67 Amyotrophic Lateral Sclerosis
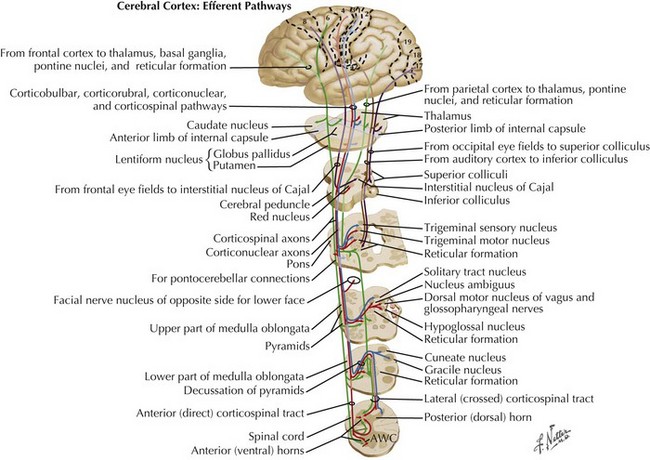
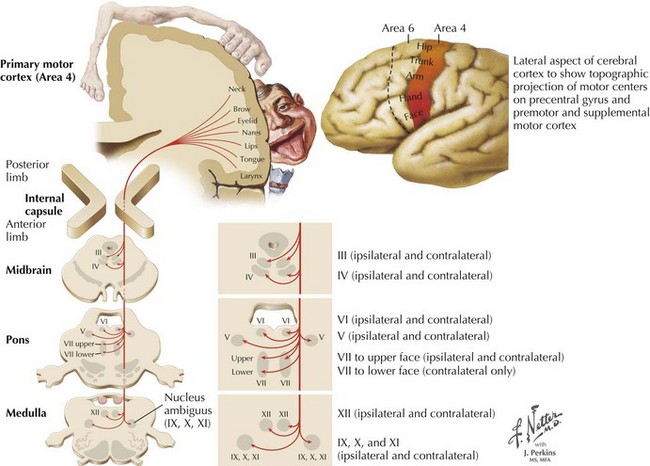
Charcot’s description was of a disorder characterized by loss of voluntary motor function, resulting from degeneration of anterior horn cells, corticospinal tracts, and motor cranial nerve nuclei and cortical motor neurons (Figs. 67-1 and 67-2). ALS is a sporadic disorder (sALS) in the majority of cases. ALS is inherited in 5–10% of cases, i.e., familial ALS (fALS), usually in an autosomal dominant fashion. In general, fALS patients have phenotypes that closely resemble sALS, although fALS may have an earlier onset. In absence of family history, the disorders are clinically indistinguishable.
Etiology, Genetics, and Pathogenesis
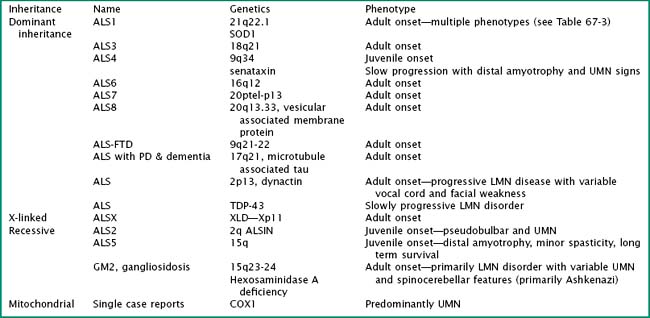
Particularly intriguing has been the recognition of the phenotypic heterogeneity in SOD1 fALS (Table 67-1). About 114 pathologic mutations have been identified within the five exons of the SOD1 gene; each of these mutations may produce a distinct phenotype. The most common mutation found in North America is an alanine for valine (A4V) substitution at codon 4; this typically produces a lower motor neuron dominant phenotype (LMN-D) with a life expectancy approximating 1 year. Table 67-1 summarizes the phenotypic heterogeneity that results from different SOD1 mutations. SOD1 mutations are not fully penetrant. It is estimated that individuals carrying the mutation have an 80% chance of developing disease by age 85 years. SOD1 mutations constitute 20–25% of all individuals with fALS. Other fALS genotypes are listed in Table 67-2. Some of these mutations produce a predominantly lower motor neuron (LMN) or upper motor neuron (UMN) disorder and more closely resemble the phenotypes of spinal muscular atrophy or hereditary spastic paraparesis, respectively.
