Chapter 16 Amenorrhea
DEFINITIONS
In common medical usage, amenorrhea refers to the abnormal cessation of menses.1 Physiologic amenorrhea exists before puberty, during pregnancy and lactation, and after menopause. However, these physiologic causes are not included in the standard amenorrhea classifications.
Amenorrhea can be divided into two major groups based on presentation: primary or secondary amenorrhea (Table 16-1).2,3 Although many of the causes of primary and secondary amenorrhea are similar, the most likely causes, and thus the diagnostic approach, are distinct.
Primary Amenorrhea
Primary amenorrhea is the absence of menstruation in a woman who has never menstruated. Because children do not normally menstruate before puberty, the age at which primary amenorrhea is diagnosed depends on the presence or absence of secondary sexual characteristics. In the absence of increased growth or development of secondary sexual characteristics, primary amenorrhea is diagnosed when the patient has no menses by age 13. In the presence of normal growth and development of secondary sexual characteristics, the diagnosis of primary amenorrhea is reserved for patients who have no menstruation by age 15. The incidence of primary amenorrhea in the United States is less than 0.1%. The majority of patients with primary amenorrhea will be found to have either gonadal dysgenesis (49%) or müllerian agenesis (16%).4
Secondary Amenorrhea
The incidence of secondary amenorrhea not due to pregnancy, lactation, or menopause is approximately 4%.5,6 Although the list of causes for amenorrhea is quite extensive (Table 16-2), it appears that this list will continue to grow or be modified as more sophisticated genetic testing becomes available and the genetic understanding of human disease expands. The majority of patients with amenorrhea will have premature ovarian failure, hyperprolactinemia, hypothalamic amenorrhea, or polycystic ovary syndrome (PCOS).
Table 16-2 Classification of Amenorrhea, Both Primary and Secondary3
Classification
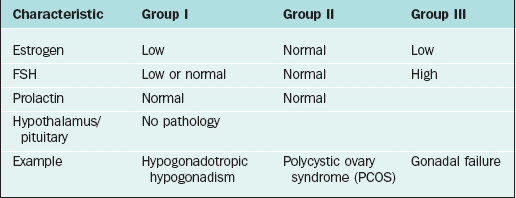
The most widely accepted classification of amenorrhea was published by the World Health Organization (WHO) and divides amenorrhea into three groups (Table 16-3).3 This WHO amenorrhea classification is designed to help the practicing clinician summarize the causes of amenorrhea to assist in evaluating the condition. Group I include individuals who lack endogenous estrogen production, in association with normal or low follicle-stimulating hormone (FSH) levels, and no evidence of hypothalamic-pituitary pathology or elevated prolactin levels. Group II is associated with evidence of estrogen production and normal levels of prolactin and FSH. Finally, Group III involves elevated serum FSH levels that indicate gonadal failure.7 Although amenorrhea can occur among patients with sexual ambiguity or virilization, it is rarely the cause for initial consultation.8
PRIMARY AMENORRHEA
The etiologies of primary amenorrhea are multiple and diverse (Tables 16-4 and 16-5). The four most common causes of primary amenorrhea have been reported to be the following:4
| Category | Frequency |
|---|---|
| Normal Secondary Sexual Development | (∼1/3 of total) |
| Müllerian agenesis | 10% |
| Androgen insensitivity | 9% |
| Constitutional delay | 8% |
| Outlet obstruction (e.g., vaginal septum, imperforate hymen) | 3% |
| Absent Secondary Sexual Development | (∼2/3 of total) |
| High FSH (gonadal dysgenesis) | |
| Abnormal karyotype (e.g., 46,XO, mosaic) | 20% |
| 46,XX | 15% |
| 46,XY | 5% |
| Low FSH | |
| Hypothalamic disorders | 8% |
| Constitutional delay | 10% |
| Hyperandrogenic conditions (e.g., PCOS, CAH) | 6% |
| Pituitary adenomas | 5% |
Adapted from Practice Committee of ASRM: Current evaluation of amenorrhea. Fertil Steril 82(Suppl 1):33-S39, 2004.
Gonadal Dysgenesis
The term gonadal dysgenesis is used globally to refer to all forms of abnormal gonads, which can occur in individuals with normal karyotypes (46,XX; 46,XY) as well as a variety of abnormal or mosaic states, most commonly Turner’s syndrome (45,XO). The gonads are usually streaks of fibrous tissue.
Mixed Gonadal Dysgenesis
Patients who present with gonadal dysgenesis and a normal karyotype need to be assessed for a variety of other conditions, such as neurosensory deafness and fragile X syndrome. These clinical associations are particularly true when familial premature ovarian failure is identified.9
General Principles of X Chromosome Genetic Disorders
Translocations of the X chromosome, although extremely rare, may cause amenorrhea depending on the location of breakpoints. In a balanced X translocation one X chromosome is normal, and the other is an X autosome translocation chromosome. X inactivation is not usually random so that the normal X is usually inactivated. If the translocated chromosome were inactivated, the autosome would also be inactivated, making the karyotype lethal. Nearly all males and half of the females with X autosome translocations are sterile.10
Turner’s Syndrome
It is known that specific genes in the X chromosome are essential for normal functioning of the ovaries.11 It appears that both X chromosomes with normally functioning genes need to be present in the oocytes to prevent the formation of a streak gonad.
The characteristic physical features common to females with Turner’s syndrome include short stature, somatic abnormalities (webbed neck, shield chest, increased angle at the elbow known as cubitus valgus, cardiovascular abnormalities), and prepubertal status associated with elevated gonadotropins.12 Patients with Turner’s syndrome require special attention to the autoimmune disorders and renal anomalies that are frequently found with the condition. All patients with Turner’s syndrome should seek expert cardiology consultation and screening, including chest X-ray and echocardiography, at the time of diagnosis. Annual cardiac examinations, including evaluation of blood pressure, and repeated screening at 3- to 5-year intervals if the initial screening reveals no abnormalities. When the cardiac echo is abnormal or the ascending aorta cannot be visualized, magnetic resonance imaging (MRI) of the chest should be performed in all patients.12
Gonadal Dysgenesis 46,XY: Swyer Syndrome
About 10% to 15% of patients with Swyer syndrome possess mutations of the SRY (sex-determining region on the Y chromosome) gene located on the distal portion of Yp.13,14 Due to the significant increased risk for tumor development in patients with a Y chromosome and streak gonads, gonadectomy is recommended at an early age.
