftriaxone | Trial in planning stages |
BDNF, brain-derived neurotrophic factor; CNTF, ciliary neurotrophic factor; GDNF, glial-derived neurotrophic factor; IGF-1, insulin-like growth factor 1.
Data from Cleveland DW, Rothstein JD: From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Natl Rev Neurosci 2001; 2:806-819; and from Carter GT, Krivickas LS, Weydt P, et al.: Drug therapy for amyotrophic lateral sclerosis: Where are we now? IDrugs 6:147-53, 2003.
Excitotoxicity
Only the glutamate antagonist riluzole has proved to alter the natural course of ALS, but its effect is small.5,158 Three large, randomized clinical trials involving patients treated with riluzole at 100 mg/day demonstrated modestly increased survival but no effect on muscle strength. An American Academy of Neurology practice advisory on the treatment of ALS recommends that riluzole be offered to patients with ALS who do not require mechanical ventilation.
Neuroinflammation and Microglial Activation
Expression of cyclooxygenase 2 is increased in the central nervous system in ALS, which led to consideration of cyclooxygenase 2 inhibitors as a potential treatment for ALS.5 One of these, celecoxib, slowed disease progression in a mouse model of ALS but showed no benefit in human patients with ALS. Minocycline is a tetracycline antibiotic with anti-inflammatory effects also shown to be beneficial in ALS mice, and a clinical trial in human ALS is ongoing. Total lymphoid irradiation, cyclophosphamide, interferon, plasmapheresis, and intravenous immune globulin demonstrated no efficacy in human ALS.1,159,160
Oxidative Stress
A variety of antioxidants have been tested for therapeutic efficacy in ALS. Vitamin C and vitamin E were beneficial in SOD1 mice. Administration of vitamin E with riluzole in human ALS may have slowed disease progression, but survival was not affected.5,160 Antioxidant supplements of possible but as yet unproved benefit in ALS include vitamin C, coenzyme Q10, betacarotene, and N-acetylcysteine. In one review of antioxidant supplements, investigators found no evidence of efficacy in ALS.161 Whether this reflects general ineffectiveness of antioxidant treatment in human ALS, flawed clinical trial design, or lack of efficacy of the antioxidants tested is unresolved. These supplements appear to have a favorable safety profile, although questions regarding safety of vitamin E in doses of 400 IU/day or higher have been raised.162 General guidelines regarding dosage in ALS have emerged (Table 66-12).5,163
TABLE 66-12 Antioxidant Supplements Used to Treat Amyotrophic Lateral Sclerosis
| Supplement | Daily Dose Range |
|---|---|
| Betacarotene | 10-25,000 IU |
| Coenzyme Q10 | 50-300 mg |
| N-acetylcysteine | 100-200 mg |
| Vitamin C | 500-1000 mg |
| Vitamin E | Up to 2000 IU |
From Carter GT, Krivickas LS, Weydt P, et al.: Drug therapy for amyotrophic lateral sclerosis: where are we now? IDrugs 2003; 6:147-153; and from Pioro EP: Antioxidant therapy in ALS. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1(Suppl 4):5-12; discussion, Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1(Suppl 4):13-15.
Trophic Factors
Neurotrophic growth factors are polypeptides shown to promote survival of motor neurons in animal models of ALS.5,160 Several have been studied in human patients with ALS, with mixed results. Insulin-like growth factor 1 yielded a modestly reduced rate of decline in a U.S. trial but not in a European study. A third trial of insulin-like growth factor 1 is under way in the United States to help resolve this discrepancy. Ciliary neurotrophic factor and brain-derived neurotrophic factor were ineffective in ALS clinical trials. Testing of glial-derived neurotrophic factor was suspended during phase 1 safety studies.
Other Pharmacological Agents
Creatine extended survival in SOD1 mice, although the mechanism of action is not established. However, in two human ALS studies, creatine at 5 to 10 g/day showed no benefit.164
The antibiotic ceftriaxone in small, uncontrolled studies appeared to have no efficacy in human ALS, but more recent animal and in vitro data have prompted a reappraisal. A multicenter trial is planned.165–167
Stem Cells
Replacement of damaged or lost neurons by stem cell therapy in ALS is intuitively attractive and holds promise as a potential treatment, but technical challenges remain.168 Anecdotal reports of successful application of this treatment in patients with ALS outside the United States have not been subject to controlled study.169
Symptomatic and Supportive Care
Patients with ALS may experience a wide range of symptoms directly or indirectly linked to progressive weakness. Manage ment of these can significantly affect quality of life. Commonly reported symptoms and their treatment are briefly reviewed as follows. Multidisciplinary ALS clinics, where available, can aid the primary neurologist in long-term care of patients with ALS. There is evidence that survival is improved for patients with ALS with whom this model of care is used.170 Hospice referral can aid patients and family members significantly in day-to-day care of patients approaching a terminal stage.4
Fatigue is common in ALS, potentially exacerbated by muscle weakness and overexertion, by sleep disturbance, and by conditions unrelated to ALS. The last contributor, such as hypothyroidism, cardiac disease, or primary pulmonary disease, should be considered. For some patients, counseling regarding energy conservation techniques may be helpful. Data suggest that modafinil may be helpful.5 The possibility of preexisting depression or depression associated with ALS should be explored; treatment is discussed later. Clinicians should inquire specifically about sleep disturbance, because patients may fail to recognize this as a cause of daytime fatigue. Management of sleep disturbance in ALS is briefly reviewed later.
Fasciculations are found in nearly all patients with ALS but often are not bothersome enough to warrant treatment. Fasciculations themselves are not harmful. For suppression of fasciculations, anecdotal recommendations include gabapentin, phenytoin, carbamazepine, or lorazepam, but none has been of proved benefit in controlled trials.171,172
Muscle cramps involving the limbs and trunk may occur in ALS either in association with activity or spontaneously. Depending on severity, nocturnal lower limb cramps may improve with stretching exercises performed before retiring and with increased fluid intake. Medications that may be effective include quinine sulfate, baclofen, and tizanidine.171
Spasticity resulting in slowness of movement, gait and limb incoordination, and limb spasms can be intrusive symptoms. Injurious falls can be a significant problem for patients with axial and limb spasticity. Gait stability may be improved with ankle-foot orthoses.172 Medications include diazepam, baclofen, and tizanidine.171,172 Intrathecal baclofen administered by an implantable pump is a consideration in patients with an extended clinical course dominated by spasticity.173
Dyspnea
The patient’s decision regarding mechanical ventilation is central to management of dyspnea in ALS.6 Counseling with regard to ventilatory support should be offered relatively early in the course of management, although timing of this discussion may be a sensitive issue for patients and caregivers. Periodic pulmonary function tests, including maximum inspiratory pressure, forced vital capacity, or vital capacity, provide important information regarding pulmonary function, and declining values offer a basis for discussion of respiratory management options. Mechanical ventilatory support includes invasive and noninvasive options (Table 66-13). Palliative approaches can be offered to patients who decline mechanical ventilatory support.
