Chapter 134 Allergy and the Immunologic Basis of Atopic Disease
Key Elements of Allergic Diseases
Allergens
Most allergens are usually proteins of 10-70 kd molecular weight; molecules < 10 kd do not bridge adjacent IgE antibody molecules on the surfaces of mast cells or basophils; most molecules > 70 kd do not pass through mucosal surfaces, a feature needed to reach antigen-presenting cells (APCs) for stimulation of the immune system. Allergens frequently contain proteases, which promote barrier dysfunction and increase allergen penetration into host tissues. Low molecular weight moieties such as drugs can become allergens by reacting with serum proteins or cell membrane proteins to be recognized by the immune system. Carbohydrate structures have also been identified as allergens. This finding is most relevant with the increasing use of biologics in clinical practice; patients with cetuximab-induced anaphylaxis have IgE antibodies in specific for galactose-α-l,3-galactose (Chapter 146).
T Cells
Everyone is exposed to potential allergens. Atopic individuals respond to allergen exposure with rapid expansion of T helper type 2 (Th2) cells that secrete cytokines, such as interleukins IL-4, IL-5, and IL-13, favoring IgE synthesis and eosinophilia. Allergen-specific IgE antibodies associated with atopic response are detectable by serum testing or positive immediate reactions to allergen extracts on prick skin testing (Chapter 135). The Th2 cytokines IL-4 and IL-13 play a key role in immunoglobulin isotype switching to IgE (Fig. 134-1). IL-5 and IL-9 are important in differentiation and development of eosinophils. The combination of IL-3, IL-4, and IL-9 contributes to mast cell activation. Th2 cytokines are important effector molecules in the pathogenesis of asthma and allergic diseases; acute allergic reactions are characterized by infiltration of Th2 cells into affected tissues. In addition, IL-25 and IL-33 contribute to Th2 response and eosinophilia.
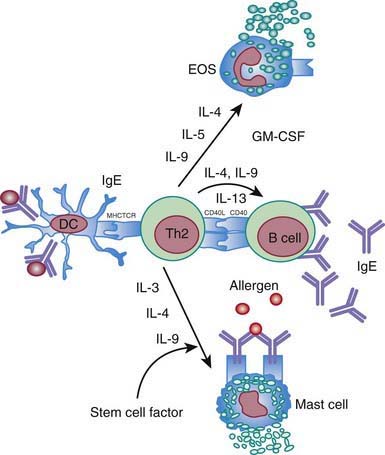
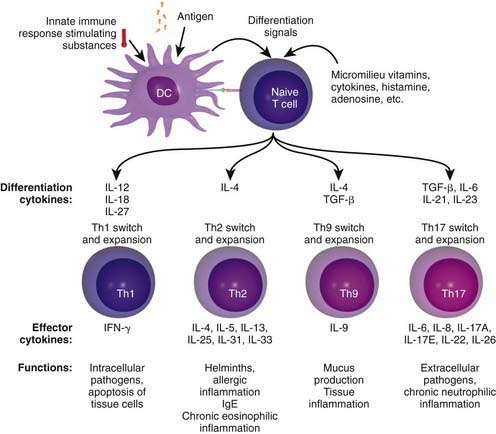
Chronic allergic reactions are characterized by infiltration of Th1 and Th17 cells. This is important because Th1 cytokines such as IFN-γ can potentiate the function of allergic inflammatory effector cells such as eosinophils and thereby contribute to disease severity. Cytokines in the IL-17 family act on multiple cell types, including epithelial cells and APCs, to cause the release of chemokines, antimicrobial peptides, and pro-inflammatory cytokines to enhance inflammation and antimicrobial responses. In addition, recently identified Th9 cells produce IL-9 but not other typical Th1, Th2 and Th17 cytokines and constitute a distinct population of effector T cells that promotes tissue inflammation. Figure 134-2 depicts the complex cytokine cascades involving Th1, Th2, Th9, and Th17 cells.