Management of Gonadal Dysgenesis
Oocyte donation offers women with Turner’s syndrome the opportunity to achieve pregnancy. However, the increased cardiovascular demands of pregnancy also may pose unique and serious risk given their high rate of cardiovascular malformations (25% to 50% prevalence). A recent Practice Committee position from the American Society for Reproductive Medicine (ASRM) has stated that because the risk for aortic dissection or rupture during pregnancy may be 2% or higher, the risk of death during pregnancy is increased as much as 100-fold.12 Any significant cardiac abnormality should be regarded as a contraindication to oocyte donation. Even those having a normal evaluation should be thoroughly counseled regarding the high risk of cardiac complications during pregnancy because aortic dissection may still occur.12
Disorders of the Genital Tract
Müllerian Agenesis
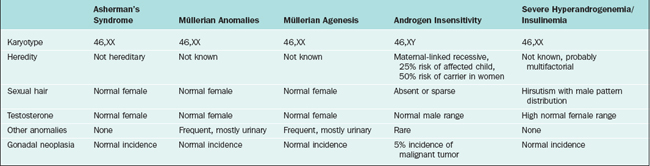
Mullerian agenesis, also referred to as Mayer-Rokitansky-Küster-Hauser syndrome, is a condition in which all or part of the uterus and vagina are absent in the presence of otherwise normal female sexual characteristics. This diagnosis accounts for approximately 10% of cases associated with primary amenorrhea.15 In Finland, the incidence was calculated to be approximately 1 of every 5000 newborn girls.16 In müllerian agenesis, the ovaries are not affected and thus ovarian function is normal. Secondary sexual development and height are in a normal range.
The differential diagnosis of patients who present with primary amenorrhea and have a genital tract anomaly is summarized in Table 16-4. Partial development of the müllerian structures can lead to obstructed menses and painful distention of a hematocolpos, hematometra, or hematoperitoneum. It is important to separate müllerian agenesis from complete androgen insensitivity syndrome, because the vagina may be absent or short in both disorders.17
It is currently unknown why müllerian agenesis occurs, but likely causes are mutations of genes that are responsible for müllerian tract maintenance. Thus far, no mutations have been reported.18 It appears that the mode of inheritance is not autosomal dominant.19
Although ultrasound could be an important aid in confirming the presence or absence of uterine structures, MRI is usually more definitive. Occasionally laparoscopic visualization is needed, because there is disagreement between MRI and laparoscopic findings.20 If persistent chronic pelvic pain and symptoms associated with endometriosis are present, laparoscopy can usually aid in determining the location and potential removal of incompletely formed müllerian structures.
Outflow Obstructions
Imperforate hymen is the most frequent obstructive female genital tract anomaly, with an estimated frequency of approximately 0.1%. Imperforate hymen usually occurs sporadically, but familial cases have been reported.21 Although this entity can present in infants or children of any age as a mucocolpos, a bulging intact hymen with hematocolpos presenting at the time of menarche is not unusual. Hymenectomy effectively alleviates this problem.
A transverse vaginal septum is somewhat less common than an imperforate hymen, occurring in fewer than 1 in 20,000 females. The presentation and surgical treatment is similar to an imperforate hymen if the transverse vaginal septa is located in the lower third of the vagina. However, more than 80% are located in the middle and upper vagina. Diagnosis is usually made by ultrasound or MRI. When located in these areas, septa tend to be thicker, and surgery is more difficult. Surgical management is described in detail in Chapter 51.
Androgen Insensitivity Syndrome
Androgen insensitivity syndrome (formerly known as testicular feminization), is an X-linked recessive condition in which genotypic males (46,XY) develop into apparently phenotypic females who have no müllerian structures and intra-abdominal testes. The underlying cause is an abnormally functioning androgen receptor, which prevents normal masculinization. This syndrome will be identified in as many as 5% of all patients presenting with primary amenorrhea.17
Evaluation of Androgen Insensitivity Syndrome
On pelvic examination, the patients will have scant or absent pubic hair and a blind vagina that is no more than a few centimeters long. More than 50% of patients have inguinal hernias and underdeveloped labia minora. Laboratory evaluation will demonstrate a 46,XY karyotype and elevated total testosterone (in the normal male range).22 This distinguishes patients with androgen insensitivity syndrome from those with müllerian agenesis, who will have an XX karyotype and total testosterone in the normal female range.
Rare Causes of Primary Amenorrhea
Isolated Gonadotropin Deficiency
This disorder is characterized by a decrease or absence in endogenous gonadotropin-releasing hormone (GnRH) secretion, which results in very low to undetectable luteinizing hormone (LH) and FSH levels. Individuals with the disorder have incomplete development of secondary sexual characteristics, primary amenorrhea, eunuchoid features, and in some cases a decreased sense of smell or anosmia (Kallmann syndrome). Male patients with Kallmann syndrome have X-linked recessive idiopathic hypogonadotropic hypogonadism accompanied by anosmia caused by mutations in the KAL1 gene, localized to the pseudoautosomal region of Xp.23,24
The protein product of KAL1, anosmin, possesses neural cell adhesion molecule properties. Anosmin provides a guide for GnRH neurons and olfactory nerves to migrate from the olfactory placode across to the olfactory bulb. When anosmin is absent or defective, GnRH and olfactory neurons fail to synapse normally. The KAL1 gene escapes X inactivation and an inactive pseudogene is present on Yq. For individuals with anosmia or hyposmia, there is evidence of hypoplasia of the olfactory bulbs on MRI. No KAL1 gene mutations have been identified in females with idiopathic hypogonadotropic hypogonadism and anosmia, suggesting that other autosomal genes may be involved.25
LH Receptor Abnormalities
Abnormalities of the LH receptor in 46,XX females will result in normal female sexual development and primary amenorrhea.26 Serum LH may be normal to increased, FSH is normal, follicular phase estradiol levels are normal, and progesterone is low. The uterus is small and the ovaries are consistent with anovulation.
Gonadotropin-Releasing Hormone Receptor Abnormalities
Abnormalities of GnRH receptors have also been identified. The phenotype of GnRH resistance ranges from complete idiopathic hypogonadotropic hypogonadism to oligo-ovulation. Baseline levels of LH and FSH may be in the prepubertal or normal range. However, levels of other pituitary hormones, such as thyrotropin, growth hormone, prolactin, and corticotropin, are normal. Due to the failure to increase gonadal sex steroid secretion during puberty, secondary sex characteristics fail to develop and closure of the epiphyseal plates of the long bones is delayed, resulting in a eunuchoid habitus in which the arm span is greater than the height. Even in patients with complete forms, pulsatile GnRH administration may increase pituitary gonadotropin response; pregnancies have been reported.27
Diagnostic Approach for Primary Amenorrhea
What Age?
An evaluation of primary amenorrhea is indicated when an adolescent fails to menstruate by age 15 in the presence of normal secondary sexual development (two standard deviations above the mean of 13 years), or within 5 years after breast development, if that occurred before age 10.2 This age has recently been adjusted to a younger age, because girls are menstruating at a younger age. If there is a failure to initiate breast development by age 13 (two standard deviations above the mean of 10 years), an investigation should also be initiated.2 These criteria are not absolute, and complete evaluation should be initiated if the patient presents with amenorrhea and obvious associated pathology such as cyclic pain or a blind vaginal pouch.