TABLE 66-13 Management of Respiratory Muscle Weakness in Amyotrophic Lateral Sclerosis
| Treatment | Description and Rationale |
|---|---|
| Nonmechanical/terminal care techniques | Body positioning to elevate head and shoulders; reduce pressure of abdominal contents on diaphragm (i.e., elevate head of bed195) |
| Treatment of reversible causes of dyspnea (i.e., pneumonia6) | |
| Oxygen by nasal cannula, 2-4 L/minute; may promote apnea if patient is retaining CO2194 | |
| For terminal dyspnea, morphine sulfate: initially 2.5 mg intravenously/subcutaneously/transdermally or oral equivalent by mouth, percutaneous endoscopic gastrostomy tube, or rectally (i.e., tablets, elixir 20 mg/mL, or rectal suppository) | |
| Chlorpromazine may be needed as antiemetic6,172,195 | |
| Mechanical techniques10 | |
| Negative pressure | Iron lung |
| Chest cuirass | |
| Pneumojacket | |
| Positive pressure | Noninvasive positive-pressure ventilation (i.e., bilevel positive airway pressure) |
| Tracheostomy ventilation |
Studies have suggested that survival and quality of life in ALS are improved with noninvasive mechanical ventilation.10,174,175 Negative- and positive-pressure options are available. Negative-pressure devices are less widely used but may be appropriate for some patients. The “iron lung” is an early example; portable equipment such as the chest cuirass is a more current alternative. Negative-pressure ventilation is not appropriate for patients with significant bulbar weakness, because negative chest pressure breathing can lead to upper airway collapse. Positive-pressure ventilation includes noninvasive and invasive methods, the latter for patients unable to tolerate noninvasive ventilation or in whom it fails. Noninvasive ventilation is administered with a mask or other interface; invasive ventilation is by tracheostomy (Fig. 66-4). Noninvasive positive-pressure ventilation in ALS typically employs an inspiratory pressure set slightly higher than expiratory pressure, referred to as bilevel positive airway pressure (BiPAP). Ventilation by tracheostomy is indicated in patients unable to control upper airway secretions, in those unable to tolerate the BiPAP interface, or when adequate oxygenation cannot be achieved with BiPAP.

Patients and caregivers tend to accept noninvasive ventilation better than ventilation by tracheostomy.6 The likelihood of accepting tracheostomy is greater in patients making an advance decision rather than during an acute episode of respiratory distress. It is appropriate in patients initiating noninvasive ventilation to consider whether they wish to proceed with tracheostomy should this become indicated. Patients opting for mechanical ventilation should have made a prior decision as to any circumstances under which they would want ventilatory support discontinued.4
Adjuncts to mechanical ventilation include management of potentially excess oral secretions, or sialorrhea, discussed later, and cough assistance techniques.176 Patients unable to generate an adequate cough may benefit from using a mechanical cough assistance device, which delivers air through the mouth, followed by rapid release, stimulating a cough.10,176
Sleep disturbance in ALS may be contributed to by a variety of factors. One of these is musculoskeletal pain from impaired mobility and improper positioning. An egg crate mattress pad, pillows to facilitate positioning, or a hospital bed may improve sleep quality. Medications potentially helpful in promoting sleep include amitriptyline, diphenhydramine, and zolpidem.171
Sleep disturbance in ALS may also be associated with disturbed sleep architecture related to periodic leg movements or oxygen desaturation. Periodic leg movements may respond to dopamine agonists.172 Oxygen desaturation from upper airway obstruction can be investigated through the use of overnight oximetry or in a formal sleep study. Suspected respiratory muscle weakness resulting in sleep disturbance is perhaps more reliably evaluated by daytime measures of respiratory function, such as pulmonary function tests and arterial blood gas measurements.174 Treatment is by noninvasive positive pressure ventilation, typically BiPAP.172
Sialorrhea, or drooling, probably caused by mechanical impairment of swallowing, can be troublesome in ALS.6 Anticholinergic medications that reduce saliva production, such as glycopyrrolate, tricyclic antidepressants, or transdermal scopolamine, may help but can produce unwanted side effects such as constipation. Botulinum toxin injection into the parotid or submandibular salivary glands inhibits saliva production but may lead to temporary weakness of adjacent muscles.177–179 Lack of systemic anticholinergic side effects is a potential advantage over standard medications. Irradiation of the salivary glands can permanently reduce saliva production, although some patients may find resulting xerostomia bothersome.180,181 A manual suction catheter offers another approach, used alone or in conjunction with other treatments.6 Portable suction machines are available.
Dysphagia can be managed with dietary modification.6 A modified barium swallow study is indicated for investigation of possible aspiration in this setting, because the history may not enable reliable determination of aspiration risk. This evaluation may be facilitated by consultation with a speech pathologist. Some degree of weight loss in ALS can be expected because of denervation-related loss of muscle mass, but weight loss despite dietary intervention or aspiration warrants consideration of a percutaneous endoscopic gastrostomy tube (PEG) or percutaneous radiological gastrostomy (PRG).182–184 Early patient education regarding the rationale for PEG or PRG tube placement is important in helping to establish patient acceptance of the procedure. Ideally, a PEG or PRG tube should be placed before forced vital capacity falls below 50% of predicted in order to reduce the risk of pulmonary complications potentially associated with the procedure. However, patients with forced vital capacity less than 50% of predicted can safely undergo PEG or PRG tube placement while supported with noninvasive mechanical ventilation (BiPAP).185
Laryngospasm is an abrupt sensation of throat closure and inability to inhale.172 The sensation may be momentary and dissipates spontaneously but can be highly distressing to patients. Hyperactive gag reflex and sensitivity to gastric acid reflux may contribute. Antireflux and antacid agents may help.171 Lorazepam oral concentrate administered sublingually may abort acute attacks.172
“Thick phlegm” refers to the sensation of secretions in the back of the throat that cannot be cleared because of compromised cough strength.172 Guaifenesin, nebulized saline, or 10% acetylcysteine can be helpful. β Blockers have been recommended but may have less efficacy.