| Phenotype | SOD 1 Mutation |
|---|---|
| Lower motor neuron predominant | A4V, L84V, D101N |
| Upper motor neuron predominant | D90A |
| Slow progression (>10-year survival) | G37R, G41D, G93C, L144S, L144F |
| Fast progression (<2-year survival) | A4T, N86S, L106V, V148G |
| Late onset | G85R, H46R |
| Early onset | G37R, L38V |
| Female predominant | G41D |
| Bulbar onset | V148I |
| Low penetrance | D90A, I113T |
| Posterior column involvement | E100G |
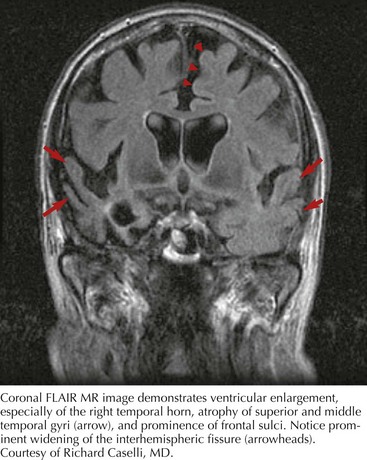
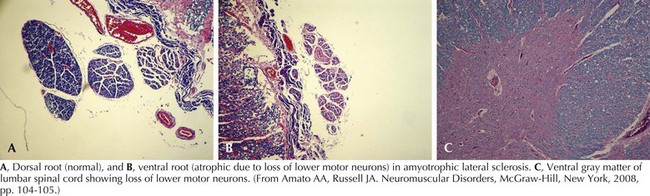
Whatever the mechanism, ALS is pathologically characterized by loss of myelinated fibers in the corticospinal and corticobulbar pathways (see Fig. 67-2) and loss of motor neurons within the anterior horns of the spinal cord and many motor cranial nerve nuclei. Even in individuals with predominantly UMN or LMN involvement clinically, pathologic involvement of both systems is seen. Patients with associated FTD have preferential lobar atrophy and neuronal loss from these portions of the brain (Fig. 67-3). As a result of anterior horn cell loss, ventral roots become atrophic in comparison to sparing of their dorsal root counterparts (Fig. 67-4). Anterior horn cell loss occurs within virtually all levels of the spinal cord with selective sparing of the third, fourth, and sixth cranial nerves, and Onuf’s nucleus within the anterior horn of sacral segments 2–4. There is also cell preservation within the intermediolateral cell columns.
Clinical Presentations
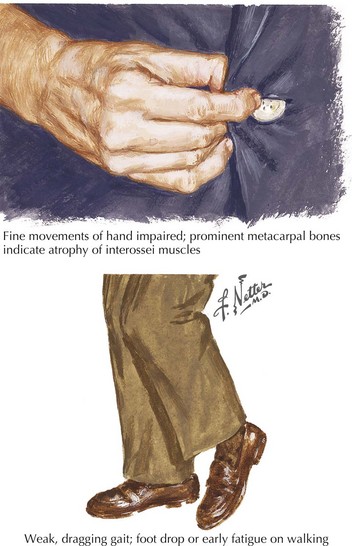
The presenting features of ALS are quite variable. Typically, the patient seeks medical care when his or her weakness begins to affect activities of daily living (Fig. 67-5). It is not uncommon for ALS to be misdiagnosed initially and the time between symptom onset and diagnosis is usually months. Unfortunately, there is a tendency to misdiagnose ALS as a potentially treatable nerve, nerve root, or spinal cord compressive syndrome or orthopedic condition. A significant percentage of ALS patients may undergo unnecessary surgeries. It should be emphasized that progressive weakness and atrophy in the absence of pain and sensory symptoms rarely represents a surgically treatable condition.