History and Physical Examination
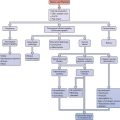
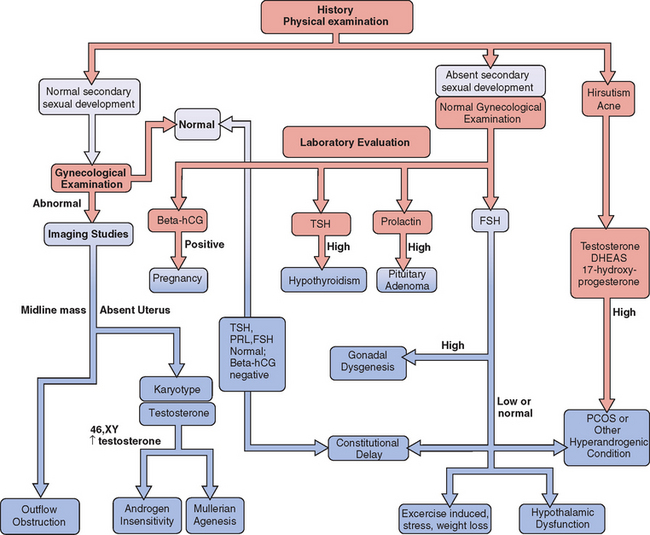
A careful history is a key component to the evaluation and planning for the treatment of amenorrhea (Fig. 16-1). Special emphasis should be focused on physical or emotional stress, nutritional status, and history of genetic inherited disorders. The family should be questioned about familial disorders such as diabetes mellitus, previous medical disorders that may have been treated with gonadotoxic agents, and surgical disorders that involve the genital tract, including abnormal sexual differentiation. The functional inquiry should include secondary sexual development, galactorrhea, and the presence of hyperandrogenic symptoms.
Imaging
In patients with primary amenorrhea, physical examination will often detect genital tract anomalies. However, the characterization of the specific anomaly often requires imaging studies. In some cases, abdominal ultrasonography is adequate to determine the presence or absence of a uterus. Probably the most effective imaging method for characterizing congenital anomalies is MRI of the pelvis. Congenital anomalies are considered in depth in Chapters 12 and 51.
Laboratory Evaluation
If hirsutism is present or if androgen insensitivity syndrome is suspected, androgens should be evaluated. Total testosterone will be elevated in this syndrome as well as in cases of ovarian tumors. Dehydroepiandrosterone sulfate (DHEAS) will be elevated in patients with adrenal tumors and Cushing’s disease. Borderline elevation of these androgens is often seen with polycycstic ovary syndrome. 17-hydroxyprogesterone is often elevated in patients with adult onset congenital adrenal hyperplasia, although a provocative test is often necessary in subtle cases.
SECONDARY AMENORRHEA
Secondary amenorrhea is a condition in which menstruation begins at the appropriate age but later stops for reasons not due to pregnancy, lactation, or menopause. For clinical purposes, the length of amenorrhea should be equal to at least 3 of the previous cycle intervals, or 6 months, although patients with oligomenorrhea (menstruation <9 times per year or bleeding intervals >40 days) often have similar underlying pathology. This condition affects at least 4% of women and is more common in those whose weight is below or above the normal range. The four most common causes of secondary amenorrhea are (1) hypothalamic amenorrhea (e.g., exercise induced), (2) hyperandrogenic states (e.g., PCOS), (3) pituitary disorders (e.g., hyperprolactinemia), and (4) premature ovarian failure (Table 16-6).
| Categories with Examples | Approximate Frequency |
|---|---|
From Herman-Giddens ME, Slora EJ, Wasserman RC, et al: Secondary sexual characteristics and menses in young girls seen in office practice: A study from the Pediatric Research in Office Settings network. Pediatr 99:505-512, 1997; and Reindollar RH, Novak M, Tho SP, McDonough PG: Adult-onset amenorrhea: A study of 262 patients. Am J Obstet Gynecol 155:531–543, 1986.
Hypothalamic Amenorrhea
Regulation of GnRH Secretion
It is not possible to directly assess GnRH secretion, because this decapeptide is rapidly metabolized within 2 to 4 minutes in the peripheral circulation. Clinical investigation relies on measurement of LH concentrations as the surrogate marker for hypothalamic GnRH secretion. In women with regular menstrual cycles, clinical studies have demonstrated a characteristic pulsatile secretion of LH at a frequency of 90 to 120 minutes during the follicular phase and a frequency of 180 to 240 minutes during the luteal phase.
Stress-Related Amenorrhea
The stress response associated with chronic stress involves activation of the HPA axis and increased secretion of a “stress response complex” of hormones such as corticotropin-releasing hormone (CRH), corticotropin, cortisol, prolactin, oxytocin, vasopressin, norepinephrine, and epinephrine (Table 16-7). These hormonal effects impact on reproductive function at several levels. For example, CRH has been shown to directly inhibit GnRH secretion in rats, monkeys, and humans at the hypothalamic level in vitro and in vivo. This inhibition can be negated by administration of a CRH receptor antagonist or by naloxone.
Table 16-7 Associated Neuroendocrine Abnormalities in Hypothalamic Amenorrhea
| Increased daytime cortisol secretion |
| Increased amplitude and duration of nocturnal melatonin secretion |
| Increased nocturnal secretion of GH |
| Elevated CRH levels in cerebrospinal fluid |
| Blunted elevation of prolactin, corticotropin, and cortisol during the noon meal |
Taken together, these observations suggest that the inhibitory effect of CRH is mediated in part by increased secretion of endogenous opioids. The increased secretion of corticotropin at the pituitary level may also suppress pituitary response to GnRH. In addition, increased cortisol levels may also dampen pituitary response to GnRH.
Exercise-Induced Hypothalamic Amenorrhea
Menstrual cycle disturbances are common among competitive athletes, including ballet dancers, marathon runners, gymnasts, and figure skaters.28 Depending on the type of activity and competition level, the incidence of amenorrhea varies from 5% to 25%. The incidence of menstrual irregularities appears to be greatest in activities that favor a low body weight, such as ballet (6% to 43%) and middle and long distance running (24% to 26%). The incidence appears to be less frequent in bicycling (12%) and swimming (12%).
Bulimia and Anorexia Nervosa
It is extremely important for the clinician to recognize early signs of these disorders so that appropriate intervention and treatment can be obtained. The mortality associated with anorexia is as high as 9%, usually secondary to cardiac arrhythmia precipitated by electrolyte abnormalities and diminished heart muscle mass. Suicide is also more common. The clinical features of bulimia and anorexia are listed in Tables 16-8 and 16-9.
| Irregular menstrual cycles |
| Dental enamel erosion |
| Enlargement of salivary glands |
| Acute irritation of esophageal mucosa |
| Esophageal or gastric rupture |
| Hypokalemia |
| Aspiration pneumonia |
| Ipecac poisoning |
Table 16-9 Common Features of Anorexia Nervosa
| Preoccupation with handling of food |
| Bulimic behavior |
| Calorie counting |
| Distortion of body self-image |
| Hyperactivity |
| Obsessive-compulsive personality |
| Increased incidence of past sexual abuse |
| Amenorrhea |
| Constipation |
| Coarse, dry skin |
| Soft, lanugo-type hair |
| Hypothermia with defective thermoregulation |
| Mild bradycardia |
| Cardiac arrhythmias |
| Hypotension |
| Hypokalemia secondary to diuretic or laxative abuse |
| Osteopenia |
| Increased serum beta-carotene levels |
| Anemia, leukopenia |
| Elevated hepatic enzymes |
Anorexia nervosa is associated with multiple neuroendocrine abnormalities (Table 16-10). As a result of decreased caloric intake, thyroxine (T4) conversion to triiodothyronine (T3) is decreased, resulting in lowered basal metabolism. As a result, thyroxine is converted to an inactive isoform, reverse triiodothyronine (reverse T3). Profound hypothalamic dysfunction is manifest by hypothermia and impaired secretion of vasopressin that can result in partial diabetes insipidus with the inability to concentrate urine. Hyperactivation of the HPA axis results in hypersecretion of cortisol, but manifestations of hypercortisolism are rarely present due to number of decreased cellular glucocorticoid receptors. These patients also have increased central opioid activity.