Dysarthria
Intelligibility with mild to moderate dysarthria can be managed by maintaining face-to-face contact and reducing background noise.186 A speech pathologist can assist the patient and family in adaptive approaches.187 With more pronounced dysarthria, patients with adequate hand control may resort to written communication. Electronic devices, handheld or operated by standard personal computers, are available with speech synthesizer and text display capabilities. These augmentive and alternative communication (AAC) devices allow effective communication but can be physically demanding for patients, and communication with them tends to be relatively slow.172
Depression, Dementia, and Anxiety
Depression should be inquired about during the course of care by direct questioning. There are no studies to indicate whether tricyclic antidepressants or selective serotonin reuptake inhibitors differ in efficacy in ALS.171 The side effect profiles of the various drugs should be considered in initiating therapy, and the selective serotonin reuptake inhibitors may be better tolerated.
Pseudobulbar affect may respond to fluvoxamine or amitriptyline.188,189 Data are suggestive of greater benefit with a combination of dextromethorphan and quinidine, 30 mg each twice daily.190
Dementia in ALS tends to be of the frontotemporal type, differing from that of Alzheimer’s disease.109 Efficacy of acetylcholinesterase inhibitors such as donezepil is uncertain. Data on rivastigmine in frontotemporal dementia raise the possibility that this agent may be beneficial, but its use in ALS dementia has not been studied.191 Cognitive dysfunction unrelated to dementia also may occur with chronic sleep disturbance caused by respiratory muscle weakness.192
Anxiety in ALS arising from psychological effects of the disease may by ameliorated by a caring, supportive relationship with the physician and by psychological or psychiatric counseling.193 If pharmacological treatment is needed, lorazepam or diazepam can help alleviate these symptoms.171 Anxiety resulting from hypoxia caused by terminal respiratory muscle weakness is discussed in the following section.
Terminal Care
Terminal care includes management of symptoms stemming from respiratory failure and severe limb and trunk muscle weakness.6 These include dyspnea and related anxiety and pain potentially exacerbated by muscle cramps, spasticity, and positional effects of immobility. Detailed reviews of terminal care in ALS have been published.6,194,195
An approach to terminal respiratory management is outlined in Table 66-13. Anxiety symptoms associated with respiratory failure can be controlled with lorazepam (i.e., 1 to 2.5 mg sublingually initially), diazepam or midazolam.6,195
Pain Management
Pain management guidelines provided in the American Academy of Neurology Practice Parameter on the care of ALS include initial use of nonnarcotic analgesic/anti-inflammatory agents and antispasticity drugs and later use of opioids for pain refractory to these medications.6,194 Hospice referral should be considered, because hospice support can significantly improve quality of life during terminal care of ALS.6
PROGNOSIS
Average survival in ALS is approximately 2.5 years after onset of limb symptoms and 1 year after onset of bulbar symptoms.5 Survival is improved if the patient opts for mechanical ventilatory support and/or a gastrostomy feeding tube.183,196,197 Decline in motor function in ALS is generally linear, although the rate of decline varies among patients.198 The practical utility of prognostic factors derived from natural history studies is limited, although available data allow some generalizations.
Better prognosis is reported with age younger than 55 years, onset of limb symptoms, manifestation with purely LMN or purely UMN signs, absence of pulmonary symptoms at onset, minimal fasciculations, and relatively mild motor impairment and/or extended time from symptom onset to diagnosis.198,199
Poor prognosis is associated with shortened time from symptom onset to diagnosis, rapid progression of early ALS symptoms, and rapid decline of pulmonary function.200–202 Familial forms of ALS in general appear to have a poorer prognosis than does sporadic ALS, although extended survival is reported with certain SOD1 mutations.198
Poor prognosis also is correlated with reduced compound muscle action potential amplitude on nerve conduction studies of the most affected limb.203 Decrement on repetitive stimulation is also suggestive of a poor prognosis.204,205
About 10% of patients with ALS survive 10 years or more.206 Survival in excess of three decades has been reported, but prolonged survival is not necessarily equated with milder disability, inasmuch as patients with severe impairment may experience extended survival.198
Patients in whom ALS develops and then appears to improve have been reported but are rare.207 LMN features predominated in these patients, and none had bulbar involvement; whether these patients in fact had ALS or a clinically similar disorder remains unresolved.198
Miller RG, Bradley WG, Gelinas D, et al. Amyotrophic Lateral Sclerosis. Continuum. American Academy of Neurology. Hagerstown, MD: Lippincott Williams & Wilkins, August 2002. 8:(4)
Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293-299. Also available at: www.wfnals.org. accessed March 23, 2006
Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Natl Rev Neurosci. 2001;2:806-819.
Forshew DA, Bromberg MB. A survey of clinicians’ practice in the symptomatic treatment of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:258-263.
Miller RG, Rosenberg JA, Gelinas DF, et al. Practice parameter: the care of the patient with amyotrophic lateral sclerosis (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology: ALS Practice Parameters Task Force. Neurology. 1999;52:1311-1323.
Mitsumoto H, Chad DA, Pioro EP, editors. Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis, 1998.
1 Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Natl Rev Neurosci. 2001;2:806-819.
2 Goldblatt D. Motor neuron disease: historical introduction. Norris FH, Kurland LT, editors. Motor Neuron Diseases. Research on Amyotrophic Lateral Sclerosis and Related Disorders: Contemporary Neurology Symposia. vol 2. New York: Grune & Stratton; 1968:3-11.
3 Strong M, Rosenfeld J. Amyotrophic lateral sclerosis: a review of current concepts. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:136-143.
4 Borasio GD, Miller RG. Clinical characteristics and management of ALS. Semin Neurol. 2001;21:155-166.
5 Carter GT, Krivickas LS, Weydt P, et al. Drug therapy for amyotrophic lateral sclerosis: where are we now? IDrugs. 2003;6:147-153.
6 Miller RG, Rosenberg JA, Gelinas DF, et al. Practice parameter: the care of the patient with amyotrophic lateral sclerosis (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology: ALS Practice Parameters Task Force. Neurology. 1999;52:1311-1323.
7 Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688-1700.
8 Desai J, Swash M. Essentials of diagnosis. In: Kuncl RW, editor. Motor Neuron Disease. Philadelphia: WB Saunders; 2002:1-20.
9 Younger DS, Chou S, Hays AP, et al. Primary lateral sclerosis. A clinical diagnosis reemerges. Arch Neurol. 1988;45:1304-1307.
10 Krvickas L. Pulmonary function and respiratory failure. In: Mitsumoto H, Chad DA, Pioro EP, editors. Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis; 1998:382-404.
11 Ince PG, Lowe J, Shaw PJ. Amyotrophic lateral sclerosis: current issues in classification, pathogenesis and molecular pathology. Neuropathol Appl Neurobiol. 1998;24:104-117.
