363 |
Alcoholic Liver Disease |
Chronic and excessive alcohol ingestion is one of the major causes of liver disease. The pathology of alcoholic liver disease consists of three major lesions, with the progressive injury rarely existing in a pure form: (1) fatty liver, (2) alcoholic hepatitis, and (3) cirrhosis. Fatty liver is present in >90% of daily as well as binge drinkers. A much smaller percentage of heavy drinkers will progress to alcoholic hepatitis, thought to be a precursor to cirrhosis. The prognosis of severe alcoholic liver disease is dismal; the mortality of patients with alcoholic hepatitis concurrent with cirrhosis is nearly 60% at 4 years. Although alcohol is considered a direct hepatotoxin, only between 10 and 20% of alcoholics will develop alcoholic hepatitis. The explanation for this apparent paradox is unclear but involves the complex interaction of facilitating factors, such as drinking patterns, diet, obesity, and gender. There are no diagnostic tools that can predict individual susceptibility to alcoholic liver disease.
GLOBAL CONSIDERATIONS
![]() Alcohol is the world’s third largest risk factor for disease burden. The harmful use of alcohol results in 2.5 million deaths each year. Most of the mortality attributed to alcohol is secondary to cirrhosis. Mortality from cirrhosis is declining in most Western countries, concurrent with a reduction in alcohol consumption, with the exceptions of the United Kingdom, Russia, Romania, and Hungary. These increases in cirrhosis and its complications are closely correlated with increased volume of alcohol consumed per capita population and are regardless of gender.
Alcohol is the world’s third largest risk factor for disease burden. The harmful use of alcohol results in 2.5 million deaths each year. Most of the mortality attributed to alcohol is secondary to cirrhosis. Mortality from cirrhosis is declining in most Western countries, concurrent with a reduction in alcohol consumption, with the exceptions of the United Kingdom, Russia, Romania, and Hungary. These increases in cirrhosis and its complications are closely correlated with increased volume of alcohol consumed per capita population and are regardless of gender.
ETIOLOGY AND PATHOGENESIS
Quantity and duration of alcohol intake are the most important risk factors involved in the development of alcoholic liver disease (Table 363-1). The roles of beverage type(s), i.e. wine, beer, or spirits, and pattern of drinking (daily versus binge drinking) are less clear. Progress beyond the fatty liver stage seems to require additional risk factors that remain incompletely defined. Although there are genetic predispositions for alcoholism (Chap. 467), gender is a strong determinant for alcoholic liver disease. Women are more susceptible to alcoholic liver injury when compared to men. They develop advanced liver disease with substantially less alcohol intake. In general, the time it takes to develop liver disease is directly related to the amount of alcohol consumed. It is useful in estimating alcohol consumption to understand that one beer, four ounces of wine, or one ounce of 80% spirits all contain ~12 g of alcohol. The threshold for developing alcoholic liver disease is higher in men, while women are at increased risk for developing similar degrees of liver injury by consuming significantly less. Gender-dependent differences result from poorly understood effects of estrogen, proportion of body fat, and the gastric metabolism of alcohol. Obesity, a high-fat diet, and the protective effect of coffee have been postulated to play a part in the development of the pathogenic process.
|
RISK FACTORS FOR ALCOHOLIC LIVER DISEASE |

Chronic infection with hepatitis C virus (HCV) (Chap. 362) is an important comorbidity in the progression of alcoholic liver disease to cirrhosis in chronic and excessive drinkers. Even moderate alcohol intake of 20–50 g/d increases the risk of cirrhosis and hepatocellular cancer in HCV-infected individuals. Patients with both alcoholic liver injury and HCV infection develop decompensated liver disease at a younger age and have poorer overall survival. Increased liver iron stores and, rarely, porphyria cutanea tarda can occur as a consequence of the overlapping injurious processes secondary to alcohol abuse and HCV infection. In addition, alcohol intake of >50 g/d by HCV-infected patients decreases the efficacy of interferon-based antiviral therapy.
The pathogenesis of alcoholic liver injury is unclear. The present conceptual foundation is that alcohol acts as a direct hepatotoxin and that malnutrition does not have a major role. Ingestion of alcohol initiates an inflammatory cascade by its metabolism to acetaldehyde, resulting in a variety of metabolic responses. Steatosis from lipogenesis, fatty acid synthesis, and depression of fatty acid oxidation appears secondary to effects on sterol regulatory transcription factor and peroxisome proliferator-activated receptor α (PPAR-α). Intestinal-derived endotoxin initiates a pathogenic process through toll-like receptor 4 and tumor necrosis factor α (TNF-α) that facilitates hepatocyte apoptosis and necrosis. The cell injury and endotoxin release initiated by ethanol and its metabolites also activate innate and adaptive immunity pathways releasing proinflammatory cytokines (e.g., TNF-α), chemokines, and proliferation of T and B cells. The production of toxic protein-aldehyde adducts, generation of reducing equivalents, and oxidative stress also contribute to the liver injury. Hepatocyte injury and impaired regeneration following chronic alcohol ingestion are ultimately associated with stellate cell activation and collagen production, which are key events in fibrogenesis. The resulting fibrosis from continuing alcohol use determines the architectural derangement of the liver and associated pathophysiology.
PATHOLOGY
The liver has a limited repertoire in response to injury. Fatty liver is the initial and most common histologic response to hepatotoxic stimuli, including excessive alcohol ingestion. The accumulation of fat within the perivenular hepatocytes coincides with the location of alcohol dehydrogenase, the major enzyme responsible for alcohol metabolism. Continuing alcohol ingestion results in fat accumulation throughout the entire hepatic lobule. Despite extensive fatty change and distortion of the hepatocytes with macrovesicular fat, the cessation of drinking results in normalization of hepatic architecture and fat content. Alcoholic fatty liver has traditionally been regarded as entirely benign, but similar to the spectrum of nonalcoholic fatty liver disease (Chap. 367e), the appearance of steatohepatitis and certain pathologic features such as giant mitochondria, perivenular fibrosis, and macrovesicular fat may be associated with progressive liver injury.
The transition between fatty liver and the development of alcoholic hepatitis is blurred. The hallmark of alcoholic hepatitis is hepatocyte injury characterized by ballooning degeneration, spotty necrosis, polymorphonuclear infiltrate, and fibrosis in the perivenular and perisinusoidal space of Disse. Mallory-Denk bodies are often present in florid cases but are neither specific nor necessary to establish the diagnosis. Alcoholic hepatitis is thought to be a precursor to the development of cirrhosis. However, like fatty liver, it is potentially reversible with cessation of drinking. Cirrhosis is present in up to 50% of patients with biopsy-proven alcoholic hepatitis, and its regression is uncertain, even with abstention.
CLINICAL FEATURES
The clinical manifestations of alcoholic fatty liver are subtle and characteristically detected as a consequence of the patient’s visit for a seemingly unrelated matter. Previously unsuspected hepatomegaly is often the only clinical finding. Occasionally, patients with fatty liver will present with right upper quadrant discomfort, nausea, and, rarely, jaundice. Differentiation of alcoholic fatty liver from nonalcoholic fatty liver is difficult unless an accurate drinking history is ascertained. In every instance where liver disease is present, a thoughtful and sensitive drinking history should be obtained. Standard, validated questions accurately detect alcohol-related problems (Chap. 467). Alcoholic hepatitis is associated with a wide gamut of clinical features. Fever, spider nevi, jaundice, and abdominal pain simulating an acute abdomen represent the extreme end of the spectrum, while many patients will be entirely asymptomatic. Portal hypertension, ascites, or variceal bleeding can occur in the absence of cirrhosis. Recognition of the clinical features of alcoholic hepatitis is central to the initiation of an effective and appropriate diagnostic and therapeutic strategy. It is important to recognize that patients with alcoholic cirrhosis often exhibit clinical features identical to other causes of cirrhosis.
LABORATORY FEATURES
Patients with alcoholic liver disease are often identified through routine screening tests. The typical laboratory abnormalities seen in fatty liver are nonspecific and include modest elevations of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and γ-glutamyl transpeptidase (GGTP), often accompanied by hypertriglyceridemia and hyperbilirubinemia. In alcoholic hepatitis and in contrast to other causes of fatty liver, AST and ALT are usually elevated two- to sevenfold. They are rarely >400 IU, and the AST/ALT ratio is >1 (Table 363-2). Hyperbilirubinemia is accompanied by modest increases in the alkaline phosphatase level. Derangement in hepatocyte synthetic function indicates more serious disease. Hypoalbuminemia and coagulopathy are common in advanced liver injury. Ultrasonography is useful in detecting fatty infiltration of the liver and determining liver size. The demonstration by ultrasound of portal vein flow reversal, ascites, and intraabdominal venous collaterals indicates serious liver injury with less potential for complete reversal.
|
LABORATORY DIAGNOSIS OF ALCOHOLIC FATTY LIVER AND ALCOHOLIC HEPATITIS |

PROGNOSIS
Critically ill patients with alcoholic hepatitis have short-term (30-day) mortality rates >50%. Severe alcoholic hepatitis is heralded by coagulopathy (prothrombin time increased >5 s), anemia, serum albumin concentrations <25 g/L (2.5 mg/dL), serum bilirubin levels >137 μmol/L (8 mg/dL), renal failure, and ascites. A discriminant function calculated as 4.6 × (the prolongation of the prothrombin time above control [seconds]) + serum bilirubin (mg/dL) can identify patients with a poor prognosis (discriminant function >32). A Model for End-Stage Liver Disease (MELD) score (Chap. 368) ≥21 also is associated with significant mortality in alcoholic hepatitis. The presence of ascites, variceal hemorrhage, deep encephalopathy, or hepatorenal syndrome predicts a dismal prognosis. The pathologic stage of the injury can be helpful in predicting prognosis. Liver biopsy should be performed whenever possible to establish the diagnosis and to guide the therapeutic decisions.
|
TREATMENT |
ALCOHOLIC LIVER DISEASE |
Complete abstinence from alcohol is the cornerstone in the treatment of alcoholic liver disease. Improved survival and the potential for reversal of histologic injury regardless of the initial clinical presentation are associated with total avoidance of alcohol ingestion. Referral of patients to experienced alcohol counselors and/or alcohol treatment programs should be routine in the management of patients with alcoholic liver disease. Attention should be directed to the nutritional and psychosocial states during the evaluation and treatment periods. Because of data suggesting that the pathogenic mechanisms in alcoholic hepatitis involve cytokine release and the perpetuation of injury by immunologic processes, glucocorticoids have been extensively evaluated in the treatment of alcoholic hepatitis. Patients with severe alcoholic hepatitis, defined as a discriminant function >32 or MELD >20, should be given prednisone, 40 mg/d, or prednisolone, 32 mg/d, for 4 weeks, followed by a steroid taper (Fig. 363-1). Exclusion criteria include active gastrointestinal bleeding, renal failure, or pancreatitis. Women with encephalopathy from severe alcoholic hepatitis may be particularly good candidates for glucocorticoids. A Lille score >0.45, at http://www.lillemodel.com, uses pretreatment variables plus the change in total bilirubin at day 7 of glucocorticoids to identify patients unresponsive to therapy.
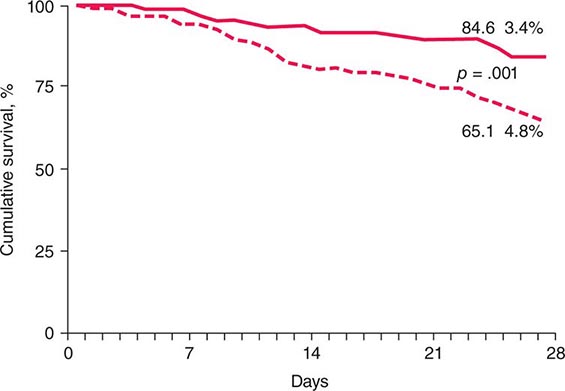
FIGURE 363-1 Effect of glucocorticoid therapy of severe alcoholic hepatitis on short-term survival: the result of a meta-analysis of individual data from three studies. Prednisolone, solid line; placebo, dotted line. (Adapted from P Mathurin et al: J Hepatol 36:480, 2002, with permission from Elsevier Science.)
The role of TNF-α expression and receptor activity in alcoholic liver injury has led to an examination of TNF inhibition as an alternative to glucocorticoids for severe alcoholic hepatitis. The nonspecific TNF inhibitor, pentoxifylline, demonstrated improved survival in the therapy of severe alcoholic hepatitis, primarily due to a decrease in hepatorenal syndrome (Fig. 363-2). Monoclonal antibodies that neutralize serum TNF-α should not be used in alcoholic hepatitis because of studies reporting increased deaths secondary to infection and renal failure.
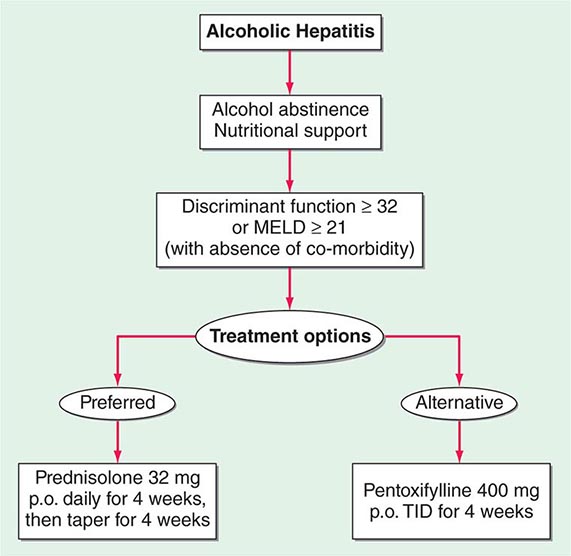
FIGURE 363-2 Treatment algorithm for alcoholic hepatitis. As identified by a calculated discriminant function >32 (see text), patients with severe alcoholic hepatitis, without the presence of gastrointestinal bleeding or infection, would be candidates for either glucocorticoids or pentoxifylline administration.
Liver transplantation is an accepted indication for treatment in selected and motivated patients with end-stage cirrhosis. Outcomes are equal or superior to other indications for transplantation. In general, transplant candidacy should be reevaluated after a defined period of sobriety. Patients presenting with alcoholic hepatitis have been largely excluded from transplant candidacy because of the perceived risk of increased surgical mortality and high rates of recidivism following transplantation. Recently, a European multidisciplinary group has reported excellent long-term transplant outcomes in highly selected patients with florid alcoholic hepatitis. General application of transplantation in such patients must await confirmatory outcomes by others.
364 |
Nonalcoholic Fatty Liver Diseases and Nonalcoholic Steatohepatitis |
INCIDENCE, PREVALENCE, AND NATURAL HISTORY
Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in many parts of the world, including the United States. Population-based abdominal imaging studies have demonstrated fatty liver in at least 25% of American adults. Because the vast majority of these subjects deny hazardous levels of alcohol consumption (defined as greater than one drink per day in women or two drinks per day in men), they are considered to have NAFLD. NAFLD is strongly associated with overweight/obesity and insulin resistance. However, it can also occur in lean individuals and is particularly common in those with a paucity of adipose depots (i.e., lipodystrophy). Ethnic/racial factors also appear to influence liver fat accumulation; the documented prevalence of NAFLD is lowest in African Americans (~25%), highest in Americans of Hispanic ancestry (~50%), and intermediate in American whites (~33%).
NAFLD encompasses a spectrum of liver pathology with different clinical prognoses. The simple accumulation of triglyceride within hepatocytes (hepatic steatosis) is on the most clinically benign extreme of the spectrum. On the opposite, most clinically ominous extreme, are cirrhosis (Chap. 365) and primary liver cancer (Chap. 111). The risk of developing cirrhosis is extremely low in individuals with chronic hepatic steatosis, but increases as steatosis becomes complicated by histologically conspicuous hepatocyte death and inflammation (i.e., nonalcoholic steatohepatitis [NASH]). NASH itself is also a heterogeneous condition; sometimes it improves to steatosis or normal histology, sometimes it remains relatively stable for years, but sometimes it results in progressive accumulation of fibrous scar that eventuates in cirrhosis. Once NAFLD-related cirrhosis develops, the annual incidence of primary liver cancer is 1%.
Abdominal imaging is not able to determine which individuals with NAFLD have associated liver cell death and inflammation (i.e., NASH), and specific blood tests to diagnose NASH are not yet available. However, population-based studies that have used elevated serum ALT as a marker of liver injury indicate that about 6–8% of American adults have serum ALT elevations that cannot be explained by excessive alcohol consumption, other known causes of fatty liver disease (Table 364-1), viral hepatitis, or drug-induced or congenital liver diseases. Because the prevalence of such “cryptogenic” ALT elevations increases with body mass index, it is presumed that they are due to NASH. Hence, at any given point in time, NASH is present in about 25% of individuals who have NAFLD (i.e., about 6% of the general U.S. adult population has NASH). Smaller cross-sectional studies in which liver biopsies have been performed on NASH patients at tertiary referral centers consistently demonstrate advanced fibrosis or cirrhosis in about 25% of those cohorts. By extrapolation, therefore, cirrhosis develops in about 6% of individuals with NAFLD (i.e., in about 1.5–2% of the general U.S. population). The risk for advanced liver fibrosis is highest in individuals with NASH who are older than 45–50 years of age and overweight/obese or afflicted with type 2 diabetes.
|
ALTERNATIVE CAUSES OF HEPATIC STEATOSIS |
To put these data in perspective, it is helpful to recall that the prevalence of hepatitis C–related cirrhosis in the United States is about 0.5%. Thus, NAFLD-related cirrhosis is about three to four times more common than cirrhosis caused by chronic hepatitis C infection. Consistent with these data, experts have predicted that NAFLD will surpass hepatitis C as the leading indication for liver transplantation in the United States within the next decade. Similar to cirrhosis caused by other liver diseases, cirrhosis caused by NAFLD increases the risk for primary liver cancer. Both hepatocellular carcinoma and intrahepatic cholangiocarcinoma (ICC) have also been reported to occur in NAFLD patients without cirrhosis, suggesting that NAFLD per se may be a premalignant condition. NAFLD, NASH, and NAFLD-related cirrhosis are not limited to adults. All have been well documented in children. As in adults, obesity and insulin resistance are the main risk factors for pediatric NAFLD. Thus, the rising incidence and prevalence of childhood obesity suggests that NAFLD is likely to become an even greater contributor to society’s burden of liver disease in the future.
PATHOGENESIS
The mechanisms underlying the pathogenesis and progression of NAFLD are not entirely clear. The best-understood mechanisms pertain to hepatic steatosis. This is proven to result when hepatocyte mechanisms for triglyceride synthesis (e.g., lipid uptake and de novo lipogenesis) overwhelm mechanisms for triglyceride disposal (e.g., degradative metabolism and lipoprotein export), leading to accumulation of fat (i.e., triglyceride) within hepatocytes. Obesity stimulates hepatocyte triglyceride accumulation by altering the intestinal microbiota to enhance both energy harvest from dietary sources and intestinal permeability. Reduced intestinal barrier function increases hepatic exposure to gut-derived products, which stimulate liver cells to generate inflammatory mediators that inhibit insulin actions. Obese adipose depots also produce excessive soluble factors (adipokines) that inhibit tissue insulin sensitivity. Insulin resistance promotes hyperglycemia. This drives the pancreas to produce more insulin to maintain glucose homeostasis. However, hyperinsulinemia also promotes lipid uptake, fat synthesis, and fat storage. The net result is hepatic triglyceride accumulation (i.e., steatosis).
Triglyceride per se is not hepatotoxic. However, its precursors (e.g., fatty acids and diacylglycerols) and metabolic by-products (e.g., reactive oxygen species) may damage hepatocytes, leading to hepatocyte lipotoxicity. Lipotoxicity also triggers the generation of other factors (e.g., inflammatory cytokines, hormonal mediators) that deregulate systems that normally maintain hepatocyte viability. The net result is increased hepatocyte death. Dying hepatocytes, in turn, release various factors that trigger wound healing responses that aim to replace (regenerate) lost hepatocytes. Such repair involves transient expansion of other cell types, such as myofibroblasts and progenitor cells, that make and degrade matrix, remodel the vasculature, and generate replacement hepatocytes, as well as the recruitment of immune cells that release factors that modulate liver injury and repair. NASH is the morphologic manifestation of lipotoxicity and resultant wound healing responses. Because the severity and duration of lipotoxic liver injury dictate the intensity and duration of repair, the histologic features and outcomes of NASH are variable. Cirrhosis and liver cancer are potential outcomes of chronic NASH. Cirrhosis results from futile repair, i.e., progressive accumulation of wound healing cells, fibrous matrix, and abnormal vasculature (scarring), rather than efficient reconstruction/regeneration of healthy hepatic parenchyma. Primary liver cancers develop when malignantly transformed liver cells escape mechanisms that normally control regenerative growth. The mechanisms responsible for futile repair (cirrhosis) and liver carcinogenesis are not well understood. Because normal liver regeneration is a very complex process, there are multiple opportunities for deregulation and, thus, pathogenic heterogeneity. To date, this heterogeneity has confounded development of both diagnostic tests and treatments for defective/deregulated liver repair (i.e., cirrhosis and cancer). Hence, current strategies focus on circumventing misrepair by preventing and/or reducing lipotoxic liver injury.
DIAGNOSIS
Diagnosing NAFLD requires demonstration of increased liver fat in the absence of hazardous levels of alcohol consumption. Thresholds for potentially dangerous alcohol ingestion have been set at more than one drink per day in women and two drinks per day in men based on epidemiologic evidence that the prevalence of serum aminotransferase elevations increases when alcohol consumption habitually exceeds these levels. In those studies, one drink was defined as having 10 g of ethanol and, thus, is equivalent to one can of beer, 4 ounces of wine, or 1.5 ounces (one shot) of distilled spirits. Other causes of liver fat accumulation (particularly exposure to certain drugs; Table 364-2) and liver injury (e.g., viral hepatitis, autoimmune liver disease, iron or copper overload, α1 antitrypsin deficiency) must also be excluded. Thus, establishing the diagnosis of NAFLD does not require invasive testing: it can be accomplished by history and physical examination, liver imaging (ultrasound is an acceptable first-line test; computed tomography [CT] or magnetic resonance imaging [MRI] enhances sensitivity for liver fat detection but adds expense), and blood tests to exclude other liver diseases. It is important to emphasize that the liver may not be enlarged, and serum aminotransferases and liver function tests (e.g., bilirubin, albumin, prothrombin time) may be completely normal, in individuals with NAFLD. Because there is yet no one specific blood test for NAFLD, confidence in the diagnosis of NAFLD is increased by identification of NAFLD risk factors. The latter include increased body mass index, insulin resistance/type 2 diabetes mellitus, and other parameters indicative of the metabolic syndrome (e.g., systemic hypertension, dyslipidemia, hyperuricemia/gout, cardiovascular disease; Chap. 422) in the patient or family members.
|
MEDICATIONS ASSOCIATED WITH HEPATIC STEATOSIS |
Establishing the severity of NAFLD-related liver injury and related scarring (i.e., staging NAFLD) is more difficult than simply diagnosing NAFLD. Staging is critically important, however, because it is necessary to define prognosis and thereby determine treatment recommendations. The goal of staging is to distinguish patients with NASH from those with simple steatosis and to identify which of the NASH patients have advanced fibrosis. The 10-year probability of developing liver-related morbidity or mortality in steatosis is negligible, and hence, this subgroup of NAFLD patients tends to be managed conservatively (see below). In contrast, more intensive follow-up and therapy are justified in NASH patients, and the subgroup with advanced fibrosis merits the most intensive scrutiny and intervention because their 10-year risk of liver-related morbidity and mortality is clearly increased.