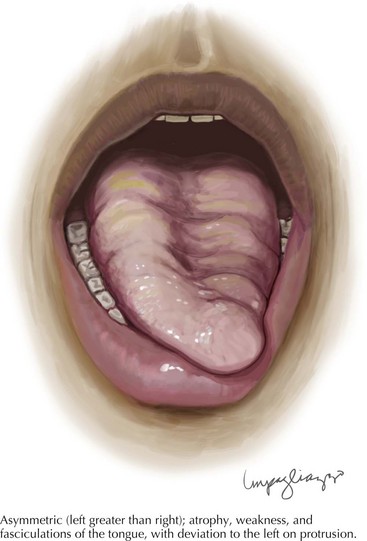
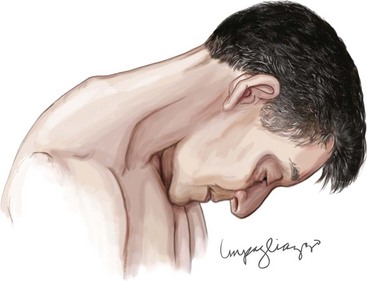
Tongue atrophy, fasciculation, and weakness are perhaps the most frequently occurring and recognized manifestations of LMN involvement of cranial nerves. It is important to observe for fasciculations when the tongue is relaxed on floor of the mouth (Fig. 67-6). Tremulousness of the tongue with attempted protrusion may be readily misinterpreted as representing fasciculations. Weakness of both facial and jaw muscles may occur in ALS but they are usually subtle. Weakness of neck extension and neck flexion is common in ALS, and head drop may be a rare presenting feature (Fig. 67-7). Neck drop is commonly associated with posterior neck discomfort and is typically relieved when the neck is supported. Notable for their absence are ptosis and ophthalmoparesis, and symptoms related to sight, hearing, taste, smell, and facial sensation.
Differential Diagnosis
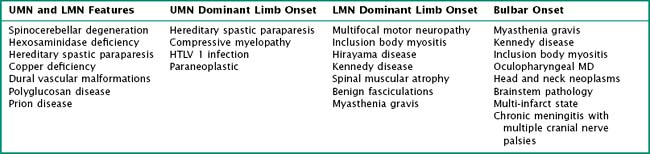
The differential diagnosis of ALS is largely that of other disorders of anterior horn cells, myopathies, disorders of neuromuscular transmission, motor predominant polyneuropathies and myelopathies (Table 67-3). With bulbar onset and predominantly LMN features, myasthenia gravis, inflammatory myopathy particularly inclusion body myositis, X-linked bulbospinal muscular atrophy (Kennedy disease), oculopharyngeal muscular dystrophy, multiple cranial neuropathies, or infiltrative head and neck cancers are the primary considerations. Of these, myasthenia gravis (MG) deserves the most attention. Clues favoring a diagnosis of MG include a weak tongue without atrophy or fasciculations, absence of UMN signs, and presence of ptosis or ophthalmoparesis. Dysphagia, usually without dysarthria, may rarely be the initial or most prominent symptom of the inflammatory myopathies. The pattern of limb weakness, the absence of fasciculations, and UMN signs in these disorders provides distinction from ALS. X-linked bulbospinal muscular atrophy or Kennedy disease may have early or prominent weakness of the throat, tongue, or jaw muscles often associated with fasciculation and may therefore easily be confused with bulbar ALS. A slower evolution of symptoms, the pattern of limb weakness, and the presence of gynecomastia and sensory abnormalities are distinctive features from ALS. Oculopharyngeal muscular dystrophy (OPMD) may be confused with bulbar ALS, although the course of OPMD is typically much longer, with a positive family history and prominent ptosis and ophthalmoplegia on examination. Multiple cranial neuropathies from chronic meningitis (e.g., cancer, sarcoidosis) are usually associated with sensory dysfunction.
Diagnostic Approach
There is no perfect algorithm in the evaluation of an ALS suspect. In a patient with LMN and UMN features that have progressed in a typical pattern and time course, the diagnosis is indisputable. In most cases where ALS is suspected, tests are ordered guided by the clinician’s index of suspicion (Table 67-4). In large part, these tests are done to exclude considerations other than ALS.