12 Norris F, Shepherd R, Denys E, et al. Onset, natural history and outcome in idiopathic adult motor neuron disease. J Neurol Sci. 1993;118:48-55.
13 Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical Limits of Amyotrophic Lateral Sclerosis” workshop contributors. J Neurol Sci. 1994;124(Suppl):96-107.
14 Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293-299.
15 Traynor BJ, Codd MB, Corr B, et al. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria: a population-based study. Arch Neurol. 2000;57:1171-1176.
16 Ince PG. Neuropathology. In: Brown RHJr, Meininger V, Swash M, editors. Amyotrophic Lateral Sclerosis. London: Martin Dunitz; 2000:83-112.
17 Gubbay SS, Kahana E, Zilber N, et al. Amyotrophic lateral sclerosis. A study of its presentation and prognosis. J Neurol. 1985;232:295-300.
18 Jokelainen M. Amyotrophic lateral sclerosis in Finland. II: clinical characteristics. Acta Neurol Scand. 1977;56:194-204.
19 Brooks BR. Design of clinical therapeutic trials in amyotrophic lateral sclerosis. Rowland LP, editor. Amyotrophic Lateral Sclerosis and Other Motor Neuron Diseases: Advances in Neurology. vol 56. New York: Raven Press; 1991:521-546.
20 Kuncl RW, Cornblath DR, Griffin JW. Assessment of thoracic paraspinal muscles in the diagnosis of ALS. Muscle Nerve. 1988;11:484-492.
21 Conradi S, Ronnevi LO, Norris FH. Motor neuron disease and toxic metals. Adv Neurol. 1982;36:201-231.
22 Roth G. Fasciculations and their F-response. Localisation of their axonal origin. J Neurol Sci. 1984;63:299-306.
23 Wettstein A. The origin of fasciculations in motoneuron disease. Ann Neurol. 1979;5:295-300.
24 Blexrud MD, Windebank AJ, Daube JR. Long-term follow-up of 121 patients with benign fasciculations. Ann Neurol. 1993;34:622-625.
25 Robbins J. Swallowing in ALS and motor neuron disorders. Neurol Clin. 1987;5:213-229.
26 Parvizi J, Anderson SW, Martin CO, et al. Pathological laughter and crying: a link to the cerebellum. Brain. 2001;124:1708-1719.
27 Gallagher JP. Pathologic laughter and crying in ALS: a search for their origin. Acta Neurol Scand. 1989;80:114-117.
28 Fromm GB, Wisdom PJ, Block AJ. Amyotrophic lateral sclerosis presenting with respiratory failure. Diaphragmatic paralysis and dependence on mechanical ventilation in two patients. Chest. 1977;71:612-614.
29 Hill R, Martin J, Hakim A. Acute respiratory failure in motor neuron disease. Arch Neurol. 1983;40:30-32.
30 Nightingale S, Bates D, Bateman DE, et al. Enigmatic dyspnoea: an unusual presentation of motor-neurone disease. Lancet. 1982;1:933-935.
31 Fallat RJ, Jewitt B, Bass M, et al. Spirometry in amyotrophic lateral sclerosis. Arch Neurol. 1979;36:74-80.
32 Kreitzer SM, Saunders NA, Tyler R, et al. Respiratory muscle function in amyotrophic lateral sclerosis. Chest. 1978;73:266-267.
33 Gibson GJ. Diaphragmatic paresis: pathophysiology, clinical features, and investigation. Thorax. 1989;44:960-970.
34 Grinman S, Whitelaw WA. Pattern of breathing in a case of generalized respiratory muscle weakness. Chest. 1983;84:770-772.
35 Goldstein RS. Hypoventilation: neuromuscular and chest wall disorders. Clin Chest Med. 1992;13:507-521.
36 Caselli RJ, Windebank AJ, Petersen RC, et al. Rapidly progressive aphasic dementia and motor neuron disease. Ann Neurol. 1993;33:200-207.
37 van den Berg-Vos RM, Visser J, Franssen H, et al. Sporadic lower motor neuron disease with adult onset: classification of subtypes. Brain. 2003;126:1036-1047.
38 Mortara P, Chio A, Rosso MG, et al. Motor neuron disease in the province of Turin, Italy, 1966–1980. Survival analysis in an unselected population. J Neurol Sci. 1984;66:165-173.
39 Ince PG, Evans J, Knopp M, et al. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology. 2003;60:1252-1258.
40 Iwanaga K, Hayashi S, Oyake M, et al. Neuropathology of sporadic amyotrophic lateral sclerosis of long duration. J Neurol Sci. 1997;146:139-143.
41 Pearn J. Autosomal dominant spinal muscular atrophy: a clinical and genetic study. J Neurol Sci. 1978;38:263-275.
42 Rudnik-Schoneborn S, Rohrig D, Morgan G, et al. Autosomal recessive proximal spinal muscular atrophy in 101 sibs out of 48 families: clinical picture, influence of gender, and genetic implications. Am J Med Genet. 1994;51:70-76.
43 Somerville MJ, Hunter AG, Aubry HL, et al. Clinical application of the molecular diagnosis of spinal muscular atrophy: deletions of neuronal apoptosis inhibitor protein and survival motor neuron genes. Am J Med Genet. 1997;69:159-165.
44 Harding AE, Thomas PK. Genetic aspects of hereditary motor and sensory neuropathy (types I and II). J Med Genet. 1980;17:329-336.
45 Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968;18:671-680.
46 Harding AE, Thomas PK, Baraitser M, et al. X-linked recessive bulbospinal neuronopathy: a report of ten cases. J Neurol Neurosurg Psychiatry. 1982;45:1012-1019.
47 La Spada AR, Wilson EM, Lubahn DB, et al. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77-79.
48 Olney RK, Aminoff MJ, So YT. Clinical and electrodiagnostic features of X-linked recessive bulbospinal neuronopathy. Neurology. 1991;41:823-828.
49 Katz JS, Wolfe GI, Andersson PB, et al. Brachial amyotrophic diplegia: a slowly progressive motor neuron disorder. Neurology. 1999;53:1071-1076.
50 Hu MT, Ellis CM, Al-Chalabi A, et al. Flail arm syndrome: a distinctive variant of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1998;65:950-951.
51 Hirayama K, Tomonaga M, Kitano K, et al. Focal cervical poliopathy causing juvenile muscular atrophy of distal upper extremity: a pathological study. J Neurol Neurosurg Psychiatry. 1987;50:285-290.
52 Hirayama K, Tokumaru Y. Cervical dural sac and spinal cord in juvenile muscular atrophy of distal upper extremity. Neurology. 2000;54:1922-1926.