Staging approaches can be separated into noninvasive testing (i.e., blood testing, physical examination, and imaging) and invasive approaches (i.e., liver biopsy). Blood test evidence of hepatic dysfunction (e.g., hyperbilirubinemia, hypoalbuminemia, prothrombin time prolongation) or portal hypertension (e.g., thrombocytopenia) and stigmata of portal hypertension on physical examination (e.g., spider angiomata, palmar erythema, splenomegaly, ascites, clubbing, encephalopathy) suggest a diagnosis of advanced NAFLD. Currently, however, liver biopsy is the gold standard for establishing the severity of liver injury and fibrosis because it is both more sensitive and specific than these other tests for establishing NAFLD severity. Although invasive, liver biopsy is seldom complicated by serious adverse sequelae such as significant bleeding, pain, or inadvertent puncture of other organs and thus is relatively safe. However, biopsy suffers from potential sampling error unless tissue cores of 2 cm or longer are acquired. Also, examination of tissue at a single point in time is not reliable for determining whether the pathologic processes are progressing or regressing. The risk of serial liver biopsies within short time intervals is generally deemed as unacceptable outside of research studies. These limitations of liver biopsy have stimulated efforts to develop noninvasive approaches to stage NAFLD. As is true for many other types of chronic liver disease, in NAFLD the levels of serum aminotransferases (aspartate aminotransferase [AST] and alanine aminotransferase [ALT]) do not reliably reflect the severity of liver cell injury, extent of liver cell death, or related liver inflammation and fibrosis. Thus, they are imperfect for determining which individuals with NAFLD have NASH. This has stimulated research to identify superior markers of liver injury. Serum levels of keratin 8 and keratin 18 appear to be promising surrogates. Keratins 8 and 18 (K8/18) are epithelial cytoskeletal proteins that undergo cleavage during programmed cell death (apoptosis). Both cleaved and full-length K8/18 are released into the blood as hepatocytes die, and studies suggest that serum levels of K8/18 differentiate individuals with NASH from those with simple steatosis or normal livers more reliably than do serum aminotransferase levels. Moreover, K8/18 levels appear to parallel the severity of liver fibrosis, with higher levels marking individuals who are likely to have worse scarring (i.e., advanced liver fibrosis or cirrhosis). While promising, testing for K8/18 has not yet become standard clinical practice. Other blood tests and imaging approaches that quantify liver fibrosis are also being developed. Recently, the U.S. Food and Drug Administration (FDA) approved an ultrasound-based test that measures liver stiffness as a surrogate marker of fibrosis (FibroScan®) (Chap. 358). This new tool will likely be used serially to monitor fibrosis progression and regression in NAFLD patients. Studies that compare the receiver operator characteristics of K8/18 plus FibroScan® versus liver biopsy for monitoring NAFLD evolution are forthcoming.
CLINICAL FEATURES OF NAFLD
Most subjects with NAFLD are asymptomatic. The diagnosis is often made when abnormal liver aminotransferases or features of fatty liver are noted during an evaluation performed for other reasons. NAFLD may also be diagnosed during the workup of vague right upper quadrant abdominal pain, hepatomegaly, or an abnormal-appearing liver at time of abdominal surgery. Obesity is present in 50–90% of subjects. Most patients with NAFLD also have other features of the metabolic syndrome (Chap. 422). Some have subtle stigmata of chronic liver disease, such as spider angiomata, palmer erythema, or splenomegaly. In a small minority of patients with advanced NAFLD, complications of end-stage liver disease (e.g., jaundice, features of portal hypertension such as ascites or variceal hemorrhage) may be the initial findings.
The association of NAFLD with obesity, diabetes, hypertriglyceridemia, hypertension, and cardiovascular disease is well known. Other associations include chronic fatigue, mood alterations, obstructive sleep apnea, thyroid dysfunction, and chronic pain syndrome. NAFLD is an independent risk factor for metabolic syndrome (Chap. 422). Longitudinal studies suggest that patients with NASH are at two- to threefold increased risk for the development of metabolic syndrome. Similarly, studies have shown that patients with NASH have a higher risk for the development of hypertension and diabetes mellitus. The presence of NAFLD is also independently associated with endothelial dysfunction, increased carotid intimal thickness, and the number of plaques in carotid and coronary arteries. Such data indicate that NAFLD has many deleterious effects on health in general.
TREATMENT OF NAFLD
Treatment of NAFLD can be divided into three components: (1) specific therapy of NAFLD-related liver disease; (2) treatment of NAFLD-associated comorbidities; and (3) treatment of the complications of advanced NAFLD. The subsequent discussion focuses on specific therapies for NAFLD, with some mention of their impact on major NAFLD comorbidities (insulin resistance/diabetes, obesity, and dyslipidemia). Treatment of the complications of advanced NAFLD involves management of the complications of cirrhosis and portal hypertension, including primary liver cancers. Approaches to accomplish these objectives are similar to those used in other chronic liver diseases and are covered elsewhere in the textbook (Chaps. 365 and 111).
At present, there are no FDA-approved therapies for the treatment of NAFLD. Thus, the current approach to NAFLD management focuses on treatment to improve the risk factors for NASH (i.e., obesity, insulin resistance, metabolic syndrome, dyslipidemia). Based on our understanding of the natural history of NAFLD, only patients with NASH or those with features of hepatic fibrosis on liver biopsy are considered currently for targeted pharmacologic therapies. This approach may change as our understanding of disease pathophysiology improves and potential targets of therapy evolve.
Diet and Exercise Lifestyle changes and dietary modification are the foundation for NAFLD treatment. Many studies indicate that lifestyle modification can improve serum aminotransferases and hepatic steatosis, with loss of at least 3–5% of body weight improving steatosis, but greater weight loss (up to 10%) necessary to improve steatohepatitis. The benefits of different dietary macronutrient contents (e.g., low-carbohydrate vs low-fat diets, saturated vs unsaturated fat diets) and different intensities of calorie restriction appear to be comparable. In adults with NAFLD, exercise regimens that improve fitness may be sufficient to reduce hepatic steatosis, but their impact on other aspects of liver histology remains unknown. Unfortunately, most NAFLD patients are unable to achieve sustained weight loss. Although pharmacologic therapies such as orlistat, topiramate, and phentermine to facilitate weight loss are available, their role in the treatment of NAFLD remains experimental.
Pharmacologic Therapies Several drug therapies have been tried in both research and clinical settings. No agent has yet been approved by the FDA for the treatment of NAFLD. Hence, this remains an area of active research. Because NAFLD is strongly associated with the metabolic syndrome and type 2 diabetes (Chaps. 417 and 418), the efficacy of various insulin-sensitizing agents has been examined. Metformin, an agent that mainly improves hepatic insulin sensitivity, has been evaluated in several small, open-label studies in adults and a recent larger, prospectively randomized trial in children (dubbed the TONIC study). Although several of the adult NASH studies suggested improvements in aminotransferases and/or liver histology, metformin did not improve liver histology in the TONIC study of children with NASH. Thus, it is not currently recommended as a treatment for NASH. Uncontrolled open-label studies have also investigated thiazolidinediones (pioglitazone and rosiglitazone) in adults with NASH. This class of drugs is known to improve systemic insulin resistance. Both pioglitazone and rosiglitazone reduced aminotransferases and improved some of the histologic features of NASH in small, uncontrolled studies. A large, National Institutes of Health–sponsored, randomized placebo-controlled clinical trial, the PIVENs Study (Pioglitazone vs Vitamin E vs Placebo for the Treatment of 247 Nondiabetic Adults with NASH), demonstrated that resolution of histologic NASH occurred more often in subjects treated with pioglitazone (30 mg/d) than with placebo for 18 months (47 vs 21%, p = .001). However, many subjects in the pioglitazone group gained weight, and liver fibrosis did not improve. Also, it should be noted that the long-term safety and efficacy of thiazolidinediones in patients with NASH has not been established. Five-year follow-up of subjects treated with rosiglitazone demonstrated no reduction in liver fibrosis, and rosiglitazone has been associated with increased long-term risk for cardiovascular mortality. Hence, it is not recommended as a treatment for NAFLD. Pioglitazone may be safer because in a recent large meta-analysis it was associated with reduced overall morality, myocardial infarction, and stroke. However, caution must be exercised when considering its use in patients with impaired myocardial function.
Antioxidants have also been evaluated for the treatment of NAFLD because oxidant stress is thought to contribute to the pathogenesis of NASH. Vitamin E, an inexpensive yet potent antioxidant, has been examined in several small pediatric and adult studies with varying results. In all of those studies, vitamin E was well tolerated, and most showed modest improvements in aminotransferase levels, radiographic features of hepatic steatosis, and/or histologic features of NASH. Vitamin E (800 IU/d) was also compared to placebo in the PIVENs and TONIC studies. In PIVENs, vitamin E was the only agent that achieved the predetermined primary endpoint (i.e., improvement in steatohepatitis, lobular inflammation, and steatosis score, without an increase in the fibrosis score). This endpoint was met in 43% of patients in the vitamin E group (p = .001 vs placebo), 34% in the pioglitazone group (p = .04 vs placebo), and 19% in the placebo group. Vitamin E also improved NASH histology in pediatric patients with NASH (TONIC trial). However, a recent population-based study suggested that chronic vitamin E therapy may increase the risk for cardiovascular mortality. Thus, vitamin E should only be considered as a first-line pharmacotherapy for nondiabetic NASH patients. Also, given its potentially negative effects on cardiovascular health, caution should be exercised until the risk-to-benefit ratio and long-term therapeutic efficacy of vitamin E are better defined. Ursodeoxycholic acid (a bile acid that improves certain cholestatic liver diseases) and betaine (metabolite of choline that raises SAM levels and decreases cellular oxidative damage) offer no histologic benefit over placebo in patients with NASH. Experimental evidence to support the use of omega-3 fatty acids in NAFLD exists; however, a recent large, multicenter, placebo-controlled study failed to demonstrate a histologic benefit. Other pharmacotherapies are also being evaluated in NAFLD (e.g., probiotics, farnesoid × receptor agonists, anticytokine agents, glucagon-like peptide agonists, dipeptidyl IV antagonists); however, sufficient data do not yet exist to justify their use as NASH treatments in standard clinical practice.
Statins are an important class of agents to treat dyslipidemia and decrease cardiovascular risk. There is no evidence to suggest that statins cause liver failure in patients with any chronic liver disease, including NAFLD. The incidence of liver enzyme elevations in NAFLD patients taking statins is also no different than that of healthy controls or patients with other chronic liver diseases. Moreover, several studies have suggested that statins may improve aminotransferases and histology in patients with NASH. Yet, there is continued reluctance to use statins in patients with NAFLD. The lack of evidence that statins harm the liver in NAFLD patients, combined with the increase risk for cardiovascular morbidity and mortality in NAFLD patients, warrants the use of statins to treat dyslipidemia in patients with NAFLD/NASH.
Bariatric Surgery Although interest in bariatric surgery as a treatment for NAFLD exists, a recently published Cochrane review concluded that lack of randomized clinical trials or adequate clinical studies prevents definitive assessment of benefits and harms of bariatric surgery as a treatment for NASH. Most studies of bariatric surgery have shown that bariatric surgery is generally safe in individuals with well-compensated chronic liver disease and improves hepatic steatosis and necroinflammation (i.e., features of NAFLD/NASH); however, effects on hepatic fibrosis have been variable. Concern lingers because some of the largest prospective studies suggest that hepatic fibrosis might progress after bariatric surgery. Thus, the Cochrane review deemed it premature to recommend bariatric surgery as a primary treatment for NASH. There is also general agreement that patients with NAFLD-related cirrhosis and portal hypertension should be excluded as candidates for bariatric surgery. However, given growing evidence for the benefits of bariatric surgery on metabolic syndrome complications in individuals with refractory obesity, it is not contraindicated in otherwise eligible patients with NAFLD or NASH.
Liver Transplantation Patients with NAFLD in whom end-stage liver disease develops should be evaluated for liver transplantation (Chap. 368). The outcomes of liver transplantation in well-selected patients with NAFLD are generally good, but comorbid medical conditions associated with NAFLD, such as diabetes mellitus, obesity, and cardiovascular disease, often limit transplant candidacy. NAFLD may recur after liver transplantation. The risk factors for recurrent or de novo NAFLD after liver transplantation are multifactorial and include hypertriglyceridemia, obesity, diabetes mellitus, and immunosuppressive therapies, particularly glucocorticoids.
GLOBAL HEALTH CONSIDERATIONS
![]() The epidemic of obesity is now a global and accelerating phenomenon. Worldwide, there are over 1 billion overweight adults, of whom at least 300 million are obese. In the wake of the obesity epidemic follow numerous comorbidities, including NAFLD. NAFLD is the most common liver disease identified in Western countries and the fastest rising form of chronic liver disease worldwide. Present understanding of NAFLD natural history is based mainly on studies in whites who became overweight/obese and developed the metabolic syndrome in adulthood. The impact of the global childhood obesity epidemic on NAFLD pathogenesis/progression is unknown. Emerging evidence demonstrates that advanced NAFLD, including cirrhosis and primary liver cancer, can occur in children, prompting concerns that childhood-onset NAFLD might follow a more aggressive course than typical adult-acquired NAFLD. Some of the most populated parts of the world are in the midst of industrial revolutions, and certain environmental pollutants seem to exacerbate NAFLD. Some studies also suggest that the risk for NASH and NAFLD-related cirrhosis may be higher in certain ethnic groups such as Asians, certain Hispanics, and Native Americans and lower in others such as African Americans, compared with whites. Although all of these variables confound efforts to predict the net impact of this obesity-related liver disease on global health, it seems likely that NAFLD will remain a major cause of chronic liver disease worldwide for the foreseeable future.
The epidemic of obesity is now a global and accelerating phenomenon. Worldwide, there are over 1 billion overweight adults, of whom at least 300 million are obese. In the wake of the obesity epidemic follow numerous comorbidities, including NAFLD. NAFLD is the most common liver disease identified in Western countries and the fastest rising form of chronic liver disease worldwide. Present understanding of NAFLD natural history is based mainly on studies in whites who became overweight/obese and developed the metabolic syndrome in adulthood. The impact of the global childhood obesity epidemic on NAFLD pathogenesis/progression is unknown. Emerging evidence demonstrates that advanced NAFLD, including cirrhosis and primary liver cancer, can occur in children, prompting concerns that childhood-onset NAFLD might follow a more aggressive course than typical adult-acquired NAFLD. Some of the most populated parts of the world are in the midst of industrial revolutions, and certain environmental pollutants seem to exacerbate NAFLD. Some studies also suggest that the risk for NASH and NAFLD-related cirrhosis may be higher in certain ethnic groups such as Asians, certain Hispanics, and Native Americans and lower in others such as African Americans, compared with whites. Although all of these variables confound efforts to predict the net impact of this obesity-related liver disease on global health, it seems likely that NAFLD will remain a major cause of chronic liver disease worldwide for the foreseeable future.
365 |
Cirrhosis and Its Complications |
Cirrhosis is a condition that is defined histopathologically and has a variety of clinical manifestations and complications, some of which can be life-threatening. In the past, it has been thought that cirrhosis was never reversible; however, it has become apparent that when the underlying insult that has caused the cirrhosis has been removed, there can be reversal of fibrosis. This is most apparent with the successful treatment of chronic hepatitis C; however, reversal of fibrosis is also seen in patients with hemochromatosis who have been successfully treated and in patients with alcoholic liver disease who have discontinued alcohol use.
Regardless of the cause of cirrhosis, the pathologic features consist of the development of fibrosis to the point that there is architectural distortion with the formation of regenerative nodules. This results in a decrease in hepatocellular mass, and thus function, and an alteration of blood flow. The induction of fibrosis occurs with activation of hepatic stellate cells, resulting in the formation of increased amounts of collagen and other components of the extracellular matrix.
Clinical features of cirrhosis are the result of pathologic changes and mirror the severity of the liver disease. Most hepatic pathologists provide an assessment of grading and staging when evaluating liver biopsy samples. These grading and staging schemes vary between disease states and have been developed for most conditions, including chronic viral hepatitis, nonalcoholic fatty liver disease, and primary biliary cirrhosis. Advanced fibrosis usually includes bridging fibrosis with nodularity designated as stage 3 and cirrhosis designated as stage 4. Patients who have cirrhosis have varying degrees of compensated liver function, and clinicians need to differentiate between those who have stable, compensated cirrhosis and those who have decompensated cirrhosis. Patients who have developed complications of their liver disease and have become decompensated should be considered for liver transplantation. Many of the complications of cirrhosis will require specific therapy. Portal hypertension is a significant complicating feature of decompensated cirrhosis and is responsible for the development of ascites and bleeding from esophagogastric varices, two complications that signify decompensated cirrhosis. Loss of hepatocellular function results in jaundice, coagulation disorders, and hypoalbuminemia and contributes to the causes of portosystemic encephalopathy. The complications of cirrhosis are basically the same regardless of the etiology. Nonetheless, it is useful to classify patients by the cause of their liver disease (Table 365-1); patients can be divided into broad groups with alcoholic cirrhosis, cirrhosis due to chronic viral hepatitis, biliary cirrhosis, and other, less common causes such as cardiac cirrhosis, cryptogenic cirrhosis, and other miscellaneous causes.
|
CAUSES OF CIRRHOSIS |
ALCOHOLIC CIRRHOSIS
Excessive chronic alcohol use can cause several different types of chronic liver disease, including alcoholic fatty liver, alcoholic hepatitis, and alcoholic cirrhosis. Furthermore, use of excessive alcohol can contribute to liver damage in patients with other liver diseases, such as hepatitis C, hemochromatosis, and fatty liver disease related to obesity. Chronic alcohol use can produce fibrosis in the absence of accompanying inflammation and/or necrosis. Fibrosis can be centrilobular, pericellular, or periportal. When fibrosis reaches a certain degree, there is disruption of the normal liver architecture and replacement of liver cells by regenerative nodules. In alcoholic cirrhosis, the nodules are usually <3 mm in diameter; this form of cirrhosis is referred to as micronodular. With cessation of alcohol use, larger nodules may form, resulting in a mixed micronodular and macronodular cirrhosis.
Pathogenesis Alcohol is the most commonly used drug in the United States, and more than two-thirds of adults drink alcohol each year. Thirty percent have had a binge within the past month, and over 7% of adults regularly consume more than two drinks per day. Unfortunately, more than 14 million adults in the United States meet the diagnostic criteria for alcohol abuse or dependence. In the United States, chronic liver disease is the tenth most common cause of death in adults, and alcoholic cirrhosis accounts for approximately 40% of deaths due to cirrhosis.
Ethanol is mainly absorbed by the small intestine and, to a lesser degree, through the stomach. Gastric alcohol dehydrogenase (ADH) initiates alcohol metabolism. Three enzyme systems account for metabolism of alcohol in the liver. These include cytosolic ADH, the microsomal ethanol oxidizing system (MEOS), and peroxisomal catalase. The majority of ethanol oxidation occurs via ADH to form acetaldehyde, which is a highly reactive molecule that may have multiple effects. Ultimately, acetaldehyde is metabolized to acetate by aldehyde dehydrogenase (ALDH). Intake of ethanol increases intracellular accumulation of triglycerides by increasing fatty acid uptake and by reducing fatty acid oxidation and lipoprotein secretion. Protein synthesis, glycosylation, and secretion are impaired. Oxidative damage to hepatocyte membranes occurs due to the formation of reactive oxygen species; acetaldehyde is a highly reactive molecule that combines with proteins to form protein-acetaldehyde adducts. These adducts may interfere with specific enzyme activities, including microtubular formation and hepatic protein trafficking. With acetaldehyde-mediated hepatocyte damage, certain reactive oxygen species can result in Kupffer cell activation. As a result, profibrogenic cytokines are produced that initiate and perpetuate stellate cell activation, with the resultant production of excess collagen and extracellular matrix. Connective tissue appears in both periportal and pericentral zones and eventually connects portal triads with central veins forming regenerative nodules. Hepatocyte loss occurs, and with increased collagen production and deposition, together with continuing hepatocyte destruction, the liver contracts and shrinks in size. This process generally takes from years to decades to occur and requires repeated insults.
Clinical Features The diagnosis of alcoholic liver disease requires an accurate history regarding both amount and duration of alcohol consumption. Patients with alcoholic liver disease can present with nonspecific symptoms such as vague right upper quadrant abdominal pain, fever, nausea and vomiting, diarrhea, anorexia, and malaise. Alternatively, they may present with more specific complications of chronic liver disease, including ascites, edema, or upper gastrointestinal (GI) hemorrhage. Many cases present incidentally at the time of autopsy or elective surgery. Other clinical manifestations include the development of jaundice or encephalopathy. The abrupt onset of any of these complications may be the first event prompting the patient to seek medical attention. Other patients may be identified in the course of an evaluation of routine laboratory studies that are found to be abnormal. On physical examination, the liver and spleen may be enlarged, with the liver edge being firm and nodular. Other frequent findings include scleral icterus, palmar erythema (Fig. 365-1), spider angiomas (Fig. 365-2), parotid gland enlargement, digital clubbing, muscle wasting, or the development of edema and ascites. Men may have decreased body hair and gynecomastia as well as testicular atrophy, which may be a consequence of hormonal abnormalities or a direct toxic effect of alcohol on the testes. In women with advanced alcoholic cirrhosis, menstrual irregularities usually occur, and some women may be amenorrheic. These changes are often reversible following cessation of alcohol.

FIGURE 365-1 Palmar erythema. This figure shows palmar erythema in a patient with alcoholic cirrhosis. The erythema is peripheral over the palm with central pallor.
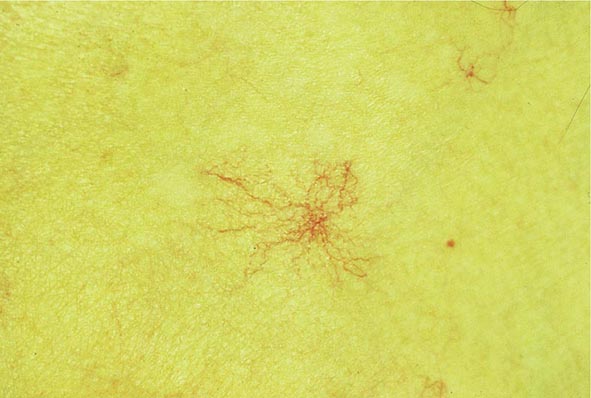
FIGURE 365-2 Spider angioma. This figure shows a spider angioma in a patient with hepatitis C cirrhosis. With release of central compression, the arteriole fills from the center and spreads out peripherally.