Table 16-10 Neuroendocrine Abnormalities Associated with Anorexia Nervosa
| Diminished GnRH LH pulsatile frequency and amplitude |
| Low blood LH and FSH levels |
| Impaired corticotropin response to CRH stimulation testing |
| Resistance to dexamethasone suppression |
| Increased corticotropin levels |
| Increased 24-hour urinary free cortisol levels |
| Normal prolactin level |
| Normal TSH levels with high reverse T3 and low T3 levels |
| Elevated GH levels |
| Decreased IGF-I levels |
| Diabetes insipidus |
Bulimia and anorexia result in a prepubertal pattern of LH similar to patients with functional hypothalamic amenorrhea secretion, presumably due to a marked decrease in GnRH secretion. With weight gain, patients with anorexia will resume normal patterns of LH secretion and may have normal or supranormal responses to GnRH. Despite return to normal body weight, up to 50% remain anovulatory.
As in all energy deprivation states, leptin plays a critical role in the disruption of the hypothalamic function that results in amenorrhea. Leptin is a protein hormone secreted by adipose tissue and has an important role in adaptation to starvation. Leptin-deficient ob/ob mice and leptin-resistant db/db mice show obesity and hypogonadotropic hypogonadism. However, the principal role of leptin is in caloric deprivation; it acts as the signal from the periphery to the brain about energy deficit. This energy deficit is signaled to the hypothalamic-pituitary axis through decreased leptin levels. Leptin may have a role in the other neuroendocrine abnormalities such as thyroid and insulin-like growth factor-I (IGF-I) seen in anorexics and in other energy-deficient states such as excessive exercise.29
Evaluation of Hypothalamic Amenorrhea
Because this is a diagnosis of exclusion, significant organic diseases must be excluded (see Table 16-2). A detailed interview may identify a stressful event or emotional crisis (divorce, relationship breakup, death of a friend or relative) preceding the amenorrhea. Other interpersonal and environmental stressors may also be present, such as academic pressures, job stresses, or psychosexual problems. A careful review of the patient’s current lifestyle, including exercise intensity, dietary choices, and the use of sedatives or hypnotics, may be helpful in characterizing the psychogenic stress components.30
Clinical Management of Hypothalamic Amenorrhea
Bone Loss
The primary cause of bone loss in these patients is hypoestrogenemia, and thus patients not desiring fertility will benefit from hormone replacement therapy. Bone loss is most sensitive to estrogen.31,32 Although several forms of hormone replacement have been described, placing young women on oral contraceptives has been the treatment of choice. Although many women prefer to have menses in association with hormone replacement, athletes who find this to be inconvenient can be safely kept on continuous hormone replacement or birth control pills.
Postcontraception Amenorrhea
Amenorrhea after long-term contraception use remains a concern, but the issues of concern have changed. In the past, use of high-dose (>50 μg ethinyl estradiol) oral contraceptives was associated with prolonged amenorrhea after discontinuation. With modern low-dose oral contraceptives, amenorrhea is uncommon, but reversible cycle disturbances after discontinuing oral contraceptives can last up to 9 months or longer.33 Fortunately, oral contraceptives do not affect fertility in the long term. Amenorrhea that continues more than 2 months after discontinuation of an oral contraceptive should trigger an investigation for other causes of amenorrhea.
A more common problem has become amenorrhea after the intramuscular use of medroxyprogesterone acetate (Depo-Provera) for contraception. Amenorrhea is common after discontinuation of this type of contraception, and the return to baseline fertility takes an average 10 months.34 For this reason, investigation for amenorrhea after discontinuation of depot medroxyprogesterone should be limited until 12 months after the last injection.
Hyperandrogenic States
Polycystic Ovary Syndrome
Polycystic ovary syndrome (PCOS) is one of the most common causes of ovulation dysfunction (see Chapter 19). Although this complex syndrome has a wide range of clinical manifestations, amenorrhea was one of the primary manifestations first described by Stein and Leventhal in 1935 along with infertility and enlarged ovaries.35 Contemporary series suggest that only about 25% of patients with PCOS will have amenorrhea.36 The pathology, diagnosis, and treatment of PCOS is discussed in depth in Chapter 15.
Other Hyperandrogenic States
The basis of the workup to exclude these conditions includes measurement of serum androgens, including total testosterone, DHEAS, and 17-hydroxyprogesterone. Imaging of the ovaries and, in some cases, adrenal glands is also important. This details of this evaluation are given in Chapter 18.
Disorders of the Anterior Pituitary
The most common pituitary tumor is the prolactin-secreting adenoma, which accounts for almost 70% of all pituitary adenomas. The second most commonly encountered pituitary tumor is a nonfunctioning pituitary adenoma, which constitutes at least 25% of all pituitary tumors. Less common pituitary tumors secrete growth hormone, corticotropin, thyrotropin, FSH, or LH. These tumors increase prolactin levels by compressing the pituitary stalk and interfering with the release of dopamine, which acts as a prolactin inhibiting factor. Amenorrhea results in addition to diseases caused by other pituitary tumors, including acromegaly (growth hormone-secreting tumors), Cushing’s disease (corticotropin-secreting tumors), or hyperthyroidism (thyrotropin-secreting tumors). Diagnosis and management of these tumors is discussed in depth in Chapter 22.
Prolactin-Secreting Adenomas
In women with hyperprolactinemia, the prevalence of pituitary tumor is 50% to 60%,37 and the likelihood of a pituitary tumor is unrelated to the level of prolactin found.37 The poor correlation between tumor size and serum prolactin indicates that MRI or computed tomography (CT) scanning of the pituitary should be performed whenever serum prolactin levels are consistently elevated.38 Although the exact incidence of these tumors is unknown, autopsy series have shown the presence of microadenomas to range from 9% to 27%.39,40 The distribution of these types of tumors appears equal among gender.
Empty Sella Syndrome
This syndrome refers to a congenital incompleteness of the diaphragm of the sella turcica, allowing for extension of the subarachnoid space into the pituitary fossa. This can also occur secondary to surgery, radiation therapy, or infarction of a pituitary tumor. This anatomic abnormality is found in approximately 5% of autopsies, with a high predominance in women. Empty sella is found in 4% to 16% of patients with amenorrhea-galactorrhea.41 Galactorrhea and elevated serum prolactin levels can be seen. Annual surveillance with serum prolactin levels is sufficient since they rarely develop prolactinomas. The purpose of treatment is alleviation of symptoms.