53 de Visser M, Ongerboer de Visser BW, et al. Amyotrophy of the hands and pyramidal features of predominantly the legs segregating within one large family. J Neurol Sci. 1988;88:241-246.
54 Rowland LP, Schneck SA. Neuromuscular disorders associated with malignant neoplastic disease. J Chronic Dis. 1963;16:777-795.
55 Gordon PH, Rowland LP, Younger DS, et al. Lymphoproliferative disorders and motor neuron disease: an update. Neurology. 1997;48:1671-1678.
56 Younger DS, Rowland LP, Latov N, et al. Lymphoma, motor neuron diseases, and amyotrophic lateral sclerosis. Ann Neurol. 1991;29:78-86.
57 Rowland LP, Sherman WH, Latov N, et al. Amyotrophic lateral sclerosis and lymphoma: bone marrow examination and other diagnostic tests. Neurology. 1992;42:1101-1102.
58 Jubelt B, Cashman NR. Neurological manifestations of the postpolio syndrome. Crit Rev Neurobiol. 1987;3:199-220.
59 Trojan DA, Cashman NR. Post-poliomyelitis syndrome. Muscle Nerve. 2005;31:6-19.
60 Dalakas MC, Elder G, Hallett M, et al. A long-term follow-up study of patients with post-poliomyelitis neuromuscular symptoms. N Engl J Med. 1986;314:959-963.
61 Johnson WG. The clinical spectrum of hexosaminidase deficiency diseases. Neurology. 1981;31:1453-1456.
62 Mitsumoto H, Sliman RJ, Schafer IA, et al. Motor neuron disease and adult hexosaminidase A deficiency in two families: evidence for multisystem degeneration. Ann Neurol. 1985;17:378-385.
63 Cashman NR, Antel JP, Hancock LW, et al. N-acetyl-β-hexosaminidase β locus defect and juvenile motor neuron disease: a case study. Ann Neurol. 1986;19:568-572.
64 Nagale SV, Bosch EP. Multifocal motor neuropathy with conduction block: current issues in diagnosis and treatment. Semin Neurol. 2003;23:325-334.
65 Nobile-Orazio E, Meucci N, Barbieri S, et al. High-dose intravenous immunoglobulin therapy in multifocal motor neuropathy. Neurology. 1993;43:537-544.
66 Bouche P, Moulonguet A, Younes-Chennoufi AB, et al. Multifocal motor neuropathy with conduction block: a study of 24 patients. J Neurol Neurosurg Psychiatry. 1995;59:38-44.
67 Lange DJ, Trojaborg W, Latov N, et al. Multifocal motor neuropathy with conduction block: is it a distinct clinical entity? Neurology. 1992;42:497-505.
68 Pestronk A, Chaudhry V, Feldman EL, et al. Lower motor neuron syndromes defined by patterns of weakness, nerve conduction abnormalities, and high titers of antiglycolipid antibodies. Ann Neurol. 1990;27:316-326.
69 Kelly JJJr. Peripheral neuropathies associated with monoclonal proteins: a clinical review. Muscle Nerve. 1985;8:138-150.
70 Ropper AH, Gorson KC. Neuropathies associated with paraproteinemia. N Engl J Med. 1998;338:1601-1607.
71 Rowland LP, Defendini R, Sherman W, et al. Macroglobulinemia with peripheral neuropathy simulating motor neuron disease. Ann Neurol. 1982;11:532-536.
72 Younger DS, Rowland LP, Latov N, et al. Motor neuron disease and amyotrophic lateral sclerosis: relation of high CSF protein content to paraproteinemia and clinical syndromes. Neurology. 1990;40:595-599.
73 Lewis RA, Sumner AJ, Brown MJ, et al. Multifocal demyelinating neuropathy with persistent conduction block. Neurology. 1982;32:958-964.
74 McKhann GM, Cornblath DR, Griffin JW, et al. Acute motor axonal neuropathy: a frequent cause of acute flaccid paralysis in China. Ann Neurol. 1993;33:333-342.
75 Barohn RJ, Kissel JT, Warmolts JR, et al. Chronic inflammatory demyelinating polyradiculoneuropathy. Clinical characteristics, course, and recommendations for diagnostic criteria. Arch Neurol. 1989;46:878-884.
76 Drachman DB. Myasthenia gravis. N Engl J Med. 1994;330:1797-1810.
77 Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve. 2004;29:484-505.
78 McConville J, Farrugia ME, Beeson D, et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. 2004;55:580-584.
79 Mareska M, Gutmann L. Lambert-Eaton myasthenic syndrome. Semin Neurol. 2004;24:149-153.
80 Lennon VA, Kryzer TJ, Griesmann GE, et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. 1995;332:1467-1474.
81 Lotz BP, Engel AG, Nishino H, et al. Inclusion body myositis. Observations in 40 patients. Brain. 1989;112(Pt 3):727-747.
82 Dalakas MC. Polymyositis, dermatomyositis and inclusion-body myositis. N Engl J Med. 1991;325:1487-1498.
83 Mitsumoto H, Chad D, Pioro E. The differential diagnosis of ALS. In: Mitsumoto H, Chad DA, Pioro EP, editors. Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis; 1998:87-121.
84 Le Forestier N, Maisonobe T, Spelle L, et al. Primary lateral sclerosis: further clarification. J Neurol Sci. 2001;185:95-100.
85 Pringle CE, Hudson AJ, Munoz DG, et al. Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain. 1992;115(Pt 2):495-520.
86 Gastaut JL, Bartolomei F. Mills’ syndrome: ascending (or descending) progressive hemiplegia: a hemiplegic form of primary lateral sclerosis? J Neurol Neurosurg Psychiatry. 1994;57:1280-1281.
87 Hudson AJ, Kiernan JA, Munoz DG, et al. Clinicopathological features of primary lateral sclerosis are different from amyotrophic lateral sclerosis. Brain Res Bull. 1993;30:359-364.
88 Kojan S, Goodwin W, Bryan WW, et al. Clinical and laboratory features of primary lateral sclerosis. J Child Neurol. 2000;15:200.
89 Brownell B, Oppenheimer DR, Hughes JT. The central nervous system in motor neurone disease. J Neurol Neurosurg Psychiatry. 1970;33:338-357.
90 Fortini D, Cricchi F, Di Fabio R, et al. Current insights into familial spastic paraparesis: new advances in an old disease. Funct Neurol. 2003;18:43-49.
91 Fink JK. Progressive spastic paraparesis: hereditary spastic paraplegia and its relation to primary and amyotrophic lateral sclerosis. Semin Neurol. 2001;21:199-207.