Laboratory tests may be completely normal in patients with early compensated alcoholic cirrhosis. Alternatively, in advanced liver disease, many abnormalities usually are present. Patients may be anemic either from chronic GI blood loss, nutritional deficiencies, or hypersplenism related to portal hypertension, or as a direct suppressive effect of alcohol on the bone marrow. A unique form of hemolytic anemia (with spur cells and acanthocytes) called Zieve’s syndrome can occur in patients with severe alcoholic hepatitis. Platelet counts are often reduced early in the disease, reflective of portal hypertension with hypersplenism. Serum total bilirubin can be normal or elevated with advanced disease. Direct bilirubin is frequently mildly elevated in patients with a normal total bilirubin, but the abnormality typically progresses as the disease worsens. Prothrombin times are often prolonged and usually do not respond to administration of parenteral vitamin K. Serum sodium levels are usually normal unless patients have ascites and then can be depressed, largely due to ingestion of excess free water. Serum alanine and aspartate aminotransferases (ALT, AST) are typically elevated, particularly in patients who continue to drink, with AST levels being higher than ALT levels, usually by a 2:1 ratio.
Diagnosis Patients who have any of the above-mentioned clinical features, physical examination findings, or laboratory studies should be considered to have alcoholic liver disease. The diagnosis, however, requires accurate knowledge that the patient is continuing to use and abuse alcohol. Furthermore, other forms of chronic liver disease (e.g., chronic viral hepatitis or metabolic or autoimmune liver diseases) must be considered or ruled out, or if present, an estimate of relative causality along with the alcohol use should be determined. Liver biopsy can be helpful to confirm a diagnosis, but generally when patients present with alcoholic hepatitis and are still drinking, liver biopsy is withheld until abstinence has been maintained for at least 6 months to determine residual, nonreversible disease.
In patients who have had complications of cirrhosis and who continue to drink, there is a <50% 5-year survival. In contrast, in patients who are able to remain abstinent, the prognosis is significantly improved. In patients with advanced liver disease, the prognosis remains poor; however, in individuals who are able to remain abstinent, liver transplantation is a viable option.
CIRRHOSIS DUE TO CHRONIC VIRAL HEPATITIS B OR C
![]() Of patients exposed to the hepatitis C virus (HCV), approximately 80% develop chronic hepatitis C, and of those, about 20–30% will develop cirrhosis over 20–30 years. Many of these patients have had concomitant alcohol use, and the true incidence of cirrhosis due to hepatitis C alone is unknown. Nonetheless, this represents a significant number of patients. It is expected that an even higher percentage will go on to develop cirrhosis over longer periods of time. In the United States, approximately 5 to 6 million people have been exposed to HCV, with about 4 million who are chronically viremic. Worldwide, about 170 million individuals have hepatitis C, with some areas of the world (e.g., Egypt) having up to 15% of the population infected. HCV is a noncytopathic virus, and liver damage is probably immune-mediated. Progression of liver disease due to chronic hepatitis C is characterized by portal-based fibrosis with bridging fibrosis and nodularity developing, ultimately culminating in the development of cirrhosis. In cirrhosis due to chronic hepatitis C, the liver is small and shrunken with characteristic features of a mixed micro- and macronodular cirrhosis seen on liver biopsy. In addition to the increased fibrosis that is seen in cirrhosis due to hepatitis C, an inflammatory infiltrate is found in portal areas with interface hepatitis and occasionally some lobular hepatocellular injury and inflammation. In patients with HCV genotype 3, steatosis is often present.
Of patients exposed to the hepatitis C virus (HCV), approximately 80% develop chronic hepatitis C, and of those, about 20–30% will develop cirrhosis over 20–30 years. Many of these patients have had concomitant alcohol use, and the true incidence of cirrhosis due to hepatitis C alone is unknown. Nonetheless, this represents a significant number of patients. It is expected that an even higher percentage will go on to develop cirrhosis over longer periods of time. In the United States, approximately 5 to 6 million people have been exposed to HCV, with about 4 million who are chronically viremic. Worldwide, about 170 million individuals have hepatitis C, with some areas of the world (e.g., Egypt) having up to 15% of the population infected. HCV is a noncytopathic virus, and liver damage is probably immune-mediated. Progression of liver disease due to chronic hepatitis C is characterized by portal-based fibrosis with bridging fibrosis and nodularity developing, ultimately culminating in the development of cirrhosis. In cirrhosis due to chronic hepatitis C, the liver is small and shrunken with characteristic features of a mixed micro- and macronodular cirrhosis seen on liver biopsy. In addition to the increased fibrosis that is seen in cirrhosis due to hepatitis C, an inflammatory infiltrate is found in portal areas with interface hepatitis and occasionally some lobular hepatocellular injury and inflammation. In patients with HCV genotype 3, steatosis is often present.
![]() Similar findings are seen in patients with cirrhosis due to chronic hepatitis B. Of adult patients exposed to hepatitis B, about 5% develop chronic hepatitis B, and about 20% of those patients will go on to develop cirrhosis. Special stains for hepatitis B core (HBc) and hepatitis B surface (HBs) antigen will be positive, and ground-glass hepatocytes signifying hepatitis B surface antigen (HBsAg) may be present. In the United States, there are about 2 million carriers of hepatitis B, whereas in other parts of the world where hepatitis B virus (HBV) is endemic (i.e., Asia, Southeast Asia, sub-Saharan Africa), up to 15% of the population may be infected, having acquired the infection vertically at the time of birth. Thus, over 300–400 million individuals are thought to have hepatitis B worldwide. Approximately 25% of these individuals may ultimately develop cirrhosis.
Similar findings are seen in patients with cirrhosis due to chronic hepatitis B. Of adult patients exposed to hepatitis B, about 5% develop chronic hepatitis B, and about 20% of those patients will go on to develop cirrhosis. Special stains for hepatitis B core (HBc) and hepatitis B surface (HBs) antigen will be positive, and ground-glass hepatocytes signifying hepatitis B surface antigen (HBsAg) may be present. In the United States, there are about 2 million carriers of hepatitis B, whereas in other parts of the world where hepatitis B virus (HBV) is endemic (i.e., Asia, Southeast Asia, sub-Saharan Africa), up to 15% of the population may be infected, having acquired the infection vertically at the time of birth. Thus, over 300–400 million individuals are thought to have hepatitis B worldwide. Approximately 25% of these individuals may ultimately develop cirrhosis.
Clinical Features and Diagnosis Patients with cirrhosis due to either chronic hepatitis C or B can present with the usual symptoms and signs of chronic liver disease. Fatigue, malaise, vague right upper quadrant pain, and laboratory abnormalities are frequent presenting features. Diagnosis requires a thorough laboratory evaluation, including quantitative HCV RNA testing and analysis for HCV genotype, or hepatitis B serologies to include HBsAg, anti-HBs, HBeAg (hepatitis B e antigen), anti-HBe, and quantitative HBV DNA levels.
CIRRHOSIS FROM AUTOIMMUNE HEPATITIS AND NONALCOHOLIC FATTY LIVER DISEASE
Other causes of posthepatitic cirrhosis include autoimmune hepatitis and cirrhosis due to nonalcoholic steatohepatitis. Many patients with autoimmune hepatitis (AIH) present with cirrhosis that is already established. Typically, these patients will not benefit from immunosuppressive therapy with glucocorticoids or azathioprine because the AIH is “burned out.” In this situation, liver biopsy does not show a significant inflammatory infiltrate. Diagnosis in this setting requires positive autoimmune markers such as antinuclear antibody (ANA) or anti-smooth-muscle antibody (ASMA). When patients with AIH present with cirrhosis and active inflammation accompanied by elevated liver enzymes, there can be considerable benefit from the use of immunosuppressive therapy.
Patients with nonalcoholic steatohepatitis are increasingly being found to have progressed to cirrhosis. With the epidemic of obesity that continues in Western countries, more and more patients are identified with nonalcoholic fatty liver disease (Chap. 364). Of these, a significant subset has nonalcoholic steatohepatitis and can progress to increased fibrosis and cirrhosis. Over the past several years, it has been increasingly recognized that many patients who were thought to have cryptogenic cirrhosis in fact have nonalcoholic steatohepatitis. As their cirrhosis progresses, they become catabolic and then lose the telltale signs of steatosis seen on biopsy. Management of complications of cirrhosis due to either AIH or nonalcoholic steatohepatitis is similar to that for other forms of cirrhosis.
BILIARY CIRRHOSIS
Biliary cirrhosis has pathologic features that are different from either alcoholic cirrhosis or posthepatitic cirrhosis, yet the manifestations of end-stage liver disease are the same. Cholestatic liver disease may result from necroinflammatory lesions, congenital or metabolic processes, or external bile duct compression. Thus, two broad categories reflect the anatomic sites of abnormal bile retention: intrahepatic and extrahepatic. The distinction is important for obvious therapeutic reasons. Extrahepatic obstruction may benefit from surgical or endoscopic biliary tract decompression, whereas intrahepatic cholestatic processes will not improve with such interventions and require a different approach.
The major causes of chronic cholestatic syndromes are primary biliary cirrhosis (PBC), autoimmune cholangitis (AIC), primary sclerosing cholangitis (PSC), and idiopathic adulthood ductopenia. These syndromes are usually clinically distinguished from each other by antibody testing, cholangiographic findings, and clinical presentation. However, they all share the histopathologic features of chronic cholestasis, such as cholate stasis; copper deposition; xanthomatous transformation of hepatocytes; and irregular, so-called biliary fibrosis. In addition, there may be chronic portal inflammation, interface activity, and chronic lobular inflammation. Ductopenia is a result of this progressive disease as patients develop cirrhosis.
PRIMARY BILIARY CIRRHOSIS
PBC is seen in about 100–200 individuals per million, with a strong female preponderance and a median age of around 50 years at the time of diagnosis. The cause of PBC is unknown; it is characterized by portal inflammation and necrosis of cholangiocytes in small- and medium-sized bile ducts. Cholestatic features prevail, and biliary cirrhosis is characterized by an elevated bilirubin level and progressive liver failure. Liver transplantation is the treatment of choice for patients with decompensated cirrhosis due to PBC. A variety of therapies have been proposed, but ursodeoxycholic acid (UDCA) is the only approved treatment that has some degree of efficacy by slowing the rate of progression of the disease.
Antimitochondrial antibodies (AMA) are present in about 90% of patients with PBC. These autoantibodies recognize intermitochondrial membrane proteins that are enzymes of the pyruvate dehydrogenase complex (PDC), the branched-chain 2-oxoacid dehydrogenase complex, and the 2-oxogluterate dehydrogenase complex. Most relate to pyruvate dehydrogenase. These autoantibodies are not pathogenic but rather are useful markers for making a diagnosis of PBC.
Pathology Histopathologic analyses of liver biopsies of patients with PBC have resulted in identifying four distinct stages of the disease as it progresses. The earliest lesion is termed chronic nonsuppurative destructive cholangitis and is a necrotizing inflammatory process of the portal tracts. Medium and small bile ducts are infiltrated with lymphocytes and undergo duct destruction. Mild fibrosis and sometimes bile stasis can occur. With progression, the inflammatory infiltrate becomes less prominent, but the number of bile ducts is reduced and there is proliferation of smaller bile ductules. Increased fibrosis ensues with the expansion of periportal fibrosis to bridging fibrosis. Finally, cirrhosis, which may be micronodular or macronodular, develops.
Clinical Features Currently, most patients with PBC are diagnosed well before the end-stage manifestations of the disease are present, and, as such, most patients are actually asymptomatic. When symptoms are present, they most prominently include a significant degree of fatigue out of proportion to what would be expected for either the severity of the liver disease or the age of the patient. Pruritus is seen in approximately 50% of patients at the time of diagnosis, and it can be debilitating. It might be intermittent and usually is most bothersome in the evening. In some patients, pruritus can develop toward the end of pregnancy, and there are examples of patients having been diagnosed with cholestasis of pregnancy rather than PBC. Pruritus that presents prior to the development of jaundice indicates severe disease and a poor prognosis.
Physical examination can show jaundice and other complications of chronic liver disease, including hepatomegaly, splenomegaly, ascites, and edema. Other features that are unique to PBC include hyperpigmentation, xanthelasma, and xanthomata, which are related to the altered cholesterol metabolism seen in this disease. Hyperpigmentation is evident on the trunk and the arms and is seen in areas of exfoliation and lichenification associated with progressive scratching related to the pruritus. Bone pain resulting from osteopenia or osteoporosis is occasionally seen at the time of diagnosis.
Laboratory Findings Laboratory findings in PBC show cholestatic liver enzyme abnormalities with an elevation in γ-glutamyl transpeptidase and alkaline phosphatase (ALP) along with mild elevations in aminotransferases (ALT and AST). Immunoglobulins, particularly IgM, are typically increased. Hyperbilirubinemia usually is seen once cirrhosis has developed. Thrombocytopenia, leukopenia, and anemia may be seen in patients with portal hypertension and hypersplenism. Liver biopsy shows characteristic features as described above and should be evident to any experienced hepatopathologist. Up to 10% of patients with characteristic PBC will have features of AIH as well and are defined as having “overlap” syndrome. These patients are usually treated as PBC patients and may progress to cirrhosis with the same frequency as typical PBC patients. Some patients require immunosuppressive medications as well.
Diagnosis PBC should be considered in patients with chronic cholestatic liver enzyme abnormalities. It is most often seen in middle-aged women. AMA testing may be negative, and it should be remembered that as many as 10% of patients with PBC may be AMA-negative. Liver biopsy is most important in this setting of AMA-negative PBC. In patients who are AMA-negative with cholestatic liver enzymes, PSC should be ruled out by way of cholangiography.
PRIMARY SCLEROSING CHOLANGITIS
As in PBC, the cause of PSC remains unknown. PSC is a chronic cholestatic syndrome that is characterized by diffuse inflammation and fibrosis involving the entire biliary tree, resulting in chronic cholestasis. This pathologic process ultimately results in obliteration of both the intra- and extrahepatic biliary tree, leading to biliary cirrhosis, portal hypertension, and liver failure. The cause of PSC remains unknown despite extensive investigation into various mechanisms related to bacterial and viral infections, toxins, genetic predisposition, and immunologic mechanisms, all of which have been postulated to contribute to the pathogenesis and progression of this syndrome.
Pathologic changes that can occur in PSC show bile duct proliferation as well as ductopenia and fibrous cholangitis (pericholangitis). Often, liver biopsy changes in PSC are not pathognomonic, and establishing the diagnosis of PSC must involve imaging of the biliary tree. Periductal fibrosis is occasionally seen on biopsy specimens and can be quite helpful in making the diagnosis. As the disease progresses, biliary cirrhosis is the final, end-stage manifestation of PSC.
Clinical Features The usual clinical features of PSC are those found in cholestatic liver disease, with fatigue, pruritus, steatorrhea, deficiencies of fat-soluble vitamins, and the associated consequences. As in PBC, the fatigue is profound and nonspecific. Pruritus can often be debilitating and is related to the cholestasis. The severity of pruritus does not correlate with the severity of the disease. Metabolic bone disease, as seen in PBC, can occur with PSC and should be treated (see above).
Laboratory Findings Patients with PSC typically are identified in the course of an evaluation of abnormal liver enzymes. Most patients have at least a twofold increase in ALP and may have elevated aminotransferases as well. Albumin levels may be decreased, and prothrombin times are prolonged in a substantial proportion of patients at the time of diagnosis. Some degree of correction of a prolonged prothrombin time may occur with parenteral vitamin K. A small subset of patients have aminotransferase elevations greater than five times the upper limit of normal and may have features of AIH on biopsy. These individuals are thought to have an overlap syndrome between PSC and AIH. Autoantibodies are frequently positive in patients with the overlap syndrome but are typically negative in patients who only have PSC. One autoantibody, the perinuclear antineutrophil cytoplasmic antibody (p-ANCA), is positive in about 65% of patients with PSC. Over 50% of patients with PSC also have ulcerative colitis (UC); accordingly, once a diagnosis of PSC is established, colonoscopy should be performed to look for evidence of UC.
Diagnosis The definitive diagnosis of PSC requires cholangiographic imaging. Over the last several years, magnetic resonance imaging (MRI) with magnetic resonance cholangiopancreatography (MRCP) has been used as the imaging technique of choice for initial evaluation. Once patients are screened in this manner, some investigators feel that endoscopic retrograde cholangiopancreatography (ERCP) should also be performed to be certain whether or not a dominant stricture is present. Typical cholangiographic findings in PSC are multifocal stricturing and beading involving both the intrahepatic and extrahepatic biliary tree. However, although involvement may be of the intrahepatic bile ducts alone or of the extrahepatic bile ducts alone, more commonly, both are involved. These strictures are typically short and with intervening segments of normal or slightly dilated bile ducts that are distributed diffusely, producing the classic beaded appearance. The gallbladder and cystic duct can be involved in up to 15% of cases. Patients with high-grade, diffuse stricturing of the intrahepatic bile ducts have an overall poor prognosis. Gradually, biliary cirrhosis develops, and patients will progress to decompensated liver disease with all the manifestations of ascites, esophageal variceal hemorrhage, and encephalopathy.
CARDIAC CIRRHOSIS
Definition Patients with long-standing right-sided congestive heart failure may develop chronic liver injury and cardiac cirrhosis. This is an increasingly uncommon, if not rare, cause of chronic liver disease given the advances made in the care of patients with heart failure.
Etiology and Pathology In the case of long-term right-sided heart failure, there is an elevated venous pressure transmitted via the inferior vena cava and hepatic veins to the sinusoids of the liver, which become dilated and engorged with blood. The liver becomes enlarged and swollen, and with long-term passive congestion and relative ischemia due to poor circulation, centrilobular hepatocytes can become necrotic, leading to pericentral fibrosis. This fibrotic pattern can extend to the periphery of the lobule outward until a unique pattern of fibrosis causing cirrhosis can occur.
Clinical Features Patients typically have signs of congestive heart failure and will manifest an enlarged firm liver on physical examination. ALP levels are characteristically elevated, and aminotransferases may be normal or slightly increased with AST usually higher than ALT. It is unlikely that patients will develop variceal hemorrhage or encephalopathy.
Diagnosis The diagnosis is usually made in someone with clear-cut cardiac disease who has an elevated ALP and an enlarged liver. Liver biopsy shows a pattern of fibrosis that can be recognized by an experienced hepatopathologist. Differentiation from Budd-Chiari syndrome (BCS) can be made by seeing extravasation of red blood cells in BCS, but not in cardiac hepatopathy. Venoocclusive disease can also affect hepatic outflow and has characteristic features on liver biopsy. Venoocclusive disease can be seen under the circumstances of conditioning for bone marrow transplant with radiation and chemotherapy; it can also be seen with the ingestion of certain herbal teas as well as pyrrolizidine alkaloids. This is typically seen in Caribbean countries and rarely in the United States. Treatment is based on management of the underlying cardiac disease.
OTHER TYPES OF CIRRHOSIS
There are several other less common causes of chronic liver disease that can progress to cirrhosis. These include inherited metabolic liver diseases such as hemochromatosis, Wilson’s disease, α1 antitrypsin (α1AT) deficiency, and cystic fibrosis. For all of these disorders, the manifestations of cirrhosis are similar, with some minor variations, to those seen in other patients with other causes of cirrhosis.
Hemochromatosis is an inherited disorder of iron metabolism that results in a progressive increase in hepatic iron deposition, which, over time, can lead to a portal-based fibrosis progressing to cirrhosis, liver failure, and hepatocellular cancer. While the frequency of hemochromatosis is relatively common, with genetic susceptibility occurring in 1 in 250 individuals, the frequency of end-stage manifestations due to the disease is relatively low, and fewer than 5% of those patients who are genotypically susceptible will go on to develop severe liver disease from hemochromatosis. Diagnosis is made with serum iron studies showing an elevated transferrin saturation and an elevated ferritin level, along with abnormalities identified by HFE mutation analysis. Treatment is straightforward, with regular therapeutic phlebotomy.
Wilson’s disease is an inherited disorder of copper homeostasis with failure to excrete excess amounts of copper, leading to an accumulation in the liver. This disorder is relatively uncommon, affecting 1 in 30,000 individuals. Wilson’s disease typically affects adolescents and young adults. Prompt diagnosis before end-stage manifestations become irreversible can lead to significant clinical improvement. Diagnosis requires determination of ceruloplasmin levels, which are low; 24-h urine copper levels, which are elevated; typical physical examination findings, including Kayser-Fleischer corneal rings; and characteristic liver biopsy findings. Treatment consists of copper-chelating medications.
α1AT deficiency results from an inherited disorder that causes abnormal folding of the α1AT protein, resulting in failure of secretion of that protein from the liver. It is unknown how the retained protein leads to liver disease. Patients with α1AT deficiency at greatest risk for developing chronic liver disease have the ZZ phenotype, but only about 10–20% of such individuals will develop chronic liver disease. Diagnosis is made by determining α1AT levels and phenotype. Characteristic periodic acid–Schiff (PAS)-positive, diastase-resistant globules are seen on liver biopsy. The only effective treatment is liver transplantation, which is curative.
Cystic fibrosis is an uncommon inherited disorder affecting whites of northern European descent. A biliary-type cirrhosis can occur, and some patients derive benefit from the chronic use of UDCA.
MAJOR COMPLICATIONS OF CIRRHOSIS
The clinical course of patients with advanced cirrhosis is often complicated by a number of important sequelae that can occur regardless of the underlying cause of the liver disease. These include portal hypertension and its consequences of gastroesophageal variceal hemorrhage, splenomegaly, ascites, hepatic encephalopathy, spontaneous bacterial peritonitis (SBP), hepatorenal syndrome, and hepatocellular carcinoma (Table 365-2).
|
COMPLICATIONS OF CIRRHOSIS |
PORTAL HYPERTENSION
Portal hypertension is defined as the elevation of the hepatic venous pressure gradient (HVPG) to >5 mmHg. Portal hypertension is caused by a combination of two simultaneously occurring hemodynamic processes: (1) increased intrahepatic resistance to the passage of blood flow through the liver due to cirrhosis and regenerative nodules, and (2) increased splanchnic blood flow secondary to vasodilation within the splanchnic vascular bed. Portal hypertension is directly responsible for the two major complications of cirrhosis: variceal hemorrhage and ascites. Variceal hemorrhage is an immediate life-threatening problem with a 20–30% mortality rate associated with each episode of bleeding. The portal venous system normally drains blood from the stomach, intestines, spleen, pancreas, and gallbladder, and the portal vein is formed by the confluence of the superior mesenteric and splenic veins. Deoxygenated blood from the small bowel drains into the superior mesenteric vein along with blood from the head of the pancreas, the ascending colon, and part of the transverse colon. Conversely, the splenic vein drains the spleen and the pancreas and is joined by the inferior mesenteric vein, which brings blood from the transverse and descending colon as well as from the superior two-thirds of the rectum. Thus, the portal vein normally receives blood from almost the entire GI tract.
The causes of portal hypertension are usually subcategorized as prehepatic, intrahepatic, and posthepatic (Table 365-3). Prehepatic causes of portal hypertension are those affecting the portal venous system before it enters the liver; they include portal vein thrombosis and splenic vein thrombosis. Posthepatic causes encompass those affecting the hepatic veins and venous drainage to the heart; they include BCS, venoocclusive disease, and chronic right-sided cardiac congestion. Intrahepatic causes account for over 95% of cases of portal hypertension and are represented by the major forms of cirrhosis. Intrahepatic causes of portal hypertension can be further subdivided into presinusoidal, sinusoidal, and postsinusoidal causes. Postsinusoidal causes include venoocclusive disease, whereas presinusoidal causes include congenital hepatic fibrosis and schistosomiasis. Sinusoidal causes are related to cirrhosis from various causes.
|
CLASSIFICATION OF PORTAL HYPERTENSION |
Cirrhosis is the most common cause of portal hypertension in the United States, and clinically significant portal hypertension is present in >60% of patients with cirrhosis. Portal vein obstruction may be idiopathic or can occur in association with cirrhosis or with infection, pancreatitis, or abdominal trauma.