Postpartum Pituitary Necrosis (Sheehan’s Syndrome)
Postpartum pituitary necrosis is usually preceded by a history of severe obstetrical hemorrhage with hypotension, circulatory collapse, and shock.42 After fluid resuscitation of the patient, this condition may be manifested by clinical evidence of partial or panhypopituitarism. Simmonds was the first to describe this clinical syndrome although the most complete description has been attributed to Sheehan. This condition constitutes an endocrine emergency that can be life-threatening.
The pathophysiology of this process is not entirely clear. With pregnancy, there is an increase in blood supply to the pituitary bed and the pituitary gland enlarges. During the period of profound hypotension, Sheehan postulated that occlusive spasm of the arteries that supply the pituitary and stalk occurs. This leads to venous stasis and thrombosis of the pituitary portal vessels, causing a variable degree of pituitary ischemia and cell death. Many patients initially present with a failure to have breast engorgement and lactation due to a deficiency in prolactin secretion. These women may also have other anterior pituitary deficiencies.
Evaluation of Pituitary Adenomas
The clinical manifestations of these adenomas are usually more obvious in women, due to the disruption of menses. The most common symptoms of a prolactin-secreting adenoma in women are galactorrhea, irregular periods, headaches, and infertility. Men can experience hypogonadism due to these tumors, but the tumors are usually large and produce high serum prolactin levels. Approximately one third of women with amenorrhea will have elevated prolactin levels; one third of women with galactorrhea and elevated prolactin levels will have normal menses and one third of women will have a high prolactin level without galactorrhea.43 As many as a third of patients with secondary amenorrhea will have a prolactinoma and when associated with galactorrhea, half of the patients will have normal findings on imaging of the sella turcica.43,44
The apparent clinical difficulty in associating clinical symptoms, laboratory values, and sella turcica imaging is the fact that there is tremendous variability in the detection of prolactin in clinical assays. The immunoreactivity of clinical assays usually detects the small form of prolactin, which also has more biologic activity. Big forms of prolactin can also be secreted by pituitary adenomas and, since this prolactin type cannot usually be detected by the immunoassays, the diagnosis of a pituitary adenoma may be missed. Therefore, whenever the clinical scenario of galactorrhea is present, particularly in a patient with irregular menstruation, imaging of the pituitary gland must be considered and clinical intervention should be instituted.45,46
Some patients with high serum prolactin levels can also have what is referred to as a “high-dose hook effect” where large amounts of prolactin prevent accurate assessment by the immunoassay. Dilutions of serum samples are able to detect the abnormality.47 The mechanism by which prolactin causes oligomenorrhea and amenorrhea is due to the inhibition of pulsatile secretion of GnRH by elevated prolactin.48,49
The measurement of thyrotropin should be included as part of the evaluation. Although prolactin levels associated with hypothyroidism are generally less than 100 ng/mL, these levels can induce galactorrhea. Hyperprolactinemia in hypothyroidism is the result of increased thyrotropin-releasing hormone (TRH) stimulation of the pituitary gland. TRH is a potent stimulant of prolactin-secreting cells. The extent of pituitary deficiencies can be characterized by provocative testing with combined intravenous injection of the hypothalamic releasing factors GnRH, TRH, growth hormone releasing hormone, and CRH.50
Management of Pituitary Adenomas
The use of dopamine agonists such as bromocriptine lowers the circulating levels of prolactin and restores the normal response of the ovary to gonadotropins and restores normal menstrual function. The treatment with bromocriptine, a dopamine agonist, inhibits pituitary prolactin secretion. This medication is associated with many side effects; 10% of patients cannot tolerate the medication and will discontinue it. Most patients complain of nausea, headache, and faintness, usually due to orthostatic hypotension. Other side effects include dizziness, fatigue, nasal congestion, vomiting, and abdominal cramps, which can be diminshed by starting the patient on a low dose of the medication and slowly increasing it. Some patients can benefit from vaginal administration of bromocriptine to avoid the gastrointestinal side effects.51 Vaginal administration is effective largely by avoiding the first pass through the liver.
There is no evidence that bromocriptine harms the fetus. However, most clinicians recommend discontinuing the medication during pregnancy.52 More than 80% of patients with amenorrhea and galactorrhea in association with hyperprolactinemia will have their menses restored 5 to 7 weeks after therapy is started.53 The cessation of galactorrhea is much slower than the restoration of menses, and complete cessation of galactorrhea occurs in half of patients after 4 months of use. Up to 75% of patients who stop the treatment will develop symptoms again.54
It is possible for macroadenomas to regress with bromocriptine treatment, but it requires higher doses and a longer period of use. Most patients have a rapid shrinkage in the first 3 months of therapy followed by a slower reduction.55 Patients who initially present with serum prolactin levels over 1000 ng/mL have tumors that invade into their cavernous sinuses. These individuals usually have inoperable tumors and require long-term suppression with dopamine agonists. Bromocriptine can also be used for patients who have failed surgery and radiation therapy.
Cabergoline is an alternative to bromocriptine. Side effects seem to develop less frequently and the medication is taken once or twice weekly. Due to a limited experience on fetal safety, this agent should be used with caution in patients who are trying to conceive.56–58
Transsphenoidal surgery has also been used to treat pituitary adenomas. Symptom resolution is achieved in 30% of patients with macroadenomas and 70% of patients with microadenomas; this is highly dependent on the experience of the neurosurgeon.59 Tumor recurrence is high, especially after surgery for macroadenoma. Potential postoperative complications include panhypopituitarism, meningitis, cerebrospinal fluid leaks, and diabetes insipidus. The high success rate of medical therapy, recurrence of disease, and potential complications of surgery have limited the use of surgical intervention to patients who have failed medical therapy. Radiation therapy is less satisfactory than surgery for the treatment of pituitary adenomas, and the response is very slow. Under specific circumstances radiation can be delivered with a gamma knife procedure.
Patients who respond to treatment of hyperprolactinemia can breast-feed if desired and usually can experience normal lactation without fear of tumor growth. There is only a small chance of growth of most pituitary tumors during pregnancy. Up to 5% of patients will have tumor enlargement, which is usually asymptomatic; less than 2% will develop symptoms.60 Most symptomatic patients present with headaches, which usually precede any visual abnormalities. During pregnancy there is normal pituitary growth, typically due to the increased size of the prolactin-secreting cells. Insufficient blood supply to the adenoma may cause this infarct. Occasionally patients may have restoration of normal menses from tumor infarction in pregnancy or postpartum.
Surveillance during pregnancy with monthly visual field examinations or serum prolactin measurements has not been clinically useful. Patients who become symptomatic should be assessed and treated. Most patients will respond to bromocriptine, and it is very uncommon to require termination of pregnancy or neurosurgery.61 Dopamine agonists used during pregnancy do not affect decidual secretion of prolactin, which is under control of estrogen and progesterone rather than dopamine.
Premature Ovarian Failure
Premature ovarian failure is defined as amenorrhea with persistent estrogen deficiency and elevated FSH levels before age 40. This affects at least 1% of women.62,63 Most causes of premature ovarian failure are easily identified, such as chemotherapy and radiation therapy for cancer. Premature ovarian failure is usually irreversible, although spontaneous recovery of ovarian function can occur. Premature ovarian failure is a common cause of secondary amenorrhea, accounting for 4% to 18% of cases.64 This topic is examined in depth in Chapter 20.