92 Fink JK. The hereditary spastic paraplegias: nine genes and counting. Arch Neurol. 2003;60:1045-1049.
93 Bouslam N, Benomar A, Azzedine H, et al. Mapping of a new form of pure autosomal recessive spastic paraplegia (SPG28). Ann Neurol. 2005;57:567-571.
94 Rowland LP. Diagnosis of amyotrophic lateral sclerosis. J Neurol Sci. 1998;160(Suppl 1):S6-S24.
95 Yamada M, Furukawa Y, Hirohata M. Amyotrophic lateral sclerosis: frequent complications by cervical spondylosis. J Orthop Sci. 2003;8:878-881.
96 Noseworthy JH, Heffernan LP. Motor radiculopathy—an unusual presentation of multiple sclerosis. Can J Neurol Sci. 1980;7:207-209.
97 Fisher M, Long RR, Drachman DA. Hand muscle atrophy in multiple sclerosis. Arch Neurol. 1983;40:811-815.
98 Matsuzaki T, Nakagawa M, Nagai M, et al. HTLV-I-associated myelopathy (HAM)/tropical spastic paraparesis (TSP) with amyotrophic lateral sclerosis–like manifestations. J Neurovirol. 2000;6:544-548.
99 MacGowan DJ, Scelsa SN, Waldron M. An ALS-like syndrome with new HIV infection and complete response to antiretroviral therapy. Neurology. 2001;57:1094-1097.
100 von Giesen HJ, Kaiser R, Koller H, et al. Reversible ALS-like disorder in HIV infection. An ALS-like syndrome with new HIV infection and complete response to antiretroviral therapy.Neurology. 2002;59:474. author reply, Neurology. 2002;59:474-475. [Erratum in Neurology 2002; 59:2009].
101 Mariani C, Cislaghi MG, Barbieri S, et al. The natural history and results of surgery in 50 cases of syringomyelia. J Neurol. 1991;238:433-438.
102 Griffin JW, Goren E, Schaumburg H, et al. Adrenomyeloneuropathy: a probable variant of adrenoleukodystrophy. I. Clinical and endocrinologic aspects. Neurology. 1977;27:1107-1113.
103 Mitsumoto H, Chad D, Pioro E. History, terminology, and classification of ALS. In: Mitsumoto H, Chad DA, Pioro EP, editors. Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis; 1998:3-17.
104 Sawaya RA. Foramen magnum meningioma presenting as amyotrophic lateral sclerosis. Neurosurg Rev. 1998;21:277-280.
105 Fan X, Rouleau GA. Progress in understanding the pathogenesis of oculopharyngeal muscular dystrophy. Can J Neurol Sci. 2003;30:8-14.
106 Noseworthy JH, Rae-Grant AD, Brown WF. An unusual subacute progressive motor neuronopathy with myasthenia-like features. Can J Neurol Sci. 1988;15:304-309.
107 Hayashi H, Kato S, Kawada A. Amyotrophic lateral sclerosis patients living beyond respiratory failure. J Neurol Sci. 1991;105:73-78.
108 Kuncl RW. Errors in diagnosis. In: Kuncl RW, editor. Motor Neuron Disease. London: WB Saunders; 2002:21-38.
109 Lomen-Hoerth C. Characterization of amyotrophic lateral sclerosis and frontotemporal dementia. Dement Geriatr Cogn Disord. 2004;17:337-341.
110 Kondo K, Hemmi I. Clinical statistics in 515 fatal cases of motor neuron disease. Neuroepidemiology. 1984;3:129-148.
111 Hosler BA, Siddique T, Sapp PC, et al. Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22. JAMA. 2000;284:1664-1669.
112 Qureshi AI, Wilmot G, Dihenia B, et al. Motor neuron disease with parkinsonism. Arch Neurol. 1996;53:987-991.
113 Liang TW, Forman MS, Duda JE, et al. Multiple pathologies in a patient with a progressive neurodegenerative syndrome. J Neurol Neurosurg Psychiatry. 2005;76:252-255.
114 Rodgers-Johnson P, Garruto RM, Yanagihara R, et al. Amyotrophic lateral sclerosis and parkinsonismdementia on Guam: a 30-year evaluation of clinical and neuropathologic trends. Neurology. 1986;36:7-13.
115 Armon C. Epidemiology of amyotrophic lateral sclerosis/motor neuron disease. In: Shaw PJ, Strong MJ, editors. Motor Neuron Disorders: Blue Books of Practical Neurology. Philadelphia: Butterworth-Heinemann; 2003:167-205.
116 Gilbert JJ, Kish SJ, Chang LJ, et al. Dementia, parkinsonism, and motor neuron disease: neurochemical and neuropathological correlates. Ann Neurol. 1988;24:688-691.
117 Hudson AJ. Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurological disorders: a review. Brain. 1981;104:217-247.
118 Schmitt HP, Emser W, Heimes C. Familial occurrence of amyotrophic lateral sclerosis, parkinsonism, and dementia. Ann Neurol. 1984;16:642-648.
119 Waring SC, Esteban-Santillan C, Reed DM, et al. Incidence of amyotrophic lateral sclerosis and of the parkinsonismdementia complex of Guam, 1950–1989. Neuroepidemiology. 2004;23:192-200.
120 Kunst CB. Complex genetics of amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:933-947.
121 Furtado S, Payami H, Lockhart PJ, et al. Profile of families with parkinsonism-predominant spinocerebellar ataxia type 2 (SCA2). Mov Disord. 2004;19:622-629.
122 Strong MJ, Lomen-Hoerth C, Caselli RJ, et al. Cognitive impairment, frontotemporal dementia, and the motor neuron diseases. Ann Neurol. 2003;54(Suppl 5):S20-S23.
123 Siddique T, Lalani I. Genetic aspects of amyotrophic lateral sclerosis. Adv Neurol. 2002;88:21-32.
124 Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59-62.
125 Andersen PM, Sims KB, Xin WW, et al. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:62-73.
126 Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet. 2004;74:1128-1135.
127 De Jonghe P, Auer-Grumbach M, Irobi J, et al. Autosomal dominant juvenile amyotrophic lateral sclerosis and distal hereditary motor neuronopathy with pyramidal tract signs: synonyms for the same disorder? Brain. 2002;125:1320-1325.
128 Hand CK, Khoris J, Salachas F, et al. A novel locus for familial amyotrophic lateral sclerosis, on chromosome 18q. Am J Hum Genet. 2002;70:251-256.
129 Chance PF, Rabin BA, Ryan SG, et al. Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am J Hum Genet. 1998;62:633-640.