Coagulation disorders that can lead to the development of portal vein thrombosis include polycythemia vera; essential thrombocytosis; deficiencies in protein C, protein S, antithrombin 3, and factor V Leiden; and abnormalities in the gene-regulating prothrombin production. Some patients may have a subclinical myeloproliferative disorder.
Clinical Features The three primary complications of portal hypertension are gastroesophageal varices with hemorrhage, ascites, and hypersplenism. Thus, patients may present with upper GI bleeding, which, on endoscopy, is found to be due to esophageal or gastric varices; with the development of ascites along with peripheral edema; or with an enlarged spleen with associated reduction in platelets and white blood cells on routine laboratory testing.
ESOPHAGEAL VARICES Over the last decade, it has become common practice to screen known cirrhotics with endoscopy to look for esophageal varices. Such screening studies have shown that approximately one-third of patients with histologically confirmed cirrhosis have varices. Approximately 5–15% of cirrhotics per year develop varices, and it is estimated that the majority of patients with cirrhosis will develop varices over their lifetimes. Furthermore, it is anticipated that roughly one-third of patients with varices will develop bleeding. Several factors predict the risk of bleeding, including the severity of cirrhosis (Child’s class, MELD score); the height of wedged-hepatic vein pressure; the size of the varix; the location of the varix; and certain endoscopic stigmata, including red wale signs, hematocystic spots, diffuse erythema, bluish color, cherry red spots, or white-nipple spots. Patients with tense ascites are also at increased risk for bleeding from varices.
Diagnosis In patients with cirrhosis who are being followed chronically, the development of portal hypertension is usually revealed by the presence of thrombocytopenia; the appearance of an enlarged spleen; or the development of ascites, encephalopathy, and/or esophageal varices with or without bleeding. In previously undiagnosed patients, any of these features should prompt further evaluation to determine the presence of portal hypertension and liver disease. Varices should be identified by endoscopy. Abdominal imaging, either by computed tomography (CT) or MRI, can be helpful in demonstrating a nodular liver and in finding changes of portal hypertension with intraabdominal collateral circulation. If necessary, interventional radiologic procedures can be performed to determine wedged and free hepatic vein pressures that will allow for the calculation of a wedged-to-free gradient, which is equivalent to the portal pressure. The average normal wedged-to-free gradient is 5 mmHg, and patients with a gradient >12 mmHg are at risk for variceal hemorrhage.
|
TREATMENT |
VARICEAL HEMORRHAGE |
Treatment for variceal hemorrhage as a complication of portal hypertension is divided into two main categories: (1) primary prophylaxis and (2) prevention of rebleeding once there has been an initial variceal hemorrhage. Primary prophylaxis requires routine screening by endoscopy of all patients with cirrhosis. Once varices that are at increased risk for bleeding are identified, primary prophylaxis can be achieved either through nonselective beta blockade or by variceal band ligation. Numerous placebo-controlled clinical trials of either propranolol or nadolol have been reported in the literature. The most rigorous studies were those that only included patients with significantly enlarged varices or with hepatic vein pressure gradients >12 mmHg. Patients treated with beta blockers have a lower risk of variceal hemorrhage than those treated with placebo over 1 and 2 years of follow-up. There is also a decrease in mortality related to variceal hemorrhage. Unfortunately, overall survival was improved in only one study. Further studies have demonstrated that the degree of reduction of portal pressure is a significant feature to determine success of therapy. Therefore, it has been suggested that repeat measurements of hepatic vein pressure gradients may be used to guide pharmacologic therapy; however, this may be cost-prohibitive. Several studies have evaluated variceal band ligation and variceal sclerotherapy as methods for providing primary prophylaxis.
Endoscopic variceal ligation (EVL) has achieved a level of success and comfort with most gastroenterologists who see patients with these complications of portal hypertension. Thus, in patients with cirrhosis who are screened for portal hypertension and are found to have large varices, it is recommended that they receive either beta blockade or primary prophylaxis with EVL.
The approach to patients once they have had a variceal bleed is first to treat the acute bleed, which can be life-threatening, and then to prevent further bleeding. Prevention of further bleeding is usually accomplished with repeated variceal band ligation until varices are obliterated. Treatment of acute bleeding requires both fluid and blood-product replacement as well as prevention of subsequent bleeding with EVL.
The medical management of acute variceal hemorrhage includes the use of vasoconstricting agents, usually somatostatin or octreotide. Vasopressin was used in the past but is no longer commonly used. Balloon tamponade (Sengstaken-Blakemore tube or Minnesota tube) can be used in patients who cannot get endoscopic therapy immediately or who need stabilization prior to endoscopic therapy. Control of bleeding can be achieved in the vast majority of cases; however, bleeding recurs in the majority of patients if definitive endoscopic therapy has not been instituted. Octreotide, a direct splanchnic vasoconstrictor, is given at dosages of 50–100 µg/h by continuous infusion. Endoscopic intervention is used as first-line treatment to control bleeding acutely. Some endoscopists will use variceal injection therapy (sclerotherapy) as initial therapy, particularly when bleeding is vigorous. Variceal band ligation is used to control acute bleeding in over 90% of cases and should be repeated until obliteration of all varices is accomplished. When esophageal varices extend into the proximal stomach, band ligation is less successful. In these situations, when bleeding continues from gastric varices, consideration for a transjugular intrahepatic portosystemic shunt (TIPS) should be made. This technique creates a portosystemic shunt by a percutaneous approach using an expandable metal stent, which is advanced under angiographic guidance to the hepatic veins and then through the substance of the liver to create a direct portocaval shunt. This offers an alternative to surgery for acute decompression of portal hypertension. Encephalopathy can occur in as many as 20% of patients after TIPS and is particularly problematic in elderly patients and in patients with preexisting encephalopathy. TIPS should be reserved for individuals who fail endoscopic or medical management or who are poor surgical risks. TIPS can sometimes be used as a bridge to transplantation. Surgical esophageal transsection is a procedure that is rarely used and generally is associated with a poor outcome.
PREVENTION OF RECURRENT BLEEDING (FIG. 365-3)
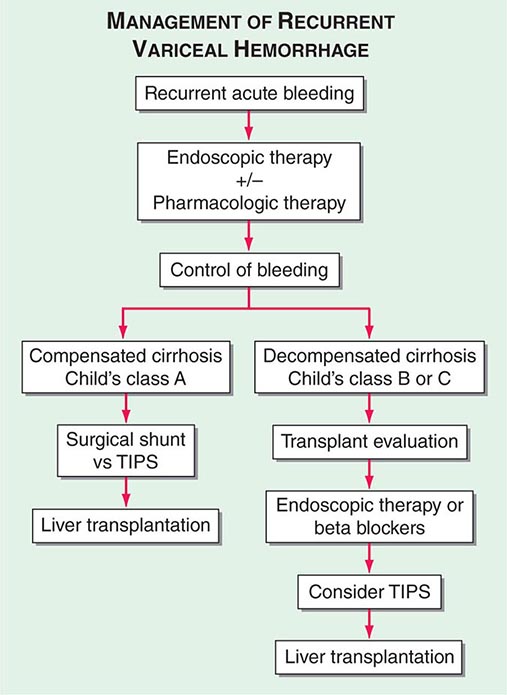
FIGURE 365-3 Management of recurrent variceal hemorrhage. This algorithm describes an approach to management of patients who have recurrent bleeding from esophageal varices. Initial therapy is generally with endoscopic therapy often supplemented by pharmacologic therapy. With control of bleeding, a decision needs to be made as to whether patients should go on to a surgical shunt or TIPS (if they are Child’s class A) and be considered for transplant, or if they should have TIPS and be considered for transplant (if they are Child’s class B or C). TIPS, transjugular intrahepatic portosystemic shunt.
Once patients have had an acute bleed and have been managed successfully, attention should be paid to preventing recurrent bleeding. This usually requires repeated variceal band ligation until varices are obliterated. Beta blockade may be of adjunctive benefit in patients who are having recurrent variceal band ligation; however, once varices have been obliterated, the need for beta blockade is lessened. Despite successful variceal obliteration, many patients will still have portal hypertensive gastropathy from which bleeding can occur. Nonselective beta blockade may be helpful to prevent further bleeding from portal hypertensive gastropathy once varices have been obliterated.
Portosystemic shunt surgery is less commonly performed with the advent of TIPS; nonetheless, this procedure should be considered for patients with good hepatic synthetic function who could benefit by having portal decompressive surgery.
SPLENOMEGALY AND HYPERSPLENISM
Congestive splenomegaly is common in patients with portal hypertension. Clinical features include the presence of an enlarged spleen on physical examination and the development of thrombocytopenia and leukopenia in patients who have cirrhosis. Some patients will have fairly significant left-sided and left upper quadrant abdominal pain related to an enlarged and engorged spleen. Splenomegaly itself usually requires no specific treatment, although splenectomy can be successfully performed under very special circumstances.
Hypersplenism with the development of thrombocytopenia is a common feature of patients with cirrhosis and is usually the first indication of portal hypertension.
ASCITES
Definition Ascites is the accumulation of fluid within the peritoneal cavity. Overwhelmingly, the most common cause of ascites is portal hypertension related to cirrhosis; however, clinicians should remember that malignant or infectious causes of ascites can be present as well, and careful differentiation of these other causes are obviously important for patient care.
Pathogenesis The presence of portal hypertension contributes to the development of ascites in patients who have cirrhosis (Fig. 365-4). There is an increase in intrahepatic resistance, causing increased portal pressure, but there is also vasodilation of the splanchnic arterial system, which, in turn, results in an increase in portal venous inflow. Both of these abnormalities result in increased production of splanchnic lymph. Vasodilating factors such as nitric oxide are responsible for the vasodilatory effect. These hemodynamic changes result in sodium retention by causing activation of the renin-angiotensin-aldosterone system with the development of hyperaldosteronism. The renal effects of increased aldosterone leading to sodium retention also contribute to the development of ascites. Sodium retention causes fluid accumulation and expansion of the extracellular fluid volume, which results in the formation of peripheral edema and ascites. Sodium retention is the consequence of a homeostatic response caused by underfilling of the arterial circulation secondary to arterial vasodilation in the splanchnic vascular bed. Because the retained fluid is constantly leaking out of the intravascular compartment into the peritoneal cavity, the sensation of vascular filling is not achieved, and the process continues. Hypoalbuminemia and reduced plasma oncotic pressure also contribute to the loss of fluid from the vascular compartment into the peritoneal cavity. Hypoalbuminemia is due to decreased synthetic function in a cirrhotic liver.
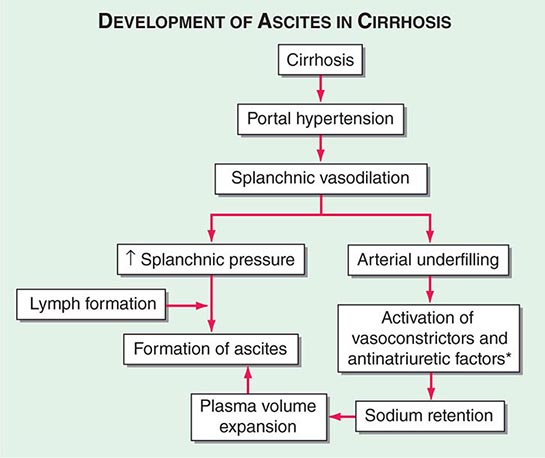
FIGURE 365-4 Development of ascites in cirrhosis. This flow diagram illustrates the importance of portal hypertension with splanchnic vasodilation in the development of ascites. *Antinatriuretic factors include the renin-angiotensin-aldosterone system and the sympathetic nervous system.
Clinical Features Patients typically note an increase in abdominal girth that is often accompanied by the development of peripheral edema. The development of ascites is often insidious, and it is surprising that some patients wait so long and become so distended before seeking medical attention. Patients usually have at least 1–2 L of fluid in the abdomen before they are aware that there is an increase. If ascitic fluid is massive, respiratory function can be compromised, and patients will complain of shortness of breath. Hepatic hydrothorax may also occur in this setting, contributing to respiratory symptoms. Patients with massive ascites are often malnourished and have muscle wasting and excessive fatigue and weakness.
Diagnosis Diagnosis of ascites is by physical examination and is often aided by abdominal imaging. Patients will have bulging flanks, may have a fluid wave, or may have the presence of shifting dullness. This is determined by taking patients from a supine position to lying on either their left or right side and noting the movement of the dullness to percussion. Subtle amounts of ascites can be detected by ultrasound or CT scanning. Hepatic hydrothorax is more common on the right side and implicates a rent in the diaphragm with free flow of ascitic fluid into the thoracic cavity.
When patients present with ascites for the first time, it is recommended that a diagnostic paracentesis be performed to characterize the fluid. This should include the determination of total protein and albumin content, blood cell counts with differential, and cultures. In the appropriate setting, amylase may be measured and cytology performed. In patients with cirrhosis, the protein concentration of the ascitic fluid is quite low, with the majority of patients having an ascitic fluid protein concentration <1 g/dL. The development of the serum ascites-to-albumin gradient (SAAG) has replaced the description of exudative or transudative fluid. When the gradient between the serum albumin level and the ascitic fluid albumin level is >1.1 g/dL, the cause of the ascites is most likely due to portal hypertension; this is usually in the setting of cirrhosis. When the gradient is <1.1 g/dL, infectious or malignant causes of ascites should be considered. When levels of ascitic fluid proteins are very low, patients are at increased risk for developing SBP. A high level of red blood cells in the ascitic fluid signifies a traumatic tap or perhaps a hepatocellular cancer or a ruptured omental varix. When the absolute level of polymorphonuclear leukocytes is >250/µL, the question of ascitic fluid infection should be strongly considered. Ascitic fluid cultures should be obtained using bedside inoculation of culture media.
|
TREATMENT |
ASCITES |
Patients with small amounts of ascites can usually be managed with dietary sodium restriction alone. Most average diets in the United States contain 6–8 g of sodium per day, and if patients eat at restaurants or fast-food outlets, the amount of sodium in their diet can exceed this amount. Thus, it is often extremely difficult to get patients to change their dietary habits to ingest <2 g of sodium per day, which is the recommended amount. Patients are frequently surprised to realize how much sodium is in the standard U.S. diet; thus, it is important to make educational pamphlets available to the patient. Often, a simple recommendation is to eat fresh or frozen foods, avoiding canned or processed foods, which are usually preserved with sodium. When a moderate amount of ascites is present, diuretic therapy is usually necessary. Traditionally, spironolactone at 100–200 mg/d as a single dose is started, and furosemide may be added at 40–80 mg/d, particularly in patients who have peripheral edema. In patients who have never received diuretics before, the failure of the above-mentioned dosages suggests that they are not being compliant with a low-sodium diet. If compliance is confirmed and ascitic fluid is not being mobilized, spironolactone can be increased to 400–600 mg/d and furosemide increased to 120–160 mg/d. If ascites is still present with these dosages of diuretics in patients who are compliant with a low-sodium diet, then they are defined as having refractory ascites, and alternative treatment modalities including repeated large-volume paracentesis or a TIPS procedure should be considered (Fig. 365-5). Recent studies have shown that TIPS, while managing the ascites, does not improve survival in these patients. Unfortunately, TIPS is often associated with an increased frequency of hepatic encephalopathy and must be considered carefully on a case-by-case basis. The prognosis for patients with cirrhosis with ascites is poor, and some studies have shown that <50% of patients survive 2 years after the onset of ascites. Thus, there should be consideration for liver transplantation in patients with the onset of ascites.
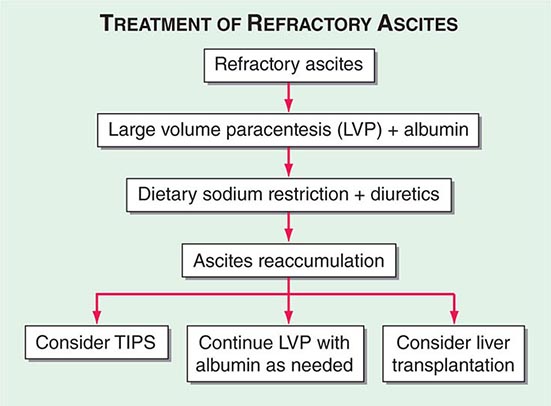
FIGURE 365-5 Treatment of refractory ascites. In patients who develop azotemia in the course of receiving diuretics in the management of their ascites, some will require repeated large-volume paracentesis (LVP), some may be considered for transjugular intrahepatic portosystemic shunt (TIPS), and some would be good candidates for liver transplantation. These decisions are all individualized.
SPONTANEOUS BACTERIAL PERITONITIS
SBP is a common and severe complication of ascites characterized by spontaneous infection of the ascitic fluid without an intraabdominal source. In patients with cirrhosis and ascites severe enough for hospitalization, SBP can occur in up to 30% of individuals and can have a 25% in-hospital mortality rate. Bacterial translocation is the presumed mechanism for development of SBP, with gut flora traversing the intestine into mesenteric lymph nodes, leading to bacteremia and seeding of the ascitic fluid. The most common organisms are Escherichia coli and other gut bacteria; however, gram-positive bacteria, including Streptococcus viridans, Staphylococcus aureus, and Enterococcus sp., can also be found. If more than two organisms are identified, secondary bacterial peritonitis due to a perforated viscus should be considered. The diagnosis of SBP is made when the fluid sample has an absolute neutrophil count >250/μL. Bedside cultures should be obtained when ascitic fluid is tapped. Patients with ascites may present with fever, altered mental status, elevated white blood cell count, and abdominal pain or discomfort, or they may present without any of these features. Therefore, it is necessary to have a high degree of clinical suspicion, and peritoneal taps are important for making the diagnosis. Treatment is with a second-generation cephalosporin, with cefotaxime being the most commonly used antibiotic. In patients with variceal hemorrhage, the frequency of SBP is significantly increased, and prophylaxis against SBP is recommended when a patient presents with upper GI bleeding. Furthermore, in patients who have had an episode(s) of SBP and recovered, once-weekly administration of antibiotics is used as prophylaxis for recurrent SBP.
HEPATORENAL SYNDROME
The hepatorenal syndrome (HRS) is a form of functional renal failure without renal pathology that occurs in about 10% of patients with advanced cirrhosis or acute liver failure. There are marked disturbances in the arterial renal circulation in patients with HRS; these include an increase in vascular resistance accompanied by a reduction in systemic vascular resistance. The reason for renal vasoconstriction is most likely multifactorial and is poorly understood. The diagnosis is made usually in the presence of a large amount of ascites in patients who have a stepwise progressive increase in creatinine. Type 1 HRS is characterized by a progressive impairment in renal function and a significant reduction in creatinine clearance within 1–2 weeks of presentation. Type 2 HRS is characterized by a reduction in glomerular filtration rate with an elevation of serum creatinine level, but it is fairly stable and is associated with a better outcome than that of type 1 HRS.
HRS is often seen in patients with refractory ascites and requires exclusion of other causes of acute renal failure. Treatment has, unfortunately, been difficult, and in the past, dopamine or prostaglandin analogues were used as renal vasodilating medications. Carefully performed studies have failed to show clear-cut benefit from these therapeutic approaches. Currently, patients are treated with midodrine, an α-agonist, along with octreotide and intravenous albumin. The best therapy for HRS is liver transplantation; recovery of renal function is typical in this setting. In patients with either type 1 or type 2 HRS, the prognosis is poor unless transplant can be achieved within a short period of time.
HEPATIC ENCEPHALOPATHY
Portosystemic encephalopathy is a serious complication of chronic liver disease and is broadly defined as an alteration in mental status and cognitive function occurring in the presence of liver failure. In acute liver injury with fulminant hepatic failure, the development of encephalopathy is a requirement for a diagnosis of fulminant failure. Encephalopathy is much more commonly seen in patients with chronic liver disease. Gut-derived neurotoxins that are not removed by the liver because of vascular shunting and decreased hepatic mass get to the brain and cause the symptoms that we know of as hepatic encephalopathy. Ammonia levels are typically elevated in patients with hepatic encephalopathy, but the correlation between severity of liver disease and height of ammonia levels is often poor, and most hepatologists do not rely on ammonia levels to make a diagnosis. Other compounds and metabolites that may contribute to the development of encephalopathy include certain false neurotransmitters and mercaptans.
Clinical Features In acute liver failure, changes in mental status can occur within weeks to months. Brain edema can be seen in these patients, with severe encephalopathy associated with swelling of the gray matter. Cerebral herniation is a feared complication of brain edema in acute liver failure, and treatment is meant to decrease edema with mannitol and judicious use of intravenous fluids.
In patients with cirrhosis, encephalopathy is often found as a result of certain precipitating events such as hypokalemia, infection, an increased dietary protein load, or electrolyte disturbances. Patients may be confused or exhibit a change in personality. They may actually be quite violent and difficult to manage; alternatively, patients may be very sleepy and difficult to rouse. Because precipitating events are so commonly found, they should be sought carefully. If patients have ascites, this should be tapped to rule out infection. Evidence of GI bleeding should be sought, and patients should be appropriately hydrated. Electrolytes should be measured and abnormalities corrected. In patients presenting with encephalopathy, asterixis is often present. Asterixis can be elicited by having patients extend their arms and bend their wrists back. In this maneuver, patients who are encephalopathic have a “liver flap”—i.e., a sudden forward movement of the wrist. This requires patients to be able to cooperate with the examiner and obviously cannot be elicited in patients who are severely encephalopathic or in hepatic coma.
The diagnosis of hepatic encephalopathy is clinical and requires an experienced clinician to recognize and put together all of the various features. Often when patients have encephalopathy for the first time, they are unaware of what is transpiring, but once they have been through the experience for the first time, they can identify when this is developing in subsequent situations and can often self-medicate to impair the development or worsening of encephalopathy.
MALNUTRITION IN CIRRHOSIS
Because the liver is principally involved in the regulation of protein and energy metabolism in the body, it is not surprising that patients with advanced liver disease are commonly malnourished. Once patients become cirrhotic, they are more catabolic, and muscle protein is metabolized. There are multiple factors that contribute to the malnutrition of cirrhosis, including poor dietary intake, alterations in gut nutrient absorption, and alterations in protein metabolism. Dietary supplementation for patients with cirrhosis is helpful in preventing patients from becoming catabolic.
ABNORMALITIES IN COAGULATION
Coagulopathy is almost universal in patients with cirrhosis. There is decreased synthesis of clotting factors and impaired clearance of anticoagulants. In addition, patients may have thrombocytopenia from hypersplenism due to portal hypertension. Vitamin K–dependent clotting factors are factors II, VII, IX, and X. Vitamin K requires biliary excretion for its subsequent absorption; thus, in patients with chronic cholestatic syndromes, vitamin K absorption is frequently diminished. Intravenous or intramuscular vitamin K can quickly correct this abnormality. More commonly, the synthesis of vitamin K–dependent clotting factors is diminished because of a decrease in hepatic mass, and, under these circumstances, administration of parenteral vitamin K does not improve the clotting factors or the prothrombin time. Platelet function is often abnormal in patients with chronic liver disease, in addition to decreases in platelet levels due to hypersplenism.