The etiology for the majority of patients with premature ovarian failure is unknown. However, for patients younger than age 30 presenting with amenorrhea, a karyotype should be obtained to rule out sex chromosome translocation, short arm deletions, or persistence of an occult Y chromosome, because these conditions are associated with an increased risk of ovarian malignancies. Some experts recommend that all patients with premature ovarian failure have a chromosomal analysis. However, in patients with amenorrhea secondary to premature ovarian failure, the single most common karyotype is XX (see Table 16-6).
Gonadal Dysgenesis
Patients who present with gonadal dysgenesis and a normal karyotype need to be assessed for a variety of other conditions, such as neurosensory deafness (Perrault’s syndrome), as well as fragile X syndrome, the most common genetic cause of developmental disorders. About 16% of women who are carriers of the premutation allele for fragile X syndrome experience premature ovarian failure, particularly when familial premature ovarian failure or mental retardation is identified.9 When women with sporadic premature ovarian failure are screened, approximately 3% will be premutation carriers. Some females with premutation alleles may be affected with mild degrees of mental deficiency or learning disability.
There is also an association of premature ovarian failure with autosomal-dominant eyelid abnormalities. This is known as blepharophimosis-ptosis-epicanthus inversus syndrome. This syndrome is caused by a mutation in FOXL2, which is a transcription gene factor found on chromosome 3.65,66 Several other autosomal disorders have been associated with ovarian failure; these will produce elevations of FSH without necessarily having oocyte depletion. Some of these include mutations of the phosphomannomutase 2 (PMM2) genes, the galactose-1-phosphate uridyltransferase (GALT) gene, the FSH receptor gene, and the autoimmune regulator gene (ARE), which is responsible for polyendocrinopathy-candidiasis-ectodermal dystrophy.67
Autoimmune Causes
A common form of premature ovarian failure is due to autoimmune disease. Up to 40% of women with premature ovarian failure may have autoimmune abnormalities, most commonly autoimmune thyroiditis resulting in hypothyroidism.68,69 Premature ovarian failure is also more common in women with insulin-dependent diabetes, myasthenia gravis, and parathyroid disease than in healthy women.70 In 10% to 60% of cases of Addison’s disease, autoimmune ovarian failure may also be present.
Because these individuals are at increased risk for endocrine autoimmune conditions, patients should be evaluated every other year for occurrence of these abnormalities so early intervention can be achieved. Patients with unexplained premature ovarian failure should have a complete evaluation to exclude other autoimmune disorders; tests to perform include tests for calcium, phosphorus, fasting glucose, adrenal antibodies to 21-hydroxylase enzyme, free T4, thyrotropin, and thyroid antibodies. This evaluation should be repeated on a yearly or every other year basis.71 Screening for antiovarian antibodies is not warranted because the assays have poor sensitivity and specificity.
Premature Ovarian Failure: Other Causes
There have been a few reports of single gonadotropin deficiency; the measurement of both LH and FSH together will uncover these unusual disorders. In premature ovarian failure you will always find elevations of both hormones; a single elevation is suspicious.72 Most of these abnormalities are due to single-gene or amino acid substitutions. In these cases, an MRI of the pituitary will also uncover a pituitary adenoma that secretes these hormones, particularly if associated with an elevated α-subunit. However, these tumors are generally not associated with amenorrhea.
There can also be mutations of the receptors for gonadotropins; these patients are diagnosed with resistant or insensitive ovary syndrome. These patients generally have secondary amenorrhea with normal secondary sexual characteristics and do not respond to gonadotropins and have small antral follicles on ultrasound.73
Mutations for the human FSH receptor gene have also been identified, both in females74 and males. Females display hypergonadotropic hypogonadism from FSH resistance. The phenotype ranges from absent to normal breast development and primary or secondary amenorrhea. This is a relatively uncommon finding, but is found predominantly in certain populations such as Finland (1% of females are heterozygotes).
Mutations of the LH receptor in 46,XX females consist of normal sexual development and amenorrhea.26 Serum LH may be normal to increased, FSH is normal, follicular phase estradiol levels are normal, and progesterone is low. The uterus is small and the ovaries are consistent with anovulation.
Management of Premature Ovarian Failure
Patients with ovarian failure should be offered estrogen and progesterone replacement to maintain their secondary sexual characteristics and reduce the risk of osteoporosis. This can be easily achieved with combined oral contraceptives until the age of natural menopause as long as no contraindications to combined oral contraceptives are met. Some women who take low-dose exogenous estrogen therapy with a progestin, who have some ovarian follicles left with premature ovarian failure, can have spontaneous ovulation and conception is possible in rare cases.75
Disorders of the Genital Tract
Asherman’s Syndrome
Asherman’s syndrome is commonly used to describe the presence of intrauterine adhesions (or synechiae), most commonly secondary to uterine surgery. Patients are found to have Asherman’s syndrome after undergoing aggressive curettage for postpartum bleeding, after removal of multiple submucosal fibroids, or after metroplasty.76 Recently uterine artery embolization has also been associated with the development of Asherman’s syndrome; although its mechanism is not well understood, it is believed to be secondary to ischemia associated with the embolization procedure.77 Although the syndrome was first described as amenorrhea secondary to intrauterine synechiae, the presence of hypomenorrhea or amenorrhea is not considered a requirement for making this diagnosis today.76,78
Diagnostic Approach for Secondary Amenorrhea
Evaluation of the woman with secondary amenorrhea begins with a careful history to detect sometimes subtle symptoms of one of a wide variety of conditions that can bring a halt to menses (Fig. 16-2). Physical examination will sometimes give hints of the most likely etiologies. Initial laboratory evaluation is an important step, not only to exclude physiologic causes of amenorrhea (e.g., pregnancy), but also to detect subtle hormonal conditions that often have no other symptoms or physical signs. Progestin challenge is then used to detect genital tract disorders and hypoestrogenemic states. By the second visit, enough information can be gathered to pursue more directed diagnostic tests to come up with a definite diagnosis.
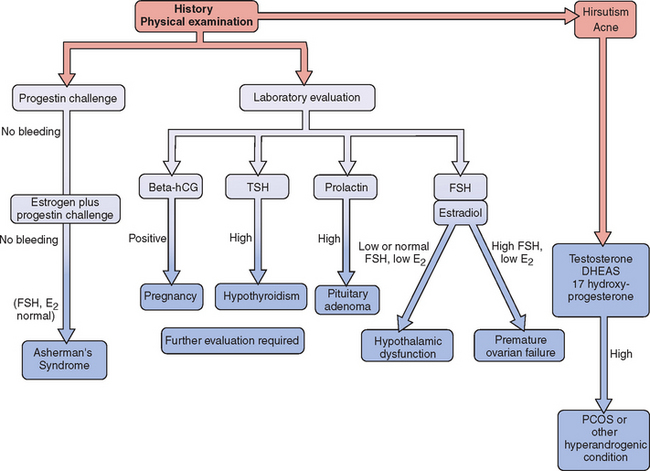
Figure 16-2 Flow diagram in the evaluation of women with secondary amenorrhea, showing major decision points.