130 Hadano S, Hand CK, Osuga H, et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet. 2001;29:166-173.
131 Yang Y, Hentati A, Deng HX, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29:160-165.
132 Hentati A, Ouahchi K, Pericak-Vance MA, et al. Linkage of a commoner form of recessive amyotrophic lateral sclerosis to chromosome 15q15-q22 markers. Neurogenetics. 1998;2:55-60.
133 Cleveland JL. A new piece of the ALS puzzle. Nat Genet. 2003;34:357-358.
134 Lambrechts D, Storkebaum E, Morimoto M, et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet. 2003;34:383-394.
135 Veldink JH, van den Berg LH, Cobben JM, et al. Homozygous deletion of the survival motor neuron 2 gene is a prognostic factor in sporadic ALS. Neurology. 2001;56:749-752.
136 Corcia P, Mayeux-Portas V, Khoris J, et al. Abnormal SMN1 gene copy number is a susceptibility factor for amyotrophic lateral sclerosis. Ann Neurol. 2002;51:243-246.
137 Veldink JH, Kalmijn S, Groeneveld GJ, et al. Physical activity and the association with sporadic ALS. Neurology. 2005;64:241-245.
138 Weisskopf MG, O’Reilly EJ, McCullough ML, et al. Prospective study of military service and mortality from ALS. Neurology. 2005;64:32-37.
139 Daube JR. Electrodiagnostic studies in amyotrophic lateral sclerosis and other motor neuron disorders. Muscle Nerve. 2000;23:1488-1502.
140 Lambert EH, Mulder DW. Electromyography in amyotrophic lateral sclerosis. Norris FH, Kurland LT, editors. Motor Neuron Diseases; Research on Amyotrophic Lateral Sclerosis and Related Disorders: Contemporary Neurology Symposia. vol 2. New York: Grune & Stratton; 1968:135-153.
141 Chaudhry V, Corse AM, Cornblath DR, et al. Multifocal motor neuropathy: electrodiagnostic features. Muscle Nerve. 1994;17:198-205.
142 Olney RK, Lewis RA, Putnam TD, et al. Consensus criteria for the diagnosis of multifocal motor neuropathy. Muscle Nerve. 2003;27:117-121.
143 Cornblath DR, Kuncl RW, Mellits ED, et al. Nerve conduction studies in amyotrophic lateral sclerosis. Muscle Nerve. 1992;15:1111-1115.
144 Henderson RD, Daube JR. Decrement in surface-recorded motor unit potentials in amyotrophic lateral sclerosis. Neurology. 2004;63:1670-1674.
145 Cornblath DR, Kuncl RW, Rechthand E, et al. The value of rectus abdominal muscle electromyography [Letter]. Muscle Nerve. 1987;10:376.
146 Gooch CL, Shefner JM. ALS surrogate markers. MUNE. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(Suppl 1):104-107.
147 Osei-Lah AD, Mills KR. Optimising the detection of upper motor neuron function dysfunction in amyotrophic lateral sclerosis—a transcranial magnetic stimulation study. J Neurol. 2004;251:1364-1369.
148 Kaufmann P, Pullman SL, Shungu DC, et al. Objective tests for upper motor neuron involvement in amyotrophic lateral sclerosis (ALS). Neurology. 2004;62:1753-1757.
149 Theys PA, Peeters E, Robberecht W. Evolution of motor and sensory deficits in amyotrophic lateral sclerosis estimated by neurophysiological techniques. J Neurol. 1999;246:438-442.
150 Mitsumoto H, Chad D, Pioro E. Electrodiagnosis. In: Mitsumoto H, Chad DA, Pioro EP, editors. Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis; 1998:78-79.
151 Kaufmann P, Levy G, Thompson JL, et al. The ALSFRSr predicts survival time in an ALS clinic population. Neurology. 2005;64:38-43.
152 Kaufmann P, Mitsumoto H. Amyotrophic lateral sclerosis: objective upper motor neuron markers. Curr Neurol Neurosci Rep. 2002;2:55-60.
153 Kalra S, Arnold D. Neuroimaging in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:243-248.
154 da Rocha AJ, Oliveira AS, Fonseca RB, et al. Detection of corticospinal tract compromise in amyotrophic lateral sclerosis with brain MR imaging: relevance of the T1-weighted spinecho magnetization transfer contrast sequence. AJNR Am J Neuroradiol. 2004;25:1509-1515.
155 Mitsumoto H, Chad D, Pioro E. Diagnostic evaluation of ALS. In: Mitsumoto H, Chad DA, Pioro EP, editors. Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis; 1998:125-128.
156 Cudkowicz M, Qureshi M, Shefner J. Measures and markers in amyotrophic lateral sclerosis. NeuroRx. 2004;1:273-283.
157 Shefner JM. Multi-drug therapy in amyotrophic lateral sclerosis: combinations of multiple, untested drugs should not be used at this time. Muscle Nerve. 2004;30:676-678.
158 Miller RG, Mitchell JD, Lyon M, et al. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:191-206.
159 Meucci N, Nobile-Orazio E, Scarlato G. Intravenous immunoglobulin therapy in amyotrophic lateral sclerosis. J Neurol. 1996;243:117-120.
160 Miller R. Pharmacology and clinical trials. Continuum: Amyotrophic Lateral Sclerosis. 2002;8:56-81.
161 Orrell RW, Lane JM, Ross MA. Antioxidant treatment for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. (4):2004. CD002829.
162 Miller ER3rd, Pastor-Barriuso R, Dalal D, et al. Metaanalysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142:37-46.
163 Pioro EP. Antioxidant therapy in ALS.Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(Suppl 4):5-12. discussion, Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(Suppl 4):13-15.
164 Shefner JM, Cudkowicz ME, Schoenfeld D, et al. A clinical trial of creatine in ALS. Neurology. 2004;63:1656-1661.
165 Carelli V, Liguori R, Cordivari C, et al. Ceftriaxone is ineffective in ALS. Ital J Neurol Sci. 1994;15:66.
166 Couratier P, Vallat JM, Merle L, et al. Report of six sporadic cases of ALS patients receiving ceftriaxone. Therapie. 1994;49:146.
167 Rothstein JD, Patel S, Regan MR, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73-77.
168 Silani V, Cova L, Corbo M, et al. Stem-cell therapy for amyotrophic lateral sclerosis. Lancet. 2004;364:200-202.
169 Watts J. Controversy in China. Lancet. 2005;365:109-110.
170 Traynor BJ, Alexander M, Corr B, et al. Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996–2000. J Neurol Neurosurg Psychiatry. 2003;74:1258-1261.