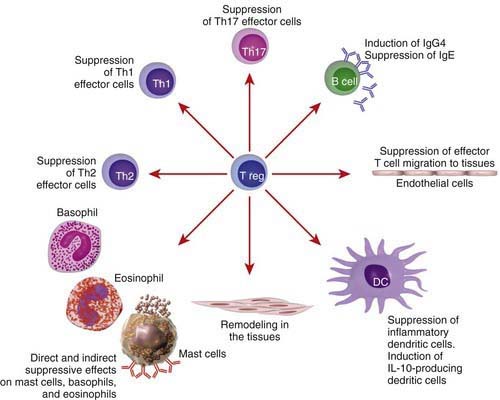
T regulatory (Treg) cells are a subset of T cells thought to play a critical role in expression of allergic and autoimmune diseases. These cells have the ability to suppress effector T cells of either the Th1 or Th2 phenotypes (Fig. 134-3). Treg cells express CD4+CD25+ surface molecules and immunosuppressive cytokines such as IL-10 and transforming growth factor-β (TGF-β1). The forkhead box/winged-helix transcription factor gene FOXP3 is expressed specifically by CD4+CD25+ Treg cells and programs their development and function. Adoptive transfer of Treg cells inhibits the development of airway eosinophilia and protects against airway hyperreactivity in animal models of asthma. T cell response to allergens in healthy individuals shows a wide range, from no detectable response to involvement of active peripheral tolerance mechanisms mediated by different subsets of Treg cells. Individuals who are not allergic even though they are exposed to high doses of allergens, such as beekeepers and cat owners, show a detectable allergen-specific IgG4 response accompanied by IL-10–producing Treg cells. It is thought that CD4+CD25+ Treg cells play an important role in mitigating the allergic immune response and that the lack of such cells may predispose to the development of allergic diseases. Patients with mutations in the human FOXP3 gene lack CD4+CD25+ Treg cells and develop severe immune dysregulation, with polyendocrinopathy, food allergy, and high serum IgE levels (XLAAD/IPEX disease) (Chapter 120).
Immunoglobulin E and its Receptors
The acute allergic response depends on IgE and its ability to bind selectively to the α chain of the high-affinity FcεRI or the low-affinity FcεRII (CD23). Cross linking of receptor-bound IgE molecules by allergen initiates a complex intracellular signaling cascade followed by the release of various mediators of allergic inflammation from mast cells and basophils. The FcεRI molecule is also found on the surface of antigen-presenting DCs (e.g., Langerhans cells), but differs from the structure found on mast cells/basophils in that the FcεRI molecule found on DCs lacks the β chain. CD23 is found on B cells, eosinophils, platelets, and dendritic cells. Cross linking and FcεRI aggregation on mast cells and basophils can also lead to anaphylaxis (Chapter 143). Differential expression of tyrosine kinases responsible for positive and negative regulation of mast cell/basophil degranulation are thought to be responsible for this aberrant allergic response.
Afshar R, Medoff BD, Luster AD. Allergic asthma: a tale of many T cells. Clin Exp Allergy. 2008;38:1847-1857.
Akdis CA, Akdis M. Mechanisms and treatment of allergic disease in the big picture of regulatory T cells. J Allergy Clin Immunol. 2009;123:735-746.
Bieber T. Atopic dermatitis. N Engl J Med. 2008;358:1483-1494.
Boyce JA, Broide D, Matsumoto K, et al. Advances in mechanisms of asthma, allergy, and immunology in 2008. J Allergy Clin Immunol. 2009;123:569-574.
De Benedetto A, Agnihothri R, McGirt LY, et al. Atopic dermatitis: a disease caused by innate immune defects? J Invest Dermatol. 2009;129:14-30.
Holgate S. Epithelium dysfunction in asthma. J Allergy Clin Immunol. 2007;120:1233-1244.
Kelly JT, Busse WW. Host immune response to rhinovirus: host mechanisms in asthma. J Allergy Clin Immunol. 2008;122:671-682.
Schauber J, Gallo R. Antimicrobial peptides and the skin immune defense system. J Allergy Clin Immunol. 2008;122:261-266.
Van Bever H, Lane B, Common J. Gene defects and allergy. BMJ. 2009;339:b1203.
Willart MAM, Lambrecht BN. The danger within: endogenous danger signals, atopy and asthma. Clin Exp Allergy. 2008;39:12-19.