Laboratory Evaluation
Evaluation of androgens is indicated in women with amenorrhea and any sign of androgen excess such as hirsutism or acne. Although many women with PCOS will have modestly elevated androgens (see Chapter 15), the most important objective of measuring androgens in women with apparent PCOS is exclusion of other causes of hyperandrogenic amenorrhea, most notably androgen-producing tumors of the ovary and adrenal glands, Cushing’s syndrome, and adult-onset adrenal hyperplasia.
Progestin Challenge Test
Failure to bleed after a progestin challenge is usually the result of estrogen deficiency or Asherman’s syndrome. To differentiate between these two, the patient should be pretreated with estrogen (e.g., conjugated estrogens 1.25 mg daily for 6 to 8 weeks) and the progestin challenge should be repeated. Patients who do not bleed after administration of both estrogen and progestin are likely to have an anatomic problem that is preventing menses.
1 Stedman’s Medical Dictionary. 27th ed. Philadelphia: Lippincott Williams & Wilkins; 2000:56.
2 Herman-Giddens ME, Slora EJ, Wasserman RC, et al. Secondary sexual characteristics and menses in young girls seen in office practice: A study from the Pediatric Research in Office Settings network. Pediatr. 1997;99:505-512.
3 Practice Committee of the American Society for Reproductive Medicine. Current evaluation of amenorrhea. Fertil Steril. 2004;82(Suppl 1):S33-S39.
4 Timmreck LS, Reindollar RH. Contemporary issues in primary amenorrhea. Obstet Gynecol Clin North Am. 2003;30:287-302.
5 Pettersson F, Frieds H, Nillius SJ. Epidemiology of secondary amenorrhea. I. Incidence and prevalence rates. Am J Obstet Gynecol. 1973;117:80-86.
6 Bachmann G, Kemmerman E. Prevalence of oligomenorrhea and amenorrhea in a college population. Am J Obstet Gynecol. 1982;144:98-102.
7 Insler V. Gonadotrophin therapy: New trends and insights. Int J Fertil. 1988;33:85-97.
8 Doody KM, Carr BR. Amenorrhea. Obstet Gynecol Clin North Am. 1990;17:361-387.
9 Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, et al. Fragile X premutation is a significant risk factor for premature ovarian failure: The International Collaborative POF in fragile-X study/preliminary data. Am J Med Genet. 1999;83:322-325.
10 Layman L. Familial ovarian failure. In: Lobo RA, editor. Perimenopause. New York: Springer-Verlag; 1997:46-77.
11 Powell CN, Taggart DT, Drumheller TC, et al. Molecular and cytogenic studies of an X: Out to some translocation in a patient with premature ovarian failure in a review of the literature. Am J Med Genet. 1994;52:19-26.
12 Practice Committee of the American Society for Reproductive Medicine. Increased maternal cardiovascular mortality associated with pregnancy in women with Turner syndrome. Fertil Steril. 2005;83:1074-1075.
13 Jager RJ, Anvret M, Hall K, Scherer G. A human XY female with a frame shift mutation in the candidate testis-determining gene SRY.. Nature. 1990;348:452-454.
14 Ostrer H. Sexual differentiation. Semin Reprod Med. 2000;18:41-49.
15 Reinhold C, Hricak H, Forstner R, et al. Primary amenorrhea: Evaluation with MR imaging. Radiology. 1997;203:383-390.
16 Aittomaki K, Eroila H, Kajanoja P. A population-based study of the incidence of müllerian aplasia in Finland. Fertil Steril. 2004;76:624-625.
17 Jagiello J. Prevalence of testicular feminization. Lancet. 1962;1:329-333.
18 Imbeaud S, Faure E, Lamarre I, et al. Insensitivity to anti-müllerian hormone due to a mutation in the human anti-müllerian hormone receptor. Nat Genet. 1995;11:382-388.
19 Petrozza JC, Gray MR, Davis AJ, Reindollar RH. Congenital absence of the uterus and vagina is not commonly transmitted as a dominant genetic trait: Outcomes of surrogate pregnancy. Fertil Steril. 1997;67:387.
20 Economy KE, Barnewolt C, Laufer MR. A comparison of MRI and laparoscopy in detecting pelvic structures in cases of vaginal agenesis. J Pediatr Adolesc Gynecol. 2002;15:101-104.
21 Stelling JR, Gray MR, Davis AJ, et al. Dominant transmission of imperforate hymen. Fertil Steril. 2000;74:1241-1244.
22 Wilson JD. Syndrome of androgen resistance. Biol Reprod. 1992;46:168-173.
23 Franco B, Guioli S, Pragliola A, et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:529-536.
24 Legouis R, Hardelin J, Levilliers J, et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell. 1991;67:423-435.
25 Layman LC. Human gene mutations causing infertility. J Med Genet. 2003;39:153-161.
26 Toledo SPA, Brunner HG, Kraaij R, et al. An inactivating mutation of the luteinizing hormone receptor causes amenorrhea in a 46,XX female. J Clin Endocrinol Metab. 1996;81:3850-3854.
27 Layman LC, Cohen DP, Jin M, et al. Mutations in the gonadotropin-releasing hormone receptor gene cause hypogonadotropic hypogonadism. Nat Genet. 1998;18:14-15.
28 Goodman LR, Warren MP. The female athlete and menstrual function. Curr Opin Obstet Gynecol. 2005;17:466-470.
29 Montague CT, Farooqi S, Whitehead FP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903-908.
30 Berga SL. Behaviorally induced reproductive compromise in women and men. Semin Reprod Endocrinol. 1997;15:47-53.
31 Drinkwater BL, Nilson K, Chesnut CH, et al. Bone mineral content of amenorrheic and eumenorrheic athletes. NEJM. 1984;311:277-281.
32 Drinkwater BL, Nilson K, Ott S, Chesnut CH. Bone mineral density after resumption of menses in amenorrheic athletes. JAMA. 1986;256:380-382.
33 Gnoth C, Frank-Herrmann P, Schmoll A, et al. Cycle characteristics after discontinuation of oral contraceptives. Gynecol Endocrinol. 2002;16:307-317.
34 Schwallie PC, Assenzo JR. The effect of depo-medroxyprogesterone acetate on pituitary and ovarian function, and the return of fertility following its discontinuation: A review. Contraception. 1974;10:181-202.
35 Stein IF, Levinthal M. Amenorrhea associated with bilateral polycystic ovaries. Am J Obstet Gynecol. 1935;29:181-191.
36 Franks S. Polycystic ovary syndrome. NEJM. 1995;333:853-861.
37 Brenner SH, Lessing JB, Quagliarello J, Weiss JG. Hyperprolactinemia in associated pituitary prolactinomas. Obstet Gynecol. 1985;65:661-664.
38 Schlechte J, Dolan K, Sherman B, et al. The natural history of untreated hyperprolactinemia: A perspective analysis. J Clin Endocrinol Metab. 1989;68:412-418.
39 Costello RT. Subclinical adenoma of the pituitary gland. Am J Pathol. 1936;12:191-197.
40 Burrow GN, Wortzman G, Rewcastle MB, et al. Microadenomas of the pituitary and abnormal cellar tomograms in an unselected autopsy series. NEJM. 1981;304:156-158.