BONE DISEASE IN CIRRHOSIS
Osteoporosis is common in patients with chronic cholestatic liver disease because of malabsorption of vitamin D and decreased calcium ingestion. The rate of bone resorption exceeds that of new bone formation in patients with cirrhosis, resulting in bone loss. Dual x-ray absorptiometry (DEXA) is a useful method for determining osteoporosis or osteopenia in patients with chronic liver disease. When a DEXA scan shows decreased bone mass, treatment should be administered with bisphosphonates that are effective at inhibiting resorption of bone and efficacious in the treatment of osteoporosis.
HEMATOLOGIC ABNORMALITIES IN CIRRHOSIS
Numerous hematologic manifestations of cirrhosis are present, including anemia from a variety of causes including hypersplenism, hemolysis, iron deficiency, and perhaps folate deficiency from malnutrition. Macrocytosis is a common abnormality in red blood cell morphology seen in patients with chronic liver disease, and neutropenia may be seen as a result of hypersplenism.
366e |
Atlas of Liver Biopsies |
Although clinical and laboratory features yield clues to the extent of inflammatory processes (disease grade), the degree of scarring and architectural distortion (disease stage), and the nature of the disease process, the liver biopsy is felt to represent the gold standard for assessing the degree of liver injury and fibrosis. Examination of liver histology provides not only a basis for quantitative scoring of disease activity and progression but also a wealth of qualitative information that can direct and inform diagnosis and management.
A normal liver lobule consists of portal (zone 1), lobular (midzonal or zone 2), and central (zone 3) zones. The portal tract contains the hepatic artery (HA) and portal vein (PV), which represent the dual vascular supply to the liver, as well as the bile duct (BD). The lobular area contains cords of liver cells surrounded by vascular sinusoids, and the central zone consists of the central vein (CV), the terminal branch of the hepatic vein (see figure below).
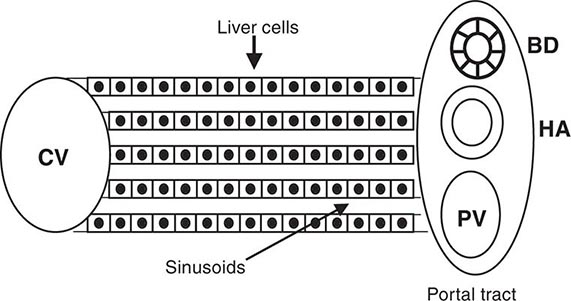
Included in this atlas of liver biopsies are examples of common morphologic features of acute and chronic liver disorders, some involving the lobular areas (e.g., the lobular inflammatory changes of acute hepatitis, apoptotic hepatocyte degeneration in acute and chronic hepatitis, virus antigen localization in hepatocyte cytoplasm and/or nuclei, viral inclusion bodies, copper or iron deposition, other inclusion bodies) and others involving the portal tracts (e.g., the portal mononuclear infiltrate that expands and spills over beyond the border of periportal hepatocytes in chronic hepatitis C, autoimmune hepatitis, and liver allograft rejection) or centrizonal areas (e.g., acute acetaminophen hepatotoxicity). Other histologic features of importance include hepatic steatosis (observed in alcoholic liver injury, nonalcoholic fatty liver disorders, and metabolic disorders–including mitochondrial injury–and in patients with chronic viral hepatitis); injury of bile ducts in the portal tract, an important diagnostic hallmark of primary biliary cirrhosis, primary sclerosing cholangitis, and liver allograft rejection; cholestasis in intrahepatic or extrahepatic biliary obstruction or in infiltrative disorders; ductular proliferation in the setting of marked hepatocellular necrosis; plasma cell infiltration common in autoimmune hepatitis; portal inflammation affecting portal veins (“endothelialitis”) in liver allograft rejection; and mild-to-severe fibrosis, in varying distribution and pattern, as a consequence of liver injury common to many disorders. (All magnifications reflect the objective lens used.)
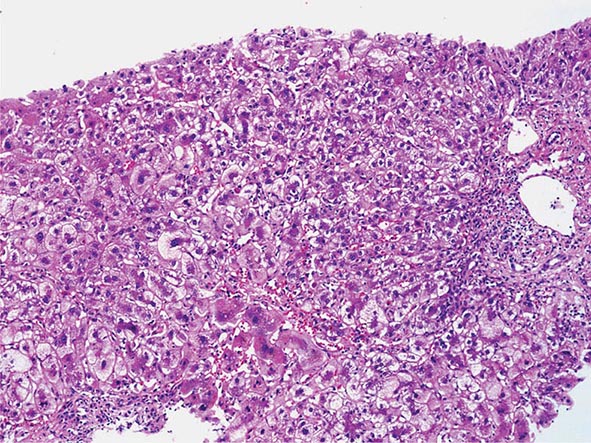
FIGURE 366e-1 Acute hepatitis with lobular inflammation and hepatocellular ballooning (hematoxylin and eosin [H&E], 10×).
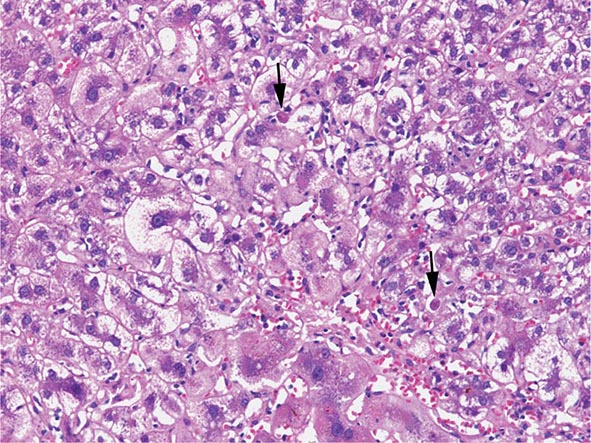
FIGURE 366e-2 Acute hepatitis, higher magnification, showing lobular inflammation, hepatocellular ballooning, and acidophilic bodies (arrows) (H&E, 20×).
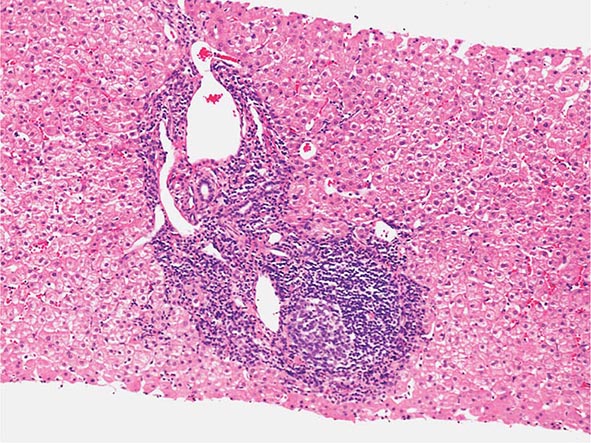
FIGURE 366e-3 Chronic hepatitis C with portal lymphoid infiltrate and lymphoid follicle containing germinal center (H&E, 10×).
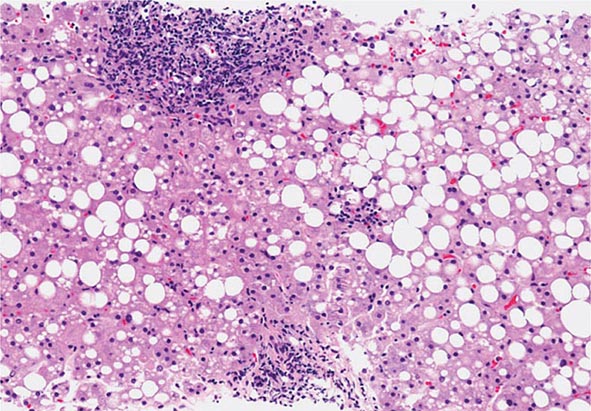
FIGURE 366e-4 Chronic hepatitis C with portal and lobular inflammation and steatosis (H&E, 10×).
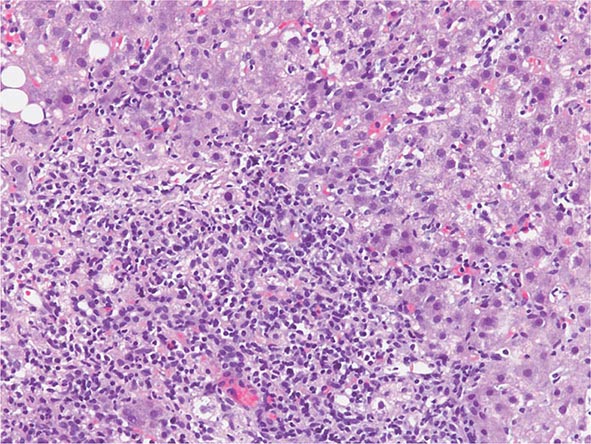
FIGURE 366e-5 Chronic hepatitis C with portal inflammation and interface hepatitis (erosion of the limiting plate of periportal hepatocytes by infiltrating mononuclear cells) (H&E, 20×).
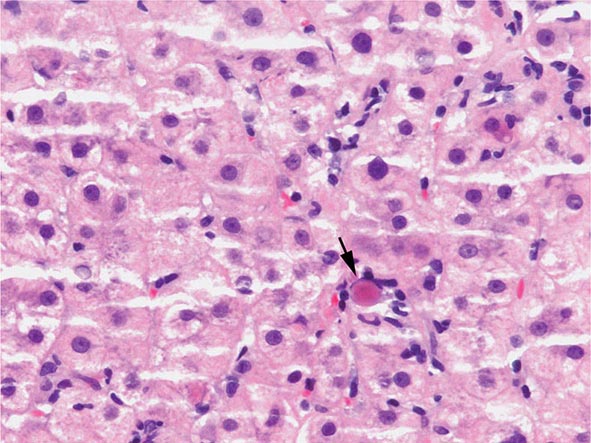
FIGURE 366e-6 Lobular inflammation with acidophilic body (apoptotic body) surrounded by lymphoid cells (H&E, 40×).
FIGURE 366e-7 Chronic hepatitis B with hepatocellular cytoplasmic staining for hepatitis B surface antigen (immunoperoxidase, 20×).
FIGURE 366e-8 Chronic hepatitis B with hepatocellular nuclear staining for hepatitis B core antigen (immunoperoxidase, 20×).
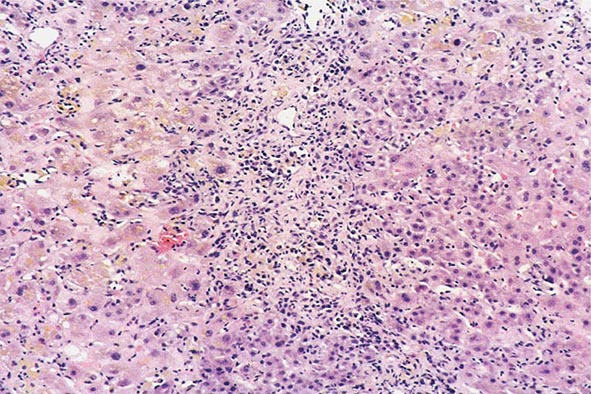
FIGURE 366e-9 Autoimmune hepatitis with portal and lobular inflammation, interface hepatitis, and cholestasis (H&E, 10×).
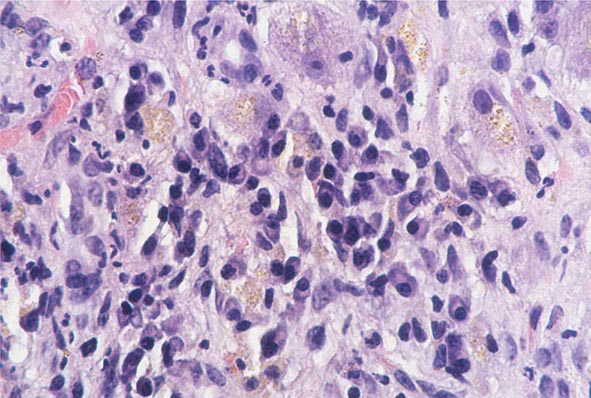
FIGURE 366e-10 Autoimmune hepatitis, higher magnification, showing dense plasma cell infiltrate in the portal and periportal regions (H&E, 40×).
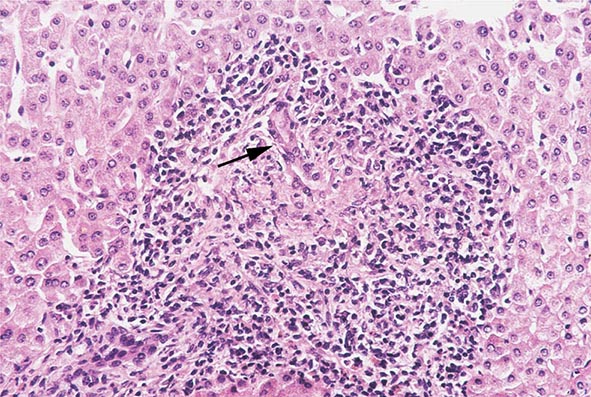
FIGURE 366e-11 Primary biliary cirrhosis with degenerating bile duct epithelium (“florid ductular lesion”) (arrow) surrounded by epithelioid granulomatous reaction and lymphoplasmacytic infiltrate (H&E, 40×).
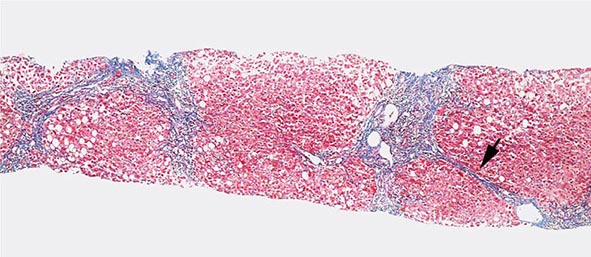
FIGURE 366e-12 Chronic hepatitis C with bridging fibrosis (arrow) (Masson trichrome, 10×).
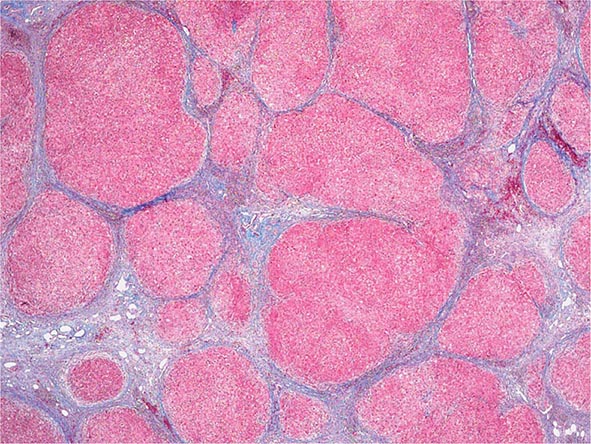
FIGURE 366e-13 Cirrhosis with architectural alteration resulting from fibrosis and nodular hepatocellular regeneration (Masson trichrome, 2×).
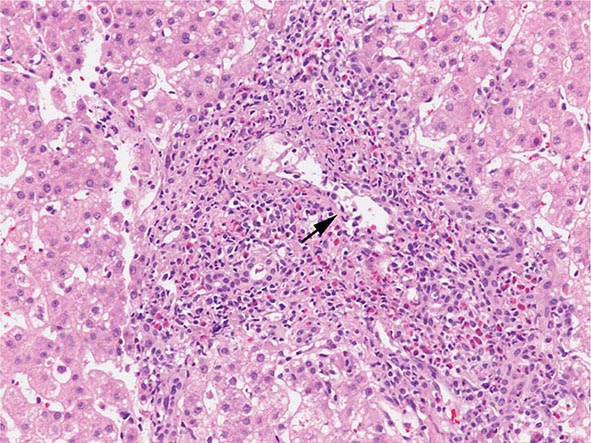
FIGURE 366e-14 Acute cellular rejection of orthotopic liver allograft demonstrating a mixed inflammatory cell infiltrate (lymphoid cells, eosinophils, neutrophils) of the portal tract as well as endothelialitis of the portal vein (arrow) and bile duct injury (H&E, 10×).
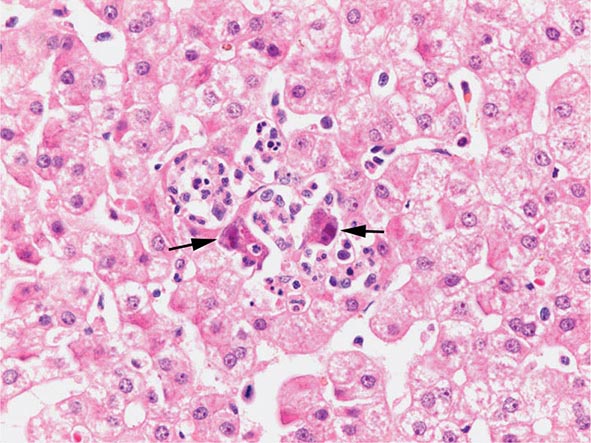
FIGURE 366e-15 Liver allograft with cytomegalovirus infection showing hepatocytes with nuclear inclusions (arrows) surrounded by a neutrophilic and lymphoid infiltrate (H&E, 10×).
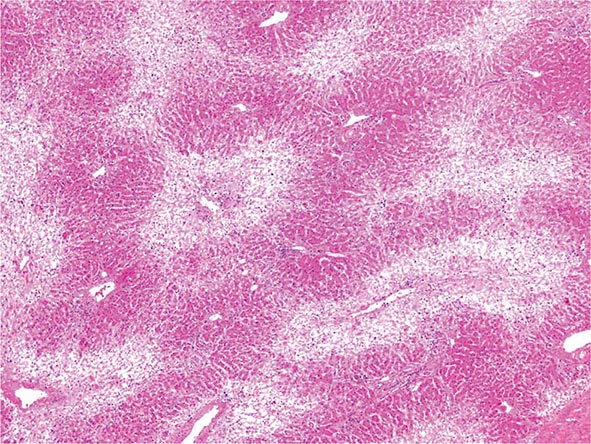
FIGURE 366e-16 Combined acetaminophen hepatotoxicity and alcoholic liver injury with extensive centrilobular areas of necrosis (H&E, 4×).
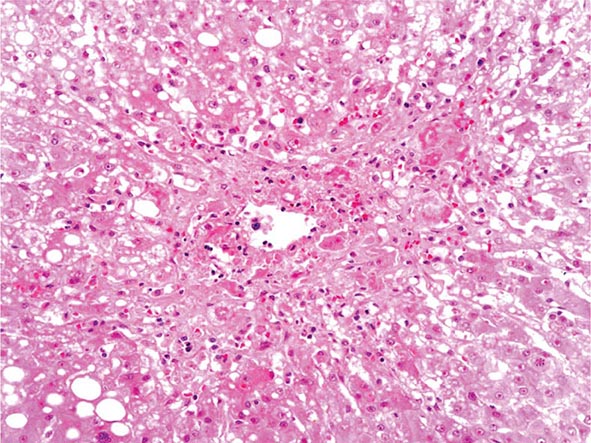
FIGURE 366e-17 Combined acetaminophen hepatotoxicity and alcoholic liver injury at higher magnification showing necrotic centrilobular area with Mallory bodies (H&E 20×).
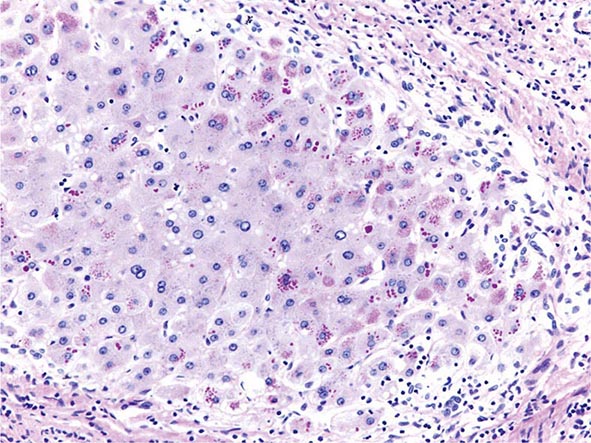
FIGURE 366e-18 α1 Antitrypsin deficiency with cytoplasmic periodic acid–Schiff (PAS)-positive, diastase-resistant globules in many hepatocytes, predominantly at the periphery of a cirrhotic nodule (PAS, 20×).
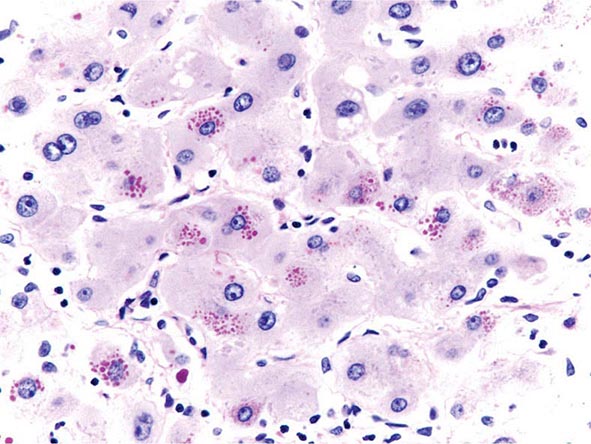
FIGURE 366e-19 α1 Antitrypsin deficiency with higher magnification of PAS-positive, diastase-resistant globules (PAS, 40×).
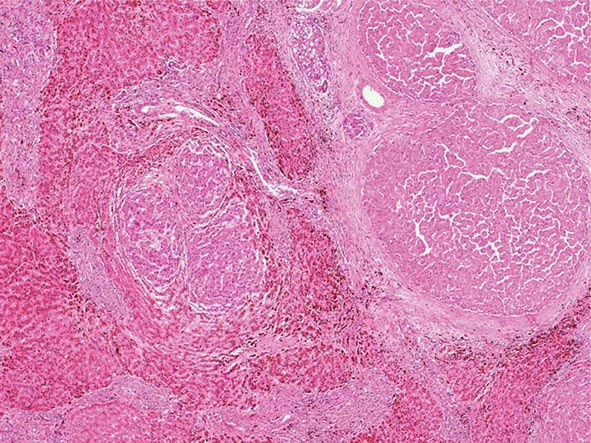
FIGURE 366e-20 Cirrhosis secondary to hemochromatosis with hepatocellular carcinoma; brown hemosiderin pigment (iron) is present in the cirrhotic liver, while the hepatocellular carcinoma nodules are hemosiderin-free (H&E, 4×).
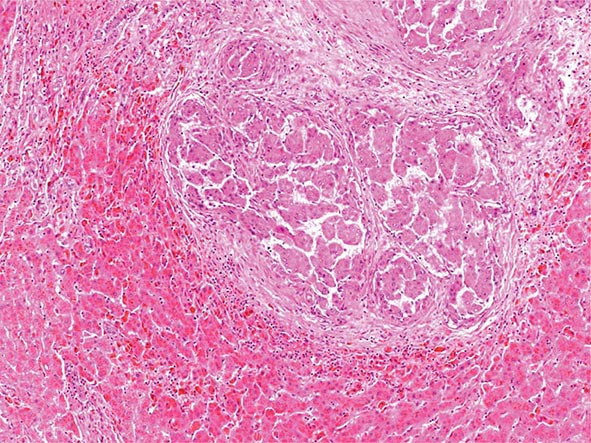
FIGURE 366e-21 Cirrhosis secondary to hemochromatosis with hepatocellular carcinoma at higher magnification, demonstrating nodules of large malignant cells with highly disorganized architecture (H&E, 10×).