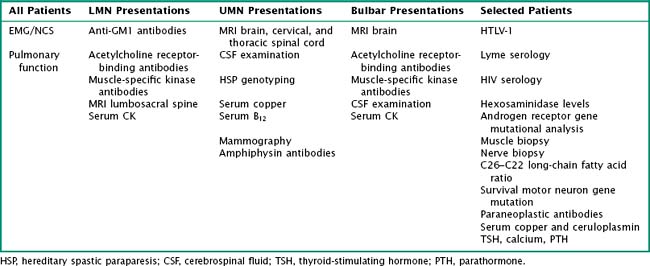
Virtually every ALS patient undergoes electromyography and nerve conduction studies, collectively referred to as electrodiagnosis (EDX) (Table 67-5). The goal of EDX in ALS is to confirm a pattern of active denervation, chronic denervation, and fasciculation potentials in multiple muscles innervated by multiple segments in multiple regions. According to the modified El Escorial EDX criteria, a definite diagnosis of ALS requires evidence of active denervation (fibrillation potentials and positive sharp waves) in at least three of the following four body regions: cranial, cervical, thoracic, and lumbosacral. In the limbs, at least two different muscles belonging to different nerve and root innervations need to be affected. Involvement in a single cranial muscle is sufficient to satisfy that region’s requirements. Thoracic paraspinal muscles are particularly helpful as they are uncommonly denervated in non-ALS disorders. Fasciculation potentials are a supportive but not mandatory electrodiagnostic feature. Features that would suggest an alternative diagnosis that might mimic ALS need to be excluded; examples include abnormal sensory conductions in Kennedy syndrome, decremental response to repetitive stimulation consistent with myasthenia, conduction block suggestive of multifocal motor neuropathy, or small motor unit potentials suggestive of a myopathy such as IBM. Finally EX may offer insight into the rate of progression, that is, active denervation without chronic denervation and reinnervation, motor unit variability, and a rapid decline in motor unit estimation being electrodiagnostic indicators of a rapidly progressive course.
| Pattern of abnormalities | Multisegmental in multiple regions (cranial–cervical–thoracoabdominal–lumbosacral) |
| Motor conductions | ↓ or normal CMAP amplitudes depending on severity, relative preservation of conduction velocities, and distal latencies |
| Sensory conductions | Normal |
| F waves and H reflexes | F waves normal or absent, M response of H reflex reduced in amplitude, absent or normal depending on severity. Latencies preserved |
| Slow repetitive stimulation | May demonstrate decremental response |
| Spontaneous activity (EMG) | Fibrillation potentials and positive waves necessary for diagnosis, complex repetitive, myotonic, myokymic, and neuromyotonic discharges uncommon and should lead to alternative considerations |
| MUP morphology | Typically long duration, high amplitude with instability if sought for |
| MUP recruitment | MUP recruitment reduced, MUP activation may be reduced as well with prominent UMN component. |
CMAP, compound muscle action potential; MUP, motor unit potential.
It is important to recognize that elevated serum creatine kinase levels are not specific for myopathy. Approximately two thirds of ALS patients will have creatine kinase elevations, typically in the 300–500 U/L range but occasionally as high as 1000 or more. Antibodies directed at the GM1 ganglioside are found in high titer in 30–80% of patients with MMN. They are typically ordered in patients with LMN syndromes without cranial nerve or UMN findings. Serologic tests for myasthenia, typically acetylcholine receptor binding, may be obtained in patients with bulbar presentations. Other tests, listed in Table 67-4, are used more judiciously in the appropriate clinical context. Many patients with ALS inquire about the possibility of Lyme disease, and Lyme serologies are frequently ordered to lessen these concerns. HIV testing is not done in suspected ALS unless the clinical context would suggest an increased probability of infection. Historically, screening for heavy metals, thyroid and parathyroid disorders, and occult neoplasia was emphasized. Serum copper, ceruloplasmin, and zinc levels should be considered in any patient with weakness and unexplained sensory symptoms.