41 Hodgson SF, Randall RV, Holman CB, MacCarty CS. Empty sella syndrome. Med Clin North Am. 1972;56:897-907.
42 Sheehan HL, Murdoch R. Postpartum necrosis of the interior pituitary: Pathological and clinical aspects. J Obstet Gynaecol Br Emp. 1938;45:456-464.
43 Schlechte J, Sherman B, Halm IN, et al. Prolactin-secreting pituitary tumors. Endocr Rev. 1980;1:295-298.
44 Kleinberg DL, Noel GL, Frantz AG. Galactorrhea: A study of 235 cases including 48 with pituitary tumors. NEJM. 1977;296:589-600.
45 Jackson RD, Wortsman J, Malarkey WB. Characterization of a large molecular weight Prolactin in women with idiopathic hyperprolactinemia and normal menses. J Clin Endocrinol Metab. 1985;61:258-264.
46 Hattori N, Inagaki C. Anti-prolactin auto-antibodies cause a symptomatic hyperprolactinemia: Bioassay and clearance studies of prolactin-immunoglobulin G complex. J Clin Endocrinol Metab. 1997;82:3107-3110.
47 Schofl C, Schofl-Siegert B, Karstens JH, et al. Falsely low serum prolactin in two cases of invasive microprolactinoma. Pituitary. 2002;5:261-265.
48 Cook CB, Nippoldt TB, Kletter GB, et al. Naloxone increases the frequency of pulsatile leuteinizing hormone secretion in women with hyperprolactinemia. J Clin Endocrinol Metab. 1991;73:1099-1105.
49 Sauder SE, Frager M, Case GD, et al. Abnormal patterns of pulsatile leuteinizing hormone secretion in women with hyperprolactinemia and amenorrhea: Responses to bromocriptine. J Clin Endocrinol Metab. 1984;59:941-948.
50 Veldhuis JD, Hammond JM. Endocrine function after spontaneous infarction of the human pituitary: Report, review, and reappraisal. Endocr Rev. 1980;1:100-107.
51 Katz E, Schran HF, Adashi EY. Successful treatment of a prolactin-producing pituitary macroadenoma with intervaginal bromocriptine mesylate: A noble approach to intolerance to oral therapy. Obstet Gynecol. 1989;73:517-520.
52 Turkalj I, Braun P, Krupp P. Surveillance of bromocriptine in pregnancy. JAMA. 1982;247:1589-1591.
53 Cuellar FG. Bromocriptine mesylate (Parlodel) in the management of amenorrhea-galactorrhea associated with hyperprolactinemia. Obstet Gynecol. 1980;55:278-284.
54 Passos PQ, Souza JJ, Musolino NR, Bronstein MD. Long-term follow up of prolactinomas: Normal prolactinemia after bromocriptine withdrawal. J Clin Endocrinol Metab. 2002;87:3578-3582.
55 Mori H, Mori S, Saitoh Y, et al. Effects of bromocriptine on prolactin-secreting pituitary adenomas. Cancer. 1985;56:230-238.
56 Rains CP, Bryson HM, Fitton A. Cabergoline. A review of its pharmacologic properties and therapeutic potential in the treatment of hyperprolactinemia and inhibition of lactation. Drugs. 1995;49:255.
57 Robert E, Musatti L, Piscitelli G, Ferrari CI. Pregnancy outcome after treatment with the ergot derivative cabergoline. Reprod Toxicol. 1996;10:333-337.
58 Webster J, Piscitelli G, Polli A, et alThe Cabergoline compared to a study group. A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. NEJM. 1994;331:904-909.
59 Schlechte JA, Sherman BM, Chapler FK, Vangilder J. Long-term follow-up of women with surgical treated prolactin-secreting tumors. J Clin Endocrinol Metab. 1986;62:1296-1301.
60 Molitch ME. Pregnancy and the hyperprolactinemic woman. NEJM. 1985;312:1364-1370.
61 Bevan JS, Webster J, Berke CW, Scanlon MF. Dopamine agonist and pituitary tumor shrinkage. Endocr Rev. 1992;13:220-240.
62 Jones GS, DeMoraes-Ruehsen M. A new syndrome of amenorrhea in association with hypergonadotropism and apparently normal ovarian follicular apparatus. Am J Obstet Gynecol. 1969;104:597-600.
63 Van Campenhout J, Vauclair R, Maraghi K. Gonadotropin-resistant ovaries in primary amenorrhea. Obstet Gynecol. 1972;40:6-12.
64 Anasti JN. Premature ovarian failure: An update. Fertil Steril. 1998;70:1-15.
65 Schlessinger D, Herrera L, Crispni L, et al. Genes and translocation involved in POF. Am J Med Genet. 2002;111:328.
66 Hundscheid RD, Smits AP, Vomis CM, et al. Female carriers for fragile-X pre-mutation have no increased risk for additional disease other than premature ovarian failure. Am J Med Genet. 2003;117:6.
67 Laml T, Preyer J, Umek W, et al. Genetic disorders in premature ovarian failure. Hum Reprod Update. 2002;8:483-491.
68 LeBarbera AR, Miller MM, Ober C, Rebar RW. Autoimmune etiology in premature ovarian failure. Am J Reprod Immunol Microbiol. 1988;16:115-122.
69 Hoek A, Schoemaker J, Drexhage HA. Premature ovarian failure and ovarian autoimmunity. Endocr Rev. 1997;18:107-134.
70 Nelson LM, Anasti JN, Flack MR. Premature ovarian failure. In: Adashi EY, Rock JA, Rosenwalks Z, editors. Reproductive Endocrinology, Surgery and Technology. Philadelphia: Lippincott-Raven; 1996:1393-1410.
71 Bakaolv VK, Vanderhoof VH, Bondy CA, Nelson LM. Adrenal antibodies detect asymptomatic auto-immune adrenal insufficiency in young women with spontaneous premature ovarian failure. Hum Reprod. 2002;17:2096.
72 Weiss J, Axelrod L, Whitcomb RW, et al. Hypogonadism caused by a single aminoacid substitution in the β-subunit of luteinizing hormone. NEJM. 1992;326:179-183.
73 Aittomaki K, Herva R, Stenman U-H, et al. Clinical features of primary ovarian failure caused by a point mutation in the follicle-stimulating hormone receptor gene. J Clin Endocrinol Metab. 1996;81:3722-3726.
74 Touraine P, Beau I, Gougeon A, et al. New natural inactivating mutations of the follicle-stimulating hormone receptor: Correlations between receptor function and phenotype. Mol Endocrinol. 1999;13:1844-1854.
75 Rebar RW, Connolly HV. Clinical features of young women with hypergonadotropic amenorrhea. Fertil Steril. 1990;53:804-810.
76 Schenker JG, Margalioth EJ. Intrauterine adhesion: An updated appraisal. Fertil Steril. 1982;37:593-610.
77 Davies C, Gibson M, Holt EM, Torrie EPH. Amenorrhea secondary to endometrial ablation and Asherman’s syndrome following uterine artery embolization. Clin Radiology. 2002;57:317-318.
78 Asherman JC. Amenorrhoea traumatica (atretica). J Obstet Gynecol Br Empire. 1948;55:23-30.