FIGURE 366e-22 Hemochromatosis with iron stain demonstrating extensive iron deposition and characteristic pattern of pericanalicular distribution of iron (iron stain, 10×).
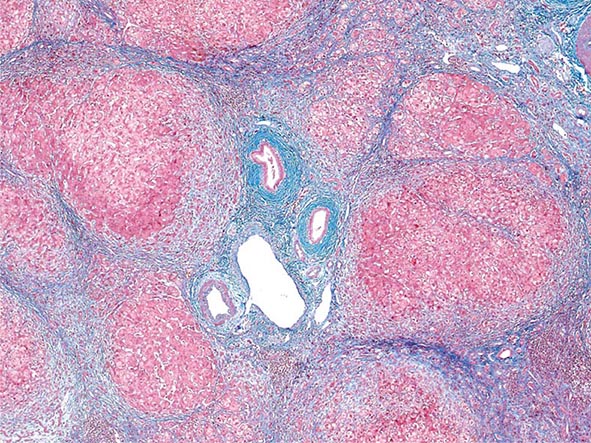
FIGURE 366e-23 Primary sclerosing cholangitis showing cirrhosis and periductular fibrosis (Masson trichrome, 4×).
FIGURE 366e-24 Primary sclerosing cholangitis showing the extrahepatic bile duct (in a liver explant obtained at the time of hepatectomy for orthotopic liver transplantation) with marked mural chronic inflammation and fibrosis as well as peribiliary glands (H&E, 2×).
FIGURE 366e-25 Primary sclerosing cholangitis showing peripheral cholestasis (green) and cytoplasmic red granular staining of hepatocytes for copper (rhodamine copper stain, 20×).
FIGURE 366e-26 Nonalcoholic steatohepatitis (NASH) showing steatosis, ballooned hepatocytes, and Mallory bodies with surrounding polymorphonuclear leukocytes (arrow) (H&E, 20×).
FIGURE 366e-27 Nonalcoholic steatohepatitis (NASH) showing steatosis with perisinusoidal and pericellular fibrosis (H&E, 20×).
FIGURE 366e-28 Acute hepatitis with submassive hepatic necrosis with marked parenchymal collapse, remnant islands of surviving hepatocytes, and a marked ductular reaction (H&E, 10×).
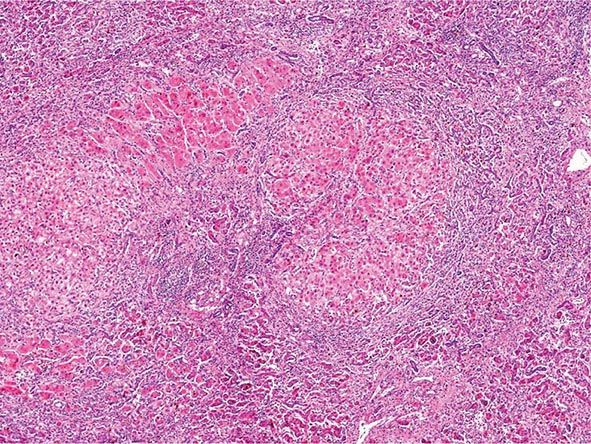
FIGURE 366e-29 Wilson’s disease showing cirrhosis, extensive collapse, and ductular reaction in a teenager with an acute presentation (H&E, 4×).
FIGURE 366e-30 Wilson’s disease showing extensive hepatocyte cytoplasmic red granular staining for copper in a cirrhotic nodule (rhodanine copper stain, 20×).
367e |
Genetic, Metabolic, and Infiltrative Diseases Affecting the Liver |
There are a number of disorders of the liver that fit within the categories of genetic, metabolic, and infiltrative disorders (Table 367e-1). Inherited disorders include hemochromatosis, Wilson’s disease, α1 antitrypsin (α1AT) deficiency, and cystic fibrosis (CF). Hemochromatosis is the most common inherited disorder affecting white populations, with the genetic susceptibility for the disease being identified in 1 in 250 individuals. Over the past 15 years, it has become increasingly apparent that nonalcoholic fatty liver disease (NAFLD) is the most common cause of elevated liver enzymes found in the U.S. population. This disorder is discussed in greater detail in Chap. 364. Infiltrative disorders of the liver are relatively rare.
|
GENETIC, METABOLIC, AND INFILTRATIVE DISEASES AFFECTING THE LIVER |
GENETIC LIVER DISEASES
Hereditary Hemochromatosis Hereditary hemochromatosis (HH) is a common inherited disorder of iron metabolism (Chap. 428). Our knowledge of the disease and its phenotypic expression has changed since 1996, when the gene for HH, called HFE, was identified, allowing for genetic testing for the two major mutations (C282Y and H63D) that are responsible for HFE-related HH. Subsequently, several additional genes/proteins involved in the regulation of iron homeostasis have been identified, contributing to a better understanding of cellular iron uptake and release and the characterization of additional causes of inherited iron overload (Table 367e-2).
|
CLASSIFICATION OF IRON OVERLOAD SYNDROMES |
Abbreviations: HAMP, hepcidin; HJV, hemojuvelin; TfR2, transferrin receptor 2.
Most patients with HH are asymptomatic; however, when patients present with symptoms, they are frequently nonspecific and include weakness, fatigue, lethargy, and weight loss. Specific, organ-related symptoms include abdominal pain, arthralgias, and symptoms and signs of chronic liver disease. Increasingly, most patients are now identified before they have symptoms, either through family studies or from the performance of screening iron studies. Several prospective population studies have shown that C282Y homozygosity is found in about 1 in 250 individuals of northern European descent, with the heterozygote frequency seen in approximately 1 in 10 individuals. It is important to consider HH in patients who present with the symptoms and signs known to occur in established HH. When confronted with abnormal serum iron studies, clinicians should not wait for typical symptoms or findings of HH to appear before considering the diagnosis. However, once the diagnosis of HH is considered, either by an evaluation of abnormal screening iron studies in the context of family studies, in a patient with an abnormal genetic test, or in the evaluation of a patient with any of the typical symptoms (Table 367e-3) or clinical findings (Table 367e-4), definitive diagnosis is relatively straightforward. Transferrin saturation (serum iron divided by total iron-binding capacity [TIBC] or transferrin, times 100%) and ferritin levels should be obtained. Both of these will be elevated in a symptomatic patient. It must be remembered that ferritin is an acute-phase reactant and can be elevated in a number of other inflammatory disorders, such as rheumatoid arthritis, or in various neoplastic diseases, such as lymphoma or other cancers. Also, serum ferritin is elevated in a majority of patients with nonalcoholic steatohepatitis (NASH), hepatitis C, and alcoholic liver disease in the absence of iron overload.
|
SYMPTOMS OF HEREDITARY HEMOCHROMATOSIS |
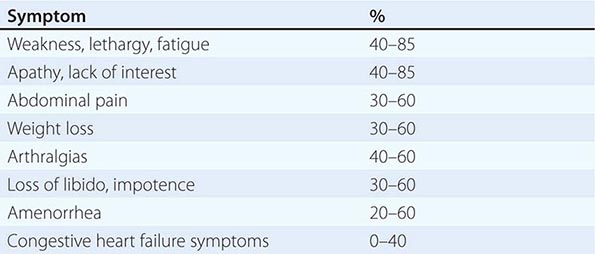
|
PHYSICAL FINDINGS IN HEREDITARY HEMOCHROMATOSIS |
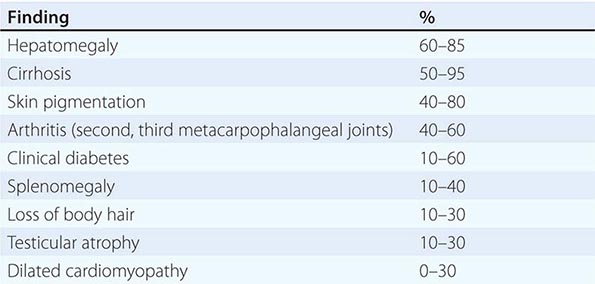
At present, if patients have an elevated transferrin saturation or ferritin level, genetic testing should be performed; if they are a C282Y homozygote or a compound heterozygote (C282Y/H63D), the diagnosis is confirmed. If liver enzymes (alanine aminotransferase [ALT], aspartate aminotransferase [AST]) are elevated or the ferritin is >1000 μg/L, the patient should be considered for liver biopsy because there is an increased frequency of advanced fibrosis in these individuals. If liver biopsy is performed, iron deposition is found in a periportal distribution with a periportal to pericentral gradient; iron is found predominantly in parenchymal cells, and Kupffer cells are spared.
Wilson’s Disease Wilson’s disease is an inherited disorder of copper homeostasis first described in 1912 (Chap. 429). The Wilson’s disease gene was discovered in 1993, with the identification of ATP7B. This P-type ATPase is involved in copper transport and is necessary for the export of copper from the hepatocyte. Thus, in patients with mutations in ATP7B, copper is retained in the liver, leading to increased copper storage and ultimately liver disease as a result.
The clinical presentation of Wilson’s disease is variable and includes chronic hepatitis, hepatic steatosis, and cirrhosis in adolescents and young adults. Neurologic manifestations indicate that liver disease is present and include speech disorders and various movement disorders. Diagnosis includes the demonstration of a reduced ceruloplasmin level, increased urinary excretion of copper, the presence of Kayser-Fleischer rings in the corneas of the eyes, and an elevated hepatic copper level, in the appropriate clinical setting. The genetic diagnosis of Wilson’s disease is difficult because >500 mutations in ATP7B have been described with different degrees of frequency and penetration in certain populations.
α1 Antitrypsin Deficiency α1AT deficiency was first described in the late 1960s in patients with severe pulmonary disease. α1AT is a 52-kDa glycoprotein produced in hepatocytes, phagocytes, and epithelial cells in the lungs, which inhibits serine proteases, primarily neutrophil elastase. In α1AT deficiency, increased amounts of neutrophil elastase can result in progressive lung injury from degradation of elastin, leading to premature emphysema. In the 1970s, α1AT deficiency was discovered as a cause of neonatal liver disease, so-called “neonatal hepatitis.” It is now known to be a cause of liver disease in infancy, early childhood, and adolescence, and in adults.
In α1AT deficiency, variants in the proteinase inhibitor (Pi) gene located on chromosome 14 alter α1AT structure, interfering with hepatocellular export. Aggregated, deformed polymers of α1AT accumulate in the hepatocyte endoplasmic reticulum. There are over 75 different α1AT variants. Conventional nomenclature identifies normal variants as PiMM; these individuals have normal blood levels of α1AT. The most common abnormal variants are called S and Z. Individuals homozygous for the Z mutation (PiZZ) have low levels of α1AT (about 15% of normal), and these patients are susceptible to liver and/or lung disease, yet only a proportion (about 25%) of PiZZ patients develop disease manifestations. Null variants have undetectable levels of α1AT and are susceptible to premature lung disease.
α1AT deficiency has been identified in all populations; however, the disorder is most common in patients of northern European and Iberian descent. The disorder affects about 1 in 1500 to 2000 individuals in North America. The natural history of α1AT deficiency is quite variable because many individuals with the PiZZ variant never develop disease, whereas others can develop childhood cirrhosis leading to liver transplantation.
In adults, the diagnosis often comes in the course of evaluation of patients with abnormal liver test abnormalities or in a workup for cirrhosis. A hint to diagnosis may be coexistent lung disease at a relatively young age or a family history of liver and/or lung disease. Patients may have symptoms of pulmonary disease with cough and dyspnea. Liver disease may be asymptomatic other than fatigue, or patients may present with complications of decompensated liver disease.
Diagnosis of α1AT deficiency is confirmed by blood tests showing reduced levels of serum α1AT, accompanied by Pi determinations. Most patients with liver disease have either PiZZ or PiSZ; occasionally, patients with PiMZ have reduced levels of α1AT, but they usually do not have a low enough level to cause disease. Liver biopsy is often performed to determine stage of hepatic fibrosis and shows characteristic PAS-positive, diastase-resistant globules in the periphery of the hepatic lobule.
Cystic Fibrosis CF should also be considered as an inherited form of chronic liver disease, although the principal manifestations of CF include chronic lung disease and pancreatic insufficiency (Chap. 313). A small percentage of patients with CF who survive to adulthood have a form of biliary cirrhosis characterized by cholestatic liver enzyme abnormalities and the development of chronic liver disease. Ursodeoxycholic acid is occasionally helpful in improving liver test abnormalities and in reducing symptoms. The disease is slowly progressive.
METABOLIC LIVER DISEASES
Nonalcoholic Fatty Liver Disease NAFLD and NASH are common liver diseases causing abnormal liver test results and progressing to cirrhosis. NAFLD and NASH are discussed in detail in Chap. 364.
Lipid Storage Diseases There are a number of rare lipid storage diseases that involve the liver, including the inherited disorders of Gaucher’s disease and Niemann-Pick disease (Chap. 433e). Other rare disorders include abetalipoproteinemia, Tangier disease, Fabry’s disease, and types I and V hyperlipoproteinemia (Table 367e–5). Hepatomegaly is present due to increased fat deposition, and increased glycogen is found in the liver.
|
LIPID STORAGES DISEASES |
Porphyrias The porphyrias are a group of metabolic disorders in which there are defects in the biosynthesis of heme necessary for incorporation into numerous hemoproteins such as hemoglobin, myoglobin, catalase, and the cytochromes (Chap. 430). Porphyrias can present as either acute or chronic diseases, with the acute disorder causing recurring bouts of abdominal pain, and the chronic disorders characterized by painful skin lesions. Porphyria cutanea tarda (PCT) is the most commonly encountered porphyria. Patients present with characteristic vesicular lesions on sun-exposed areas of the skin, principally the dorsum of the hands, the tips of the ears, or the cheeks. About 40% of patients with PCT have mutations in the gene for hemochromatosis (HFE), and ~50% have hepatitis C; thus, iron studies and HFE mutation analysis as well as hepatitis C testing should be considered in all patients who present with PCT. PCT is also associated with excess alcohol use and some medications, most notably estrogens.
INFILTRATIVE DISORDERS
Amyloidosis Amyloidosis is a metabolic storage disease that results from deposition of insoluble proteins that are aberrantly folded and assembled and then deposited in a variety of tissues (Chap. 136). Amyloidosis is divided into two types, primary and secondary, based on the broad concepts of association with myeloma (primary) or chronic inflammatory illnesses (secondary). The disease is generally considered rare, although, in certain disease states or in certain populations, it can be more common. For example, when associated with familial Mediterranean fever, it is seen in high frequency in Sephardic Jews and Armenians living in Armenia and less frequently in Ashkenazi Jews, Turks, and Arabs. Amyloidosis frequently affects patients suffering from tuberculosis and leprosy and can be seen in upwards of 10–15% of patients with ankylosing spondylitis, rheumatoid arthritis, or Crohn’s disease. In one surgical pathology series, amyloid was found in <1% of cases. The liver is commonly involved in cases of systemic amyloidosis, but it is frequently not clinically apparent and only documented at autopsy. Pathologic findings in the liver include positive staining with the Congo red histochemical stain where there is an apple-green birefringence noted under polarizing light.
Granulomas Granulomas are frequently found in the liver when patients are being evaluated for cholestatic liver enzyme abnormalities. Granulomas can be seen in primary biliary cirrhosis, but there are other characteristic clinical (e.g., pruritus, fatigue) and laboratory findings (cholestatic liver tests, antimitochondrial antibody) that allow for a definitive diagnosis of that disorder. Granulomatous infiltration can also be seen as the principal hepatic manifestation of sarcoidosis, and this is the most common presentation of hepatic granulomas (Chap. 390). The vast majority of these patients do not require any specific treatment other than what would normally be used for treatment of their sarcoidosis. A small subset, however, can develop a particularly bothersome desmoplastic reaction with a significant increase in fibrosis, which can progress to cirrhosis and liver failure. These patients may require treatment with immunosuppressive therapy and may require liver transplantation. In patients who have granulomas in the liver not associated with sarcoidosis, treatment is rarely needed.
Diagnosis requires liver biopsy, and it is important to establish a diagnosis so that a cause for the elevated liver enzymes is carefully identified. Some medications can cause granulomatous infiltration of the liver, the most notable of which is allopurinol.
Lymphoma Involvement of the liver with lymphoma can sometimes be with bulky mass lesions but can also be as a difficult-to-diagnose infiltrative disorder that does not show any characteristic findings on abdominal imaging studies (Chap. 134). Patients may present with severe liver disease, jaundice, hypoalbuminemia, mild to moderately elevated aminotransferases, and an elevated alkaline phosphatase.
A liver biopsy is required for diagnosis and should be considered when routine blood testing does not lead to a diagnosis of the liver dysfunction.
368 |
Liver Transplantation |
Liver transplantation—the replacement of the native, diseased liver by a normal organ (allograft)—has matured from an experimental procedure reserved for desperately ill patients to an accepted, lifesaving operation applied more optimally in the natural history of end-stage liver disease. The preferred and technically most advanced approach is orthotopic transplantation, in which the native organ is removed and the donor organ is inserted in the same anatomic location. Pioneered in the 1960s by Thomas Starzl at the University of Colorado and, later, at the University of Pittsburgh and by Roy Calne in Cambridge, England, liver transplantation is now performed routinely worldwide. Success measured as 1-year survival has improved from ~30% in the 1970s to >90% today. These improved prospects for prolonged survival resulted from refinements in operative technique, improvements in organ procurement and preservation, advances in immunosuppressive therapy, and, perhaps most influentially, more enlightened patient selection and timing. Despite the perioperative morbidity and mortality, the technical and management challenges of the procedure, and its costs, liver transplantation has become the approach of choice for selected patients whose chronic or acute liver disease is progressive, life-threatening, and unresponsive to medical therapy. Based on the current level of success, the number of liver transplants has continued to grow each year; in 2012, 6256 patients received liver allografts in the United States. Still, the demand for new livers continues to outpace availability; as of mid-2013, 15,806 patients in the United States were on a waiting list for a donor liver. In response to this drastic shortage of donor organs, many transplantation centers supplement cadaver-organ liver transplantation with living-donor transplantation.
INDICATIONS
Potential candidates for liver transplantation are children and adults who, in the absence of contraindications (see below), suffer from severe, irreversible liver disease for which alternative medical or surgical treatments have been exhausted or are unavailable. Timing of the operation is of critical importance. Indeed, improved timing and better patient selection are felt to have contributed more to the increased success of liver transplantation in the 1980s and beyond than all the impressive technical and immunologic advances combined. Although the disease should be advanced, and although opportunities for spontaneous or medically induced stabilization or recovery should be allowed, the procedure should be done sufficiently early to give the surgical procedure a fair chance for success. Ideally, transplantation should be considered in patients with end-stage liver disease who are experiencing or have experienced a life-threatening complication of hepatic decompensation or whose quality of life has deteriorated to unacceptable levels. Although patients with well-compensated cirrhosis can survive for many years, many patients with quasi-stable chronic liver disease have much more advanced disease than may be apparent. As discussed below, the better the status of the patient prior to transplantation, the higher will be its anticipated success rate. The decision about when to transplant is complex and requires the combined judgment of an experienced team of hepatologists, transplant surgeons, anesthesiologists, and specialists in support services, not to mention the well-informed consent of the patient and the patient’s family.
TRANSPLANTATION IN CHILDREN
Indications for transplantation in children are listed in Table 368-1. The most common is biliary atresia. Inherited or genetic disorders of metabolism associated with liver failure constitute another major indication for transplantation in children and adolescents. In Crigler-Najjar disease type I and in certain hereditary disorders of the urea cycle and of amino acid or lactate-pyruvate metabolism, transplantation may be the only way to prevent impending deterioration of central nervous system function, despite the fact that the native liver is structurally normal. Combined heart and liver transplantation has yielded dramatic improvement in cardiac function and in cholesterol levels in children with homozygous familial hypercholesterolemia; combined liver and kidney transplantation has been successful in patients with primary hyperoxaluria type I. In hemophiliacs with transfusion-associated hepatitis and liver failure, liver transplantation has been associated with recovery of normal factor VIII synthesis.
|
INDICATIONS FOR LIVER TRANSPLANTATION |
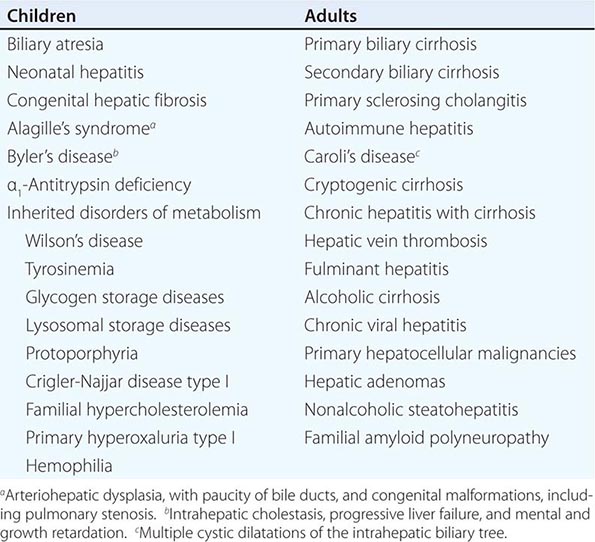
TRANSPLANTATION IN ADULTS
Liver transplantation is indicated for end-stage cirrhosis of all causes (Table 368-1). In sclerosing cholangitis and Caroli’s disease (multiple cystic dilatations of the intrahepatic biliary tree), recurrent infections and sepsis associated with inflammatory and fibrotic obstruction of the biliary tree may be an indication for transplantation. Because prior biliary surgery complicates and is a relative contraindication for liver transplantation, surgical diversion of the biliary tree has been all but abandoned for patients with sclerosing cholangitis. In patients who undergo transplantation for hepatic vein thrombosis (Budd-Chiari syndrome), postoperative anticoagulation is essential; underlying myeloproliferative disorders may have to be treated but are not a contraindication to liver transplantation. If a donor organ can be located quickly, before life-threatening complications—including cerebral edema—set in, patients with acute liver failure are candidates for liver transplantation. Routine candidates for liver transplantation are patients with alcoholic cirrhosis, chronic viral hepatitis, and primary hepatocellular malignancies. Although all three of these categories are considered to be high risk, liver transplantation can be offered to carefully selected patients. Currently, chronic hepatitis C and alcoholic liver disease are the most common indications for liver transplantation, accounting for over 40% of all adult candidates who undergo the procedure. Patients with alcoholic cirrhosis can be considered as candidates for transplantation if they meet strict criteria for abstinence and reform; however, these criteria still do not prevent recidivism in up to a quarter of cases. In highly selected cases in a limited number of centers, transplantation for severe acute alcoholic hepatitis has been performed with success; however, because patients with acute alcoholic hepatitis are still actively using alcohol, and because continued alcohol abuse remains a concern, acute alcoholic hepatitis is not a routine indication for liver transplantation. Patients with chronic hepatitis C have early allograft and patient survival comparable to those of other subsets of patients after transplantation; however, reinfection in the donor organ is universal, recurrent hepatitis C is insidiously progressive, allograft cirrhosis develops in 20–30% at 5 years, and cirrhosis and late organ failure occur at a higher frequency beyond 5 years. With the introduction of highly effective direct acting antiviral agents targeting HCV, it is expected that allograft outcomes will improve significantly in the coming years. In patients with chronic hepatitis B, in the absence of measures to prevent recurrent hepatitis B, survival after transplantation is reduced by approximately 10–20%; however, prophylactic use of hepatitis B immune globulin (HBIg) during and after transplantation increases the success of transplantation to a level comparable to that seen in patients with nonviral causes of liver decompensation. Specific oral antiviral drugs (e.g., entecavir, tenofovir disoproxil fumarate) (Chap. 362) can be used both for prophylaxis against and for treatment of recurrent hepatitis B, facilitating further the management of patients undergoing liver transplantation for end-stage hepatitis B; most transplantation centers rely on antiviral drugs with or without HBIg to manage patients with hepatitis B. Issues of disease recurrence are discussed in more detail below. Patients with nonmetastatic primary hepatobiliary tumors—primary hepatocellular carcinoma (HCC), cholangiocarcinoma, hepatoblastoma, angiosarcoma, epithelioid hemangioendothelioma, and multiple or massive hepatic adenomata—have undergone liver transplantation; however, for some hepatobiliary malignancies, overall survival is significantly lower than that for other categories of liver disease. Most transplantation centers have reported 5-year recurrence-free survival rates in patients with unresectable HCC for single tumors <5 cm in diameter or for three or fewer lesions all <3 cm comparable to those seen in patients undergoing transplantation for nonmalignant indications. Consequently, liver transplantation is currently restricted to patients whose hepatic malignancies meet these criteria. Expanded criteria for patients with HCC continue to be evaluated. Because the likelihood of recurrent cholangiocarcinoma is very high, only highly selected patients with limited disease are being evaluated for transplantation after intensive chemotherapy and radiation.