171 Forshew DA, Bromberg MB. A survey of clinicians’ practice in the symptomatic treatment of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:258-263.
172 Sufit R, Miller R. Symptomatic management. Continuum: Amyotrophic Lateral Sclerosis. 2002;8:82-94.
173 Marquardt G, Lorenz R. Intrathecal baclofen for intractable spasticity in amyotrophic lateral sclerosis. J Neurol. 1999;246:619-620.
174 Bourke SC, Bullock RE, Williams TL, et al. Noninvasive ventilation in ALS: indications and effect on quality of life. Neurology. 2003;61:171-177.
175 Gelanis DF. Respiratory failure or impairment in amyotrophic lateral sclerosis. Curr Treat Options Neurol. 2001;3:133-138.
176 Perrin C, Unterborn JN, Ambrosio CD, et al. Pulmonary complications of chronic neuromuscular diseases and their management. Muscle Nerve. 2004;29:5-27.
177 Giess R, Naumann M, Werner E, et al. Injections of botulinum toxin A into the salivary glands improve sialorrhoea in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2000;69:121-123.
178 Winterholler MG, Erbguth FJ, Wolf S, et al. Botulinum toxin for the treatment of sialorrhoea in ALS: serious side effects of a transductal approach. J Neurol Neurosurg Psychiatry. 2001;70:417-418.
179 Tan EK, Lo YL, Seah A, et al. Recurrent jaw dislocation after botulinum toxin treatment for sialorrhoea in amyotrophic lateral sclerosis. J Neurol Sci. 2001;190:95-97.
180 Andersen PM, Gronberg H, Franzen L, et al. External radiation of the parotid glands significantly reduces drooling in patients with motor neurone disease with bulbar paresis. J Neurol Sci. 2001;191:111-114.
181 Harriman M, Morrison M, Hay J, et al. Use of radiotherapy for control of sialorrhea in patients with amyotrophic lateral sclerosis. J Otolaryngol. 2001;30:242-245.
182 Mitsumoto H, Davidson M, Moore D, et al. Percutaneous endoscopic gastrostomy (PEG) in patients with ALS and bulbar dysfunction. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:177-185.
183 Chio A, Galletti R, Finocchiaro C, et al. Percutaneous radiological gastrostomy: a safe and effective method of nutritional tube placement in advanced ALS. J Neurol Neurosurg Psychiatry. 2004;75:645-647.
184 Thornton FJ, Fotheringham T, Alexander M, et al. Amyotrophic lateral sclerosis: enteral nutrition provision—endoscopic or radiologic gastrostomy? Radiology. 2002;224:713-717.
185 Rio A, Leigh N. Noninvasive ventilation allows gastrostomy tube placement in patients with advanced ALS.Neurology. 2001;57:1351. discussion, Neurology. 2001;57:1351-1352.
186 Mitsumoto H, Chad D, Pioro E. Speech and communication management. In: Mitsumoto H, Chad DA, Pioro EP, editors. Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis; 1998:405-420.
187 Yorkston KM. Treatment efficacy: dysarthria. J Speech Hear Res. 1996;39:S46-S57.
188 Schiffer RB, Herndon RM, Rudick RA. Treatment of pathologic laughing and weeping with amitriptyline. N Engl J Med. 1985;312:1480-1482.
189 Iannaccone S, Ferini-Strambi L. Pharmacologic treatment of emotional lability. Clin Neuropharmacol. 1996;19:532-535.
190 Brooks BR, Thisted RA, Appel SH, et al. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial. Neurology. 2004;63:1364-1370.
191 Moretti R, Torre P, Antonello RM, et al. Rivastigmine in frontotemporal dementia: an open-label study. Drugs Aging. 2004;21:931-937.
192 Newsom-Davis IC, Lyall RA, Leigh PN, et al. The effect of noninvasive positive pressure ventilation (NIPPV) on cognitive function in amyotrophic lateral sclerosis (ALS): a prospective study. J Neurol Neurosurg Psychiatry. 2001;71:482-487.
193 Mitsumoto H, Chad D, Pioro E. Symptomatic treatment. In: Mitsumoto H, Chad DA, Pioro EP, editors. Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis; 1998:321-328.
194 Bradley WG, Anderson F, Bromberg M, et al. Current management of ALS: comparison of the ALS CARE Database and the AAN Practice Parameter. The American Academy of Neurology. Neurology. 2001;57:500-504.
195 Borasio GD, Voltz R, Miller RG. Palliative care in amyotrophic lateral sclerosis. Neurol Clin. 2001;19:829-847.
196 Mazzini L, Corra T, Zaccala M, et al. Percutaneous endoscopic gastrostomy and enteral nutrition in amyotrophic lateral sclerosis. J Neurol. 1995;242:695-698.
197 Aboussouan LS, Khan SU, Banerjee M, et al. Objective measures of the efficacy of noninvasive positive-pressure ventilation in amyotrophic lateral sclerosis. Muscle Nerve. 2001;24:403-409.
198 Mitsumoto H, Chad D, Pioro E. Course and prognosis. In: Mitsumoto H, Chad DA, Pioro EP, editors. Amyotrophic Lateral Sclerosis: Contemporary Neurology Series 49. Philadelphia: FA Davis; 1998:151-163.
199 Magnus T, Beck M, Giess R, et al. Disease progression in amyotrophic lateral sclerosis: predictors of survival. Muscle Nerve. 2002;25:709-714.
200 del Aguila MA, Longstreth WTJr, McGuire V, et al. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. 2003;60:813-819.
201 Chio A, Mora G, Leone M, et al. Early symptom progression rate is related to ALS outcome: a prospective population-based study. Neurology. 2002;59:99-103.
202 Turner M, Al-Chalabi A. Early symptom progression rate is related to ALS outcome: a prospective population-based study. Neurology. 2002;59:2012-2013. author reply, Neurology 2002; 59:2013.
203 Daube JR. Electrophysiologic studies in the diagnosis and prognosis of motor neuron diseases. Neurol Clin. 1985;3:473-493.
204 Bernstein LP, Antel JP. Motor neuron disease: decremental responses to repetitive nerve stimulation. Neurology. 1981;31:204-207.
205 Wang FC, De Pasqua V, Gerard P, et al. Prognostic value of decremental responses to repetitive nerve stimulation in ALS patients. Neurology. 2001;57:897-899.
206 Mulder DW, Howard FMJr. Patient resistance and prognosis in amyotrophic lateral sclerosis. Mayo Clin Proc. 1976;51:537-541.
207 Tucker T, Layzer RB, Miller RG, et al. Subacute, reversible motor neuron disease. Neurology. 1991;41:1541-1544.