Management and Therapy
Management of ALS includes disease-specific treatments, symptomatic and supportive treatments, as well as adequate education and counseling. These are summarized in Table 67-6 and are elaborated on in two of the reviews listed in the bibliography. Rilutek is the only FDA-approved and effective pharmacologic agent identified to date. Unfortunately, it prolongs life expectancy on average by 10% without noticeable improvement in function or sense of well-being. Its cost is substantial, and it should be prescribed only after the patient has been informed of its benefits and drawbacks. Patients should be encouraged to participate in clinical trials when available. Many ALS patients utilize alternative health measures. Patients should be informed that any treatment biologically active enough to help is also biologically capable of harm. It is not uncommon for patients to ask for medications that have been touted, but unproven to be effective. If these agents are to be studied in clinical trials, clinicians should discourage their use outside of the trial so as to not subvert enrollment in and/or the integrity of the trial.
| Problem | Potential Prescription |
|---|---|
| ALS | Riluzole 50 mg bid |
| ALS | Clinical trials |
| Sialorrhea (excessive thin secretions) |
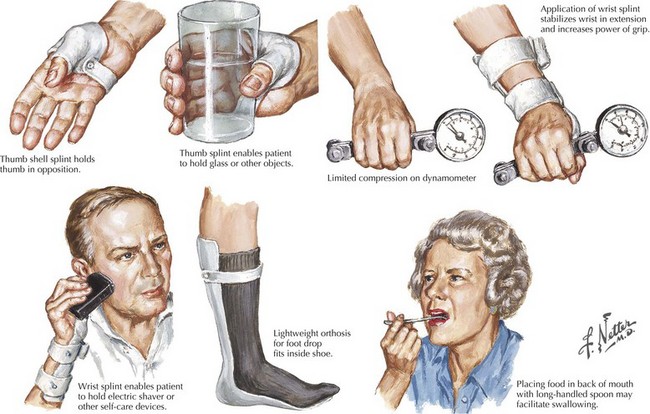
An important aspect of the management of ALS patients and their families is the provision of reliable education. It is important to discuss end-of-life issues with a patient at some point before a ventilatory crisis occurs or the ability to communicate is lost. Until an effective treatment is found, the primary goals in ALS management are to provide symptom relief and to maintain independent and safe function. In the later stages of disease, the primary goal shifts to maintenance of comfort. Table 67-6 provides a list of many of these potential interventions (Fig. 67-8). In our clinic, we focus on symptoms referable to the following domains: pain, sleep, psychosocial issues, speech and swallowing, ventilation, motor function, and miscellaneous issues. As mentioned above, pain occurs commonly in this disease. Prophylactic range of motion should be applied to immobilized body parts. Analgesics including opioids may be required. Impaired sleep has many potential causes in an ALS patient, including discomfort secondary to immobility or cramping, depression, impaired ventilation and bathroom requirements, each of which may be have to be identified and addressed separately.
Amato AA, Russell JA. Neuromuscular Disorders. New York: McGraw Hill; 2008.
Bensimon G, Lacomblez L, Meininger V, et al. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med. 1994;330:585-591.
Chaudhuri KR, Crump SJ, Al-Sarraj S, et al. The validation of El Escorial criteria for the diagnosis of amyotrophic lateral sclerosis: a clinical-pathological study. J Neurol Sci. 1995;129(Suppl.):11-12.
Hand C, Rouleau GA. Familial amyotrophic lateral sclerosis. Muscle Nerve. 2002;25:135-159.
Miller RG, Jackson CE, Kasarskis EJ, et al. Practice Parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1218-1226.
Miller RG, Jackson CE, Kasarskis EJ, et al. Practice Parameter update: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1227-1233.
Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nature Reviews. 2006;7:710-723.
Rowland L. Diagnosis of amyotrophic lateral sclerosis. J Neurol Sci. 1998;160(Suppl.):6-24.
Shaw CE, Al-Chalabi A. Susceptibility genes in sporadic ALS: separating the wheat from the chaff by international collaboration. Neurology. 2006;67:738-739.
Strong MJ, Lomen-Hoerth C, Caselli RJ, et al. Cognitive impairment, frontotemporal dementia and the motor neuron diseases. Ann Neurol. 2003;54(5):S20-S23.