CONTRAINDICATIONS
Absolute contraindications for transplantation include life-threatening systemic diseases, uncontrolled extrahepatic bacterial or fungal infections, preexisting advanced cardiovascular or pulmonary disease, multiple uncorrectable life-threatening congenital anomalies, metastatic malignancy, and active drug or alcohol abuse (Table 368–2). Because carefully selected patients in their sixties and even seventies have undergone transplantation successfully, advanced age per se is no longer considered an absolute contraindication; however, in older patients a more thorough preoperative evaluation should be undertaken to exclude ischemic cardiac disease and other comorbid conditions. Advanced age (>70 years), however, should be considered a relative contraindication—that is, a factor to be taken into account with other relative contraindications. Other relative contraindications include portal vein thrombosis, HIV infection, preexisting renal disease not associated with liver disease (which may prompt consideration of combined liver and kidney transplantation), intrahepatic or biliary sepsis, severe hypoxemia (PO2 <50 mmHg) resulting from right-to-left intrapulmonary shunts, portopulmonary hypertension with high mean pulmonary artery pressures (>35 mmHg), previous extensive hepatobiliary surgery, any uncontrolled serious psychiatric disorder, and lack of sufficient social supports. Any one of these relative contraindications is insufficient in and of itself to preclude transplantation. For example, the problem of portal vein thrombosis can be overcome by constructing a graft from the donor liver portal vein to the recipient’s superior mesenteric vein. Now that highly active antiretroviral therapy has dramatically improved the survival of persons with HIV infection (Chap. 226), and because end-stage liver disease caused by chronic hepatitis C and B has emerged as a serious source of morbidity and mortality in the HIV-infected population, liver transplantation has now been performed successfully in selected HIV-positive persons who have excellent control of HIV infection. Selected patients with CD4± T cell counts >100/μL and with pharmacologic suppression of HIV viremia have undergone transplantation for end-stage liver disease. HIV-infected persons who have received liver allografts for end-stage liver disease resulting from chronic hepatitis B have experienced survival rates compared to those of HIV-negative persons undergoing transplantation for the same indication. In contrast, recurrent hepatitis C virus (HCV) in the allograft has limited long-term success in persons with HCV-related end-stage liver disease. Again, it is expected that the availability of direct acting antiviral agents targeting HCV, will significantly improve allograft outcomes.
|
CONTRAINDICATIONS TO LIVER TRANSPLANTATION |
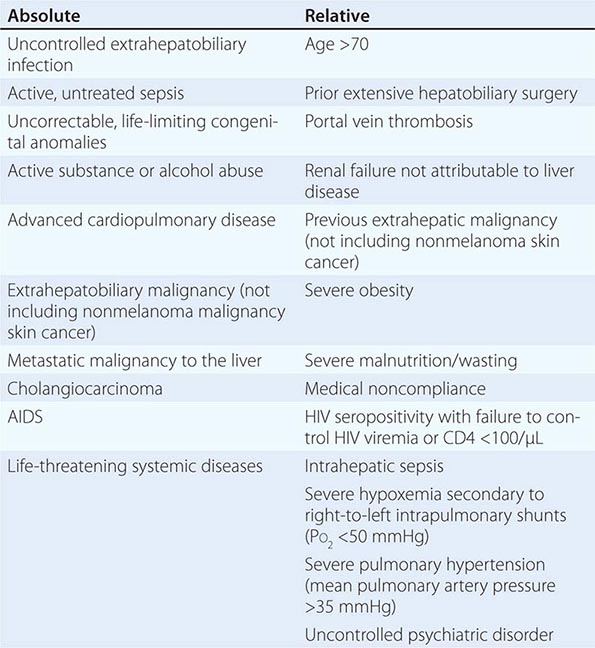
TECHNICAL CONSIDERATIONS
CADAVER DONOR SELECTION
Cadaver donor livers for transplantation are procured primarily from victims of head trauma. Organs from brain-dead donors up to age 60 are acceptable if the following criteria are met: hemodynamic stability, adequate oxygenation, absence of bacterial or fungal infection, absence of abdominal trauma, absence of hepatic dysfunction, and serologic exclusion of hepatitis B (HBV) and C viruses and HIV. Occasionally, organs from donors with hepatitis B and C are used (e.g., for recipients with prior hepatitis B and C, respectively). Organs from donors with antibodies to hepatitis B core antigen (anti-HBc) can also be used when the need is especially urgent, and recipients of these organs are treated prophylactically with antiviral drugs. Cardiovascular and respiratory functions are maintained artificially until the liver can be removed. Transplantation of organs procured from deceased donors who have succumbed to cardiac death can be performed successfully under selected circumstances, when ischemic time is minimized and liver histology preserved. Compatibility in ABO blood group and organ size between donor and recipient are important considerations in donor selection; however, ABO-incompatible, split liver, or reduced-donor-organ transplants can be performed in emergencies or marked donor scarcity. Tissue typing for human leukocyte antigen (HLA) matching is not required, and preformed cytotoxic HLA antibodies do not preclude liver transplantation. Following perfusion with cold electrolyte solution, the donor liver is removed and packed in ice. The use of University of Wisconsin (UW) solution, rich in lactobionate and raffinose, has permitted the extension of cold ischemic time up to 20 h; however, 12 h may be a more reasonable limit. Improved techniques for harvesting multiple organs from the same donor have increased the availability of donor livers, but the availability of donor livers is far outstripped by the demand. Currently in the United States, all donor livers are distributed through a nationwide organ-sharing network (United Network for Organ Sharing [UNOS]) designed to allocate available organs based on regional considerations and recipient acuity. Recipients who have the highest disease severity generally have the highest priority, but allocation strategies that balance highest urgency against best outcomes continue to evolve to distribute cadaver organs most effectively. Allocation based on the Child-Turcotte-Pugh (CTP) score, which uses five clinical variables (encephalopathy stage, ascites, bilirubin, albumin, and prothrombin time) and waiting time, has been replaced by allocation based on urgency alone, calculated by the Model for End-Stage Liver Disease (MELD) score. The MELD score is based on a mathematical model that includes bilirubin, creatinine, and prothrombin time expressed as international normalized ratio (INR) (Table 368-3). Neither waiting time (except as a tie breaker between two potential recipients with the same MELD scores) nor posttransplantation outcome is taken into account, but use of the MELD score has been shown to reduce waiting list mortality, to reduce waiting time prior to transplantation, to be the best predictor of pretransplantation mortality, to satisfy the prevailing view that medical need should be the decisive determinant, and to eliminate both the subjectivity inherent in the CTP scoring system (presence and degree of ascites and hepatic encephalopathy) and the differences in waiting times among different regions of the country. Recent data indicate that liver recipients with MELD scores <15 experienced higher posttransplantation mortality rates than similarly classified patients who remained on the wait list. This observation led to the modification of UNOS policy to allocate donor organs to candidates with MELD scores exceeding 15 within the local or regional procurement organization before offering the organ to local patients whose scores are <15. In addition, serum sodium, another important predictor of survival in liver transplantation candidates, is taken into consideration in allocating donor livers.
|
UNITED NETWORK FOR ORGAN SHARING (UNOS) LIVER TRANSPLANTATION WAITING LIST CRITERIA |

The highest priority (status 1) continues to be reserved for patients with fulminant hepatic failure or primary graft nonfunction. Because candidates for liver transplantation who have HCC may not be sufficiently decompensated to compete for donor organs based on urgency criteria alone, and because protracted waiting for cadaver donor organs often results in tumor growth beyond acceptable limits for transplantation, such patients are assigned disease-specific MELD points (Table 368–3). Other disease-specific MELD exceptions include portopulmonary hypertension, hepatopulmonary syndrome, familial amyloid polyneuropathy, primary hyperoxaluria (necessitating liver-kidney transplantation), cystic fibrosis liver disease, and highly selected cases of hilar cholangiocarcinoma.
LIVING DONOR TRANSPLANTATION
Occasionally, especially for liver transplantation in children, one cadaver organ can be split between two recipients (one adult and one child). A more viable alternative, transplantation of the right lobe of the liver from a healthy adult donor into an adult recipient, has gained increased popularity. Living donor transplantation of the left lobe (left lateral segment), introduced in the early 1990s to alleviate the extreme shortage of donor organs for small children, accounts currently for approximately one-third of all liver transplantation procedures in children. Driven by the shortage of cadaver organs, living donor transplantation involving the more sizable right lobe is being considered with increasing frequency in adults; however, living donor liver transplantation cannot be expected to solve the donor organ shortage; 246 such procedures were done in 2012, representing only about 4% of all liver transplant operations done in the United States.
Living donor transplantation can reduce waiting time and cold-ischemia time; is done under elective, rather than emergency, circumstances; and may be lifesaving in recipients who cannot afford to wait for a cadaver donor. The downside, of course, is the risk to the healthy donor (a mean of 10 weeks of medical disability; biliary complications in ~5%; postoperative complications such as wound infection, small-bowel obstruction, and incisional hernias in 9–19%; and even, in 0.2–0.4%, death) as well as the increased frequency of biliary (15–32%) and vascular (10%) complications in the recipient. Potential donors must participate voluntarily without coercion, and transplantation teams should go to great lengths to exclude subtle coercive or inappropriate psychological factors as well as outline carefully to both donor and recipient the potential benefits and risks of the procedure. Donors for the procedure should be 18–60 years old; have a compatible blood type with the recipient; have no chronic medical problems or history of major abdominal surgery; be related genetically or emotionally to the recipient; and pass an exhaustive series of clinical, biochemical, and serologic evaluations to unearth disqualifying medical disorders. The recipient should meet the same UNOS criteria for liver transplantation as recipients of a cadaver donor allograft. Comprehensive outcome data on adult-to-adult living donor liver transplantation are being collected (www.nih-a2all.org).
SURGICAL TECHNIQUE
Removal of the recipient’s native liver is technically difficult, particularly in the presence of portal hypertension with its associated collateral circulation and extensive varices and especially in the presence of scarring from previous abdominal operations. The combination of portal hypertension and coagulopathy (elevated prothrombin time and thrombocytopenia) may translate into large blood product transfusion requirements. After the portal vein and infrahepatic and suprahepatic inferior vena cavae are dissected, the hepatic artery and common bile duct are dissected. Then the native liver is removed and the donor organ inserted. During the anhepatic phase, coagulopathy, hypoglycemia, hypocalcemia, and hypothermia are encountered and must be managed by the anesthesiology team. Caval, portal vein, hepatic artery, and bile duct anastomoses are performed in succession, the last by end-to-end suturing of the donor and recipient common bile ducts (Fig. 368-1) or by choledochojejunostomy to a Roux-en-Y loop if the recipient common bile duct cannot be used for reconstruction (e.g., in sclerosing cholangitis). A typical transplant operation lasts 8 h, with a range of 6–18 h. Because of excessive bleeding, large volumes of blood, blood products, and volume expanders may be required during surgery; however, blood requirements have fallen sharply with improvements in surgical technique, blood-salvage interventions, and experience.
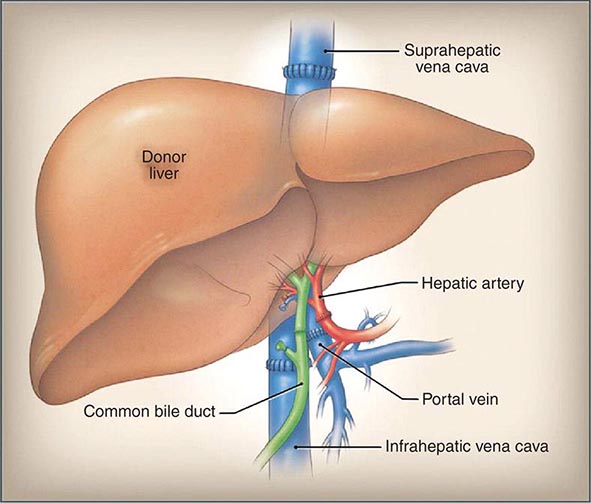
FIGURE 368-1 The anastomoses in orthotopic liver transplantation. The anastomoses are performed in the following sequence: (1) suprahepatic and infrahepatic vena cava, (2) portal vein, (3) hepatic artery, and (4) common bile duct-to-duct anastomosis. (Adapted from JL Dienstag, AB Cosimi: N Engl J Med 367:1483, 2012.)
As noted above, emerging alternatives to orthotopic liver transplantation include split-liver grafts, in which one donor organ is divided and inserted into two recipients; and living donor procedures, in which part of the left (for children), the left (for children or small adults), or the right (for adults) lobe of the liver is harvested from a living donor for transplantation into the recipient. In the adult procedure, once the right lobe is removed from the donor, the donor right hepatic vein is anastomosed to the recipient right hepatic vein remnant, followed by donor-to-recipient anastomoses of the portal vein and then the hepatic artery. Finally, the biliary anastomosis is performed, duct-to-duct if practical or via Roux-en-Y anastomosis. Heterotopic liver transplantation, in which the donor liver is inserted without removal of the native liver, has met with very limited success and acceptance, except in a very small number of centers. In attempts to support desperately ill patients until a suitable donor organ can be identified, several transplantation centers are studying extracorporeal perfusion with bioartificial liver cartridges constructed from hepatocytes bound to hollow fiber systems and used as temporary hepatic-assist devices, but their efficacy remains to be established. Areas of research with the potential to overcome the shortage of donor organs include hepatocyte transplantation and xenotransplantation with genetically modified organs of nonhuman origin (e.g., swine).
POSTOPERATIVE COURSE AND MANAGEMENT
IMMUNOSUPPRESSIVE THERAPY
The introduction in 1980 of cyclosporine as an immunosuppressive agent contributed substantially to the improvement in survival after liver transplantation. Cyclosporine, a calcineurin inhibitor, blocks early activation of T cells and is specific for T cell functions that result from the interaction of the T cell with its receptor and that involve the calcium-dependent signal transduction pathway. As a result, the activity of cyclosporine leads to inhibition of lymphokine gene activation, blocking interleukins 2, 3, and 4, tumor necrosis factor a, and other lymphokines. Cyclosporine also inhibits B cell functions. This process occurs without affecting rapidly dividing cells in the bone marrow, which may account for the reduced frequency of posttransplantation systemic infections. The most common and important side effect of cyclosporine therapy is nephrotoxicity. Cyclosporine causes dose-dependent renal tubular injury and direct renal artery vasospasm. Following renal function is therefore important in monitoring cyclosporine therapy, perhaps even a more reliable indicator than blood levels of the drug. Nephrotoxicity is reversible and can be managed by dose reduction. Other adverse effects of cyclosporine therapy include hypertension, hyperkalemia, tremor, hirsutism, glucose intolerance, and gingival hyperplasia.
Tacrolimus, a macrolide lactone antibiotic isolated from a Japanese soil fungus, Streptomyces tsukubaensis, has the same mechanism of action as cyclosporine but is 10–100 times more potent. Initially applied as “rescue” therapy for patients in whom rejection occurred despite the use of cyclosporine, tacrolimus was shown to be associated with a reduced frequency of acute, refractory, and chronic rejection. Although patient and graft survival are the same with these two drugs, the advantage of tacrolimus in minimizing episodes of rejection, reducing the need for additional glucocorticoid doses, and reducing the likelihood of bacterial and cytomegalovirus (CMV) infection has simplified the management of patients undergoing liver transplantation. In addition, the oral absorption of tacrolimus is more predictable than that of cyclosporine, especially during the early postoperative period when T-tube drainage interferes with the enterohepatic circulation of cyclosporine. As a result, in most transplantation centers, tacrolimus has now supplanted cyclosporine for primary immunosuppression, and many centers rely on oral rather than IV administration from the outset. For transplantation centers that prefer cyclosporine, a better-absorbed microemulsion preparation is available.
Although more potent than cyclosporine, tacrolimus is also more toxic and more likely to be discontinued for adverse events. The toxicity of tacrolimus is similar to that of cyclosporine; nephrotoxicity and neurotoxicity are the most commonly encountered adverse effects, and neurotoxicity (tremor, seizures, hallucinations, psychoses, coma) is more likely and more severe in tacrolimus-treated patients. Both drugs can cause diabetes mellitus, but tacrolimus does not cause hirsutism or gingival hyperplasia. Because of overlapping toxicity between cyclosporine and tacrolimus, especially nephrotoxicity, and because tacrolimus reduces cyclosporine clearance, these two drugs should not be used together. Because 99% of tacrolimus is metabolized by the liver, hepatic dysfunction reduces its clearance; in primary graft nonfunction (when, for technical reasons or because of ischemic damage prior to its insertion, the allograft is defective and does not function normally from the outset), tacrolimus doses have to be reduced substantially, especially in children. Both cyclosporine and tacrolimus are metabolized by the cytochrome P450 IIIA system, and, therefore, drugs that induce cytochrome P450 (e.g., phenytoin, phenobarbital, carbamazepine, rifampin) reduce available levels of cyclosporine and tacrolimus; and drugs that inhibit cytochrome P450 (e.g., erythromycin, fluconazole, ketoconazole, clotrimazole, itraconazole, verapamil, diltiazem, danazol, metoclopramide, the HIV protease inhibitor ritonavir, and the HCV protease inhibitors telaprevir and boceprevir) increase cyclosporine and tacrolimus blood levels. Indeed, itraconazole is used occasionally to help boost tacrolimus levels. Like azathioprine, cyclosporine and tacrolimus appear to be associated with a risk of lymphoproliferative malignancies (see below), which may occur earlier after cyclosporine or tacrolimus than after azathioprine therapy. Because of these side effects, combinations of cyclosporine or tacrolimus with prednisone and an antimetabolite (azathioprine or mycophenolic acid, see below)—all at reduced doses—are preferable regimens for immunosuppressive therapy.
Mycophenolic acid, a nonnucleoside purine metabolism inhibitor derived as a fermentation product from several Penicillium species, is another immunosuppressive drug being used for patients undergoing liver transplantation. Mycophenolate has been shown to be better than azathioprine, when used with other standard immunosuppressive drugs, in preventing rejection after renal transplantation and has been adopted widely as well for use in liver transplantation. The most common adverse effects of mycophenolate are bone marrow suppression and gastrointestinal complaints.
In patients with pretransplantation renal dysfunction or renal deterioration that occurs intraoperatively or immediately postoperatively, tacrolimus or cyclosporine therapy may not be practical; under these circumstances, induction or maintenance of immunosuppression with antithymocyte globulin (ATG, thymoglobulin) or monoclonal antibodies to T cells, OKT3, may be appropriate. Therapy with these agents has been especially effective in reversing acute rejection in the posttransplantation period and is the standard treatment for acute rejection that fails to respond to methylprednisolone boluses. Available data support the use of thymoglobulin induction to delay calcineurin inhibitor use and its attendant nephrotoxicity. IV infusions of thymoglobulin may be complicated by fever and chills, which can be ameliorated by premedication with antipyretics and a low dose of glucocorticoids. Infusions of OKT3 may be complicated by fever, chills, and diarrhea, or by pulmonary edema, which can be fatal. Because OKT3 is such a potent immunosuppressive agent, its use is also more likely to be complicated by opportunistic infection or lymphoproliferative disorders; therefore, because of the availability of alternative immunosuppressive drugs, OKT3 is now used sparingly.
Sirolimus, an inhibitor of the mammalian target of rapamycin (mTOR), blocks later events in T cell activation, is approved for use in kidney transplantation, but is not approved for use in liver transplant recipients because of the reported association with an increased frequency of hepatic artery thrombosis in the first month posttransplantation. In patients with calcineurin inhibitor–related nephrotoxicity, conversion to sirolimus has been demonstrated to be effective in preventing rejection with accompanying improvements in renal function. Because of its profound antiproliferative effects, sirolimus has also been suggested to be a useful immunosuppressive agent in patients with a prior or current history of malignancy, such as HCC. Side effects include hyperlipidemia, peripheral edema, oral ulcers, and interstitial pneumonitis. Everolimus is a hydroxyethyl derivative of sirolimus that, when used in conjunction with low-dose tacrolimus, also provides successful protection against acute rejection, with decreased renal impairment compared to that associated with standard tacrolimus dosing. Everolimus and sirolimus share a similar adverse events profile; therefore, neither of these agents is approved for routine use in liver allograft recipients.
The most important principle of immunosuppression is that the ideal approach strikes a balance between immunosuppression and immunologic competence. In general, given sufficient immunosuppression, acute liver allograft rejection is nearly always reversible. On one hand, incompletely treated acute rejection predisposes to the development of chronic rejection, which can threaten graft survival. On the other hand, if the cumulative dose of immunosuppressive therapy is too large, the patient may succumb to opportunistic infection. In hepatitis C, pulse glucocorticoids or OKT3 use accelerate recurrent allograft hepatitis. Further complicating matters, acute rejection can be difficult to distinguish histologically from recurrent hepatitis C. Therefore, immunosuppressive drugs must be used judiciously, with strict attention to the infectious consequences of such therapy and careful confirmation of the diagnosis of acute rejection. In this vein, efforts have been made to minimize the use of glucocorticoids, a mainstay of immunosuppressive regimens, and steroid-free immunosuppression can be achieved in some instances. Patients who undergo liver transplantation for autoimmune diseases such as primary biliary cirrhosis, autoimmune hepatitis, and primary sclerosing cholangitis are less likely to achieve freedom from glucocorticoids.
POSTOPERATIVE COMPLICATIONS
Complications of liver transplantation can be divided into nonhepatic and hepatic categories (Tables 368-4 and 368-5). In addition, both immediate postoperative and late complications are encountered. As a rule, patients who undergo liver transplantation have been chronically ill for protracted periods and may be malnourished and wasted. The impact of such chronic illness and the multisystem failure that accompanies liver failure continue to require attention in the postoperative period. Because of the massive fluid losses and fluid shifts that occur during the operation, patients may remain fluid-overloaded during the immediate postoperative period, straining cardiovascular reserve; this effect can be amplified in the face of transient renal dysfunction and pulmonary capillary vascular permeability. Continuous monitoring of cardiovascular and pulmonary function, measures to maintain the integrity of the intravascular compartment and to treat extravascular volume overload, and scrupulous attention to potential sources and sites of infection are of paramount importance. Cardiovascular instability may also result from the electrolyte imbalance that may accompany reperfusion of the donor liver as well as from restoration of systemic vascular resistance following implantation. Pulmonary function may be compromised further by paralysis of the right hemidiaphragm associated with phrenic nerve injury. The hyperdynamic state with increased cardiac output that is characteristic of patients with liver failure reverses rapidly after successful liver transplantation.
|
NONHEPATIC COMPLICATIONS OF LIVER TRANSPLANTATION |
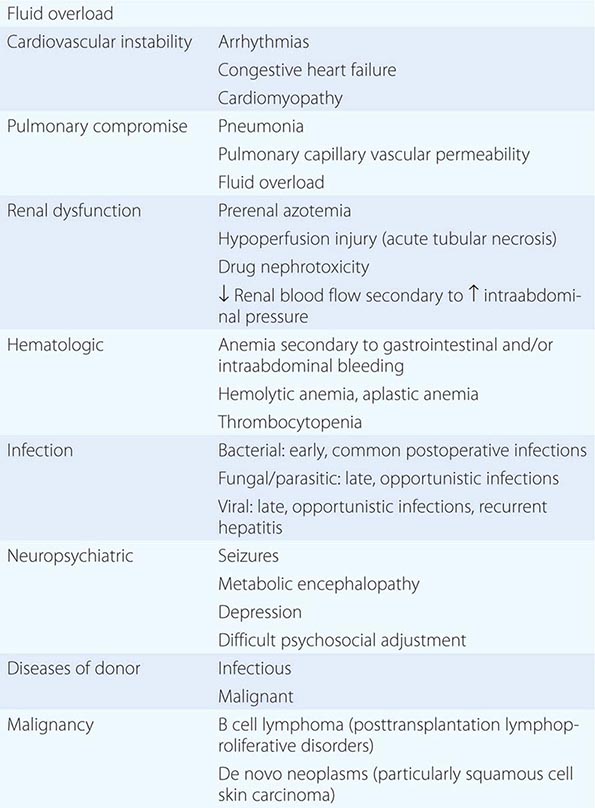
|
HEPATIC COMPLICATIONS OF LIVER TRANSPLANTATION |
Other immediate management issues include renal dysfunction. Prerenal azotemia, acute kidney injury associated with hypoperfusion (acute tubular necrosis), and renal toxicity caused by antibiotics, tacrolimus, or cyclosporine are encountered frequently in the postoperative period, sometimes necessitating dialysis. Hemolytic-uremic syndrome can be associated with cyclosporine, tacrolimus, or OKT3. Occasionally, postoperative intraperitoneal bleeding may be sufficient to increase intraabdominal pressure, which, in turn, may reduce renal blood flow; this effect is rapidly reversible when abdominal distention is relieved by exploratory laparotomy to identify and ligate the bleeding site and to remove intraperitoneal clot.
Anemia may also result from acute upper gastrointestinal bleeding or from transient hemolytic anemia, which may be autoimmune, especially when blood group O livers are transplanted into blood group A or B recipients. This autoimmune hemolytic anemia is mediated by donor intrahepatic lymphocytes that recognize red blood cell A or B antigens on recipient erythrocytes. Transient in nature, this process resolves once the donor liver is repopulated by recipient bone marrow–derived lymphocytes; the hemolysis can be treated by transfusing blood group O red blood cells and/or by administering higher doses of glucocorticoids. Transient thrombocytopenia is also commonly encountered. Aplastic anemia, a late occurrence, is rare but has been reported in almost 30% of patients who underwent liver transplantation for acute, severe hepatitis of unknown cause.
Bacterial, fungal, or viral infections are common and may be life-threatening postoperatively. Early after transplant surgery, common postoperative infections predominate—pneumonia, wound infections, infected intraabdominal collections, urinary tract infections, and IV line infections—rather than opportunistic infections; these infections may involve the biliary tree and liver as well. Beyond the first postoperative month, the toll of immunosuppression becomes evident, and opportunistic infections—CMV, herpes viruses, fungal infections (Aspergillus, Candida, cryptococcal disease), mycobacterial infections, parasitic infections (Pneumocystis, Toxoplasma), bacterial infections (Nocardia, Legionella, Listeria)—predominate. Rarely, early infections represent those transmitted with the donor liver, either infections present in the donor or infections acquired during procurement processing. De novo viral hepatitis infections acquired from the donor organ or, almost unheard of now, from transfused blood products occur after typical incubation periods for these agents (well beyond the first month). Obviously, infections in an immunosuppressed host demand early recognition and prompt management; prophylactic antibiotic therapy is administered routinely in the immediate postoperative period. Use of sulfamethoxazole with trimethoprim reduces the incidence of postoperative Pneumocystis carinii pneumonia. Antiviral prophylaxis for CMV with ganciclovir should be administered in patients at high risk (e.g., when a CMV-seropositive donor organ is implanted into a CMV-seronegative recipient).
Neuropsychiatric complications include seizures (commonly associated with cyclosporine and tacrolimus toxicity), metabolic encephalopathy, depression, and difficult psychosocial adjustment. Rarely, diseases are transmitted by the allograft from the donor to the recipient. In addition to viral and bacterial infections, malignancies of donor origin have occurred. Posttransplantation lymphoproliferative disorders, especially B cell lymphoma, are a recognized complication associated with immunosuppressive drugs such as azathioprine, tacrolimus, and cyclosporine (see above). Epstein-Barr virus has been shown to play a contributory role in some of these tumors, which may regress when immunosuppressive therapy is reduced. De novo neoplasms appear at increased frequency after liver transplantation, particularly squamous cell carcinomas of the skin. Routine screening should be performed.
Long-term complications after liver transplantation attributable primarily to immunosuppressive medications include diabetes mellitus and osteoporosis (associated with glucocorticoids and calcineurin inhibitors) as well as hypertension, hyperlipidemia, and chronic renal insufficiency (associated with cyclosporine and tacrolimus). Monitoring and treating these disorders are routine components of posttransplantation care; in some cases, they respond to changes in immunosuppressive regimen, while in others, specific treatment of the disorder is introduced. Data from a large U.S. database showed that the prevalence of renal failure was 18% at year 5 and 25% at year 10 after liver transplantation. Similarly, the high frequency of diabetes, hypertension, hyperlipidemia, obesity, and the metabolic syndrome renders patients susceptible to cardiovascular disease after liver transplantation; although hepatic complications account for most of the mortality after liver transplantation, renal failure and cardiovascular disease are the other leading causes of late mortality after liver transplantation.
HEPATIC COMPLICATIONS
Hepatic dysfunction after liver transplantation is similar to the hepatic complications encountered after major abdominal and cardiothoracic surgery; however, in addition, hepatic complications include primary graft failure, vascular compromise, failure or stricture of the biliary anastomoses, and rejection. As in nontransplantation surgery, postoperative jaundice may result from prehepatic, intrahepatic, and posthepatic sources. Prehepatic sources represent the massive hemoglobin pigment load from transfusions, hemolysis, hematomas, ecchymoses, and other collections of blood. Early intrahepatic liver injury includes effects of hepatotoxic drugs and anesthesia; hypoperfusion injury associated with hypotension, sepsis, and shock; and benign postoperative cholestasis. Late intrahepatic sources of liver injury include exacerbation of primary disease. Posthepatic sources of hepatic dysfunction include biliary obstruction and reduced renal clearance of conjugated bilirubin. Hepatic complications unique to liver transplantation include primary graft failure associated with ischemic injury to the organ during harvesting; vascular compromise associated with thrombosis or stenosis of the portal vein or hepatic artery anastomoses; vascular anastomotic leak; stenosis, obstruction, or leakage of the anastomosed common bile duct; recurrence of primary hepatic disorder (see below); and rejection.
TRANSPLANT REJECTION
Despite the use of immunosuppressive drugs, rejection of the transplanted liver still occurs in a proportion of patients, beginning 1–2 weeks after surgery. Clinical signs suggesting rejection are fever, right upper quadrant pain, and reduced bile pigment and volume. Leukocytosis may occur, but the most reliable indicators are increases in serum bilirubin and aminotransferase levels. Because these tests lack specificity, distinguishing among rejection, biliary obstruction, primary graft nonfunction, vascular compromise, viral hepatitis, CMV infection, drug hepatotoxicity, and recurrent primary disease may be difficult. Radiographic visualization of the biliary tree and/or percutaneous liver biopsy often help to establish the correct diagnosis. Morphologic features of acute rejection include a mixed portal cellular infiltrate, bile duct injury, and/or endothelial inflammation (“endothelialitis”); some of these findings are reminiscent of graft-versus-host disease, primary biliary cirrhosis, or recurrent allograft hepatitis C. As soon as transplant rejection is suspected, treatment consists of IV methylprednisolone in repeated boluses; if this fails to abort rejection, many centers use thymoglobulin or OKT3. Caution should be exercised when managing acute rejection with pulse glucocorticoids or OKT3 in patients with HCV infection, because of the high risk of triggering recurrent allograft hepatitis C.
Chronic rejection is a relatively rare outcome that can follow repeated bouts of acute rejection or that occurs unrelated to preceding rejection episodes. Morphologically, chronic rejection is characterized by progressive cholestasis, focal parenchymal necrosis, mononuclear infiltration, vascular lesions (intimal fibrosis, subintimal foam cells, fibrinoid necrosis), and fibrosis. This process may be reflected as ductopenia—the vanishing bile duct syndrome, which is more common in patients undergoing liver transplantation for autoimmune liver disease. Reversibility of chronic rejection is limited; in patients with therapy-resistant chronic rejection, retransplantation has yielded encouraging results.
OUTCOME
SURVIVAL
The survival rate for patients undergoing liver transplantation has improved steadily since 1983. One-year survival rates have increased from ~70% in the early 1980s to 85–90% from 2003 to the present time. Currently, the 5-year survival rate exceeds 60%. An important observation is the relationship between clinical status before transplantation and outcome. For patients who undergo liver transplantation when their level of compensation is high (e.g., still working or only partially disabled), a 1-year survival rate of >85% is common. For those whose level of decompensation mandates continuous in-hospital care prior to transplantation, the 1-year survival rate is ~70%, whereas for those who are so decompensated that they require life support in an intensive care unit, the 1-year survival rate is ~50%. Since the adoption by UNOS in 2002 of the MELD system for organ allocation, posttransplantation survival has been found to be affected adversely for candidates with MELD scores >25, considered high disease severity. Thus, irrespective of allocation scheme, high disease severity before transplantation corresponds to diminished posttransplantation survival. Another important distinction in survival has been drawn between high- and low-risk patient categories. For patients who do not fit any “high-risk” designations, 1-year and 5-year survival rates of 85 and 80%, respectively, have been recorded. In contrast, among patients in high-risk categories—cancer, fulminant hepatitis, age >65, concurrent renal failure, respirator dependence, portal vein thrombosis, and history of a portacaval shunt or multiple right upper quadrant operations—survival statistics fall into the range of 60% at 1 year and 35% at 5 years. Survival after retransplantation for primary graft nonfunction is ~50%. Causes of failure of liver transplantation vary with time. Failures within the first 3 months result primarily from technical complications, postoperative infections, and hemorrhage. Transplant failures after the first 3 months are more likely to result from infection, rejection, or recurrent disease (such as malignancy or viral hepatitis).
RECURRENCE OF PRIMARY DISEASE
Features of autoimmune hepatitis, primary sclerosing cholangitis, and primary biliary cirrhosis overlap with those of rejection or posttransplantation bile duct injury. Whether autoimmune hepatitis and sclerosing cholangitis recur after liver transplantation is controversial; data supporting recurrent autoimmune hepatitis (in up to one-third of patients in some series) are more convincing than those supporting recurrent sclerosing cholangitis. Similarly, reports of recurrent primary biliary cirrhosis after liver transplantation have appeared; however, the histologic features of primary biliary cirrhosis and chronic rejection are virtually indistinguishable and occur as frequently in patients with primary biliary cirrhosis as in patients undergoing transplantation for other reasons. The presence of a florid inflammatory bile duct lesion is highly suggestive of the recurrence of primary biliary cirrhosis, but even this lesion can be observed in acute rejection. Hereditary disorders such as Wilson’s disease and α1-antitrypsin deficiency have not recurred after liver transplantation; however, recurrence of disordered iron metabolism has been observed in some patients with hemochromatosis. Hepatic vein thrombosis (Budd-Chiari syndrome) may recur; this can be minimized by treating underlying myeloproliferative disorders and by anticoagulation. Because cholangiocarcinoma recurs almost invariably, few centers now offer transplantation to such patients; however, a few highly selected patients with operatively confirmed stage I or II cholangiocarcinoma who undergo liver transplantation combined with neoadjuvant chemoradiation may experience excellent outcomes. In patients with intrahepatic HCC who meet criteria for transplantation, 1- and 5-year survivals are similar to those observed in patients undergoing liver transplantation for nonmalignant disease. Finally, metabolic disorders such as nonalcoholic steatohepatitis recur frequently, especially if the underlying metabolic predisposition is not altered. The metabolic syndrome occurs commonly after liver transplantation as a result of recurrent nonalcoholic fatty liver, immunosuppressive medications, and/or, in patients with hepatitis C related to the impact of HCV infection on insulin resistance, diabetes and fatty liver.
Hepatitis A can recur after transplantation for fulminant hepatitis A, but such acute reinfection has no serious clinical sequelae. In fulminant hepatitis B, recurrence is not the rule; however, in the absence of any prophylactic measures, hepatitis B usually recurs after transplantation for end-stage chronic hepatitis B. Before the introduction of prophylactic antiviral therapy, immunosuppressive therapy sufficient to prevent allograft rejection led inevitably to marked increases in hepatitis B viremia, regardless of pretransplantation levels. Overall graft and patient survival were poor, and some patients experienced a rapid recapitulation of severe injury—severe chronic hepatitis or even fulminant hepatitis—after transplantation. Also recognized in the era before availability of antiviral regimens was fibrosing cholestatic hepatitis, rapidly progressive liver injury associated with marked hyperbilirubinemia, substantial prolongation of the prothrombin time (both out of proportion to relatively modest elevations of aminotransferase activity), and rapidly progressive liver failure. This lesion has been suggested to represent a “choking off” of the hepatocyte by an overwhelming density of HBV proteins. Complications such as sepsis and pancreatitis were also observed more frequently in patients undergoing liver transplantation for hepatitis B prior to the introduction of antiviral therapy. The introduction of long-term prophylaxis with HBIg revolutionized liver transplantation for chronic hepatitis B. Preoperative hepatitis B vaccination, preoperative or postoperative interferon (IFN) therapy, or short-term (≤2 months) HBIg prophylaxis has not been shown to be effective, but a retrospective analysis of data from several hundred European patients followed for 3 years after transplantation has shown that long-term (≥6 months) prophylaxis with HBIg is associated with a lowering of the risk of HBV reinfection from ~75 to 35% and a reduction in mortality from ~50 to 20%.
As a result of long-term HBIg use following liver transplantation for chronic hepatitis B, similar improvements in outcome have been observed in the United States, with 1-year survival rates between 75% and 90%. Currently, with HBIg prophylaxis, the outcome of liver transplantation for chronic hepatitis B is indistinguishable from that for chronic liver disease unassociated with chronic hepatitis B; essentially, medical concerns regarding liver transplantation for chronic hepatitis B have been eliminated. Passive immunoprophylaxis with HBIg is begun during the anhepatic stage of surgery, repeated daily for the first 6 postoperative days, and then continued with infusions that are given either at regular intervals of 4–6 weeks or, alternatively, when anti-hepatitis B surface (HBs) levels fall below a threshold of 100 mIU/mL. The current approach in most centers is to continue HBIg indefinitely, which can add approximately $20,000 per year to the cost of care; some centers are evaluating regimens that shift to less frequent administration or to IM administration in the late posttransplantation period or, in low-risk patients, maintenance with antiviral therapy (see below) alone. Still, “breakthrough” HBV infection occasionally occurs.
Further improving the outcome of liver transplantation for chronic hepatitis B is the current availability of such antiviral drugs as lamivudine, adefovir, entecavir, and tenofovir disoproxil fumarate (Chap. 362). When these drugs are administered to patients with decompensated liver disease, a proportion improve sufficiently to postpone imminent liver transplantation. In addition, antiviral therapy can be used to prevent recurrence of HBV infection when administered prior to transplantation; to treat hepatitis B that recurs after transplantation, including in patients who break through HBIg prophylaxis; and to reverse the course of otherwise fatal fibrosing cholestatic hepatitis. Clinical trials have shown that lamivudine antiviral therapy reduces the level of HBV replication substantially, sometimes even resulting in clearance of hepatitis B surface antigen (HBsAg); reduces alanine aminotransferase (ALT) levels; and improves histologic features of necrosis and inflammation. Long-term use of lamivudine is safe and effective, but after several months, a proportion of patients become resistant to lamivudine, resulting from YMDD (tyrosine-methionine-aspartate-aspartate) mutations in the HBV polymerase motif (Chap. 362). In approximately one-half of such resistant patients, hepatic deterioration may ensue. Fortunately, adefovir and tenofovir disoproxil fumarate are available as well and can be used to treat lamivudine-associated YMDD variants, effectively “rescuing” patients experiencing hepatic decompensation after lamivudine breakthrough. Currently, most liver transplantation centers combine HBIg plus lamivudine, adefovir, entecavir, or tenofovir disoproxil fumarate. In low-risk patients with no detectable hepatitis B viremia at the time of transplantation, a number of clinical trials have suggested that antiviral prophylaxis can suffice, without HBIg or with a finite duration of HBIg, to prevent recurrent HBV infection of the allograft. Antiviral prophylactic approaches applied to patients undergoing liver transplantation for chronic hepatitis B are being used as well for patients without hepatitis B who receive organs from donors with antibody to hepatitis B core antigen (anti-HBc). Patients who undergo liver transplantation for chronic hepatitis B plus D are less likely to experience recurrent liver injury than patients undergoing liver transplantation for hepatitis B alone; still, such co-infected patients would also be offered standard posttransplantation prophylactic therapy for hepatitis B.
Accounting for up to 40% of all liver transplantation procedures, the most common indication for liver transplantation is end-stage liver disease resulting from chronic hepatitis C. Recurrence of HCV infection after liver transplantation can be documented in almost every patient. The clinical consequences of recurrent hepatitis C are limited during the first 5 years after transplantation. Nonetheless, despite the relative clinical benignity of recurrent hepatitis C in the early years after liver transplantation, and despite the negligible impact on patient survival during these early years, histologic studies have documented the presence of moderate to severe chronic hepatitis in more than one-half of all patients and cirrhosis in ~20% at 5 years. Allograft cirrhosis is even more common, occurring in up to two-thirds of patients at 5 years, if moderate hepatitis is detected in a 1-year liver biopsy. Not surprisingly, then, for patients undergoing liver transplantation for hepatitis C, allograft and patient survival are diminished substantially between 5 and 10 years after transplantation.
In a proportion of patients, even during the early posttransplantation period, recurrent hepatitis C may be sufficiently severe biochemically and histologically to merit antiviral therapy. Treatment with pegylated IFN can suppress HCV-associated liver injury but rarely leads to sustained benefit. Sustained virologic responses are the exception, and reduced tolerability is often dose-limiting. Preemptive combination antiviral therapy with pegylated IFN and the nucleoside analogue ribavirin immediately after transplantation does not appear to provide any advantage over therapy introduced after clinical hepatitis has occurred. Similarly, although IFN-based antiviral therapy is not recommended for patients with decompensated liver disease, some centers have experimented with pretransplantation antiviral therapy in an attempt to eradicate HCV replication prior to transplantation; preliminary results are promising, but IFN treatment of patients with end-stage liver disease can lead to worsening of hepatic decompensation, and HCV infection has recurred after transplantation in some of these recipients. Trials of hepatitis C immune globulin preparations to prevent recurrent hepatitis C after liver transplantation have not been successful. Similarly, a trial of a high-dose monoclonal antibody to the HCV E2 envelope glycoprotein delayed but did not prevent reappearance of viremia.
Although the current standard-of-care treatment of allograft hepatitis C is pegylated IFN and ribavirin, in a number of studies, the safety and efficacy of the addition of the approved HCV protease inhibitors telaprevir or boceprevir to pegylated IFN and ribavirin in genotype 1–infected patients with recurrent hepatitis C have been examined. Because of the profound inhibitory effects of the HCV protease inhibitors on the metabolism of the calcineurin inhibitors (increasing cyclosporine levels almost 5-fold and tacrolimus levels 70-fold), calcineurin inhibitor doses must be reduced to safe levels in these patients. In one multicenter study, treatment with a telaprevir- or boceprevir-based triple-drug regimen (with pegylated IFN and ribavirin) achieved rates of HCV clearance similar to those achieved in patients with chronic hepatitis C who had not undergone transplantation. Unfortunately, tolerability of these protease inhibitor–based regimens remains problematic in this population, particularly in persons with allograft cirrhosis, in whom the frequency of hepatic decompensation is increased. The approval of several new direct-acting antiviral (DAA) agents and of IFN-free DAA regimens against HCV will have a major impact on the management and outcome of both pretransplantation and posttransplantation HCV infection. Such therapeutic approaches (1) permit the clearance of viremia in a substantial proportion of decompensated cirrhotics, thereby preventing recurrent allograft infection and, possibly, even improving the clinical status of these patients, delaying or obviating the need for liver replacement; and (2) achieve sustained virologic responses in a much higher proportion of persons with allograft HCV infection, because of improvements in antiviral treatment efficacy and tolerability.
A small number of allograft recipients succumb to early HCV-associated liver injury, and a syndrome reminiscent of fibrosing cholestatic hepatitis (see above) has been observed rarely. Because patients with more episodes of rejection receive more immunosuppressive therapy, and because immunosuppressive therapy enhances HCV replication, patients with severe or multiple episodes of rejection are more likely to experience early recurrence of hepatitis C after transplantation. Both high viral levels and older donor age have been linked to recurrent HCV-induced liver disease and to earlier disease recurrence after transplantation.
Patients who undergo liver transplantation for end-stage alcoholic cirrhosis are at risk of resorting to drinking again after transplantation, a potential source of recurrent alcoholic liver injury. Currently, alcoholic liver disease is one of the more common indications for liver transplantation, accounting for 20–25% of all liver transplantation procedures, and most transplantation centers screen candidates carefully for predictors of continued abstinence. Recidivism is more likely in patients whose sobriety prior to transplantation was <6 months. For abstinent patients with alcoholic cirrhosis, liver transplantation can be undertaken successfully, with outcomes comparable to those for other categories of patients with chronic liver disease, when coordinated by a team approach that includes substance abuse counseling.
POSTTRANSPLANTATION QUALITY OF LIFE
Full rehabilitation is achieved in the majority of patients who survive the early postoperative months and escape chronic rejection or unmanageable infection. Psychosocial maladjustment interferes with medical compliance in a small number of patients, but most manage to adhere to immunosuppressive regimens, which must be continued indefinitely. In one study, 85% of patients who survived their transplant operations returned to gainful activities. In fact, some women have conceived and carried pregnancies to term after transplantation without demonstrable injury to their infants.